

Abstract
Central nervous system (CNS) diseases are the leading cause of morbidity and mortality; their treatment, however, remains constrained by the blood–brain barrier (BBB) that impedes the access of most therapeutics to the brain. We report herein a CNS–delivery platform for protein therapeutics, which is achieved by encapsulating the proteins within nanocapsules that contain choline and acetylcholine analogues. Mediated by nicotinic acetylcholine receptors and choline transporters, such nanocapsules can effectively penetrate the BBB and deliver the therapeutics to the CNS, as demonstrated in mice and non–human primates. This universal platform, in general, enables the delivery of any protein therapeutics of interest to the brain, opening a new avenue for the treatment of CNS diseases.
Keywords: nanomedicine, central nervous system delivery, blood brain barrier penetration, nanocapsules
A universal CNS-delivery platform for protein therapeutics is developed enabling the delivery of any protein therapeutics of interest to the brain via choline transporters and acetylcholine receptors.
Graphical Abstract

The central nervous system (CNS) diseases, such as Alzheimer’s disease, Parkinson’s disease, stroke and brain tumors, have become the most prevalent illness but remain poorly treated[1,2]. A key obstacle that constrains the treatment for the CNS diseases is the blood–brain barrier (BBB), a highly restrictive barrier separating the circulating blood from the brain and extracellular fluid in the CNS, which impedes approximately 98% of the small molecule therapeutics and almost all macromolecules (e.g., enzymes, growth factors, and monoclonal antibodies)[3,4].
Extensive efforts have been undertaken to improve the BBB penetration of therapeutic proteins; representative strategies include the conjugation of proteins with poly(ethylene glycol) (PEG), modification of proteins with charged or lipophilic moieties, and fusion of proteins with BBB–specific molecules. Conjugating proteins with PEG prolongs their circulation time but still with poor BBB penetration ability[5,6]. Modifying proteins with charged or lipophilic moieties enhances their adsorptive transcytosis[7,8] but also promotes their immune clearance and accumulation in peripheral tissues[9]. Fusing proteins with BBB–specific molecules may enhance their receptor–mediated transcytosis while the efficacy remains to be validated.[10] In addition, immune cells that can cross the BBB via extravasation[11], as well as invasive physical methods (e.g., ultrasonic[12] and microwave radiation[13]) that can open the tight junctions of the BBB, were also explored. Despite the decades of efforts, effective delivery of protein therapeutics to the CNS remains challenging.
To achieve effective brain delivery of therapeutic proteins, we foresee that the ultimate vectors should be the ones that can be uptaken actively by the brain. For example, acetylcholine transporters and choline transporters (ChTs) are extensively expressed in the nervous system and luminal brain capillary endothelial cells[14,15]. Through ChTs, choline is actively transported from the circulating blood to the brain for the synthesis of acetylcholine and other molecules[16,17]. Through acetylcholine transporters, as–synthesized acetylcholine is stored and transported within the vesicles of neuron cells[18], serving as an essential neurotransmitter and neuromodulator. Inspired by the active transport of choline and acetylcholine, we envision that therapeutic proteins can be effectively delivered to the CNS by encapsulating them within nanocapsules that contain acetylcholine and choline analogues.
As illustrated in Figure 1, acetylcholine and choline interact with their receptors mainly through electrostatic interaction (the choline moiety) and hydrogen bonding (the acetyl moiety)[17,19,20]. 2–methacryloyloxyethyl phosphorylcholine (MPC), a molecule that contains a choline and acetylcholine analogue, may interact with nicotinic acetylcholine receptors (nAChRs) and ChTs in a similar fashion to acetylcholine and choline. Using MPC as the monomer, along with a degradable poly(lactide)–based crosslinker, we grow a thin shell of polymer network around an individual protein molecule by in situ polymerization[21,22], which creates protein nanocapsules with abundant choline and acetylcholine analogues. Mediated by nAChRs and ChTs, such nanocapsules can be effectively transported through the BBB and release the protein cargoes in the brain, upon the cleavage of the crosslinkers that breaks down the shells of the nanocapsules. Note that the gene of enhanced green fluorescent protein (EGFP) was delivered to the CNS using a choline–derivate–polylysine vector[23]. Small interference RNA (siRNA) was also delivered to the CNS using a peptide derived from the rabies virus glycoprotein, a vector that binds to nAChRs[15]. The expression of the genes in the CNS was confirmed, yet the delivery efficiency of such approaches has not been reported. This universal platform, in general, enables the delivery of any protein of interest to the CNS, simply by encapsulating the protein within cleavable nanocapsules that are made from monomers with choline and acetylcholine analogues.
Figure 1. A Schematic illustration of the design of the bioinspired delivery system for effective delivery of therapeutic proteins to the CNS.
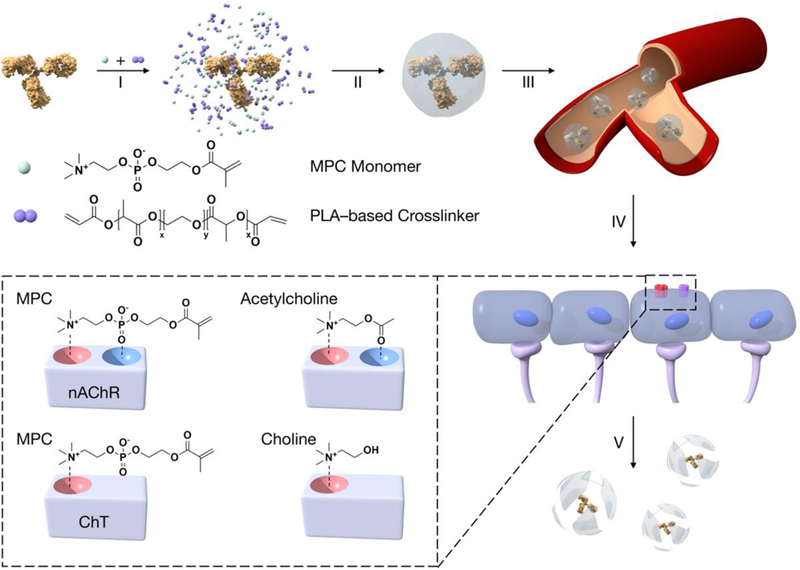
2–methacryloyloxyethyl phosphorylcholine (MPC monomer) and the degradable crosslinker, poly(lactide)–b–poly(ethylene glycol)–b–poly(lactide)–diacrylate triblock copolymer (PLA-based crosslinker), are enriched around the protein molecule (I). A thin layer of polymer shell is then grown around the protein molecule by in situ polymerization, forming protein nanocapsules (II). Administered via intravenous injection (III), the protein nanocapsules that contain the acetylcholine and choline analogues (inset), are translocated crossing the BBB mediated by nAChRs and ChTs (IV), and release the protein cargos within the CNS, upon cleaving the crosslinkers that breakdowns the nanocapsule shells (V).
For demonstration, protein nanocapsules of horseradish peroxidase (HRP, Mw=44 kDa), bovine serum albumin (BSA, Mw=66.5 kDa), nerve growth factor (NGF, Mw=26 kDa) and rituximab (RTX, Mw=145 kDa), were synthesized, denoted herein as n(Protein). BSA and HRP were adopt as model proteins to investigate the pharmaceutical properties of nanocapsules due to their stability, manipulability and low cost. The nanocapsules of BSA and HRP were synthesized using a non-degradable crosslinker. In addition, NGF is a promising candidate for neurodegenerative disorders due to its role to prevent the death of cholinergic neurons in the human brain[24,25]. RTX is a well-established monoclonal antibody proved for systemic cancer treatment[26]. Both NGF and RTX exhibit poor CNS bioavailability with limited therapeutic effects[24,27,28]. The nanocapsules of NGF and RTX, n(NGF) and n(RTX), were synthesized using a degradable crosslinker, poly(lactide)–b–poly(ethylene glycol)–b–poly(lactide)–diacrylate triblock copolymer. Degradation of the crosslinkers enables their release from the nanocapsules in the CNS to execute their biological functions.
These nanocapsules exhibit a similar size of ~30 nm and zeta potential of ~ 0 mV, which are significantly different from their native counterparts, indicating successful encapsulation of the proteins (Figure 2a, b). Figure 2c shows representative transmission electron microscopy (TEM) images of n(BSA) and n(NGF), exhibiting a uniform spherical morphology with a similar average diameter of 30 nm. It is worth noting that, the neutral charge and zwitterionic structure prolong the circulation time of the nanoparticles[29]. In addition, for intracellular delivery of nanoparticles, it takes the least energy to deform the cell membranes when the size of the nanoparticles is ~ 30 nm[30]; while the extracellular space of the brain generally has a dimension between 40 nm to 60 nm[31]. The ability to synthesize nanocapsules with such optimum size allows their effective transport crossing the BBB and within the CNS. In addition, the encapsulating process well retains the activity of the proteins (e.g., n(HRP) retains more than 96% of the HRP activity, Supplementary Figure S1), and as-synthesized nanocapsules also exhibit extremely low cytotoxicity. For example, mouse brain endothelial cells (bEnd.3) treated with 1 μM of n(BSA), n(HRP), n(RTX) or n(NGF) still retain more than 96% of the viability (Figure S2).
Figure 2. Structure, morphology, and sustainable release of the protein nanocapsules.
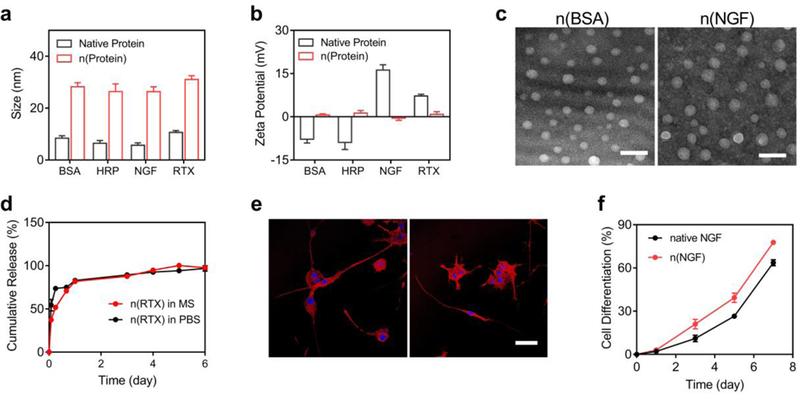
a, The average size and b, zeta potential of n(BSA), n(HRP), n(NGF), n(RTX), and their native counterparts. c, TEM images of n(BSA) and n(NGF) showing a uniform size around 30 nm. Scale bar, 100 nm. d, RTX releasing profiles of n(RTX) in mouse serum (MS) and PBS (25 μg/mL) at 37 ˚C as determined by ELISA. e, Confocal microscopy images of PC12 cells treated with 100 ng/mL native NGF (left) or n(NGF) (right), respectively. The nuclei and actin were stained by Hoechst 33342 and Texas Red–Phalloidin. Scale bar, 50 μm. f, The percentage of differentiated PC12 cells induced by native NGF or NGF released from n(NGF). The percentage was calculated by counting the number of cells, with at least one neurite of which the length is equal to or longer than the diameter of the cell body, vs. the total cell population. Error bars represent the s.e.m. of triplicate samples (n=3).
The ability to release the protein cargos from the nanocapsules was demonstrated by n(RTX) and n(NGF). Figure 2d presents the accumulative release profiles of RTX from n(RTX) in PBS and mouse plasma at 37 ˚C as determined by the enzyme–linked–immunosorbent assay (ELISA), showing the release of RTX sustainably for over one week. Meanwhile, the size of n(RTX) was reduced from 30 to 8 nm (Figure S3a), and the zeta potential was increased from 0 to 5 mV (Figure S3b) during one-week incubation in PBS at 37 ˚C. Similar results were also observed during the degradation of n(NGF) (Figure S3a,b). For another demonstration, after incubating PC12 cells with n(NGF) for 5 days, as–released NGF promotes the growth of neurites to a level similar to native NGF (Figure 2e). Figure 2f further plots the percentage of the PC12 cells, of which the length of one neurite is equal to or longer than the diameter of the cell body, vs. the incubation time, indicating as–released NGF promotes the differentiation of PC12 cells more effectively than the native NGF in the culture media.
The ability of the nanocapsules to penetrate the BBB was first examined in mouse brain endothelial cells, bEnd.3 cell line. After incubating with TAMRA–labelled n(BSA) (TAMRA–n(BSA)) at 37 ˚C and 4 ˚C for 2 h, fluorescence–activated cell sorting (FACS) shows that 6.3% and 0.4% of the cells are positive (containing TAMRA–n(BSA)), respectively, suggesting an energy–dependent transcytosis process (Figure 3a). The BBB penetration was then examined using a widely adopted in vitro BBB model, which was constructed by growing a dense layer of bEnd.3 cells on the apical surface of a transwell, of which the integrity was confirmed via fluorescence microscopy and the transendothelial electrical resistance (TEER) (Figure S4, S5). TAMRA–n(BSA) was added to the apical compartment, and the percentage of the nanocapsules penetrated (penetration efficiency) was monitored based on the fluorescent intensity. Figure 3b shows the penetration efficiency of n(BSA), which increases with incubation time and reaches ~ 20% after 8 hr. For comparison, PEG–containing n(BSA) (PEG–n(BSA)), which has similar size and surface charge as n(BSA), shows a much lower penetration efficiency of 4%. Figure 3c plots the average penetration rate of n(BSA) in the initial 3 hr vs. the n(BSA) concentration, exhibiting an enzyme–reaction–like profile that well fits the Michaelis–Menten equation with a fitting coefficient R2 of 0.99, an apparent association constant of 19 μM, and a maximum penetration rate of 1.2 nmol‧h–1‧cm–2. Enzymatic reactions generally involve the formation of the enzyme–substrate reversible complex; this finding implying the transcytosis involves the reversible interactions between the nanocapsules and the receptors.
Figure 3. Translocation of the nanocapsules through mouse brain endothelial cells (bEnd.3) in a transwell model.
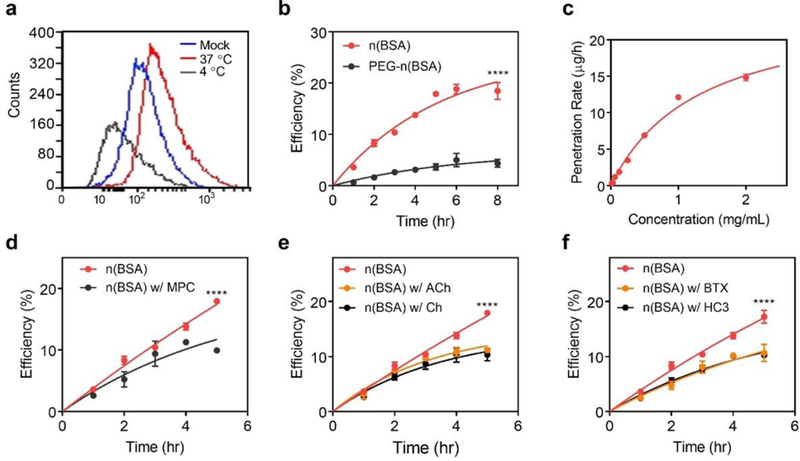
a, Fluorescence–activated cell sorting (FACS) of bEnd.3 cells in a 24–well plate incubated with TAMRA–n(BSA) (0.5 mg/mL) for 2 hr at 37 ˚C and 4 ˚C. b, Accumulative penetration efficiency of n(BSA)and PEG–n(BSA) labelled with TAMRA through a monolayer bEnd.3 cells in a transwell. The fluorescence intensity in the basolateral compartment was monitored. c, The initial penetration rates of n(BSA) through the bEnd.3 layer at different concentrations of nBSA. d–f, The penetration efficiency of n(BSA) through the bEnd.3 layer with or without d, MPC monomer, e, acetylcholine (ACh) or choline (Ch), and f, α–BTX or HC3. n(BSA) were incubated with α–BTX inhibited or HC3 for 1 hr before the measurement. Error bars represent the s.e.m. of triplicate samples (n=3), ****P < 0.0001.
To examine the receptors that may involve in the transcytosis, we first measured the penetration efficiency of n(BSA) in the presence of the MPC at a molar ratio of 1:1000. As shown in Figure 3d, the penetration efficiency of n(BSA) is reduced from 17.9 % to 9.9 % after 5 hr of incubation, confirming the transcytosis involves the MPC moiety of the nanocapsules. Given the similarity between MPC and acetylcholine and choline, we also measured the penetration efficiency of n(BSA) in the presence of acetylcholine (ACh) or choline (Ch) at a molar ratio of 1:100. As shown in Figure 3e, the penetration efficiency is reduced from 17.9 % to 11 % (with ACh) and 10 % (with Ch) after 5 hr incubation, confirming nAChRs and ChTs do mediate the transcytosis. For further confirmation, bEnd.3 cells were pre–incubated with 30 μM of α–bungarotoxin (α–BTX), an irreversible inhibitor for nAChRs[32]. The penetration efficiency is reduced to 10.6 % after 5 hr incubation, a similar level observed in the presence of ACh. The similar inhibiting effect was observed when incubating bEnd.3 cells with hemicholinium–3 (HC3), an inhibitor for ChTs[17] (Figure 3f). These studies collectively confirm the transcytosis of the nanocapsules is medicated by nAChRs and ChTs. Compared with the apparent binding affinity of acetylcholine (3–30 μM)[33,34] and choline (30–450 μM)[35,36], n(BSA) exhibits a similar binding affinity (~19 μM). Considering the concentration of the nanocapsules in the CNS is low (~10 nM, Figure 4e), we do not expect the delivery of the nanocapsules would perturb the normal neuronal activity. To examine the biocompatibility of the nanocapsules in the CNS, the inflammatory activation of microglia and astrocytes following an administration of n(BSA) was investigated by immunohistochemical staining of ionized calcium-binding adapter molecule 1 (Iba1, a biomarker of microglia) and glia fibrillary acidic proteins (GFAP, a biomarker of astrocytes). Compared with the mice treated by PBS, there is no significant elevation of Iba1 nor GFAP observed in the brains of mice intravenously injected with 1 mg/mL n(BSA) (Figure S6 a–d).
Figure 4. Pharmacokinetics, biodistribution, delivery efficiency and biocompatibility of protein nanocapsules to the CNS of mice and non–human primates.

a, Pharmacokinetic profiles of mice after intravenous injection of 1 mg/mL of native BSA or n(BSA) labelled with TAMRA. Mean ± s.e.m., n=3. b, Ex vivo images and c, normalized fluorescence intensity of dissected tissues including brain, heart, liver, spleen, lung, kidney, and lymph nodes of the mice administered with TAMRA–labelled BSA and n(BSA). The mice were perfused with PBS and organs were harvested 24 hr post–intravenous injection. Mean ± s.e.m., n=3, ****P < 0.0001. d, RTX plasma and e, CSF concentration of the mice intravenously injected with 5 mg/kg of native RTX or n(RTX) measured by ELISA. Mean ± s.e.m., n=3. f, TEM image of the CSF of rhesus macaques 24 hr after intravenous administration of 10 mg/kg n(HRP). g, The plasma and h, CSF concentration of n(HRP), as well as i, the ratios of the CSF concentration vs. the plasma concentration of rhesus macaques after intravenous administration of 2.5, 5.0, and 10 mg/kg of n(HRP). Mean ± s.e.m., n=2. j, Hepatic and k, Renal function, as well as l, hematology assessment of the rhesus macaques administrated with n(HRP). ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase. Mean ± s.e.m., n=3.
BBB penetration of the nanocapsules in vivo was further examined in mice. Encapsulating the protein with the MPC-based shells avoids the adsorption of serum proteins to the nanocapsules (Figure S7), resulting in prolonged circulation time. Figure 4a shows the relative plasma fluorescence intensity of mice intravenously injected with 1 mg/mL TAMRA–n(BSA). Based on one–compartment model, n(BSA) shows a half–life of 48.8 hr, which is 6.3–fold longer than that of the native BSA (7.8 hr). Figure 4b shows the fluorescent images of the main organs; high accumulation of n(BSA) in the brain is observed, which is 42–fold higher than that with the native BSA (Figure 4c). The localization of n(BSA) in the brain parenchyma was demonstrated by immunofluorescent histopathology analysis (Figure S8). Efficient penetration of the nanocapsules through BBB and their uniform distribution in the extracellular space of neurons is observed one day after intravenous administration of n(BSA) (1 mg/mL).
Figure 4d shows the pharmacokinetics of mice intravenously injected with native RTX and n(RTX). The mice with native RTX shows a plasma–concentration profile, of which the RTX concentration exponentially decreases with a long half–life attributed to its well–engineered protein structure[37]. The mice with n(RTX) show a concentration profile with a similar half–life, indicating the nanocapsules are long circulating. It is worth noting that the RTX concentration maintains nearly constant for the 1st week, which is consistent with the sustainable–release capability of n(RTX) (Figure 2e). Figure 4e shows the RTX concentration in the cerebrospinal fluid (CSF). The mice injected with native RTX show a CSF concentration of 0.18 μg/mL and 0.025 μg/mL at day 1 and day 4, corresponding to 0.08% and 0.01% of the plasma concentration. In contrast, the mice treated with n(RTX) shows 5 and 29–fold higher CSF concentration at day 1 (0.87 μg/mL) and day 4 (0.73 μg/mL), corresponding to 0.7% and 0.6% of the plasma concentration, respectively. Even at day 14, the CSF concentration of RTX still remains as high as 0.6% of the plasma concentration, which is 7–fold higher than that with native RTX at day 1.
The ability to deliver proteins to the CNS was further validated in non-human primates. Due to its low cost, high stability and ease of quantification, n(HRP) was used as a model therapeutic protein, which was intravenously injected to rhesus macaques (Macaca mulatta) at 2.5, 5.0, and 10.0 mg per kg of body weight. The CSF collected (day 1, 10 mg/kg) was investigated by TEM (Figure 4f). The presence of spherical particles in the CSF, which is similar to n(HRP) in PBS (Figure S9), is observed. Since CSF is nearly protein–free and the proteins in the CSF is less than 10 nm in size, this observation directly indicates successful delivery of n(HRP) to the CNS. Figure 4g and 4h show the concentrations of n(HRP) in the plasma and CSF estimated by their activity, respectively. The concentration of n(HRP) in the plasma is dose–dependent, which shows an initial concentration of 0.5, 0.9, and 3.4 μg/mL and a half–life of 0.7, 2.0, and 2.1 days, respectively. The CSF concentration of n(HRP) at day 1 is 5.57, 24.17, and 68.21 ng/mL, corresponding to 1.12%, 2.63%, and 1.99% of the plasma concentration, respectively (Figure 4i). The CSF concentration reaches 5.3 % of the plasma concentration at day 4 (Figure 4i) and decreases gradually within two weeks. Even at day 14, the CSF concentration for the rhesus macaques injected with 10 mg/kg n(HRP) (3.9 ng/mL) still remains as high as 2.3% of the plasma concentration (168.6 ng/mL). This study proves the effectiveness of this delivery system for non-human primates with dosage control.
The biocompatibility of the nanocapsules was evaluated in rhesus macaques intravenously injected with n(HRP) at 10 mg per kg of the body weight. The hepatic function was assessed via the plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) (Figure 4j), as well as the levels of globulin, albumin, total protein and gamma-glutamyl transferase (GGT). Slightly increased levels of ALT were observed in Day 2 post the injection (83.1 U/L), which were recovered to a normal level from Day 3 (5.5 ~ 63.4 U/L, Supplementary Table S3). Meanwhile, the AST levels were slightly elevated during the first two days post the injection, nevertheless, the AST levels were maintained within the normal range (18.4 ~ 76.2 U/L, Supplementary Table S3) during the whole process. These results indicate an absence of obvious hepatic toxicity of the nanocapsules upon intravenous administration. Other biochemistry indices related to liver function, including globulin level, albumin level, albumin to globulin (A/G) ratio, total protein concentration and GGT level, were also measured. Consistently, the levels of such indices are within the normal ranges (Supplementary Figure S10, Table S3). In addition, the renal function was assessed via the levels of creatinine, uric acid and urea, which also maintained unchanged and fall within the normal ranges (Figure 4k, Supplementary Table S3), indicating an absence of kidney damage.
The systemic toxicity of the nanocapsules were further investigated by the complete blood count tests. The cell counts of the white blood cells, lymphocytes, monocytes and neutrophils (Figure 4l), as well as eosinophils and basophils (Supplementary Fig S11, Table S4) remained unchanged, indicating no inflammation was induced by the nanocapsules administrated. Other hematological parameters, including the count of red blood cells, platelets, levels of hemoglobin and hematocrit, and mean corpuscular volume, were also quantified. These parameters are consistently within the normal ranges, suggesting an absence of hematopoietic malfunction (Supplementary Figure S11, Table S4). These findings collectively suggest that no obvious systematic toxicity could be observed in the treated rhesus macaques, indicating an excellent biocompatibility of the nanocapsules.
To summarize, we have developed a delivery platform enabling the effective delivery of therapeutic proteins to the CNS, as demonstrated in mice and non–human primates. Through encapsulation within the nanocapsules that contain choline and acetylcholine analogues, protein therapeutics can be effectively delivered to the CNS through intravenous injection for the treatment of CNS diseases. This delivery strategy may be extended to deliver therapeutics beyond proteins, bring benefits for the treatment of various CNS diseases, such as Alzheimer’s disease, Parkinson’s disease, stroke, spinal cord injury and brain tumors, which remain poorly treated but devastatingly affect the quality of life.
Supplementary Material
Acknowledgements
The authors would like to thank the California NanoSystem Institute and the Translational Pathology Core Laboratory for technical assistance. The authors would also like to thank Yaqing Huang and Jun Ma for the assistance of experiment and statistical analysis. Mice research described in the study was conducted under the University of California, Los Angeles (UCLA)’s Chancellor’s Animal Research Committee (Institutional Animal Care and Use Committee [IACUC]) in accordance with guidelines for housing and care of laboratory animals of the National Institutes of Health (NIH) and the Association for the Assessment and Accreditation of Laboratory Animal Care (AALAC) International. The study of non-human primates was performed at the Institute of Medical Biology, Chinese Academy of Medical Sciences. The animal protocol was approved by the local animal care committee. This work was supported by the UCLA AIDS Institute HIV extinction project funded from the McCarthy Family Foundation (M.K.), NIH/NIAID RO1AI110200 (M.K.), NIH/NCI RO1CA232015 (M.K.), NIH/NIAID U19AI117941 (I.S.Y.C), R21 AI114433 (I.S.Y.C), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-2–001) and National Nature Science Foundation of China (Grant No. 81602208). Equipment located in the UCLA AIDS Institute is supported by the James B Pendleton Charitable Trust. Cell sorting was performed in the CFAR Flow Cytometry Core Facility supported by NIH P30 CA016042 and P30 AI028697.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Di Wu, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
Meng Qin, College of Life Science and Technology, and Beijing Advanced Innovation Center for Soft Matter Science and Engineering Beijing University of Chemical Technology Beijing, 100029, China.
Duo Xu, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
Lan Wang, Department of Microbiology, Immunology and Molecular Genetics University of California Los Angeles, CA 90095, USA.
Chaoyong Liu, College of Life Science and Technology, and Beijing Advanced Innovation Center for Soft Matter Science and Engineering Beijing University of Chemical Technology Beijing, 100029, China.
Jie Ren, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
George Zhou, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
Chen Chen, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
Fengmei Yang, Institute of Medical Biology Chinese Academy of Medical Sciences and Peking Union Medical College Kunming, Yunnan 650118, China.
Yanyan Li, Institute of Medical Biology Chinese Academy of Medical Sciences and Peking Union Medical College Kunming, Yunnan 650118, China.
Yuan Zhao, Institute of Medical Biology Chinese Academy of Medical Sciences and Peking Union Medical College Kunming, Yunnan 650118, China.
Ruyi Huang, Department of Neurosurgery University of California Los Angeles, CA 90095, USA.
Sina Pourtaheri, Department of Neurosurgery University of California Los Angeles, CA 90095, USA.
Chunsheng Kang, Tianjin Neurological Institute, MOE Key Laboratory of Post–Neuroinjury Neuro–Repair and Regeneration in Central Nervous System The General Hospital of Tianjin Medical University Tianjin, 300052, China.
Masakazu Kamata, UCLA AIDS Institute Los Angeles, CA 90095, USA; Division of Hematology–Oncology David Geffen School of Medicine at UCLA Los Angeles, CA 90095, USA.
Irvin S.Y. Chen, Department of Microbiology, Immunology and Molecular Genetics University of California Los Angeles, CA 90095, USA UCLA AIDS Institute Los Angeles, CA 90095, USA.
Zhanlong He, Institute of Medical Biology Chinese Academy of Medical Sciences and Peking Union Medical College Kunming, Yunnan 650118, China.
Jing Wen, Department of Microbiology, Immunology and Molecular Genetics University of California Los Angeles, CA 90095, USA; ; UCLA AIDS Institute Los Angeles, CA 90095, USA.
Wei Chen, Beijing Institute of Biotechnology Beijing, 100029, China.
Yunfeng Lu, Department of Chemical and Biomolecular Engineering University of California Los Angeles, CA 90095, USA.
Reference
- [1].W. H. Organization, Neurological Disorders: Public Health Challenges, World Health Organization, 2006. [Google Scholar]
- [2].Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS, Scientific Advisory Board and the Executive Committee of the Grand Challenges on Global Mental Health, Anderson W, Dhansay MA, Phillips A, Shurin S, Walport M, Ewart W, Savill SJ, Bordin IA, Costello EJ, Durkin M, Fairburn C, Glass RI, Hall W, Huang Y, Hyman SE, Jamison K, Kaaya S, Kapur S, Kleinman A, Ogunniyi A, Otero-Ojeda A, Poo M-M, Ravindranath V, Sahakian BJ, Saxena S, Singer PA, Stein DJ, Nature 2011, 475, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Obermeier B, Daneman R, Ransohoff RM, Nat. Med. 2013, 19, 1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pardridge WM, NeuroRx 2005, 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Y, Liu L, Adv. Drug Deliv. Rev. 2012, 64, 640. [DOI] [PubMed] [Google Scholar]
- [6].Nance EA, Woodworth GF, Sailor KA, Shih T-Y, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J, Sci. Transl. Med. 2012, 4, 149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Banks WA, Nat. Rev. Drug Discov. 2016, 15, 275. [DOI] [PubMed] [Google Scholar]
- [8].Liu L, Xu K, Wang H, Tan PKJ, Fan W, Venkatraman SS, Li L, Yang Y-Y, Nat. Nanotechnol. 2009, 4, 457. [DOI] [PubMed] [Google Scholar]
- [9].Hervé F, Ghinea N, Scherrmann J-M, AAPS J. 2008, 10, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pardridge WM, Nat. Rev. Drug Discov. 2002, 1, 131. [DOI] [PubMed] [Google Scholar]
- [11].Batrakova EV, Gendelman HE, Kabanov AV, Expert. Opin. Drug Deliv. 2011, 8, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aryal M, Arvanitis CD, Alexander PM, McDannold N, Adv. Drug Deliv. Rev. 2014, 72, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moriyama E, Salcman M, Broadwell RD, Surg Neurol 1991, 35, 177. [DOI] [PubMed] [Google Scholar]
- [14].Gotti C, Clementi F, Prog. Neurobiol. 2004, 74, 363. [DOI] [PubMed] [Google Scholar]
- [15].Kumar P, Wu H, McBride JL, Jung K-E, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N, Nature 2007, 448, 39. [DOI] [PubMed] [Google Scholar]
- [16].Abbott NJ, Rönnbäck L, Hansson E, Nat. Rev. Neurosci. 2006, 7, 41. [DOI] [PubMed] [Google Scholar]
- [17].Lockman PR, Allen DD, Drug Dev Ind Pharm 2002, 28, 749. [DOI] [PubMed] [Google Scholar]
- [18].Misawa H, Nakata K, Matsuura J, Nagao M, Okuda T, Haga T, Neuroscience 2001, 105, 87. [DOI] [PubMed] [Google Scholar]
- [19].Unwin N, J. Mol. Biol. 2005, 346, 967. [DOI] [PubMed] [Google Scholar]
- [20].Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK, Nature 2001, 411, 269. [DOI] [PubMed] [Google Scholar]
- [21].Yan M, Du J, Gu Z, Liang M, Hu Y, Zhang W, Priceman S, Wu L, Zhou ZH, Liu Z, Segura T, Tang Y, Lu Y, Nat. Nanotechnol. 2010, 5, 48. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y, Du J, Yan M, Lau MY, Hu J, Han H, Yang OO, Liang S, Wei W, Wang H, Li J, Zhu X, Shi L, Chen W, Ji C, Lu Y, Nat. Nanotechnol. 2013, 8, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li J, Zhou L, Ye D, Huang S, Shao K, Huang R, Han L, Liu Y, Liu S, Ye L, Lou J, Jiang C, Adv. Mater. 2011, 23, 4516. [DOI] [PubMed] [Google Scholar]
- [24].Tuszynski MH, Yang JH, Barba D, U H-S, Bakay RAE, Pay MM, Masliah E, Conner JM, Kobalka P, Roy S, Nagahara AH, JAMA Neurol. 2015, 72, 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anand P, Terenghi G, Warner G, Kopelman P, Williams-Chestnut RE, Sinicropi DV, Nat. Med. 1996, 2, 703. [DOI] [PubMed] [Google Scholar]
- [26].Abès R, Gélizé E, Fridman WH, Teillaud J-L, Blood 2010, 116, 926. [DOI] [PubMed] [Google Scholar]
- [27].Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J, Nat. Med. 2005, 11, 551. [DOI] [PubMed] [Google Scholar]
- [28].Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, Ignoffo R, Aldape K, Shen A, Lee D, Grillo-Lopez A, Shuman MA, Blood 2003, 101, 466. [DOI] [PubMed] [Google Scholar]
- [29].Duan X, Li Y, Small 2013, 9, 1521. [DOI] [PubMed] [Google Scholar]
- [30].Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S, Adv. Mater. 2009, 21, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Thorne RG, Nicholson C, Proc. Natl. Acad. Sci. USA 2006, 103, 5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Couturier S, Bertrand D, Matter J-M, Hernandez M-C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M, Neuron 1990, 5, 847. [DOI] [PubMed] [Google Scholar]
- [33].Yu W, Yang F, Hao X, Ma J, Zhao Y, Li Y, Wang J, Xu H, Chen L, Liu Q, Duan S, Huang F, He Z, Manuscript submitted [Google Scholar]
- [34].Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J, Mol. Pharmacol. 1995, 48, 774. [PubMed] [Google Scholar]
- [35].Allen DD, Smith QR, J. Neurochem. 2001, 76, 1032. [DOI] [PubMed] [Google Scholar]
- [36].Allen DD, Lockman PR, Roder KE, Dwoskin LP, Crooks PA, J. Pharmacol. Exp. Ther. 2003, 304, 1268. [DOI] [PubMed] [Google Scholar]
- [37].Kimby E, Cancer Treat. Rev. 2005, 31, 456. [DOI] [PubMed] [Google Scholar]
- [38].Yu W, Yang F, Hao X, Ma J, Zhao Y, Li Y, Wang J, Xu H, Chen L, Liu Q, Duan S, Huang F, He Z, Manuscript submitted. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.