

Abstract
Astrocytic differentiation is developmentally impaired in patients with childhood-onset schizophrenia (SCZ). To determine why, we used genetic gain- and loss-of-function studies to establish the contributions of differentially-expressed transcriptional regulators to the defective differentiation of glial progenitor cells (GPCs) produced from SCZ patient-derived iPSCs. Negative regulators of the BMP pathway were upregulated in SCZ GPCs, including BAMBI, FST and GREM1, whose over-expression retained SCZ GPCs at the progenitor stage. SMAD4 knock-down suppressed the production of these BMP inhibitors by SCZ GPCs, and rescued normal astrocytic differentiation. In addition, the BMP-regulated transcriptional repressor REST was upregulated in SCZ GPCs, and its knock-down similarly restored normal glial differentiation. REST knockdown also rescued potassium-transport associated gene expression and K+ uptake, which were otherwise deficient in SCZ glia. These data suggest that the glial differentiation defect in childhood-onset SCZ, and its attendant disruption in K+ homeostasis, may be rescued by targeting BMP/SMAD4- and REST-dependent transcription.
INTRODUCTION
Schizophrenia (SCZ) affects roughly 1% of the population worldwide, and has been the focus of increasingly fruitful efforts to understand its genetic foundations; nonetheless, its cellular basis remains poorly understood (Allen et al., 2008; Sawa and Snyder, 2002). Yet recent reports, including both genome-wide association and differential expression studies, have linked a number of candidate loci as well as specific differentially-expressed genes to schizophrenic pathogenesis (Fromer et al., 2016; Marshall et al., 2017; Schizophrenia Working Group of the Psychiatric Genomics, 2014). Over the past decade in particular, it has become clear that a number of these schizophrenia-associated genes are involved in the development and physiology of glial cells (Takahashi and Sakurai, 2013; Takahashi et al., 2011a; Takahashi et al., 2011b; Yin et al., 2012). Accordingly, both astrocytic and oligodendrocytic dysfunction has been implicated in the etiology of SCZ (Aberg et al., 2006; Fields, 2008; Goudriaan et al., 2014; Tkachev et al., 2003; Voineskos et al., 2013; Wang et al., 2015). Astrocytes in particular have essential roles in both the structural development of neural networks as well as the coordination of neural circuit activity, the latter through their release of glial transmitters, maintenance of synaptic density, and regulation of synaptic potassium and neurotransmitter levels (Christopherson et al., 2005; Chung et al., 2013; Rangroo Thrane et al., 2013). As such, defective astrocytic maturation may be an important contributor to the neural circuit disruption characteristic of schizophrenia (Singh et al., 2016; Verkhratsky and Parpura, 2015; Xia et al., 2014).
To investigate the role of glial pathology in schizophrenia as well as other neuropsychiatric disorders, we first established a protocol for generating human glial progenitor cells (hGPCs) from patient-derived induced pluripotent cells (iPSCs) (Wang et al., 2013). This protocol permits us to generate hGPCs and their derived astrocytes and oligodendrocytes from patients with schizophrenia, in a manner that preserves their genetic integrity and functional repertoires. As such, this method has provided us a means by which to assess the differentiation, gene expression and functional competence of astrocytes derived from patients with schizophrenia, both in vitro and in vivo, the latter after engraftment into immunodeficient mice (Windrem et al., 2017). We found that such glial chimeric mice, when neonatally colonized with iPSC-derived hGPCs produced from patients with childhood-onset schizophrenia, exhibited profound abnormalities in both astrocytic differentiation and structure; these were associated with significant behavioral abnormalities in the engrafted mice, relative to controls implanted with normal human hGPCs. Importantly, RNA sequence analysis revealed that the maturational defects in these SCZ hGPCs were associated with the down-regulation of a core set of differentiation-associated genes, whose transcriptional targets included a host of transporters, channels and synaptic modulators that were themselves deficient in SCZ glia (Windrem et al., 2017).
In the present study, we sought to capitalize upon these findings by identifying targetable signaling nodes at which schizophrenia-associated glial pathology might be moderated. To that end, we generated iPSC hGPCs from patients with childhood-onset SCZ, or from normal control (CTR) subjects, and then compared both the patterns of gene expression and extent of astrocytic functional differentiation by these cells. We found that excessive bone morphogenetic protein (BMP)-dependent signaling plays a critical role in the disrupted astroglial differentiation of SCZ hGPCs, that BMP’s actions in this context are signaled through SMAD4, and that aspects of normal phenotype may be restored to SCZ glia by SMAD4 knock-down. Since the preservation of K+ homeostasis is a critical element of astrocytic function, and our RNA-seq data indicated the down-regulation of a number of potassium channels, pumps and transporters (hereafter referred to simply as potassium transporters) by SCZ glia, we then assessed the uptake of K+ by SCZ astrocytes. We found that these cells indeed manifested significantly impaired K+ uptake relative to control astrocytes, and moreover, that aberrant expression of the REST repressor was responsible for their diminished transcription of the potassium transporter genes. Together, these studies highlight the dysregulation of glial BMP/SMAD4- and REST-dependent transcription as important contributors to the pathogenesis of schizophrenia, and identify these as intriguing new targets for rescuing both glial phenotype and function in this disorder.
RESULTS
Astrocytic differentiation was impaired in SCZ GPCs
iPSCs were produced from skin samples obtained from patients with childhood-onset schizophrenia, as well as healthy young adult controls free of known mental illness, as previously described (Windrem et al., 2017). Patient identifiers were not available to investigators besides the treating psychiatrist, although age, gender, race, diagnosis and medication history accompanied cell line identifiers. Briefly, fibroblasts were isolated from each sample; from these, 8 hiPSC lines were derived from patient samples and normal controls (5 juvenile-onset schizophrenia patients and 3 healthy gender-matched and age-analogous controls (Supplemental Table S1). iPSCs were generated using excisable floxed polycistronic hSTEMCCA lentivirus (Somers et al., 2010; Zou et al., 2012) encoding Oct4, Sox2, Klf4 and c-Myc (Takahashi et al., 2007; Welstead et al., 2008). A fourth hiPSC control line, C27 (Chambers et al., 2009), was also used, to ensure that all genomic and phenotypic data were consistent with prior work in our lab (Wang et al., 2013). All lines were validated as pluripotent using RNA sequencing and immunolabeling to assess pluripotent gene expression. The identity of each iPSC line was confirmed to match the parental donor fibroblasts using short tandem repeat (STR)-based DNA fingerprinting, and each line was karyotyped and arrayed for comparative genomic hybridization to confirm genomic integrity. In addition, these iPSC lines were arrayed for genome-wide methylation to compare their methylation state.
We first compared the glial differentiation efficiency of cells derived from SCZ patients and control subjects (n=4 lines from 4 different patients, each with ≥ 3 repeats/patient, each versus paired control), by instructing these iPSC cells to GPC fate as previously described (Wang et al., 2013), and assessing their expression of stage-specific markers of maturation as a function of time. We found that all tested iPSCs exhibited typical colonies, and expressed markers of pluripotency by flow cytometry, including SSEA4 (Figure S1A). At the neural progenitor cell (NPC) stage, both ICC and flow cytometry revealed that the expression levels of the stage-selective markers paired box protein pax-6 (PAX6), sex determining region Y-box 1 (SOX1) and the cell surface marker prominin-1/CD133, were no different between CTR- and SCZ-derived lines (Figures 1A–D; Figures S1B). At the GPC stage, their expression of the GPC-selective platelet-derived growth factor receptor alpha (PDGFRα/CD140a) (Sim et al., 2011) was then assessed, which revealed that the efficiencies of GPC generation did not differ significantly between SCZ- and CTR-derived NPCs (Figures 1E–G; Figures S1C). At the astrocytic progenitor stage, the flow cytometry confirmed that the expression levels of cell surface marker CD44 was no different between CTR- and SCZ-derived lines (Figure S1D). Thus, no differences in the differentiation of SCZ and CTR iPSCs were noted through the GPC and astrocytic progenitor stages.
Figure 1. Astrocytic differentiation was impaired in SCZ GPCs.

A–D, At the neural progenitor cell (NPC) stage, both SCZ and CTR (4 distinct patients and derived lines each, n ≥ 3/each line) hNPCs highly expressed both SOX1 and PAX6. E–G, Similarly, the efficiency of PDGFRA/CD140a-defined hGPC generation did not differ between SCZ and CTR lines (4 different patient-specific lines each, n ≥ 3/each line). H–J, In contrast, the proportion of GFAP+ astrocytes was significantly higher in CTR lines (4 CTR lines, n ≥ 3/each line [70.1 ± 2.4%]) vs. SCZ lines (4 SCZ lines, n ≥ 3/each line, [39.9 ± 2.0%]).
Scale: 50 μm; ***p<0.001 by two tailed t-test; NS: not significant; mean ± SEM.
At that point, the SCZ- and CTR-derived GPCs were further differentiated into astrocytes, by incubating in M41 medium supplemented with 20 ng/ml BMP4 for 4 weeks. Immunolabeling revealed that the proportion of GFAP+ astrocytes was significantly higher in control lines (4 CTR lines, n ≥ 3 per line, mean of 4 CTR lines = 70.1 ± 2.4%) than in SCZ lines (4 SCZ lines, n ≥ 3 per line, mean of 4 SCZ lines = 39.9 ± 2.0%; p<0.001, 2-tailed t-test) (Figures 1H–J). In addition to GFAP, the percentage of S100β+ astrocytes was also significantly higher in CTR lines relative to SCZ lines (Figure S1F). In contrast, the proportion of PDGFαR+ GPCs was significantly higher in BMP4-treated SCZ glia (4 SCZ lines, n ≥ 3 per line) relative to BMP4-treated CTR glia (4 CTR lines, n ≥ 3 per line) (Figure S1E). This defect of astrocytic differentiation was consistently observed in all SCZ GPCs relative to CTR cells, and comprised an in vitro correlate to previously described astroglial differentiation defects in vivo (Windrem et al., 2017).
SCZ GPCs upregulated expression of BAMBI, an inhibitor of BMP signaling
To identify the molecular concomitants to the defective astrocytic differentiation of SCZ GPCs, we had earlier performed RNA-seq on FACS-sorted CD140a+ GPCs from 3 different CTR- and 4 SCZ-derived lines at time points ranging from 154 to 242 days in vitro (Windrem et al., 2017). mRNA was isolated from these cells with polyA-selection for RNA sequencing on an Illumina HiSeq 2500 platform for approximately 45 million 1×100 bp reads per sample. The original counts were analyzed to determine disease-dysregulated genes at 5% FDR and log2 fold change > 1. By that means, we had identified 118 mRNAs that were consistently and significantly differentially expressed by CD140a-sorted SCZ hGPCs relative to their control iPSC hGPCs (Windrem et al., 2017). Among these, a number of genes involved in glial lineage progression were downregulated in SCZ hGPCs, relative to their normal controls, suggesting that astroglial differentiation was impaired in SCZ in a cell-autonomous fashion, due to intrinsic defects in SCZ-derived glial progenitor cells.
Capitalizing upon these earlier data, in this study we first used Ingenuity Pathway Analysis (IPA) to identify pathways that were significantly differentially regulated in SCZ hGPCs. We found that among these, BMP signaling-related transcripts were upregulated in SCZ hGPCs, compared to CTR hGPCs (Figure S2A). qPCR then validated that the expression of a number of TGFβ pathway regulators, including BAMBI, was indeed significantly elevated in SCZ GPCs (Figure S2B). In contrast, these BMP signaling-related transcripts did not differ between SCZ and CTR lines at the NPC stage (Figure S2C). Moreover, the methylation states of CTR- and SCZ-derived iPSCs were similar (Figure S2D); the little variability noted across lines in iPSC methylation state appeared due to sex and line, but not to disease state or subject age (Figure S2E). Thus, the upregulation of BAMBI and other TGFβ and BMP pathway regulators that we noted in SCZ hGPCs was not due to any systematic, disease-dependent difference in methylation pattern between CTR and SCZ cells at the pluripotent stem cell stage.
BMP4 is a strong stimulus for astrocytic differentiation by human GPCs, and BAMBI is a strong antagonist to BMP4-induced glial induction, acting as a pseudo-receptor and hence dominant-negative inhibitor of BMP signaling (Sim et al., 2006). Yet BAMBI expression may be activated by TGFβ and BMP receptor-dependent signaling, as a compensatory negative feedback response (Onichtchouk et al., 1999). Accordingly, our RNA-seq, qPCR, data revealed that both BMP signaling-dependent transcripts and BAMBI were upregulated in SCZ hGPCs, but not in SCZ hNPCs (Figure S2B–C). These data suggest that the upregulation of BMP signaling was specific to SCZ glia and first appeared at the glial progenitor stage, and that this process was associated with the upregulated expression of BAMBI, which in turn suppressed the astrocytic differentiation of SCZ hGPCs.
On that basis, we asked whether BAMBI over-expression in normal control subject-derived hGPCs might mimic or reproduce the SCZ GPC phenotype, by suppressing the differentiation of these hGPCs. To that end, we genetically modulated the expression of BAMBI in hGPCs, both SCZ and CTR-derived GPCs (Figure S3). We found that overexpression of BAMBI in CTR GPCs significantly decreased their efficiency of astrocytic transition (4 CTR lines with 3 repeats/each line, means of 4 CTR lines/36.4% ± 4.3%), yielding cells that resembled SCZ hGPCs in their refractoriness to terminal astrocytic maturation (4 SCZ lines with 3 repeats/each line, means of 4 SCZ lines/45.5% ± 3.6%; p=0.12 by two-tailed t test) (Figures 2A–B). However, BAMBI knockdown in SCZ GPCs did not rescue astrocytic differentiation in the latter, suggesting that BAMBI overexpression contributed to the resistance of SCZ hGPCs to maturation, but was not sufficient in this regard (Figure 2A–B). Accordingly, when we used qPCR to assess the expression of alternative inhibitors of BMP signaling, we found that the mRNAs encoding both follistatin (FST) and gremlin1 (GREM1), two potent antagonists of BMPs and BMP-dependent signaling, were both significantly upregulated by SCZ GPCs (SCZ vs CTR; 4 SCZ and 4 CTR lines, 3 repeats/each line; ddCt of FST=2.45 ± 0.39, p<0.05; GREM1=3.38 ± 0.53, p<0.01; two-tailed t test) (Figure 2C).
Figure 2. BAMBI expression in normal hGPCs phenocopied the glial differentiation defect of SCZ.

A–B, Overexpression of the membrane-bound BMP antagonist BAMBI in CTR hGPCs (4 CTR lines, 3 repeats/each line) significantly decreased their efficiency of astrocytic transition. B, However, BAMBI knockdown in SCZ hGPCs (4 SCZ lines, 3 repeats/each line) was not sufficient to restore astrocytic differentiation. C, Besides BAMBI, the BMP antagonists follistatin (FST) and gremlin1 (GREM1) were also upregulated in SCZ hGPCs, relative to controls. Scale: 50 μm; ***p<0.001, 1-way ANOVA for B; **P<0.001 by 2-tailed t-test for C; NS: not significant; mean ± SEM.
Astrocytic differentiation by SCZ GPCs may be rescued by SMAD4 knockdown
SMAD4 is necessary for canonical BMP signaling, in that it acts as a common effector for multiple upstream signals, in response to which it translocates to the nucleus, where it activates both BMP and TGFB-regulated genes (Herhaus and Sapkota, 2014). These include BAMBI as well as FST and GREM1, all acting in concert as negative feedback regulators of pro-gliogenic BMP signals (Brazil et al., 2015; Onichtchouk et al., 1999) (Figure 3A). On that basis, we posited that SMAD4 knockdown in hGPCs, by inhibiting the early expression of BAMBI, FST and GREM1, might potentiate astrocytic differentiation from hGPCs. Furthermore, to the extent that the differentiation block in SCZ hGPCs was due to the SMAD4-mediated over-expression of endogenous BMP inhibitors, we postulated that SMAD4 knock-down would therefore differentially potentiate astroglial differentiation by SCZ hGPCs. To test this possibility, we used doxycycline (DOX) induction of SMAD4 shRNAi to conditionally knock-down SMAD4 expression in both SCZ and CTR hGPCs, and then assessed their expression of BMP-regulated genes by qPCR (Figures S4A–C). We found that SMAD4 knockdown indeed repressed the expression of BMP signaling-dependent genes, including BAMBI, FST, and GREM1 (SCZ-LV-Scrambled vs SCZ-LV-SMAD4-shRNA; 4 different patient iPSC lines/group, 3 repeats/line; ddCt of BAMBI: 2.56±0.35, p<0.05; FST: 2.38±0.24, p<0.01; GREM1: 3.04±0.45, p<0.05; all comparisons by ANOVA with post hoc t tests) (Figure 3B). Importantly, transient DOX-induced SMAD4 knockdown, in which shRNAi expression was limited to the progenitor stage, robustly promoted the astrocytic differentiation of the SCZ GPCs, overcoming their relative block in glial differentiation to effectively rescue astrocytic phenotype (Figures 3C–D). In particular, SMAD4 knockdown (KD) in SCZ GPCs restored their efficiency of GFAP-defined astrocytic differentiation to that of CTR GPCs (SCZ-SMAD4-shRNA at the GPC stage: 56.8% ± 3.8%; CTR lines: 62.2% ± 4.0%; p>0.05, one-way ANOVA; means ± SEs of 4 distinct patient lines/group, n ≥3 replicates/line) (Figures 3C–D). In contrast, continuous SMAD4 knock-down after astrocytic induction, as mediated via continuous DOX exposure (as outlined in Figure S4B), caused a diminution of GFAP-defined astrocytes in both SCZ and CTR groups (Figures 3C–D). Thus, maintenance of mature astrocytic phenotype appeared to require ongoing SMAD4 signaling, in SCZ and CTR astrocytes alike.
Figure 3. SMAD4 regulated the astrocytic differentiation of SCZ GPCs.

A, SMAD4 regulates the expression of TGFβ and BMP pathway signaling through: 1) phosphorylation of both SMAD2/3 and SMAD1/5/8; 2) SMAD nuclear translocation and activation of target promoters, including the early induction of the endogenous BMP inhibitors BAMBI, follistatin (FST) and gremlin1 (GREM1); and 3) their subsequent feedback inhibition of BMP signals. B, BAMBI, FST and GREM1 were all significantly over-expressed in SCZ CD140a-sorted hGPCs relative to control-derived hGPCs. SMAD4 knockdown in SCZ hGPCs (4 SCZ lines, 3 repeats/line) then repressed the expression of BAMBI, FST and GREM1 to control levels. C, SMAD4 knockdown in SCZ hGPCs restored astrocytic differentiation to that of CTR hGPCs (4 SCZ lines, 3 repeats/each line). DOX (−)/(+) means short-term/long-term culture with DOX. D, SMAD4 knock-down after astrocytic induction, as mediated via continuous doxycycline exposure, caused a loss of GFAP-defined astrocytes in both SCZ and CTR groups. DOX (−)/(+) means short-term/long-term culture with DOX. Scale: 50 μm; *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA; NS: not significant; mean ± SEM.
Together, these data indicate that aberrant BMP signaling in SCZ GPCs, by driving the excessive expression of inhibitors of BMP signaling, suppresses astrocytic differentiation, and that this differentiation defect can be rescued by SMAD4 knock-down. Nonetheless, once SCZ GPCs have progressed to astrocytic differentiation, SMAD4 expression is then required for maintenance of the astrocytic phenotype in CTR and SCZ astrocytes alike, consistent with its previously described function as the effector of BMP-mediated astrocytic maturation (Kohyama et al., 2010). These data indicate that pathological BMP-dependent signaling in SCZ GPs may delimit their astrocytic maturation, and suggest that this cellular pathology may arise in part from the SMAD4-dependent over-expression of endogenous inhibitors of pro-gliogenic BMP signaling by GPCs.
SCZ astrocytes exhibit reduced potassium uptake
Together with the impaired astrocytic differentiation of SCZ GPCs, our RNA-seq data suggested that those astrocytes that do successfully differentiate might nonetheless be functionally impaired. In particular, the RNA-seq revealed the downregulated transcription in SCZ GPCs of a broad set of potassium channel (KCN)-encoding genes, including the Na+-K+ ATPase, Na+-K+/2Cl− cotransporter (NKCC), and Kir-family inwardly rectifying potassium channels (Figure 4A) (Windrem et al., 2017), all of which play important roles in potassium uptake by astrocytes (Larsen et al., 2014; Macaulay and Zeuthen, 2012) (Figure 5A). Among these dysregulated KCN genes, ATP1A2, SLC12A6, and KCNJ9, which respectively encode the Na+/K+-ATPase pump, NKCC1 Na+/K+/2Cl− cotransporter, and the Kir3.3 voltage-gated K+ channel (Bottger et al., 2016; Gamba and Friedman, 2009; Lesage et al., 1995), were substantially down-regulated in SCZ lines, compared to the 4 control lines; these findings suggested a broad-based impairment of K+ uptake by SCZ glia.
Figure 4. REST represses KCN-associated gene expression in SCZ hGPCs.

A, Differentially expressed potassium channel genes in SCZ-derived hGPC lines. Each SCZ-derived hGPC line was individually compared against three pooled CTR-derived hGPC lines (FDR 5%, FC > 2.00 [if applicable]). Genes shown were found differentially expressed in at least three out of four assessed SCZ-derived hGPC lines. B, qPCR confirmed that potassium channel-associated genes including ATP1A2, SLC12A6, and KCNJ9 were all significantly downregulated in SCZ hGPCs (4 SCZ lines, 3 repeats/each line), relative to CTR cells (4 CTR lines, 3 repeats/each line). C, Biobase-Transfac revealed that the majority of downregulated potassium channel-associated genes in SCZ hGPCs from our RNA-seq data were targets of the REST repressor.
**p<0.01 by two tailed t-test; mean ± SEM.
Figure 5. Potassium uptake was decreased by SCZ astrocytes.

A, The Na+/K+-ATPase pump, NKCC1 Na+/K+/2Cl− cotransporter, and inwardly rectifying K+ channels are all involved in the regulation of potassium uptake by astrocytes. B, qPCR confirmed that several K+ channel-associated genes were down-regulated in SCZ CD44+ astrocyte-biased GPCs relative to CTR cells. C, SCZ and CTR CD44+ GPCs were cultured in FBS with BMP4 to produce mature GFAP+ astrocytes, which were then assessed for K+ uptake; the results were normalized to both total protein and cell number. K+ uptake by SCZ astrocytes was significantly reduced (4 SCZ lines, 5 repeats/each line), compared to K+ uptake by CTR astrocytes (4 CTR lines, 5 repeats/each line). D-E, Astrocytes were treated with ouabain, bumetanide, and tertiapin to assess which potassium transporter classes were functionally impaired in SCZ astrocytes relative to control (4 lines of each, 4 repeats/line). Both ouabain and bumetanide efficiently decreased K+ uptake by CTR astrocytes (D, gray bars), whereas neither affected K+ uptake by SCZ astrocytes (E, purple bars).
*P<0.05, **P<0.01, ***P<0.001 by two tailed t-test for B and C; ***P<0.001 by one-way ANOVA for D; NS: not significant; mean ± SEM.
On the basis of these genomic data, we next asked whether K+ uptake was actually impaired in SCZ astrocytes. To address this hypothesis, we first used qPCR to confirm whether these K+ channel-associated genes were dysregulated in SCZ glia. They were indeed significantly down-regulated, thus validating the RNA-seq analysis (Figures 4B and 5B). We next assessed functional K+ uptake directly, in cultured SCZ- and CTR-derived astrocytes. To obtain mature SCZ and CTR astrocyte cultures, CD44-sorted glial progenitors were cultured in base media supplemented with 10% fetal bovine serum (FBS) and 20 ng/ml BMP4 for 4 weeks, so as to potentiate the differentiation of mature, glial fibrillary acidic protein (GFAP)-expressing, fiber-bearing astrocytes (Figure S5). Under these highly astrogliogenic conditions, and using cells already sorted for the early astrocytic marker CD44, astrocytic maturation was achieved by both SCZ- as well as CTR-derived progenitors (Figure S5). Astrocytes from 4 different SCZ and 4 different CTR lines were then incubated with 86Rb, a surrogate monovalent cation for K+ uptake (Larsen et al., 2014), and rubidium uptake measured as a function of both cell number and total protein. We found the K+ uptake in SCZ glia (4 SCZ cell lines, 5 repeats/each line) was sharply decreased relative to CTR glia (4 CTR cell lines, 5 repeats/each line), normalized by both cell number and total protein (Figure 5C; P<0.001 by two tailed t-test).
Since genes encoding different potassium Na+/K+-ATPase pumps, transporters, and inwardly rectifying channels were dysregulated in SCZ glia, the drugs ouabain, bumetanide, and tertiapin were used to respectively block these three K+ uptake mechanisms. We first tested different concentrations of each drug to determine its optimal dose range for modulating human astroglial K+ uptake. We found that ouabain and bumetanide, respectively targeting the Na+/K+-ATPase pump and NKCC1-encoded Na+/K+/2Cl− cotransporter, significantly inhibited K+ uptake in CTR glia, while tertiapin, which targets Kir channels, did not (Figures 5D–E). In marked contrast, neither ouabain nor bumetanide affected K+ uptake by SCZ astrocytes (Figures 5D–E). This suggests that the functional decrement in K+ uptake by SCZ-derived astrocytes may be primarily due to down-regulated Na+/K+-ATPase and Na+/K+/2Cl− cotransporter function, rendering these cells refractory to ouabain and bumetanide treatment.
REST regulates potassium uptake by SCZ astrocytes
Since a large number of potassium channel-encoding genes are dysregulated in SCZ glia, it is difficult to modulate glial K+ uptake through genetic means targeting individual potassium channels alone. To address this issue, we used Biobase-Transfac analysis, which was developed to identify regulatory regions common to different genes, as a means of defining their shared upstream regulators (Hu et al., 2014). By this means, we attempted to pinpoint shared regulatory elements within 1 kb of the transcription start sites (TSS) of the SCZ-associated glial genes in our data sets; our intent was to identify upstream transcription factors able to modulate these genes as a group. Using a 13 nucleotide consensus sequence (CCNNGGTGCTGAA), we determined that the majority of all down-regulated potassium channel genes were targets of the neuron restrictive silencing factor (NRSF) REST (Figure 4C), a potent transcriptional repressor that acts in neural cells to repress non-neural gene expression (Hirabayashi and Gotoh, 2010). REST levels are relatively high in rodent oligodendrocyte progenitor cells and rise with terminal oligodendroglial maturation (Dewald et al., 2011); similarly, REST levels rise with astrocytic differentiation from uncommitted neural progenitor cells (Kohyama et al., 2010). Importantly, REST transcription may be positively modulated by SMAD1/5/8 through BMP signaling (Kohyama et al., 2010), such that the pathologically up-regulated BMP signaling we observed in SCZ hGPCs might be expected to drive similarly high levels of REST. On that basis, we queried our RNA-seq data, which confirmed that REST was indeed significantly upregulated in SCZ GPCs relative to CTR GPCs (Windrem et al., 2017). We then used qPCR to confirm that the upregulation of REST expression by SCZ glia was consistent across all of the patients in our series (Figure 6A). Interestingly, while REST levels have been previously linked to terminal glial differentiation (Kohyama et al., 2010), the SCZ hGPCs we evaluated were decidedly refractory to glial differentiation, whether as astrocytes or oligodendrocytes, and this observation was consistent across all patient samples assessed. Thus, the disease context informed the cellular response to REST activity.
Figure 6. REST knock-down restored potassium uptake by SCZ astrocytes.
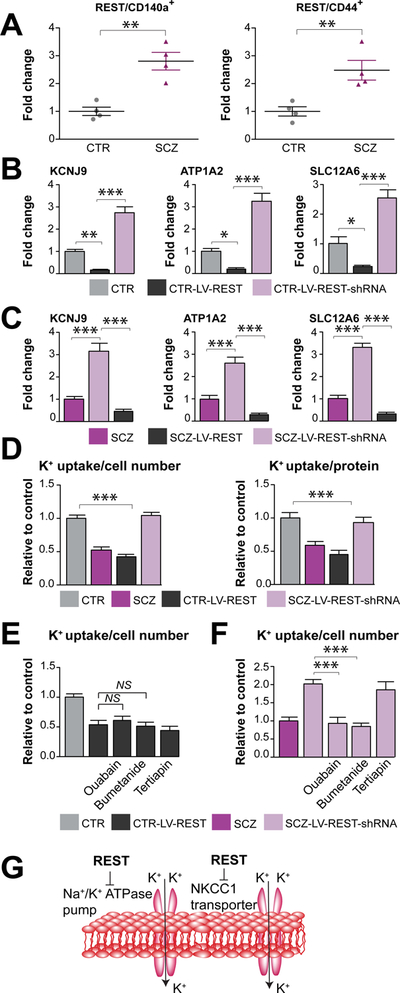
A, qPCR confirmed that REST was upregulated in both CD140a-sorted SCZ hGPCs relative to their controls, and in CD44-sorted SCZ astrocytic progenitor cells relative to CTR cells. B–C, The expression of several K+ transport-associated genes, including SLC12A6, KCNJ9, and ATP1A2, was significantly repressed in CTR and SCZ glia in which REST was overexpressed via lentiviral-REST transduction (4 CTR and SCZ lines, 3 repeats/each line). In contrast, their expression was robustly upregulated in CTR and SCZ lines in which REST was knocked down via lentiviral-REST shRNAi transduction. D, K+ uptake by REST-transduced CTR astrocytes fell, mimicking that SCZ glia, whereas K+ uptake was rescued in SCZ lines subjected to REST knockdown. E–F, Both ouabain and bumetanide significantly reduced K+ uptake by SCZ glia with REST knockdown (4 SCZ lines, 3 repeats/each line). In contrast, they failed to reduce K+ uptake by CTR glia with REST overexpression (4 CTR lines, 3 repeats/each line). G, These data indicate that upregulated REST in SCZ glia may decrease K+ uptake in part by suppressing the expression and function of the Na+/K+-ATPase pump and NKCC1 transporter.
**P<0.01 by two tailed t-test for A; *P<0.05, **P<0.01, ***P<0.001 by one-way ANOVA for B–F; NS: not significant; mean ± SEM.
On the basis of these data, we postulated that the upregulation of REST in schizophrenia-derived glia, and their attendant epigenetic modification, might be sufficient to repress potassium transporter and channel-associated gene expression. To test this hypothesis, we used lentivirus to overexpress REST in CTR glial cells, and assessed K+ uptake by the transduced cells. In parallel, we knocked down REST expression in SCZ glia through lentiviral shRNAi transduction, and then similarly assessed K+ uptake by these cells. qPCR validation confirmed that REST was significantly modulated as intended in both CTR and SCZ glial cells (Figure S6A). By this means, we found that in both CTR and SCZ glia subjected to REST overexpression, that the expression of a number of K+ transport genes, including SLC12A6, KCNJ9, and ATP1A2, was indeed significantly repressed (Figures 6B–C). In contrast, the expression of these genes was strongly upregulated in both CTR and SCZ cells subjected to REST knock-down (Figures 6B–C). In addition, we next found that BMP4 teatment of CTR GPCs significantly upregulated REST expression and downregulated K+ transport gene expression, relative to untreated cells, which was restored with BMP signaling inhibitor DMH1 (Figure S6B), and that lentiviral-SMAD4-shRNAi transduction of SCZ GPCs robustly repressed REST expression and enhanced K+ transport gene expression (Figure S6C).
Importantly, we noted that REST-overexpressing CTR astrocytes mimicked the functional potassium dysregulation of SCZ glia, in that their K+ uptake was significantly reduced compared to that of untreated CTR glia (Figure 6D). Neither ouabain nor bumetanide further decreased K+ uptake by these cells, suggesting that their targeted pumps and transporters were down-regulated to the point of functional irrelevance (Figures 6E). Conversely, K+ uptake by SCZ astrocytes subjected to REST knock-down was strongly increased, to levels no different from those of CTR astrocytes (Figure 6D); both ouabain and bumetanide were then able to significantly reduce K+ uptake by these REST shRNAi-transduced SCZ astrocytes (Figures 6F). Together, these data suggest that REST knock-down in SCZ glia promoted the restoration of normal K+ uptake by these cells, by rescuing their expression of both Na+/K+-ATPase and the NKCC1 Na+/K+/2Cl− cotransporter, thereby restoring a salient feature of astroglial potassium homeostasis (Figure 6G). As such, these observations indicate that REST operates to suppress K+ uptake by SCZ glia by broadly suppressing the expression and hence function of multiple mechanisms of glial potassium transport.
DISCUSSION
Our data indicate that astrocytic differentiation is impaired in GPCs derived from childhood-onset schizophrenics, and that this maturational defect may be rescued by the suppression of either BMP signaling via SMAD4 knock-down, or by the de-repression of glial differentiation-associated transcription via REST knock-down. Importantly, astrocytic depletion has been recently noted in both cortical and subcortical regions of patients with schizophrenia, and this might be especially prominent in the white matter, (Rajkowska et al., 2002; Steffek et al., 2008; Williams et al., 2013). Astrocytes play key contributions to neural circuit formation and stability (Christopherson et al., 2005; Clarke and Barres, 2013). Thus, any such developmental defect of astrocytic differentiation in SCZ GPCs might lead to profound defects in the initial formation or stability of neural circuits, a defect that is one of the hallmarks of schizophrenia (Penzes et al., 2011). In this regard, our RNA-seq data suggested upregulated TGFBR and BMP signaling in SCZ GPCs, which was associated with the activation of downstream BMP-regulated genes that included BAMBI, a competitive inhibitor of pro-gliogenic BMP signaling (Onichtchouk et al., 1999). We have previously noted that high-expression of BAMBI in adult human GPCs significantly inhibits their astrocytic differentiation as induced by BMP4 (Sim et al., 2006), suggesting that the pathological elevation of BMP signaling-induced BAMBI expression in SCZ hGPCs, relative to normal control hGPCs, might be sufficient to suppress their differentiation as mature astrocytes. Besides BAMBI, several other inhibitors of TGFβ/BMP signaling, including FST and GREM1 (Brazil et al., 2015), were also upregulated by SCZ GPCs; these may have permitted SCZ hGPCs to avoid astrocytic fate even after BAMBI knockdown.
Of note, the activation of canonical TGFβ signaling is dependent upon either SMAD2/3 activation via the TGFβ pathway, or SMAD1/5/8 via BMP receptor-dependent signals; each of these effectors needs to combine with SMAD4 for nuclear translocation prior to the activation of their downstream genetic targets (Hata and Chen, 2016; Herhaus and Sapkota, 2014). Accordingly, we found that SMAD4 knockdown efficiently suppressed BMP signaling-induced expression of endogenous BMP inhibitors, and by so doing robustly promoted the astrocytic differentiation of otherwise differentiation-resistant SCZ GPCs. Importantly, this differentiative response of hGPCs to SMAD4 inhibition was only noted at the hGPC stage, and only in SCZ hGPCs; control patient-derived hGPCs showed no such potentiated differentiation in response to SMAD4 suppression. Thus, the modulation of SMAD4 might represent an appropriate strategy towards relieving the glial differentiation defect in schizophrenia.
Glial maturation is precisely regulated in human brain development (Goldman and Kuypers, 2015; Molofsky et al., 2012). Astrocytes have a multitude of roles in the CNS, including energy support to both neurons and oligodendrocytes, potassium buffering, neurotransmitter recycling, and synapse formation and maturation; as such, astrocytes play critical roles in neural circuit formation and maintenance (Blanco-Suarez et al., 2017; Clarke and Barres, 2013; Verkhratsky et al., 2015). Astrocytes also contribute to the glymphatic system, through the regulation of cerebral spinal fluid flow through the brain interstitium (Xie et al., 2013). Thus, the delayed differentiation of SCZ astrocytes may have significant effects on neural network formation, organization and mature function alike.
We found that a number of potassium transporters were down-regulated in SCZ glia. Interestingly, prior genome wide association studies (GWAS) have identified an association of potassium pump, transport and channel genes with schizophrenia. For instance, the chromosome 1q21–q22 locus, containing KCNN3, has a significant linkage to familial schizophrenia (Brzustowicz et al., 2000). KCNN3 is widely expressed in the human brain, and selectively regulates neuronal excitability and neurotransmitter release in monoaminergic neurons (O’Donovan and Owen, 1999). In addition to KCNN3, a number of other potassium channel genes have been associated with schizophrenia, including KCNQ2 and KCNAB1 (Lee et al., 2013). More recently, a novel de novo mutation in ATP1A3, a subunit of the sodium-potassium pump, has been specifically associated with childhood-onset schizophrenia (Smedemark-Margulies et al., 2016).
The down-regulation or dysfunction of these potassium transporters in GPCs and their derived astrocytes may contribute significantly to disease phenotype in schizophrenia. Potassium channel, pump and transport genes are widely expressed in both GPCs (Coppi et al., 2013; Maldonado et al., 2013) and astrocytes (Larsen et al., 2014; Zhang and Barres, 2010), in which they regulate not only proliferation, migration, and differentiation, but also the relationship of glia to neurons (Coppi et al., 2013; Maldonado et al., 2013). In regards to the latter, astrocytes also regulate synaptic K+ uptake through all three major K+ transport mechanisms, including the Na+/K+-ATPase, the NKCC1 cotransporter, and inwardly rectifying Kir channels (Larsen et al., 2014; Zhang and Barres, 2010), thereby establishing neuronal firing thresholds over broad regional domains.
Accordingly, dysregulated K+ transport and potassium channel gene expression have been associated with a broad variety of neurological and psychiatric diseases. Several Kir genes, including Kir4.1, are involved in astrocytic potassium buffering and glutamate uptake, and deletion of these genes has been noted in both Huntington’s disease and multiple sclerosis (Seifert et al., 2006; Tong et al., 2014). In addition, mutation of astrocytic ATP1A2, the α2-isoform of the sodium-potassium pump, may be causally associated with familial hemiplegic migraine (Bottger et al., 2016; Swarts et al., 2013). In all of these examples, glial K+ uptake is impaired, just as in SCZ glia, and all are associated with elements of phenotypic hyperexcitability. Indeed, eleverated extracellular K+ has been shown to alter the neuronal excitability and neural circuit stability in a mouse model of schizophrenia (Crabtree et al., 2017). Thus, the decreased K+ uptake of SCZ glia may be a significant contributor to schizophrenia pathogenesis, especially in regards to those schizophrenic phenotypes associated with hyperexcitability and seizure disorders, which would be potentiated in the setting of disrupted potassium homeostasis.
We next established that the upregulated REST in SCZ glia appears both necessary and sufficient for their suppression of both potassium channel gene expression and potassium uptake. REST, as a transcriptional repressor, regulates neural gene expression in both neurons and glia (Bruce et al., 2004; Dewald et al., 2011). In broad terms, REST represses neural genes through its recruitment of CoREST and mSIN3a, the complex of which further recruits HDAC1/2 and methyltransferase G9a to promote concurrent histone deacetylation and methylation, which together serve to block transcription (Hirabayashi and Gotoh, 2010). As such, the misdirected upregulation of REST inhibits potassium channel gene expression, and thereby contributes to the disease phenotype of those disorders associated with dysregulated potassium homeostasis and its attendant neuronal hyperexcitability. For instance, up-regulated REST in peripheral sensory neurons induces the down-regulation of KCNQ2, which in turn potentiates the hyperexcitability of sensory neurons and hence the maintenance of neuropathic pain (Rose et al., 2011). REST similarly represses KCNQ3 gene expression, and the down-regulation of KCNQ3 by REST provokes neuronal hyperexcitability in specific neonatal epilepsies (Mucha et al., 2010).
Furthermore, REST is involved in schizophrenia through its modulation of miR137 (Warburton et al., 2015), which regulates multiple schizophrenia-associated genes, including CACNA1C, TCF4, and ANK3 (Kwon et al., 2013; Schizophrenia Psychiatric Genome-Wide Association Study, 2011). REST has been reported to be highly expressed in postmortem brain tissues from patients with schizophrenia (Loe-Mie et al., 2010). Other investigators have reported that REST can repress the expression of potassium channel-associated genes (Bruce et al., 2004), and can suppress oligodendroglial differentiation within the glial lineage (Dewald et al., 2011). We thus hypothesized that pathologically high levels of REST might repress K+ transporter and channel-associated gene expression, and thereby decrease K+ uptake, in schizophrenia-derived glia. This would be expected to lead to high interstitial K+, and hence to relative neuronal hyperexcitability and network desynchronization. That said, the role of REST in disordered glial potassium homeostasis has not hitherto been reported. Our data suggest the likelihood that some fraction of schizophrenic patients might suffer high interstitial K+ levels, as a function of diminished glial potassium uptake. This would be expected to yield neuronal hyperexcitability, as has been reported in Huntington disease, another disorder characterized by glial potassium channel dysfunction (Benraiss et al., 2016). As such, our observation of a REST-dependent impairment of K+ uptake by SCZ-derived glia suggests that REST might be an effective target for the treatment of schizophrenia. In that regard, several REST-targeted drugs have been developed for epilepsy and Huntington disease, including valproic acid and X5050 (Charbord et al., 2013; Graff and Tsai, 2013); our data suggest that these agents may have therapeutic utility in schizophrenia as well.
Thus, our data reveal the defective differentiation into astrocytes by SCZ GPCs, the potential reversibility of that defect by SMAD4 knockdown, and the differentiation-coupled, REST-dependent suppression of potassium channel gene expression and concomitant defective uptake of K+ by SCZ glia. The resultant deficiencies in synaptic potassium homeostasis may be expected to significantly lower neuronal firing thresholds while accentuating network desynchronization (Benraiss et al., 2016). As such, one might expect that positive modulators of glial K+ uptake may have real value in the treatment of schizophrenia (Calcaterra et al., 2016; He et al., 2013; Rahmanzadeh et al., 2017). Together, these findings identify a causal contribution of astrocytic pathology to the neuronal dysfunction of SCZ, and in so doing suggest a set of tractable molecular targets for its treatment.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Steve Goldman. He may be contacted at: (steven_goldman@urmc.rochester.edu or goldman@sund.ku.dk).
Experimental Model and Subject Details
Patient identification, protection and sampling
Patients from which these lines were derived were diagnosed with disabling degrees of schizophrenia with onset in early adolescence; all patients and their guardians were consented/assented by a child and adolescent psychiatrist working under the supervision of one of us (RLF), and under the auspices of an approved protocol of the University Hospitals Case Medical Center Institutional Review Board, blinded as to subsequent line designations. No study investigators had access to patient identifiers.
Cell sources and lines
Schizophrenia-derived iPSC lines were produced from subjects with childood-onset schizophrenia, and control lines were produced from age- and gender-appropriate control subjects; all iPSC lines were derived as previously reported (Windrem et al., 2017). An additonal control line (C27; Wang et al., 2013) was graciously provided by Dr. Lorenz Studer (Memorial Sloan-Kettering). Control-derived lines included: CWRU-22 (26 year-old male), −37 (32 year-old female), −208 (25 year-old male), and C27; SCZ-derived lines included CWRU-8 (10 year-old female), −51(16 year-old male), −52 (16 year-old male), −193 (15 year-old female), −164 (14 year-old female), −29 (12 year-old male), −30 (12 year-old male), and −31 (12 year-old male) (Windrem et al., 2017; see Supplemental Table 1). CWRU-51/52 and CWRU-29/30/31 comprised different lines from the same patients, and were assessed to estimate inter-line variability from single patients. All iPSCs were generated from fibroblasts by retroviral expression of Cre-excisable Yamanaka factors (Oct4, Sox2, Klf4, c-Myc) (Takahashi et al., 2007), with validation of pluripotency and karyotypic stability as described (Windrem et al., 2017).
Method Details
hiPSC culture and passage
hiPSCs were cultured on irradiated mouse embryonic fibroblasts (MEFs), in 0.1% gelatin (Sigma G1890–100G)-coated 6-well plates with 1–1.2 million cells/well in hESC medium (see below) supplemented with 10 ng/ml bFGF (Invitrogen, 13256–029). Media changes were performed daily, and cells passaged at 80% confluence, after 4–7 days of culture. For hiPSC passage, cells were first incubated with 1ml collagenase (Invitrogen, 17104–019) at 37°C for 3–5 minutes, and then cells were transferred into a 15 ml tube for centrifuge with 3 minutes. The pellet was re-suspended with ES medium with bFGF, and was plated onto new irradiated MEFs at 1:3–1:4.
GPC and astrocytic generation from hiPSCs
When hiPSCs reached 80% confluence, they were incubated with 1 ml Dispase (Invitrogen, 17105–041) to permit the generation of embryoid bodies (Ebs); these were cultured in ES medium without bFGF for 5 days. At DIV6, Ebs were plated onto poly-ornithine (Sigma, P4957) and laminin (VWR, 47743)-coated dishes, and cultured in neural induction media (NIM; see below) (Wang et al., 2013), supplemented with 20 ng/ml bFGF, 2 μg/ml heparin and 10 μg/ml laminin for 10 days.
At DIV 25, the Ebs were gently scraped with a 2 ml glass pipette, then cultured in NIM plus 1 μM purmorphamine (Calbiochem, 80603–730) and 0.1 μM RA (Sigma, R2625). At DIV 33, NPCs appeared and were serially switched to NIM with 1 μM purmorphamine and 10 ng/ml bFGF for 7 days, followed by glial induction medium (GIM) (Wang et al., 2013), with 1 μM purmorphamine for another 15 days. At DIV 56, the resultant glial spheres were mechanically cut with microsurgical blades under a dissection microscope, and switched to GIM with 10 ng/ml PDGF, 10 ng/ml IGF, and 10 ng/ml NT3, with media changes every 2 days. At DIV 80–100, CTR GPCs were cultured with 10 ng/ml BMP4 (PeproTech, AF-120–05ET) and 0.5 μM DMH1 (Sigma, D8946–5MG) for 2 weeks, and SCZ GPCs were transduced with lentiviral-SMAD4-shRNAi for 2 weeks, both of which were used for validation of REST and K+ transport gene expression. At DIV 150–180, GPCs were incubated with mouse anti-CD44 microbeads (1:50), and then incubated with rabbit anti-mouse IgG2a+b micro-beads (1:100) and further sorted by magnetic cell sorting (MACS) with a magnetic stand column. The CD44+ cells were then matured as astrocytes in M41 supplemented with 10% FBS (VWR, 16777–014) plus 20 ng/mL BMP4 for 4 weeks.
Media recipes are listed in Supplemental Table S2 (hESC and neural media) and Supplemental Table S3 (Glial and Astrocyte induction media).
FACS/MACS sorting
Cells were incubated with Accutase (Fisher Scientific, SCR005) for 5 minutes at 37°C to obtain a single cell suspension, and then spun down at 200RCF for 10 minutes. These GPCs were re-suspended in cold Miltenyi Wash buffer with primary antibody (phycoerythrin (PE)-conjugated mouse anti-CD140a, 1:50, for FACS; mouse anti-CD140a, 1:100, for MACS), and incubated on ice for 30 min, gently swirling every 10 minutes. After primary antibody incubation, these cells were then washed and either incubated with a secondary antibody (rabbit anti-mouse IgG2a+b micro-beads, 1:100) followed by sorting on a magnetic stand column for MACS, or directly sorted by FACS on a FACSAria IIIu (Becton-Dickinson). The sorted cells were counted and plated onto poly-ornithine- and laminin-coated 24-well plate for further experiments. Antibodies and dilutions used see Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Nanog, 1:200 | Millipore | Cat # AB9220; RRID: AB_570613 |
| Mouse monoclonal anti-human TRA1-60, 1:200 | Millipore | Cat # MAB4360; RRID: AB_2119183 |
| Rabbit polyclonal anti-PAX6, 1:400 | Covance Research Products Inc | Cat # PRB-278p; RRID: AB_291612 |
| Rabbit monoclonal anti-PDGF Receptor alpha, clone D13C6, 1:300 | Cell Signaling Technology | Cat #5241S; RRID: AB_10692773 |
| Mouse monoclonal anti-human GFAP, clone SMI 21R, 1:500 | Covance Research Products | Cat#SMI-21R-500; RRID: AB_509979 |
| Mouse monoclonal anti-S100 beta, clone SH-B1, 1:500 | Abcam | Cat #ab11178; RRID: AB_297817 |
| Goat polyclonal anti-human SOX1, 1:200 | R&D Systems | Cat # AF3369; RRID: AB_2239897 |
| Donkey anti-mouse IgG (H+L) Alexa Fluor 594,1:400 | ThermoFisher Scientific | Cat # A-21203; RRID: AB_2535789 |
| Donkey anti-mouse IgG (H+L) Alexa Fluor 488, 1:400 | ThermoFisher Scientific | Cat #A-21202; RRID: AB_141607 |
| Donkey anti-Rabbit IgG (H+L) Alexa Fluor 594,1:400 | ThermoFisher Scientific | Cat #A-21207; RRID: AB_141637 |
| Donkey anti-Rabbit IgG (H+L) Alexa Fluor 488, 1:400 | ThermoFisher Scientific | Cat #A-21206; RRID: AB_2535792 |
| Donkey anti-Goat IgG (H+L) Alexa Fluor 594,1:400 | ThermoFisher Scientific | Cat #A-11058; RRID: AB_2534105 |
| Mouse monoclonal anti-SSEA4 FITC Conjugate, 1:100 | Life Technologies | Cat # MC-813-70; |
| APC-conjugated mouse IgG1, Isotype Control, 1:10 | Miltenyi Biotec | Cat#130-092-214 |
| APC-mouse IgM, Isotype Control, 1:40 | Miltenyi Biotec | Cat#130-093-176 |
| Rat anti-mouse IgG2a+b, microbeads, 1:100 | Miltenyi Biotec | Cat #130-095-194 |
| Mouse anti-CD44 microbeads, 1:100 | Miltenyi Biotec | Cat #130-095-194 |
| APC-conjugated anti-CD133/1, 1:10 | Miltenyi Biotec | Cat#130-090-826 |
| Mouse monoclonal anti-CD140a Unconjugated, 1:100 | BD Biosciences | Cat #556001; RRID: AB_396285 |
| PE-conjugated anti-CD140a, 1:10 | BD Biosciences | Cat#556002 |
| APC-conjugated anti-CD44, 1:10 | Miltenyi Biotec | Cat #130-095-177; RRID: AB_10839563 |
| PE-conj. anti-mouse IgG2a, isotype control, 1:10 | BD Pharmingen | Cat#555574 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle Medium | Invitrogen | Cat #11965-092 |
| Fetal Bovine Serum | Invitrogen | Cat #16000-044 |
| Non-Essential Amino Acid | Invitrogen | Cat # 11140-050 |
| Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 | Invitrogen | Cat #11330-032 |
| KnockOut Serum Replacement | Invitrogen | Cat #10828-028 |
| FBS | VWR | Cat#16777-014 |
| Donkey serum | Millipore | Cat#5058837 |
| Goat serum | Invitrogen | Cat#16210-072 |
| DPBS | Invitrogen | Cat# 14190-250 |
| Thimerosal | Sigma | T5125 |
| L-glutamine | Invitrogen | Cat #25030-081 |
| Gelatin | Sigma | Cat#G1890-100G |
| 2-Mercaptoethanol | Sigma | Cat #M7522 |
| Saponin | Fluka Analytical | Cat#47036 |
| B27 Supplement | Invitrogen | Cat #12587-010 |
| N1 Supplement | Sigma | Cat # N6530 |
| Selenite | Sigma | Cat #S9133 |
| Progesterone | Sigma | Cat #P6149 |
| Rubidium-86 | PerkinElmer | Cat#NEZ072001MC |
| Ouabain | Sigma | Cat # O3125 |
| Bumetanide | R&D Systems | Cat #3108 |
| Tertiapin | R&D Systems | Cat #1316 |
| NaOH | Fisher Scientific | Cat#MSX0607H6 |
| NaCl | Invitrogen | Cat#AM9760G |
| KCl | Invitrogen | Cat#AM9640G |
| CaCl2 | Sigma | Cat#21108-500g |
| vitamin C | Sigma | Cat#A4034-100G |
| NaHCO3 | Sigma | Cat#S-8875 |
| MgCl2 | Sigma | Cat#M8266-IKG |
| NaH2PO4 | Sigma | Cat#S3264-500G |
| Glucose | Sigma | Cat#G8769-100ML |
| Cocktail liquid | Fisher Scientific | Cat#509050575 |
| N2 Supplement | Invitrogen | Cat #17502-048 |
| bFGF | Sigma | Cat #F0291 |
| bFGF | Invitrogen | Cat#13256-029 |
| Collagenase | Invitrogen | Cat#17104-019 |
| Dispase | Invitrogen | Cat#17105-041 |
| Poly-ornithine | Sigma | Cat# P4957 |
| Laminin | VWR | Cat#47743 |
| Biotin | Sigma | Cat #B4639 |
| dibutyryl cAMP | Sigma | Cat #D0260 |
| Heparin | Fisher | Cat#NC9484621 |
| IGF-1 | R&D Systems | Cat #291-G1-050 |
| Laminin | Corning | Cat#354232 |
| NT3 | R&D Systems | Cat #267-N3-025 |
| PDGFaa | R&D Systems | Cat #221-AA-50 |
| BMP4 | PeproTech | Cat#AF-120-05ET |
| Accutase | Fisher Scientific | Cat# SCR005 |
| Purmorphamine | Calbiochem | Cat #80603-730 |
| Retinoic acid | Sigma | Cat #R2625 |
| DMH1 | Sigma | Cat#D8946-5MG |
| T3 | Sigma | Cat #T5516-1MG |
| 4% paraformaldehyde | Fisher Scientific | Cat#NC9245948 |
| X-tremeGENE | Roche | Cat#06366236001 |
| Doxycycline | Fisher Scientific | Cat#ICN19895510 |
| Critical Commercial Assays | ||
| RNeasy mini kit | QIAGEN | Cat#74104 |
| QIAamp DNA micro kit | QIAGEN | Cat#56304 |
| Taqman Reverse Transcription kit | Fisher Scientific | Cat#N8080234 |
| BCA Protein Assay kit | Fisher Scientific | Cat#23227 |
| Perkin Elmer LLC ULTIMA-GOLD LITERS | Fisher Scientific | Cat#509050575 |
| Deposited Data | ||
| All raw data | Mendeley data | doi:10.17632/wvnxgw7xzf.1 |
| RNA expression data | GEO | accession no. GSE86906 |
| Data processing and analytic routines | Github | https://github.com/cbtncph/GoldmanetalSCZ2016 |
| Experimental Models: Cell Lines | ||
| C27 | L. Studer, SKI | N/A |
| CWRU8, female, age 10 | P. Tesar, Case Western | N/A |
| CWRU208, male, age 25 | P. Tesar, Case Western | N/A |
| CWRU22, male, age 26 | P. Tesar, Case Western | N/A |
| CWRU29, male, age 12 (same person as line 30 and 31) | P. Tesar, Case Western | N/A |
| CWRU30, male, age 12 (same person as line 29 and 31) | P. Tesar, Case Western | N/A |
| CWRU31, male, age 12 (same person as line 29 and 30) | P. Tesar, Case Western | N/A |
| CWRU37, female, age 32 | P. Tesar, Case Western | N/A |
| CWRU51 male, age 16 (same person as line 52) | P. Tesar, Case Western | N/A |
| CWRU52 male, age 16 (same person as line 51) | P. Tesar, Case Western | N/A |
| CWRU164, female, age 14 | P. Tesar, Case Western | N/A |
| CWRU193, female, age 15 | P. Tesar, Case Western | N/A |
| 293T | Fisher Scientific | Cat# R70007 |
| Oligonucleotides | ||
| ShRNA targeting sequence: REST #1: GCAGTGGCAACATTGGAATGG; #2: CGGCTACAATACTAATCGA | This paper | N/A |
| ShRNA targeting sequence: SMAD4 #1: TGGTCAGCCAGCTACTTAC; #2: ATGAATATGACTCACTTCT | GE Healthcare | Cat#V3SH11252 |
| shScramble: AAGTTGCAAATCGCGTCTCTA | This paper | N/A |
| Recombinant DNA | ||
| human cDNA of REST | Westbrook et al., 2008 | Addgene plasmid #41903 |
| human cDNA of SMAD4 | GE Healthcare | Cat#MHS6278 |
| Plasmid: pTANK-EF1a-coGFP-P2a-Puro-WPRE | This paper | N/A |
| Plasmid: pTANK-EF1a-IRES-mCherry-WPRE | Benraiss et al., 2016 | N/A |
| Bacterial and Virus Strains | ||
| TOP10 Chemically Competent E.coli | Invitrogen | Cat#K4600-01 |
| Software and Algorithms | ||
| Photoshop CS6 | Adobe | N/A |
| Illustrator CS6 | Adobe | N/A |
| FlowJo | TreeStar | N/A |
| Ingenuity Pathway Analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| TRANSFAC | Genexplain | https:/www.genexplain.com/transfac/ |
| minfi (version 1.28.2) | (Aryee et al., 2014) | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| Other | ||
| Agilent Bioanalyzer | Agilent | N/A |
| BD FACS Aria IIIU | BD Biosciences | N/A |
| Ultracentrifuge | Beckman | Cat #L8-70 |
| Becksman Coulter | Beckman | Cat #LS6500 |
| Hemocytometer | Fisher Scientific | Cat#02-671-54 |
| HiSeq 2500 | Illumina | N/A |
| Nanodrop 1000 spectrophotometer | Nanodrop | N/A |
| Olympus IX71 Inverted Microscope | Olympus | N/A |
| QuantStudio 12K Flex Real-Time PCR system | Applied Biosystems | N/A |
| Orca-R2 Digital CCD Camera | Hamamatsu | Cat #C10600-10B |
RT-PCR
Total RNA was extracted from cell lines with miRNeasy mini kit (Qiagen, 217004), and then was reversely transcribed into cDNA with Taqman Reverse Transcription kit (Fisher Scientific, N8080234). The relative expression of mRNA was measured by the Bio-RAD S6048, which was further normalized to the expression of 18S mRNA.
The primer sequences are listed in Supplemental Table S4.
Methylation
DNA was extracted from iPSC lines with the QIAamp DNA micro kit (Qiagen, 56304), and then whole genome methylation analysis was performed using Illumina Methylation Epic arrays; this was done at the UCLA Neuroscience Genomics Core. Raw data from Intensity Data (IDAT) files were imported into R and normalized with the preprocessQuantile function from the package minfi (Aryee et al., 2014). Probes with poor quality signal were eliminated based on set threshold of detection p values (> 0.01). Probes were also eliminated if they map to the sex chromosomes, to multiple genomic locations, or if they contain single nucleotide polymorphisms at the CpG site. Following preprocessing, samples were assessed by principal component analysis (native R functions, https://www.R-project.org) based on their features of methylated intensities (M-values). To determine if a covariate (sex, age, cell line, etc.) could explain variation in our samples’ methylation landscape, a linear regression model was fit for covariates and each principal component. Covariates with significant p values (< 0.05) were highlighted, indicating meaningful relationship between changes in the covariate (predictor variable) and changes in the principal component values (response variable).
In vitro immunocytochemistry
Cells were first fixed with 4% paraformaldehyde for 5 minutes at room temperature. After washing with D-PBS (Invitrogen, 14190–250) with thimerosal (Sigma, T5125) for 3 times, cells were penetrated with 0.1% saponin (Fluka Analytical, 47036) plus 1% of either goat or donkey serum for 15 minutes at room temperature. Cells were further blocked with 5% of either goat or donkey serum plus 0.05% saponin for 15 minutes at RT. After incubation with primary antibodies at 4°C overnight, the cells were incubated with secondary antibodies for 30 min at RT. The counts of immunofluorescent cells were taken from 10 random fields per each replicate, and each sample had three replicates.
Antibodies and dilutions used see Key Resources Table.
Molecular cloning and viral construction
REST shRNAs of human REST (target sequences: GCAGTGGCAACATTGGAATGG or CGGCTACAATACTAATCGA) were cloned into the vector pTANK-EF1α-CoGFP-Puro-WPRE immediately downstream puromycin. The human cDNAs encoding REST (a gift of Stephen Elledge, Addgene 41903) (Westbrook et al., 2008) and SMAD4 (GE Healthcare, MHS6278) were cloned downstream of the EF1α promoter in pTANK-EF1a-IRES-mCherry-WPRE (Benraiss et al., 2016). The lentiviral vector allowed for expression of REST and SMAD4 in tandem with the reporter mCherry. SMAD4 Doxycycline-inducible shRNAs of human SMAD4 (Gene target sequence: TGGTCAGCCAGCTACTTAC or ATGAATATGACTCACTTCT) in pSMART-TRE3G-EGFP-Puro-WPRE were ordered from GE Healthcare (V3SH11252). BAMBI The human shRNA and cDNA of BAMBI were generated previously (Sim et al., 2006). The final constructs were validated for the correct insertion by sequencing. The plasmids were then co-transfected with pLP-VSV (Invitrogen, K497500) and psPAX2 (a gift from Didier Trono, Addgene 12260) into 293FT cells (Fisher Scientific, R70007) through X-tremeGENE (Roche, 06366236001) for lentiviral generation. The supernatants of 293T cells were then collected and spun at 76000 RCF for 3 hrs to concentrate virus (Beckman L8–70, Ultracentrifuge). A 10-fold serial dilution of virus was then prepared and transduced into 293T cells, and fluorescent colonies counted to estimate viral titer.
Cell transduction
CD140a+ hGPCs were isolated by MACS and then transduced with either lenti-TRE3G-SMAD4-shRNAi or lenti-EF1α-BAMBI-shRNAi, or their respective scrambled control viruses, MACS-sorted CD44+ cells were transduced with either lenti-EF1α-REST-shRNAi or control virus, each at 1 MOI (multiplicities of infection) for 4 hours. Both lenti-EF1α-REST-shRNAi and lenti-EF1a-BAMBI-shRNAi efficiently inhibited the expression of their target genes (Figures S3 and S6A). Cells infected with lenti-TRE3G-SMAD4-shRNAi were treated with 0.5 μg/ml doxycycline (Fisher, CN19895510) beginning 4 days after viral infection; this was maintained for 1 week prior to experiment initiation; during this period, the cells were maintained in glial induction media. Under doxycycline, SMAD4 mRNA expression fell to <30% of control; no inhibition was noted in the absence of doxycycline (Figures S4A–C).
Potassium uptake
Astrocytes were plated onto poly-ornithine- and laminin-coated 24-well plates with 30,000 cells/well. For the potassium uptake assay, astrocytes were incubated with 86Rb (1.0–3.3uCi/well) for 15min, and then they were washed three time with ice-cold artificial cerebrospinal fluid (aCSF, 500uL/well). 0.5N NaOH (200uL/well) was put into each well for cell lysis, which was put into 5ml cocktail liquid (Ultima Gold, Fisher Scientific, 509050575) and measured by scintillation counter (Beckman Coulter, LS6500), and the results were normalized to both total protein (BCA Protein Assay Kit, Fisher Scientific, 23227) and cell number (Hemocytometer, Fisher Scientific, 02-671-54). The aCSF solution contained (in mM): 124 NaCl, 2.5 KCl, 1.75 NaH2PO4, 2 MgCl2, 2 CaCl2, 0.04 vitamin C, 10 glucose and 26 NaHCO3, pH 7.4.
Quantification and Statistical analysis
Statistical parameters including the exact n, the center, dispersion, precision measures (mean ± SEM), and statistical significance are reported in the Figures and Figure Legends. All analyses were done with GraphPad PRISM 6 using one-way ANOVA and two tailed t-test. Statistical significance was considered as P-values less than 0.05. Significances were represented as *p < 0.05, **p < 0.01 and ***p < 0.001. Graphs and figures were made and assembled with Prism 6.
Data and Software Availability
All raw data have been uploaded to Mendeley data (https://data.mendeley.com), at https://data.mendeley.com/datasets/wvnxgw7xzf/draft?a=7661a24e-3cb1-;4a1b-a072-ba6ac169d909.
Previous data processing and analysis routines from Windrem et al., 2017 are available from https://github.com/cbtncph/GoldmanetalSCZ2016, while the genomic data from that study have been deposited to GEO: GSE86906.
Supplementary Material
Acknowledgements
Supported by NIMH, the Novo Nordisk Foundation, the Lundbeck Foundation, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and the Mathers Charitable Foundation. We thank Lorenz Studer (Memorial Sloan-Kettering) for the C27 hiPSC cell line, and Didier Trono for the psPAX2 lentiviral vector. All genomic data have been deposited to GEO, accession number GSE86906.
Footnotes
Declaration of Interests: The authors declare no competing interests in regards to this study. In regards to other, non-overlapping studies, Dr. Goldman is a co-founder and officer of Oscine Corp., a cell therapy company, and Drs. Goldman and Windrem are co-inventors on US and EU patents describing human glial chimeric mice and their use in modeling glial disorders. None of the other authors have any known conflicts of interest in regards to this work.
References
- Aberg K, Saetre P, Lindholm E, Ekholm B, Pettersson U, Adolfsson R, and Jazin E (2006). Human QKI, a new candidate gene for schizophrenia involved in myelination. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 141B, 84–90. [DOI] [PubMed] [Google Scholar]
- Allen NC, Bagade S, McQueen MB, Ioannidis JPA, Kavvoura FK, Khoury MJ, Tanzi RE, and Bertram L (2008). Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nature Genetics 40, 827–834. [DOI] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, and Irizarry RA (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, Burm HB, Toner M, Osipovitch M, Jim Xu Q, et al. (2016). Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nature communications 7, 11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Suarez E, Caldwell AL, and Allen NJ (2017). Role of astrocyte-synapse interactions in CNS disorders. J Physiol 595, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottger P, Glerup S, Gesslein B, Illarionova NB, Isaksen TJ, Heuck A, Clausen BH, Fuchtbauer EM, Gramsbergen JB, Gunnarson E, et al. (2016). Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci Rep 6, 22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Church RH, Surae S, Godson C, and Martin F (2015). BMP signalling: agony and antagony in the family. Trends Cell Biol 25, 249–264. [DOI] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, and Buckley NJ (2004). Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A 101, 10458–10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, and Bassett AS (2000). Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science 288, 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcaterra NE, Hoeppner DJ, Wei H, Jaffe AE, Maher BJ, and Barrow JC (2016). Schizophrenia-Associated hERG channel Kv11.1–3.1 Exhibits a Unique Trafficking Deficit that is Rescued Through Proteasome Inhibition for High Throughput Screening. Sci Rep 6, 19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbord J, Poydenot P, Bonnefond C, Feyeux M, Casagrande F, Brinon B, Francelle L, Auregan G, Guillermier M, Cailleret M, et al. (2013). High throughput screening for inhibitors of REST in neural derivatives of human embryonic stem cells reveals a chemical compound that promotes expression of neuronal genes. Stem Cells 31, 1816–1828. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, and Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, and Barres BA (2013). Emerging roles of astrocytes in neural circuit development. Nature reviews. Neuroscience 14, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppi E, Maraula G, Fumagalli M, Failli P, Cellai L, Bonfanti E, Mazzoni L, Coppini R, Abbracchio MP, Pedata F, et al. (2013). UDP-glucose enhances outward K(+) currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 61, 1155–1171. [DOI] [PubMed] [Google Scholar]
- Crabtree GW, Sun Z, Kvajo M, Broek JA, Fenelon K, McKellar H, Xiao L, Xu B, Bahn S, O’Donnell JM, et al. (2017). Alteration of neuronal excitability and short-term synaptic plasticity in the prefrontal cortex of a mouse model of mental illness. J Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald LE, Rodriguez JP, and Levine JM (2011). The RE1 binding protein REST regulates oligodendrocyte differentiation. J Neurosci 31, 3470–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD (2008). White matter in learning, cognition and psychiatric disorders. Trends in neurosciences 31, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, et al. (2016). Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G, and Friedman PA (2009). Thick ascending limb: the Na(+):K (+):2Cl (−) co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflugers Arch 458, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, and Kuypers NJ (2015). How to make an oligodendrocyte. Development 142, 3983–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudriaan A, de Leeuw C, Ripke S, Hultman CM, Sklar P, Sullivan PF, Smit AB, Posthuma D, and Verheijen MH (2014). Specific glial functions contribute to schizophrenia susceptibility. Schizophrenia bulletin 40, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, and Tsai LH (2013). The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol 53, 311–330. [DOI] [PubMed] [Google Scholar]
- Hata A, and Chen YG (2016). TGF-beta Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He FZ, McLeod HL, and Zhang W (2013). Current pharmacogenomic studies on hERG potassium channels. Trends Mol Med 19, 227–238. [DOI] [PubMed] [Google Scholar]
- Herhaus L, and Sapkota GP (2014). The emerging roles of deubiquitylating enzymes (DUBs) in the TGFbeta and BMP pathways. Cell Signal 26, 2186–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, and Gotoh Y (2010). Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 11, 377–388. [DOI] [PubMed] [Google Scholar]
- Hu J, Wang D, Li J, Jing G, Ning K, and Xu J (2014). Genome-wide identification of transcription factors and transcription-factor binding sites in oleaginous microalgae Nannochloropsis. Sci Rep 4, 5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama J, Sanosaka T, Tokunaga A, Takatsuka E, Tsujimura K, Okano H, and Nakashima K (2010). BMP-induced REST regulates the establishment and maintenance of astrocytic identity. The Journal of cell biology 189, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E, Wang W, and Tsai LH (2013). Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol Psychiatry 18, 11–12. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, and MacAulay N (2014). Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Kim JH, and Song GG (2013). Pathway analysis of a genome-wide association study in schizophrenia. Gene 525, 107–115. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, and Lazdunski M (1995). Molecular properties of neuronal G-protein-activated inwardly rectifying K+ channels. J Biol Chem 270, 28660–28667. [DOI] [PubMed] [Google Scholar]
- Loe-Mie Y, Lepagnol-Bestel AM, Maussion G, Doron-Faigenboim A, Imbeaud S, Delacroix H, Aggerbeck L, Pupko T, Gorwood P, Simonneau M, et al. (2010). SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Human molecular genetics 19, 2841–2857. [DOI] [PubMed] [Google Scholar]
- Macaulay N, and Zeuthen T (2012). Glial K(+) clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res 37, 2299–2309. [DOI] [PubMed] [Google Scholar]
- Maldonado PP, Velez-Fort M, Levavasseur F, and Angulo MC (2013). Oligodendrocyte precursor cells are accurate sensors of local K+ in mature gray matter. J Neurosci 33, 2432–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 49, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Krenick R, Ullian E, Tsai HH, Deneen B, Richardson WD, Barres BA, and Rowitch DH (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes & development 26, 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, Gamper N, and Wood IC (2010). Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci 30, 13235–13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan MC, and Owen MJ (1999). Candidate-gene association studies of schizophrenia. Am J Hum Genet 65, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J, and Niehrs C (1999). Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 401, 480–485. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, and Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmanzadeh R, Shahbazi A, Ardakani MK, Mehrabi S, Rahmanzade R, and Joghataei MT (2017). Lack of the effect of bumetanide, a selective NKCC1 inhibitor, in patients with schizophrenia: A double-blind randomized trial. Psychiatry Clin Neurosci 71, 72–73. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J, and Stockmeier C (2002). Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res 57, 127–138. [DOI] [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, et al. (2013). Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19, 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K, Ooi L, Dalle C, Robertson B, Wood IC, and Gamper N (2011). Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 152, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa A, and Snyder SH (2002). Schizophrenia: Diverse approaches to a complex disease. Science 296, 692–695. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study, C. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Schilling K, and Steinhauser C (2006). Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7, 194–206. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Lang JK, Waldau B, Roy NS, Schwartz TE, Pilcher WH, Chandross KJ, Natesan S, Merrill JE, and Goldman SA (2006). Complementary patterns of gene expression by human oligodendrocyte progenitors and their environment predict determinants of progenitor maintenance and differentiation. Ann Neurol 59, 763–779. [DOI] [PubMed] [Google Scholar]
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, and Goldman SA (2011). CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nature biotechnology 29, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, Kim IH, Manhaes AC, Rodrigues WS Jr., Pamukcu A, et al. (2016). Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell 164, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedemark-Margulies N, Brownstein CA, Vargas S, Tembulkar SK, Towne MC, Shi J, Gonzalez-Cuevas E, Liu KX, Bilguvar K, Kleiman RJ, et al. (2016). A novel de novo mutation in ATP1A3 and childhood-onset schizophrenia. Cold Spring Harb Mol Case Stud 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers A, Jean JC, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, et al. (2010). Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 28, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffek AE, McCullumsmith RE, Haroutunian V, and Meador-Woodruff JH (2008). Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr Res 103, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts HG, Weigand KM, Venselaar H, van den Maagdenberg AM, Russel FG, and Koenderink JB (2013). Familial hemiplegic migraine mutations affect Na,K-ATPase domain interactions. Biochim Biophys Acta 1832, 2173–2179. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Takahashi N, and Sakurai T (2013). Roles of glial cells in schizophrenia: possible targets for therapeutic approaches. Neurobiology of disease 53, 49–60. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Sakurai T, Bozdagi-Gunal O, Dorr NP, Moy J, Krug L, Gama-Sosa M, Elder GA, Koch RJ, Walker RH, et al. (2011a). Increased expression of receptor phosphotyrosine phosphatase-beta/zeta is associated with molecular, cellular, behavioral and cognitive schizophrenia phenotypes. Translational psychiatry 1, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Sakurai T, Davis KL, and Buxbaum JD (2011b). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Progress in neurobiology 93, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, and Bahn S (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362, 798–805. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nature neuroscience 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, and Hertz L (2015). Why are astrocytes important? Neurochemical research 40, 389–401. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, and Parpura V (2015). Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiology of disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineskos AN, Felsky D, Kovacevic N, Tiwari AK, Zai C, Chakravarty MM, Lobaugh NJ, Shenton ME, Rajji TK, Miranda D, et al. (2013). Oligodendrocyte genes, white matter tract integrity, and cognition in schizophrenia. Cereb Cortex 23, 2044–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Aleksic B, and Ozaki N (2015). Glia-related genes and their contribution to schizophrenia. Psychiatry and clinical neurosciences. [DOI] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, et al. (2013). Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton A, Breen G, Rujescu D, Bubb VJ, and Quinn JP (2015). Characterization of a REST-Regulated Internal Promoter in the Schizophrenia Genome-Wide Associated Gene MIR137. Schizophr Bull 41, 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welstead GG, Brambrink T, and Jaenisch R (2008). Generating iPS cells from MEFS through forced expression of Sox-2, Oct-4, c-Myc, and Klf4. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, Leng Y, Maehr R, Shi Y, Harper JW, et al. (2008). SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 452, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Hampton T, Pearce RK, Hirsch SR, Ansorge O, Thom M, and Maier M (2013). Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur Arch Psychiatry Clin Neurosci 263, 41–52. [DOI] [PubMed] [Google Scholar]
- Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH, et al. (2017). Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208 e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Abazyan S, Jouroukhin Y, and Pletnikov M (2014). Behavioral sequelae of astrocyte dysfunction: focus on animal models of schizophrenia. Schizophrenia research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin DM, Chen YJ, Sathyamurthy A, Xiong WC, and Mei L (2012). Synaptic dysfunction in schizophrenia. Adv Exp Med Biol 970, 493–516. [DOI] [PubMed] [Google Scholar]
- Zhang Y, and Barres BA (2010). Astrocyte heterogeneity: an underappreciated topic in neurobiology. Current opinion in neurobiology 20, 588–594. [DOI] [PubMed] [Google Scholar]
- Zou XY, Yang HY, Yu Z, Tan XB, Yan X, and Huang GT (2012). Establishment of transgene-free induced pluripotent stem cells reprogrammed from human stem cells of apical papilla for neural differentiation. Stem Cell Res Ther 3, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data have been uploaded to Mendeley data (https://data.mendeley.com), at https://data.mendeley.com/datasets/wvnxgw7xzf/draft?a=7661a24e-3cb1-;4a1b-a072-ba6ac169d909.
Previous data processing and analysis routines from Windrem et al., 2017 are available from https://github.com/cbtncph/GoldmanetalSCZ2016, while the genomic data from that study have been deposited to GEO: GSE86906.