

Abstract
A series of platinum (II) metallacycles were prepared via the coordination-driven self-assembly of a phenazine-cored dipyridyl donor with a 90° Pt(II) acceptor and various dicarboxylate donors in a 1:1:2 ratio. While the metallacycles display similar absorption profiles, they exhibit a trend of blue-shifted fluorescence emission with the decrease in the bite angles between the carboxylate building blocks. Comprehensive spectroscopic and dynamic studies, as well as a computational approach were conducted, revealing that the difference in the degree of constraint imposed on the excited-state planarization of the phenazine core within these metallacycles results in the distinct photophysical behaviors. As such, a small initial difference in the dicarboxylate building blocks is amplified into distinct photophysical properties of the metallacycles, which is reminiscent of the efficient functional tuning observed in natural systems. In addition to the pre-assembly approach, the photophysical properties of a metallacycle can also be modulated using a post-assembly modification to the dicarboxylate building block, suggesting another strategy for functional tuning. This research illustrated the potential of coordination-driven self-assembly for the preparation of materials with precisely tailored functionalities at the molecular level.
Graphical Abstract
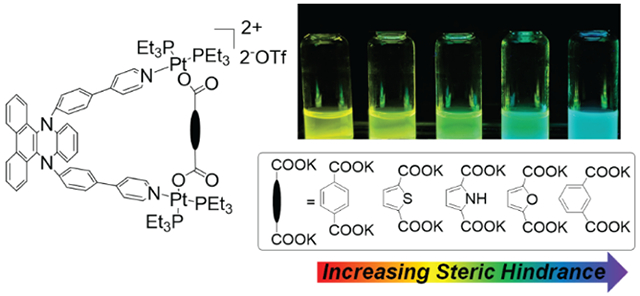
INTRODUCTION
In natural systems, large biomolecules such as proteins and nucleic acids are constructed via the covalent linking of small molecular building blocks and the subsequent self-assembly of these components.1 Subtle changes in these molecular building blocks can have a profound influence on the structure of the resulting biomolecules and consequently, their biological function as well.1 This serves as the basis for the structural and functional diversity of biomolecules that are involved in innumerable biological processes.
Coordination-driven self-assembly is a powerful strategy for the synthesis of supramolecular coordination complexes (SCCs) based on the spontaneous formation of metal-ligand bonding between Lewis basic organic donors and Lewis acidic metal acceptors.2 By the careful selection of the precursors, SCCs with pre-designed, well-defined sizes and shapes, ranging from simple rectangles to complex polyhedral, were obtained via this methodology.2-3 In addition to efforts on the design and synthesis of complexes with high structural complexity, the development of functional systems is an emerging area of interest.2, 4 However, the properties of SCCs are typically directly inherited from the precursor building blocks. Hence, covalent modifications/alterations to the functional moieties (which may require tedious synthesis and purification) are necessary to tune the properties of the resulting assemblies. Based on nature’s elegant strategy for the customization of functions and activities via the self-assembly of small molecular building blocks, we expect that the precise control over the structures of SCCs enabled by coordination-driven self-assembly can be further translated into facile functional tunability when molecules with environment-based property change are used as building blocks. We chose photoluminescence property due to its diverse responsiveness towards the local environments and widespread applications.5 The photophysical properties of SCCs have been an emerging area of interest.4b, 6 However, covalent modification to the luminophore6c, 7 or directed aggregation of SCCs8 is required to modulate the properties. The facile and precise control over the photophysical properties using the SCC platform is still challenging.
N, N‑disubstituted dihydrodibenzo[a,c]phenazines are a class of molecules that exhibit unique red and blue dual fluorescence emission. Previous studies used 9,14-diphenyl-9,14-dihydrodibenzo[a,c]phenazine (DPAC) as a prototype and indicated that the property could be attributed to the vibrational motions of the two aromatic moieties along the N–N axis.9 Excitation of the molecule results in an initial bent excited state due to the steric hindrance caused by the N-substitutes. Subsequent planarization of the dibenzo[a,c]phenazine moiety along the N–N axis extends the overall π-conjugation, and a planar excited state is attained at the global minimum.9a, 10 Thus, the emission color of the molecules is tuned from red to deep blue by varying the degree of inhibition of the planarization process. This phenomenon is applied for the development of probes,11 imaging agents,12 and light-emitting materials.13
We expect that coordination-driven self-assembly can be employed to regulate the excited-state deformation of DPAC molecule when it was used as a structural element in the preparation of SCCs. This resulted in efficient and precise tuning of photophysical properties, which is reminiscent of efficient functional diversification observed in natural systems. Therefore, a series of DPAC-cored metallacycles with varying sizes were designed and synthesized to modulate the excited-state conformation of the DPAC core chromophore within the macrocycles via regulating the constraint imposed on its planarization process. There are three main strategies as follows: (i) To gain further insights into the effect of structural hindrance on planarization by formation of metallacycles via the heteroligation of the Pt(II) center with the DPAC-containing precursor and complementary dicarboxylate donors with varying geometries using dynamic experiments and computational studies. (ii) To explore the association between the reversible assembly/reassembly process of the metallacycles and their fluorescence properties. (iii) A post-assembly modification strategy was employed on a metallacycle to reveal that post-assembly structural changes can also induce fluorescence emission changes. Comprehensive spectroscopic and dynamic studies and a computational approach exhibit a correlation between the size-dependent fluorescence emission and kinetics of planarization versus structural changes in the metallacycles. The reversible assembling/disassembling process of metallacycles and post-assembly modification experiments further supported the proposed origin of changes in the photophysical properties of the metallacycles and provided an alternative route for switching the functions expressed within a supramolecular system.
RESULTS AND DISCUSSION
Synthesis and Structural Characterization.
In order to be exploited as a donor precursor for coordination-driven self-assembly, the DPAC fluorophore was functionalized with two pyridyl groups at the para sites of the 9,14-diphenyl groups. Given the flexibility of the DPAC moiety, a multi-component self-assembly strategy using Pt(II) heteroligation was employed for the synthesis of the metallacycles.14 DPAC-containing ligand 1, dicarboxylate donors 2a–2e with different bite angles between the carboxylate groups and a 90° di-Pt(II) acceptor 3 were mixed in a stoichiometry of 1:1:2 to yield metallacycles 4a–4e (Scheme 1). The structure of the metallacycles was characterized via multinuclear NMR and electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS). The 31P{1H} spectra of the metallacycles (Figures 1a, S9, S13, S17, S21, and S25) exhibited two sets of coupled doublets of similar intensity with concomitant 195Pt satellites that corresponded to two distinct phosphorous environments. This indicated that the heteroligation of the Pt(II) center with pyridyl and carboxylate moieties broke the symmetry of the two capping phosphine ligands.14 In the 1H NMR spectra of the metallacycles (Figures 1c, S7, S11, S15, S19, and S23), downfield shifts (ca. 0.05 ppm for Ha and 0.25 ppm for Hb), and broadening were observed for the resonances of the pyridyl groups when compared with those of free donor 1 (Figure 1b). This indicated the formation of Pt-pyridyl coordination bonds that reduced the electron density of the pyridyl moieties.2 The ESI-TOF-MS data (Figures 1d, S10, S14, S18, S22, and S26) further supported the formation of discrete metallacycles with the expected stoichiometry. For all metallacycles, ESI-TOF-MS exhibited two peaks ([M-OTf]+ and [M-2OTf]2+) that corresponded to the assigned [1+1+2] assembly with charge states resulting from the loss of the OTf− counterions. All the peaks were isotopically resolved and in good agreement with their theoretical distributions.
Scheme 1.

Self-Assembly of Metallacycles 4a–4e.
Figure 1.

(a) 31P{1H} NMR spectrum of metallacycle 4a in CD2Cl2. (b) Partial 1H NMR spectrum of 1 in CD2Cl2. (c) Partial 1H NMR spectrum of 4a in CD2Cl2. (d) Full ESI-MS spectrum of 4a.
Steady-State Photophysical Properties.
The normalized absorption and emission spectra of ligand 1 and metallacycles 4a–4e in CH2Cl2 are shown in Figures 2a-2f. Ligand 1 exhibited the lowest lying absorption band maximized at 411 nm and dual emission bands centered at 421 and 613 nm that originated from the unrelaxed structure and planarized species, respectively, in the excited state. The results were similar to those previously reported in the DPAC-chromophore, exhibiting the absorption onsets at approximately 400 nm and dual emission at approximately 400 and 610 nm.9 Conversely, all metallacycles displayed nearly identical absorption bands with a peak wavelength at 360 nm. The result indicated that the metallacycles exhibited similar ground-state characteristics and differed from 1 due to the formation of Pt-pyridyl coordination bond. Despite their similar absorption profiles, the metallacycles exhibited differences in the fluorescence properties with each other. For 4a that contained a 180° dicarboxylate donor, the fluorescence peak wavelength was observed at 570 nm with a shoulder at 500 nm. A trend of blue-shifted emission for 4b (528 nm), 4c (519 nm), 4d (511 nm), and 4e (499 nm) was observed and related to a decrease in the bite angle between the dicarboxylate donor (vide infra). This result indicated that the degree of structural relaxation for the metallacycles was in the order of 1 < 4a < 4b < 4c < 4d < 4e and was induced by steric hindrance. The blue shift of the emission is easily visualized as shown in the fluorescence images of 4a–4e (inset in Figure 2g). The changes in Stokes shifts (calculated from peak to peak between absorption and emission in energy) ranged from 1.27 eV (10234 cm−1) of 4a to 0.97 eV (7816 cm−1) of 4e in the order of 4a > 4b > 4c > 4d > 1 > 4e as shown in Figure 2h. The metallacycles exhibited the opposite trend in Stokes shifts when compared with the degree of structural relaxation among each other, and this affirmed the proposed steric hindrance on the formation of the Pt-pyridyl coordination. When compared to the metallacycles, ligand 1 exhibited the most red-shifted emission although it was ranked second last in Stokes shifts of 2.02 eV (8019 cm-1). This type of difference was attributed to changes in the absorption peak in 1, i.e., the structural variation in its ground state.
Figure 2.

Steady-state absorption (dashed line) and photoluminescence (solid line) spectra of 1 (a), 4a (b), 4b (c), 4c (d), 4d (e), and 4e (f) in CH2Cl2 at room temperature. Q.Y. = emission quantum yield. (g) Plots of the energy of absorption and emission peaks of ligand 1 and metallacycles 4a–4e. λabs and λem denote wavelengths of the lowest-lying absorption and emission peaks, respectively, and the values (nm) are converted into energy units (eV). Insets: photographs of the metallacycles obtained under irradiation of 365-nm UV lamp. (h) Plots of the Stokes shifts in energy versus the compounds.
Time-Resolved Emission Spectroscopy.
In order to gain insights into the photophysical properties of the compounds, comprehensive spectral/dynamic analyses were conducted using nanosecond time-correlated single photon counting (TCSPC) and femtosecond fluorescence up-conversion techniques. The dynamics of the excited-state relaxation of 1 and metallacycles 4a–4e were recorded in solutions at 298 K and are shown in Figures 3 and 4 for 4a and 4e, respectively, and Figures S35 to S39 for the remaining compounds while the pertinent data are tabulated in Table 1 and S1. For ligand 1 in CH2Cl2, with respect to monitoring at the normal Stokes-shifted emission band (420 nm), the relaxation dynamics exhibited a fast decay component (τ ~17 ps, Figure S35) while the decay of the large Stokes-shifted emission band (monitored at 610 nm) was fitted as 235 ps. When compared to DPAC, a similar fast decay component (τ ~15 ps) was observed for the short-wavelength band (410 nm) while the population decay time of the 610-nm emission reached 11.6 ns.13 This explained the differences in the ratiometric emission intensity between 1 and DAPC where the intensity of the 420-nm emission band (quantum yield, Q.Y. ~ 0.1%) was significantly weaker than that of the emission at 610 nm (Q.Y. ~ 22%) for DAPC. Conversely, an almost equal emission intensity was observed for the 420 nm emission band (Q.Y. ~0.24%) and 610 nm emission (Q.Y. ~0.45%) in compound 1 (Figure 2). The weaker 610 nm emission for 1 (cf. DAPC) mainly resulted from the torsional motion of the two 4-phenylpyridine substituents that induced dominant radiationless deactivation.
Figure 3.

Femtosecond fluorescence transients (open circles) of 4a in CH2Cl2 at room temperature observed at 460 nm (a, f), 500 nm (b, g), 540 nm (c, h), 580 nm (d, i), and 620 nm (e, j) as depicted in 10 ps and 30 ps time scales. The solid lines in the figure denote the fitting curves. The excitation wavelength is 380 nm. The fitting results are tabulated in Table 1.
Figure 4.

Femtosecond fluorescence transients (open circles) of 4e in CH2Cl2 at room temperature observed at 460 nm (a, f), 500 nm (b, g), 540 nm (c, h), 580 nm (d, i), and 620 nm (e, j) as depicted in 10 ps and 30 ps time scales. The solid lines in the figure denote the fitting curves. The excitation wavelength is 380 nm. The fitting results are tabulated in Table 1.
Table 1.
Fitting results of femtosecond up-conversion measurements for 4a and 4e in CH2Cl2 at room temperature.
| Metallacycles | λprobe (nm) | τobs (pre-exponential factor) b |
|---|---|---|
| 4a a | 460 | τ1: 0.52 ps (0.04); τ2: 6.07 ps (0.92); τp: 621 ps (0.04) c |
| 500 | τ1: 0.52 ps (0.03); τ2: 6.07 ps (0.90); τp: 621 ps (0.07) c | |
| 540 | τ1: 0.52 ps (−0.42); τ2: 6.07 ps (0.43); τp: 621 ps (0.14) c | |
| 580 | τ1: 0.52 ps (−0.44); τ2: 6.07 ps (0.40); τp: 621 ps (0.16) c | |
| 620 | τ1: 0.52 ps (−0.39); τ2: 6.07 ps (−0.02); τp: 621 ps (0.59) c | |
| 4e a | 460 | τ1: 0.52 ps (−0.17); τ2: 45.01 ps (0.78); τp: 1517 ps (0.05) c |
| 500 | τ1: 0.52 ps (−0.29); τ2: 45.01 ps (0.64); τp: 1517 ps (0.07) c | |
| 540 | τ1: 0.52 ps (−0.43); τ2: 45.01 ps (0.40); τp: 1517 ps (0.17) c | |
| 580 | τ1: 0.52 ps (−0.48); τ2: 45.01 ps (0.28); τp: 1517 ps (0.24) c | |
| 620 | τ1: 0.52 ps (−0.15); τ2: 45.01 ps (−0.30); τp: 1517 ps (0.55) c |
Lifetime is measured by using an up-conversion system with femtosecond excitation pulses. (λex = 380 nm)
Numbers in parenthesis are normalized pre-exponential factors of the decay expressed in percentage.
Time constant τp is determined from the TCSPC result and used for the fitting of up-conversion signals
Based on the design strategy, the dicarboxylate donors between the two Pt atoms introduced various degrees of steric effect that affected planarization kinetics. Using excitation at 380 nm and monitoring at the blue edge of the steady-state emission (460 nm), the relaxation dynamics of 4a exhibited a fast decay component (0.52 ps) (Figure 3). This was followed by a fast decay of 6.07 ps and a long decay component with a considerably low intensity, which could not be resolved in the time window of approximately 30 ps that was applied for the femtosecond fluorescence up-conversion measurement. The long decay was further fitted as 621 ps by TCSPC (Figure S35). With respect to monitoring from 500 to 520 nm, both the 0.52 and 6.07 ps decay components as shown in Table 1 gradually decreases their pre-exponential factor, and thereby the weighing ratio. This was accompanied by an increase in the intensity of the long 621 ps decay component. When the monitored emission wavelength extended beyond 540 nm, the pre-exponential factors for both 0.52 ps and 6.07 ps components started to become negative, indicating rising kinetics. With respect to monitoring at the red edge of the emission (e.g., 620 nm), the kinetic trace was fitted by a rise component of 6.07 ps and a long population decay of 621 ps while the 0.52 ps became vague due to its considerably low contribution in the weighing percentage. The same fitted time constant of 6.07 ps between 460 nm (decay) and 620 nm (rise) emissions revealed a precursor-successor type of kinetic relationship. Based on the previous studies on DPAC-type systems9-10 and computational studies (vide infra), the dynamics of the emission are qualitatively described by a process that is expressed as: , where R* represents the initial Franck–Condon excited state that possesses a charge transfer character and P* denotes the global minimum mainly after the planarization. The parent DPAC molecule exhibited an additional intermediate excited state during the planarization.13 However, 4a–4e only displayed R* and P* states due to the constraint imposed by their macrocyclic structures and as is supported by our computational studies (vide infra). This was similar to a few of the reported locked DPAC chromophores that lacked an intermediate excited state in the planarization process.15 Hence, it was reasonable to assign the first decay time constant τ1 (0.52 ps) to the solvent relaxation that responded to the charge transfer character of R* of 4a and thus channeled into the reaction coordinates because a time constant of sub- to few picosecond time scales was commonly reported for polar and low viscous solvents such as CH2Cl2.15 Given the sparse solubility, the time-dependent structural planarization studies for 4a–4e could not be performed in nonpolar solvents to avoid (or diminish) the solvent relaxation process.9-10 The gradual disappearance of the contribution of solvent relaxation given increases in emission wavelength monitored was rationalized by the structural planarization that increased the ππ* character and thereby reduced the charge transfer properties. Therefore, the 6.07 ps was assigned as the time constant for the planarization of 4a in CH2Cl2.
Similar kinetic patterns were observed for 4e in CH2Cl2 (Figure 4), in which the early fast decay component of 0.52 ps is ascribed to the solvent relaxation dynamics. The other time constants were fitted as 45 ps for the planarization and 1.5 ns for the population decay time. The significantly longer planarization time constant for 4e relative to 4a was attributed to the decrease in the bite angle between the dicarboxylate donor, and this enhanced the steric hindrance in the planarization process (vide infra). As shown in Table 1, the planarization time constant for the metallacycles is in the order of 4a (6.07 ps) < 4b (8.20 ps) < 4c (9.08 ps) < 4d (11.10 ps) < 4e (45.01 ps), and this was correlated (in a reverse manner) with the magnitude of the emission Stokes shift in the order of 4a > 4b > 4c > 4d > 4e. Therefore, both the kinetics and steady-state emission spectrum revealed that the R* → P* process for all the metallacycles was subject to different degrees of constraint. All metallacycles 4a–4e exhibited a similar 0.52 ps fast relaxation time constant, thereby supporting its assignment to the solvent relaxation time in CH2Cl2 due to the charge transfer character of R*.
Computational Studies.
Further insights into the structure-relaxation relationship for the metallacycles were obtained by a computational approach. Ground state geometries of the compounds were optimized via the density functional theory (DFT) method associated with a LANL2DZ basis set for the Pt atom and a 6–31+G(d,p) basis set for all other atoms.16 The electronically excited structures with relevant photophysical properties were computed by a time-dependent density functional theory (TD-DFT) method (see the computational approaches section in SI for details). All calculated geometries and associated frontier orbitals are shown in Figures 5-6 and Tables S6-S8. The results for reference compound 1 indicated that the S0 → S1 electronic transition was from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) in which the electron density distributions in the HOMO and LUMO were mainly localized in the middle of the DPAC core and nearby phenylene-fused rings, respectively. This type of flow of electron density supported the excited-state charge transfer character (see details in Table S6-S7). With the introduction of the Pt-pyridyl coordination bonds, the orbital energy on the 9,14-diphenyl substituents decreased due to the electron deficient nature of the transition metal. Thus, the HOMOs of the metallacycles extended to the pyridyl groups at the para sites of the 9,14-diphenyl groups. In contrast, the LUMOs of the metallacycles were mainly localized at the two pyridyl groups. The computation results indicated that the initial states for the metallacycles exhibit relatively less charge-transfer character when compared with that of ligand 1.17
Figure 5.
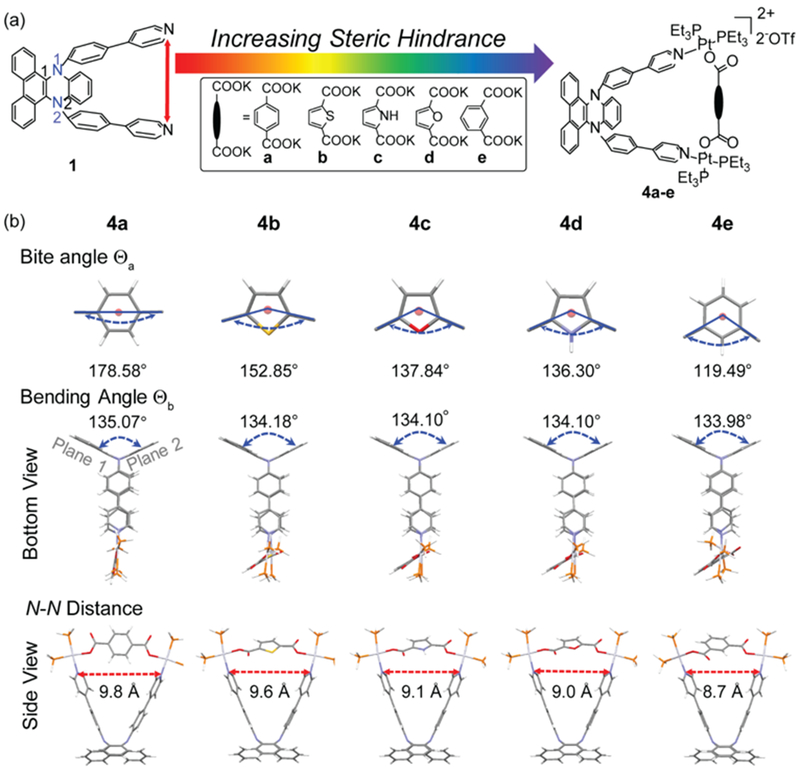
(a) Schematic illustration for the tunable fluorescence controlled by increasing steric hindrance from metallacycles 4a to 4e based on the proposed molecular design. (b) DFT-optimized ground state structures of titled molecules with the recorded values for the bite angle (Θa), bending angle (Θb), and N-N distance (Å).
Figure 6.
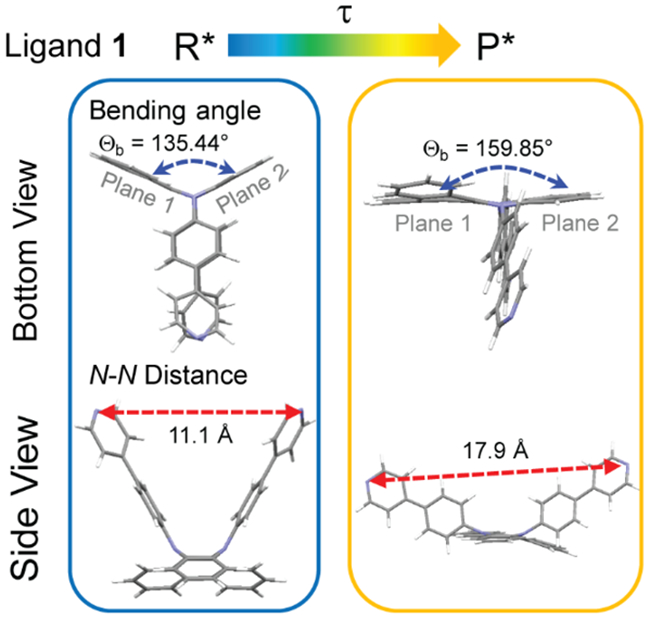
DFT-optimized excited state structures of ligand 1 with recorded values for the bending angle (Θa) and N-N distance (Å) in R* and P* states, respectively.
In the ground state, ligand 1 and all DPAC-cored metallacycles were fully optimized in geometry to exhibit a global energy minimum (Figures 5b and 6). The angle between the two carboxylate groups of the 5 or 6- member ring in dicarboxylate donor moieties (Θa), dihedral angle∠C1−N1−N2−C2 that represents the bending angle between planes 1 and 2 (Θb), and distance between the two N atoms in the pyridyl groups for ligand 1 and 4a-4e were measured (Figures 5-6). Specifically, Θa exhibited an order of 4a (178.58°) > 4b (152.85°) > 4c (137.84°) > 4d (136.30°) > 4e (119.49°), and this was in agreement with our molecular design. Ligand 1 exhibited the largest bending angle Θb = 135.44° and the longest N-N distance (11.1 Å) when compared with those of the metallacycles. The degree of the bending angles (Θb) and N-N distance (in the order of 1 > 4a > 4b > 4c > 4d > 4e) suggested that the degree of steric hindrance (i.e., the formation of Pt-pyridyl coordination and smaller Θa between the dicarboxylate donor) exhibited an order of 1 < 4a < 4b < 4c < 4d < 4e. This result was consistent with steady-state emission profiles and associated relaxation kinetics as discussed above.
The excited-state properties of the metallacycles are illustrated via calculation studies on ligand 1. The vertical electronic transition from R → R* of 1 was determined as 391 nm in CH2Cl2 and agreed with the experimental results (Figure 2a). Only a global minimum in its S1 state at Θb = 158.85° with an N-N distance of 17.9 Å was obtained for 1 in the computational results, which was denoted as P* (Figure 6). Hence, given optical excitation to the charge transfer state R*, fast solvent relaxation occurred and led to the solvent stabilized R* state. Additionally, the structural relaxation process R* → P* proceeded with the conformational change in the DPAC core via planarization, and this was accompanied by the motion of the two pyridyl substituents, i.e., an increase in Θb and N-N distance. The dynamics of the structural relaxation in 1 (although inaccessible at this stage) are prohibited in 4a-4e via the formation of Pt-pyridyl coordination bonds.
In order to further illustrate the structure-property relationship for the metallacycles with different dicarboxylate donors, a 3-D plot that comprises of Θa, the N-N distances, and emission Stokes shifts are plotted (Figure 7). The Θa (X axis in Figure 7b) are correlated with the N-N distance (Y axis) based on the projections on XY plane as denoted by red circles, and this indicated that the decrease in Θa constrained the N-N distance. On the YZ plane, the emission Stokes shift (Z axis) exhibited a correlation with the N-N distance (Y axis) (see the blue circle), thereby indicating that the decreased constraint imposed by the metallacycles results in a larger degree of structural relaxation. The Θa (X axis) also exhibited a linear relationship with the emission Stokes shift (Z axis). The results suggest that fine-tuning of fluorescence emission of metallacycles was achieved via the selection of dicarboxylate donors.
Figure 7.
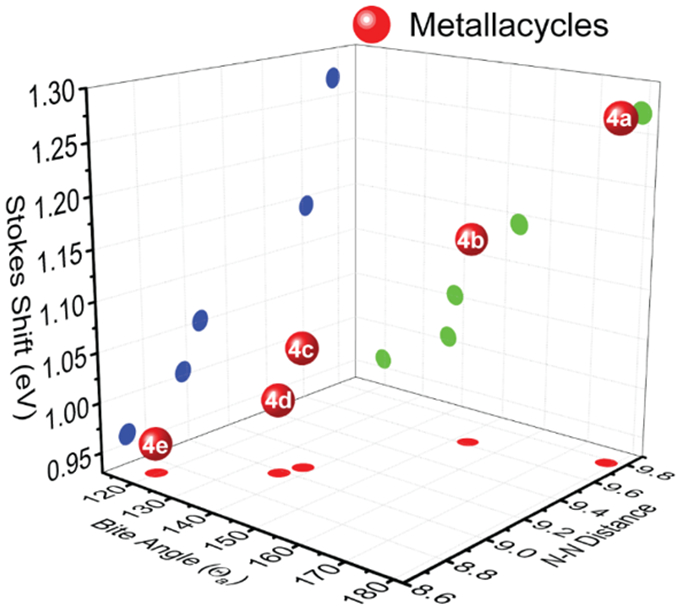
3-D plot figure of the DPAC-cored metallacycles, in which bite angle (Θa), N-N distance (Å), and Stokes shift (eV) were recorded as X, Y and Z axis, respectively. Red, blue, and green circles denote the projections on XY, YZ, and XZ planes, respectively.
Reversible Fluorescence Tuning by Assembling/Disassembling the Metallacycles.
The reversibility of the assembling/disassembling of the metallacycles is demonstrated by their emitting properties. Four equivalents of halides (i.e., Cl−, Br− or I−) are required for the coordination sites of the Pt atoms to disassemble the metallacycles and release free ligand 1. As for the re-assembling process, silver cation (Ag+) is required to precipitate the silver halide salts, so that the Pt(II) complexes can coordinate ligand 1, reforming the metallacycles. In the study, tetrabutylammonium chloride and silver trifluoromethanesulfonate (AgOTf) were employed.
The photoluminescence spectra of 4a under several assembling/disassembling cycles are recorded in CH2Cl2 and shown in Figure 8. Results on 4b–4e are shown in Figures S41-S44. The fluorescence intensity of 4a decreased with the introduction of tetrabutylammonium chloride into the solution, and the mixture gradually exhibited dual emission peaks at 421 and 613 nm, which corresponded to the characteristic emission of ligand 1 (Figure 8a). Furthermore, an isoemissive point was observed (inset of Figure 8a) and indicated the existence of an equilibrium during the disassembling process. After the completion of the disassembling process by adding four equivalents of tetrabutylammonium chloride, AgOTf was added to proceed with the re-assembling process. The emission band at 570 nm emission spectrum gradually reappeared (Figure 8b). In a manner different from the disassembling process, the re-assembly required a certain incubation time to achieve equilibrium. The reversibility of the assembling/disassembling process was evidenced by five switching cycles while maintaining 75% fluorescence intensity (Figure 8c). The decrease in fluorescence intensity in the first cycle was more significant than the following cycles, and this was attributed to emission quenching induced by the tetrabutylammonium salts. Metallacycles 4b–4e exhibited the same properties, thereby suggesting that the structure-dependent emission properties can be utilized to monitor the reversible assembling/disassembling process of supramolecular systems.
Figure 8.

(a) Photoluminescence spectra of 4a (purple), addition of one equivalence of Cl− (blue), addition of two equivalence of Cl− (green), addition of three equivalence of Cl− (yellow), and addition of four equivalence of Cl− (red) in CH2Cl2 −. (b) Photoluminescence spectra of 4a and four equivalents of Cl− (purple), addition of three equivalence of Ag+ (blue), addition of four equivalence of Ag+ after 0 min (teal), addition of four equivalence of Ag+ after 5 min (green), addition of four equivalence of Ag+ after 15 min (yellow), and addition of four equivalence of Ag+ after 30 min (red). Insets: the enlarged fluorescence spectra to exhibit the isoemissive points recorded from 380 to 475 nm. (c) Normalized Photoluminescence intensity of 4a (blue open circle) monitored at the emission maximum (570 nm) under the reversible emission tuning experiments. Black solid and red dash lines denote that the solution is added with four equivalents of Cl− and four equivalents of Ag+ after a duration time of one hour, respectively. λex = 360 nm.
Post-Assembly Modification for Tuning Fluorescence.
A post-assembly modification (PAM) strategy was employed to provide an alternative route for the tuning of the fluorescence emission properties of the metallacycles. PAM reactions are powerful tools in natural systems to regulate functions and activities of biomolecules. In non-biological self-assembled systems, PAM reactions provide a new approach for preparing complexes with tailored functionalities and inducing structural transformations between ensembles.18 Metallacycle 5a that is comprised of an alkenyl dicarboxylate building block was synthesized, and its PAM by hydrogenation of the alkenyl group afforded 5b (Scheme 2). Both 5a and 5b were characterized via multinuclear NMR and ESI-TOF-MS. The 1H NMR spectrum of 5b revealed that the resonance at 6.11 ppm which originated from the protons on the alkenyl group of 5a disappeared (Figure 9a), and a new peak was observed at 1.84 ppm (Figure 9b), thereby suggesting the reduction of the alkenyl group to alkyl group. In a manner similar to metallacycles 4a–4e, 5a and 5b exhibited absorption bands centered at 360 nm albeit with distinctive emission profiles. A red-shifted emission maximum at 533 nm (c.f. 5a at 502 nm) and marked decrease in the fluorescence intensity was observed for 5b relative to 5a (Figure 9c) due to a decrease in the constraint imposed on the planarization of the DPAC-cored chromophore given the reduction of the alkenyl building block to a more flexible alkyl building block. The computed results also support the less constraint for 5b relative to 5a by its larger bending angle and longer N-N distance (Figure S40). As discussed above, the change in the emissive properties due to PAM is an example where structural changes are translated into photoluminescence signals.
Scheme 2.
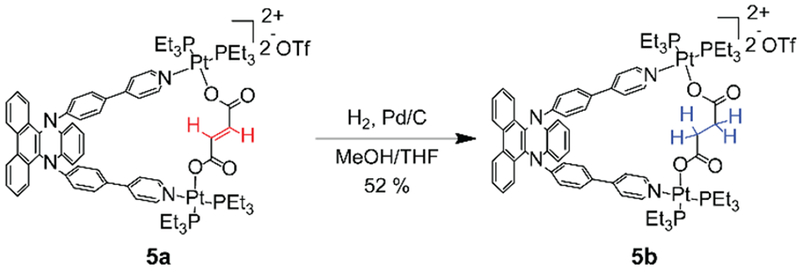
Post-assembly Modification of Metallacycle 5a.
Figure 9.
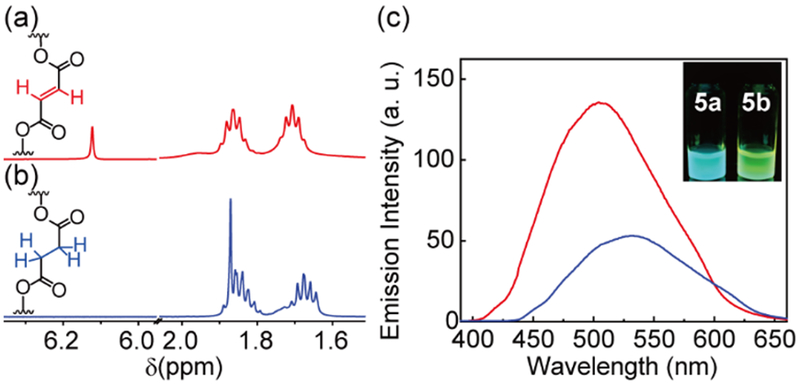
(a) Partial 1H NMR spectrum of 5a in CD2Cl2. (b) Partial 1H NMR spectrum of 5b in CD2Cl2. (c) Emission spectra of 5a (red) and 5b (blue) in CH2Cl2. Insert: Photoluminescence images of 5a and 5b in CH2Cl2 under irradiation corresponding to 365-nm UV light.
CONCLUSION
By systematically selecting the dicarboxylate building block used for the self-assembly, a series of phenazine-cored metallacycles with tunable fluorescence spanning the visible region was obtained. With decreases in the size of the metallacycles, the emission exhibits a trend of blue shifts from red-orange (570 nm) to blue (499 nm). The spectroscopic, dynamic, and computational studies indicated that the size of the metallacycles dictated the constraint imposed on the excited-state planarization of the 9,14-diphenyl-9,14-dihydrodibenzo[a,c]phenazine core, thereby leading to diversity in the photophysical properties. The empirical correlation among the emission Stokes shift, bite angles of the dicarboxylate building block, and pyridyl N-N distance was established for the metallacycles. Reversible assembling/disassembling processes of the metallacycles were probed via structure-dependent emission properties. A PAM strategy was also developed and provided an alternative approach for functional tuning. Overall, the results of the study indicated that coordination-driven self-assembly can be adapted for the precise and systematic control over functions expressed within a system by utilizing either pre-assembly selection of building blocks or PAMs.
Supplementary Material
ACKNOWLEDGMENT
P.J.S. thanks NIH (Grant R01 CA215157) for financial support. X.L. thanks the NSF (CHE-1506722) and NIH (R01GM128037) for their support. This work was supported by the funding from Ministry of Science and Technology (MOST), featured areas research program within the framework of the Higher Education Sprout Project administrated by Ministry of Education (MOE) of Taiwan. We are grateful to the National Center for the High-performance Computing (NCHC) of Taiwan for the valuable computer time and facilities.
Footnotes
Supporting Information. Supporting Information. The supporting information is available free of charge via the Internet at http://pubs.acs.org.
Synthetic methods for the compounds, NMR, ESI-TOF-MS, details for photophysical measurements, computational approaches and reversible emission tuning of the metallacycles.
The authors declare no competing financial interests.
REFERENCES
- 1. (a).Branden C; Tooze J Introduction to Protein Structure; Garland Science: New York, 1999. [Google Scholar]; (b) Lodish H; Berk A; Kaiser AC; Krieger M; Scoot MP; Brescher A; Ploegh H; Matsudaira P Molecular Cell Biology; Freeman WH: New York, 2007. [Google Scholar]
- 2.Cook TR; Stang PJ Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages Via Coordination. Chem. Rev. 2015, 115, 7001. [DOI] [PubMed] [Google Scholar]
- 3. (a).Chakrabarty R; Mukherjee PS; Stang PJ Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011, 111, 6810. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zarra S; Wood DM; Roberts DA; Nitschke JR Molecular Containers in Complex Chemical Systems. Chem. Soc. Rev. 2015, 44, 419. [DOI] [PubMed] [Google Scholar]; (c) Fujita D; Ueda Y; Sato S; Mizuno N; Kumasaka T; Fujita M Self-Assembly of Tetravalent Goldberg Polyhedra from 144 Small Components. Nature 2016, 540, 563. [DOI] [PubMed] [Google Scholar]; (d) Zhang Z; Wang H; Wang X; Li Y; Song B; Bolarinwa O; Reese RA; Zhang T; Wang X-Q; Cai J; Xu B; Wang M; Liu C; Yang H-B; Li X Supersnowflakes: Stepwise Self-Assembly and Dynamic Exchange of Rhombus Star-Shaped Supramolecules. J. Am. Chem. Soc. 2017, 139, 8174. [DOI] [PubMed] [Google Scholar]; (e) Pan M; Wu K; Zhang J-H; Su C-Y Chiral Metal–Organic Cages/Containers (Mocs): From Structural and Stereochemical Design to Applications. Coord. Chem. Rev. 2019, 378, 333. [Google Scholar]
- 4. (a).Brown CJ; Toste FD; Bergman RG; Raymond KN Supramolecular Catalysis in Metal–Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012. [DOI] [PubMed] [Google Scholar]; (b) Saha ML; Yan X; Stang PJ Photophysical Properties of Organoplatinum(Ii) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and Its Applications. Acc. Chem. Res. 2016, 49, 2527. [DOI] [PubMed] [Google Scholar]; (c) Szalóki G; Croué V; Carré V; Aubriet F; Alévêque O; Levillain E; Allain M; Aragó J; Ortí E; Goeb S; Sallé M Controlling the Host-Guest Interaction Mode through a Redox Stimulus. Angew. Chem. Int. Ed. 2017, 56, 16272. [DOI] [PubMed] [Google Scholar]; (d) Jansze SM; Severin K Clathrochelate Metalloligands in Supramolecular Chemistry and Materials Science. Acc. Chem. Res. 2018, 51, 2139. [DOI] [PubMed] [Google Scholar]; (e) Zhang D; Ronson TK; Nitschke JR Functional Capsules Via Subcomponent Self-Assembly. Acc. Chem. Res. 2018, 51, 2423. [DOI] [PubMed] [Google Scholar]; (f) Pullen S; Clever GH Mixed-Ligand Metal–Organic Frameworks and Heteroleptic Coordination Cages as Multifunctional Scaffolds—a Comparison. Acc. Chem. Res. 2018, 51, 3052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wu G-Y; Chen L-J; Xu L; Zhao X-L; Yang H-B Construction of Supramolecular Hexagonal Metallacycles Via Coordination-Driven Self-Assembly: Structure, Properties and Application. Coord. Chem. Rev. 2018, 369, 39. [Google Scholar]
- 5. (a).Ostroverkhova O Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279. [DOI] [PubMed] [Google Scholar]; (b) Mako TL; Racicot JM; Levine M Supramolecular Luminescent Sensors. Chem. Rev. 2018, 119, 322. [DOI] [PubMed] [Google Scholar]; (c) Mei J; Leung NLC; Kwok RTK; Lam JWY; Tang BZ Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718. [DOI] [PubMed] [Google Scholar]
- 6. (a).Xu L; Wang Y-X; Yang H-B Recent Advances in the Construction of Fluorescent Metallocycles and Metallocages Via Coordination-Driven Self-Assembly. Dalton Trans. 2015, 44, 867. [DOI] [PubMed] [Google Scholar]; (b) Pan M; Liao W-M; Yin S-Y; Sun S-S; Su C-Y Single-Phase White-Light-Emitting and Photoluminescent Color-Tuning Coordination Assemblies. Chem. Rev. 2018, 118, 8889. [DOI] [PubMed] [Google Scholar]; (c) Jing X; He C; Zhao L; Duan C Photochemical Properties of Host–Guest Supramolecular Systems with Structurally Confined Metal–Organic Capsules. Acc. Chem. Res. 2018, 52, 100. [DOI] [PubMed] [Google Scholar]
- 7. (a).Pollock JB; Schneider GL; Cook TR; Davies AS; Stang PJ Tunable Visible Light Emission of Self-Assembled Rhomboidal Metallacycles. J. Am. Chem. Soc. 2013, 135, 13676. [DOI] [PubMed] [Google Scholar]; (b) Neelakandan PP; Jiménez A; Nitschke JR Fluorophore Incorporation Allows Nanomolar Guest Sensing and White-Light Emission in M4l6 Cage Complexes. Chem. Sci. 2014, 5, 908. [Google Scholar]; (c) Yamashina M; Sartin MM; Sei Y; Akita M; Takeuchi S; Tahara T; Yoshizawa M Preparation of Highly Fluorescent Host–Guest Complexes with Tunable Color Upon Encapsulation. J. Am. Chem. Soc. 2015, 137, 9266. [DOI] [PubMed] [Google Scholar]
- 8. (a).Sun Y; Yao Y; Wang H; Fu W; Chen C; Saha ML; Zhang M; Datta S; Zhou Z; Yu H; Li X; Stang PJ Self-Assembly of Metallacages into Multidimensional Suprastructures with Tunable Emissions. J. Am. Chem. Soc. 2018, 140, 12819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chang X; Zhou Z; Shang C; Wang G; Wang Z; Qi Y; Li Z-Y; Wang H; Cao L; Li X; Fang Y; Stang PJ Coordination-Driven Self-Assembled Metallacycles Incorporating Pyrene: Fluorescence Mutability, Tunability, and Aromatic Amine Sensing. J. Am. Chem. Soc. 2019, 141, 1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. (a).Zhang Z; Wu Y-S; Tang K-C; Chen C-L; Ho J-W; Su J; Tian H; Chou P-T Excited-State Conformational/Electronic Responses of Saddle-Shapedn,N′-Disubstituted-Dihydrodibenzo[a,C]Phenazines: Wide-Tuning Emission from Red to Deep Blue and White Light Combination. J. Am. Chem. Soc. 2015, 137, 8509. [DOI] [PubMed] [Google Scholar]; (b) Zhang ZY; Chen CL; Chen YA; Wei YC; Su JH; Tian H; Chou PT Tuning the Conformation and Color of Conjugated Polyheterocyclic Skeletons by Installing Ortho-Methyl Groups. Angew. Chem. Int. Ed. 2018, 57, 9880. [DOI] [PubMed] [Google Scholar]
- 10.Chen W; Chen CL; Zhang ZY; Chen YA; Chao WC; Su JH; Tian H; Chou PT Snapshotting the Excited-State Planarization of Chemically Locked N,N ‘-Disubstituted Dihydrodibenzo[a,C]Phenazines. J. Am. Chem. Soc. 2017, 139, 1636. [DOI] [PubMed] [Google Scholar]
- 11. (a).Zhou H; Mei J; Chen Y-A; Chen C-L; Chen W; Zhang Z; Su J; Chou P-T; Tian H Phenazine-Based Ratiometric Hg2+Probes with Well-Resolved Dual Emissions: A New Sensing Mechanism by Vibration-Induced Emission (Vie). Small 2016, 12, 6542. [DOI] [PubMed] [Google Scholar]; (b) Humeniuk HV; Rosspeintner A; Licari G; Kilin V; Bonacina L; Vauthey E; Sakai N; Matile S White-Fluorescent Dual-Emission Mechanosensitive Membrane Probes That Function by Bending Rather Than Twisting. Angew. Chem. Int. Ed. 2018, 57, 10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dou W-T; Chen W; He X-P; Su J; Tian H Vibration-Induced-Emission (Vie) for Imaging Amyloid Β Fibrils. Faraday Discuss. 2017, 196, 395. [DOI] [PubMed] [Google Scholar]
- 13. (a).Huang W; Sun L; Zheng Z; Su J; Tian H Colour-Tunable Fluorescence of Single Molecules Based on the Vibration Induced Emission of Phenazine. Chem. Commun. 2015, 51, 4462. [DOI] [PubMed] [Google Scholar]; (b) Chen D-G; Lin T-C; Chen C-L; Chen Y-T; Chen Y-A; Lee G-H; Chou P-T; Liao C-W; Chiu P-C; Chang C-H; Lien Y-J; Chi Y Optically Triggered Planarization of Boryl-Substituted Phenoxazine: Another Horizon of Tadf Molecules and High-Performance Oleds. Acs Appl Mater Inter 2018, 10, 12886. [DOI] [PubMed] [Google Scholar]; (c) Chen D-G; Lin T-C; Chen Y-A; Chen Y-H; Lin T-C; Chen Y-T; Chou P-T Revisiting Dual Intramolecular Charge-Transfer Fluorescence of Phenothiazine-Triphenyltriazine Derivatives. J Phys Chem C 2018, 122, 12215. [Google Scholar]
- 14.Zheng Y-R; Zhao Z; Wang M; Ghosh K; Pollock JB; Cook TR; Stang PJ A Facile Approach toward Multicomponent Supramolecular Structures: Selective Self-Assembly Via Charge Separation. J. Am. Chem. Soc. 2010, 132, 16873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. (a).Scherer POJ; Seilmeier A; Kaiser W Ultrafast Intra-Molecular and Intermolecular Energy-Transfer in Solutions after Selective Infrared Excitation. J. Chem. Phys. 1985, 83, 3948. [Google Scholar]; (b) Laermer F; Elsaesser T; Kaiser W Ultrashort Vibronic and Thermal Relaxation of Dye Molecules after Femtosecond Ultraviolet Excitation. Chem. Phys. Lett. 1989, 156, 381. [Google Scholar]; (c) Jurkiewicz P; Sykora J; Olzynska A; Humplickova J; Hof M Solvent Relaxation in Phospholipid Bilayers: Principles and Recent Applications. J. Fluoresc. 2005, 15, 883. [DOI] [PubMed] [Google Scholar]
- 16. (a).Frisch MJT, G. W.; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01. Gaussian 09, Revision D.01; Gaussian Inc. 2013, Wallingford. [Google Scholar]; (b) Yang Y; Weaver MN; Merz KM Assessment of the “6–31+G**+Lanl2dz” Mixed Basis Set Coupled with Density Functional Theory Methods and the Effective Core Potential: Prediction of Heats of Formation and Ionization Potentials for First-Row-Transition-Metal Complexes. J. Phys. Chem. A. 2009, 113, 9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. (a).Gierschner J; Mack HG; Luer L; Oelkrug D Fluorescence and Absorption Spectra of Oligophenylenevinylenes: Vibronic Coupling, Band Shapes, and Solvatochromism. J Chem Phys 2002, 116, 8596. [Google Scholar]; (b) Bredas JL; Beljonne D; Coropceanu V; Cornil J Charge-Transfer and Energy-Transfer Processes in Pi-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971. [DOI] [PubMed] [Google Scholar]
- 18. (a).Roberts DA; Pilgrim BS; Nitschke JR Covalent Post-Assembly Modification in Metallosupramolecular Chemistry. Chem. Soc. Rev. 2018, 47, 626. [DOI] [PubMed] [Google Scholar]; (b) Roberts DA; Pilgrim BS; Cooper JD; Ronson TK; Zarra S; Nitschke JR Post-Assembly Modification of Tetrazine-Edged (Fe4l6)-L-Ii Tetrahedra. J. Am. Chem. Soc. 2015, 137, 10068. [DOI] [PubMed] [Google Scholar]; (c) Pilgrim BS; Roberts DA; Lohr TG; Ronson TK; Nitschke JR Signal Transduction in a Covalent Post-Assembly Modification Cascade. Nat. Chem. 2017, 9, 1276. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.