

Abstract
Online citizen science projects such as GalaxyZoo1, Eyewire2 and Phylo3 have been very successful for data collection, annotation, and processing, but for the most part have harnessed human pattern recognition skills rather than human creativity. An exception is the game EteRNA4, in which game players learn to build new RNA structures by exploring the discrete two-dimensional space of Watson-Crick base pairing possibilities. Building new proteins, however, is a more challenging task to present in a game, as both the representation and evaluation of a protein structure are intrinsically three-dimensional. We posed the challenge of de novo protein design in the online protein folding game Foldit5. Players were presented with a fully extended peptide chain and challenged to craft a folded protein structure with an amino acid sequence encoding that structure. After many iterations of player design, analysis of the top scoring solutions, and subsequent game improvement, Foldit players can now, starting from an extended polypeptide chain, generate a diversity of protein structures and sequences which encode them in silico. 146 Foldit player designs with sequences unrelated to naturally occurring proteins were encoded in synthetic genes; 56 were found to be expressed in E. coli with good solubility and to adopt stable monomeric folded structures in solution. The diversity of these structures is unprecedented in de novo protein design, representing 20 different folds—including a new fold not observed in natural proteins. High resolution structures were determined for four of the designs, and are nearly identical to the player models. This work makes explicit the considerable implicit knowledge contributing to success in de novo protein design, and shows that citizen scientists can discover creative new solutions to outstanding scientific challenges, such as the protein design problem.
The principle underlying de novo protein design is that proteins fold to their lowest free energy state6; hence, designing a new protein structure requires finding an amino acid sequence whose lowest energy state is the prescribed structure. In practice, this challenge can be divided into two subproblems: first, crafting a protein backbone that is designable (i.e. that could be the lowest energy state of some sequence); and second, finding a sequence whose lowest energy state is the crafted structure. One of the challenges of protein design is the exponentially increasing number of conformations available to a polypeptide chain, which is astronomical even for a modestly-sized protein of 60–100 residues. Thus, the first subproblem of crafting a plausible backbone is extremely open-ended, and the second subproblem is difficult because it is not tractable to explicitly check that a designed sequence has lower energy in the crafted structure than in any other structure. There has been considerable progress in de novo protein design in recent years7–10, but it is unclear whether all of the contributions to this success have been made explicit in the protocols used to design proteins, and how much implicit knowledge resides in the expertise of the designers. Disentangling the role of expert knowledge is particularly difficult for the extremely open-ended challenge posed by the first subproblem (i.e. crafting a plausible backbone), for which there are a practically unlimited number of solutions. Because full computer enumeration of backbones is not possible, there is considerable room for human creativity and intuition in generating and designing new protein structures.
To investigate how crowd-based creativity could contribute to solving the de novo protein design problem, we incorporated de novo design tools into the protein folding game Foldit. Foldit is a free online computer game developed to crowdsource problems in protein modeling, and provides full control over the three-dimensional structure of a protein model5 (Figure 1). Players compete to build a model with the lowest free energy, as calculated by the Rosetta energy function11. In the past, Foldit has been primarily applied to protein structure prediction problems, in which Foldit players were presented with an unstructured amino acid sequence and challenged to determine its native conformation5,12. Foldit players in one case redesigned a loop region of an already folded structure13, but the de novo design of an entire protein is a far more expansive challenge.
Figure 1. The Foldit user interface.

a, The Foldit score is the Rosetta energy with a negative multiplier, so that better models yield higher scores. b, The design palette allows players to change the amino-acid residue identity at any position of the model. c, The Pull tool allows players to manipulate the three-dimensional structure of the model. d, The Undo graph tracks the score as a model is developed, and allows players to backtrack and load previous versions of a model. e, Additional Foldit tools (from left to right): full structure minimization, sidechain minimization, backbone minimization, auto-design sidechains, repack sidechains, translate/rotate model, secondary structure assignment, idealize secondary structure, manually design sidechains, delete residues, insert residues, insert cutpoint, idealize peptide bond geometry. f, Foldit players explore diverse structures that have no sequence or structural homology to natural proteins.
We repeatedly challenged Foldit players to design stably folded proteins from scratch, and iteratively improved the game based on their results. In each challenge, players were provided with a poly-isoleucine backbone in a fully extended conformation (60–100 residues in length), and were given seven days to fold the backbone into a compact structure and identify a sequence specifying this backbone. Initially, most top-scoring (low energy) Foldit player designs were highly extended, lacked a solvent-inaccessible core, and were composed entirely of polar residues (Extended Data Figure 1). Such extended, fully α-helical structures have more favorable hydrogen bonding, electrostatic, and local torsional energies than collapsed structures, which must contort to create a buried core. While poly-lysine and other extended polar sequences resembling these initial Foldit solutions are often α-helical in solution15,16, the lack of long-range interactions precludes specific folding into a single stable structure17. This highlights a limitation of using absolute energy as an optimization criterion for protein design: a low energy design does not guarantee structural specificity, which arises only if all other alternative conformations have higher energy. To favor the design of globular solvent-excluding protein folds, with sequences that uniquely encode them, we introduced three supplementary design rules into Foldit: a “Core Exists” rule that requires a minimum proportion of residues (e.g. 30%) to be solvent-inaccessible in the designed structure; a “Secondary Structure Design” rule that prohibits glycine and alanine in all secondary structure elements; and a “Residue Interaction Energy” rule to penalize large residues that make insufficient intramolecular interactions in the designed structure. With the addition of these rules to Foldit, subsequent top-scoring designs from Foldit players were compact globular proteins.
We obtained custom synthetic genes encoding 12 player designs for which structure prediction calculations converged on the player designed conformation14. The sequences of these proteins have no homology to any known protein (Supplementary Table 1). The de novo designs were expressed in E. coli and purified by metal affinity and size exclusion chromatography. Chromatography and circular dichroism (CD) spectroscopy indicated that 6 of the 12 designs were monomeric and folded in solution, with helical secondary structure consistent with the players’ models (Supplementary Figure 1). All of the experimentally tested proteins described in this paper are entirely the work of Foldit players.
During gameplay, the Foldit application uploads the player’s latest model to the Foldit server every 2–5 minutes; from these snapshots we can reconstruct the process by which a Foldit player develops a protein design (Figure 2). Foldit players employ more varied and complex exploration strategies than standard Rosetta automated design protocols, and frequently revert to a previous iteration of their model to explore an alternative path, resulting in a highly-branched search tree. A typical automated design protocol, by contrast, includes only two branch points18. In addition, Foldit players regularly sample much higher energy states than the automated protocol, which has only a limited ability to escape local energy minima.
Figure 2. Comparison of Foldit player and automated design sampling strategies.

a, Single trajectories (ignoring abandoned branches) for three Foldit player-designed proteins in red (Foldit1), blue (Peak6), and green (Ferredog-Diesel); and design trajectories for four Rosetta-designed proteins in gray. The y-axis is the Rosetta energy rescaled so that the final design has a value of −1.00, and positive energies are shown as zero. Foldit players are willing to undergo large increases in energy to explore new regions; the Rosetta protocol in contrast has a limited ability to escape local energy minima. Red circles correspond to structures shown in (b). b, Snapshots from the design trajectory of Foldit1: (i) the initial extended chain of poly-isoleucine; (ii) development of secondary structure; (iii) development of folded tertiary structure; (iv) sequence design of folded structure, with inset showing favorable packing at positions 13 and 45; (v) high-energy intermediate design, with inset showing redesign at positions 13 and 45, which results in steric clashes with the protein backbone; (vi) the final refined design, with inset showing renewed favorable interactions at positions 13 and 45. c, The design strategy for Foldit1 represented as a graph, showing all branch points where multiple design trajectories were spawned from a single intermediate. The final design was reached after 17 branch points. Node colors correspond to five different cooperating Foldit players, and the final design is marked as a star. d, Similar representation of a Rosetta design trajectory; there are only two branch points.
Encouraged by the success of Foldit players in designing stable proteins from scratch, we made additions to the game encouraging players to explore more diverse protein structures. Up until this point, all top-scoring Foldit designs had consisted of either three or four α-helices connected by minimal loops. Indeed, Foldit players had determined that designs with β-sheets did not score on par with α-helical bundles (Extended Data Figure 2), and competitive players had abandoned any attempt to design more varied folds. (This has an interesting parallel to protein design by practicing scientists, which has also focused much more on helical bundles than other classes of protein folds19–22.) To encourage the design of a wider variety of folds, we introduced a “Secondary Structure” rule stipulating that no more than 50% of residues may form α-helices. Foldit players responded by designing a multitude of mixed α/β proteins, which were indistinguishable from expert designs upon visual inspection. However, structure prediction calculations for these α/β design sequences showed poor sampling close to the target design structure, suggesting that the designed sequences did not strongly encode their local structures14. Further analysis showed that these player designs contained many residues with locally strained backbone conformations (backbone phi and psi torsions in unfavored regions of the Ramachandran plot23,24). That such designs had very low energies revealed a problem in the Rosetta energy function at the time: since Rosetta users typically sampled backbones starting from fragments of native proteins, unfavorable local conformations were rarely encountered—hence it had not been discovered that the energies associated with local backbone strain were being underestimated. We addressed this flaw in the Rosetta model by increasing the steepness of the energetic penalties associated with strained local backbone geometry; this is now standard in the latest Rosetta energy function11. We also added to Foldit an “Ideal Loops” rule restricting players to a set of 19 unstrained reverse-turn conformations7, and incorporated new tools to aid generation of unstrained backbones: a fragment lookup-based loop-closure tool, an interactive Ramachandran map, and a protein Blueprint scheme for drag-and-drop assembly of secondary structure elements and common loop conformations (Extended Data Figure 3). Together, these upgrades brought about a marked improvement in the local backbone quality of Foldit player-designed proteins (Extended Data Figure 4).
The importance of reducing local backbone strain was borne out in experimental characterization. Prior to the backbone modeling improvements described in the previous paragraph, only 4 of 37 Foldit α/β designs tested (11%) were monomeric and structured in solution. Following the backbone modeling additions, 46 of 97 (47%) were monomeric and exhibited the expected secondary structure in solution. Most showed exceptional stability in thermal and chemical denaturation experiments, with free energies of unfolding (ΔGunf) up to >20 kcal/mol; indeed, 32 designed proteins remained completely folded at 95°C (Figure 3; Supplementary Figure 1). This success rate surpasses previous reports of designed α/β proteins7,12.
Figure 3. Structural characterization of Foldit player designed proteins.

a, Dendrogram showing all 56 folded Foldit player designs clustered by structural similarity (TM-align26), with colored circles highlighting the four designs characterized in (b-f). The stacked bars show the 20 different folds among the clustered designs (Extended Data Figure 5). Fold XX (see design 2003594_S028) is a new fold, previously unobserved in natural proteins. b, Cartoon depiction of four select Foldit designs. c, Rosetta@home ab initio calculations show that the sequence for each design has an energy landscape that is strongly funneled toward the design structure. Rosetta energy is on the y-axis and Cα-RMSD to the designed structure on the x-axis; points represent lowest energy structures sampled starting from an extended chain (red points), and starting from the Foldit design model (green points). d, Size-exclusion chromatography (SEC) traces of elution absorbance at 280 nm show that designs are monomeric in solution. e, Circular dichroism (CD) spectra indicate that the designs adopt the expected secondary structure content in solution at 25°C (blue trace), when heated to 95°C (red trace), and when cooled again to 25°C (green trace). f, CD mean residue ellipticity at 220 nm as temperature is increased from 25°C to 95°C; the designs do not temperature denature. g, Cooperative unfolding during titration with guanidinium hydrochloride. Blue circles show CD mean residue ellipticity at 220 nm with increasing concentration of denaturant, and the black curve shows a two-state unfolding model fit to the data. ΔGunf values were determined by linear extrapolation using the fit model parameters27.
Overall, the 56 successful Foldit designs are diverse in structure, representing 20 different protein folds (Figure 3; Extended Data Figure 5)—one of which is a new fold previously unobserved in natural proteins. The success of Foldit designs is not attributed to just one or two exceptional Foldit players, but is shared broadly by the Foldit community (Supplementary Table 1). The 56 successful designs were created by 36 different Foldit players (the most prolific player authored 10 successful designs); 19 designs were created collaboratively by at least two cooperating players; and 5 successful designs were not top-scoring, but regardless were flagged by players as personal favorites.
We succeeded in solving high-resolution structures of four Foldit player-designed proteins. X-ray crystal structures of three designed proteins (named by their designers Foldit1, Peak6, and Ferredog-Diesel) closely match the designed conformations, with Cα-RMSD of 1.1, 0.9, and 1.7 Å, respectively (Figure 4). Well-resolved electron density in the protein core of Foldit1 and Peak6 shows that most sidechains adopt the intended rotamers and preserve the designed packing interactions. The electron density of Ferredog-Diesel is less clear, but the protein backbone adopts the designed fold, and many core sidechains appear to pack as intended. The solution NMR structure of a fourth design, Foldit3, also closely matches the design conformation, with a Cα-RMSD of 1.1 Å between the design model and a representative structure (i.e., the medoid conformer25) of the ensemble.
Figure 4. High-resolution structures of Foldit player-designed proteins.

a, The Foldit1 design (fold V in Fig 3: 3 β-strands with sheet order 1–2–3) model backbone (rainbow) aligns to the crystal structure (gray) with Cα-RMSD of 1.1 Å. b, The Peak6 design (fold III: 4 strands, sheet order 1-2-4-3) model backbone aligns to the crystal structure with Cα-RMSD of 0.9 Å. c, The Ferredog-Diesel design (fold I: 4 strands, sheet order 4-1-3-2) model backbone aligns to the crystal structure with Cα-RMSD of 1.7 Å. Cross-sections show core residue sidechains, with the composite omit 2mFo-DFc map contoured at 2.0 σ (a-b) or 1.0 σ (c). d, The Foldit3 design model (fold XVIII: 4 strands, sheet order 2-1-3-4) and NMR ensemble. The design model aligns to the representative (medoid) NMR model with a Cα-RMSD of 1.1 Å. Cross sections compare core side chains in the design model (rainbow) and representative NMR model (gray).
We can draw several general conclusions from these results about scientific models, citizen science, and the interplay between the two. First, a scientific model which holds within the domain space considered by practicing scientists may not hold outside of this domain. This is most vividly illustrated by the highly extended structures generated by Foldit players in their first de novo design efforts, and later by the structures with strained local geometry not previously sampled by Rosetta users. Second, for citizen scientists to make essential and creative scientific contributions through online gaming, the scoring function of the game must be an accurate representation of the science. In our initial iterations, Foldit did not present to players a sufficiently accurate and general model to allow them to robustly design new proteins, even though the underlying Rosetta software had been used for protein design by practicing scientists. Third and most important, citizen science offers a powerful way to systematically improve a scientific model, through iterations of model trial and model improvement. Human game players are exceptionally capable at finding and exploiting unanticipated solutions that are otherwise unexplored by experienced scientists, whose focus is not on getting a high score, but rather on solving their specific scientific problem.
We have demonstrated that non-expert citizen scientists, playing the online computer game Foldit, can accurately design completely new protein structures from scratch. Locally, players’ solutions are physically plausible and resemble natural proteins, but globally, they are creative and diverse. Proteins designed by citizen-scientist Foldit players are by no measure inferior to those of expert protein designers: they fold accurately to the intended conformation, show exceptional folding stability, and span a wide diversity of structures. This result is all the more impressive given that de novo protein design was an almost completely unsolved problem just a few years ago, and the diversity in protein folds spanned by the successful Foldit players’ models considerably exceeds that in any previous protein design report. The sustained success of Foldit players over a wide diversity of protein folds highlights the power of human creativity when guided by scientific understanding presented in a readily comprehensible form.
Methods
Foldit protein design puzzles
Foldit puzzles were set up with a model poly-isoleucine in fully extended conformation, with fixed length ranging from 60 to 100 residues. Each puzzle was posted online for seven days, during which Foldit players competed to develop a protein model with the lowest energy, as calculated by the Rosetta energy function. Foldit puzzles used the talaris2013_cart scorefunction with the following modifications: (1) the cart_bonded scoreterm was upweighted (increased from 0.5 to 2.0) to ensure realistic bond lengths and angles as players cut and splice the backbone chain; (2) a penalty-only envsmooth scoreterm (weighted at 2.0) was added to supplement the Rosetta solvation treatment, and to discourage the design of buried polar and exposed nonpolar residues; (3) the reference energy of alanine was modified (increased to 3.0) to discourage the excessive design of alanine. See Supplementary Data for configuration files for all Foldit puzzles. Each Foldit puzzle was accompanied by a brief description, along with an explanation of any supplementary rules enforced in the puzzle. Design puzzles were accessible to all Foldit users; Foldit user registration is free and open to the public, at http://fold.it/http://fold.it. Models were collected continuously as Foldit players worked on the puzzles, since the Foldit application automatically uploads the user’s latest model to a server every 2–5 minutes. This study was approved by the University of Washington Institutional Review Board, and informed consent for this research was obtained from all Foldit users at the time of user registration.
Protein design selection
After the end of each puzzle, we selected player models for further analysis as follows: First we selected the lowest-energy model from each of the 10 top-ranked groups, where independent players were treated as individual groups (designs named with suffix “0000–9”). Second, we selected the lowest-energy model from the 10 top-ranked solo players, which includes independent players as well as group members that developed a model without assistance from their group (suffix “s000–9”). Third, we visually inspected models that were flagged by Foldit players for special consideration, and selected any models that appeared plausible (suffix “S***”). Last, we ranked and pruned the set of remaining models, by removing any models that align to a better-scoring model with Cα-RMSD less than 2.5 Å. We visually inspected the 50 top-ranked models in the pruned set and selected any models that appeared plausible (suffix “1001–50”). Models deemed “implausible” typically lacked secondary structure, contained buried polar residues, or included long stretches of completely polar residues. At each step, we used TM-align26 to eliminate duplicate models (TM-score > 0.98) that had already been selected (e.g. models that were top-ranking and flagged by players). In Rounds 2 and 3, the top-ranked group and solo models were automatically selected for further analysis, without visual inspection. The sequences of selected models were subjected to Rosetta ab initio structure prediction14, using the distributed computing platform Rosetta@home. If ab initio predictions identified any decoy structures with energy comparable to (or lower than) the designed structure, or if ab initio predictions were unable to sample the designed structure, the design was rejected. All other designs were selected for experimental characterization. The majority of experimentally tested designs (96 of 146) were top-ranked group or solo designs, which were selected “blindly” (without visual inspection). See Supplementary Data for models and FASTA sequences of all tested designs.
Protein expression and purification
A 6x-His tag with TEV-cleavable linker (sequence ‘MGHHHHHHGWSENLYFQGS’) was prepended to the N-terminus of each design selected for experimental characterization. Plasmids containing the encoded genes were ordered from Genscript in pET15 (designs with prefix between 997258 and 1998925), or in pET21 (1998555–2002990), or from Twist in pET29 (2003048–2003594) vectors. Plasmids were transformed into E. coli BL21 Star (DE3) cells (Invitrogen), and grown overnight in 4 mL Luria-Bertani medium (LB) with 50 μg/mL carbenicillin (for pET15, pET21 vectors) or 30 μg/mL kanamycin (for pET29). Overnight cultures were used to inoculate 0.5 L auto-induction media, and grown at 37 °C for 18 hours. Cultures were pelleted and resuspended in 25 mL lysis buffer (20 mM Tris pH 8.0, 300 mM NaCl, 1 mg/mL lysozyme, 0.1 mg/mL DNase, 1 mM PMSF), and lysed by microfluidization. The cell lysate was pelleted and supernatant was filtered with a 0.22 μm filter before loading onto a 2 mL nickel affinity gravity column. Protein bound to the column was washed with 20 mL wash buffer (20 mM Tris pH 8.0, 500 mM NaCl, 30 mM imidazole) and eluted in 10 mL elution buffer (20 mM Tris pH 8.0, 500 mM NaCl, 250 mM imidazole). Purified protein was dialyzed into TBS (20 mM Tris pH 8.0, 300 mM NaCl) at 4°C overnight to remove imidazole and further purified by size exclusion chromatography on an AKTAxpress (GE Healthcare) with a Superdex S75 10/300 GL column (GE Healthcare). For proteins containing cysteine, dialysis and gel filtration were carried out in TBS with 1 mM TCEP. Protein expression and solubility was determined from SDS-PAGE and mass spectrometry. Oligomeric state was determined by size exclusion chromatography.
Circular dichroism
Purified protein was dialyzed into 50 mM sodium phosphate pH 7.4 at 4°C overnight (plus 500 μM TCEP for proteins containing cysteine). All circular dichroism data were collected on an AVIV Model 420 spectrometer. Far UV spectra and temperature melts were measured with 11–62 μM protein in a quartz cuvette with path length of 1 mm. Protein concentration was determined by absorbance at 280 nm using a NanoDrop spectrophotometer (Thermo Scientific), using predicted extinction coefficients. Wavelength spectra were measured between 195 and 260 nm at 25°C, 95°C, and again after cooling to 25°C. For temperature melts, ellipticity at 220 nm was monitored as temperature increased from 25°C to 95°C, in increments of 2°C. Chemical titrations were carried out with 1.0–21 μM protein in a quartz cuvette with path length of 10 mm. Ellipticity at 220 nm was monitored at concentrations of guanidinium chloride increasing from 0 to 7 M, in increments of 0.25 M. Denaturation curves were fitted with non-linear regression to two-state unfolding model with six parameters: the folding free energy, m-value, and slope and y-intercept for baseline curves27.
X-ray crystallography
Prior to x-ray crystallography, the N-terminal 6x-His tag was cleaved from protein samples by incubation with 250 μg TEV protease at 25°C for four hours in 20 mM Tris pH 8.0, 300 mM NaCl, 1 mM DTT. The reaction product was dialyzed into TBS overnight at 4°C to remove DTT and flowed over a 2 mL metal affinity gravity column to remove TEV protease and residual histidine tag. The cleaved protein was further purified by gel filtration as described above. Purified protein was concentrated to 20–100 mg/mL in 20 mM Tris pH 8.0, 300 mM NaCl. Crystallization screening was carried out with a variety of 96-condition spare matrix suites available from Qiagen or Hampton Research. A Mosquito Crystal nanoliter robot (TTP Labtech) was used to prepare screens in 3-well sitting drop plates, with 200 nL drops and protein:precipitant ratios of 1:1, 1:2, and 2:1.
Foldit1 (2002949_0000) was crystallized at 20 mg/mL in 50 mM HEPES pH 7.5, 0.2 M potassium chloride, 35% v/v pentaerythritol propoxylate. Crystals were flash-frozen in liquid nitrogen without further cryo-protection. X-ray diffraction was collected to a resolution of 1.18 Å.
Peak6 (2003333_0006) was crystallized at 40 mg/mL in 0.1 M sodium acetate pH 4.5, 0.2 M lithium sulfate, 50% w/v PEG 400. Crystals were briefly soaked in mother liquor plus 20% PEG 200, then flash frozen in liquid nitrogen. X-ray diffraction was collected to a resolution of 1.54 Å.
Ferredog-Diesel (2003169_S953) was crystallized with 6x-His tag intact, at 80 mg/mL in 0.1 M citrate pH 4.0, 3.0 M NaCl. Crystals were dehydrated by soaking in 5 μL mother liquor in open air for 10 minutes, then flash frozen in liquid nitrogen. X-ray diffraction was collected to a resolution of 1.92 Å.
X-ray diffraction datasets were collected at the Advanced Light Source (Berkeley, CA). Data was processed with HKL200031. Crystal structures were solved by molecular replacement with Phaser29, using the backbone of the original designed model with sidechains truncated to the beta carbon (Foldit1 and Peak6), or using the backbone of a model predicted ab initio from the design sequence (Ferredog-Diesel). Models were built and refined in iterative cycles using Coot and PHENIX30,31. Diffraction data and refinement statistics are listed Supplementary Table 2.
NMR spectroscopy
NMR studies were performed using uniformly15N,13C-enriched protein samples. A synthetic gene for Foldit3 (2003265_s008) was obtained from Genscript already incorporated into plasmid pET15TEV_NESG, which includes a N-terminal 6xHis purification tag, followed by a TEV protease cleavage site (sequence ‘MGHHHHHHGWSENLYFQGS’). E. coli BL21(DE3) cells harboring plasmid pET15TEV_NESG-Foldit3 were grown in 1L MJ9 minimal media32, supplemented with 100 μg/ml ampicillin at 37 °C. In order to produce uniformly15N and 13C enriched protein samples, 1g / L 15NH4-salts and 2g / L U-13C glucose were added as sole a nitrogen and a carbon sources, respectively. When O.D.600 reached around 0.5 units, the culture was transferred to 18 °C, and the protein production was induced by addition of 1 mM IPTG. After overnight incubation, the cells were collected and resuspended in 20 ml binding buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl and 20 mM imidazole). After passing the cells through 900–1000 psi French press twice, cell debris were removed by 10,000 rpm for 30 min. The supernatant was further spun down at 40,000 rpm for 1hr. The obtained supernatant (soluble fraction) was mixed with 1 ml of Ni-resin and incubated at 4 °C for 1 hr. The non-specific binding proteins were removed by 20 mL binding buffer and washing buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl and 50 mM imidazole) and the target protein was eluted by 5 mL elution buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl and 300 mM imidazole). The protein was dialyzed against GF buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl) for overnight and gel filtration was carried out using AKTA express with high-load 26/600 Superdex 200 pg column. Homogeneity (> 97%) was validated by SDS polyacrylamide gel electrophoresis. The purified protein was dialyzed against 20 mM potassium phosphate (pH 6.5), and the protein concentration was adjusted to between 0.3–0.4 mM for NMR studies.
All NMR spectra were recorded at 25 °C using cryogenic NMR probes. All NMR data were collected on the Bruker AVANCE III 600 MHz spectrometers and processed using the program NMRPipe33, and analyzed using the programs SPARKY and XEASY34. Spectra were referenced to external DSS. Sequence-specific resonance assignments were determined using AutoAssign software together with interactive manual analysis, as described previously35. Backbone dihedral angle constraints were derived from the chemical shifts using the program TALOS_N36 for residues located in well-defined secondary structure elements. The programs ASDP37 and CYANA38,39 were used to automatically assign NOEs and to calculate structures. RPF analysis37,40 was used in parallel to guide iterative cycles of noise/artifact peak removal, peak picking, and NOESY peak assignments. The 20 conformers with the lowest target CYANA function value were then refined in explicit water41 using the program CNS42. The structural statistics and global structure quality factors (Supplementary Table 3) including Verify3D43, ProsaII44, PROCHECK45, and MolProbity46 raw and statistical Z-scores were computed using the PSVS47 1.5 and PDBStat48 software packages. The global goodness-of-fit of the final structure ensembles with the NOESY peak list data, the NMR DP score, was determined using the RPF analysis program40.
Extended Data
Extended Data Figure 1. Initial top-ranking Foldit player designs.

When challenged to design a protein with only the talaris2013 score function (and no additional rules), Foldit players discovered low-energy models that are unlikely to fold as designed. a, An extended α-helix, composed entirely of lysine and glutamate, has very favorable energies for hydrogen-bonding, electrostatic, and backbone torsions, but is unlikely to fold cooperatively into a single stable structure. This type of design is discouraged with the “Core Exists” rule. b, Due to their greater surface area, large aromatic sidechains can make more interactions than smaller aliphatic sidechains, even when under-packed or solvent-exposed. This type of design is discouraged with the “Residue Interaction Energy” rule. c, A design with an alanine- and glycine-saturated core can make favorable van der Waals interactions between closely packed backbone atoms; however, the burial of these small sidechains is associated with a weaker hydrophobic effect, and the lack of interdigitation allows exchange between multiple conformations with similar core packing energies (i.e. “molten globule” behavior). These designs are discouraged with the “Secondary Structure Design” rule.
Extended Data Figure 2. Rosetta energy of top Foldit player designs.

Rosetta energy of top-ranking designs was calculated with the talaris2013 score function and normalized by residue count. a, Energy of top 10-ranked designs from: initial Foldit puzzles (Round 0; n = 30 designs), Round 1 puzzles (n = 170), Round 2 puzzles (n = 510), Round 3 puzzles (n = 250). The introduction of supplementary rules in Round 1 and Round 2 resulted in higher-energy designs (p < 1e-6 and p < 0.01, respectively; Wilcoxon rank-sum test). The backbone modeling improvements in Round 3 resulted in lower-energy designs (p < 1e-15; Wilcoxon rank-sum test). b, Energy of top 10-ranked designs from Round 3 all-α puzzles (n = 30) or α/β puzzles using the Secondary Structure rule (n = 220). All-α designs tend to have lower energy than α/β designs (p < 1e-10; Wilcoxon rank-sum test). Boxplots show: center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers.
Extended Data Figure 3. New backbone modeling tools in Foldit.

a, The Remix tool allows players to select a region of the model and search a library of backbone fragments for a conformation that can be substituted. b, An interactive Ramachandran map allows players to easily identify residues with outlier backbone conformations. Players can also click-and-drag points on the Ramachandran map to set the backbone torsions of individual residues. c, A Blueprint panel shows the primary sequence and secondary structure content of the model. Residues are colored according to the ABEGO quadrants of the Ramachandran plot7. d, Players can drag-and-drop modular Building Blocks onto the Blueprint panel to insert common turn conformations into their model.
Extended Data Figure 4. Improvement of backbone quality in Round 3 Foldit designs.

MolProbity24 was used to calculate the proportion of residues with “unfavored” or “outlier” backbone torsions in: high-resolution crystal structures of native proteins (n = 6342), de novo design models by Lin et al.7 (n = 72), and top-ranking Foldit player-designs from before (n = 680) and after (n = 250) improvements to Foldit backbone modeling tools. Initial Foldit player designs contained significantly more unfavored torsions than native proteins or other de novo designs by Lin et al. (p < 1e-15, two-tailed t-test). Improvements to Foldit’s backbone modeling tools led Foldit players to produce designs with fewer unfavored torsions (p < 1e-15, two-tailed t-test). Boxplots show: center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers.
Extended Data Figure 5. Protein folds represented by successful Foldit player designs.

Each fold has a unique arrangement and connectivity of secondary structure elements, depicted in cartoon diagrams. Diagrams are labeled with Roman numerals as in Figure 3. Fold XX is a new fold, previously unobserved in natural proteins; TM-align26 and DALI55 alignments of design 2003594_S028 against the entire PDB found no structural homologs with this fold.
Extended Data Figure 6. Foldit player demographics.

All players who participated in Foldit protein design puzzles and who had not opted out of Foldit-related email were solicited for survey questions. Data is shown for n = 324 responding Foldit players.
Extended Data Figure 7. Category rankings of Foldit players.

Foldit player rankings are strongly correlated in the Design and Prediction categories (Spearman’s rank correlation coefficient of 0.84). This suggests that skills developed playing Foldit structure prediction puzzles carry over to design puzzles, and vice versa.
Extended Data Table 1.
Success rates of Foldit player-designed proteins.
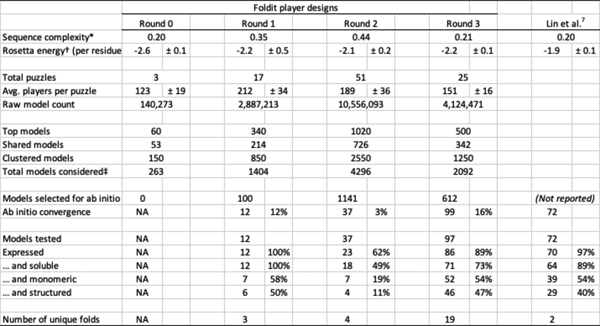
|
Linguistic sequence complexity52 was calculated from the top 10-ranked models in all puzzles, using word lengths of 1, 2, and 3.
Rosetta energy is the talaris2013 energy normalized by residue count. Values shown are mean and standard deviation for the 10 top-ranked models in all puzzles. See Extended Data Figure 6 for sample sizes.
Includes redundant models, since very similar models can appear in two or more categories (top, shared, and clustered). See Methods for details on model selection.
Extended Data Table 2.
X-ray crystallography data and refinement statistics
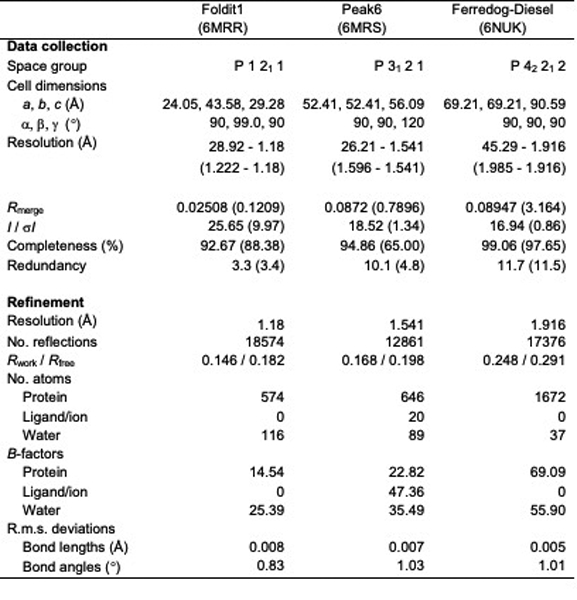
|
Extended Data Table 3.
NMR and refinement statistics for protein structures
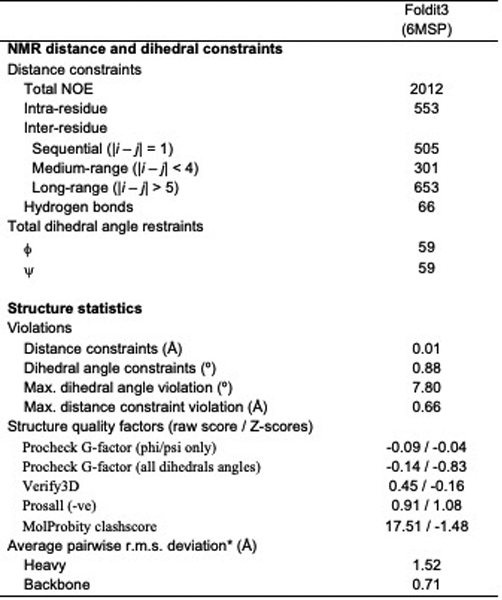
|
Supplementary Material
Acknowledgements
We thank all Foldit players for their gameplay contributions, and for feedback offered on the https://fold.it website. We thank A. Kang, S.A. Rettie, C. Chow, and L. Carter for help with experiments; D. Alonso, L. Goldschmidt, P. Vecchiato, D. Kim for computer support; and Rosetta@home (https://boinc.bakerlab.org) volunteers for computing resources. We thank G. Rocklin, V. Mulligan, and other members of the Baker lab for discussions. This material is based upon work supported by the National Science Foundation (NSF) Graduate Research Fellowship under Grant No. DGE-1256082, NSF Grant No. 1629879, National Institutes of Health (NIH) Grant 1UH2CA203780, and NIH Grant 5R01 GM120574 (to G.T.M.). The ALS-ENABLE beamlines are supported in part by the NIH, National Institute of General Medical Sciences, grant P30 GM124169–01. The Advanced Light Source is a DOE User Facility under Contract No. DE-AC02–05CH11231. Foldit3 was a nominated target of the CASP COMMONS Community Outreach program.
Footnotes
The authors declare the following competing interests: G.T.M. is a co-founder of Nexomics Biosciences, Inc.
Code Availability
Because Foldit crowdsourcing relies on regulated, fair competition between participants, the source code of the Foldit user interface is not open. The underlying Rosetta macromolecular modeling suite (https://www.rosettacommons.org) is freely available to academic and non-commercial users, and commercial licenses are available via the University of Washington CoMotion Express License Program. Analysis scripts used in this paper are available in the Supplementary Information.
Data Availability
The atomic coordinates of Foldit1, Peak6, and Ferredog-Diesel crystal structures, and the Foldit3 NMR structure, have been deposited in the RCSB Protein Database with accession numbers 6MRR, 6MRS, 6NUK and 6MSP, respectively. Chemical shift and NOESY peak list data for Foldit3 were deposited in the Biological Magnetic Resonance Bank (BMRB ID 30527).
Supplementary information is linked to the online version of the paper at http://www.nature.com/nature.
References
- 1.Lintott CJ et al. Galaxy Zoo: morphologies derived from visual inspection of galaxies from the Sloan Digital Sky Survey. Monthly Notices of the Royal Astronomical Society 389, 1179–1189 (2008). [Google Scholar]
- 2.Kim JS et al. Space-time wiring specificity supports direction selectivity in the retina. Nature 509, 331–336 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawrykow A et al. Phylo: A Citizen Science Approach for Improving Multiple Sequence Alignment. PLoS ONE 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J et al. RNA design rules from a massive open laboratory. Proceedings of the National Academy of Sciences 111, 2122–2127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper S et al. Predicting protein structures with a multiplayer online game. Nature 466, 756–760 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epstein CJ, Goldberger RF & Anfinsen CB The Genetic Control of Tertiary Protein Structure: Studies With Model Systems. Cold Spring Harbor Symposia on Quantitative Biology 28, 439–449 (1963). [Google Scholar]
- 7.Lin Y-R et al. Control over overall shape and size in de novo designed proteins. Proceedings of the National Academy of Sciences (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang P-S, Boyken SE & Baker D The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Marcos E et al. Principles for designing proteins with cavities formed by curved β sheets. Science 355, 201–206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou J et al. De novo design of a fluorescence-activating β-barrel. Nature (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alford RF et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khatib F et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat Struct Mol Biol 18, 1175–1177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eiben CB et al. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players. Nature Biotechnology 30, 190–192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohl CA, Strauss CEM, Misura KMS & Baker D Protein structure prediction using Rosetta. Meth. Enzymol 383, 66–93 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Blout ER & Idelson M Compositional Effects on the Configuration of Water-soluble Polypeptide Copolymers of L-Glutamic Acid and L-Lysine. Journal of the American Chemical Society 80, 4909–4913 (1958). [Google Scholar]
- 16.Doty P, Imahori K & Klemperer E The solution properties and configurations of a polyampholytic polypeptide: copoly-L-lysine-L-glutamic acid. Proceedings of the National Academy of Sciences 44, 424–431 (1958). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh K & Dill KA Theory for Protein Folding Cooperativity: Helix Bundles. J. Am. Chem. Soc 131, 2306–2312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koga N et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Regan L & DeGrado W Characterization of a helical protein designed from first principles. Science 241, 976–978 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Harbury PB, Plecs JJ, Tidor B, Alber T & Kim PS High-resolution protein design with backbone freedom. Science 282, 1462–1467 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Thomson AR et al. Computational design of water-soluble alpha-helical barrels. Science 346, 485–488 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Jacobs TM et al. Design of structurally distinct proteins using strategies inspired by evolution. Science 352, 687–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran GN & Sasisekharan V Conformation of Polypeptides and Proteins. Advances in Protein Chemistry 23, 283–437 (1968). [DOI] [PubMed] [Google Scholar]
- 24.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montelione GT et al. Recommendations of the wwPDB NMR Validation Task Force. Structure 21, 1563–1570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y & Skolnick J TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santoro MM & Bolen DW Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 (1988). [DOI] [PubMed] [Google Scholar]
Additional References
- 28.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jansson M, Li YC, Jendeberg L, Anderson S, Montelione GT, Nilsson B. High-level production of uniformly ¹⁵N- and ¹³C-enriched fusion proteins in Escherichia coli. J Biomol NMR 7, 131–141 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F et al. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Bartels C, Xia TH, Billeter M, Guntert P & Wuthrich K The Program Xeasy for Computer-Supported Nmr Spectral-Analysis of Biological Macromolecules. J Biomol NMR 6, 1–10 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Liu GH et al. NMR data collection and analysis protocol for high-throughput protein structure determination. Proceedings of the National Academy of Sciences of the United States of America 102, 10487–10492 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y, Delaglio F, Cornilescu G & Bax A TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44, 213–223, doi: 10.1007/s10858-009-9333-z (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang YJ, Tejero R, Powers R & Montelione GT A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins 62, 587–603, doi: 10.1002/prot.20820 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Guntert P, Mumenthaler C & Wuthrich K Torsion angle dynamics for NMR structure calculation with the new program DYANA. Journal of Molecular Biology 273, 283–298 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Herrmann T, Guntert P & Wuthrich K Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. Journa l of Molecular Biology 319, 209–227 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Huang YJ, Powers R & Montelione GT Protein NMR recall, precision, and F-measure scores (RPF scores): Structure quality assessment measures based on information retrieval statistics. Journal of the American Chemical Society 127, 1665–1674 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Linge JP, Williams MA, Spronk CA, Bonvin AM & Nilges M Refinement of protein structures in explicit solvent. Proteins 50, 496–506, doi: 10.1002/prot.10299 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica Section D-Biological Crystallography 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Luthy R, Bowie JU & Eisenberg D Assessment of protein models with three-dimensional profiles. Nature 356, 83–85, doi: 10.1038/356083a0 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Sippl MJ Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362, doi: 10.1002/prot.340170404 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Laskowski RA, Macarthur MW, Moss DS & Thornton JM Procheck - a Program to Check the Stereochemical Quality of Protein Structures. Journal of Applied Crystallography 26, 283–291 (1993). [Google Scholar]
- 46.Word JM, Bateman RC, Presley BK, Lovell SC & Richardson DC Exploring steric constraints on protein mutations using MAGE/PROBE. Protein Science 9, 2251–2259 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharya A, Tejero R & Montelione GT Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795, doi: 10.1002/prot.21165 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Tejero R, Snyder D, Mao B, Aramini JM, Montelione GT PDBStat: A universal restraint converter and restraint analysis software package for protein NMR. J. Biomol. NMR 56, 337–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocklin GJ et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357, 168–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W & Sander C Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 51.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 52.Trifonov EN in Structure and Methods, Vol. 1: The Proceedings of the Sixth Conversation held at The University-SUNY, Albany NY, June 6–10, 1989 69–77 (Adenine Press, 1990). [Google Scholar]
- 53.Tejero R, Snyder D, Mao B, Aramini JM, Montelione GT PDBStat: A universal restraint converter and restraint analysis software package for protein NMR. J. Biomol. NMR 56, 337–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhattacharya A, Tejero R, Montelione GT Evaluating protein structures determined by structural genomics consortia. PROTEINS: Struct. Funct. Bioinformatics 66, 778–795 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Holm L & Laakso LM Dali server update. Nucleic Acids Res. 44, W351–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.