

Abstract
Post-translational modification by small ubiquitin-like modifier (SUMO) has emerged as a global mechanism for the control and integration of a wide variety of biological processes through the regulation of protein activity, stability and intracellular localization. As SUMOylation is examined in greater detail, it has become clear that the process is at the root of several pathologies including heart, endocrine, and inflammatory disease, and various types of cancer. Moreover, it is certain that perturba-tion of this process, either globally or of a specific protein, accounts for many instances of congenital birth defects. In order to be successful, practical strategies to ameliorate conditions due to disruptions in this post-translational modification will need to consider the multiple components of the SUMOylation machinery and the extraordinary number of proteins that undergo this modification.
Keywords: SUMO, SUMO ligase, SENP protease, heart development, heart failure, cancer, neurodegenerative disease, congenital birth defects, spina bifida, neural tube
1. INTRODUCTION
1.1. The SUMO Pathway
Small ubiquitin-like modifier (SUMO), an 11 kDa polypeptide, is present in four isoforms in higher eukaryotes (reviewed in [1-3]). SUMO2 and SUMO3 share 97% identity and are frequently represented as SUMO2/3; they share only about 50% identity with SUMO1. To a large degree, SUMO1 and SUMO2/3 are functionally distinct, exhibiting different target specificity in most, but not all, cases [4, 5]. SUMO isoforms 1 to 3 are expressed ubiquitously, whereas the occurrence of SUMO4 appears to be restricted to renal, immune, placental, and pancreatic cells [6-8].
The mechanism of SUMO conjugation to target proteins is similar to that of the ubiquitination pathway (Fig. 1). The precursor forms of SUMO are activated through proteolytic processing by members of the sentrin-specific proteases (SENP) to generate a C-terminal di-glycine, which is a substrate for the first enzyme in the pathway, the E1 SUMO-activating enzyme. This heterodimer, comprised of SAE1 and UBA2 subunits, uses ATP to form a thioester linkage between SUMO and a cysteine in UBA2 [9]. The SUMO moiety is then passed to the E2 SUMO conjugating enzyme (UBC9). Although E2 is capable of acting directly on target proteins, in the majority of cases an E3 ligase is required for efficient transfer of the SUMO polypeptide to a specific target. Whereas only single E1 and E2 enzymes have been identified to date, there are multiple proteins with E3 activity that confer specificity and, thus, regulation on this process. Similarly, there are multiple members of the SENP family that remove SUMO from modified proteins and, thereby, add an additional level of control over this reversible post-translational modification [10].
Fig. (1).
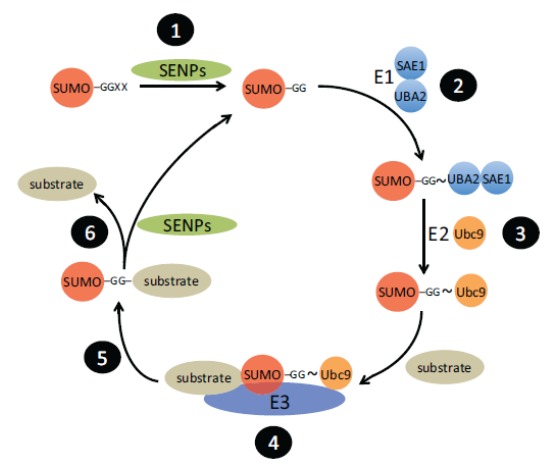
The SUMO pathway. SUMO precursor polypeptide undergoes cleavage by a SENP protease to reveal a C-terminal di-glycine sequence (1) which is the substrate for the ATP-dependent formation of the covalent intermediate with the E1 SUMO activating enzyme (2). The SUMO polypeptide is transferred to the E2 SUMO conjugating enzyme (3). In most cases, an E3 SUMO ligase brings together the E2 enzyme and substrate protein (4) to promote the transfer SUMO to the target (5). SUMO moieties can be removed by members of the SENP family of proteases (6).
The SUMO modification of proteins appears to occur primarily in the nucleus. The expression of epitope-tagged SUMO2 in human cells combined with flow cytometry enabled the identification of more than 1,600 proteins conjugated to this SUMO isoform [11], the majority of which are nuclear proteins or those that are nucleocytoplasmic. This observation reflects the large number of transcription factors, RNA binding proteins, chromatin-associated proteins, and cell cycle regulators that are documented targets of SUMOylation [12, 13]. However, a bioinformatics analysis of Xenopus egg extract revealed that an appreciable number (39.5%) of the identified SUMOylated proteins were linked to metabolic processes and protein synthesis (translation), indicating the importance of this post-translational modification in cytoplasmic processes as well [14]. Subcellular compartmentalization of the SUMO machinery has also been reported. The E3 ligase, PIAS2b, is confined to the cytoplasm of Xenopus oocytes, but then immediately relocates to the nucleus after fertilization [15].
1.2. Target Specificity
The steady-state level of SUMOylated protein in any given instance is typically low and has made detection of SUMO-modified proteins difficult [16]. A number of strategies have been implemented in order to identify targets and a recent compilation that encompassed all available proteomic data revealed that more than 3,500 human proteins undergo SUMOylation [17]. Formation of the isopeptide bond to target lysines usually occurs within the consensus sequence ΨKX(D/E) where Ψ is a hydrophobic side chain. However, with the accumulation of more proteomic data, it is clear that lysines not embedded in this consensus sequence are also targeted. There is excellent evidence that SUMOylation of a particular site can be influenced by adjacent post-translational modifications that include ubiquitination, acetylation, and methylation [18]. Notwithstanding these modifications, phosphorylation appears to be the most frequently used regulator of SUMOylation. Phosphorylation-dependent SUMO motifs (ΨKX(D/E)XXSP) are targets of proline-directed kinases such as the cyclin-dependent kinases that likely account for the role of SUMO in progression through the cell cycle [12]. However, regulation of SUMOylation by phosphorylation in the absence of this sequence motif has also been documented [3]. The SUMOylation of lysines that do not occur in a consensus sequence has been documented, however, it is not clear how specificity is achieved in these cases [19, 20].
1.3. SUMOylation Products
Like ubiquitin, SUMO2/3 adducts can occur in the form of polymeric chains whose formation is reversible due to the action of SENP6, SENP7, and Ulp2 proteases [21]. On the other hand, SUMO1 lacks a consensus sequence for E2, which presumably accounts for its presence as a monomer or a cap on SUMO2/3 chains. Interestingly, the SUMO family members can undergo a variety of post-translational modifications that include phosphorylation, acetylation, and ubiquitination [17]. The variety of possible polymeric structures combined with different post-translational modifications of individual subunits can generate molecules that rival glycoproteins in structural complexity and the ability to produce intricate signaling codes.
1.4. SUMO Interaction Motifs
SUMO conjugation can affect the target protein in a number of ways: stability, intracellular localization, conformation, or as a substrate for other post-translational modifications (often as a mutually exclusive alternative to ubiquitination). However, the primary role of SUMOylation is the control of protein-protein interactions. While the addition of the polypeptide could block an interaction simply by steric exclusion, SUMO often mediates contact to proteins that contain SUMO-interacting motifs (SIMs). β-strand structures in the SIM and SUMO interact chiefly by hydrophobic contacts with electrostatic contacts also contributing to binding [22, 23]. For some SIMs, phosphorylation of an adjacent serine residue can enhance interaction with SUMO [24]. It is particularly notable that just as transcription factors are frequent targets of SUMOylation, many also possess SIMs, explaining the major role of SUMO in the regulation of transcription and chromatin structure.
1.5. The SUMO Pathway is Essential for Viability
Knockout and knockdown mutations have shown that the SUMO pathway is essential in eukaryotes from yeast to vertebrates [12, 25-28]. Mice deficient in the E2-conjugating enzyme die at an early post-implantation stage [27]. The loss of E2 enzyme results in a wide variety of nuclear phenotypes that include nuclear envelope dysmorphy, disruption of nuclear bodies, and failure of RanGAP1 to incorporate into the nuclear pore complex [27]. Similarly, SUMO2 deficiency is embryonic lethal, whereas, SUMO3 is not [29]. SUMO1 knockout is not lethal and loss of this isoform appears to be compensated by SUMO2/3 [30, 31]. Gene knockout or other methods of gene inactivation that target other components of the SUMOylation machinery (i.e., E3 ligases and SENP proteases) result in a variety of phenotypes [26].
2. SUMO and DISEASE
Given the large number of proteins regulated by SUMOylation and the variety of outcomes, it is not surprising that this post-translational modification has now been implicated in a number of disparate diseases. We present a brief summary of pathologies where a connection to SUMOylation is firmly established and note those where a practical drug target may exist. We also call attention to those cases where a therapeutic strategy is less obvious.
2.1. Cancer
SUMO targets proteins involved in the cell cycle [12, 32, 33], chromatin structure [34, 35], DNA repair [36], as well as a variety of transcription regulatory factors that act as oncogenes and tumor suppressors [37, 38], accounting for the frequent connection of various cancers to this post-translational modification. Indeed, among the first SUMO targets identified were promyelocytic leukemia protein (PML), a nuclear phosphoprotein, associated with acute promyelocytic leukemia [39] and p53 tumor suppressor [40, 41].
Correlations of SUMOylation activity with cancer can involve components of the SUMO machinery (i.e., E1, E2, E3, or SENP proteases), transcription factors, or signaling molecules. In the first instance, altered levels of E1 (SAE1/SAE2) have been implicated in lung [42, 43] and gastric [44] cancers and lymphoma [45]. A much greater number of cancers have been linked to E2 (Ubc9) activity [46-53] and E3 (ligase) activity [52, 54-62]. An equally large number of carcinoma cells have exhibited changes in the expression of SENP proteases (see reviews in [12, 37, 63]). The mutations that have been associated with various cancers can result in either an increase or decrease in the levels of SUMO-modified proteins, underscoring the fact that a simple imbalance in this post-translational modification is enough to be carcinogenic. SUMOylation itself is neither pro- nor anti-oncogenic.
The wide range of SUMO targets and the disparate effects of under or overexpression of constituents of the SUMO pathway make them challenging drug targets. Nonetheless, several small molecule inhibitors of E1, E2, and SENP proteases have been reported [63, 64]. Therapeutic success may be realized only in cases where the drug can be delivered specifically to cancer cells. However, if such specificity can be realized, these molecules could be highly effective cytotoxins, given the appreciable number of cell cycle regulators and DNA repair enzymes that are regulated by SUMOylation [12, 36, 65-67]. Thus, cell death could be initiated through a number of disparate pathways working in parallel.
The epithelial-to-mesenchymal transition (EMT) is an essential process for cancer cell invasion and metastasis; moreover, the underlying changes in gene expression are also necessary for the establishment of cancer stem cell properties. The reprogramming of the transcriptome during the EMT is obviously controlled by the activity of an appreciable number of transcription factors, many of which are regulated by SUMOylation [68]. Again, the consequences of changes in SUMOylation activity are likely to be complex, since several of the transcription factors that promote EMT and its converse, MET, are targeted by SUMO. And while SUMOylation of this group of factors generally activates transcription that favors the mesenchymal phenotype, this outcome is not absolute. Whereas inhibition of SUMOylation of FOXM1 and TFAP2A should promote an epithelial phenotype [69, 70], loss of SUMOylation should promote a TGFβ-induced mesenchymal phenotype [68]. Thus, many factors such as cell type, metabolic status, and microenvironment likely would determine how a change in SUMOylation activity impacts EMT in any particular instance. In Xenopus embryos in which SUMOylation activity is knocked down, there is a decreased expression of twist, zeb2, vimentin, and laminb1, and increased expression of zo2 that together indicate an epithelial phenotype [71]. The overall balance between mesenchymal and epithelial states may be tipped by SUMOylation, but in a therapeutic regimen, other cellular factors and processes may also need to be simultaneously manipulated along with any drug that targets the SUMOylation machinery.
Several proteins that are classified as tumor suppressors or oncogenes are targeted by SUMOylation. Regulation of p53 [72], Ras [73], and Myc [64] by SUMO have been reviewed. Myc controls the expression of several components of the SUMO pathway and overexpression of the oncogene can account for a hyper-SUMOylation state that promotes proliferation of breast cancer cells and B-cell lymphoma [45, 74]. When E1 activity is knocked down, Myc-driven cell growth is halted and cells ultimately undergo cell death. Importantly, small molecule inhibitors of E1 (anacardic acid) or E2 (2-D08) were effective in inducing growth arrest and apoptosis in B-cell lymphoma cells, but not in cells that overexpress other oncogenes. This differential effect indicates that these small molecules are promising candidates for cancers driven by Myc. The usual caveat still applies concerning specificity and the complexity of the SUMO regulation [74]. Transcription factor network building demonstrated that Myc is highly connected to genes affected by knock down of SUMOylation, indicating that dosing and duration of action will also be aspects of any therapeutic regiment [71]. Nonetheless, preliminary studies in mice are promising [45].
Other examples of synthetic lethality or “non-oncogene addition” to SUMOylation have been reported for NOTCH1 [75] and Ras [76]. In the latter case, the mutant cells did not exhibit a hyper-SUMOylation state, but rather a more limited number of proteins whose degree of modification was changed [76]. Thus, there is an expanding number of malignant transformations that require and depend on an elevated level of SUMOylation activity. Success at exploiting this phenotype will, in the first instance, require the development of drugs that target the SUMO pathway at considerably lower IC50 concentrations and, in the second instance, have minimal impact on the steady-state SUMOylation activity of normal cells.
There has been less examination of p53 most likely because regulation of p53 activity by SUMOylation is complex with the modification having a positive, negative, or no effect depending on the target gene in question [72]. In addition, regulators and co-activators of p53 such as MDM2 are also targets of SUMO [77]. However, it is reasonable to expect that, given the role of p53 in DNA repair, inhibition of SUMOylation activity would compromise its role in this process and, thus, contribute to cell death.
The exceptional number of cases in which a particular cancer exhibits dysregulation of SUMOylation, either globally or of a specific protein, makes a powerful case for the development of new drugs with improved IC50 and/or cell specificity. General inhibitors of the SUMO pathway, i.e., those that target E1, E2, or SENP proteases, would be particularly useful in those cases where the malignant transformation depends on highly elevated levels of SUMOylation activity, provided these drugs do not perturb the activity of normal cells. In cases that arise from a mutation in a single protein, the challenges are considerably greater, since the drug would likely need to be targeted specifically to the aberrant protein.
2.2. Cardiovascular Disease
SUMO plays multiple and varied roles in the development and function of the cardiovascular system [78-83]. Decreased levels of SUMOylated proteins have been detected in cardiac injury due to ischemia [84, 85], dilated cardiomyopathy [86], and cardiac failure [87, 88], while increased levels are observed in some conduction system defects [89]. These cardiac phenotypes can, in some instances, be ameliorated by restoration of SUMOylated protein levels or SUMOylation activity.
Dysfunction of individual components of the SUMO pathway has been specifically attributed to particular cardiovascular abnormalities. Elevated levels of SENP5 occur in some instances of human idiopathic cardiac failure. Transgenic mice overexpressing SENP5 under control of the cardiac specific α-MHC promoter exhibited dilated cardiomyopathy and heart failure due to decreased cell proliferation and increased apoptosis [90]. The hearts of these transgenic mice presented with diminished levels of SUMOylated Drp1, which is a factor critical for mitochondrial fission during steady state and apoptosis [90]. Knockdown of E2 expression using coronary artery infusion of short hairpin RNA lentiviral vectors significantly reduced the progression of cardiac fibrosis in mice that had undergone transverse aortic constriction [91]. Alternatively, transgenic mice overexpressing E2 in cardiomyocytes exhibited increased cardiac autophagy, which was associated with decreased fibrosis, reduced hypertrophy, and improved cardiac function and survival [92]. These studies reflect a discordant role for E2 in cardiac disease that is likely due to widespread disruption of SUMOylation activity in cardiomyopathy and illustrates the need for targeted therapies that spare global SUMOylation machinery and instead modify the specific factors perturbed during the disease state.
The ATPase controlling calcium reuptake in the membrane of the sarcoplasmic reticulum of cardiac muscle, SERCA2a, is activated and stabilized by SUMOylation [87]. Reduction of SERCA2a activity compromises cardiac contractility and results in cardiac failure in a mouse model [87]. Restoration of SUMOylated SERCA2a via SUMO-1 overexpression rescued the cardiac failure phenotype by improving hemodynamic function and reducing mortality. Both SUMO-1 and SERCA2a gene delivery using an Adeno-Associated Vector type 1 (AAV1) administered by antegrade coronary artery infusion significantly improved cardiac function and stabilized left ventricular volumes in a swine model of ischemic cardiac failure [85]. Post-mortem examination of the AAV1-treated swine two months following gene delivery did not reveal pathologies associated with off target effects. However, it will be critical to monitor for long term sequela of these therapies considering the systemic drug access afforded by coronary artery infusion and the compelling evidence of SUMOylation anomalies in many malignancies [37].
The small molecule, N-(4-methoxybenzo[d]thiazol-2-yl)-5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-amine (N106), increases the level of SUMOylated SERCA2a and improves contractility in a model of heart failure [93]. This drug targets the E1 enzyme of the SUMO pathway and increases its activity. However, N106 increases the overall level of SUMOylated proteins in general, which may restrict its use to acute treatment strategies. Luteolin, a plant-derived flavone, can partially reverse myocardial ischemia and reperfusion injury. This natural product appears to work on many levels, decreasing expression of miR-208b-3p [94], stimulating signaling through the PI3K/Akt pathway [95], and increasing expression of SUMO1 and concomitant SUMOylation of SERCA2 [84]. Moreover, this drug can have a protective effect, with pretreatment reducing myocardial infarct size after myocardial ischemia and reperfusion injury [84]. While N106 and luteolin represent promising therapeutic targets that improve cardiac contractility by increasing SERCA2a SUMOylation, studies of long-term dosing and possible side effects, particularly increased incidents of malignancies, will determine whether they lead to a practical treatment for predispositions to heart failure.
Lamin A is a nuclear structural protein that plays a critical role in maintaining the cell nucleus and is a target of SUMO2 modification. Two mutations identified in naturally occurring human familial dilated cardiomyopathy alter a region of the protein necessary for SUMO modification; patients bearing these mutations presented lower levels of SUMO-conjugated lamin A [86]. Therapeutic strategies aimed at increasing levels of SUMOylated lamin A in patients presenting with these mutations represent a promising drug target that could, in part, rescue these dilated cardiomyopathies.
SUMOylation has also been implicated in defects of the cardiac conduction system. Mutations in TRPM4, a calcium-activated nonselective cation channel, prevent deSUMOylation of TRPM4 [89]. This gain of function mutation results in impaired endocytosis and increased levels of cation channels at the cell surface. The resulting increase in membrane leak conductance prevents action potential propagation through Purkinje fibers, which are cardiac cells specialized for conduction. Phenotypically, this mutation results in progressive familial heart block (type 1), a disease characterized by diminished function of the cardiac electrical system [89]. In this instance, the disease state results from increased levels of SUMOylated TRPM4. This is in contrast to the cardiac injuries due to ischemia, cardiac failure, and dilated cardiomyopathy described above, which are characterized by reductions in SUMOylation. These studies further iterate the need for therapeutic strategies that target the SUMOylation status of a specific protein rather than those that globally modify SUMOylation activity.
2.3. Neurodegenerative Disease
A number of proteins implicated in a variety of neurodegenerative diseases are targets of SUMOylation ([96] for a recent review of this topic). Indeed, multiple targets of SUMO may contribute to a particular neurological disorder with a noteworthy number of these proteins being involved in stress responses. In the case of Parkinson’s Disease (PD), an example is DJ-1, a transcription co-activator that becomes SUMOylated as part of the antioxidant response to ROS (Reactive Oxygen Species) [97] and to UV irradiation [98]. The loss of DJ-1 function is seen as a marker for early stage PD and two different mutations in the gene have been observed in PD patients. One mutant removes the site of SUMO addition at K130 [98], while a second mutation that involves a substitution of proline for leucine at position 166 (L166P) actually enhances SUMOylation of the protein and seemingly leads to its degradation [99]. In the latter case, a drug specific for the L166P mutant protein that makes it a poorer substrate for either the E2 conjugating enzyme or its cognate E3 ligase could potentially restore the correct balance in DJ-1 solubility and, thus, be an effective treatment to lessen or delay the onset of PD pathology.
Another causative agent of PD is α-synuclein, which is also implicated in certain dementias, because of its presence in Lewy bodies. SUMOylation of α-synuclein appears to decrease its propensity to form aggregates [100], although contradictory data, i.e., SUMO-induced aggregation of α-synuclein, has also been presented [101]. Until there is a better understanding of the normal function of α-synuclein and the effects of SUMOylation on its activity, it is not a promising drug target for PD.
Parkin is a ubiquitin E3 ligase that is mutated in a significant number of instances of PD, consistent with the idea that deficits in the ubiquitin-proteasome system contribute to this disease [102]. Parkin associates with SUMO1 and promotes the ubiquitination and degradation of the SUMO E3 ligase, RanBP2 as well as α-synuclein [103]. Information to date suggests that the noncovalent interaction of Parkin with SUMO1 has an allosteric effect on the former and its nuclear localization. Thus, screens for drugs that alter this protein-protein interaction would appear to be a promising starting point for a PD therapeutic.
Multiple targets of SUMO are at the heart of Alzheimer’s disease (AD) etiology, including Amyloid Precursor Protein (APP), β-secretase (BACE1), and tau. It is widely held that the proteolytic cleavage of APP by BACE1 to generate amyloid-beta (Aβ) peptide is a major causative factor in AD [104]. Deposition of Aβ plaques is a critical feature of AD even at early stages of the disease. Increased SUMOylation of APP is correlated with decreased levels of Aβ aggregation [105]. Consistent with this observation, increased expression of the SUMO E2 enzyme along with SUMO1 has the same mitigating effect. Thus, one can envision a therapeutic strategy directed towards APP that is similar to the employment of the small molecules, N106 and luteolin, used to increase SUMOylation of SERCA2 to treat predispositions to heart failure. Such a targeted approach, as opposed to simple overexpression of SUMO1, circumvents the problem that increased SUMOylation of BACE1 leads to increased levels of Aβ [106].
Defective variants of tau, a microtubule associated protein abundant in neurons, are implicated in both PD and AD. The protein undergoes three post-translational modifications that impact its activity: phosphorylation, ubiquitination, and SUMOylation. There is evidence that ubiquitination and SUMOylation of tau is mutually exclusive [107], while SUMOylation and phosphorylation have a mutually positive effect on each other that prevent ubiquitin-dependent degradation of tau [108]. Therapeutically, combined inhibition of SUMOylation and phosphorylation could have a synergistic effect on the accumulation of tau that, in turn, would counteract tau aggregation and the formation of neurofibrillary tangles.
An apparent competition between ubiquitination and SUMOylation has also been established for the Huntingtin protein (HTT), the causative agent of Huntington’s Disease (HD) [109]. SUMOylation of mutant HTT, which has an expanded poly-glutamine sequence, increases its stability, but decreases aggregation, that together may contribute to cytotoxicity [109]. In a Drosophila model of HD, decreased levels of SUMO reduced neurodegeneration, suggesting that targeting mutant HTT to reduce its SUMOylation could be an effective means to stop HD. However, a far better target may be the G protein, Rhes (Ras homolog enriched in striatrum), that stimulates the SUMOylation of mutant HTT, but not the wild type protein [110]. In this case, Rhes is acting like an E3 ligase, specifically for the mutant protein. Thus, inhibiting Rhes activity would allow for exquisite specificity with no perturbation of the SUMO proteome. Since Rhes expression is limited to the corpus striatum, a drug targeted to this protein would act in a tissue-specific fashion. Given the many adverse consequences of globally increasing or decreasing SUMOylation activity, it is essential to identify targets, as exemplified by Rhes, that will alter the modification of one or a limited number of proteins.
Other SUMOylated proteins that have been implicated in neurodegerative diseases include ataxin-1, hypoxia-inducible factor 1α (HIF-1α), PML, superoxide dismutase 1 (SOD1) and various ataxin variants [96].
2.4. Congenital Birth Defects
General disruption of SUMOylation activity has been tied to developmental defects. For example, SUMO1 knockout mice exhibit ventricular and atrial septal defects [79]. A 16-base pair substitution mutation in the upstream promoter region of the SUMO1 gene was identified in a patient displaying both oral-facial clefts and atrial septal defects, suggesting that diminished SUMOylation activity during gestation can often be the source of certain congenital defects [79]. This notion was underscored when a cohort of 87 newborns with atrial septal defects and oral-facial clefts were compared to 100 control newborns. Five members of the former group had mutations in the promoter region of the SUMO1 gene that resulted in decreased levels of the protein, while no mutations at SUMO1 were found in the control group [79]. SUMO1 hypomorphic mice exhibit reduced expression of several cell proliferation genes and increased expression of liver enriched genes, reflecting the extremely wide range of gene regulation by SUMO and the challenges of developing therapeutics for either a deficiency or excess of this protein, especially for in utero treatment.
Cardiac specific overexpression of SENP2, a deSUMOylation enzyme, results in septal defects and cardiomyopathy in adult mice [111]. A decrease in cardiomyocytes was also observed. Since the SENP2 enzyme is specific for proteins conjugated to the SUMO1 isoform, it is likely that the remaining SUMO isoforms partially compensate for this loss [29, 31]. Furthermore, a SENP2 knockout in mice caused defects in the embryonic heart, which were correlated to an accumulation of SUMOylated forms of Pc2/CBX4 that led to a repression of GATA4 expression [82].
Experiments with Xenopus embryos in which all SUMOylation activity was temporarily knocked down following fertilization up to early neuralization exhibited two prominent phenotypes later in development: heart defects and failure of neural tube closure (spina bifida) [71]. The former phenotype is consistent with earlier work [82, 112]; whereas, other than a report of SUMOylation of thymidylate synthase and dihydrofolate reductase [113], there has been no proposed causative link between SUMO and spina bifida. However, the knockdown of SUMOylation activity in Xenopus embryos disrupted planar cell polarity and supports the proposed relationship between this pathway and risk for spina bifida [114]. Perhaps the most notable observation in this case is that the temporary disruption of SUMOylation activity during the earliest stages of embryogenesis can have phenotypic consequences long after the activity has returned to normal levels, emphasizing the challenges of treating instances of SUMOylation dysfunction that give rise to congenital defects. It is noteworthy that developmental abnormalities of the heart and neural tube are the two most commonly occurring birth defects in humans.
Transcription factors represent the majority of SUMO targets. Thus, it is not surprising that the post-translational modification has been implicated in congenital birth defects. While the most prominent are cleft lip/palate [115, 116] and heart defects [78, 79, 111, 112, 117], other developmental processes are likely disrupted due to aberrant SUMOylation [26]. Transcription factors that serve as master regulators of heart development and are modified by SUMO include serum response factor (SRF) [118], myocardin [81], GATA4 [80], Nkx2.5 [83], mef2 [119], YY1 [120], and prox1 [121]. Consequently, it is not surprising that proper development of the cardiovascular system is exceptionally sensitive to any perturbations of SUMOylation activity.
SRF is a highly conserved transcription factor required for mesoderm formation [122] and for the emergence of cardiac sarcomeres [123]. Conditional SRF knockout in cardiac progenitor cells blocks the appearance of rhythmic beating myocytes [123]. Rescue of SRF–/– null embryonic stem cells with a virally expressed SRF lysine 147 to alanine substitution mutation, which prevents SUMOylation, did not restore the expression of several SRF target genes, demonstrating the necessity of this post-translational modification for proper cardiovascular development [123]. Myocardin is a co-activator of SRF that must also be SUMOylated in order to turn on critical cardiac genes such as MLC2 and BMP10 [81].
The homeobox gene Nkx2.5 is required for normal cardiac development and is highly conserved among vertebrates. It is expressed both in early cardiac progenitor cells before cardiogenic differentiation and in adult cardiac cells. Nkx2.5 is modified by SUMOylation at lysine 51 and mutation of this site to an arginine residue prevents addition of SUMO and reduces the ability of Nkx2.5 to bind DNA [83]. Homozygous knockout mice develop hearts that fail to undergo looping morphogenesis, exhibit poor blood circulation, and develop pericardial edema [124]. These embryos die during embryogenesis due to early hemodynamic insufficiency.
YY1 is a ubiquitously expressed transcription factor that regulates genes involved in embryonic development, apoptosis, and cell cycle regulation; as such, it has been associated with a number of cancers [125, 126]. YY1 is necessary for cardiomyocyte differentiation; knocking out YY1 during early development (before formation of the precardiac mesoderm) prevents cardiomyocyte formation [127]. YY1 activity is regulated by SUMOylation on lysine 288 [120, 126].
GATA4, a zinc-finger transcription factor, regulates expression of several cardiac genes such as α-myosin heavy chain (α-MHC) and Atrial Natriuretic Factor (ANF). Mutation of lysine 366 to arginine blocks SUMOylation of GATA4 and alters its nuclear localization [80]. In mice, point mutations in GATA4 that impair its interaction with FOG-2, an essential cardiac co-factor, result in septation and coronary vasculature defects [128] similar to those seen in human patients with congenital heart defects linked to GATA4 mutations [129].
SUMOylation also impacts development through its regulation of autophagy [130, 131], which is critical for many morphological processes, including cardiac development [132-134]. Morpholino knockdowns of essential autophagy genes (atg5, atg7, becn1) in zebrafish embryos resulted in numerous cardiac abnormalities including failure in looping and valve defects [133].
While there is overwhelming evidence that mutations that disrupt SUMOylation, either globally or of a specific protein, are frequently the basis for congenital defects that occur during early development, how they can be identified and corrected is much less obvious. The experiments with Xenopus revealed that even temporary repression of SUMOylation activity during early embryogenesis can engender acute morphological defects at later points in development [71], meaning preventative intervention would likely have to occur prior to fertilization. At this time, genetic screening combined with gene editing (of eggs) appears to be the most feasible, albeit complex, means to this end.
CONCLUSION AND FUTURE DIRECTIONS
The activity, stability, and intracellular location of a considerable number of proteins, implicated in a spectrum of diseases, are regulated by SUMOylation. This has prompted considerable interest in the development of drugs that target members of the SUMOylation pathway. However, the scope of the SUMO network and the huge number of biological processes under its control presents seemingly insurmountable challenges. The association of various cancers with either increased or decreased levels of SUMOylation suggests that any perturbation of this activity can have a negative outcome. Notwithstanding this caveat, general inhibitors may still hold promise in cases of synthetic lethality where the cancer relies on a hyper-SUMOylation state. This therapeutic strategy will require dosing that leaves the SUMOylation activity of normal cells unchanged while reducing the activity of cancer cells sufficiently to prevent oncogene-driven malignant transformation. Another reasonable approach is to deliver an inhibitor of the SUMOylation machinery directly to cancer cells.
Similar concerns apply to the small molecule drug, N106, which increases E1 activity and, thereby, increases SUMOylation of SERCA2, and has been proposed as a treatment for heart failure [93]. Long-term studies will be needed to determine whether there are increased incidents of cancers or neurodegenerative diseases with prolonged treatment.
In cases where the disease can be traced to a single protein, different strategies can be envisioned. If a mutation does not eliminate the site of SUMOylation, but only changes the degree, efforts can be made to identify a molecule that targets and restores normal levels of modification specifically of this protein. It is worth noting that only a fraction of any given protein appears to be SUMOylated in vivo, suggesting that there may be considerable latitude in identifying molecules that change the degree of SUMOylation of any individual protein [16]. Between drugs that globally affect SUMOylation by targeting the SUMO pathway and drugs that bind to an individual protein and change its activity as a substrate for SUMO modification, there could potentially be a class of drugs that target one of the E3 SUMO ligases. Since the ligases promote SUMOylation of a subset of proteins, targeting a specific E3 ligase would be less disruptive to the SUMO-proteome, reducing the potential for inducing other diseases associated with SUMOylation dysfunction. An excellent example is the aforementioned Rhes protein that acts as an E3 ligase, specifically for the mutant HTT protein [110]. Drugs that target E3 ligases of the ubiquitin system have, in fact, shown some success [135].
There is accumulating evidence that an appreciable number of incidents of congenital birth defects are due to mutations that either reduce SUMOylation activity in general [71, 79, 114] or prevent modification of a specific protein [117, 128, 136]. These cases will require identification of mutations and corrective measures (i.e., gene therapy or gene editing) prior to fertilization. While there are no examples of this type of intervention at present, its ultimate realization gives additional urgency to the development of CRISPR methods for gene repair [137].
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
Research in the authors’ laboratory was supported by the National Institutes of Health (GM38200) and the University of Notre Dame. We thank Drs. Mariam Malik (National Cancer Institute) and Michelle Bertke (University of Maryland) for initiating our work on SUMO and Kyle Dubiak and Chao Yang for careful reading of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Geiss-Friedlander R., Melchior F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 2.Zhao J. Sumoylation regulates diverse biological processes. Cell. Mol. Life Sci. 2007;64:3017–3033. doi: 10.1007/s00018-007-7137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gareau J.R., Lima C.D. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saitoh H., Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000;275:6252–6258. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 5.Vertegaal A.C., Andersen J.S., Ogg S.C., Hay R.T., Mann M., Lamond A.I. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteomics. 2006;5:2298–2310. doi: 10.1074/mcp.M600212-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Baczyk D., Audette M.C., Drewlo S., Levytska K., Kingdom J.C. SUMO-4: A novel functional candidate in the human placental protein SUMOylation machinery. PLoS One. 2017;12:e0178056. doi: 10.1371/journal.pone.0178056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong-Yi W., Jin-Xiong S. SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metab. Res. Rev. 2008;24:93–102. doi: 10.1002/dmrr.797. [DOI] [PubMed] [Google Scholar]
- 8.Chen S., Yang T., Liu F., et al. Inflammatory factor-specific sumoylation regulates NF-kB signalling in glomerular cells from diabetic rats. Inflamm. Res. 2014;63:23–31. doi: 10.1007/s00011-013-0675-3. [DOI] [PubMed] [Google Scholar]
- 9.Olsen S.K., Capili A.D., Lu X., Tan D.S., Lima C.D. Active site remodeling accompanies thioester bond formation in the SUMO E1. Nature. 2010;463:906–912. doi: 10.1038/nature08765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunz K., Piller T., Muller S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018:131. doi: 10.1242/jcs.211904. [DOI] [PubMed] [Google Scholar]
- 11.Hendriks I.A., D’Souza R.C., Yang B., et al. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014;21:927–936. doi: 10.1038/nsmb.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eifler K., Vertegaal A.C. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem. Sci. 2015;40:779–793. doi: 10.1016/j.tibs.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iñiguez-Lluhi J.A. in: wilson vg, editor. sumo regulation of cellular processes: Springer Netherlands; 2009. SUMO modification and transcriptional regulation. pp. 13–40. [Google Scholar]
- 14.Ma L., Aslanian A., Sun H., et al. Identification of small ubiquitin-like modifier substrates with diverse functions using the Xenopus egg extract system. Mol. Cell. Proteomics. 2014;13:1659–1675. doi: 10.1074/mcp.M113.035626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malik M.Q., Bertke M.M., Huber P.W. Small ubiquitin-like modifier (SUMO)-mediated repression of the Xenopus oocyte 5 S rRNA genes. J. Biol. Chem. 2014;289:35468–35481. doi: 10.1074/jbc.M114.609123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay R.T. SUMO: a history of modification. Mol. Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 17.Hendriks I.A., Vertegaal A.C.O. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016;17:581–595. doi: 10.1038/nrm.2016.81. [DOI] [PubMed] [Google Scholar]
- 18.Hendriks I.A., Lyon D., Young C., Jensen L.J., Vertegaal A.C., Nielsen M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017;24:325–336. doi: 10.1038/nsmb.3366. [DOI] [PubMed] [Google Scholar]
- 19.Karvonen U., Jaaskelainen T., Rytinki M., Kaikkonen S., Palvimo J.J. ZNF451 is a novel PML body- and SUMO-associated transcriptional coregulator. J. Mol. Biol. 2008;382:585–600. doi: 10.1016/j.jmb.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Figueroa-Romero C., Iniguez-Lluhi J.A., Stadler J., et al. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009;23:3917–3927. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vertegaal A.C. SUMO chains: polymeric signals. Biochem. Soc. Trans. 2010;38:46–49. doi: 10.1042/BST0380046. [DOI] [PubMed] [Google Scholar]
- 22.Reverter D., Lima C.D. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435:687–692. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song J., Zhang Z., Hu W., Chen Y. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J. Biol. Chem. 2005;280:40122–40129. doi: 10.1074/jbc.M507059200. [DOI] [PubMed] [Google Scholar]
- 24.Stehmeier P., Muller S. Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell. 2009;33:400–409. doi: 10.1016/j.molcel.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Eifler K., Vertegaal A.C. Mapping the SUMOylated landscape. FEBS J. 2015;282:3669–3680. doi: 10.1111/febs.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lomeli H., Vazquez M. Emerging roles of the SUMO pathway in development. Cell. Mol. Life Sci. 2011;68:4045–4064. doi: 10.1007/s00018-011-0792-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nacerddine K., Lehembre F., Bhaumik M., et al. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell. 2005;9:769–779. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Nowak M., Hammerschmidt M. Ubc9 regulates mitosis and cell survival during zebrafish development. Mol. Biol. Cell. 2006;17:5324–5336. doi: 10.1091/mbc.E06-05-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L., Wansleeben C., Zhao S., et al. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014;15:878–885. doi: 10.15252/embr.201438534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evdokimov E., Sharma P., Lockett S.J., Lualdi M., Kuehn M.R. Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell Sci. 2008;121:4106–4113. doi: 10.1242/jcs.038570. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F.P., Mikkonen L., Toppari J., et al. Sumo-1 function is dispensable in normal mouse development. Mol. Cell. Biol. 2008;28:5381–5390. doi: 10.1128/MCB.00651-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S.J., Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 33.Cuijpers S.A.G., Vertegaal A.C.O. Guiding mitotic progression by crosstalk between post-translational modifications. Trends Biochem. Sci. 2018;43:251–268. doi: 10.1016/j.tibs.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Wotton D., Pemberton L.F., Merrill-Schools J. SUMO and Chromatin Remodeling. Adv. Exp. Med. Biol. 2017;963:35–50. doi: 10.1007/978-3-319-50044-7_3. [DOI] [PubMed] [Google Scholar]
- 35.Cubeñas-Potts C., Matunis M.J. SUMO: a multifaceted modifier of chromatin structure and function. Dev. Cell. 2013;24:1–12. doi: 10.1016/j.devcel.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson S.P., Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Seeler J.S., Dejean A. SUMO and the robustness of cancer. Nat. Rev. Cancer. 2017;17:184–197. doi: 10.1038/nrc.2016.143. [DOI] [PubMed] [Google Scholar]
- 38.Lee J.S., Choi H.J., Baek S.H. Sumoylation and Its Contribution to Cancer. Adv. Exp. Med. Biol. 2017;963:283–298. doi: 10.1007/978-3-319-50044-7_17. [DOI] [PubMed] [Google Scholar]
- 39.Duprez E., Saurin A.J., Desterro J.M., et al. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J. Cell Sci. 1999;112:381–393. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]
- 40.Gostissa M., Hengstermann A., Fogal V., et al. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999;18:6462–6471. doi: 10.1093/emboj/18.22.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez M.S., Desterro J.M., Lain S., et al. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inamura K., Shimoji T., Ninomiya H., et al. A metastatic signature in entire lung adenocarcinomas irrespective of morphological heterogeneity. Hum. Pathol. 2007;38:702–709. doi: 10.1016/j.humpath.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 43.Liu X., Xu Y., Pang Z., et al. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J. Hematol. Oncol. 2015;8:67. doi: 10.1186/s13045-015-0164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shao D.F., Wang X.H., Li Z.Y., et al. High-level SAE2 promotes malignant phenotype and predicts outcome in gastric cancer. Am. J. Cancer Res. 2015;5:140–154. [PMC free article] [PubMed] [Google Scholar]
- 45.Hoellein A., Fallahi M., Schoeffmann S., et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 2014;124:2081–2090. doi: 10.1182/blood-2014-06-584524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S.F., Gong C., Luo M., et al. Ubc9 expression predicts chemoresistance in breast cancer. Chin. J. Cancer. 2011;30:638–644. doi: 10.5732/cjc.011.10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moschos S.J., Jukic D.M., Athanassiou C., et al. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum. Pathol. 2010;41:1286–1298. doi: 10.1016/j.humpath.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Yang W., Wang L., Roehn G., et al. Small ubiquitin-like modifier 1-3 conjugation is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci. 2013;104:70–77. doi: 10.1111/cas.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDoniels-Silvers A.L., Nimri C.F., Stoner G.D., Lubet R.A., You M. Differential gene expression in human lung adenocarcinomas and squamous cell carcinomas. Clin. Cancer Res. 2002;8:1127–1138. [PubMed] [Google Scholar]
- 50.Tomasi M.L., Tomasi I., Ramani K., et al. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology. 2012;56:982–993. doi: 10.1002/hep.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moschos S.J., Smith A.P., Mandic M., et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene. 2007;26:4216–4225. doi: 10.1038/sj.onc.1210216. [DOI] [PubMed] [Google Scholar]
- 52.Driscoll J.J., Pelluru D., Lefkimmiatis K., et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115:2827–2834. doi: 10.1182/blood-2009-03-211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu F., Zhu S., Ding Y., Beck W.T., Mo Y.Y. MicroRNA-mediated regulation of Ubc9 expression in cancer cells. Clin. Cancer Res. 2009;15:1550–1557. doi: 10.1158/1078-0432.CCR-08-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu B., Tahk S., Yee K.M., et al. PIAS1 regulates breast tumorigenesis through selective epigenetic gene silencing. PLoS One. 2014;9:e89464. doi: 10.1371/journal.pone.0089464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coppola D., Parikh V., Boulware D., Blanck G. Substantially reduced expression of PIAS1 is associated with colon cancer development. J. Cancer Res. Clin. Oncol. 2009;135:1287–1291. doi: 10.1007/s00432-009-0570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei J., Costa C., Ding Y., et al. mRNA expression of BRCA1, PIAS1, and PIAS4 and survival after second-line docetaxel in advanced gastric cancer. J. Natl. Cancer Inst. 2011;103:1552–1556. doi: 10.1093/jnci/djr326. [DOI] [PubMed] [Google Scholar]
- 57.Rabellino A., Carter B., Konstantinidou G., et al. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res. 2012;72:2275–2284. doi: 10.1158/0008-5472.CAN-11-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J., Xu Y., Long X.D., et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 2014;25:118–131. doi: 10.1016/j.ccr.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Sun L., Li H., Chen J., et al. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J. Cell Sci. 2013;126:3939–3947. doi: 10.1242/jcs.127381. [DOI] [PubMed] [Google Scholar]
- 60.Chien W., Lee K.L., Ding L.W., et al. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br. J. Cancer. 2013;109:1795–1804. doi: 10.1038/bjc.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoefer J., Schafer G., Klocker H., et al. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012;180:2097–2107. doi: 10.1016/j.ajpath.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 62.Wang L., Banerjee S. Differential PIAS3 expression in human malignancy. Oncol. Rep. 2004;11:1319–1324. [PubMed] [Google Scholar]
- 63.Yang Y., Xia Z., Wang X., et al. Small-molecule inhibitors targeting protein sumoylation as novel anticancer compounds. Mol. Pharmacol. 2018;94:885–894. doi: 10.1124/mol.118.112300. [DOI] [PubMed] [Google Scholar]
- 64.Licciardello M.P., Kubicek S. Pharmacological treats for SUMO addicts. Pharmacol. Res. 2016;107:390–397. doi: 10.1016/j.phrs.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 65.Schimmel J., Eifler K., Sigurethsson J.O., et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell. 2014;53:1053–1066. doi: 10.1016/j.molcel.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 66.Eifler K., Cuijpers S.A.G., Willemstein E., et al. SUMO targets the APC/C to regulate transition from metaphase to anaphase. Nat. Commun. 2018;9:1119. doi: 10.1038/s41467-018-03486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarangi P., Zhao X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 2015;40:233–242. doi: 10.1016/j.tibs.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bogachek M.V., De Andrade J.P., Weigel R.J. Regulation of epithelial-mesenchymal transition through SUMOylation of transcription factors. Cancer Res. 2015;75:11–15. doi: 10.1158/0008-5472.CAN-14-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bogachek M.V., Chen Y., Kulak M.V., et al. Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell. 2014;25:748–761. doi: 10.1016/j.ccr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang C.M., Liu R., Wang L., Nascimento L., Brennan V.C., Yang W.H. SUMOylation of FOXM1B alters its transcriptional activity on regulation of MiR-200 family and JNK1 in MCF7 human breast cancer cells. Int. J. Mol. Sci. 2014;15:10233–10251. doi: 10.3390/ijms150610233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bertke MM, Cronin L, Zeng E, Huber PW. PW. A deficiency in SUMOylation activity disrupts multiple pathways leading to neural tube and heart defects in Xenopus embryos. Bio Rxiv. 2019;3:0. doi: 10.1186/s12864-019-5773-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hock A., Vousden K.H. Regulation of the p53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 2010;42:1618–1621. doi: 10.1016/j.biocel.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 73.Zhang H., Luo J. SUMO wrestling with Ras. Small GTPases. 2016;7:39–46. doi: 10.1080/21541248.2016.1161698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kessler J.D., Kahle K.T., Sun T., et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Sci. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Licciardello M.P., Mullner M.K., Durnberger G., et al. NOTCH1 activation in breast cancer confers sensitivity to inhibition of SUMOylation. Oncogene. 2015;34:3780–3790. doi: 10.1038/onc.2014.319. [DOI] [PubMed] [Google Scholar]
- 76.Yu B., Swatkoski S., Holly A., et al. Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc. Natl. Acad. Sci. USA. 2015;112:E1724–E1733. doi: 10.1073/pnas.1415569112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miyauchi Y., Yogosawa S., Honda R., Nishida T., Yasuda H. sumoylation of mdm2 by protein inhibitor of activated stat (pias) and ranbp2 enzymes. J. Biol. Chem. 2002;277:50131–50136. doi: 10.1074/jbc.M208319200. [DOI] [PubMed] [Google Scholar]
- 78.Costa M.W., Lee S., Furtado M.B., et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One. 2011;6:e24812. doi: 10.1371/journal.pone.0024812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J., Chen L., Wen S., et al. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res. A Clin. Mol. Teratol. 2011;91:468–476. doi: 10.1002/bdra.20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang J., Feng X.H., Schwartz R.J. SUMO-1 modification activated GATA4-dependent cardiogenic gene activity. J. Biol. Chem. 2004;279:49091–49098. doi: 10.1074/jbc.M407494200. [DOI] [PubMed] [Google Scholar]
- 81.Wang J., Li A., Wang Z., et al. Myocardin sumoylation transactivates cardiogenic genes in pluripotent 10T1/2 fibroblasts. Mol. Cell. Biol. 2007;27:622–632. doi: 10.1128/MCB.01160-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J., Schwartz R.J. Sumoylation and regulation of cardiac gene expression. Circ. Res. 2010;107:19–29. doi: 10.1161/CIRCRESAHA.110.220491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J., Zhang H., Iyer D., Feng X.H., Schwartz R.J. Regulation of cardiac specific nkx2.5 gene activity by small ubiquitin-like modifier. J. Biol. Chem. 2008;283:23235–23243. doi: 10.1074/jbc.M709748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Du Y., Liu P., Xu T., et al. Luteolin modulates SERCA2a leading to attenuation of myocardial ischemia/ reperfusion injury via sumoylation at lysine 585 in mice. Cell. Physiol. Biochem. 2018;45:883–898. doi: 10.1159/000487283. [DOI] [PubMed] [Google Scholar]
- 85.Tilemann L., Lee A., Ishikawa K., et al. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013;5:211ra159. doi: 10.1126/scitranslmed.3006487. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Y.Q., Sarge K.D. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J. Cell Biol. 2008;182:35–39. doi: 10.1083/jcb.200712124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kho C., Lee A., Jeong D., et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee A., Jeong D., Mitsuyama S., et al. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid. Redox Signal. 2014;21:1986–2001. doi: 10.1089/ars.2014.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kruse M., Schulze-Bahr E., Corfield V., et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 2009;119:2737–2744. doi: 10.1172/JCI38292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim E.Y., Zhang Y., Beketaev I., et al. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J. Mol. Cell. Cardiol. 2015;78:154–164. doi: 10.1016/j.yjmcc.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 91.Liu Y., Zhao D., Qiu F., et al. Manipulating PML SUMOylation via silencing ubc9 and rnf4 regulates cardiac fibrosis. Mol. Ther. 2017;25:666–678. doi: 10.1016/j.ymthe.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupta M.K., McLendon P.M., Gulick J., et al. UBC9-Mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ. Res. 2016;118:1894–1905. doi: 10.1161/CIRCRESAHA.115.308268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kho C., Lee A., Jeong D., et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015;6:7229. doi: 10.1038/ncomms8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bian C., Xu T., Zhu H., et al. Luteolin inhibits ischemia/reperfusion-induced myocardial injury in rats via downregulation of microrna-208b-3p. PLoS One. 2015;10:e0144877. doi: 10.1371/journal.pone.0144877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang F., Li D., Pan H., et al. Luteolin inhibits apoptosis and improves cardiomyocyte contractile function through the PI3K/Akt pathway in simulated ischemia/reperfusion. Pharmacol. 2011;88:149–158. doi: 10.1159/000330068. [DOI] [PubMed] [Google Scholar]
- 96.Anderson D.B., Zanella C.A., Henley J.M., Cimarosti H. Sumoylation: implications for neurodegenerative diseases. In: Wilson V.G., editor. SUMO regulation of cellular processes. Cham: Springer International Publishing; 2017. pp. 261–281. [DOI] [PubMed] [Google Scholar]
- 97.Zhong N., Xu J. Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: regulation by SUMOylation and oxidation. Hum. Mol. Genet. 2008;17:3357–3367. doi: 10.1093/hmg/ddn230. [DOI] [PubMed] [Google Scholar]
- 98.Shinbo Y., Niki T., Taira T., et al. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 2006;13:96–108. doi: 10.1038/sj.cdd.4401704. [DOI] [PubMed] [Google Scholar]
- 99.Moore D.J., Zhang L., Dawson T.M., Dawson V.L. A missense mutation (L166P) in DJ-1, linked to familial Parkinson’s disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 100.Abeywardana T., Pratt M.R. Extent of inhibition of alpha-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochem. 2015;54:959–961. doi: 10.1021/bi501512m. [DOI] [PubMed] [Google Scholar]
- 101.Kim Y.M., Jang W.H., Quezado M.M., et al. Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. J. Neurol. Sci. 2011;307:157–161. doi: 10.1016/j.jns.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tan E.K., Skipper L.M. Pathogenic mutations in Parkinson disease. Hum. Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 103.Um J.W., Chung K.C. Functional modulation of parkin through physical interaction with SUMO-1. J. Neurosci. Res. 2006;84:1543–1554. doi: 10.1002/jnr.21041. [DOI] [PubMed] [Google Scholar]
- 104.Vassar R., Bennett B.D., Babu-Khan S., et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Sci. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 105.Zhang Y.Q., Sarge K.D. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 2008;374:673–678. doi: 10.1016/j.bbrc.2008.07.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yun S.M., Cho S.J., Song J.C., et al. SUMO1 modulates Abeta generation via BACE1 accumulation. Neurobiol. Aging. 2013;34:650–662. doi: 10.1016/j.neurobiolaging.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 107.Dorval V., Fraser P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- 108.Luo H.B., Xia Y.Y., Shu X.J., et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA. 2014;111:16586–16591. doi: 10.1073/pnas.1417548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Steffan J.S., Agrawal N., Pallos J., et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Sci. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 110.Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Sci. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim E.Y., Chen L., Ma Y., et al. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J. Mol. Cell. Cardiol. 2012;52:638–649. doi: 10.1016/j.yjmcc.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mendler L., Braun T., Muller S. The Ubiquitin-Like SUMO system and heart function: from development to disease. Circ. Res. 2016;118:132–144. doi: 10.1161/CIRCRESAHA.115.307730. [DOI] [PubMed] [Google Scholar]
- 113.Anderson D.D., Woeller C.F., Stover P.J. Small ubiquitin-like modifier-1 (SUMO-1) modification of thymidylate synthase and dihydrofolate reductase. Clin. Chem. Lab. Med. 2007;45:1760–1763. doi: 10.1515/CCLM.2007.355. [DOI] [PubMed] [Google Scholar]
- 114.Wen S., Zhu H., Lu W., et al. Planar cell polarity pathway genes and risk for spina bifida. Am. J. Med. Genet. A. 2010;152A:299–304. doi: 10.1002/ajmg.a.33230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pauws E., Stanier P. FGF signalling and SUMO modification: new players in the aetiology of cleft lip and/or palate. Trends Genet. 2007;23:631–640. doi: 10.1016/j.tig.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 116.Alkuraya F.S., Saadi I., Lund J.J., et al. SUMO1 haploinsufficiency leads to cleft lip and palate. Sci. 2006;313:1751. doi: 10.1126/science.1128406. [DOI] [PubMed] [Google Scholar]
- 117.Kim E.Y., Chen L., Ma Y., et al. Expression of sumoylation deficient Nkx2.5 mutant in Nkx2.5 haploinsufficient mice leads to congenital heart defects. PLoS One. 2011;6:e20803. doi: 10.1371/journal.pone.0020803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Matsuzaki K., Minami T., Tojo M., et al. Serum response factor is modulated by the SUMO-1 conjugation system. Biochem. Biophys. Res. Commun. 2003;306:32–38. doi: 10.1016/s0006-291x(03)00910-0. [DOI] [PubMed] [Google Scholar]
- 119.Grégoire S., Tremblay A.M., Xiao L., et al. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J. Biol. Chem. 2006;281:4423–4433. doi: 10.1074/jbc.M509471200. [DOI] [PubMed] [Google Scholar]
- 120.Deng Z., Wan M., Sui G. PIASy-mediated sumoylation of Yin Yang 1 depends on their interaction but not the RING finger. Mol. Cell. Biol. 2007;27:3780–3792. doi: 10.1128/MCB.01761-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pan M.R., Chang T.M., Chang H.C., et al. Sumoylation of Prox1 controls its ability to induce VEGFR3 expression and lymphatic phenotypes in endothelial cells. J. Cell Sci. 2009;122:3358–3364. doi: 10.1242/jcs.050005. [DOI] [PubMed] [Google Scholar]
- 122.Arsenian S., Weinhold B., Oelgeschlager M., Ruther U., Nordheim A. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 1998;17:6289–6299. doi: 10.1093/emboj/17.21.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Niu Z., Iyer D., Conway S.J., et al. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc. Natl. Acad. Sci. USA. 2008;105:17824–1789. doi: 10.1073/pnas.0805491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lyons I., Parsons L.M., Hartley L., et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 125.Khachigian L.M. The Yin and Yang of YY1 in tumor growth and suppression. Int. J. Cancer. 2018;143:460–465. doi: 10.1002/ijc.31255. [DOI] [PubMed] [Google Scholar]
- 126.Deng Z., Cao P., Wan M.M., Sui G. Yin Yang 1: a multifaceted protein beyond a transcription factor. Transcription. 2010;1:81–84. doi: 10.4161/trns.1.2.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gregoire S., Karra R., Passer D., et al. Essential and unexpected role of Yin Yang 1 to promote mesodermal cardiac differentiation. Circ. Res. 2013;112:900–910. doi: 10.1161/CIRCRESAHA.113.259259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Crispino J.D., Lodish M.B., Thurberg B.L., et al. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev. 2001;15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garg V., Kathiriya I.S., Barnes R., et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 130.Naidu S.R., Lakhter A.J., Androphy E.J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle. 2012;11:2717–2728. doi: 10.4161/cc.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang Y., Fiskus W., Yong B., et al. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA. 2013;110:6841–6846. doi: 10.1073/pnas.1217692110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb. Symp. Quant. Biol. 2011;76:397–402. doi: 10.1101/sqb.2011.76.011023. [DOI] [PubMed] [Google Scholar]
- 133.Lee E., Koo Y., Ng A., et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10:572–587. doi: 10.4161/auto.27649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Terman A., Brunk U.T. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc. Res. 2005;68:355–365. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 135.Nalepa G., Rolfe M., Harper J.W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 136.Bartlett H.L., Sutherland L., Kolker S.J., et al. Transient early embryonic expression of Nkx2-5 mutations linked to congenital heart defects in human causes heart defects in Xenopus laevis. Dev. Dyn. 2007;236:2475–2484. doi: 10.1002/dvdy.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]