

ABSTRACT
The mammalian nervous system is invaded by a number of intracellular bacterial pathogens which can establish and progress infection in susceptible individuals. Subsequent clinical manifestation is apparent with the impairment of the functional units of the nervous system, i.e., the neurons and the supporting glial cells that produce myelin sheaths around axons and provide trophic support to axons and neurons. Most of these neurotrophic bacteria display unique features, have coevolved with the functional sophistication of the nervous system cells, and have adapted remarkably to manipulate neural cell functions for their own advantage. Understanding how these bacterial pathogens establish intracellular adaptation by hijacking endogenous pathways in the nervous system, initiating myelin damage and axonal degeneration, and interfering with myelin maintenance provides new knowledge not only for developing strategies to combat neurodegenerative conditions induced by these pathogens but also for gaining novel insights into cellular and molecular pathways that regulate nervous system functions. Since the pathways hijacked by bacterial pathogens may also be associated with other neurodegenerative diseases, it is anticipated that detailing the mechanisms of bacterial manipulation of neural systems may shed light on common mechanisms, particularly of early disease events. This chapter details a classic example of neurodegeneration, that caused by Mycobacterium leprae, which primarily infects glial cells of the peripheral nervous system (Schwann cells), and how it targets and adapts intracellularly by reprogramming Schwann cells to stem cells/progenitor cells. We also discuss implications of this host cell reprogramming by leprosy bacilli as a model in a wider context.
INTRODUCTION
Bacterial Infection of the Adult Nervous System
The nervous system comprises the central nervous system (CNS) and the peripheral nervous system (PNS). Although many bacterial pathogens are known to invade the CNS and cause associated neuropathologies, much less is known about their intracellular manipulation of neural cells, particularly early events of bacterial infections, and how such bacterium-induced neural cell alterations could lead to bacterial survival, persistence, and the progression of infection as well as pathogenesis. A majority of the studies with these bacterial pathogens are immune-centric and focused on inflammatory aspects of nervous system diseases, and many reviews are available elsewhere with more detail on inflammatory and immune mechanisms of this bacteria-induced neurodegeneration (1–3).
Bacterial Infections of the Adult PNS
Because the PNS connects CNS communication with the organs and limbs in order to effectively coordinate the body functions, the PNS is as important as the CNS when it comes to motor, sensory, and autonomous neuronal functions (Fig. 1). Thus, the bacterial pathogens that preferentially invade the PNS provide a model to dissect how they naturally target nerves and initiate and induce nerve degeneration by deregulating neural cell functions, most of which are yet to be identified.
FIGURE 1.
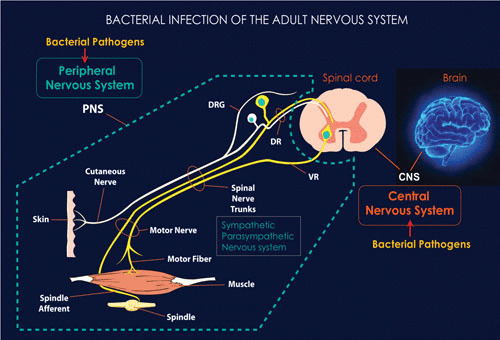
The adult nervous system comprises the PNS and CNS. The CNS is connected to the organs and limbs by the PNS, which also includes a sympathetic and parasympathetic nervous system. Infection of both the PNS and CNS by bacterial pathogens often leads to neurodegenerative diseases. Understanding how such bacterial pathogens target the nervous system and naturally cause disease not only provides insights into combating infectious neurodegenerative diseases but also sheds light on common themes of how neurodegenerative diseases are initiated. Some details of the adult PNS with innervation of skin and muscles are shown; these nerves are usually affected during PNS infections, leading to sensory loss and muscle atrophy, as in leprosy neuropathy.
Virtually all tissues of the body are innervated by peripheral nerves, supplied with a neuronal network along with the supporting glial cells (i.e., Schwann cells), which form myelin sheaths around larger axons and do not form myelin sheaths around smaller-diameter axons; the nerves and glial cells collectively serve as the functional units of the PNS (4, 5). Their peripheral location and ubiquitous presence give rise to a potential susceptibility of the peripheral nerves to invading pathogens; nerve terminals are present close to external sites on the body, including the skin and nasal cavity, and nerves frequently run close to blood vessels carrying systemic infectious agents. Considering this unprotected nature and close proximity of the PNS to the exterior, peripheral nerves are expected to be vulnerable even to environmental pathogens. Surprisingly, however, only a few bacterial pathogens have the capacity to invade the PNS and establish a productive infection. This level of defense against pathogens may be due to the privileged nature of the peripheral nerves, which are protected and surrounded by a connective tissue-rich perineurium and the blood-nerve barrier (BNB), akin to the blood-brain barrier of the CNS (6, 7).
MYCOBACTERIUM LEPRAE AS A MODEL FOR NEUROPATHOGENESIS
A classic example of an intracellular bacterial pathogen that breaches the BNB and preferentially enters the glial cells of the peripheral nerves (Schwann cells) is Mycobacterium leprae. Leprosy causes one of the most common infectious neuropathies, leprosy neuropathy, which is currently prevalent in low- and middle-income countries on three continents (8, 9). With the number of new cases detected exceeding 200,000 annually, leprosy remains a major public health problem in countries where it is endemic. Due to a lack of early diagnosis and an extremely long incubation period, most newly diagnosed leprosy patients clinically present with some form of neuropathy (8, 9). However, it remains unknown how this pathogen causes neuropathy.
M. leprae, the causative organism of human leprosy, preferentially invades the glial cells of the PNS (Schwann cells). This unique capacity for Schwann cell invasion by M. leprae and the subsequent neuropathy with sensory and motor neuronal impairment in humans demonstrate that M. leprae is an excellent model to dissect how bacteria initiate neuropathic conditions by targeting differentiated glial cells in the adult PNS. Such studies will provide new insights which will be useful for developing early intervention not only for leprosy neuropathy but also perhaps for other neuropathic diseases with unknown etiologies but similar neuropathological features whose early events are completely unknown.
Pathology Caused by Leprosy Bacilli
Leprosy has been known to humankind since biblical times, having been described since about 600 BCE in texts originating from India, China, and Egypt (10). It is perhaps the most feared archetype of stigmatizing disease, to the extent that the term “leper” has become a generic term for a person shunned by others. The disease was highly prevalent in Europe, and the Norwegian physician Armauer Hansen was the first to identify the causative agent, M. leprae, in 1873 (11). Leprosy was the first human disease known to be caused by a microorganism (11). The first effective antibacterial therapies for leprosy were introduced in the 1940s, and the current standard therapy involves multidrug treatment regimens with dapsone, clofazimine, and rifampin (12). However, leprosy remains endemic in low- and middle-income countries on three continents, with a stable annual rate of new-case detection (13). If not diagnosed or treated early, M. leprae infection in humans develops into neuropathic conditions and stigmatizing skin conditions as a result of uncontrolled bacterial propagation in the skin and the peripheral nerves. The hallmark of leprosy is its effect on sensory and motor neurons; loss of sensation is associated with complications in the extremities and subsequent formation of ulcers, known as neuropathic ulcers, which can proceed to destroy other structures underlying the skin, including cartilage and bones, if untreated, causing severe disability. Indeed, at the time of diagnosis, most patients manifest some form of disability due to neurological injuries as a result of extremely long incubation. Facial disfiguration and bone loss are common in the late stage of the disease, and damage to the nerves controlling blinking can lead to blindness. Interestingly, most of these unusual pathologies occur in multibacillary leprosy patients (i.e., those harboring high loads of bacteria in the tissues), suggesting that direct host tissue responses to M. leprae may be associated with pathogenesis.
M. leprae Infection in Animal Models and Humans
Entirely dependent on host cells for bacterial survival, M. leprae is a strictly obligately intracellular bacterium (9, 14). This strictly intracellular lifestyle is directly linked to the bacterial genome, which comprises more pseudogenes and noncoding genes than protein-coding genes, rendering M. leprae dependent on host cell functions and metabolism for bacterial survival and replication (15, 16). In addition to humans, two animal hosts which are susceptible to naturally occurring systemic infections are known so far; M. leprae causes multibacillary leprosy-like disease in nine-banded armadillos (17, 18) and red squirrels in the wild (19), as well as in experimentally infected armadillos. The majority of the human population generally does not develop clinical leprosy following M. leprae infection (20, 21). Human leprosy could also be acquired by zoonotic transmission via nine-banded armadillos infected in the wild in the southern United States (22). The association of genetic factors with human leprosy was exemplified by the findings that genes PARK2 and PACRG are risk factors for leprosy (23–25). Interestingly, mutation of the PARK2 gene, which encodes a ubiquitin E3 ligase, has been shown to be the cause of autosomal recessive early-onset Parkinson’s disease, and this finding connects leprosy susceptibility to other neurodegenerative diseases with unknown etiology (26). Interestingly, potential environmental factors like bacterial and viral infections have been implicated in triggering Parkinson’s and other neurodegenerative diseases (27, 28). Therefore, a better understanding of early molecular events during M. leprae infection could provide new insights into how other neurodegenerative diseases with unknown etiology might be triggered.
Temperature Sensitivity in Intracellular Bacterial Growth
Apart from the strictly obligate intracellular lifestyle, leprosy bacilli also require lower body temperature for survival and replication, and thus, in humans, preferential bacterial growth can be seen in peripheral tissues like peripheral nerves, skin, and testis, where peripheral body temperature is relatively low (20, 29, 30). Experimental evidence has shown that a temperature of 37°C is unfavorable for intracellular M. leprae viability, while in the footpads of immunocompromised nude mice, prolific bacterial growth can be achieved locally without disseminated infection due to the lower temperature in the extremities (31). The latter provides a valuable resource for the provision of in vivo-grown bacteria for basic leprosy research (31, 32). Interestingly, in nine-banded armadillos, the core body temperature is 33 to 35°C, and M. leprae propagates in both peripheral tissues and internal organs, such as the liver (33, 34).
M. leprae Infection in Humans
In humans, M. leprae infection of Schwann cells in the PNS is primarily responsible for developing neuropathic conditions. In multibacillary leprosy, a large number of intracellular M. leprae organisms can be seen in Schwann cells in peripheral nerves (20, 35). M. leprae rarely infects the axons, although axonal degeneration eventually contributes to neuropathic conditions which cause not only disability but also stigmatized conditions, including bone loss and muscle atrophy due to loss of sensation and associated complications that affect tissues underneath the skin. These conditions have caused leprosy to be one of the most feared and stigmatized diseases known to humankind. Humans show a wide spectrum of clinical immunological and histological presentations of leprosy, from tuberculoid leprosy, which entails a strong immune response and minimal detectable bacilli (paucibacillary leprosy), to lepromatous leprosy, with more widespread bacterial presence in peripheral tissues like the PNS and skin (multibacillary leprosy) and low or absent cell-mediated immune responses (9, 29, 36, 37), with intermediate classifications between these polar groups. Regardless of the clinical spectrum, nerve damage is widely seen in all groups of leprosy patients.
Although nerve damage in tuberculoid leprosy is expected to be caused by immune-mediated tissue destruction due to strong cell-mediated immune responses (38, 39), it is unknown how multibacillary leprosy with high numbers of bacteria in Schwann cells and minimal or no cell-mediated immune response causes neurological damage. In this context, previous research on the biology of multibacillary leprosy was designed to address several important questions. (i) How does M. leprae target and manipulate the functions of Schwann cells in the adult peripheral nerves? (ii) How does it hijack Schwann cell properties once inside the cells? (iii) How does it propagate and disseminate infection? (iv) How does preferential M. leprae infection in Schwann cells initiate and cause neurological injury?
SIGNIFICANCE OF MODELING M. LEPRAE INTERACTION
During human infection, M. leprae preferentially resides and replicates within adult Schwann cells for a long period before immune cell recruitment and immune-mediated attack, which over a long incubation period eventually manifests clinically as sensory or sensorimotor loss (29, 35, 40–42). We know nothing about what occurs early in human infection, from initial infection of Schwann cells to the first symptoms of nerve damage. This is a critical initial phase for the propagation of M. leprae within this privileged niche, including evasion of immune surveillance and the establishment of productive infection within the PNS. Although host immune responses to M. leprae play a decisive role in developing the clinical state of leprosy, it is the leprosy bacillus itself and its propagation in the preferred peripheral tissue niches, such as the PNS and the skin, that establish the productive infection that sets the stage for subsequent infectious processes. However, how M. leprae establishes infection in the PNS and what strategies it uses to achieve this are largely unknown. Also, tissue cell responses directly to high bacterial load, independent of or under the partial influence of immune responses, could give rise to pathological conditions, especially when bacilli reside in privileged niches like Schwann cells, where immune cell trafficking is minimal. This is the case in patients with multibacillary leprosy, who harbor a high load of bacteria in the tissues which cause the pathology independent of cell-mediated immune responses. Thus, multibacillary leprosy provides an excellent disease model to study how a neurotrophic bacterial pathogen directly causes nervous system pathology. Therefore, mimicking multibacillary leprosy in model systems and searching for detailed mechanisms of how leprosy bacilli target the PNS are warranted.
M. leprae Targeting of the Peripheral Nerves
Molecular details of specific M. leprae-peripheral nerve interaction require model systems that mimic the unique PNS niche environment, since the anatomy of the PNS is critical for studying the specific interaction of M. leprae with cells in the PNS. This is because the way in which M. leprae might interact with the structural PNS components is distinct from the way in which it might interact with the CNS, both the brain and spinal cord. M. leprae interacts specifically with the mature glia of the human PNS (Schwann cells) (43), and clinical presentations involve peripheral nerves and tissues (9, 29, 42) and not the glia of the CNS (oligodendrocytes or astrocytes). Although some phenotypic similarities exist between myelin-producing Schwann cells and oligodendrocytes, these two cell types are distinct in terms of functional and signaling properties, developmental origin, and their interactions with axons (4, 44–48). Most importantly, the anatomical differences between the PNS and CNS are critical for the initial in vivo interaction of M. leprae with Schwann cells, which are the specific target of leprosy bacilli in the nervous system of humans.
Differences between glia of the adult PNS and CNS
The functional units that facilitate rapid nerve conduction of the PNS and CNS comprise glia-axon units: Schwann cell–axon units in the PNS and oligodendrocyte-axon units in the CNS. In the adult PNS, there are two functional units; some Schwann cells wrap around a single larger-diameter axon to form myelinated Schwann cell–axon units, and other Schwann cells make nonmyelinating Schwann cells by enclosing multiple smaller-diameter axons. Depending on the type of nerves, i.e., motor or sensory neurons, the ratio of myelinated to nonmyelinated axons varies (4, 5). In both cases, Schwann cell–axon units in the PNS are completely surrounded by the basal lamina (also called basement membrane when associated with epithelial cells, e.g., skin and intestine), which is composed of tissue-specific isoforms of matrix components secreted by Schwann cells (Fig. 2). These extracellular matrix (ECM) proteins on the outer surface of Schwann cells are anchored to the cell membrane via ECM receptors on the Schwann cell membrane (Fig. 2).
FIGURE 2.
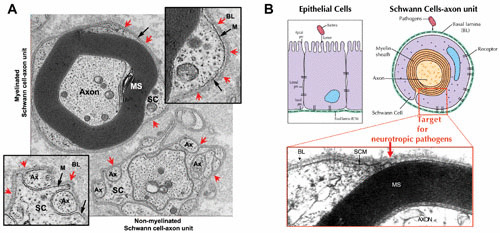
(A) Functional units of the adult human peripheral nerves, i.e., myelinated and nonmyelinated Schwann cell–axon units, depicting the distinct basal lamina that surrounds each Schwann cell–axon unit in situ. Red arrows indicate the basal lamina (BL) completely surrounding both myelinated (top inset) and nonmyelinated (bottom inset) Schwann cell–axon units. The Schwann cell membrane (M) is shown by black arrows. SC, Schwann cells; Ax, axons; MS, myelin sheath. (B) Sites of bacterial pathogens’ targets and entry into epithelia and peripheral nerves. Pathogenic bacteria enter epithelia at the apical side of the cells which anchor the basal lamina, whereas neurotrophic bacterial pathogens (e.g., M. leprae) must cross the basal lamina barrier, and thus attach to the basal lamina matrix proteins deposited around Schwann cell–axon units. The micrograph (adapted from reference 70) shows myelinated Schwann cell–axon units with the basal lamina (BL), Schwann cell membrane (SCM), and the axons ensheathed by the myelin sheath (MS).
This characteristic feature of the basement membrane around each glia-axon unit is not present in the oligodendrocytes, astrocytes, or microglia in the CNS. Oligodendrocytes form highly complex network-like connections with multiple axons by extending oligodendrocyte processes, enclosing multiple axons and forming myelin sheaths with each encountered axon, thus elaborating a highly complex myelinated nerve network in the CNS (48, 49). On the other hand, in non-neural tissues, cell types such as epithelia anchor to the basal lamina and/or basement membrane only through the basal side of the cells and are not surrounded completely by basal lamina or basement membranes (Fig. 2B). Also, macrophages in the PNS and both microglia and macrophages in the CNS, which usually engulf many bacterial and viral pathogens in the nervous system, are not surrounded by basal lamina or basement membranes. These facts indicate that the basal lamina that completely surrounds Schwann cell–axon units is a distinct anatomical feature in the PNS, and leprosy bacilli must cross the structural components of the basal lamina in order to invade Schwann cells. Perhaps the tropism of this bacterium to the adult peripheral nerves lies within this distinct bacterium–host component interaction, as immune cells such as ubiquitous macrophages are unlikely to exhibit such specific neural tropism.
DRG neuron and Schwann cell coculture model system
A model of the initial interaction of M. leprae with the native basal lamina components that surround both myelinated and nonmyelinated nerve fibers has been established using an ex vivo coculture system of purified primary rodent Schwann cells with purified dorsal root ganglion (DRG) neurons (50–54). In this model, as in peripheral nerve development, Schwann cells naturally wrap around DRG neuronal axons and establish a 1:1 relationship with larger-diameter axons and produce a myelin sheath, whereas other Schwann cells form nonmyelinated Schwann cell–axon units on smaller-diameter axons. As in adult peripheral nerves, both Schwann cell–axon units in these cultures are surrounded by the basal lamina comprising ECM components. The major components of the basal lamina are laminin-2 isoform, collagen IV, heparin sulfate proteoglycan, and nidogen (also known as entactin) (55–57). Among them, the tissue restriction of ECM components lies within the laminin-2 isoform, which is formed by assembling three subunits of laminin chains, the β1, γ1, and α2 chains (51).
Role of neural laminin α2 chain
Laminins are large glycoproteins composed of three polypeptide chains, α, β, and γ, which are assembled in different combinations to form asymmetrical cruciform structures to give rise to various laminin isoforms with restricted tissue distribution (58–60). These laminin isoforms are major structural and functional components of the basal lamina and/or basement membrane to which cells from different tissue types anchor via various extracellular ligands and cellular receptors (43, 61–64). Such specific cell-ECM component interactions are critical for cell differentiation and survival within specialized tissue niches; for example in the gut, cells anchor to the basal lamina that is secreted specifically by gut epithelial cells, and in the Schwann cell–axon units, the basal lamina that surrounds the units is secreted by Schwann cells (43, 65).
Likewise, combinations of laminin α, β, and γ chains that are assembled to form different isoforms vary in different tissues. It is believed the α chain determines this restricted tissue distribution, because the same α chain is rarely found in different tissues; for example, the α chain of laminin-2 is expressed predominantly in the basal lamina of Schwann cells and muscle cells, whereas the laminin β1 and γ1 chains have a wider tissue distribution (43, 61, 63, 64). Importantly, the tissue specificity of the laminin α chain is relevant to its characteristic larger globular (G) domain at the carboxyl terminus, and this G domain is responsible for the binding through which cells anchor to the basal lamina (43) (Fig. 3B and 4). In the basal lamina of Schwann cell–axon units, the tissue specificity lies within the laminin α2 chain and not within the β1 and γ1 chains, which are shared with other laminin isoforms, such as laminin-1, with a much broader tissue distribution (43, 51, 61, 63, 64). In the DRG Schwann cell–neuron coculture system, this tissue-restricted laminin α2 chain is specifically secreted by Schwann cells, particularly under the influence of axons, and thus contributes to the basal lamina that surrounds each nerve fiber, as in peripheral nerves in vivo.
FIGURE 3.
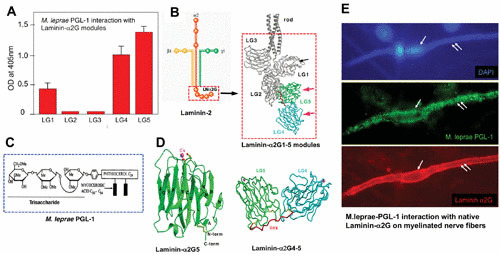
Molecular basis of neural tropism of M. leprae. Interaction of M. leprae-specific PGL-1 on the bacterial cell wall with the tissue-specific α2LG domain on the basal lamina. (A) PGL-1 binding to the recombinant G modules of the α2LG domain. OD, optical density. (B) Subunits of the laminin-2 isoform comprising α, β, and γ chains with the cell-binding α2G domain and its modules α2LG1 to α2LG5. (C) Composition of M. leprae PGL-1. (D) Crystal structure of PGL-1-binding α2LG5 and α2LG4-5 modules of the α2LG domain. (E) M. leprae PGL-1 binding (green) to the native α2LG domain (red) on the basal lamina surrounding a myelinated Schwann cell–axon unit (outer surface of nerve fiber is labeled in red to demarcate the α2G domain) colocalized with PGL-1 (green) when cultures were incubated with a PGL-1 suspension.
FIGURE 4.
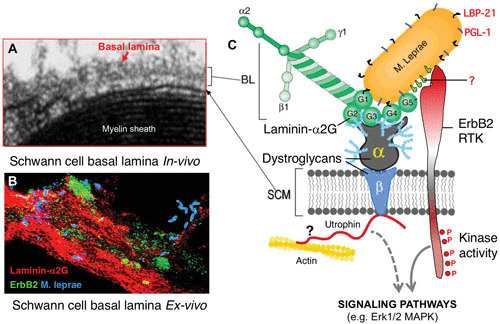
Schwann cell receptors α/β-dystroglycan and receptor tyrosine kinase ErbB2 serve as receptors for M. leprae on the Schwann cell membrane (SCM) in an α2LG domain-dependent and -independent manner. (A) Basal lamina (BL) and cell membrane of myelinated Schwann cells in vivo. Their molecular assembly is shown in the schematic (C). (B) M. leprae infection in an ex vivo Schwann cell–neuron coculture system, where M. leprae (blue) associates with α2LG in the basal lamina (red) and ErbB2 on the Schwann cell membrane (green). (C) Schematic showing the molecular basis of M. leprae interaction with α2LG, α/β-dystroglycan, and ErbB2 of Schwann cell–axon units in the peripheral nerves and potential activation of kinase domain of ErbB2, which initiate signaling cascades like phosphorylation of the Erk1/2 mitogen-activated protein kinase pathway.
Role of globular (G) domain of laminin α2 in neural tropism
Molecular analysis of the laminin α2 chain in relation to the tissue specificity of the basal lamina of Schwann cell–axon units in the PNS provides evidence that the neural tropism of M. leprae is determined by the G domain of the laminin α2 chain (α2LG), which is also the cell binding site of the laminin-2 isoform (43, 51). The α2LG modules are integral components of the basal lamina that surrounds the Schwann cell–axon units and interacts with host cell receptors such as α-dystroglycan, a laminin-2 receptor in both PNS and muscles (64, 66) (Fig. 3 and 4). Several of these receptor binding sites have been mapped to various α2LG modules (67). The resolved crystal structures of α2LG modules reveal a compact β-sandwich fold and a novel calcium-binding site architecture (68, 69) (Fig. 3). The fact that M. leprae interacts specifically with α2LG modules but cannot bind to the G domain of the α1 chain of the laminin-1 isoform, which in contrast shows wider tissue distribution, is consistent with the high sequence divergence between the G domains of the α1 and α2 chains (43, 51) (Fig. 3 and 4). In terms of tissue tropism, the limited sequence identity of the G domains of different laminin α chains (20 to 40%) appears to contribute to the restricted tissue distribution of a given laminin isoform (reference 51 and references therein). Based on the findings that α2LG specifically mediates the M. leprae–Schwann cell interaction and that in vivo, M. leprae failed to adhere to peripheral nerves of laminin-α2-deficient mice, which also lack α2LG, it was concluded that the tissue-restricted α2LG of the laminin-2 isoform is largely responsible for the peripheral-nerve tropism of M. leprae (51, 70).
It should be noted that the neural tropism of M. leprae could be demonstrated only by using α2LG modules and laminin α2-deficient mice, not with total laminin-2, simply because of the fact that total laminin-2 contains both laminin β1 and γ1 chains with wider tissue distribution (43, 51), and many bacterial pathogens, including mycobacterial species, have been shown to bind to these common laminin isoforms. Therefore, the use of commercially available total laminin-2 or -4 alone containing several common laminin chains does not confirm the tissue specificity; previous studies using such total laminin without molecular details showed binding of mycobacterial species other than M. leprae (71). Considering these crucial factors determining general versus specific binding, it is important to clarify these molecular details for future studies of tissue-specific interaction of M. leprae with the peripheral nerves.
CONTRIBUTION OF BACTERIAL FACTORS TO NEURAL TROPISM
Although the peculiar affinity of leprosy bacilli for the peripheral nerve has been known since the histopathological study of leprosy began after the discovery by Armauer Hansen of M. leprae as the causative organism of leprosy (11), and early scientists were curious about the basis for the neural affinity of this organism, it remained unexplained until studies published in 1997 and 2000 which detailed a mechanism for neural tropism of leprosy bacilli (50, 51). Once the host molecules responsible for neural tropism in leprosy bacilli had been identified, the next studies were launched to delineate the M. leprae-specific molecules that determined such host-pathogen interaction at the molecular level.
Role of PGL-1 and Proteins in the M. leprae Cell Wall
The overall tissue tropism of a bacterial pathogen is determined by both host and bacterial components. In the case of pathogenic mycobacteria, it has long been proposed that the bacterial cell wall contains most of the elements that are associated with pathogenesis (72). These elements may include the specific cell wall components that direct these pathogens to their favored niches. The cell wall of M. leprae contains an extensive electron-transparent outer layer that is largely composed of phthiocerol dimycocerosic acid and related glycolipids, which mainly comprise M. leprae-specific phenolic glycolipid-1 (PGL-1) (73–76). These complex cell wall lipid structures, including PGL-1, are commonly thought of in the context of resistance to intracellular killing by macrophages (77), serological analysis (78), and complement fixation (79). Detailed analysis related to the role of bacterial cell wall components showed evidence that PGL-1 is involved in determining the unique affinity of M. leprae for peripheral nerves (50). PGL-1 is specific for M. leprae because it contains an antigenically distinct trisaccharide, which consists of 3,6-di-O-methylglucose linked α1→4 to 2,3-di-O-methyl-rhamnose, linked β1→2 to 3-O-methylrhamnose, that has not been found in any other bacterium (73, 74). Purified PGL-1 specifically binds to the laminin α2 chain in the basal lamina of Schwann cell–axon units in ex vivo tissue cultures, and this binding is mediated by the M. leprae-specific trisaccharide portion of PGL-1 (50).
Contribution of M. leprae PGL-1 to Neural Tropism
The finding that PGL-1 binding to the basal lamina of Schwann cell–axon units is mediated by the naturally cleaved fragments of the peripheral nerve laminin α2 chain encouraged the concept of neural tropism, as this is a rare bacterium-specific and host tissue-restricted interaction (43, 50, 80). In peripheral nerves, the proteolytic cleavage of the α2 chain occurs at the carboxy-terminal G domain, resulting in an 80-kDa fragment and a large (∼300-kDa) amino-terminal fragment (81–83). PGL-1 binds to both peripheral-nerve-derived 80-kDa and 300-kDa fragments (50). Characterization of this binding using individual recombinant α2LG modules demonstrated that the activity to the 80-kDa fragment is associated with PGL-1 binding to the tissue-restricted α2LG4 and α2LG5 modules (50) (Fig. 3A and E). On the other hand, the lack of binding of lipoarabinomannan, another carbohydrate-containing cell wall component in M. leprae and Mycobacterium tuberculosis, to α2LG modules underscores the specific interaction of M. leprae PGL-1 trisaccharides with α2LG modules (50). The specificity of this interaction may be explained by the limited sequence identity of the LG modules of the laminin α2 and α1 chains. Sequence identities of the LG1, LG4, and LG5 modules of the laminin α2 and α1 chains are 45.7%, 36.1%, and 49.7%, respectively (84, 85). Thus, PGL-1 binding to α2LG modules corresponding to a highly divergent region of the α2 chain (α2LG1, α2LG4, and α2LG5) was found be the key to the neural affinity of M. leprae and thus establishes that these host and bacterial factors are responsible for neural tropism (50, 61, 86, 87) (Fig. 3).
SCHWANN CELL SIGNALING PATHWAYS ACTIVATED BY M. LEPRAE
α-Dystroglycan as a Receptor for M. leprae
In Schwann cell–axon units, the basal lamina anchors to the Schwann cell membrane via ECM receptors (61, 63, 64). One such receptor is dystroglycan, a highly glycosylated component of the dystrophin-glycoprotein complex (DGC) which is involved in the pathogenesis of muscular dystrophies (70, 88). The DGC is encoded by a single gene and serves as a receptor for Schwann cell–laminin-2 interaction and signaling (89). The encoded gene product is cleaved into two proteins, peripheral membrane α-dystroglycan and transmembrane β-dystroglycan, by posttranslational processing (90). Yamada et al. (91) have shown that the laminin α2 chain in the Schwann cell basal lamina binds peripheral membrane α-dystroglycan and that laminin α2 links to the Schwann cell cytoskeleton via the DGC (92). M. leprae binds to native α-dystroglycan purified from peripheral nerves only in the presence of α2LG (89), suggesting that α2LG has two binding sites, one for M. leprae and the other for α-dystroglycan; i.e., α2LG forms a bridge between M. leprae and the α-dystroglycan (89) (Fig. 3 and 4).
ErbB2 in Activation of Erk1/2 Mitogen-Activated Protein Kinase Signaling
Schwann cells, both during development and after acquisition of terminal differentiation, interact with neuronal (axonal) ligands for communication, as they are interdependent for maintaining their survival and functional properties (49, 93). This is mediated mainly by neuregulins, the axonal ligands, by binding to the receptor tyrosine kinase complex, ErbB2/ErbB3, on the Schwann cell membrane, which leads to the initiation of intracellular signaling pathways to drive proliferation and differentiation of Schwann cells, particularly the myelination of axons (94, 95). The ErbB family receptor ErbB2 is one of the major receptor tyrosine kinases expressed on Schwann cells and plays an important role in glial cell functions (95–98). Among the members of the ErbB family, ErbB2 is considered a “ligandless” receptor that transduces strong signals by avid dimerization with other ErbB members (99, 100). In Schwann cells, ErbB2 is known to process the signaling by dimerization with ErbB3 after neuregulin binds to ErbB3, which lacks a kinase domain (97). Strikingly, M. leprae directly binds to and activates ErbB2 without ErbB3 heterodimerization (53). This binding is sufficient to induce early demyelination, following activation of ErbB2 by a novel route that bypasses the classical signaling ErbB2-ErbB3 heterodimerization induced by growth factors including neuregulin, and subsequently induces a downstream MEK-dependent Erk1/2 signaling pathway, leading to myelin breakdown (53, 97) (Fig. 4 and 5). It is likely that M. leprae takes advantage of the existing Erk1/2 signaling that is involved in both nerve degeneration and regeneration (101).
FIGURE 5.
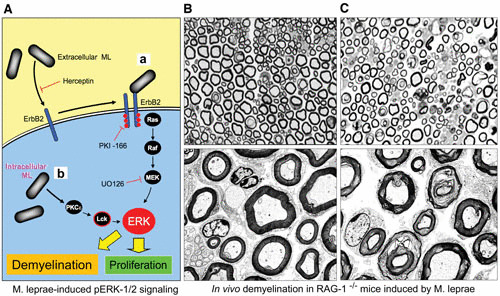
In vivo induction of demyelination by direct M. leprae injection into sciatic nerves of adult Rag-1 knockout mice, suggesting that early demyelination can be caused by the activation of signaling pathways in the absence of immune responses. (A) Schematic showing the activation of Erk1/2 MAPK signaling pathways by extracellular (a) and intracellular (b) M. leprae via two different pathways and their role in proliferation and demyelination. M. leprae binds to the ErbB2 receptor to induce Schwann cell demyelination and proliferation. (a) The binding of M. leprae (ML) to ErbB2 on the surface of myelinated Schwann cells triggers demyelination through the Ras-Raf-MEK-ERK pathway. ErbB2 inhibitors such as herceptin, PKI166, and U0126 block activation of this pathway in response to M. leprae. (b) Intracellular M. leprae induces proliferation of nonmyelinated Schwann cells through a different route to ERK that involves PKCε and Lck and that is independent of signaling through the Ras-Raf-MEK pathway. (B and C) Direct injection of M. leprae into sciatic nerves of Rag–/– knockout mice induces demyelination (C), in contrast to injection of phosphate-buffered saline alone, which shows almost intact myelinated Schwann cell–axon units (B).
M. LEPRAE-INDUCED DEMYELINATION OF SCHWANN CELLS
Myelin damage is known to be caused by immune-mediated inflammatory responses, as in many neurodegenerative diseases (102). However, whether such demyelination events also occur in the earliest stage of neurodegenerative diseases where inflammation is minimal or absent is unknown. M. leprae is known to initiate neurodegenerative conditions in humans and in susceptible animal models like nine-banded armadillos (9, 29, 33). Therefore, underlying mechanisms by which M. leprae initiates neurodegenerative conditions provide valuable insights into early events of neurodegeneration.
The initial interaction of M. leprae with the basal lamina of Schwann cell–axon units appears to deregulate the delicate Schwann cell–axon communication system, leading to the breakdown of the myelin sheath (80, 101). Although such early demyelination in vivo may not initially lead to clinical manifestation, as peripheral nerves possess a remarkable capacity to regenerate following injury, it may lead to activation of additional signaling from Schwann cells similar to nerve injury (101). Functional consequences like demyelination provide a survival advantage for M. leprae, as it induces dedifferentiation and proliferation and generates myelin-free Schwann cells with high plasticity, which naturally promote remyelination and nerve regeneration and are also highly susceptible to M. leprae invasion (103). This suggests that initial interactions with and activation of Schwann cells are crucial, as these events set the stage for subsequent intracellular survival and replication within the peripheral nerves.
From Signaling-Mediated Demyelination to Nerve Damage
Although nerve degeneration in the early phase of M. leprae infection in both in vitro and in vivo mouse models does not involve immune cells or macrophages, such nerve injury is likely to cause the destabilization of the neural microenvironment. This might subsequently lead to a cascade of cellular responses that eventually recruit immune cells. Evidence from studies with Rag-1–/– knockout mice, which lack mature B and T cells and are thus unable to mount an adaptive immune response, showed the induction of significant demyelination in sciatic nerves 3 days after intraneural administration of M. leprae and its cell wall fraction (52) (Fig. 5). It is likely that the sequential recruitment and propagation of immune cell populations, preceded by non-immune-mediated demyelination and axonal damage at the site of infection, could eventually cause further aggravation of neurological injury to the peripheral nerves during M. leprae infection.
Zebrafish Spinal Cord Model
Recent studies have attempted to recapitulate myelin damage in response to M. leprae in zebrafish larvae using spinal cord injection of M. leprae as a CNS model (104). Since the study of spinal cord injection of M. leprae examines the initial events in CNS cells, it is unlikely that a zebrafish CNS model exhibits the specific peripheral nerve pathology which is so distinctive of M. leprae infection in humans (9, 29, 42) due to the nervous systems’ fundamental and functional differences as described above (Fig. 2). The underlying pathology of PNS diseases is distinct from that of CNS diseases, and as such, no studies to our knowledge use CNS models for insights into PNS damage.
M. leprae infects Schwann cell–axon units in the PNS, but not the oligodendrocyte-axon units in the spinal cord. Schwann cell–axon units are completely surrounded by the basal lamina (43, 51, 55, 65) (Fig. 2 and 4), providing barriers to M. leprae (Fig. 2). In contrast, the axon-oligodendrocyte and astrocyte units of the CNS are not surrounded by such a basal lamina, so it is not surprising that M. leprae could bind directly and cause subsequent demyelination when it is injected directly into the zebrafish larvae with immature myelin sheaths (93). Thus, the bacterial tropism for the PNS is difficult to evaluate with such a CNS model. Furthermore, without the PNS barriers, macrophages may have more straightforward access to oligodendrocytes in the zebrafish CNS model, causing the defects seen in the immature larval myelin sheath (104). Such findings indicating that an innate macrophage reaction to bacterial PGL-1 causes early nerve damage and neural tropism should be interpreted with caution, as the macrophage-mediated innate immune responses are ubiquitous and are unlikely to determine neural tropism.
Hijacking of Nerve Injury Responses by Leprosy Bacilli
Unlike CNS neurons, the neurons in the PNS can regenerate effectively when damaged; this differential response to injury is due largely to the proliferative and regenerative properties of adult Schwann cells (105, 106). Following demyelination and nerve injury, terminally differentiated Schwann cells undergo proliferation, and one of the key regulators of the cell cycle, cyclin D1, plays a role in this re-entry into the cell division cycle (107, 108). These nerve injury responses lead to the generation of new dedifferentiated Schwann cells, which serve as repair cells to facilitate the remyelination and regeneration of the damaged axons by initiating a Schwann cell redifferentiation program (106, 108, 109). M. leprae appears to mimic the events following nerve injury, since intracellular M. leprae induces the accumulation of cyclin D1 in infected human Schwann cells and subsequently increases the cell division (107). When newly generated Schwann cells or similar dedifferentiated Schwann cells are infected by M. leprae, they turn off myelination-associated genes (110) and myelin protein expression, the integral components of the compact myelin sheath. These findings suggest that intracellular M. leprae turns off the Schwann cell differentiation program and maintains the infected cells in an undifferentiated stage.
M. leprae appears to hijack multiple pathways of injury repair processes during infection. At the signaling level, mitogen-activated protein kinase pathway Erk1/2 signaling is strongly activated in Schwann cells undergoing dedifferentiation during nerve injury in vivo (111). Similarly, once inside the cells, M. leprae also activates Erk1/2 signaling in dedifferentiated human Schwann cells (107). However, M. leprae activates Erk1/2 intracellularly, not via the Ras-MEK-dependent canonical pathway, but by a novel signaling pathway using a noncanonical MEK-independent and protein kinase Cε- and lymphocyte kinase (Lck)-dependent pathway (107). Therefore, it is possible that sustained activation of noncanonical Erk1/2 signaling in infected Schwann cells maintains cells in a dedifferentiated state and thus prevents remyelination.
M. LEPRAE REPROGRAMMING OF SCHWANN CELLS
The extremely long incubation period of M. leprae within Schwann cells during human infection provides M. leprae with ample time to alter host cell behavior for its strictly obligate intracellular lifestyle. This is likely to occur in peripheral nerves in multibacillary leprosy, where a high load of bacteria resides in Schwann cells for a long period (9, 29). What the fate of these bacteria-laden Schwann cells is and whether these cells retain or change Schwann cell identity were unknown until recently. It was also unknown how these bacteria disseminate from this privileged niche to other tissues. Intriguingly, in a study by Masaki et al. (110), it was discovered that leprosy bacteria hijack the notable plasticity and regenerative properties of adult Schwann cells for the establishment of infection within the PNS (52, 53, 101).
Masaki et al. (110) demonstrated that M. leprae can transcriptionally reprogram Schwann cells to a progenitor stem-like cell (pSLC) state by changing epigenetic modification of key genes (110). By reprogramming adult Schwann cells to stem cell-like cells with migratory properties, M. leprae facilitates its spread to other tissues (110) (Fig. 6). This strategy is highly favorable for spread of a strictly obligately intracellular pathogen like M. leprae via cell-to-cell transfer, as it was shown that bacteria-laden reprogrammed cells effectively redifferentiate to other tissues like muscles and directly transfer infection to muscle fibers, another host niche for leprosy bacilli in humans (110, 112, 113). Moreover, reprogrammed cells also have the capacity to transfer infection more effectively to fibroblasts than nonreprogrammed Schwann cells (114), permitting dissemination of infection to multiple tissues through intermediate cells like fibroblasts, which are abundant in most of the tissue milieu.
FIGURE 6.
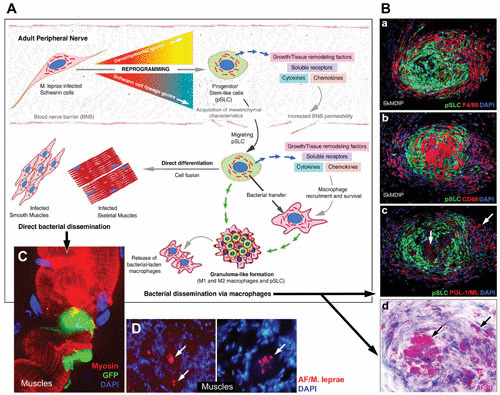
Proposed model of how adult Schwann cell reprogramming to stem cell-like cells by intracellular M. leprae promotes dissemination of infection. (A) Infected Schwann cells in the adult peripheral nerves undergo a reprogramming process whereby Schwann cell differentiation and myelination program-associated genes are turned off and embryonic genes of mesenchymal and neural crest development are turned on. The resulting pSLC acquire migratory properties and immunomodulatory characteristics and thus release immune factors, chemokines, cytokines, and growth and remodeling factors, which not only increase the permeability of the BNB but also recruit macrophages. (B) Acquired migratory properties promote M. leprae-laden pSLC to exit by breaching the BNB and disseminate to other preferred tissue niches, such as smooth muscles and skeletal muscles, where they are exposed to respective tissue microenvironments and undergo direct differentiation and thus transfer bacteria passively to these tissues. (C) By recruiting macrophages, pSLC can transfer M. leprae and form typical granuloma-like structures, which then release bacterium-laden macrophages, a mechanism by which reprogrammed cells may channel bacterial dissemination via systemic routes. DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein-tagged pSLC; AF, acid-fast labeling of M. leprae.
Bacterial Usage of Transcription Factors for Reprogramming
By recapitulating an injury-like response where M. leprae dedifferentiates adult Schwann cells following demyelination of infected nerves at the early stage of infection, Masaki et al. (110) purified dedifferentiated Schwann cells from wild-type adult mouse peripheral nerves and then infected them with M. leprae at a high bacterial load, mimicking the conditions in multibacillary leprosy. Over time, it was found that the transcriptional and proteomic profile underwent radical alterations, including loss of Schwann cell-specific lineage markers such as Sox10 and ErbB3 and myelin genes including that encoding myelin basic protein, suggesting turning off of the differentiation-myelination program (95, 115–118). Concordantly, infection upregulated a range of early developmental and embryonic markers, particularly mesenchymal and neural crest stem cell transcripts (110). A key early event appears to be the loss of the Sox10 transcription factor from the nuclei of Schwann cells after infection (110). As a master regulator of Schwann cell identity and lineage specificity, Sox10 is present throughout Schwann development and the adult stage (4, 115, 116, 119), and thus the bacterial removal of Sox10 clears the way for loss of Schwann cell lineage identity and the ability of these reprogrammed cells to convert to other cell types. Change in Sox10 gene expression appears to be regulated epigenetically, as M. leprae causes DNA methylation at the Sox10 locus (110), which is associated with gene silencing.
On the other hand, the transcription factor Sox2, which is activated in dedifferentiated Schwann cells during injury but not in intact adult peripheral nerves and is known to be a repressor of myelination (120–122), is retained after reprogramming by M. leprae. Sox2 is an embryonic stem cell marker known to function in regulating gene networks and phenotype in embryonic stem and neural progenitor cells (123–140). Indeed, a transgenic mouse model in which overexpression of Sox2 was sustained in adult nerves was shown to inhibit myelination and cause subsequent neuropathic conditions (122). Taken together, these observations indicate that it is possible that the bacteria use this stem cell factor in the switch between a Schwann cell program and a progenitor cell program, aided by the loss of Sox10. However, such mechanistic possibilities need further investigation.
Epithelial-Mesenchymal Transition during Schwann Cell Reprogramming
Schwann cell reprogramming by M. leprae also involves the epithelial-mesenchymal transition (EMT) program, a key developmental program which also occurs during neural crest cell specification and epithelial and endothelial cell transition to mesenchymal phenotypes as well as in metastatic epithelial cancers (110, 141). EMT genes upregulated after infection, including Twist, Snail1/2, and Msx2, may enforce such inherent phenotypic plasticity to change Schwann cell fate, as dedifferentiated Schwann cells appear to behave as mesenchymal-type cells. Upregulation of these EMT genes is also regulated by epigenetic mechanisms, as further analysis of Schwann cell reprogramming revealed that there are widespread changes in DNA methylation status arising from bacterial influence (110). More research will undoubtedly yield further knowledge of how this pathogen uses sophisticated approaches to alter the host cell transcriptional and epigenetic networks.
Schwann Cell Reprogramming and Neuropathy
Reprogramming of functional adult Schwann cells means that these cells reactivate their developmental program to convert to an immature-like state that lacks mature-cell properties necessary to maintain functional nerves. Certainly, such reprogramming would contribute to the pathology observed in patients, since disruption of the well-structured Schwann cell–axon unit would undoubtedly lead to Schwann cell dedifferentiation. These events lead to reduced conduction of action potentials and exposition of the axon to damage and degeneration over time during extremely long-term infection in multibacillary leprosy patients, consistent with sensory and motor loss of leprosy neuropathy. Although ultimately detrimental to the host, reprogramming of Schwann cells appears to bring a range of survival advantages to the bacteria, as it might provide a favorable microenvironment for bacterial propagation within the PNS.
Schwann Cell Reprogramming and Bacterial Dissemination
The reprogrammed Schwann cells have expression patterns resembling a neural crest/stem cell-like phenotype and display multipotency to differentiate not only to muscles but also to bone and adipose tissues (110). This, alongside acquired migratory and immunomodulatory properties as in mesenchymal stem cells (142), may enable the bacteria to exit peripheral nerves, potentially breaching the BNB by immunomodulatory agents that reprogrammed cells can release and that can thus enter the bloodstream, recruit macrophages, or directly transdifferentiate into desired tissue cell types once reaching other preferred target cells like muscles, bone, and adipose tissues (Fig. 6) (103, 110). An additional bacterial advantage is that reprogrammed Schwann cells, but not normal Schwann cells, show more efficient cell-cell transfer of the bacteria to fibroblasts from both skin and peripheral nerve origins (114). As such, bacterial retention in Schwann cells during early infection may permit a period of survival and growth of the extremely slowly dividing bacteria prior to onward invasion of surrounding nerve tissue and beyond.
Granuloma formation
As another route of bacterial spread, reprogrammed cells, which release chemokines/cytokines, also contribute to granuloma formation by recruiting macrophages, which in turn contribute to bacterial propagation and dissemination. Indeed, granulomas are a commonly observed pathological feature in leprosy and other mycobacterial diseases (110, 143–145). Such granuloma formation is facilitated by many innate immune chemokines and cytokines produced by reprogrammed Schwann cells that can recruit macrophages to produce granulomatous formations (Fig. 6) (110, 142, 146, 147). So, by invading the Schwann cells and subsequent reprogramming, M. leprae has acquired a number of advantages to promote its survival, persistence, and dissemination. The combination of these strategies is quite distinct from the strategies of many other bacterial pathogens that have shorter life cycles and cause more acute damage to host cells and tissues. It is possible that the long evolutionary history of M. leprae within human and animal hosts, the complete reliance on host cells for replication, and the extremely long bacterial replication time have contributed to the special coexistence of this intracellular bacterium with long-lived host cells, particularly the nervous system cells.
CONCLUDING REMARKS
Pathogen interactions with nervous system cells are crucial in the establishment and pathogenesis of infectious diseases affecting the nervous system. We describe, using leprosy bacilli as an example, how intracellular bacterial pathogens can make use of or subvert host cell molecules and pathways and their functions for bacterial invasion with selective tropisms with underlying mechanisms, and how they can hijack the unique properties of cell plasticity once they are inside the nervous system cells for survival, establishment of infection, and dissemination. The sustained abilities of the bacterial pathogens to coexist with nervous system cells and develop subsequent innate and inflammatory responses can lead to neuropathologies causing neurodegeneration. Thus, neurotrophic pathogen interactions with nerve cells can be used as models to gain insights into how neurodegeneration is initiated following infectious triggers and to translate such knowledge to the understanding of potential common themes of early events in neurodegenerative diseases whose causes or triggers are unknown. Despite limited available information for many of these changes due to the short period of studies of neurotrophic bacteria, the long life of M. leprae demonstrates how bacteria can have a long-term influence on their host cells and manipulate cellular plasticity to reprogram cells in a way that is beneficial for their persistence and that helps achieve the most difficult task of dissemination despite their strictly obligately intracellular lifestyle. This unexpected link between host cell reprogramming and natural bacterial infection presents a previously unseen degree of sophistication in cell manipulation by the hijacking of the genomic plasticity of host cells by a human bacterial pathogen and thus opens up a new premise in host-pathogen interactions as well as a novel theme of intersection of infection biology with stem cell and regeneration biology. Understanding detailed mechanisms regarding these bacterial processes will help us to develop new tools for cellular reprogramming in stem cell biology and regenerative medicine and provide us with new targets not only for combating bacterial pathogens but also for developing treatments and cures for neurodegenerative diseases. In particular, this understanding can help us learn how neurotrophic pathogens naturally manipulate the nervous system cells by hijacking endogenous pathways and thus understand better how the nervous system works normally.
ACKNOWLEDGMENTS
We thank present and past members of the Rambukkana laboratory at the University of Edinburgh and the Rockefeller University in New York and collaborators who contributed for many years of work, which are described and cited here. Research presented was funded in part by grants from NINDS (NS45187), NIAID (AI45816), the Rockefeller University, the University of Edinburgh, and the Wellcome Trust Institutional Strategic Support Funds.
Contributor Information
Samuel Hess, Medical Research Council (MRC) Centre for Regenerative Medicine.
Anura Rambukkana, Medical Research Council (MRC) Centre for Regenerative Medicine; Centre for Edinburgh Infectious Diseases, University of Edinburgh, Edinburgh, United Kingdom.
Pascale Cossart, Institut Pasteur, Paris, France.
Craig R. Roy, Yale University School of Medicine, New Haven, Connecticut
Philippe Sansonetti, Institut Pasteur, Paris, France.
REFERENCES
- 1.Drevets DA, Leenen PJM, Greenfield RA. 2004. Invasion of the central nervous system by intracellular bacteria. Clin Microbiol Rev 17:323–347 10.1128/CMR.17.2.323-347.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deigendesch N, Stenzel W. 2018. Acute and chronic bacterial infections and sarcoidosis, p 217–226. In Handbook of Clinical Neurology. Elsevier, New York, NY. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.Suthar R, Sankhyan N. 2018. Bacterial infections of the central nervous system. Indian J Pediatr 86:60–69. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Jessen KR, Mirsky R. 2005. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci 6:671–682 10.1038/nrn1746. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Jessen KR, Mirsky R, Lloyd AC. 2015. Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol 7:a020487 10.1101/cshperspect.a020487. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peltonen S, Alanne M, Peltonen J. 2013. Barriers of the peripheral nerve. Tissue Barriers 1:e24956 10.4161/tisb.24956. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhold AK, Rittner HL. 2017. Barrier function in the peripheral and central nervous system—a review. Pflugers Arch 469:123–134 10.1007/s00424-016-1920-8. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. 2017. Global leprosy update, 2016: accelerating reduction of disease burden. Wkly Epidemiol Rec 92:501–519. [PubMed] [Google Scholar]
- 9.van Brakel WH, Post E, Saunderson PR, Gopal PK. 2017. Leprosy, p 391–401. In Quah SR (ed), International Encyclopedia of Public Health, 2nd ed. Academic Press, New York, NY. [Google Scholar]
- 10.Browne SG. 1985. The history of leprosy, p 1–14. In Hastings RC (ed), Leprosy. Churchill Livingstone, Edinburgh, United Kingdom. [Google Scholar]
- 11.Hansen GHA. 1874. Undersøgelser angående spedalskhedens årsager (investigations concerning the etiology of leprosy). Nor Mag Laegervidenskaben 4:1–88. [Google Scholar]
- 12.Suzuki K, Akama T, Kawashima A, Yoshihara A, Yotsu RR, Ishii N. 2012. Current status of leprosy: epidemiology, basic science and clinical perspectives. J Dermatol 39:121–129 10.1111/j.1346-8138.2011.01370.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.World Health Organization. 2017. Global Leprosy Strategy 2016-2020. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 14.Cruz RCDS, Bührer-Sékula S, Penna MLF, Penna GO, Talhari S. 2017. Leprosy: current situation, clinical and laboratory aspects, treatment history and perspective of the uniform multidrug therapy for all patients. An Bras Dermatol 92:761–773 10.1590/abd1806-4841.20176724. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole ST, et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 16.Singh P, Cole ST. 2011. Mycobacterium leprae: genes, pseudogenes and genetic diversity. Future Microbiol 6:57–71 10.2217/fmb.10.153. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truman R. 2005. Leprosy in wild armadillos. Lepr Rev 76:198–208. [PubMed] [Google Scholar]
- 18.Balamayooran G, Pena M, Sharma R, Truman RW. 2015. The armadillo as an animal model and reservoir host for Mycobacterium leprae. Clin Dermatol 33:108–115 10.1016/j.clindermatol.2014.07.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Avanzi C, Del-Pozo J, Benjak A, Stevenson K, Simpson VR, Busso P, McLuckie J, Loiseau C, Lawton C, Schoening J, Shaw DJ, Piton J, Vera-Cabrera L, Velarde-Felix JS, McDermott F, Gordon SV, Cole ST, Meredith AL. 2016. Red squirrels in the British Isles are infected with leprosy bacilli. Science 354:744–747 10.1126/science.aah3783. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Scollard DM, Truman RW, Ebenezer GJ. 2015. Mechanisms of nerve injury in leprosy. Clin Dermatol 33:46–54 10.1016/j.clindermatol.2014.07.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Lázaro FP, Werneck RI, Mackert CCO, Cobat A, Prevedello FC, Pimentel RP, Macedo GMM, Eleutério MAM, Vilar G, Abel L, Xavier MB, Alcaïs A, Mira MT. 2010. A major gene controls leprosy susceptibility in a hyperendemic isolated population from north of Brazil. J Infect Dis 201:1598–1605 10.1086/652007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Truman RW, Singh P, Sharma R, Busso P, Rougemont J, Paniz-Mondolfi A, Kapopoulou A, Brisse S, Scollard DM, Gillis TP, Cole ST. 2011. Probable zoonotic leprosy in the southern United States. N Engl J Med 364:1626–1633 10.1056/NEJMoa1010536. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chopra R, Ali S, Srivastava AK, Aggarwal S, Kumar B, Manvati S, Kalaiarasan P, Jena M, Garg VK, Bhattacharya SN, Bamezai RNK. 2013. Mapping of PARK2 and PACRG overlapping regulatory region reveals LD structure and functional variants in association with leprosy in unrelated Indian population groups. PLoS Genet 9:e1003578 10.1371/journal.pgen.1003578. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chopra R, Kalaiarasan P, Ali S, Srivastava AK, Aggarwal S, Garg VK, Bhattacharya SN, Bamezai RNK. 2014. PARK2 and proinflammatory/anti-inflammatory cytokine gene interactions contribute to the susceptibility to leprosy: a case-control study of North Indian population. BMJ Open 4:e004239 10.1136/bmjopen-2013-004239. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mira MT, Alcaïs A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, Mai CP, Nguyen TH, Nguyen NB, Pham XK, Sarno EN, Alter A, Montpetit A, Moraes ME, Moraes JR, Doré C, Gallant CJ, Lepage P, Verner A, Van De Vosse E, Hudson TJ, Abel L, Schurr E. 2004. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427:636–640 10.1038/nature02326. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Domingo A, Klein C. 2018. Genetics of Parkinson disease, p 211–227. In Handbook of Clinical Neurology. Elsevier, New York, NY. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Meng L, Shen L, Ji HF. 2019. Impact of infection on risk of Parkinson’s disease: a quantitative assessment of case-control and cohort studies. J Neurovirol 25:221–228 10.1007/s13365-018-0707-4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT. 2012. Infectious agents and neurodegeneration. Mol Neurobiol 46:614–638 10.1007/s12035-012-8320-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masaki T, Rambukkana A. 2014. Neurodegeneration in leprosy: insights from model systems and patients, p 217–232. In Bentivoglio M, Cavalheiro EA, Kristensson K, Patel NB (ed), Neglected Tropical Diseases and Conditions of the Nervous System. Springer, New York, NY. 10.1007/978-1-4614-8100-3_12 [DOI] [Google Scholar]
- 30.Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. 2006. The continuing challenges of leprosy. Clin Microbiol Rev 19:338–381 10.1128/CMR.19.2.338-381.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Truman RW, Krahenbuhl JL. 2001. Viable M. leprae as a research reagent. Int J Lepr Other Mycobact Dis 69:1–12. [PubMed] [Google Scholar]
- 32.Colston MJ, Hilson GRF. 1976. Growth of Mycobacterium leprae and M. marinum in congenitally athymic (nude) mice. Nature 262:399–401 10.1038/262399a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Truman RW, Ebenezer GJ, Pena MT, Sharma R, Balamayooran G, Gillingwater TH, Scollard DM, McArthur JC, Rambukkana A. 2014. The armadillo as a model for peripheral neuropathy in leprosy. ILAR J 54:304–314 10.1093/ilar/ilt050. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma R, Lahiri R, Scollard DM, Pena M, Williams DL, Adams LB, Figarola J, Truman RW. 2013. The armadillo: a model for the neuropathy of leprosy and potentially other neurodegenerative diseases. Dis Model Mech 6:19–24 10.1242/dmm.010215. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Job CK. 1989. Nerve damage in leprosy. Int J Lepr Other Mycobact Dis 57:532–539. [PubMed] [Google Scholar]
- 36.Jennekens FG, van Brakel WH. 1998. Neuropathy in leprosy, p 319–339. In Latov N, Wokke JHJ, Kelly JJ Jr (ed), Immunological and Infectious Diseases of the Peripheral Nerves. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 37.Rodrigues LC, Lockwood DN. 2011. Leprosy now: epidemiology, progress, challenges, and research gaps. Lancet Infect Dis 11:464–470 10.1016/S1473-3099(11)70006-8. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L. 2017. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9:7204–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ransohoff RM. 2016. How neuroinflammation contributes to neurodegeneration. Science 353:777–783 10.1126/science.aag2590. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Stoner GL. 1979. Importance of the neural predilection of Mycobacterium leprae in leprosy. Lancet ii:994–996 10.1016/S0140-6736(79)92564-9. [DOI] [PubMed] [Google Scholar]
- 41.Miko TL, Le Maitre C, Kinfu Y. 1993. Damage and regeneration of peripheral nerves in advanced treated leprosy. Lancet 342:521–525 10.1016/0140-6736(93)91647-5. [DOI] [PubMed] [Google Scholar]
- 42.Ooi WW, Srinivasan J. 2004. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve 30:393–409 10.1002/mus.20113. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Rambukkana A. 2001. Molecular basis for the peripheral nerve predilection of Mycobacterium leprae. Curr Opin Microbiol 4:21–27 10.1016/S1369-5274(00)00159-4. [DOI] [PubMed] [Google Scholar]
- 44.Nave K-A, Werner HB. 2014. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol 30:503–533 10.1146/annurev-cellbio-100913-013101. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Pereira JA, Lebrun-Julien F, Suter U. 2012. Molecular mechanisms regulating myelination in the peripheral nervous system. Trends Neurosci 35:123–134 10.1016/j.tins.2011.11.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Taveggia C, Feltri ML, Wrabetz L. 2010. Signals to promote myelin formation and repair. Nat Rev Neurol 6:276–287 10.1038/nrneurol.2010.37. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahrendsen JT, Macklin W. 2013. Signaling mechanisms regulating myelination in the central nervous system. Neurosci Bull 29:199–215 10.1007/s12264-013-1322-2. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitew S, Hay CM, Peckham H, Xiao J, Koenning M, Emery B. 2014. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 276:29–47 10.1016/j.neuroscience.2013.11.029. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Nave KA. 2010. Myelination and support of axonal integrity by glia. Nature 468:244–252 10.1038/nature09614. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Ng V, Zanazzi G, Timpl R, Talts JF, Salzer JL, Brennan PJ, Rambukkana A. 2000. Role of the cell wall phenolic glycolipid-1 in the peripheral nerve predilection of Mycobacterium leprae. Cell 103:511–524 10.1016/S0092-8674(00)00142-2. [DOI] [PubMed] [Google Scholar]
- 51.Rambukkana A, Salzer JL, Yurchenco PD, Tuomanen EI. 1997. Neural targeting of Mycobacterium leprae mediated by the G domain of the laminin-α2 chain. Cell 88:811–821 10.1016/S0092-8674(00)81927-3. [DOI] [PubMed] [Google Scholar]
- 52.Rambukkana A, Zanazzi G, Tapinos N, Salzer JL. 2002. Contact-dependent demyelination by Mycobacterium leprae in the absence of immune cells. Science 296:927–931 10.1126/science.1067631. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Tapinos N, Ohnishi M, Rambukkana A. 2006. ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat Med 12:961–966. CORRIGENDUM Nat Med 12:1100 10.1038/nm1433. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Einheber S, Milner TA, Giancotti F, Salzer JL. 1993. Axonal regulation of Schwann cell integrin expression suggests a role for alpha 6 beta 4 in myelination. J Cell Biol 123:1223–1236 10.1083/jcb.123.5.1223. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cornbrooks CJ, Carey DJ, McDonald JA, Timpl R, Bunge RP. 1983. In vivo and in vitro observations on laminin production by Schwann cells. Proc Natl Acad Sci USA 80:3850–3854 10.1073/pnas.80.12.3850. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaakkola S, Peltonen J, Riccardi V, Chu ML, Uitto J. 1989. Type 1 neurofibromatosis: selective expression of extracellular matrix genes by Schwann cells, perineurial cells, and fibroblasts in mixed cultures. J Clin Invest 84:253–261 10.1172/JCI114148. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanes JR, Engvall E, Butkowski R, Hunter DD. 1990. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol 111:1685–1699 10.1083/jcb.111.4.1685. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burgeson RE, Chiquet M, Deutzmann R, Ekblom P, Engel J, Kleinman H, Martin GR, Meneguzzi G, Paulsson M, Sanes J, Timpl R, Tryggvason K, Yamada Y, Yurchenco PD. 1994. A new nomenclature for the laminins. Matrix Biol 14:209–211 10.1016/0945-053X(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 59.Engvall E, Wewer UM. 1996. Domains of laminin. J Cell Biochem 61:493–501 . [DOI] [PubMed] [Google Scholar]
- 60.Timpl R, Brown JC. 1994. The laminins. Matrix Biol 14:275–281 10.1016/0945-053X(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 61.Leivo I, Engvall E. 1988. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA 85:1544–1548 10.1073/pnas.85.5.1544. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yurchenco PD, O’Rear JJ. 1994. Basal lamina assembly. Curr Opin Cell Biol 6:674–681 10.1016/0955-0674(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 63.Aumailley M, Smyth N. 1998. The role of laminins in basement membrane function. J Anat 193:1–21 10.1046/j.1469-7580.1998.19310001.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasaki T, Timpl R. 1999. Laminins, p 434–443. In Kreis T, Vale R (ed), Guidebook to the Extracellular Matrix, Anchor and Adhesion Proteins, 3rd ed. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 65.Bunge MB, Bunge RP. 1986. Linkage between Schwann cell extracellular matrix production and ensheathment function. Ann N Y Acad Sci 486(1 Neurofibromat):241–247 10.1111/j.1749-6632.1986.tb48077.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Henry MD, Campbell KP. 1999. Dystroglycan inside and out. Curr Opin Cell Biol 11:602–607 10.1016/S0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 67.Talts JF, Andac Z, Göhring W, Brancaccio A, Timpl R. 1999. Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J 18:863–870 10.1093/emboj/18.4.863. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hohenester E, Tisi D, Talts JF, Timpl R. 1999. The crystal structure of a laminin G-like module reveals the molecular basis of α-dystroglycan binding to laminins, perlecan, and agrin. Mol Cell 4:783–792 10.1016/S1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- 69.Tisi D, Talts JF, Timpl R, Hohenester E. 2000. Structure of the C-terminal laminin G-like domain pair of the laminin α2 chain harbouring binding sites for α-dystroglycan and heparin. EMBO J 19:1432–1440 10.1093/emboj/19.7.1432. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rambukkana A. 2000. How does Mycobacterium leprae target the peripheral nervous system? Trends Microbiol 8:23–28 10.1016/S0966-842X(99)01647-9. [DOI] [PubMed] [Google Scholar]
- 71.Marques MAM, Antônio VL, Sarno EN, Brennan PJ, Pessolani MCV. 2001. Binding of α2-laminins by pathogenic and non-pathogenic mycobacteria and adherence to Schwann cells. J Med Microbiol 50:23–28 10.1099/0022-2615-50-1-23. [PubMed] [DOI] [PubMed] [Google Scholar]
- 72.Anderson RJ. 1932. The chemistry of the lipoids of tubercle bacilli. Physiol Rev 12:166–189 10.1152/physrev.1932.12.2.166. [DOI] [Google Scholar]
- 73.Hunter SW, Brennan PJ. 1981. A novel phenolic glycolipid from Mycobacterium leprae possibly involved in immunogenicity and pathogenicity. J Bacteriol 147:728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunter SW, Fujiwara T, Brennan PJ. 1982. Structure and antigenicity of the major specific glycolipid antigen of Mycobacterium leprae. J Biol Chem 257:15072–15078. [PubMed] [Google Scholar]
- 75.Brennan PJ. 1989. Structure of mycobacteria: recent developments in defining cell wall carbohydrates and proteins. Rev Infect Dis 11(Suppl 2):S420–S430 10.1093/clinids/11.Supplement_2.S420. [DOI] [PubMed] [Google Scholar]
- 76.Rastogi N, Frehel C, David HL. 1986. Triple-layered structure of mycobacterial cell wall: evidence for the existence of a polysaccharide-rich outer layer in 18 mycobacterial species. Curr Microbiol 13:237–242 10.1007/BF01568645. [DOI] [Google Scholar]
- 77.Neill MA, Klebanoff SJ. 1988. The effect of phenolic glycolipid-1 from Mycobacterium leprae on the antimicrobial activity of human macrophages. J Exp Med 167:30–42 10.1084/jem.167.1.30. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young DB, Buchanan TM. 1983. A serological test for leprosy with a glycolipid specific for Mycobacterium leprae. Science 221:1057–1059 10.1126/science.6348948. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Schlesinger LS, Horwitz MA. 1991. Phenolic glycolipid-1 of Mycobacterium leprae binds complement component C3 in serum and mediates phagocytosis by human monocytes. J Exp Med 174:1031–1038 10.1084/jem.174.5.1031. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rambukkana A. 2004. Mycobacterium leprae-induced demyelination: a model for early nerve degeneration. Curr Opin Immunol 16:511–518 10.1016/j.coi.2004.05.021. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Ehrig K, Leivo I, Argraves WS, Ruoslahti E, Engvall E. 1990. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci USA 87:3264–3268 10.1073/pnas.87.9.3264. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Talts JF, Timpl R. 1999. Mutation of a basic sequence in the laminin α2LG3 module leads to a lack of proteolytic processing and has different effects on β1 integrin-mediated cell adhesion and α-dystroglycan binding. FEBS Lett 458:319–323 10.1016/S0014-5793(99)01180-1. [DOI] [PubMed] [Google Scholar]
- 83.Talts JF, Mann K, Yamada Y, Timpl R. 1998. Structural analysis and proteolytic processing of recombinant G domain of mouse laminin α2 chain. FEBS Lett 426:71–76 10.1016/S0014-5793(98)00312-3. [DOI] [PubMed] [Google Scholar]
- 84.Bernier SM, Utani A, Sugiyama S, Doi T, Polistina C, Yamada Y. 1995. Cloning and expression of laminin α2 chain (M-chain) in the mouse. Matrix Biol 14:447–455 10.1016/0945-053X(95)90002-0. [DOI] [PubMed] [Google Scholar]
- 85.Sasaki M, Kleinman HK, Huber H, Deutzmann R, Yamada Y. 1988. Laminin, a multidomain protein. The A chain has a unique globular domain and homology with the basement membrane proteoglycan and the laminin B chains. J Biol Chem 263:16536–16544. [PubMed] [Google Scholar]
- 86.Engvall E, Earwicker D, Haaparanta T, Ruoslahti E, Sanes JR. 1990. Distribution and isolation of four laminin variants; tissue restricted distribution of heterotrimers assembled from five different subunits. Cell Regul 1:731–740 10.1091/mbc.1.10.731. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patton BL, Miner JH, Chiu AY, Sanes JR. 1997. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol 139:1507–1521 10.1083/jcb.139.6.1507. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Campbell KP. 1995. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80:675–679 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 89.Rambukkana A, Yamada H, Zanazzi G, Mathus T, Salzer JL, Yurchenco PD, Campbell KP, Fischetti VA. 1998. Role of alpha-dystroglycan as a Schwann cell receptor for Mycobacterium leprae. Science 282:2076–2079 10.1126/science.282.5396.2076. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. 1992. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355:696–702 10.1038/355696a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 91.Yamada H, Denzer AJ, Hori H, Tanaka T, Anderson LVB, Fujita S, Fukuta-Ohi H, Shimizu T, Ruegg MA, Matsumura K. 1996. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J Biol Chem 271:23418–23423 10.1074/jbc.271.38.23418. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Saito F, Masaki T, Kamakura K, Anderson LVB, Fujita S, Fukuta-Ohi H, Sunada Y, Shimizu T, Matsumura K. 1999. Characterization of the transmembrane molecular architecture of the dystroglycan complex in Schwann cells. J Biol Chem 274:8240–8246 10.1074/jbc.274.12.8240. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Nave K-A, Trapp BD. 2008. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci 31:535–561 10.1146/annurev.neuro.30.051606.094309. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Nave K-A, Salzer JL. 2006. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol 16:492–500 10.1016/j.conb.2006.08.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 95.Lemke G. 2006. Neuregulin-1 and myelination. Sci STKE 2006:pe11. [PubMed] [DOI] [PubMed] [Google Scholar]
- 96.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. 1997. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389:725–730 10.1038/39593. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Garratt AN, Britsch S, Birchmeier C. 2000. Neuregulin, a factor with many functions in the life of a Schwann cell. BioEssays 22:987–996 . [DOI] [PubMed] [Google Scholar]
- 98.Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA. 2005. Microanatomy of axon/glial signaling during Wallerian degeneration. J Neurosci 25:3478–3487 10.1523/JNEUROSCI.3766-04.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yarden Y, Sliwkowski MX. 2001. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137 10.1038/35052073. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Hynes NE, Lane HA. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5:341–354 10.1038/nrc1609. [PubMed] [DOI] [PubMed] [Google Scholar]
- 101.Rambukkana A. 2010. Usage of signaling in neurodegeneration and regeneration of peripheral nerves by leprosy bacteria. Prog Neurobiol 91:102–107 10.1016/j.pneurobio.2009.12.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Waxman SG. 1998. Demyelinating diseases—new pathological insights, new therapeutic targets. N Engl J Med 338:323–325. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Hess S, Rambukkana A. 2015. Bacterial-induced cell reprogramming to stem cell-like cells: new premise in host-pathogen interactions. Curr Opin Microbiol 23:179–188 10.1016/j.mib.2014.11.021. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Madigan CA, Cambier CJ, Kelly-Scumpia KM, Scumpia PO, Cheng TY, Zailaa J, Bloom BR, Moody DB, Smale ST, Sagasti A, Modlin RL, Ramakrishnan L. 2017. A macrophage response to Mycobacterium leprae phenolic glycolipid initiates nerve damage in leprosy. Cell 170:973–985.E10 10.1016/j.cell.2017.07.030. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim HA, Mindos T, Parkinson DB. 2013. Plastic fantastic: Schwann cells and repair of the peripheral nervous system. Stem Cells Transl Med 2:553–557 10.5966/sctm.2013-0011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jessen KR, Mirsky R. 2016. The repair Schwann cell and its function in regenerating nerves. J Physiol 594:3521–3531 10.1113/JP270874. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tapinos N, Rambukkana A. 2005. Insights into regulation of human Schwann cell proliferation by Erk1/2 via a MEK-independent and p56Lck-dependent pathway from leprosy bacilli. Proc Natl Acad Sci USA 102:9188–9193 10.1073/pnas.0501196102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim HA, Pomeroy SL, Whoriskey W, Pawlitzky I, Benowitz LI, Sicinski P, Stiles CD, Roberts TM. 2000. A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron 26:405–416 10.1016/S0896-6273(00)81173-3. [DOI] [PubMed] [Google Scholar]
- 109.Stoll G, Jander S, Myers RR. 2002. Degeneration and regeneration of the peripheral nervous system: From Augustus Waller’s observations to neuroinflammation. J Peripher Nerv Syst 7:13–27. [PubMed] [DOI] [PubMed] [Google Scholar]
- 110.Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A. 2013. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 152:51–67 10.1016/j.cell.2012.12.014. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harrisingh MC, Perez-Nadales E, Parkinson DB, Malcolm DS, Mudge AW, Lloyd AC. 2004. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J 23:3061–3071 10.1038/sj.emboj.7600309. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pearson JM, Rees RJ, Weddell AG. 1970. Mycobacterium leprae in the striated muscle of patients with leprosy. Lepr Rev 41:155–166 10.5935/0305-7518.19700023. [PubMed] [DOI] [PubMed] [Google Scholar]
- 113.Werneck LC, Teive HA, Scola RH. 1999. Muscle involvement in leprosy. Study of the anterior tibial muscle in 40 patients. Arq Neuropsiquiatr 57(3B):723–734 10.1590/S0004-282X1999000500001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Masaki T, McGlinchey A, Tomlinson SR, Qu J, Rambukkana A. 2013. Reprogramming diminishes retention of Mycobacterium leprae in Schwann cells and elevates bacterial transfer property to fibroblasts. F1000 Res 2:198 10.12688/f1000research.2-198.v1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Finzsch M, Schreiner S, Kichko T, Reeh P, Tamm ER, Bösl MR, Meijer D, Wegner M. 2010. Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J Cell Biol 189:701–712 10.1083/jcb.200912142. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bremer M, Fröb F, Kichko T, Reeh P, Tamm ER, Suter U, Wegner M. 2011. Sox10 is required for Schwann-cell homeostasis and myelin maintenance in the adult peripheral nerve. Glia 59:1022–1032 10.1002/glia.21173. [PubMed] [DOI] [PubMed] [Google Scholar]
- 117.Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. 2001. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev 15:66–78 10.1101/gad.186601. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mirsky R, Woodhoo A, Parkinson DB, Arthur-Farraj P, Bhaskaran A, Jessen KR. 2008. Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J Peripher Nerv Syst 13:122–135. [PubMed] [DOI] [PubMed] [Google Scholar]
- 119.Fröb F, Bremer M, Finzsch M, Kichko T, Reeh P, Tamm ER, Charnay P, Wegner M. 2012. Establishment of myelinating Schwann cells and barrier integrity between central and peripheral nervous systems depend on Sox10. Glia 60:806–819 10.1002/glia.22310. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Le N, Nagarajan R, Wang JYT, Araki T, Schmidt RE, Milbrandt J. 2005. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci USA 102:2596–2601 10.1073/pnas.0407836102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jessen KR, Mirsky R. 2008. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia 56:1552–1565 10.1002/glia.20761. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Roberts SL, Dun XP, Doddrell RDS, Mindos T, Drake LK, Onaitis MW, Florio F, Quattrini A, Lloyd AC, D’Antonio M, Parkinson DB. 2017. Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 144:3114–3125 10.1242/dev.150656. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chambers I, Tomlinson SR. 2009. The transcriptional foundation of pluripotency. Development 136:2311–2322 10.1242/dev.024398. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH. 2006. A protein interaction network for pluripotency of embryonic stem cells. Nature 444:364–368 10.1038/nature05284. [PubMed] [DOI] [PubMed] [Google Scholar]
- 125.Miyagi S, Masui S, Niwa H, Saito T, Shimazaki T, Okano H, Nishimoto M, Muramatsu M, Iwama A, Okuda A. 2008. Consequence of the loss of Sox2 in the developing brain of the mouse. FEBS Lett 582:2811–2815 10.1016/j.febslet.2008.07.011. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Favaro R, Valotta M, Ferri ALM, Latorre E, Mariani J, Giachino C, Lancini C, Tosetti V, Ottolenghi S, Taylor V, Nicolis SK. 2009. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci 12:1248–1256 10.1038/nn.2397. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Komitova M, Eriksson PS. 2004. Sox-2 is expressed by neural progenitors and astroglia in the adult rat brain. Neurosci Lett 369:24–27 10.1016/j.neulet.2004.07.035. [PubMed] [DOI] [PubMed] [Google Scholar]
- 128.Cavallaro M, Mariani J, Lancini C, Latorre E, Caccia R, Gullo F, Valotta M, DeBiasi S, Spinardi L, Ronchi A, Wanke E, Brunelli S, Favaro R, Ottolenghi S, Nicolis SK. 2008. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development 135:541–557 10.1242/dev.010801. [PubMed] [DOI] [PubMed] [Google Scholar]
- 129.Gómez-López S, Wiskow O, Favaro R, Nicolis SK, Price DJ, Pollard SM, Smith A. 2011. Sox2 and Pax6 maintain the proliferative and developmental potential of gliogenic neural stem cells in vitro. Glia 59:1588–1599 10.1002/glia.21201. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.Cimadamore F, Fishwick K, Giusto E, Gnedeva K, Cattarossi G, Miller A, Pluchino S, Brill LM, Bronner-Fraser M, Terskikh AV. 2011. Human ESC-derived neural crest model reveals a key role for SOX2 in sensory neurogenesis. Cell Stem Cell 8:538–551 10.1016/j.stem.2011.03.011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. 2006. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev 20:1187–1202 10.1101/gad.1407906. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Surzenko N, Crowl T, Bachleda A, Langer L, Pevny L. 2013. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Muller glia. Development 140:1445–1456 10.1242/dev.071878. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jaenisch R, Young R. 2008. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132:567–582 10.1016/j.cell.2008.01.015. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim J, Chu J, Shen X, Wang J, Orkin SH. 2008. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132:1049–1061 10.1016/j.cell.2008.02.039. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Young RA. 2011. Control of the embryonic stem cell state. Cell 144:940–954 10.1016/j.cell.2011.01.032. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.De Los Angeles A, Ferrari F, Xi R, Fujiwara Y, Benvenisty N, Deng H, Hochedlinger K, Jaenisch R, Lee S, Leitch HG, Lensch MW, Lujan E, Pei D, Rossant J, Wernig M, Park PJ, Daley GQ. 2015. Hallmarks of pluripotency. Nature 525:469–478. CORRIGENDUM Nature 531:400 10.1038/nature15515. [PubMed] [DOI] [PubMed] [Google Scholar]
- 137.Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, Boyer LA. 2013. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9:e1003288 10.1371/journal.pgen.1003288. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. 2003. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17:126–140 10.1101/gad.224503. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K. 2011. Sox2+ adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9:317–329 10.1016/j.stem.2011.09.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, Pevny L. 2004. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci 26:148–165 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 141.Lim J, Thiery JP. 2012. Epithelial-mesenchymal transitions: insights from development. Development 139:3471–3486 10.1242/dev.071209. [PubMed] [DOI] [PubMed] [Google Scholar]
- 142.Masaki T, McGlinchey A, Cholewa-Waclaw J, Qu J, Tomlinson SR, Rambukkana A. 2014. Innate immune response precedes Mycobacterium leprae-induced reprogramming of adult Schwann cells. Cell Reprogram 16:9–17 10.1089/cell.2013.0064. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Modlin RL, Rea TH. 1988. Immunopathology of leprosy granulomas. Springer Semin Immunopathol 10:359–374 10.1007/BF02053846. [PubMed] [DOI] [PubMed] [Google Scholar]
- 144.Chan J, Flynn J. 2004. The immunological aspects of latency in tuberculosis. Clin Immunol 110:2–12 10.1016/S1521-6616(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 145.Bold TD, Ernst JD. 2009. Who benefits from granulomas, mycobacteria or host? Cell 136:17–19 10.1016/j.cell.2008.12.032. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qiu B, Frait KA, Reich F, Komuniecki E, Chensue SW. 2001. Chemokine expression dynamics in mycobacterial (type-1) and schistosomal (type-2) antigen-elicited pulmonary granuloma formation. Am J Pathol 158:1503–1515 10.1016/S0002-9440(10)64101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chiu B-C, Freeman CM, Stolberg VR, Hu JS, Komuniecki E, Chensue SW. 2004. The innate pulmonary granuloma: characterization and demonstration of dendritic cell recruitment and function. Am J Pathol 164:1021–1030 10.1016/S0002-9440(10)63189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]