

Abstract
The pre-melanosomal protein (Pmel17) aggregates within melanosomes to form functional amyloid fibrils that facilitate melanin polymerization. The repeat domain (RPT) of Pmel17 fibrillates under strict acidic melanosomal pH. Alternative splicing results in a shortened repeat domain (sRPT), which also forms amyloid fibrils. Here, we explored the effects of pH and protein concentration on sRPT aggregation by monitoring the intrinsic fluorescence of the sole tryptophan at position 381 (381W). 381 W emission properties revealed changes of local environment polarity for sRPT fibrils formed at different pH. At pH 4, fibrils formed rapidly with no lag phase. A high 381W intensity was observed with a slight blue shift (10 nm). These fibrils underwent further structural rearrangements at intermediate pH (5–6), mirroring that of melanosome maturation, which initiates at pH 4 and increases to near neutral pH. In contrast, typical sigmoidal kinetics were observed at pH 6 with slower rates and 381W exhibited quenched emission. Interestingly, biphasic kinetics were observed at pH 5 in a protein concentration-dependent manner. A large 381W blue shift (23 nm) was measured, indicating a more hydrophobic environment for fibrils made at pH 5. Consistent with 381W fluorescence, Raman spectroscopy revealed molecular level perturbations in sRPT fibrils that were not evident from circular dichroism, transmission electron microscopy, or limited proteolysis analysis. Finally, sRPT fibrils did not form at pH ≥7 and preformed fibrils rapidly disaggregated under these solution conditions. Collectively, this work yields mechanistic insights into pH-dependent sRPT aggregation in the context of melanosome maturation.
Keywords: functional amyloid, melanosome, transmission electron microscopy, circular dichroism, Raman spectroscopy
1. Introduction
The aggregation of soluble proteins into β-sheet-rich insoluble fibrils, called amyloid, is implicated in the etiology of ~50 human diseases [1]. While this has largely led to an association of amyloids with pathology, the discovery of “functional amyloids” over the past two decades has challenged this notion [2]. In human melanocytes, the pre-melanosomal protein (Pmel17, also known as SILV or gp100) forms functional amyloid fibrils that promote melanin biosynthesis [3]. This occurs within the melanosome, an acidic organelle similar to the lysosome, which undergoes four distinct stages of maturation. In vivo, Pmel17 fibrils form during the early stages of maturation (I and II), upon which melanin is synthesized during the later stages (III and IV) [4, 5]. It has been shown that the intralumenal pH increases during melanosome maturation, beginning at ~pH 4 in stages I and II and reaching near neutral pH upon full maturation [6]. Pmel17 fibrils serve as a scaffold for melanin polymerization, which also offer the benefit of sequestering toxic intermediates of melanin biosynthesis [3].
Pmel17 is a 668 amino-acid protein containing a single transmembrane domain, which undergoes several stages of processing prior to fibril formation within early stage melanosomes (Figure S1) [7–9]. The luminal domain of Pmel17, otherwise known as Mα, contains the amyloid forming region [10] and is made up of an N-terminal domain, a polycystic kidney disease-like domain, and a repeat (RPT) domain (Figure S1). While the exact domain(s) within Mα that is responsible for amyloid formation is not clearly understood [11–14], previous work has established that the RPT domain forms amyloid fibrils in vitro under pH solution conditions found in early stages of melanosomes [12, 13, 15–20].
The RPT domain comprises 10 imperfect repeats of 13 amino acids, each containing a high degree of proline, threonine, serine, and glutamic acid residues. Nichols et al. [21] have shown that alternative splicing of a “cryptic intron” within RPT leads to a shorter sequence (sRPT, Figure 1a) which lacks the 6th and 7th repeats and portions of the 5th and 8th repeats. This shorter sRPT domain has also been shown to form amyloid fibrils in acidic conditions in vitro. Notably, when exposed to neutral pH, sRPT fibrils disassemble [16], displaying reversible aggregation/disaggregation. This unique feature of RPT has not yet been documented for pathological amyloids, which are known to withstand harsh solution conditions. It is clear that the protonation/deprotonation of acidic residues (of which there are 10 Glu and 1 Asp, colored red in Figure 1a) plays an important role in the mechanism of amyloid formation of RPT [15].
Fig. 1.

Effect of pH on sRPT aggregation. (a) The one-letter amino acid sequence of sRPT. The underlined region represents the amyloidogenic core as defined in this work. Acidic and basic residues are colored red and blue, respectively. 381W is colored in purple. (b) sRPT (30 µM) aggregation kinetics at pH 4 (red), 5 (yellow), 6 (green), 7 (cyan), and 8 (blue), monitored by 381W fluorescence at 37 °C (n = 2 microplate experiments, 5 replicates each). (c) 381W fluorescence at t0 and tend. (d) ThT binding (10 µM) to sRPT at t0 and tend. (e-j) Representative TEM images of sRPT aggregates formed at pH 4 (e, h), 5 (f, i), and 6 (g, j).
Here, the effects of pH on sRPT amyloid formation were examined in detail. Using the intrinsic fluorescence of the sole tryptophan residue at position 381 (381 W) within the C-terminus of sRPT (Figure 1a), we showed that rates of aggregation increase with protein concentration and are highly dependent on pH (pH 4 > 5 > 6). Atypical for amyloid formation, sRPT fibrils formed quickly at pH 4 with no apparent lag phase, which upon a solution pH shift to 5 or 6, the fibrils reorganize at the C-terminus as indicated by 381W spectral changes. A similar fibril maturation process, albeit at a much faster timescale and only at protein concentration ≥ 30 µM, also was observed at pH 5. Aggregation at pH 6 exhibited the expected sigmoidal kinetics trend. Corresponding secondary structural changes upon amyloid formation were investigated by circular dichroism (CD) and Raman spectroscopy. Fibril morphology was characterized by transmission electron microscopy (TEM). Both 381W emission and Raman spectral features revealed molecular level perturbations in sRPT fibrils as a function of pH that were not evident from CD, TEM, or limited proteolysis analysis. In light of these results, we discuss the potential biological implications of the pH-dependent sRPT aggregation kinetics and fibril stability as they relate to the melanosome maturation process.
2. Materials and Methods
2.1. Materials
Unless otherwise noted, all reagents were purchased from Sigma-Aldrich.
2.2. Methods
2.2.1. sRPT protein expression and purification.
An sRPT construct containing a C-terminal hexa-histidine tag was expressed in E. coli BL21(DE3) cells and purified using affinity chromatography as described previously [16], with the following minor modifications to buffer conditions. The lysis buffer (pH 7.6) contained 6 M guanidine hydrochloride (GuHCl; Invitrogen), 100 mM NaCl, 100 mM Na2HPO4, and 10 mM imidazole. The Ni-NTA (Qiagen) column was equilibrated in 8 M urea, 100 mM NaCl, 100 mM Na2HPO4, 20 mM imidazole, pH 7.6 buffer and sRPT was eluted with the same buffer containing 200 mM imidazole. Purified protein was dialyzed against 20 mM tris(hydroxymethyl)aminomethane (Tris; Invitrogen), pH 8.0 and stored at −80 °C until use. The protein purity was first confirmed by SDS-PAGE analysis, which migrated with an apparent mass of ~50 kDa. The monoisotopic mass of 9694.5 Da was further confirmed by mass spectrometry (NHLBI Biochemistry Core), which displayed a purity greater than 95%.
2.2.2. Aggregation kinetics.
Stock solutions of sRPT were first buffer exchanged (pH 4 and 5: 20 mM sodium acetate, 100 mM NaCl; pH 6: 20 mM 2-(N-morpholino)ethanesulfonic acid, 100 mM NaCl; pH 7: 20 mM sodium phosphate, 100 mM NaCl; pH 8: 20 mM Tris, 100 mM NaCl) using a Sephadex G-25 PD-10 column (GE Healthcare) following the manufacturer’s protocol. After passing through a 100 kDa Amicon filter (Millipore), the protein concentrations were determined on a Cary 300 Series UV-Vis Spectrometer (Agilent Technologies) using the intrinsic 381W absorbance at 280 nm (ε = 5,500 M−1 cm−1). Reactions of 70 µL were then made into black 384-well polypropylene flat bottom plates (Greiner Bio-One) supplemented with a 2 mm borosilicate glass bead, and sealed with a MicroAmp optical adhesive film (Thermo Fisher Scientific). Reactions were monitored using a SPARK Multimode Microplate reader (Tecan) maintained at 37 °C with constant linear shaking (6 mm, 330 rpm). Fluorescence measurements were collected from the top using excitation and emission wavelengths of 280 and 350 nm, respectively.
2.2.3. Fluorescence spectroscopy.
Fluorescence measurements were collected on a Fluorolog FL-3 instrument (Horiba Scientific). For thioflavin-T (ThT) fluorescence, the dye was added to a final concentration of 10 µM. Emission was monitored from 430–600 nm (2 nm slit width) while exciting at 415 nm (1 nm slit width). For 381W measurements, emission was monitored from 290– 450 nm (2 nm slit width) while exciting at 280 nm (1 nm slit width). All data have their respective buffer backgrounds subtracted. In addition, 381W data were further scaled by relative protein concentration.
2.2.4. Transmission electron microscopy.
Transmission electron microscopy (TEM) images were collected on a JEM 1200 EXII microscope (JEOL USA) equipped with an XR-60 digital camera (Advanced Microscopy Techniques) operating at 80 kV (NHBLI Electron Microscopy Core). Grids were prepared by depositing 5 µL of solution onto a 400-mesh copper grid with a formvar/carbon film (Electron Microscopy Sciences) and allowing it to adhere for 1 min. Excess solution was wicked away using grade 1 Whatman filter paper (GE Healthcare). Grids were then stained for 30 s using 5 µL of 2% (w/v) uranyl acetate, followed by wicking. Finally, grids were dried overnight at RT before collecting images.
2.2.5. Limited cathepsin L proteolysis.
Reactions were prepared in sterile, 1.5 mL microcentrifuge tubes by adding cathepsin L (CtsL; 216 nM, Sigma #C6854) to a solution of preformed sRPT aggregates (30 µM, generated at pH 4, 5, or 6 as described in section 2.2.2) in reducing buffer (DTT, 5 mM). After shaking (575 rpm) at 37 °C overnight, reactions were transferred to LC-MS vials and TFA was added to a final amount of 0.5% (w/v), followed by GuHCl to a final concentration of 4 M. Samples were then subjected to LC-MS analysis using a Agilent 6200 series ESI-TOF LC-MS instrument (NHLBI Biochemistry Core). Deconvolution of the primary eluting species was then performed using MassHunter software (Agilent Technologies) to identify peptide fragments.
2.2.6. Circular dichroism spectroscopy.
Circular dichroism (CD) measurements were collected on a JASCO J-715 instrument (NHLBI Biophysics Core) in a 1-mm quartz cuvette. Scans from 260–195 nm (1 nm data pitch) were collected at a rate of 50 nm/min with a 1-s response time. A total of three spectra were averaged. Mean residue ellipticity (θMRE) was calculated using the following equation, as described previously [22]:
where MRW represents the mean residue weight (molecular weight/number of peptide bonds), θdeg represents the ellipticity (degrees) as measured by the instrument, d and c represent the pathlength (cm) and protein concentration (g/mL), respectively.
2.2.7. Raman spectroscopy.
Raman spectra were collected using a home-built instrument as previously described [23]. Samples were concentrated by centrifuging at 17,000 g for 30 min, followed by deposition of a 20 µL droplet onto an 8-well #1 coverglass Nunc LabTek chamber. Spectra represent an average of 5 collections from varying spatial locations, with each collection constituting 30 accumulations (10 s integration time). Data were processed first by buffer subtraction and then by first-polynomial baseline correction using LabSpec 6 software (Horiba Scientific). For comparison, the amide I (1665 cm−1) intensity was normalized to 1.
2.2.8. Dilution/disaggregation kinetics.
For dilution experiments, aggregates were diluted 6-fold into the specified buffer and aliquoted into 384-well microplates. 381W fluorescence was then monitored as described in section 2.2.2 above. For disaggregation experiments, measurements were collected on a Fluorolog FL-3 instrument (Horiba Scientific). A magnetic stir bar was added to a 10-mm quartz cuvette, followed by the appropriate buffer. Measurements were then initiated under constant stirring (setting = 4) by monitoring the emission at 350 nm (2 nm slit width) while exciting at 280 nm (1 nm slit width, 100 ms integration) using the kinetics mode (FluorEssence V3.8, Horiba Scientific). After ~10 s, sRPT was added directly into the cuvette, such that the final dilution was 6-fold. Disaggregation kinetics were monitored for a total of 5 min.
3. Results
3.1. Effect of pH on sRPT aggregation.
The effect of pH on sRPT aggregation kinetics were evaluated by monitoring 381W fluorescence, which has been previously shown to directly report the aggregation state of the longer RPT isoform (lRPT) [18]. We considered this to be a superior approach to the commonly utilized amyloid-binding dye thioflavin-T (ThT) [24], since 381W is a native probe that directly reports on aggregation. ThT fluorescence was compared with 381W, which showed similar kinetic profiles (data not shown). At pH 4, the 381W fluorescence rapidly increased within the first two hours with no lag phase observed, then plateaued for the remainder of the reaction (Figure 1b). A similar initial increase in 381W fluorescence was observed at pH 5, albeit a bit slower, which was then followed by a second transition before plateauing at a significantly lower fluorescence intensity (Figure 1b). We note that there was a small decrease in fluorescence intensity from 0 to 1 h as a result of temperature equilibration. The initial increase in 381W fluorescence at pH 5 may represent the formation of an oligomeric intermediate, which was investigated and is described in detail below. In contrast, after a lag phase of ~6 h, a single transition was observed at pH 6 before plateauing at a final fluorescence intensity similar to that observed at pH 5 (Figure 1b). No changes in the 381W fluorescence was observed over the course of the reaction at pH 7 or pH 8 (Figure 1b), which was also evident by comparing 381W emission scans from the beginning to end of the reaction (Figure 1c). Clearly, sRPT aggregation is highly pH dependent from pH 4–6 and no aggregation occurs beyond pH 7.
Fluorescence of 381W at the end of reactions at pH 4, 5, and 6 revealed blue shifts in the emission maxima (Figure 1c), indicating the sidechain resides in a more hydrophobic environment. This is expected for sRPT aggregation, as the protein is intrinsically disordered (IDP) in its soluble form [16] and would therefore have a solvent-exposed 381 W at t0. It is important to note that the magnitude of this blue-shift varies from pH 5 (23 nm, 346 to 323 nm) to pH 4 and 6 (both 10 nm, 346 to 336 nm). We attribute this observation to the formation of different fibril polymorphs, which has been shown previously for other amyloidogenic proteins as a function of pH [25–28]. In addition, the quenched emission intensity observed at pH 5 and 6 may be due to π-stacking of aromatic rings within the parallel-in-register alignment of β-strands in the final amyloid structure, indicating that fibrils formed at pH 4 may have a distinct fibril architecture from those of pH 5 and 6.
The absence of fibril formation at pH 7 and 8 was further confirmed by the lack of ThT fluorescence at the end of reactions (Figure 1d). However, ThT enhancement was seen for pH 4, 5, and 6 samples, albeit with varying intensities (Figure 1d). Interestingly, the ThT binding intensity seemed to correlate with the amount of aggregates observed by transmission electron microscopy (TEM, Figure 1e–g), with pH 4 grids being most densely populated with filamentous aggregates. Magnified images (Figure 1h–j) revealed that pH 4, 5, and 6 aggregates had a similar morphology containing twisted protofilaments. Thus, despite having ultrastructural similarities, the local environments of 381W are distinct in these fibrils.
To investigate if the observed differences in the local 381W environment would manifest in the protease-resistant amyloidogenic core, cathepsin L (CtsL) was used to digest fibrils formed at pH 4, 5, or 6 (Figure S2). CtsL was chosen due to it being ubiquitous in lysosomes, an acidic organelle similar to the melanosome, as well as previous reports detailing its potency to proteolyze α-synuclein amyloid fibrils [29, 30]. Upon digestion, a large fragment (7.7 kDa) constituting residues 336 to the C-terminus was identified in all samples, suggesting that the amyloid core of pH 4, 5, and 6 fibrils does not vary. This result is consistent with the previously reported proteinase K-resistant core of residues 333 to the C-terminus for sRPT fibrils formed at pH 5 [16].
3.2. Secondary structural characterization of sRPT fibrils.
To further probe whether there are other measurable differences in the sRPT fibrils formed at different pH, their secondary structures were examined using CD and Raman spectroscopies (Figure 2). As mentioned prior, sRPT is an IDP in its soluble form, which was reflected in the CD spectra collected at t0 (Figure 2a). No transition from this disordered structure was observed at pH 7 and 8 after incubation (48 h), further corroborating that no amyloid formation has occurred (Figure 2b). At pH 4, a broad minimum was observed at ~208 nm with a shoulder near 220 nm. While the latter absorption is indicative of β-sheet formation, the former band suggests the presence of α-helical conformation. A similar spectral profile was observed for pH 5 and 6 samples, with the signal intensity seeming to correlate with the amount of aggregates observed by TEM (Figure 1e–g). A nearly identical CD spectrum for sRPT was previously reported, where additional electron diffraction and solid-state nuclear magnetic resonance data suggested the formation of cross-β structure [16].
Fig 2.

Secondary structural characterization of sRPT fibrils. (a-b) CD spectra of sRPT (30 µM) aggregation reactions at pH 4 (red), 5 (yellow), 6 (green), 7 (cyan), and 8 (blue) at the beginning (t0, a) and end (tend, b) of the reactions. Θ is defined as the mean residue ellipticity. (c) Raman spectra of sRPT aggregates at tend. (d) Expanded view of 1200–1380 cm−1 region of the spectrum.
To bring more clarity to the secondary structure of sRPT, fibrils were characterized by Raman spectroscopy (Figure 2c). Raman spectroscopy has been previously shown to provide unique molecular fingerprints of different amyloids, including lRPT [31]. The Raman fingerprint of sRPT aggregates formed at pH 4, 5, and 6 were largely similar. A sharp peak at 1665 cm−1 was observed, which corresponds to the amide I band arising from C=O stretching vibration in the protein amide backbone. This is characteristic of β-sheet secondary structure [31, 32], confirming the formation of β-sheets in sRPT fibrils. Similar bands also were observed at 1552 and 1454 cm−1, which represented vibrations from Trp sidechains and C–H deformation, respectively [32]. However, upon closer inspection of the 1200–1380 cm−1 region of the spectrum, differences in the relative intensities of the 1320 and 1348 cm −1 peaks could be discerned (Figure 2d). The intensity ratio (I1320/I1348) at pH 4 (0.9) was closer to 1 as compared to pH 5 and 6, which were both lower (~0.3). These vibrations have been assigned to C–H deformation frequencies of the sidechains [23]. We interpret that while the overall secondary structures of the fibrils are similar, the sidechain packing in the pH 4 fibrils are different.
3.3. Disaggregation kinetics of sRPT fibrils.
381W fluorescence was employed to observe the kinetics of sRPT disaggregation upon dilution into pH 7 buffer (Figure 3). Fibrils (30 µM) grown at pH 4 (FpH 4, Figure 3a), 5 (FpH 5, Figure 3b), or 6 (FpH 6, Figure 3c) were diluted 6-fold into their corresponding buffer (red, yellow, and green curves, respectively) or into pH 7 buffer (cyan curves). The vertical dashed line represents the moment when aggregates were added to a cuvette containing buffer, which was subjected to constant mixing using a magnetic stir bar. Upon self-dilution (i.e. FpH 4 into pH 4 buffer), no changes in 381W kinetics were observed over the five minute reaction period for FpH 4, FpH 5, or FpH 6. However, diluting all aggregates into pH 7 buffer resulted in disaggregation, as evident by a lack of ThT emission increase at the end of the reaction (discussed in more detail below). For FpH 5 and FpH 6, 381W intensity increased upon mixing and reached a plateau by the end of the reaction period. In contrast, for FpH 4, the signal decreased upon disaggregation. For ease of comparison, changes in 381W were normalized and are shown in Figure 3d. Clearly, disaggregation of FpH 5 (yellow) and FpH 6 (green) occurred immediately and proceeded with similar rates, while there was a ~7 s lag time prior to FpH 4 (red) disaggregation (Figure 3d inset).
Fig. 3.

Disaggregation kinetics of sRPT fibrils. Fibrils formed at pH 4 (a), 5 (b), or 6 (c) were diluted 6-fold into their corresponding buffers (red, yellow, and green curves) or into pH 7 buffer (cyan curves). The vertical dashed line represents the timepoint when sRPT aggregates were introduced. (d) Normalized 381W fluorescence change after diluting into pH 7 buffer for fibrils formed initially at pH 4 (red), 5 (yellow), and 6 (green). (Inset) an expanded view of the first 30 s. The data are representative of two independent experiments.
3.4. Effect of protein concentration on sRPT aggregation.
The pH-dependent aggregation reactions mentioned thus far were all measured at a single protein concentration (30 µM). To investigate the concentration-dependence of the unique pH-dependent behavior, aggregation kinetics reactions were performed at lower protein concentrations (Figure 4a–c). The 30 µM reactions from Figure 1b are reproduced in Figure 4d for comparison. Similar kinetic trends were observed at pH 4 (red curves), where the 381 W fluorescence rapidly increased within a few hours. Similar to what was observed at 30 µM for pH 5 (Figure 1a), a subsequent decrease in the 381W intensity was observed for lower concentrations at pH 4 (Figure 4a–b), revealing a second kinetic phase. Plotting the time at which the signal intensity reached half the maximum value (t½) revealed that at pH 4, aggregation rates increased with increasing protein concentration from 3–15 µM (Figure 4e). However, the magnitude of this concentration-dependent difference was negligible compared to that observed at pH 5 and 6 (Figure 4e).
Fig. 4.

Effect of protein concentration on sRPT aggregation. (a-d) sRPT aggregation at pH 4 (red), 5 (yellow), and 6 (green) at 3 (a), 7.5 (b), 15 (c), and 30 µM (d, reproduced from Fig 1b), respectively. (e) Mean (<t1/2>) values for each concentration at the specified pH. Error bars are indicative of the standard deviation from the mean. n = 10, two independent microplate experiments. No aggregation was observed for 3 µM sRPT at pH 6. (f) Normalized 381W fluorescence for sRPT aggregation at 30 (light yellow), 60 (gold), and 150 µM (black) in pH 5 buffer.
At pH 6 (green curves), from 7.5–30 µM (Figure 4b–d), the observed t½ values decreased with increasing concentration (Figure 4e). No aggregation was observed at 3 µM using 381 W as a probe (Figure 4a). Excluding 30 µM, aggregation at pH 5 (yellow curves) displayed a single sigmoidal decay (Figure 4a–c) similar to that observed at pH 6. To ensure this concentration-dependent behavior at pH 5 was not the result of specific aggregation conditions, reactions were performed in quiescent conditions in the absence of a glass bead (Figure S3). Similar results were observed, where biphasic kinetic trends were only observed at 30 µM (Figure S3d). While the apparent biphasic kinetics vary with protein concentration at pH 5, the t½ displayed a linear trend (Figure 4e; the initial phase was considered when calculating t½ at 30 µM). Interestingly, the second phase (from high to low 381W intensity) was inhibited by increasing the protein concentration beyond 30 µM (Figure 4f), which may suggest the formation of kinetically-trapped oligomeric species. Further characterization is needed to understand this behavior at high protein concentration.
3.5. Characterization of an sRPT intermediate.
To explore the possibility of an sRPT oligomeric intermediate being formed at pH 5, 30 µM aggregation reactions were initiated as described before (Figure 5a). As a control, a reaction was started concomitantly at 10 µM, a protein concentration in which biphasic kinetics was not observed. As expected, the 381W fluorescence of the 30 µM reaction began to increase after ~1 h and continued to increase for an additional ~1 h. No changes in the 381W fluorescence of the 10 µM control reaction was observed in this time frame. At this point, reactions were stopped and individual samples of the 30 µM reaction were pooled together (tint) for further characterization. Collecting 381W emission scans showed that fluorescence intensity increased and a blue-shifted maximum was observed at tint compared to the soluble protein at t0 (Figure 5b). It is interesting that the blue-shift from t0 to tint (6 nm, 346 to 340 nm) was smaller than that of fibrils formed after several days of agitation (tend), which had a final blue-shift of 23 nm (346 to 323 nm; Figure 5c). Moreover, a difference in ThT fluorescence was observed with the tend point having ~4-fold higher intensity compared to tint (Figure 5d). Samples at tint were also visualized by TEM (Figure 5e–g), which surprisingly showed elongated fibrillar aggregates with twisted protofilaments similar to that observed at tend. Contrary to our initial thought, the prompt increase in 381W fluorescence does not represent oligomer formation, but rather is due to the formation of fibrils.
Fig. 5.

Characterization of an sRPT intermediate. (a) sRPT aggregation at 10 (light gray) and 30 µM (dark gray) at pH 5. After ~2 h, reactions were stopped and 381W (b-c) and ThT (d) fluorescence measurements of the 30 µM reaction (tint) were collected. sRPT fibrils formed at pH 5 (tend, black) and soluble sRPT at pH 5 (t0, light gray) are also shown for comparison. (d-f) TEM images of tint samples. Scale bars indicate 200 nm. (g) CD spectra of sRPT samples, as described in (b-c). (h) CD spectra of the total (dark gray), supernatant (light gray), and pellet (black) fractions of tint samples upon centrifugation at 17,000 g for 30 min. (i) Raman spectra of the tint pellet fraction (dark gray) and tend (black) samples.
To assess the secondary structure of samples at tint, CD and Raman spectroscopies were employed (Figure 5h–j). As shown earlier, soluble sRPT (t0) was initially disordered (Figure 5h). However, at tint a distinct CD spectrum with a minimum at ~205 nm was observed compared to that of the spectrum measured at tend, which had characteristic minima at 209 and 218 nm, respectively (Figure 5h). To ascertain if the unique CD spectra of sRPT taken at tint could be due to a mixture of fibrillar and soluble material, the sample was subjected to centrifugation at 17,000 g, and the supernatant and pellet fractions were independently measured (Figure 5i). We found that the pellet fraction had a similar CD spectrum to that taken at tend, indicating it contained vprimarily fibrillar aggregates. The soluble material in the supernatant remained disordered, likely still monomeric (Figure 5i).
Raman spectroscopy was used to investigate any changes in the molecular vibrations of sRPT at tint (Figure 5j). Compared to the spectrum taken at tend (reproduced from Figure 2c), a mostly similar fingerprint region was observed for sRPT at tint (Figure 5j). However, the ratio of the 1320 and 1348 cm−1 peaks at tint (0.9) was closer to the value reported for FpH 4 (Figure 2c). While this would indicate that the structure at tint may be more similar to FpH 4, the 1200–1300 cm−1 region at tint was different from that observed for FpH 4 (Figure 1c). Based on these results, we conclude that the initial increase in 381W fluorescence at high sRPT concentrations at pH 5 does not represent an oligomeric intermediate, but rather an “immature” fibrillar state.
3.6. pH-dependent stability of sRPT fibrils.
To investigate the stability of sRPT fibrils, the pH of the buffer was altered (by dilution) and 381W fluorescence kinetics were monitored over a period of 3 days (Figure 6). Dilution of FpH 4 into pH 4 buffer did not result in any significant kinetics change in 381W fluorescence intensity (Figure 6a). However, dilution of FpH 4 into pH 5 and 6 buffer showed a steady decrease in 381W fluorescence intensity. Collecting 381W emission scans of FpH 4 at the end of 3 days revealed a concomitant blue-shift and intensity decrease in emission as the pH increased from 4 to 6 (Figure 6a inset), suggesting that the environment of 381W for FpH 4 can slowly transition into a more FpH 5- or FpH 6-like state. We confirmed that this observation was not a result of fibril dissociation, as all samples still showed increased ThT fluorescence intensity (Figure S4), except for pH 7 in which disaggregation occurred as described earlier in Figure 3. No changes in FpH 5 or FpH 6 were observed upon dilution at pH 4, 5, or 6 (Figure 6b–c), indicating that these fibrils are stable to pH changes.
Fig. 6.
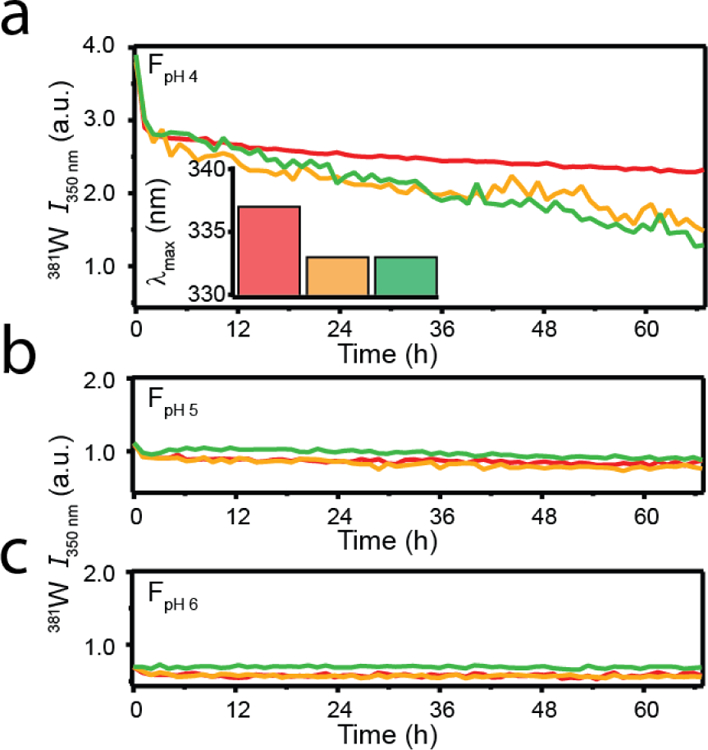
pH-dependent stability of sRPT fibrils. sRPT fibrils (30 µM) formed at pH 4 (a), 5 (b), or 6 (d) were diluted 6-fold into buffers at pH 4 (red), 5 (yellow), and 6 (green). Averaged data are shown (n ≥ 3). (Inset) λmax values extracted from 381W emission scans at the end of the reaction.
4. Discussion
4.1. sRPT aggregation and fibril maturation mirror melanosomal conditions.
Described in this work are the pH- and concentration-dependent aggregation of the Pmel17 sRPT domain, which are summarized schematically in Figure 7. At pH 4, sRPT rapidly formed immature fibrils (FI) at concentrations as low as 3 µM. This highly-acidic environment mimics that of stages I–II melanosomes, where Pmel17 fibril formation is thought to initiate [4, 5]. A protein concentration-dependent process was observed at pH 5 for sRPT, where FI were formed at high concentrations (≥30 µM) at a rate similar to that observed at pH 4. Aggregation at lower concentrations (≤15 µM) directly led to mature fibril (FM) formation, albeit slower. At pH 6, FM formation proceeded much slower than those observed at pH 4 and 5.
Fig. 7.
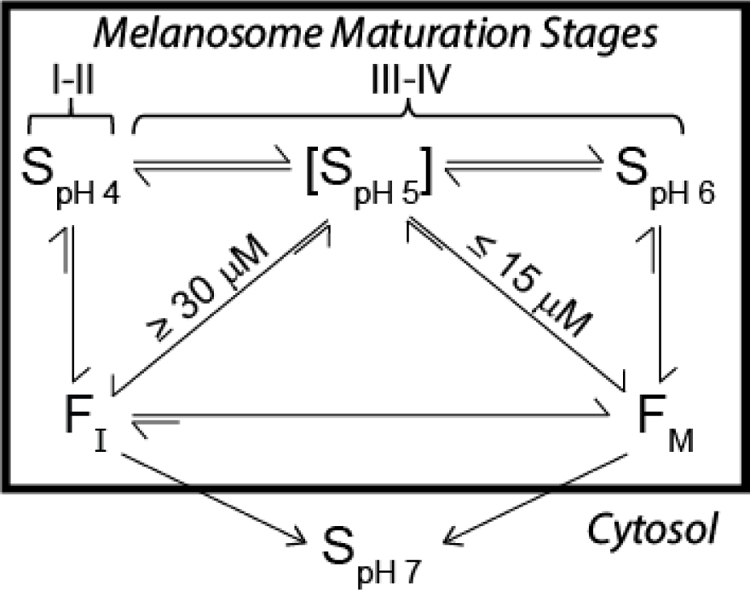
sRPT aggregation and fibril maturation mirror melanosomal conditions. S represents soluble sRPT at pH 5, 6, or 7. FI and FM represent immature and mature sRPT fibrils, respectively. At pH 4, sRPT rapidly forms FI, which converts into FM at pH 5 and 6. This process mimics melanosome maturation, which is at ~pH 4 in stages I–II, and reaches near neutral pH upon full maturation (stage IV). sRPT displays concentration-dependent behavior at pH 5, forming FI at concentrations above 30 µM and FM at concentrations below 15 µM. At pH 6, sRPT forms FM. Both FI and FM dissolve into their soluble form at pH 7.
To mimic the maturation process of melanosomes, sRPT fibrils made at pH 4 were diluted into buffers of increasing pH. This revealed that FI can mature into FM, but diluting FM (generated at pH 5 and 6) into more acidic buffers had no effect on the environment of 381W. Collectively, a picture emerges where fibrils are rapidly formed in early stages at pH 4, which undergo conformational dynamics as melanosome maturation proceeds to pH 6. A similar mechanism has previously been proposed for lRPT [18]. While no obvious morphological differences in the fibrils formed could be discerned by TEM under the solution conditions examined here, reorganization was revealed in the C-terminus by 381W fluorescence, which was corroborated by the differences in C–H deformation frequencies in the Raman spectra. In the context of melanogenesis, it remains to be investigated if similar aggregation mechanisms would be observed in the lipid-rich environment of the melanosome, or if melanin would bind to one fibril polymorph (FI or FM) more strongly than the other. However, it is noteworthy that FI formed at pH 4 has the highest ThT response which we inferred as having a stronger affinity for ThT, which Fowler et al. have suggested to resemble the molecular features of a melanin precursor [3].
4.2. The importance of the C-terminus in sRPT aggregation/disaggregation.
McGlinchey et al. have previously shown that sRPT fibrils dissolve upon exposure to neutral pH [16], which may be an evolutionary mechanism designed to prevent toxicity should Pmel17 fibrils escape the melanosome and enter the cytosol [17]. Here, 381W was used to monitor the disaggregation process in real-time. This revealed that FM immediately disassembled with no observable lag time, while a short 7-s lag time was observed for FI at pH 4, indicating that dissolution is more difficult for FI compared to FM. The reverse was observed for the formation of the fibrils, where FI is faster than FM.
The pH-dependent aggregation/disaggregation process of sRPT further substantiates the importance of protonation/deprotonation events, which has been described in detail for lRPT [15]. Specifically, the residue 422E (380E in sRPT) was the primary (de)protonation site driving aggregation/disaggregation of lRPT. While we do not directly investigate this for sRPT, it is noteworthy that 381W lies directly on the C-terminal side of this residue. Given the similar spectral changes in 381W observed during aggregation reported here, it is likely that sRPT aggregation proceeds in a similar fashion, where the protonation of 380E facilitates sRPT fibril formation at acidic pH. However, because we observe a protein concentration-dependent aggregation kinetics for sRPT at pH 5, the mechanism may be more nuanced than lRPT and further investigation is needed.
4.3. Biological implications for sRPT in melanogenesis.
The conserved pH-dependent aggregation/disaggregation observed for both sRPT and lRPT, along with work from Nichols et al. showing the two isoforms can be expressed independently in MNT-1 cells [21], leads us to speculate an interplay between the two isoforms in vivo. In bacterial functional amyloid systems, specific genes within the operon serve as nucleators of amyloid formation. In E. coli, the CsgB protein rapidly forms a fibrillar template upon which CsgA binds and elongates to form the biofilm matrix [33, 34]. Likewise, a similar role for FapB has been put forward for amyloid formation in P. aeruginosa [35, 36]. Because sRPT rapidly forms fibrils at pH 4, it is reasonable to propose that it could serve as a nucleator of lRPT (and therefore, Pmel17) amyloid formation within the melanosome. Future work testing this hypothesis could reveal an evolutionary-conserved mechanism of functional amyloid formation from bacteria to humans.
Supplementary Material
Acknowledgements
This work was supported by the Intramural Research Program at the National Institutes of Health, National Heart, Lung, and Blood Institute. We thank the NHLBI Biochemistry (LC-MS), Biophysics (CD), and Electron Microscopy (TEM) Core Facilities for the use of instrumentation and expert advice.
Abbreviations:
- Pmel17
pre-melanosomal protein
- sRPT
short repeat domain
- FI
immature fibril
- FM
mature fibril
ABOUT THE AUTHORS

Dr. Dexter Dean joined the Lee lab as a postdoctoral fellow after completing his Ph.D. in Biochemistry at The University of Southern Mississippi in 2018. Prior to his Ph.D., he received a B.A. in Biochemistry from Huntingdon College in 2013. He is interested in using biophysical tools to understand mechanisms of amyloid formation as it relates to both function and disease.

Dr. Jennifer C. Lee is a Senior Investigator at the National Heart, Lung, and Blood Institute, National Institutes of Health. Her research efforts are dedicated towards the elucidation of mechanisms of amyloid formation through biophysical and biochemical approaches. She is interested in understanding the chemical nature of intermolecular interactions that drive amyloid formation in a cellular environment. Prior to joining the NIH, she was a Beckman Senior Research Fellow at the Beckman Institute Laser Resource Center at the California Institute of Technology. Jennifer obtained a Ph.D. in Chemistry from Caltech in 2002 working in the laboratory of Harry Gray and has undergraduate degrees in Chemistry and Economics from the University of California at Berkeley.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
REFERENCES
- [1].Knowles TP, Vendruscolo M, Dobson CM, The amyloid state and its association with protein misfolding diseases, Nat. Rev. Mol. Cell Biol, 15 (2014) 384–396. [DOI] [PubMed] [Google Scholar]
- [2].Fowler DM, Koulov AV, Balch WE, Kelly JW, Functional amyloid – from bacteria to humans, Trends Biochem. Sci, 32 (2007) 217–224. [DOI] [PubMed] [Google Scholar]
- [3].Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW, Functional Amyloid Formation within Mammalian Tissue, PLoS Biol, 4 (2005) e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Berson JF, Harper DC, Tenza D, Raposo G, Marks MS, Pmel17 initiates premelanosome morphogenesis within multivesicular bodies, Mol. Biol. Cell, 12 (2001) 3451–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hurbain I, Geerts WJ, Boudier T, Marco S, Verkleij AJ, Marks MS, Raposo G, Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets, Proc. Natl. Acad. Sci. U. S. A, 105 (2008) 19726–19731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS, Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells, J. Cell Biol, 152 (2001) 809–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hoashi T, Muller J, Vieira WD, Rouzaud F, Kikuchi K, Tamaki K, Hearing VJ, The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers, J. Biol. Chem, 281 (2006) 21198–21208. [DOI] [PubMed] [Google Scholar]
- [8].Valencia JC, Rouzaud F, Julien S, Chen KG, Passeron T, Yamaguchi Y, Abu-Asab M, Tsokos M, Costin GE, Yamaguchi H, Jenkins LM, Nagashima K, Appella E, Hearing VJ, Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils, J. Biol. Chem, 282 (2007) 11266–11280. [DOI] [PubMed] [Google Scholar]
- [9].Harper DC, Theos AC, Herman KE, Tenza D, Raposo G, Marks MS, Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes, J. Biol. Chem, 283 (2008) 2307–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS, Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis, J. Cell Biol, 161 (2003) 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Watt B, van Niel G, Fowler DM, Hurbain I, Luk KC, Stayrook SE, Lemmon MA, Raposo G, Shorter J, Kelly JW, Marks MS, N-terminal domains elicit formation of functional Pmel17 amyloid fibrils, J. Biol. Chem, 284 (2009) 35543–35555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McGlinchey RP, Lee JC, Reversing the Amyloid Trend: Mechanism of Fibril Assembly and Dissolution of the Repeat Domain from a Human Functional Amyloid, Isr. J. Chem, 57 (2017) 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].McGlinchey RP, Lee JC, Why Study Functional Amyloids? Lessons from the Repeat Domain of Pmel17, J. Mol. Biol, (2018). [DOI] [PMC free article] [PubMed]
- [14].Hee JS, Mitchell SM, Liu X, Leonhardt RM, Melanosomal formation of PMEL core amyloid is driven by aromatic residues, Sci. Rep, 7 (2017) 44064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McGlinchey RP, Jiang Z, Lee JC, Molecular origin of pH-dependent fibril formation of a functional amyloid, Chembiochem, 15 (2014) 1569–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McGlinchey RP, Shewmaker F, Hu KN, McPhie P, Tycko R, Wickner RB, Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH, J. Biol. Chem, 286 (2011) 8385–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McGlinchey RP, Shewmaker F, McPhie P, Monterroso B, Thurber K, Wickner RB, The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis, Proc. Natl. Acad. Sci. U. S. A, 106 (2009) 13731–13736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pfefferkorn CM, McGlinchey RP, Lee JC, Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17, Proc. Natl. Acad. Sci. U. S. A, 107 (2010) 21447–21452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jiang Z, Lee JC, Lysophospholipid-containing membranes modulate the fibril formation of the repeat domain of a human functional amyloid, pmel17, J. Mol. Biol, 426 (2014) 4074–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dogra P, Bhattacharya M, Mukhopadhyay S, pH-Responsive Mechanistic Switch Regulates the Formation of Dendritic and Fibrillar Nanostructures of a Functional Amyloid, J. Phys. Chem. B, 121 (2017) 412–419. [DOI] [PubMed] [Google Scholar]
- [21].Nichols SE, Harper DC, Berson JF, Marks MS, A novel splice variant of Pmel17 expressed by human melanocytes and melanoma cells lacking some of the internal repeats, J. Invest. Dermatol, 121 (2003) 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kelly SM, Jess TJ, Price NC, How to study proteins by circular dichroism, Biochim. Biophys. Acta, 1751 (2005) 119–139. [DOI] [PubMed] [Google Scholar]
- [23].Flynn JD, McGlinchey RP, Walker RL 3rd, Lee JC, Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy, J. Biol. Chem, 293 (2018) 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].LeVine H, Quantification of β-sheet amyloid fibril structures with thioflavin T, Methods Enzymol, 309 (1999) 274–284. [DOI] [PubMed] [Google Scholar]
- [25].Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R, Solid state NMR reveals a pH-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide, J. Mol. Biol, 335 (2004) 247–260. [DOI] [PubMed] [Google Scholar]
- [26].Elgersma RC, Kroon-Batenburg LMJ, Posthuma G, Meeldijk JD, Rijkers DTS, Liskamp RMJ, pH-controlled aggregation polymorphism of amyloidogenic Aβ(16–22): Insights for obtaining peptide tapes and peptide nanotubes, as function of the N-terminal capping moiety, Eur. J. Med. Chem, 88 (2014) 55–65. [DOI] [PubMed] [Google Scholar]
- [27].Moriarty GM, Olson MP, Atieh TB, Janowska MK, Khare SD, Baum J, A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues, J. Biol. Chem, 292 (2017) 16368–16379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu W, Zhang C, Morozova-Roche L, Zhang JZH, Mu Y, pH-Dependent Conformational Ensemble and Polymorphism of Amyloid-β Core Fragment, J. Phys. Chem. B, 117 (2013) 8392–8399. [DOI] [PubMed] [Google Scholar]
- [29].McGlinchey RP, Lee JC, Cysteine cathepsins are essential in lysosomal degradation of α-synuclein, Proc. Natl. Acad. Sci. U. S. A, 112 (2015) 9322–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McGlinchey RP, Dominah GA, Lee JC, Taking a Bite Out of Amyloid: Mechanistic Insights into α-Synuclein Degradation by Cathepsin L, Biochemistry, 56 (2017) 3881–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Flynn JD, Lee JC, Raman fingerprints of amyloid structures, Chem. Commun. (Camb.), 54 (2018) 6983–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Movasaghi Z, Rehman S, Rehman IU, Raman Spectroscopy of Biological Tissues, Appl. Spectrosc. Rev, 42 (2007) 493–541. [Google Scholar]
- [33].Hammer ND, Schmidt JC, Chapman MR, The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization, Proc. Natl. Acad. Sci. U. S. A, 104 (2007) 12494–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deshmukh M, Evans ML, Chapman MR, Amyloid by Design: Intrinsic Regulation of Microbial Amyloid Assembly, J. Mol. Biol, 430 (2018) 3631–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rouse SL, Matthews SJ, Dueholm MS, Ecology and Biogenesis of Functional Amyloids in Pseudomonas, J. Mol. Biol, 430 (2018) 3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dueholm MS, Otzen D, Nielsen PH, Evolutionary Insight into the Functional Amyloids of the Pseudomonas, PLoS One, 8 (2013) e76630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.