

SUMMARY
Successful anti-viral response requires the sustained activation and expansion of CD8+ T cells for periods that far exceed the time limit of physical T cell interaction with antigen-presenting cells (APCs). The expanding CD8+ T cell pool generates the effector and memory cell populations that provide viral clearance and long-term immunity, respectively. Here, we demonstrate that 3BP2 is recruited in cytoplasmic microclusters and nucleates a signaling complex that facilitates MHC:peptide-independent activation of signaling pathways downstream of the TCR. We show that induction of the adaptor molecule 3BP2 is a sensor of TCR signal strength and is critical for sustaining CD8+ T cell proliferation and regulating effector and memory differentiation.
In Brief
Dimitriou et al. show that the adaptor protein 3BP2 lowers the threshold of T cell activation and that the induction of the 3BP2 signaling module at later time points may serve to recapitulate and prolong the biochemical signals emanating from the TCR required for sustained MHC:peptide-independent T cell proliferation.
Graphical Abstract

INTRODUCTION
CD8+ T cell priming, expansion, and differentiation into effector and long-lived memory cells are controlled by the input of multiple stimuli. These consist of T cell receptor (TCR) signals along with environmental influences including antigen levels, antigen-presenting cell (APC) type and activation status, engagement with costimulation pathways, and availability of CD4 T helper cell-derived cytokines (Cui and Kaech, 2010; Zhang and Bevan, 2011). The generation of CD8+ T cell effector function requires CD8+ T cell programming, whereby an initial, brief encounter with the major histocompatibility complex (MHC):antigen complex commits the naive CD8+ T cells to complete the developmental program that directs proliferation and effector differentiation, in the absence of further antigenic stimulation (Kaech and Ahmed, 2001; van Stipdonk et al., 2001). This process is choreographed in three distinct phases (Mempel et al., 2004). The initial interaction of dendritic cells (DCs) with T cells is random and characterized by high frequency and short duration, leading to initial CD8+T cell activation. The DC-T cell engagement then becomes more sustained and results in full T cell activation associated with cytokine production. Within 24 hr after the initial interaction, T cells disengage from their DC partners, regain a state of high motility, and enter into a highly proliferative blastogenic phase. The initial, brief serial contacts of T cells with the antigen-loaded DCs provide the cognate antigenic and costimulatory signals that allow T cells to exceed a critical strength of signal threshold required to progress into the cell cycle and effector differentiation.
Antigen recognition by the TCR complex in concert with costimulatory receptors and integrins delivers the necessary biochemical information required for survival, proliferation, and differentiation programs (Conley et al., 2016). The TCRα and TCRβ polymorphic chains recognize the peptide-MHC complex and convey antigenic stimulation by coupling to a core signaling complex of enzymes that involves the SRC family kinase LCK, ZAP-70 family protein kinases, PLCγ, and the guanine nucleotide exchange factor for RAC-1 GTPase, VAV. The output of these enzymes activates the MAPK and NFκB pathways, hydrolyzing phosphoinositol bis-phosphate (PIP2) involved in gating calcium flux and the activation of PKC. Activation of small GTPases induces the polymerization of the actin cytoskeleton involved in the control of cell morphology, adhesion, and migration. Although the assembly of this TCR signaling complex is an early event upon TCR engagement, it is not yet well understood how persistence of these signaling pathways is sustained during the expansion and differentiation phases of CD8+ T cells, following the termination of the MHC:antigen-TCR engagement.
The temporal and spatial control of the TCR-induced signaling module is mediated by the adaptor and scaffold proteins, including LAT, GAD, GRB-2, SH2-leukocyte protein-76 (SLP-76), ADAP, and SKAP1 (Sherman et al., 2013). These adaptor molecules contain protein interaction domains, which direct the combinatorial assembly of multiprotein and protein-lipid interactions (Ménasché et al., 2007). Confocal microscopy has revealed that many components of the TCR-induced signaling pathway are assembled in microclusters that are recruited to the activated TCR complex (Bunnell et al., 2002). These signaling clusters are dynamically transported either to the center of the cell (Grakoui et al., 1999) or near to the microtubule organizing center (Bunnell et al., 2002). The composition of the microclusters changes overtime by a rapid exchange with cytosolic components, and the whole microcluster is constantly reforming with new molecules (Douglass and Vale, 2005). These peripheral microclusters are critical for sustained signals, as they contain activated forms of most signaling molecules, and defects in their formation and persistence abolishes T cell activation (Varma et al., 2006). The assembly of signaling proteins in microclusters increases the local concentration of enzymes with their substrates and increases the speed of information flow in the cell.
3BP2 (SH3 binding protein-2) is an adaptor molecule initially identified as the binding partner of the Abl Src-homology 3 (SH3) domain (Ren et al., 1993). 3BP2 is highly expressed in hematopoietic cells and is important for growth and differentiation of monocyte-lineage cells, including osteoclasts, osteoblasts, granulocytes, and mast cells (Ainsua-Enrich et al., 2012; Chen et al., 2012; Levaot et al., 2011a; Ueki et al., 2007). 3BP2 is also required for the activation of marginal zone B cells (Chen et al., 2007) and natural killer (NK) cells (Jevremovic et al., 2001). We and others have shown that 3BP2 forms a complex with SRC family kinases, SYK family kinases, and the Rho-gua-nine nucleotide exchange factor VAV (Deckert and Rottapel, 2006). 3BP2 is mutated in an autosomal disease affecting the development of cranial facial bones called cherubism (Ueki et al., 2001). Cherubism mutations cluster within a span of six amino acids that define the binding site for tankyrase, a poly-ADP ribosyl polymerase (PARP) family member. 3BP2 is post-translationally modified by tankyrase through the addition of ADP-ribosyl chains. ADP ribosylation marks 3BP2 for recognition by the E3-ubiquitin ligase RNF146, which leads to ubiquity-lation and proteasomal-mediated destruction of 3BP2 (Levaot et al., 2011b). A genetically engineered mouse that carries two copies of a cherubism mutation knocked into the sh3bp2 locus (3BP2kI/kI) are severely osteopenic and die of a lethal autoinflam-matory disorder characterized by high levels of TNFα and myeloid monocytic infiltration of visceral organs by 3 months of age (Ueki et al., 2007). Cherubism mutations uncouple 3BP2 from the negative regulation of tankyrase and RNF146, leading to increased steady-state 3BP2 protein levels, thus explaining the autosomal-dominant mode of inheritance of the disease (Levaot et al., 2011b). Basal levels of phosphorylated SRC, VAV, and SYK are elevated in macrophages and osteoclasts derived from the cherubism knockin mice, whereas 3BP2-deficient osteoclasts fail to form normally and do not activate SRC in response to osteopontin (Levaot et al., 2011a). These data demonstrate that 3BP2 is required for activation of SRC family kinases in myeloid cells and is a key regulator of macrophage and osteoclast function.
In addition to these functions within the myeloid compartment, we report that endogenous 3BP2 is required for the proliferation and differentiation of CD8+ T cells. 3BP2-deficient CD8+ T cells are unable to undergo proliferative expansion and mount normal effector and memory responses. We show that 3BP2 is induced upon CD8+ T cell activation and nucleates a signaling complex composed of LCK, ZAP-70, and VAV proteins, which is recruited to microclusters following TCR stimulation. 3BP2 is part of a time-dependent positive feedback loop required for sustained ERK and NFAT activity at late time points during CD8+ T cell activation.
RESULTS
Endogenous 3BP2 Is Required for the Activation and Proliferation of CD8+ T Cells In Vitro
To examine the requirement of endogenous 3BP2 during T cell activation, we purified CD4+ and CD8+ T cells from 3BP2+/+ and 3BP2−/− mice and stimulated them in vitro using limiting amounts of plate-bound anti-CD3 or anti-CD3 plus anti-CD28 antibodies for 24 hr. The surface expression of the T cell activation markers CD69, CD25, CD44, and CD62L was reduced in the absence of 3BP2, particularly at the lower anti-CD3 concentrations (Figure 1A). This defect could be overcome by CD28-medi-ated costimulation (Figure 1A). The in vitro proliferation of the 3BP2-deficient CD8+ T cells was significantly compromised as measured by 3H-thymidine incorporation or carboxyfluorescein succinimidyl ester (CFSE) dilution (Figures 1B and 1C). In distinction to CD8+ T cells, the activation and proliferation of CD4+ T cells derived from wild-type or 3BP2−/− mice was comparable (Figures 1D and S1). We next examined the dynamics of T cell activation from cells derived from 3BP2KI/KI mice. Endogenous 3BP2 protein levels were increased in 3BP2KI/KI CD8+ T cells (Figure 1E). Ex vivo analysis of splenocytes from wild-type and 3BP2kI/kI mice showed that CD8+ T cells were intrinsically activated under basal conditions, as indicated by the surface expression levels of CD69, CD44, and CD62L (Figure 1F), and were proliferating even in the absence of CD28-mediated costi-mulation (Figure 1G). CD4+ T cells from 3BP2KI/KI mice demonstrated no evidence of basal activation or enhanced in vitro pro-liferation rates compared with the wild-type cells (Figures 1H and S2). These data show that 3BP2 manifests distinct functions within the two MHC-restricted T cell lineages and is required for optimal CD8+ T cell activation and proliferation.
Figure 1. CD8+ T Cells, Unlike CD4+ T Cells, Require 3BP2 for Optimal In Vitro Activation and Proliferation.
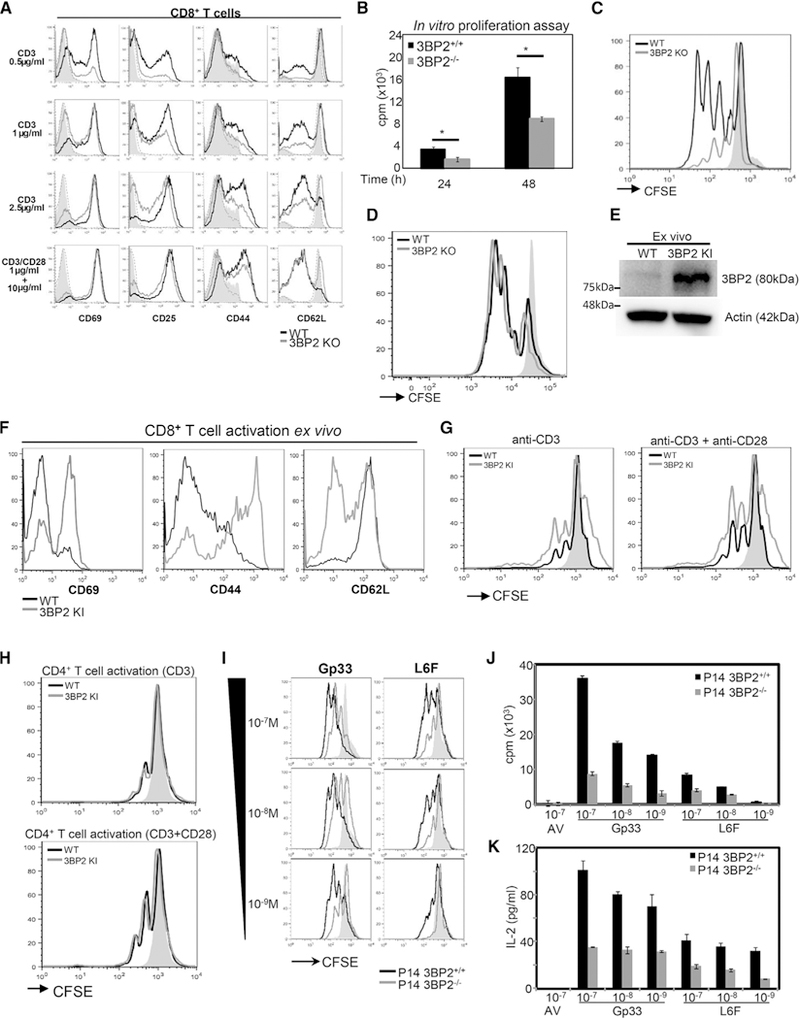
(A) Flow cytometry analysis of CD8+ T cell activation markers in 3BP2+/+ and 3BP2−/− mice after 24 hr of in vitro stimulation with varying concentrations of anti-CD3 alone or with anti-CD3 plus anti-CD28 antibody. Representative plots from at least three independent experiments are shown.
(B and C) In vitro proliferation of CD8+ T cells from 3BP2+/+ and 3BP2−/− mice was measured by either 3H-thymidine incorporation (B) or CFSE dilution (C). In (B), data are shown as mean ± SE from three independent experiments. In (C), a representative flow cytometry histogram is shown at 48 hr upon in vitro stimulation.
(D) The proliferation of CD4+ T cells from 3BP2+/+ and 3BP2−/− mice was measured by flow cytometry analysis at 48 hr upon in vitro stimulation (anti-CD3 + anti-CD28).
(E) Western blotanalysisofthe3BP2 protein expression in CD8+ T cellsfrom 3BP2+/+and 3BP2KI/KI mice ex vivo. Three independent experimentswere performed.
(F) Flow cytometry analysis of the ex vivo expression of surface activation markers In CD8+T cells from 3BP2+/+ and 3BP2kI/kI mice. Representative plots from at least three independent experiments are shown.
(G) Representative flow cytometry histograms of CFSE dilution analysis are shown from 3BP2+/+ and 3BP2kI/kI CD8+ T cells at 48 hr upon in vitro stimulation.
(H) The proliferation of CD4+ T cells from 3BP2+/+ and 3BP2kI/kI mice was measured by flow cytometry analysis at 48 hr upon in vitro stimulation (anti-CD3 + anti-CD28).
(I) CFSE dilution analysis at 48 hr upon P14+T cell stimulation in vitro with increasing concentrations of theGp33and the L6F antigenic peptides. Representative of more than three experiments is shown.
(J) In vitro proliferation of 3BP2+/+ or 3BP2-deficient P14+ T cells was determined by 3H-thymidine incorporation at 48 hr upon antigenic peptide stimulation.
(K) IL-2 production by peptide stimulated P14+ T cells was determined using an ELISA assay.
Gray areas and dotted lines in all histograms indicate unstimulated cells from 3BP2+/+ and 3BP2−/− mice or 3BP2+/+ and 3BP2ki/ki mice, respectively.
To delineate further the role of 3BP2 in the regulation of TCR signaling in CD8+ T cells, we created antigen-specific CD8+ T cells by crossing P14 transgenic mice, which express an MHC H-2Db class I restricted TCR specific for a peptide (KAVYNFATM) derived from the lymphocytic choriomeningitis virus (LCMV) glycoprotein Gp33 (Pircher et al., 1989), with 3BP2-deficient mice. To address the hypothesis that 3BP2 is required for modulating TCR signal strength, we examined the signaling properties of the P14 TCR in both the wild-type and 3BP2-defi-cient mice using Gp33-derived variant peptides that bind to the MHC:TCR complex with lower affinity (Gronski et al., 2004). Similar to our results observed with dose-dependent antibody stimulation, peptide activated P14 transgenic CD8+ T cells derived from 3BP2−/− mice are defective in upregulating CD69, CD25, and CD44 surface expression (Figure S3) and have impaired in vitro proliferation (Figures 1I and 1J). Notably, this activation and proliferation defect is dependent on peptide concentration and peptide affinity. 3BP2-deficient CD8+ T cells are less sensitive to activation at low peptide concentrations or to the lower affinity peptide compared with wild-type T cells. IL-2 production was also significantly reduced in the 3BP2−/− P14 transgenic T cells, showing that 3BP2 is required to trigger IL-2 production necessary for in vitro proliferation (Figure 1K). 3BP2+/+ and 3BP2−/− P14+ T cell effector differentiation was similar upon in vitro peptide stimulation, as evaluated by the induction of CD122, IL-7R, and KLRG1 cell surface proteins, the expression of the transcription factors T-bet, eomesoderm, and IRF-4, and the expression of the cytotoxic components granzyme B, CD107a, and perforin (Figures S4A and S4B). Collectively, these data demonstrate a requirement of 3BP2 for the in vitro activation and expansion but not differentiation of CD8+ T cells.
3BP2 Is Required for the In Vivo Expansion of Antigen-Specific P14+ T Cells and the Generation of Memory CD8+ T Cell Responses
To determine the role of 3BP2 during T cell activation and expansion in vivo, we investigated the development of 3BP2−/− cytotoxic CD8+ T cell responses using an in vivo model of viral infection. 3BP2+/+ or 3BP2−/− P14+ T cells (CD45.2+) were purified and transferred into wild-type recipient mice (CD45.1+), respectively. The two groups of mice were inoculated with 2000 plaque-forming units (PFU) of the Armstrong LCMV strain through intravenous administration. At the peak of the primary anti-viral response on day 8 post-infection, we harvested the spleens of the infected mice, and the antigen-specific P14+ T cell responses were quantified by flow cytometry using Gp33–41 tetramers (Figure 2A). We observed that the in vivo expansion of the transferred 3BP2−/− P14+ T cells was markedly compromised compared to the wild-type P14 cells. The total numbers of 3BP2−/− P14+ T cells were significantly reduced compared to the 3BP2+/+ P14+ T cells (0.75 ± 0.16 × 106 and 3.85 ± 0.84 × 106, respectively; n = 11, p = 0.005) (Figure 2B). The frequency of IFN-γ-pro-ducing splenic-derived CD8+ T cells following in vitro stimulation of spleen lymphocytes with the Gp33–41 peptide was also reduced in 3BP2−/− P14+ T cells compared to wild-type transgenic T cells (0.41 ± 0.09 × 106 and 2.22 ± 0.5 × 106, respectively; n = 11, p = 0.005) (Figure 2C). There was no difference in the expansion of the host CD45.1+ P14+ T cells in the two groups of mice, which underscores the intrinsic defect in the 3BP2-deficient CD8+ T cells (Figures 2B and 2C). The differentiation of 3BP2+/+or3BP2−/− P14+T cells into effector cells during the anti-viral response was not affected by the absence of the adaptor protein (Figure 2D). These data demonstrate that 3BP2 is necessary for the expansion of anti-viral specific CD8+ T cells in vivo.
Figure 2. 3BP2 Is Critical for the Expansion, Memory Generation, and Memory Response of P14+ T Cells upon Antigen Stimulation In Vivo.
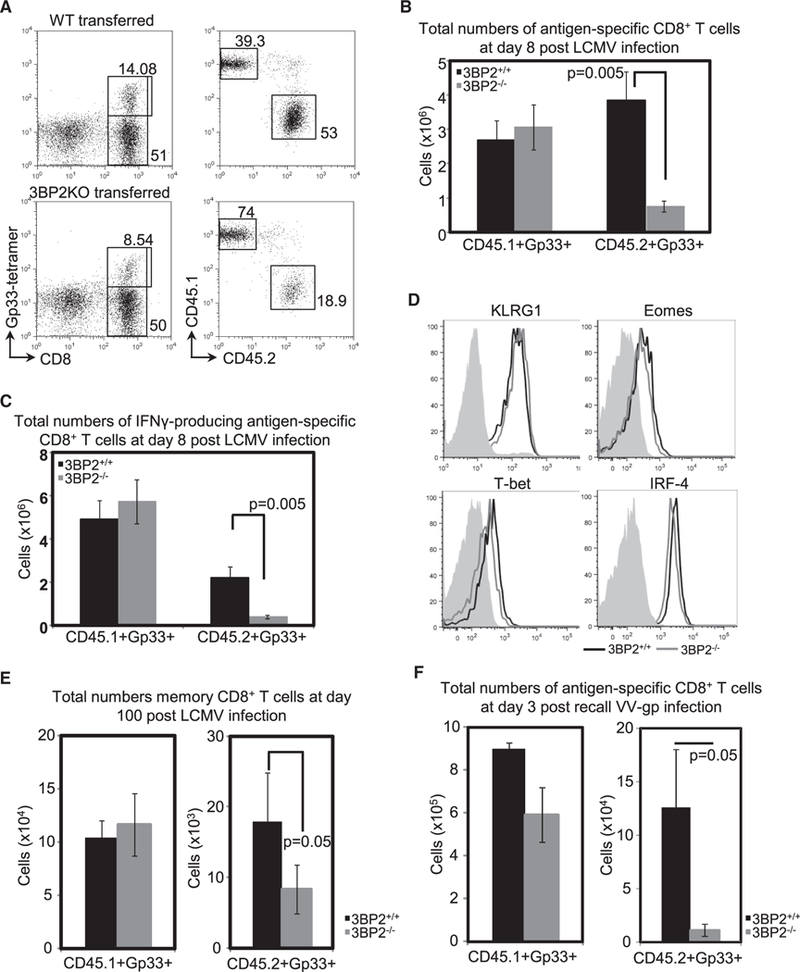
WT (CD45.2+) or 3BP2 (CD45.2+) deficient P14+ T cells were adoptively transferred In WT (CD45.1+) mice, and splenocytes were harvested 8 days post-lnfectlon with LCMV virus.
(A) Representative fluorescence-activated cell sorting (FACS) plots showing percentage of CD8+Gp33+ tetramer-stained cells in the spleens of virus infected mice (left) and percentages of endogenous (CD45.1+) or transferred (CD45.2+) antigen-specific cells (right).
(B) Pooled data from three independent experiments (n = 11 mice per group) showing total numbers of endogenous and adoptively transferred Gp33-specific CD8+ T cells in the spleens of infected animals.
(C) Absolute numbers of IFN-γ-producing CD8+ T cells from infected spleens, determined after in vitro stimulation with Gp33 peptide. Pooled data from three independent experiments are shown (n = 11 mice per group).
(D) In vivo effector differentiation of CD8+ T cells was examined by flow cytometry analysis of expression of effector markers. The gray areas indicate the expression on naive P14+ T cells.
(E) Splenocytes from LCMV infected mice were harvested 100 days post-primary infection and total numbers of endogenous or transferred memory Gp33-specific CD8+ T cells were determined.
(F) Spleen lymphocytes were harvested on day 3 post-challenge with vaccinia virus-Gp. Total numbers of endogenous (CD45.1+) and adoptively transferred (CD45.2+) Gp33-specific CD8+ T cells in the spleens of infected animals are shown.
Brackets and p values indicate statistically significant differences.
To examine whether 3BP2 deficiency can influence the generation of secondary antigen-specific CD8+ T cell response, 3BP2+/+ or 3BP2−/− P14+ T cell recipient mice were infected with the Armstrong LCMV strain, and on day 100 following the initial infection with LCMV, the memory P14+ T cell responses were quantified by flow cytometry using Gp33–41 tetramers (Figure 2E). We observed that the absolute numbers of memory 3BP2−/− P14+ T cells in the spleens of the infected mice were significantly reduced compared with the 3BP2+/+ P14+ T cells (8.3 ± 3.5 × 103 and 18 ± 6.8 × 103, respectively; n = 11, p = 0.05) (Figure 2E). Similar to the primary response, there was an inherent defect of the 3BP2-deficient CD8+ T cells, as the generation of the endogenous memory pool was normal in the two groups of mice (Figure 2E). We next sought to determine the effect of 3BP2 in the generation of secondary CD8+ T cell responses. For recall responses, mice that had received either wild-type or 3BP2−/− P14+ T cells were in-fected with the Armstrong LCMV strain and 60 days post-primary infection were re-challenged by intravenous injection of a strain of vaccinia virus (VV) that is engineered to express the Gp33–4i epitope of LCMV (Manjunath et al., 2001). At the peak of the response, on day 3 post-secondary infection, the total numbers of 3BP2−/− P14+ T cells were dramatically reduced compared with the wild-type P14+ T cells (1.1 ± 0.6 × 104 and 12.6 ± 5.4 × 104, respectively; n = 6, p = 0.05) (Figure 2F). Notably, the reduction in the absolute numbers of antigen-specific P14+ T cells during the secondary response was 5 times greater than the reduction observed in the memory P14+ T cell population (Figure 2E), which indicates an essential role of 3BP2 in the expansion of memory CD8+ T cells. Collectively, these data underscore the critical role of 3BP2 to mediate the activation, proliferation, and expansion of anti-viral antigen-specific CD8+ T cells in vivo.
3BP2 Is Induced upon CD8+ T Cell Activation and Nucleates a Signaling Module Downstream of TCR
To identify the mechanistic basis for 3BP2 in mediating T cell activation, CD8+ T cells were isolated and stimulated in vitro with either plate-bound antibodies or soluble antigenic peptides over a period of 24 hr. 3BP2 mRNA and protein expression were assayed by RT-PCR and western blot, respectively (Figures 3A–3C). Resting CD8+ T cells expressed low levels of 3BP2 mRNA and protein (Figures 3A–3C). The levels of 3BP2 mRNA and protein were induced upon CD28 costimulation or with high-affinity P14-derived peptides (Figures 3B and 3C), reaching maximum levels at 24 hr following stimulation. These findings show that 3BP2 is not required for initial T cell activation but is required at later time points following its induction by strong TCR and costimulatory signals.
Figure 3. 3BP2 Is Induced in CD8+ T Cells upon TCR and CD28 Activation and Nucleates an LCK, ZAP-70, SLP-76, and VAV Signaling Module.

(A) Spleen CD8+ T cells were isolated and activated in vitro. Samples were collected at 4,15, and 24 hr post-activation and 3BP2 mRNA levels were quantified by RT-PCR relative to GADPH levels.
(B and C) Samples of CD8+ T cells activated with either plate-bound antibodies (B) or soluble anti-genic peptides (C) were collected and 3BP2 protein levels were visualized by western blot. B-actin levels were used as protein loading control. The numbers represent the fold increase of protein levels over the untreated control.
(D and E) Purified spleen CD8+ T cells were antibody (D) or peptide (E) activated, and 3BP2 protein was immunoprecipitated from precleared soluble fractions. 3BP2-coimmunoprecipitated complexes were separated by SDS-PAGE and identified by western blot.
To interrogate the molecular interactions of the endogenous 3BP2 adaptor protein in the activated T cells, we activated wild-type CD8+ T cells with plate-bound antibodies or soluble antigenic peptides for 24 hr, a time point corresponding to maximum 3BP2 protein expression. 3BP2 was immunoprecipi-tated from cell lysates and probed for associated proteins by western blotting. We observed that LCK, the SYK family kinase ZAP-70, the Rho-family guanine nucleotide exchange factor VAV-1, and SLP-76 inducibly associated with 3BP2 following P14 peptide or antibody-mediated costi-mulation conditions (Figures 3D and 3E). Neither the p85/p110 PI3K complex nor AKT was associated with 3BP2 following TCR stimulation (Figures 3D and 3E). These data demonstrate that following its induction, 3BP2 forms a physical complex at late time points with the signaling components that recapitulate the core proximal signaling proteins activated following antigen-mediated engagement of the TCR.
3BP2 Is Recruited to Microclusters upon TCR Stimulation
We next examined the spatial organization of 3BP2 in T cells following TCR activation by live-cell imaging (Sherman et al., 2013). Wild-type (E6.1) Jurkat T cells were transiently transfected with a GFP-tagged 3BP2 protein. Cells expressing GFP-3BP2 were dropped onto glass slides covered with anti-CD3 or anti-CD45 antibodies and imaged (Figure 4A; Videos S1 and S2). We observed that immediately upon TCR engagement, 3BP2 was recruited to distinct, punctate intracellular structures that were coalesced into microclusters and moved toward the nucleus at the center of the cells (Figure 4A; Video S1). In distinction, in Jurkat cells engaged with anti-CD45 antibodies, there was minimal formation of punctate 3BP2 structure and absent microcluster formation (Video S2). These observations suggest that 3BP2 is part of a multimeric protein complex that forms in response to TCR stimulation and are consistent with previous work showing that effective TCR signaling is accomplished by the assembly and persistence of microclusters (Bunnell et al., 2006; Campi et al., 2005). These multiprotein complexes mediate TCR signaling and include LAT, SLP-76, VAV-1, ADAP, and GRB-2 (Balagopalan et al., 2013; Coussens et al., 2013; Houtman et al., 2006; Sylvain et al., 2011; Yokosuka et al., 2005). We examined whether 3BP2 is localized into microclusters containing these proteins. 3BP2-GFP-expressing Jurkat cells were activated on coverslips with anti-CD3 or anti-CD45 bound antibodies for 3 min, fixed, permeabilized, and stained with specific antibodies. 3BP2 localization was enriched at the plasma membrane with minimal microcluster formation in unstimulated cells (Figure 4B). Upon TCR stimulation, 3BP2 localized into phos-photyrosine-containing microclusters together with phospho-ZAP-70, LAT, SRC, and SLP-76 (Figures 4C–4E). Our findings uncover the in situ interactions of 3BP2 with critical kinases of the TCR signaling pathway and further underscore the importance of this adaptor protein in the organization of multiprotein complexes that are indispensable for the initiation and sustaining of T cell activation.
Figure 4. The Adaptor Protein 3BP2 Is Enriched in Microcluster Signaling Complexes and Translocates to the Nucleus upon TCR Stimulation.

(A) Live-cell imaging of Jurkat E6.1 cells transfected with WT 3BP2 GFP and plated on anti-CD3 coated coverslips. Stills taken from videos are presented as maximum Z projections.
(B) Jurkat E6.1 cells transfected with WT 3BP2 GFP were stimulated for 3 min on anti-CD45 coated coverslips as negative control, fixed in 2.4% para-formaldehyde, and stained for the localization of 3BP2, phospho-LAT, phospho-SRC, and phosphotyrosine residues (pY).
(C–E) Jurkat E56.1 cells transfected with WT 3BP2 GFP were stimulated for 3 min on anti-CD3 coated coverslips, fixed in 2.4% paraformaldehyde, and stained for the localization of 3BP2, phospho-ZAP-70 (C), pospho-SLP-76, phospho-LAT (E), phospho-SRC (D), and phosphotyrosine residues (pY) (E).
3BP2 Is Required for Optimal MAPK and NFAT Activation Downstream of the TCR in CD8+ T Cells
We examined the dependency of early signaling events in CD8+ T cells on 3BP2. Following anti-CD3 antibody ligation, we measured the absolute concentration of intracellular calcium using the fluorescent dye Indo-1 and observed no difference in calcium flux between the 3BP2+/+ and 3BP2−/− CD8+ T cells (Figure 5A). In cells lacking 3BP2, we observed no defect in the total phosphotyrosine content in cells following activation with plate-bound antibodies compared with wild-type cells (Figure 5B). Similarly, the phosphorylation of LCK and ZAP-70 was unaffected by the lack of 3BP2 in CD8+ T cells stimulated by plate-bound antibodies or soluble antigenic peptides (Figures 5C and 5D). CD8+ T cells were briefly stimulated with plate-bound antibodies and were then transferred in new, uncoated plates, and their proliferation was monitored by CFSE dilution at 48 hr upon transfer. Although 2 hr in vitro stimulation did not result in CD8+ T cell proliferation, 6 hr antibody stimulation was adequate to induce TCR-independent proliferation in wild-type (WT) CD8+ T cells but not in the 3BP2-deficient cells (Figure 5E). These data confirm that 3BP2 does not contribute significantly to early TCR activation signaling events but is required for the sustained proliferation of CD8+ T cells.
Figure 5. 3BP2 Is Not Required for Priming and Early Activation Signaling in CD8+ T Cells.

(A) Indo-1-loaded splenic CD8+ T cells were analyzed for ~120 s before stimulation with soluble anti-CD3 at 10 μg/mL. Changes in [Ca2+]i following anti-CD3 ligation are shown, with arrows indicating the addition of the stimulus. 3BP2+/+ cells are represented by black lines, and 3BP2−/− cells are represented by gray lines. The data shown are representative of three independent experiments.
(B) The phosphorylation profile of short-term-activated CD8+ T cells was detected by western blot using specific anti-phosphotyrosine (pY) antibodies.
(C) Splenic CD8+ T cells from 3BP2+/+ and 3BP2−/− mice were activated in vitro for 5, 15, 30, and 60 min with plate-bound antibodies. Cells were lysed and protein extracts were separated by SDS-PAGE and immunoblotted for phosphory-lated and total LCK.
(D) Induction of ZAP-70 phosphorylation was measured in CD8+ T cells by flow cytometry. Cells were activated by plate-bound antibodies (average of mean fluorescence intensity from three experiments is shown) or soluble peptides (representative histograms from three independent experiment) for 5, 15, and 30 min.
(E) CD8+ T cells from 3BP2+/+ and 3BP2−/− mice were briefly stimulated in vitro for 2 and 6 hr and transferred in new wells, and their proliferation was measured by CFSE dilution at 48 hr after transfer. Gray areas and dotted lines in (D) and (E) indicate unstimulated cells from 3BP2+/+ and 3BP2−/− mice, respectively.
We next examined the requirement of 3BP2 to initiate phospho-tyrosine-dependent signaling events at 24 hr following TCR activation. CD8+ T cells were stimulated by antibody ligation or soluble P14-derived peptides for 24 hr in vitro, and cell lysates were probed with phosphotyrosine-specific antibodies. T cells lacking 3BP2 demonstrated a generalized defect in total cellular phos-photyrosine content compared with WT control cells following TCR ligation or peptide induced activation (Figures 6A and 6B). The failure of 3BP2-deficient CD8+ T cells to induce phosphotyro-syl modification of cellular proteins at this time was accentuated when cells were activated with low-affinity L6F peptides. In contrast, CD8+ T cells derived from 3BP2KI/KI mice express higher basal 3BP2 protein levels and manifest enhanced global phos-photyrosine levels in unstimulated cells and following TCR stimulation compared with WT cells (Figure 6C).
Figure 6. 3BP2 Modulates Phosphotyrosine Signaling Downstream of the TCR.

The phosphorylation profile of activated CD8+ T cells from 3BP2+/+, 3BP2−/−, and 3BP2KI/KI mice was determined by western blotting at 24 hr upon in vitro stimulation with either plate-bound antibodies (A and C) or soluble antigenic peptides (B). B-actin levels were used as protein loading control. All lysates were subjected to SDS-PAGE and immunoblotted concurrently. The order of the samples was subsequently rearranged to conform with the text (boxes define WT and 3BP2 KO samples, respectively).
We next investigated the impact on LCK, Zap-70, and PLCγ activation in cells lacking 3BP2 at 24 hr following CD8+ T cell stimulation. The phosphorylation of LCK, ZAP-70, and PLCγ was significantly reduced in the 3BP2−/− CD8+ T cells compared to WT cells (Figures 7A, 7B, and S5). Similarly, the activation of NFAT and the induction of ERK phosphorylation were significantly reduced in the absence of 3BP2 at late activation time points compared to WT cells (Figures 7C, 7D, and S5). NFAT nuclear translocation was also significantly reduced in activated CD8+ T cells from 3BP2−/− compared to the WT cells (Figures 7E and S5). The difference in the intensity of the bands between 3BP2+/+ and 3BP2−/− CD8+ T cells at 0 hr time point, particularly in the higher, hyperphosphorylated fractions, likely reflects the deep state of quiescence of naive CD8+ T cells lacking 3BP2. Notably, phosphorylation of IkB was not significantly affected by 3BP2 deficiency, suggesting that activation of the NFκB pathway is not dependent on 3BP2 in CD8+ T cells.
Figure 7. The Adaptor Protein 3BP2 Is Required for Optimal ERK and NFAT Activation.

CD8+ T cells from 3BP2+/+ and 3BP2−/− mice were activated in vitro for 24 hr. Cells were lysed, and protein extracts were separated by SDS-PAGE and im-munoblotted for phosphorylated and total proteins.
(A and C) Plate-bound antibodies stimulation.
(B and D) Antigenic peptide stimulation.
(E) NFAT nuclear translocation was monitored in CD8+ T cells from 3BP2+/+ and 3BP2−/− mice activated in vitro with plate-bound antibodies.
The numbers in all panels represent average fold increase in the protein phosphorylation levels compared with the untreated or the AV-treated control, respectively. Significance levels are shown in Figure S5. All lysates were subjected to SDS-PAGE and immunoblotted concurrently. The order of the samples was subsequently rearranged to conform with the text (boxes define WT and 3BP2 KO samples, respectively).
DISCUSSION
CD8+ T cells rapidly undergo blastogenesis and proliferate in response to activation signals required for controlling the dissemination of highly replicating infectious pathogens. CD8+ T cells integrate antigen-specific and costimulatory signals, along with other environmental cues, through an elaborate signaling network of kinases, phosphatases, exchange factors, and adaptor molecules, which translate those signals into transcriptional and conformational triggers to drive CD8+ T cell activation, proliferation, and effector function differentiation. Here, we demonstrate a critical role of 3BP2 in the activation, proliferation, and expansion of CD8+ T cells at late activation time points. 3BP2 serves to sensitize TCR signaling at lower peptide binding affinities and is required for optimal activation of CD8+ T cells under suboptimal conditions. 3BP2 induction is sensitive to the strength and duration of the TCR signal and serves as a time-regulated feedback loop that reinforces and sustains T cell activation in an MHC:peptide-independent manner. Positive feedback through 3BP2 is necessary to induce proliferation, IL-2 production, and the induction of a potent anti-viral response. Moreover, the 3BP2 pathway is necessary for the efficient induction of CD8+ T cell memory population. The expansion of memory CD8+ T cells upon secondary antigen challenge was also strikingly compromised in the absence of 3BP2. We conjecture that 3BP2 feedback may have evolved to counter pathogen load by maximizing late-stage CD8+ T cell proliferative capacity at all levels of stimulation. This is further supported by results indicating 3BP2 involvement in the phosphorylation of canonical mitogenic signaling pathways at a later time after initial antigenic stimulation. We have shown that 3BP2 is dispensable for the initial phase of T cell activation mediated through TCR stimulation.
Our results clearly demonstrated that the endogenous 3BP2 protein is localized proximal to the TCR at the plasma membrane under basal conditions and is coupled to LCK, ZAP-70, VAV, and SLP-76. 3BP2 expression in T cells is transcriptionally regulated over a period of 24 hr following TCR activation. We have shown that a hypermorphic mutant form of 3BP2 uncoupled from negative regulation through ADP ribosylation leads to highly active T cells. These data demonstrate that 3BP2 in T cells is additionally repressed through post-translational modification.
3BP2 nucleates a signaling module that includes LCK, ZAP-70, PLCγ, and VAV. The induction of 3BP2 potentiates the phos-phophorylation of these key signaling proteins required for CD8+ T cell activation. 3BP2-deficient CD8+ T cells fail to efficiently activate LCK required for the downstream phosphorylation and activation of ZAP-70 and PLC-γ1 at late time points when T cells and APC conjugates have disassembled. During late-stage T cell activation 3BP2 is required to sustain activation of calcineurin and MAPK pathways. We conjecture that 3BP2 functions as an adaptor protein to assemble critical signaling proteins that recapitulate the assembly of early TCR signaling during late stages of activation but functions autonomously from immediate downstream TCR signals. Consistent with a role of 3BP2 in nucleating a signaling complex that sustains TCR-induced signals, we have observed that 3BP2 is recruited into microcluster complexes immediately upon TCR engagement.
We propose a working model for the function of 3BP2 during CD8+ T cell activation and proliferation (Figure S6). During the early phase of DC-T cell interaction in the lymphoid organs, T cells briefly interact with the APC as they rapidly sample large numbers of MHC:peptide complexes in search of conjugates that will efficiently activate the TCR. Prolonged engagement with APCs will reinforce productive TCR signaling. During this initial phase, which lasts up to 8 hr, CD8+ T cells lose their high motility rate and establish more persistent interactions with the DCs through additional costimulatory links. This second phase of T cell activation, which can last up to 24 hr after the initial MHC:peptide-TCR encounter, results in the induction of 3BP2 transcription and protein expression, among numerous biochemical and transcriptional changes. We propose that this cryptic third step is required for CD8+ T cell activation and occurs near or at the time of T cell disengagement with the APCs and the MHC:peptide complex, which corresponds to maximum 3BP2 induction. We show that 3BP2 protein induction is proportional to the strength and duration of TCR activation and that elevated, steady-state levels of 3BP2 (a feature of the cherubism mutation) are sufficient to activate T cells in the absence of TCR stimulation. Once this contingency has been fulfilled, then T cells continue to signal productively, autonomously from DC engagement necessary to drive the production of functional CD8+T cells (Chakraborty and Weiss, 2014). A similar bimodal interaction between DCs and CD4+ T cells was recently proposed by Pasqual et al. (2018), whereby CD4+ T cells are primed in two phases: an early, cognate stage and a later, TCR-independent phase.
The ability of 3BP2 to lower the threshold of T cell activation is realized during late TCR stimulation when 3BP2 protein accumulates in response to the initial strength of signal by the TCR. 3BP2 nucleates a signaling complex that recapitulates the principal signaling events activated by the TCR as part of a time-regulated feedback mechanism that is MHC:peptide complex independent and operates at late time points to reinforce the signaling activity necessary for CD8+ T cell effector and memory generation.
EXPERIMENTAL PROCEDURES
3BP2 Gene-Targeted Mice
3BP2+/+ and 3BP2−/− mice were generated as described previously (Chen et al., 2007). Mice used in all experiments were in F10 backcross generation. 3BP2ki/ki mice were a kind gift from Dr. Bjorn Olsen (Harvard School of Medicine, Boston, MA, USA). The P14 transgenic mice were kindly provided by Dr. P. Ohashi’s group. 3BP2−/− mice were crossed with P14 transgenic mice, bred with heterozygous mating, genotyped for both P14 TCR and 3BP2 expression by PCR, and confirmed for proper protein expression by SDS-PAGE and western blot. Age-matched littermates were used in subsequent experiments. All mice were maintained at the animal facilities of the Princess Margaret Cancer Institute under specific pathogen-free conditions according to University Health Network Animal Research Committee (ARC) guidelines.
T Cell Purification and In Vitro Activation
CD4+ and CD8+ T cell were isolated by negative selection from spleen preparations using the Easy Sep Enrichment Kit (Stem Cell Technologies, Vancouver, Canada) according to the manufacturer’s instructions. Purified T cells were activated by plate-bound anti-CD3 (concentrations ranging from 0.5 to 2.5 μg/mL in certain experiments; clone 2C11; eBioscience, San Diego, CA, USA) and/or anti-CD28 (10 μg/mL; clone 37.51; eBioscience, San Diego, CA, USA) at 37°C in a CO2 incubator. P14+ transgenic CD8+ T cells were also isolated by negative selection as above and were activated by the addition of soluble LCMV peptides of high (Gp33, KAVYNFATM) or low (L6F, KAVYNLATM) affinity to the P14 TCR, at concentrations of 10−7 M unless otherwise indicated. An adenovirus (AV) peptide was used as a non-relevant, non-activating negative control.
Primary and Secondary Anti-viral Responses
3BP2+/+ or 3BP2−/− CD45.2+ P14+ T cells were isolated, and 2,000 cells of each type were transferred into respective CD45.1+ WT (B6.SJL; The Jackson Laboratory, Bar Harbor, ME, USA) recipient mice by intravenous injection. The following day, the two groups of mice were inoculated with 2,000 PFU of the Armstrong LCMV strain through intravenous administration. To assess the antigen-specific CD8+Tcells at the peak of the anti-viral response on day 8 post-infection, we harvested the spleens of the infected mice, and the P14 anti-viral CD8+ T cell responses were determined by flow cytometry using gp33–41 tetramers (MHC Tetramer Production Laboratory, Baylor College of Medicine, kindly provided by Dr. P. Ohashi’s group) and antibodies for the CD8, CD45.1, and CD45.2 surface markers (eBioscience, San Diego, CA, USA). To study the recall response, mice of both groups that received either WT or 3BP2 KO P14cells and were infected with LCMV were re-challenged by intravenous injection of a strain of VV that is engineered to express the gp33–41 epitope of LCMV (Manjunath et al., 2001). The re-challenge was performed at least 60 days after the initial infection to ensure the generation of true memory P14 T cells. The kinetics of antigen-specific CD8+ T cell responses were monitored at 1, 3, 5, 7, and 10 days post-infection in the spleens and lymph nodes of the infected mice. Donor and endogenous P14 transgenic cells were tracked by flow cytometry using tetramers and anti-mouse CD45.1, CD45.2, and CD8 antibodies (eBioscience, San Diego, CA, USA).
Flow Cytometry Analysis of Activation and Proliferation
3BP2+/+ or 3BP2−/− CD8+ T cells were isolated and activated as described above. At 24 hr after activation, samples were collected, washed with ice-cold flow wash (Hank’s balanced salt solution [BSS] with 3% horse serum and 0.02% sodium azide), and stained for cell surface expression of CD8+ T cell activation markers using specific fluorophore-conjugated antibodies (BD Biosciences, San Jose, CA, USA,; eBioscience, San Diego, CA, USA). For proliferation assessment, cells were stained with 1 μM CFSE (Thermo Fisher Scientific, Waltham, MA, USA) for 10 min in the dark at room temperature (RT) and were activated for 24 and 48 hr. Cells were collected at respective time points, washed with flow wash, and stained with CD8-specific fluoro-phore-conjugated antibodies (BD Biosciences, San Jose, CA, USA). All samples were analyzed in a FACSCalibur cell analyzer and expression plots were generated using FlowJo software (NIH).
3BP2 mRNA Relative Quantification
CD8+T cells were isolated and activated as described. Samples were taken at 4, 15, and 24 hr time points, washed with ice-cold PBS, and used immediately or kept frozen at –80°C. RNA was isolated using the RNeasy Plus Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocols, cDNA was prepared using a cDNA synthesis kit (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocols, and 3BP2 mRNA quantities were determined by RT-PCR using specific primers (TaqMan; Applied Biosystems, Foster City, CA, USA). RT-PCR was performed in 10 μL reaction volumes in triplicate 384-well plates using an ABI 7900 RT-PCR thermocycler.
Immunoprecipitation and Western Blot Analysis
CD8+ T cells were isolated and activated as described. For immunoprecipitation, samples were collected at 24 hr, washed with ice-cold PBS, and used immediately or kept frozen at –80°C. Cell pellets were lysed with non-denaturing NP-40-based lysis buffer (Abcam formulation, Cambridge, MA, USA) containing protease inhibitor (100× Pierce Halt cocktail) for 30 min at 4°C, and soluble fractions were separated by centrifugation at 10,000 × g for 10 min at 4°C. Lysates were precleared at 4°C with pre-immune sheep serum for 30 min and subsequently with 20 μL of a 50% slurry of protein G-coated Sepharose beads for 1 hr. After preclearing, 3BP2 was immune precipitated with 5 μL of sheep 3BP2-immune serum for 12 hr and protein G-coated Se-pharose beads for 4 hr at 4°C. Bound protein was released with 100 μL of boiling SDS-loading buffer and separated on a 10% acrylamide gel by SDS-PAGE. Protein was transferred onto polyvinylidene difluoride (PVDF) and blocked in TBS-T with 5% BSA for western blotting with 3BP2 (Abcam, Cambridge, MA, USA), PI3K (rabbit polyclonal), AKT (rabbit polyclonal), LCK (rabbit polyclonal), ZAP-70 (rabbit polyclonal), VAV-1 (clone D45G3), and SLP-76 (rabbit polyclonal) specific antibodies (Cell Signaling Technologies, Beverly, MA, USA) using manufacturer-recommended conditions. For phosphosignal-ing, cells were collected and washed with ice-cold PBS, then lysed with denaturing SDS-based RIPA lysis buffer (Abcam formula, Cambridge, MA, USA) containing benzonase (Cell SignalingTechnologies, Beverly, MA, USA), protease, and phosphatase inhibitors (Pierce Halt cocktail) for 30 min at 4°C. The soluble fraction was separated on a 10% acrylamide gel for SDS-PAGE and transferred/blocked for western blotting by ZAP-70, pZAP-70 (rabbit polyclonal), LCK, PLC-γ (clone D9H10), pIkB (clone I4D4), IκB (rabbit polyclonal), pERK (rabbit polyclonal), ERK (rabbit polyclonal), pSRC (clone D49G4), pPLC-γ (Santa Cruz Biotechnologies, Limerick, PA, USA), pNFAT (Ser54) (Biosource), and pY (clone 4G10; Santa Cruz Biotechnologies, Limerick, PA, USA) antibodies using manufacturer-recommended conditions. For cytoplasmic and nuclear fractionation, cells were collected and washed with ice-cold PBS, then lysed with hypotonic buffer (10 mM, Tris-HCl [pH 7.4], 10 mM NaCl, 10 mM EDTA, and 0.5% Triton X-100), and the cytoplasmic extract was kept in a separate tube. The remaining nuclei were washed and lysed with RIPA lysis buffer and mild sonication. The two fractions were separated on a 10% acrylamide gel for SDS-PAGE and transferred/blocked for western blotting by GAPDH (clone 14C10), H3 histone (clone D2B12) (Cell Signaling Technologies, Beverly, MA, USA), and NFATc1 (BD PharMingen, San Jose, CA, USA).
Protein quantitation was performed using ImageJ software (NIH). The ratio of protein phosphorylation levels over total protein levels was measured and normalized to the ratio of the untreated or AV-treated CD8+ T cells from 3BP2+/+ or 3BP2−/− mice, respectively.
Cell Culture, Transfections, Immunostaining, and Imaging
WT (E6.1) Jurkat T cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics. Transient transfections were performed using the Amaxa electroporation system kit T (Amaxa Biosystems, Gaithersburg, MD, USA). Tissue culture reagents were from BioFluids (Rockville, MD, USA). Cells were allowed to spread on coverslips as described earlier (Bunnell etal., 2003). Briefly, poly-lysine-covered four-chambered glass coverslips (LabTek II; Nunc/Nalgene, Rochester, NY, USA) were coated with 10 μg/mL antibody (anti-CD3, clone UCHT1, anti-CD3, clone HIT3a, or anti-CD45; eBioscience, San Diego, CA, USA). The chambers were loaded with 300 μL normal media without phenol red supplemented with 25 mM HEPES (pH 7.0) and warmed. For the fixed-cell immunofluorescence experiments, cells were resus pended in the same buffer, plated into the bottom of the chamber, and incubated at 37°C. After 3 min, cells were fixed in 2.4% para formal-dehydefor30 min. The cells were permeabilized with Triton X-100, incubated with blocking bufferfor30 min, and then incubated with primary antibodies for 60 min, followed by washes and 60 min incubation with Alexa-conjugated secondary antibodies. The antibodies used were anti-phospho-LAT (pTyr171) (Sigma-Aldrich, St Louis, MO, USA), anti-phosphotyrosine (clone 4G10; EMD Millipore, Billerica, MA, USA), Anti-SLP-76 (pY128) (BD PharMingen, San Jose, CA, USA), goat anti-rabbit IgG (H+L)-Alexa Fluor 568 conjugate, and goat anti-mouse IgG2b-Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Waltham, MA, USA). Images from fixed cells were collected using a Leica SP8 laser scanning confocal microscope, using a 63×, 1.4 NA objective (Leica Microsystems, Buffalo Grove, IL, USA). Z stacks (2–3 nm), with a spacing of 0.3 nm, were taken of the area contacting the coverslip. Images from live cells were collected with a Yokogawa CSU-22 spinning-disk confocal system containing Borealis2optics, usinga63×,1.4 NA objective. Images were captured using an ImagEMX2 electron-multiplying charged-coupled device (EMCCD) camera (Hamamatsu Photonics, Middlesex, NJ, USA). A Tokai Hit stage incubator and objective warmer (Tokai Hit Co, Shizuoka-ken, Japan) were used to maintain live samples at 37°C. Z stacks (2–3 nm), with a spacing of 0.5 nm, were taken of the area contacting the coverslip at the interval specified in the figure legend. Videos and still images taken from videos are presented as maximum Z projections. Leica AF software was used to produce images of fixed cells, while MetaMorph was used to produce live-cell images and videos. Adobe Photoshop and Illustrator (Adobe Systems Inc, San Jose CA) were used to prepare composite figures. Scale bars were cut from the original images and then were pasted in a more visible position on the final composite images.
Statistical Analysis
All p values were determined using two-way Student’s t tests. Statistical significance was defined as p < 0.05.
Supplementary Material
Highlights.
The adaptor protein 3BP2 is induced by CD28 costimulation in CD8+ T cells
The adaptor protein 3BP2 lowers the threshold of T cell activation
3BP2 nucleates a signaling complex that sustains MHC:peptide-independent TCR signaling
3BP2 operates at later time points to sustain CD8 T cell effector and memory generation
ACKNOWLEDGMENTS
This study was funded by the CIHR Foundation grant number 143230.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and two videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.075.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ainsua-Enrich E,Alvarez-Errico D, Gilfillan AM, Picado C, Sayós J, Rivera J, and Martín M (2012). The adaptor 3BP2 is required for early and late events in FcεRI signaling in human mast cells. J. Immunol. 189, 2727–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopalan L, Barr VA, Kortum RL, Park AK, and Samelson LE (2013). Cutting edge: cell surface linker for activation of T cells is recruited to microclusters and is active in signaling. J. Immunol. 190, 3849–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, Barr VA, and Samelson LE (2002). T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 158, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell SC, Barr VA, Fuller CL, and Samelson LE (2003). High-resolu-tion multi color imaging of dynamic signaling complexes in T cells stimulated by planar substrates. Sci. STKE 2003, PL8. [DOI] [PubMed] [Google Scholar]
- Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario MC, Barr VA, Koretzky GA, and Samelson LE (2006). Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol. Cell. Biol. 26,7155–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campi G,Varma R, and Dustin ML (2005). Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 202, 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AK, and Weiss A (2014). Insights into the initiation of TCR signaling. Nat. Immunol. 15, 798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Dimitriou ID, La Rose J, Ilangumaran S, Yeh WC, Doody G, Turner M, Gommerman J, and Rottapel R (2007). The 3BP2 adapter protein is required for optimal B-cell activation and thymus-independent type 2 humoral response. Mol. Cell. Biol. 27, 3109–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Dimitriou I, Milne L, Lang KS, Lang PA, Fine N, Ohashi PS, Kubes P, and Rottapel R (2012). The 3BP2 adapter protein is required for chemoattractant-mediated neutrophil activation. J. Immunol. 189,2138–2150. [DOI] [PubMed] [Google Scholar]
- Conley JM, Gallagher MP, and Berg LJ (2016). T cells and gene regulation: the switching on and turning up of genes after T cell receptor stimulation in CD8T cells. Front. Immunol. 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens NP, Hayashi R, Brown PH, Balagopalan L, Balbo A, Akpan I, Houtman JC, Barr VA, Schuck P, Appella E, and Samelson LE (2013). Multipoint binding of the SLP-76 SH2 domain to ADAP is critical for oligomerization of SLP-76 signaling complexes in stimulated T cells. Mol. Cell. Biol. 33,4140–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, and Kaech SM (2010). Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol. Rev. 236, 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert M, and Rottapel R (2006). The adapter 3BP2: how it plugs into leukocyte signaling. Adv. Exp. Med. Biol. 584, 107–114. [DOI] [PubMed] [Google Scholar]
- Douglass AD, and Vale RD (2005). Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 121, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, and Dustin ML (1999). The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- Gronski MA, Boulter JM, Moskophidis D, Nguyen LT, Holmberg K, El-ford AR, Deenick EK, Kim HO, Penninger JM, Odermatt B, et al. (2004). TCR affinity and negative regulation limit autoimmunity. Nat. Med. 10, 1234–1239. [DOI] [PubMed] [Google Scholar]
- Houtman JC, Yamaguchi H, Barda-Saad M, Braiman A, Bowden B, Appella E, Schuck P, and Samelson LE (2006). Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 13, 798–805. [DOI] [PubMed] [Google Scholar]
- Jevremovic D, Billadeau DD, Schoon RA, Dick CJ, and Leibson PJ (2001). Regulation of NK cell-mediated cytotoxicity by the adaptor protein 3BP2. J. Immunol. 166, 7219–7228. [DOI] [PubMed] [Google Scholar]
- Kaech SM, and Ahmed R (2001). Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levaot N, Simoncic PD, Dimitriou ID, Scotter A, La Rose J, Ng AH, Willett TL, Wang CJ, Janmohamed S, Grynpas M, et al. (2011a). 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J. Clin. Invest. 121, 3244–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deck-ert M, Krzyzanowski PM, Scotter A, Gu S, Janmohamed S, et al. (2011b). Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 147, 1324–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath N, Shankar P, Wan J, Weninger W, Crowley MA, Hieshima K, Springer TA, Fan X, Shen H, Lieberman J, and von Andrian UH (2001). Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 108, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, and Von Andrian UH (2004). T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159. [DOI] [PubMed] [Google Scholar]
- Ménasché G, Kliche S, Bezman N, and Schraven B (2007). Regulation of T-cell antigen receptor-mediated inside-out signaling by cytosolic adapter proteins and Rap1 effector molecules. Immunol. Rev. 218, 82–91. [DOI] [PubMed] [Google Scholar]
- Pasqual G, Chudnovskiy A, Tas JMJ, Agudelo M, Schweitzer LD, Cui A, Hacohen N, and Victora GD (2018). Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Bürki K, Lang R, Hengartner H, and Zinkernagel RM (1989). Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561. [DOI] [PubMed] [Google Scholar]
- Ren R, Mayer BJ, Cicchetti P, and Baltimore D (1993). Identification ofa ten-amino acid proline-rich SH3 binding site. Science 259, 1157–1161. [DOI] [PubMed] [Google Scholar]
- Sherman E, Barr V, and Samelson LE (2013). Super-resolution characterization of TCR-dependent signaling clusters. Immunol. Rev. 251, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvain NR, Nguyen K, and Bunnell SC (2011).Vav1-mediated scaffolding interactions stabilize SLP-76 microclusters and contribute to antigen-dependent T cell responses. Sci. Signal. 4, ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y,Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, Ninomiya C, doAmaral C, Peters H, Habal M, et al. (2001). Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 28, 125–126. [DOI] [PubMed] [Google Scholar]
- Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, Saheki Y, Ka-wai T, Mukherjee PM, Reichenberger E, and Olsen BR (2007). Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell 128, 71–83. [DOI] [PubMed] [Google Scholar]
- van Stipdonk MJ, Lemmens EE, and Schoenberger SP (2001). Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2, 423–429. [DOI] [PubMed] [Google Scholar]
- Varma R, Campi G, Yokosuka T, Saito T, and Dustin ML (2006). T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25,117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, and Saito T (2005). Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Zhang N, and Bevan MJ (2011). CD8(+) T cells: foot soldiers ofthe immune system. Immunity 35, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.