

SUMMARY
CKIα ablation induces p53 activation, and CKIα degradation underlies the therapeutic effect of lenalidomide in a pre-leukemia syndrome. Here we describe the development of CKIα inhibitors, which co-target the transcriptional kinases CDK7 and CDK9, thereby augmenting CKIα-induced p53 activation and its anti-leukemic activity. Oncogene-driving super-enhancers (SEs) are highly sensitive to CDK7/9 inhibition. We identified multiple newly gained SEs in primary mouse acute myeloid leukemia (AML) cells and demonstrate that the inhibitors abolish many SEs and preferentially suppress the transcription elongation of SE-driven oncogenes. We show that blocking CKIα together with CDK7 and/or CDK9 synergistically stabilize p53, deprive leukemia cells of survival and proliferation-maintaining SE-driven oncogenes, and induce apoptosis. Leukemia progenitors are selectively eliminated by the inhibitors, explaining their therapeutic efficacy with preserved hematopoiesis and leukemia cure potential; they eradicate leukemia in MLL-AF9 and Tet2−/−;Flt3ITD AML mouse models and in several patient-derived AML xenograft models, supporting their potential efficacy in curing human leukemia.
Graphical Abstract
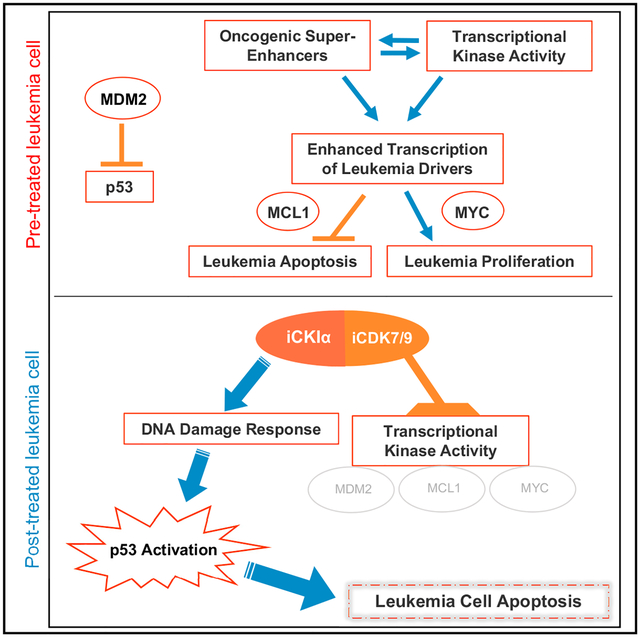
In Brief
Combined stabilization of p53 and inactivation of oncogene-driving super enhancers offers a therapeutic approach for acute myeloid leukemia.
INTRODUCTION
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy, caused by the accumulation of immature leukemic blasts in the blood and bone marrow (Löwenberg et al., 1999). Recent studies revealed the genomic landscape of the disease and pointed out its clinical and biological heterogeneity (Papaemmanuil et al., 2016). Despite advances in our understanding of the disease, there has been no major progress in the standard therapy of AML in the past 40 years (Coombs et al., 2016; Shafer and Grant, 2016). New avenues being developed for improving AML therapy include epigenetic modification of the leukemia cells by targeting chromatin regulators, such as IDH, HDAC, and BRD4 (Bradner et al., 2017; Ntziachristos et al., 2016).
A different approach to AML treatment is by means of activating the tumor suppressor p53, which is mostly non-mutated in certain malignancies including leukemia (Papaemmanuil et al., 2016). However, in many hematological malignancies including AML, p53 activity is suppressed due to multiple antagonistic mechanisms, most prominent of which is the activation of its major antagonist MDM2 (Kojima et al., 2016; Vogelstein et al., 2000). Thus, major drug development efforts for p53 activation in cancer has been focused on MDM2 inhibitors (Levine and Oren, 2009). Another robust way to activate p53 may be achieved by casein kinase 1A1 (CKIα) inhibition; this mode has been documented in vivo in mouse models of intestinal cancer (Elyada et al., 2011; Pribluda et al., 2013) and skin pigmentation (Chang et al., 2017), in cultured leukemia cells (Jaärås et al., 2014), and was recently proposed as a mechanism of action of the thalidomide derivative lenalidomide, in the pre-leukemic human myelodysplastic syndrome (MDS) (Krönke et al., 2015). Lenalidomide, functioning as a molecular glue between the human E3 ubiquitin ligase cereblon and CKIα was shown to induce the ubiquitination and degradation of this kinase (Petzold et al., 2016), thus presumably killing leukemic cells by p53 activation. While this approach indicates the therapeutic value for CKIα inhibition, it is limited to a small set of patients harboring one active allele of CK1α, such as in MDS of the del(5q) type (Krönke et al., 2015). Selective CKIα inhibitors should however reveal the full therapeutic potential of CKIα inhibition in cancer.
In an effort to develop selective small molecule CKIα inhibitors, we identified a unique class of kinase inhibitors, which as expected from knockout studies, stabilized β-catenin and p53. Unexpectedly, however, these inhibitors abolished the expression of the Wnt/β-catenin target gene Myc and the p53 master regulator and target gene Mdm2. Looking for an explanation, we found that the CKIα inhibitors co-target CDK9, the catalytic subunit of the transcription elongation factor P-TEFb, and CDK7, the catalytic subunit of the transcription factor TFIIH. Consequently, these inhibitors disrupt super-enhancers and thereby block the transcription elongation of multiple oncogenes. Robust p53 activation via CKIα inhibition and Mdm2 downregulation in combination with transcriptional shutdown of leukemia oncogenes, such as Myc, Myb, and Mcl1 provided an unprecedented therapeutic effect in different genetically engineered AML mouse models, as well as in patient-derived xenografts (PDX) of various AML subtypes.
RESULTS
Development of Small Molecule Pan-CKI Inhibitors
It has been reported that Csnk1A1 (CKIα) ablation compromises normal hematopoiesis and hypothesized that CKIα-deficient hematopoietic stem and progenitor cells (HSPCs) are eliminated via p53 activation (Schneider et al., 2014). Here, we confirmed this hypothesis, showing that specific ablation of CKIα in the hematopoietic system via Mx1-Cre eliminated all the HSPCs, thus vacating the bone marrow (BM) niches for successful donor cells engraftment (Figures 1A and S1A–S1D). This stem cell extinction function required wild-type (WT) p53, as co-ablation of CKIα and Tp53 (DKO) failed to eliminate the HSPCs (Figure 1A), nor did it allow hematopoietic rescue by bone marrow transplantation (BMT) (Figure S1D). This is likely due to occupation of the BM stem cell niches by persisting, yet non-functional DKO hematopoietic stem/progenitor cells, highlighting the importance of p53 activation.
Figure 1. Development of Small Molecule CKI Inhibitors.

(A) FACS analysis of BM HSC populations from CKIαfl/fl Mx1-Cre (CKIα KO), CKIαfl/fl Mx1-Cre/p53KI/KI (CKIα/p53, DKO), or CKIαfl/+ Mx1-Cre (Control), 7 days post initial knockout induction with pIpC. LT-HSC, long term HSCs; ST-HSC, short term HSCs; MPP, multipotent progenitor cells. Graph shows individual mouse data and mean; statistical analysis by Mann-Whitney test, N ≥ 6 for all groups.
(B) Structures of selected A-series CKI inhibitors.
(C) Binding constant (Kd) values of selected A-series inhibitors to members of the CKI (CSNK1), CK2 (CSNK2), and GSK3 (GSK3) families, measured Kd are shown in red and estimated Kd values (calculated based on percent control [%Ctrl]) are shown in black.
(D) Crystal structure of CKIα in complex with A86. CKIα shown as a cartoon representation, A86 as a stick representation (green, C and H atoms; blue, N atoms; gray, F atoms). Inset: ATP binding pocket showing the interacting residues Leu93 and Asp99 forming hydrogen bonds (blue dashed lines) with A86.
(E) Western blot (WB) analysis of β-catenin (β-Cat) phosphorylation in RKO cells, CKIα depleted (shCKIα), or shControl cells treated with 1 μM A51 for 18 hr. Proteasome inhibitor (MG132, 20 μM) was added 4 hr before cell harvesting.
(F) WB analysis of RKO cells treated for 18 hr with A-series inhibitors at the indicated concentrations or with DMSO (control). PP2Ac is a loading control.
See also Figure S1.
Small molecule inhibitors may provide a therapeutic window to target leukemic cells despite lethal or adverse effects of a genetic deletion of the same target, a recent example being MCL1 inhibitors (Kotschy et al., 2016). We therefore hypothesized that incomplete, intermittent CKIα inhibition with small molecules may spare normal HSPCs, based on the assumption that leukemia progenitors are more sensitive to CKIα inhibition (Järås et al., 2014). Of the myriad of small molecule CKI inhibitors identified, none have been proven to possess the capacity to efficiently block CKIα activity (Knippschild et al., 2014). We therefore attempted to identify selective CKIα inhibitors via cell-based screening. These screens were based on a presumption derived from mouse CKIα ablation studies (Elyada et al., 2011) that small molecules found to stabilize both β-catenin and p53 in a responsive cell line might have CKIα blocking activity. Following massive cell-based screening of kinase inhibitor libraries in a colorectal cancer cell line (RKO), we focused on a small class of pyrazole-pyrimidine scaffold molecules (A-series), which, through intensive medicinal chemistry, yielded molecules with potent CKIα inhibitory activity (Figures 1B–1F). To confirm CKIα targeting, we measured compound binding to CKI family members and to the closely related CK2 family by kinome affinity matrix assay (DiscoverX Kinome Scan) and found that these compounds target all CKI members with sub-nanomolar affinity with the exception of A64 displaying high affinity only to CKIδ and CKIε; none of the compounds bound to CK2 (Figure 1C). We determined the structure of one of the potent CKI inhibitors, A86, with CKIα at 2.3Å resolution (Figure 1D). A86 binds to the hinge backbone of the kinase via two hydrogen bonds between N1 and carbonyl of Leu93, N2 of 5-methylpyrimidine, and amide of Leu93. The cyclohexanamine and cyclopropane moieties are located at the entrance of the pocket and make hydrophobic interactions with Ile23 and Ile31, respectively. Moreover, the amine group of cyclohexanamine is hydrogen-bonded to OD2 of Asp99. Amino acids that form hydrogen bonds with the ligand in CKIα-A86 crystal structure (Leu93 and Asp99) are conserved over all the CKI family (but not in CK2) (Figure S1E). Indeed, co-crystallography of A86 with CKIδ (88% sequence homology with CKIα) indicated similar interactions at the active site (Figure S1F).
CKIα phosphorylates β-catenin on Ser45, priming its sequential phosphorylation by GSK3 on a set of conserved Ser and Thr residues in its N terminus, ultimately leading to β-catenin ubiquitination and degradation (Amit et al., 2002). Similar to CKIα depletion, inhibitor treatment of RKO cells abolished most of the Ser45 phosphorylation signal and the consecutive GSK3 phosphorylation cascade resulting in stabilization of β-catenin (Figures 1E and 1F). As CKIα modifies Ser45 uniquely (Amit et al., 2002), it attests to the CKIα-inhibitory capacity of the molecules in live cells, a property that has not been documented before for other reported CKI inhibitors. CKIα ablation stabilizes p53 (Chang et al., 2017; Elyada et al., 2011) and induces DNA damage response (DDR), evident by H2AX phosphorylation, yet without evidence of DNA damage (Pribluda et al., 2013; unpublished data). These features are reproduced by the A series inhibitors, in a dose-dependent manner, in correlation to their CKIα affinity (Figures 1C and 1F). A64, unlike other A-series members, has low CKIα affinity, but high GSK3α/β affinity, and only induces p53 at a high concentration, possibly due to GSK3-inhibition-mediated β-catenin stabilization (Damalas et al., 1999) (Figures 1C and 1F); therefore, it is often used in our functional assays as a negative control. Together, our findings affirm that these A-series molecules are pan-specific CKI inhibitors with discernible CKIα inhibitory activity.
Selective Anti-leukemic Activity of CKIα Inhibitors in the MLL-AF9 AML Mouse Model
We then asked whether the inhibitors, unlike genetic deletion of CKIα (Figure 1A), are able to distinguish leukemic from normal hematopoietic stem cells. We first tested A-series inhibitors for apoptotic killing ability against primary leukemic BM cells isolated from MLL-AF9-induced AML mice, representing a poor-prognosis human acute leukemia (Barabé et al., 2007). Of the six compounds, four (A86, A51, A75, and A14) were found to be highly effective in inducing leukemia cell apoptosis at 160 nM or lower (Figure 2A), mostly in correlation to their capacity to stabilize p53 (Figure 1F). We tested the selectivity of one of these apoptosis-inducing inhibitors in a methylcellulose colony-forming unit (CFU) assay. At the nanomolar concentration range tested, A51 did not affect normal hematopoietic CFUs. However, leukemic CFUs were highly sensitive to the inhibitors with an IC50 below 9 nM (Figure 2B), indicating a selective anti-leukemic activity of the inhibitor. Treatment of cKit+ leukemia progenitors, highly enriched in leukemic stem cells (LSCs) (Somervaille and Cleary, 2006), indicated that this population is as sensitive to inhibitor killing as the cKit− cells (Figure S2A).
Figure 2. Selective Anti-leukemic Activity of Prototype CKI Inhibitors.

(A) Primary BM cells from MLL-AF9- AML sick mice treated for 18 hr with the indicated inhibitors. FACS analysis; shown is the percentage of Annexin V-positive apoptotic cells in response to the indicated inhibitor concentration (log10). y axis intersection corresponds to DMSO control. Experiment performed in triplicates; for each inhibitor concentration, data are presented as mean ± SD.
(B) Images and quantification of colony forming unit (CFU) assay using primary BM cells isolated from WT or advanced MLL-AF9-driven AML mice, treated ex vivo with DMSO or A51 at the indicated concentrations. Shown is a representative experiment (N = 3), all performed in duplicates. Quantification of CFUs (mean ± SD) is shown below.
(C) WB analysis of BM cells isolated from leukemic mice treated for 6 hr with A51 (10 mg/kg) or vehicle. PP2Ac, a loading control.
(D) Representative images of spleen (top), tibia, and femur (bottom) from AML mice treated with vehicle or a single dose of A51 (20 mg/kg) for 16 hr.
(E) Absolute numbers of GFP+ leukemia blasts in BM from AML mice treated with a single oral dose of A51 (20 mg/kg) or vehicle for 16 hr. Graphs show mean ± SD values, N ≥ 3 for all groups.
(F and G) Representative H&E-stained tissue sections (scale bar, 200 μm for F and 50 μm for G) and blood smear (scale bar, 30 μm) of affected (F) and unaffected (G) tissue from AML mice treated with a single dose of A51 (20 mg/kg) or vehicle for 16 hr.
See also Figure S2.
Acute in vivo anti-leukemic effects of the inhibitors were first tested in heavy burden MLL-AF9 leukemic mice by single oral dose treatment. Remarkably, 5–6 hr post treatment, robust DDR, p53, and cleaved caspase 3 activation were already evident in leukemic BM cells (Figures 2C and S2J). 16 hr post treatment with A51 or A86, leukemic cell counts (GFP+) in BM and peripheral blood (PB) were down to 5%–10%, accompanied by massive reduction of the spleen mass and reduced cellularity and color change of the bone marrow due to leukemia cell apoptosis (Figures 2D, 2E, and S2B–S2I). Tissue survey showed that whereas the treatment relieved most of the leukemia signs from the BM, spleen, liver, and PB, non-leukemia-involved tissues, such as the intestine, heart, and lungs, were unaffected by this short-term treatment (Figures 2F, 2G, S2K, and S2L). These findings attest to a selective and abrupt in vivo sensitivity of AML cells to the CKI inhibitors.
Pharmacokinetic studies of the inhibitors A14, A51, and A86 at 20 mg/kg revealed rapid oral absorption with a Tmax of 0.2–2 hr, Cmax of 1,060–1,610 ng/mL, T1/2 of 1.9–4.3 hr, and area under the curve (AUC) values of 2,510–3,680 (ng*hr/mL) (Figure 3A). These values represent moderate plasma clearance, on which basis we chose A51 and A14 for a long-term treatment study. To determine the maximal tolerated dose (MTD), we first tested the 7-day effects of daily A51 and A14 treatment at 20 mg/kg or below in normal C57BL/6 mice. While treatment doses of both inhibitors above 10 mg/kg transiently reduced the white blood cells (WBC) and platelet counts (not shown), at 5–10 mg/kg, there was no adverse effect on hematopoiesis (Figure S3A; data not shown) nor did we observe any signs of toxicity in hematopoietic and other tissues by histology (Figure S3B). Treatment of a comparable therapeutic dose range in rats and dogs was also well-tolerated (Figures S3C and S3D). Furthermore, hERG inhibition assay of A51 indicates no cardiotoxicity at the therapeutic dose range and is non-mutagenic in AMES test (Figure S3E).
Figure 3. Long-Term Therapeutic Effects of CKI Inhibitors in AML Mouse Model.

(A) Pharmacokinetic (PK) analysis in CD-1 mice treated orally (20 mg/kg) with the indicated CKI inhibitors.
(B) Percentage of GFP+°leukemic blasts in PB over time of individual leukemia-inoculated mice treated with A51 (N = 15) or vehicle (N = 15). A51 treatment (5 mg/kg/day) started at day 8 post leukemia inoculation and continued once daily for 3 weeks (6 days/week).
(C) White blood cells (WBC) counts in PB over time post leukemia inoculation of A14 (N = 10) or vehicle-treated mice (N = 8). A14 treatment (10 mg/kg/day) started at day 4 post leukemia inoculation and continued once daily for 4 weeks. Graphs show mean ± SD values.
(D) Kaplan-Meier survival curve of AML mice, treated with A51 (5 mg/kg/day), or vehicle. Statistical analysis was done with log-rank (Mantel-Cox) test; p value < 0.0001.
(E) Kaplan-Meier survival curve of A14-treated (N = 10) or vehicle-treated (N = 8) AML mice. A14 treatment (10 mg/kg/day) started at day 4 post leukemia inoculation and continued once daily for 4 weeks. Statistical analysis was done with log-rank (Mantel-Cox) test; p value < 0.0001.
(F) Representative H&E stained tissue sections of A51-treated, long-term surviving mice (same experiment as B and C, tissue section taken 5 months post AML inoculation). Scale bars, 200 μm (BM and spleen); 100 μm (Intestine and lung).
(G) Kaplan-Meier survival curve of lethally irradiated WT recipient mice transplanted with BM pooled from A51-treated cured donor mice (N = 10 recipients) or from sick mice (N = 8 recipients); Donor BM cells were pooled from three A51-treated, cured (green), or sick (blue) mice.
(H) %CD45.2+ donor-derived cells in the PB of lethally irradiated WT recipient mice (N = 10) transplanted with BM from A51-cured mice, 16 weeks post transplantation. Graphs show mean ± SD values.
(I) %GFP+ leukemia blasts in PB of lethally irradiated recipient mice transplanted with BM isolated from A51-cured mice 16 weeks post transplantation. N = 10 and graphs show mean ± SD values.
See also Figure S3.
We then tested long-term anti-leukemic effects of the inhibitors in MLL-AF9-driven AML mouse model. Oral treatment was initiated at either 8 days (for A51) or 4 days (for A14) after leukemia cell inoculation, at which the percentage of leukemia cells in the BM was more than 1.5% of all cells (data not shown). Whereas all vehicle-treated mice showed leukemic cell infiltration into the peripheral blood (PB) within 2–3 weeks post-inoculation and most succumbed to the disease within a month, no leukemic cells were observed in PB of treated mice before 3 weeks (Figures 3B and 3C). Forty to fifty percent of the A51-or A14-treated mice survived with no signs of disease up to 5 months observation (Figures 3D and 3E) nor had any sequela of long-term treatment; all A51-treated mice had normal organ morphology and histology (Figure 3F) and normal blood counts (Figure S3F).
Long-term leukemia-free survival with possible cure depends on elimination of leukemia-propagating cells, or LSCs, which are often resistant to treatment (Abraham et al., 2016). Functional assessment of residual LSCs following leukemia treatment is possible by transplantation of BM cells from treated mice to lethally irradiated normal mice. This procedure also assesses preservation of normal HSPCs after treatment. Transplantation of BM pooled from several long-term A51-treated surviving AML mice into lethally irradiated WT mice led to mouse rescue with full reconstitution of the hematopoietic system (Figures 3G and 3H). None of the transplanted mice showed any evidence of leukemia at an observation period of 4 months, attesting to selective elimination of LSCs (Figure 3I).
The CKI Inhibitors Co-target CDK7 and CDK9
Observing a potent and selective effect of the inhibitors in leukemic cells, but not in normal cells at the treatment dose, prompted us to elucidate the molecular basis for this distinction. We speculated that this might stem either from partial CKIα inhibition, which does not compromise the normal function of HSPCs, or from additional properties of the CKI inhibitors, which selectively affect leukemia cells. Lenalidomide, a thalidomide analog, which functions as a protein homeostatic modulator (PHM) linking CKIα to the human E3 ligase cereblon, induces CKIα degradation exclusively in human cells (Krönke et al., 2015). We developed a thalidomide analog, BTX161, which mediates degradation of CKIα better than lenalidomide in human AML cells and activates DDR and p53, while stabilizing the p53 antagonist MDM2 (Figures 4A and 4B). In contrast, the catalytic CKIα inhibitors revealed a fundamental difference—A51 and A86 abolished the expression of MYC, MDM2, and the anti-apoptotic oncogene MCL1 (Figure 4B). Suppression of MDM2 and MYC protein levels by A51 was also reproduced in RKO cells, even on top of CKIα knockdown and proteasome inhibition, which stabilize both proteins (Figure S4A). mRNA analysis echoed the protein assay, showing that A51 and A86 induced a marked reduction in mRNA expression of MYC and MDM2, yet upregulated the expression of the Wnt targets AXIN2 and CCND1 (Cyclin D1). In contrast, BTX161 upregulated all the Wnt targets including MYC and did not affect MDM2 mRNA expression (Figure 4C). The observed disparity of CKIα-degrading PHM and the catalytic inhibitors in regulating MYC and MDM2 expression suggest that the catalytic inhibitors have additional targets, a common property of many small molecule kinase inhibitors (Klaeger et al., 2017; Knight and Shokat, 2005). Based on observing the effect on MYC and MDM2 at the mRNA level, we hypothesized that these CKIα kinase inhibitors might also target a transcriptional kinase. To identify relevant targets, we measured the binding of leukemia-active and inactive A-series inhibitors to the major transcriptional kinases. Interestingly, the active compounds A51 and A86 targeted 2 of 7 known transcriptional kinases, showing high inhibition of CDK7 (TFIIH) and CDK9 (P-TEFb) but hardly any inhibition of CDK8, CDK13, CDK11a, CDK11b, and CDK19 (Figure S4B). CDK7 phosphorylates the carboxy-terminal domain (CTD) of RNA polymerase II (RNA Pol II) at Ser5 and Ser7, enabling transcriptional initiation. Then, transcription is paused until positive transcription elongation factor (P-TEFb)/CDK9 phosphorylates RNA Pol II CTD at Ser2, facilitating transcription elongation (Zaborowska et al., 2016). Direct binding affinities showed that the active A-series inhibitors, A51 and A86, target both CDK7 and CDK9 with low nM Kd values, whereas the inactive one, A64, binds CDK7 moderately, but CDK9 poorly (Figure 4D).
Figure 4. The CKI Inhibitors Co-target CDK7 and CDK9.

(A) Structure of the novel CKIα-PHM, BTX161.
(B) WB analysis of MV4–11 cells treated for 6.5 hr with lenalidomide, BTX161, A51, or A86 at the indicated concentrations or DMSO (−). PP2Ac, loading control.
(C) qPCR analysis of MYC, MDM2, AXIN2, and CCND1 (cyclin D1) in MV4–11 cells treated for 4 hr with A51 (2μM), A86 (2μM), BTX161 (25μM), or DMSO (N = 3, Student’s t test). Graphs show mean ± SD values.
(D) CDK7 and CDK9 Kd values of A-series selected inhibitors.
(E) WB analysis of primary AML spleen cells treated ex vivo for 6 hr with A51, A86, or A64 at the indicated concentrations or DMSO (−). PP2Ac, loading control.
(F) qPCR analysis of Myc, Mdm2, Mcl1, and Axin2 of primary BM AML cells treated 4 hr ex vivo with 1 μM of A51, A86, or A64, or with DMSO (N = 3, Student’s t test). Graphs show mean ± SD values.
(G and H) WB analysis of AML spleen cells of mice treated with vehicle (Veh) or with A51 (20 mg/kg) for 3 and 6 hr (G), or with A86 (10 mg/kg) for 5 hr (H). PP2Ac, loading control.
(I) Structure of A86 in complex with P-TEFb (CDK9-CycT1) shown as a cartoon, the ligand as a stick (green, C and H atoms; blue, N atoms; gray, F atoms). Inset: ATP binding pocket showing the interacting residues Cys106 and Asp109 forming hydrogen bonds (blue dotted line) with A86.
(J) Overlay of CKIα (pink) and CDK9 (light blue) with A86 (red, bound to CKIα; blue, bound to CDK9). Hydrogen bonds in black dotted line and solvent accessible cavities as mesh.
See also Figure S4.
CDK7 (catalytic subunit of TFIIH), and in particular CDK9 (catalytic subunit of the transcription elongation factor P-TEFb), are considered as gatekeepers of the transcriptional machinery (Winter et al., 2017), and clinical trials with small molecule inhibitors of these kinases are underway (Ferguson and Gray, 2018). To implicate CDK7 and CDK9 in leukemia repression, we tested the inhibitory activity of A51, A86, and the inactive analog A64 on Pol II CTD phosphorylation at Ser2 and Ser5 in primary mouse and MV4–11 human AML cells (Figures 4E and S4C). We observed that A51 and A86, but not A64, induced a marked reduction in the corresponding phosphorylation signals, in correlation with their potency of repressing MCL1 and MYC expression (Figures 4E and S4C). qPCR analysis in mouse primary AML BM cells revealed a significant reduction of Myc, Mdm2, and Mcl1 mRNA expression post A51 and A86 treatment, but not by A64 (Figure 4F). In contrary, the mRNA expression of the Wnt target gene Axin2 was enhanced, signifying there is no global shutdown of transcription. We therefore presumed that blocking the transcriptional kinases CDK7 and CDK9 might be a major contributing factor in the downregulation of MYC, MDM2, and MCL1. Importantly, phospho-CTD inhibition with downregulation of MCL1 and activation of p53, DDR, and caspase 3 was observed in vivo after single oral treatment of leukemic mice with A51 or A86 (Figures 4G and 4H), suggesting that CDK7 and CDK9 inhibition plays an essential role in leukemic cell apoptosis. In common with most other small molecule kinase inhibitory drugs (Klaeger et al., 2017), the CKIα-CDK7/9 inhibitors target other protein kinases. Compilation of all the protein kinase hits at 100 nM reveals an aggregated list of 34 targets for all six A-series derivatives (Figure S4D). However, upon correlating the hits with the potency of these derivatives to induce leukemia cell killing and apoptosis-related signaling, CKIα and the transcriptional kinases CDK7 and CDK9 stand out among the most relevant targets. Kinases hit by both active and inactive compounds, such as the JNK family and ERK8, are likely of secondary importance.
To understand the structural basis for co-targeting CKIα and CDK9, we determined the structure of P-TEFb (CDK9-cyclin T1 complex) with A86 at 3.2Å resolution (Figure 4I). An overlay of the crystal structures of CKIα and CDK9 with the same ligand shows that both the configuration of the inhibitor within the ATP binding pockets of these two kinases and the hydrogen bond array, which holds the inhibitor in the two ATP pockets, are very similar (Figure 4J). A86 binds at the ATP pocket of CDK9 with the N2 and N1 of the fluoro-pyrimidinamine moiety, which participate in hydrogen bonding to the hinge region main chain oxygen and main chain nitrogen of residue Cys106. The spatially adjacent amino acid, Asp109, stabilizes the cyclohexanamine moiety via a hydrogen bond between N atoms (Figure 4I).
The CKIα-CDK7/9 Inhibitors Abolish Super-Enhancers and Preferentially Block Super-Enhancer-Driven Transcription Elongation
Considering that the transcription of many oncogenes, like Myc, is amplified in leukemia due to a strongly activated enhancer denoted super-enhancer (SE) (Bradner et al., 2017; Lovén et al., 2013), and SEs are sensitive both to CDK7 and CDK9 inhibition (Ferguson and Gray, 2018), we thought that part of the anti-leukemic effect of the inhibitors might be via SEs disruption. A common tool for assessing SE regulation is the acetylation of H3K27, which is highly enriched in SEs (Lovén et al., 2013). Global SE assessment of MLL-AF9-driven AML BM cells (Figure 5A) versus normal mouse BM cells (Figure S5A), revealed robust SE gains (de novo SE generation) (Bradner et al., 2017; Ntziachristos et al., 2016) in mouse AML BM cells. Many of these gains are associated with common leukemia drivers, among which are Myb, Myc, Runx2, and Meis1, scoring high in the AML hockey stick plot (Figure 5A). Of the 342 SE-associated genes identified in AML BM, 37% (125) were specifically gained in AML (AML-specific) (Figures 5A, S5A, and S5B). Upon A51 treatment, H3K27Ac in SE-associated genes was reduced, and many SEs were virtually abolished (Figures 5B and 5C). Of note, although certain enhancers like Cdk6 and Baz2b pass the threshold to be considered as SEs in WT BM (Figure S5A), their acetylation densities and corresponding scores on the “hockey stick” plots are undoubtedly amplified by at least 30- to 50-fold in MLL-AF9 leukemic cells (see Figures 5C and S5C for a few detailed examples). Interestingly, our global analysis also revealed a few SE gains linked to genes that are not commonly associated with leukemia, among which is the IKAROS family gene Ikzf2/Helios (Figure 5C) and the bromodomain-encoding gene Baz2b (Figure S5C), both fully abolished by A51 treatment. The transcriptional shutdown of these SE-associated genes in A51-treated AML cells was confirmed by mRNA analysis for major leukemia drivers such as Myb, Erg, and Cxcr4 (Figure 5D).
Figure 5. Blocking CDK7&9 Disrupts Leukemia SEs and Halts the Transcription Elongation of SE-Driven Leukemia Oncogenes.

(A) Ranking plot of enhancers identified in AML BM cells, ranked by increasing H3K27Ac signal (units: rpm). Super-enhancers (SE) are defined as enhancer clusters, excluding signals at the promoter, ranked above the inflection point of the curve. Absolute SE gains are represented in red.
(B) Quantification of log2 fold changes of H3K27Ac ChIP-seq signals of all the SEs identified in A51 (1 μM, 4hr) versus DMSO-treated AML cells.
(C) Gene tracks of H3K27Ac ChIP-seq signals at selective loci from BM cells isolated from WT and from AML mice treated ex vivo with DMSO or A51 (1 μM) for 4 hr. Red bars indicate SEs identified in DMSO-treated AML cells. The Cdc16-Upf3a locus represents a typical enhancer. y axis shows ChIP-seq signal (rpm/bp). x axis depicts genomic position.
(D) qPCR mRNA expression analysis of indicated genes in primary AML cells treated ex vivo with A51 (1 μM, 4 hr) or DMSO (N = 3, Student’s t test). Graphs show mean ± SD values.
(E) pSer2 CTD ChIP-seq analysis of SEs versus typical enhancers (TE) in primary AML cells treated with A51 (1 μM, 4 hr) relative to DMSO. p values according to Welch’s two-tailed t test is <2.2e – 16. Central rectangle shows the first quartile to the third quartile with median.
(F) Individual gene tracks of pSer2 CTD signals at indicated loci of DMSO- or A51 (1 μM, 4 hr)-treated cells (same experiment as E). y axis shows ChIP-seq signal (rpm/bp). x axis depicts genomic position.
See also Figure S5.
Super enhancers forcefully drive the transcriptional elongation of their target genes (Henriques et al., 2018), which may be curtailed by P-TEFb/CDK9 inhibition. To further test the effect of the inhibitors on transcription elongation, we ran a genome-wide analysis of pSer2 CTD chromatin immunoprecipitation sequencing (ChIP-seq) in ex vivo treated (A51 and control DMSO) primary BM cells isolated from MLL-AF9-driven AML mice in biological duplicates. Indeed, upon A51 treatment, we observed a marked downregulation of Pol II Ser2 CTD phosphorylation at SEs versus typical enhancers (TE) (Figure 5E). A few examples demonstrating pSer2 suppression downstream of the transcriptional start sites of SE-regulating genes (Figures 5F and S5D) attest to a block in transcription elongation.
Combinatorial Action and a Rapid Hit Mechanism of the Inhibitors
A significant number of the AML-specific SE gains abolished by the inhibitors treatment are involved in apoptosis control, suggesting that blocking CKIα together with either CDK7 or CDK9 may augment the apoptotic effect of p53 in leukemia cells. To test this presumption, we treated MV4–11 cells with CKIα degrader BTX161 in combination with the specific CDK9 inhibitor iCDK9 (Lu et al., 2015), with THZ1, a potent, specific CDK7 inhibitor (Kwiatkowski et al., 2014), or with both iCDK9 and THZ1. As expected, the transcriptional inhibitors THZ1 and iCDK9 reduced the pSer2/5 CTD, but at the tested concentration, had individually little effect on DDR, p53, and caspase 3 activation. BTX161, on its own, augmented p53 and MDM2 protein expression, yet in combination with THZ1, and particularly with both THZ1 and CDK9, further augmented p53 and induced maximal caspase 3 activation (Figure 6A). Remarkably, mono-treatment with A51 surpassed all the combination effects, underscoring the value of intense co-targeting CKIα and the transcriptional kinases CDK7/9 by the A-series inhibitors for optimal anti-leukemic effect.
Figure 6. Synergistic Induction of p53 and Apoptosis by Combination Treatment with CKIα and CDK7 and CDK9 Inhibitors and Demonstration of a Rapid Hit Treatment Mechanism.

(A) WB analysis of MV4–11 cells treated with BTX161 (6 hr), iCDK9 (4 hr), or THZ1 (4 hr) at the indicated concentrations in different combinations as indicated. PP2Ac is a loading control.
(B) Rapid hit action of the A-series inhibitors in primary mouse splenic AML cells treated with selected inhibitors at the indicated concentrations for 5 min, followed by inhibitor washout and further incubation for 6 hr (360 min). Continuous 6hrs treatment is a maximal activity control. PP2Ac, loading control.
(C) Rapid hit action of A51 in RKO cells treated with A51 at the indicated concentration for short time intervals, followed by inhibitor washout and further incubation for 16 hr. 16 hr continuous treatment (O.N) is a maximal activity control. Tubulin, loading control.
(D) Rapid hit action of A51 in mouse splenic AML cells treated as in (B). FACS analysis; graph shows percentage of Annexin-V positive apoptotic cells. Experiment performed in triplicates, data presented as mean ± SD values. A51 and A86 show statistically significant apoptotic values in comparison to DMSO (p < 0.05, t test).
Typical administration of kinase inhibitors for the treatment of cancer requires a protracted, continuous daily schedule to achieve above-threshold therapeutic plasma concentration. This is reflected in the AUC value, which correlates with the efficacy and toxicity of the treatment (Widmer et al., 2014). Whereas PK studies of our inhibitors show only moderate AUC values with T1/2 up to 4 hr (Figure 3A), the therapeutic efficacy was very high. This implies that unlike most kinase inhibitors, the CKIα-CDK7/9 inhibitors may only require intermittent short exposures in order to achieve long-term therapeutic efficacy. To study this presumption, we exposed primary mouse MLL-AF9-driven AML cells to a range of inhibitor concentrations for limited time intervals, followed by washout, and analyzed the signaling effects of the inhibitors 6 hr after application. Elimination of Pol II Ser2/Ser5 CTD phosphorylation and the leukemia oncogenes MCL1 and MDM2 was almost complete, whether treated for 5 min or 6 hr (Figure 6B). These rapid-hit data were also reproduced in a human colorectal RKO cell line (Figure 6C). In contrast to SEs targets, maximal β-catenin stabilization in these cells was only achieved upon continuous inhibitor treatment (Figure 6C), ruling out prolonged intracellular persistence of the inhibitor after short exposure. The efficacy of this rapid-hit action was dose-dependent: higher doses of A51 were effective within a few minutes, whereas lower doses required longer times (Figures 6B and 6C). Importantly, a similar rapid-hit effect was also observed in an ex vivo AML killing assay, when 5 mins exposure to A51 and A86 was sufficient to induce significant AML cell killing (Figure 6D). Protracted shutdown of AML-gained SEs upon brief inhibitor exposure likely contributes to the in vivo therapeutic effects—leukemia eradication with no adverse effects under the MTD.
Anti-leukemic Activity of A51 in AML Mouse Models of Tet2−/−;Flt3ITD-Driven and Patient-Derived Xenografts
AML is a heterogeneous disease ignited by a variety of mutations and bearing different vulnerabilities. FLT3 is the most abundant AML driver mutation; ~ 30% of AML patients harbor this mutation (Papaemmanuil et al., 2016). Hence, we further tested our inhibitors in a mouse model in which Flt3ITD mice were crossed with Tet2−/− mice, inducing lethal AML (Shih et al., 2015). Daily oral A51 treatment (6 days a week) for 3 weeks ameliorated the disease profoundly, similarly, or better than the FLT3-multikinase inhibitor midostaurin, reducing the total peripheral WBC, spleen mass, and leukemia blasts occupancy of the bone marrow and spleen while preserving almost normal levels of RBC and platelets (Figures 7A–7C and S6A–S6D). Moreover, blood and tissue infiltration with leukemia cells were strongly reduced by the inhibitor (Figure S6E). Notably, the BM cKitHiCD11bLow leukemic progenitor cells were almost completely eliminated by the inhibitor (Figure 7D). To pinpoint the molecular basis of the leukemia inhibitory effect in the Tet2−/−;Flt3ITD mouse model, we tested the ex vivo signaling and killing effect of A51, A86, and the inactive analog A64 as well as midostaurin in primary leukemia cells from Tet2−/−;Flt3ITD mice. Treatment of Tet2−/−;Flt3ITD leukemic cells with A51 and A86 reproduced the signaling effects observed in leukemic cells from MLL-AF9 mice (Figures 7E and S6F). This was accompanied by ex vivo apoptotic killing of the cKitHiCD11bLow progenitors 16 hr after treatment, superior to midostaurin (Figure 7F).
Figure 7. Long-Term Therapeutic Effects of A51 in Tet2−/−;Flt3IT and PDX Mouse Models.

(A–F) Tet2−/−;Flt3ITD AML mouse data. (A) WBC counts in PB from A51-, midostaurin (Mido)-, or vehicle (Veh)-treated Tet2−/−;Flt3ITD AML mice at the day of sacrifice; N = 6 for each group, graphs show mean ± SD values.
(B) Spleen images of representative three mice from each group.
(C) Representative H&E images of BM (scale bar, 50 μm) and spleen (scale bar, 1 mm) from the same mice.
(D) Abundance of cKitHiCD11bLow cells in the BM from Tet2−/−;Flt3ITDAML-treated mice; N = 6 for each group, graphs show mean ± SD values.
(E) WB analysis of AML cells from mouse spleen of Tet2−/−;Flt3ITD AML mice treated ex vivo with A-series inhibitors A51, A86, A64, and midostaurin at the indicated concentrations or with DMSO (–) for 5 hr. PP2Ac, loading control.
(F) Annexin V+ in cKitHi CD11bLow BM cells from Tet2−/−;Flt3ITD AML mice treated ex vivo with A51, A86, A64, and Mido at the indicated concentrations or with DMSO for 18 hr. Experiment done in triplicates, y axis intersection corresponds to DMSO control and graphs show mean ± SD values.
(G and H) PDX-1 mouse data (of secondary AML). (G) Ratios of PB hCD33+ cells during the 5-week treatment period with A51, cytarabine, or vehicle. (H) PB WBC counts from PDX-1 mice treated for 5 weeks. N = 6 and graphs show mean ± SD values.
(I–K) PDX-2 mouse data (of normal karyotype, NPM1-, FLT3-ITD-, FLT3-TKD-). Ratios of PB hCD33+ cells (I) and mCD45.1+ (J) cells of A51- or vehicle-treated mice at the time of sacrifice. N = 4 for veh, and 5 for A51 and graphs show mean ± SD values. (K) WB analysis of AML cells from BM of PDX-2 mice treated in duplicate ex vivo with A51 or with DMSO for 5 hr. PP2Ac, loading control.
See also Figures S6 and S7.
After demonstrating a profound effect of our inhibitors in two different AML mouse models, we sought to check the effect in human AML. First, we tested the ex vivo killing effect in primary human leukemia cells from 5 individual patients with distinct genotypes. Our results show leukemia cell apoptosis by A51 and A86, but not by A64 or midostaurin at comparable inhibitor concentrations (Figure S6G). Killing effect was achieved at a low IC50 of 20–50 nM in all 5 samples. Next, we went on to test the effect of lead compound A51 in patient-derived xenograft (PDX) mouse models. Three xenograft models representing different human AML subtypes were established in NSG mice and treated for2.5–5 weeks with A51 or vehicle, then sacrificed and assessed for anti-leukemic effects. PDX-1 model established from secondary AML patient was also treated for comparison with the standard AML chemotherapy cytarabine, and the leukemia burden was monitored weekly by flow cytometry analysis of PB human myeloid leukemia cells (hCD33). Leukemia was well-controlled in A51-treated mice, whereas all vehicle-control mice, as well as cytarabine-treated mice succumbed to leukemia at 3–5 weeks of treatment (Figure 7G). Moreover, human leukemia-derived WBCs were profoundly reduced, accompanied with mouse WBC enrichment (Figure 7H and data not shown) and normalized counts of RBCs and platelets in 5-week A51-treated mice (Figures S7A and S7B). Analysis of hCD33 showed a strong anti-leukemia effect in whole blood and the spleen, yet not in the BM, possibly due to a late phase relapse (Figure S7C). PDX-2 mice also developed myeloid leukemia. Following A51 treatment for 3 weeks, all hCD33+, hCD45+, and hCD38+ cells were eliminated, whereas mCD45+ cells were enriched up to 90% in the BM of all treated mice (Figures 7I, 7J, S7D, and S7E). Ex-vivo treatment of BM cells from PDX-2 inoculated mice shows the typical molecular response pattern observed in the non-human leukemia mouse models, including downregulation of MCL1 and MYC and activation of p53 and caspase 3 (Figure 7K). PDX-3 grew as a myelosarcoma at the injected leg muscle with typical morphology (Wilson and Medeiros, 2015) (Figure S7F). Histology and IHC staining for hCD45 and hcKit show human granulocytic leukemia cells (Figure S7G). All treated mice demonstrated a good response to A51 therapy (Figures S7F and S7H–S7K) with evidence of apoptosis in H&E and cleaved caspase 3 immunostaining (Figures S7J and S7K). Together, our PDX data consistently indicate a strong therapeutic effect of A51 in human leukemia samples of different disease subtypes.
DISCUSSION
To date, a notable disadvantage of all drug treatment modalities in human AML is either poor response or rapid relapse following short remission (Dombret and Gardin, 2016). Our studies show that small molecules with a capacity of targeting both CKIα and the transcriptional kinases CDK7 and CDK9 have a striking therapeutic effect in different AML mouse models, with high potential of curing rates. We observed elimination of leukemic stem cells without affecting normal hematopoietic stem cells, evident by secondary BM transplantation from treated MLL-AF9 AML mice. In addition, we show profound anti-leukemic effects of our kinase inhibitors in Tet2−/−;Flt3ITD-driven AML mice. Remarkably, the lead inhibitor A51 performed also well in multiple AML PDXs, including a highly aggressive human secondary leukemia, which is a prominent therapeutic challenge (Boddu et al., 2017). The therapeutic advantage of our inhibitors stems from a synergistic pro-apoptotic activity of two main targeted pathways, CKIα and CDK7/9 SE-regulated. We have shown that CKIα ablation provokes a DNA damage response and p53 activation, yet it also induces pro-leukemic Wnt activation and upregulation of Myc and Mdm2, some of the most established driver oncogenes in various hematological malignancies (Del-more et al., 2011; Kojima et al., 2016). Fortuitously, triple inhibition of CKIα and the transcriptional kinases CDK7 and CDK9 cancels these pro-leukemic effects, while accentuating synergistic p53 activation and its pro-apoptotic activities. This synergism was also documented by combination treatment of our newly developed CKIα-degrading molecule BTX161 with either one of the selective inhibitors of the transcriptional kinases CDK7 and CDK9, and even better with both transcriptional kinase inhibitors. It underscores the advantage of the dual-purpose inhibitors, in comparison to p53 exclusively based leukemia treatment (Kojima et al., 2016) as the latter are likely moderated by SE-driven antagonism. In line with this presumption, p53 activation has been shown to cooperate with Myc inhibition in eliminating chronic myelogenous leukemia-initiating cells (Abraham et al., 2016). Notably, in common with all the clinically tested kinase inhibitors (Klaeger et al., 2017), including the most selective ones, our A-series inhibitors target more than 3 kinases, and although these triple specificity correlates very well with the anti-leukemic potency of the inhibitors, we cannot exclude the contribution of other inhibitor targets to the therapeutic activity. A particularly attractive target is DYRK1A, which has a high affinity to the active inhibitors. This kinase was recently shown to phosphorylate RNA Pol II CTD Ser2/5 alternatively to CDK7/9 and to complement these two kinases in RNA Pol II phase separation, which augments transcription rates (Lu et al., 2018). In fact, having moderate rather than strict selectivity and targeting multiple pathways may overcome target redundancy, preventing the emergence of resistant leukemia clones in line with the enhanced cure rates (Klaeger et al., 2017). In addition, in a heterogeneous disease like AML, kinase-related vulnerabilities may variably synergize with each other in different AML types.
SEs have recently been highlighted as attractive targets for epigenetic therapy of cancer, particularly in hematological malignancies, and are subject to bromodomain-BET, CDK7, and CDK9 inhibitors (Bradner et al., 2017; Dawson et al., 2011; Del-more et al., 2011; Ferguson and Gray, 2018). Whereas SEs drive the expression of multiple genes in normal and leukemic stem cells, our analysis revealed many SEs that were newly gained in AML versus normal BM. Many other leukemia SEs that have counterparts in normal BM are markedly amplified in the primary AML cells. Among the newly gained and highly amplified SE-controlled genes, we identified the well-recognized leukemia oncogene drivers Myc, Myb, and Runx2, as well as genes that are not commonly associated with leukemia. Examples include the IKAROS family gene Helios, which was previously implicated in leukemic stem cell control and cancer immunity evasion in mouse models (Park et al., 2015; Nakagawa et al., 2016) and a bromodomain-encoding protein Baz2b, which could possibly function as a new SE component. These, similarly to most other identified leukemia SEs, were nearly abolished or highly suppressed by the CKIα-CDK7/9 inhibitor treatment. In addition, genome-wide analysis of primary AML cells through pSer2 CTD ChIP revealed that the transcriptional elongation of SE-regulated genes was far more repressed by the inhibitor treatment than elongation regulated by typical enhancers, again demonstrating a particular vulnerability of SE-controlled transcription that prevails in leukemia cells.
A major challenge to the development of new anticancer drugs is finding a therapeutic window (i.e., dose-dependent therapeutic efficacy with manageable toxicity). This window depends on the relative vulnerabilities of cancer cells versus normal cells of different tissues. Two related parameters seem to favor leukemia vulnerability to our triple inhibitors: leukemia addiction to SE-associated oncogenes, which sensitizes it to the inhibitors far more than normal cells, and the rapid-hit mechanism of action of the CKIα-CDK7/9 inhibitors, at variance with the occupancy-driven pharmacology paradigm that prevails in the kinase-inhibitor arena (Salami and Crews, 2017). These two parameters are likely interdependent: the ability to disrupt SEs and eliminate their associated oncogenes for many hours after a few minutes of drug exposure enables short intermittent therapy with long pauses, which may minimize generalized toxicity. This is substantially different from ordinary chemotherapy, which may also kill cancer cells instantly via different mechanisms, yet is far less selective in discriminating malignant from normal cells. In addition, unlike chemotherapy, our inhibitors induce DNA damage response and p53 activation without overt signs of DNA damage (Pribluda et al., 2013; unpublished data), a further explanation to the paucity of undesired effects within the therapeutic range. Indeed, at the therapeutic dose, we did not observe in our AML mice any significant adverse treatment effects even at daily treatment of 45 days. Likewise, our data show that the MTD and safety profile of the inhibitors at a mouse-comparable therapeutic dose range in rats and dogs are similar to those in mice; other toxicity indices, such as the cardiotoxicity index hERG and performance at the AMES mutagenesis assay score are well within the acceptable safety range. Inhibitors with similar properties may therefore prove suitable for the treatment of AML patients and possibly other human cancer types, particularly those that similarly to AML show SE-linked transcriptional addiction and likely to benefit from p53 activation.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yinon Ben-Neriah (yinonb@ekmd.huji.ac.il).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were performed in accordance with the guidelines of the institution ethics committee (AAALAC standard). For human samples, the institutional review board of Hadassah Hospital Medical Center approved the use of these samples (0416–16-HMO), according to the principles of the Helsinki Declaration. Informed consent was obtained from all patients prior to performing these experiments.
Mice
Mice with a genetically modified Csnk1a1 locus (floxed exons 1 and 2 of the mouse Csnk1a1 gene) (Elyada et al., 2011) were crossed with Mx1-cre inducible transgenic mice CKIαfl/flMx1-Cre. Mice were backcrossed for 7 generations with C57BL/6 mice to generate a pure genetic background. For conditional double KO, CKIαfl/flMx1-Cre mice were crossed with p53KI/KI (Junttila et al., 2010). p53KI/KI, Mx1-cre transgenic mice and CD45.1 WT C57BL/6 were purchased from Jackson lab. All experiments were performed on male or female mice in similar numbers as our initial data did not indicate significant differences in disease progression or response to treatment between females or males.
Health/immune status
All mice were kept under SPF conditions at the Hadassah Medical School animal facility unit of the Hebrew University and were regularly screened for standard pathogens. All experiments were performed in accordance with the guidelines of the institution ethics committee (AAALAC standard). All mice were drug and test naive prior to initiating the experimental procedures performed and described in this paper.
Cell cultures
RKO cells were grown in DMEM supplemented with 10% Serum. Cells were transduced with shRNA lentiviral vector for 4–7 days. MG132 (20 μM; Calbiochem) was added for 4 h before cell harvesting. MV4–11 human AML cells were grown with RPMI supplemented with 10% Serum. Both cell lines were obtained from ATCC collection (RKO: ATCC CRL-2577, MV4–11: ATCC CRL-9591) without further authentication. RKO sample obtained from a female patient, MV4–11 cells are from a male patient. All other sample specifications are found in the ATCC database.
Human primary cells
All human primary cells were obtained from newly diagnosed AML patients prior to treatment. Following BM biopsy, bone marrow derived mononuclear cells (BM-MNC) were extracted by Ficoll and either used directly or frozen for later usage. For defrosting cells were thawed directly into Iscove’s Modified Dulbecco’s Medium (IMDM) with 50% FBS and DNaseI (10 mg/ml). Cells were spun and resuspended in RPMI supplemented with 10% FBS (GIBCO, #12657), L-Glutamine, Pen/Strep (Biological Industries, Israel), IL-6 (20 ng/ml), IL-3 (10 ng/ml), GM-CSF (100 ng/ml) and TPO (10 ng/ml) (Peprotech). Cells were incubated with the inhibitors for 18–20 hr, and then analyzed for cell apoptosis by FACS. For blast cell identification anti-human CD45 antibody (eBioscience #304022) was added before analysis. For mRNA expression analysis, cells (sample #5546) were incubated with the inhibitors for 4 hr and analyzed by RT-PCR.
5 human samples with distinct genotypes were used for estimating the inhibitor cytotoxicity:
Sample #014: Translocation 2–14, FLT3 TKD positive, ITD negative, NPM1 negative
Sample #270: FLT3 positive for ITD, TKD negative, NPM1 positive
Sample #546: Chromosome 8 Trisomy, FLT3 positive for ITD, NPM1 positive
Sample #005: FLT3 positive for ITD, TKD negative, NPM1 positive
Sample #365: Translocations 1–19 and 9–11, FLT3 ITD and TKD negative, NPM1 negative
ITD-internal tandem duplication, TKD-tyrosine kinase domain.
METHOD DETAILS
Development of small molecule CKI inhibitors (SMI)
SMI were developed by an initial cell-based screening followed by extensive medicinal chemistry (see Compound Synthesis). Primary high throughput screens were run in RKO colorectal cancer cells, looking for β-catenin stabilization. RKO cells were plated onto 394 well plates, treated with the compounds at 10 μM for 16hrs. Treated cells were tested for accumulation of nuclear β-catenin by immunostaining (IF) using mouse anti-β-catenin antibodies (BD, Cat# 610154) and Donkey anti Mouse Alexa 488 as a secondary antibody (ThermoFisher, Cat# A21202). Cells treated with the proteasome inhibitor MG132 (10μM) served as the β-catenin stabilization high control and DMSO (the vehicle for compounds and for MG132) as a basal level control. Cell viability was monitored using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Cat# G7570). Compounds scoring positive for accumulation of nuclear β-catenin were selected for a secondary screen. The secondary assay was performed by western blot analysis of treated RKO cells, looking for compounds stabilizing both β-catenin and p53 and inducing histone H2AX phosphorylation (indicators of CKIα deficiency; see Elyada et al., 2011).
Kd determination
The binding affinity of all compounds was determined using KINOMEscan at DiscoverX. Test compounds were prepared in DMSO and Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. KINOMEscan Kd were generated as described previously (Fabian et al., 2005). Similarly, primary binding interaction screens were performed to evaluate ‘% Ctrl’, which were further used to estimate Kds.
AML mouse models and inhibitor treatment
1. MLL-AF9 AML mouse model
To generate the MLL-AF9 driven AML mouse model we followed the protocol generated by (Barabé et al., 2007). BM from WT C57BL/6 was extracted and enriched for c-Kit expressing cells (EasySep) and incubated overnight in RPMI supplemented with 15% FCS, L-Glutamine, Pen/Strep (Biological Industries, Israel), stem cell factor (SCF), IL-3, IL-6 and TPO (Peprotech). BM cell culture was infected with retrovirus MLL-AF9-IRES-GFP for 4 hr and then incubated with culture medium for 24 hr. Infected BM cells were then injected I.V. into sublethally irradiated mice (500 rad; X-RAD 225 from PXi precision X-ray). Moribund mice were sacrificed and their BM was extracted. 50,000 BM GFP+ cells were injected I.V. into C57BL/6 WT (8 – 12 weeks old, males and females, approximately in similar numbers) irradiated (250 rad) mice.
To test acute effects of the inhibitors, mice with advanced disease (> 80% GFP+ cells in PB) were treated with a single oral administration of the specific inhibitor and sacrificed at several short time points. For long-term therapeutic effects, mice were orally treated by gavage daily with either A51 (Di-HCL salt) (starting day 8-post inoculation, 5mg/kg per day, 6 days a week for 25 days) or with A14 (Di-HCL salt) (starting day 4 post leukemia inoculation, 10mg/kg per day, 6 days a week for 25 days) or vehicle.
To assess possible leukemia cure following treatment, BM (CD45.2) from rescued mice (135 days post leukemia inoculation) were pooled and 5 × 106 cells were transplanted I.V. into lethally irradiated (950 rad) CD45.1 recipient WT mice. Mice were followed for up to 5-month post bone marrow transplantation (BMT) for successful engraftment and survival. This experiment was repeated twice, each time with BM pooled from 3 rescued mice derived from two different experiments. Control lethally irradiated (950 rad) CD45.1 recipient WT mice were transplanted with BM pooled from 3 mice that developed leukemia after cessation of treatment.
2. Tet2−/−;Flt3ITD AML mouse model:
To generate the Tet2−/−;Flt3ITD mouse model, C57BL/6 WT mice were irradiated with sub-lethal (500 rad) irradiation and one day later mice were injected (via tail vein) with 5 × 105 spleen cells isolated from 4th generation Tet2−/−;Flt3ITD mice (Shih et al., 2015). Seven days post leukemia inoculation, mice were orally (gavage) treated with either A51 (Di-HCL salt) (5mg/kg), midostaurin (35mg/kg) or vehicle for 18 days (6 days a week) and were sacrificed 9 days after stopping the treatment.
3. Patient Derived Xenograft (PDX) models:
PDX-1.
NSG-SGM3 mice were sub-lethally irradiated and engrafted I.V. with AML cells harboring Trisomy11, isolated from patient with secondary AML (Model J000106124). Mice were tested for engraftments by whole blood FACS analysis for human CD45+/hCD33+ cells. Successful engraftment was observed following 8 weeks post inoculation showing a level of 2%–5% human CD33+ cells in PB. Mice were treated orally for 5 weeks, 6d/week with A51 (Di-PTSA [p-Toluenesulfonic acid] salt) (7.5mg/kg), cytarabine (30 mg/kg) or vehicle.
PDX-2.
NSG-hSCF mice were irradiated (225 rad) and transplanted intra-femoral (IF) with 1 × 106 leukemic cells with normal karyotype, NPM1-, FLT3-ITD-, FLT3-TKD-
PDX-3.
NSG-SMG3 mice were irradiated (225 rad) and transplanted intra-femoral (IF) with 1 × 106 leukemic cells with normal karyotype, NPM1+, FLT3-ITD+, FLT3-TKD-5 weeks after transplantation, PDX-2 and PDX-3 mice were treated orally for 2.5–3 weeks, 5d/week with A51 (Di-PTSA salt) (7.5mg/kg) or vehicle. Following treatment, mice were sacrificed and analyzed for leukemia spread to the bone marrow, spleen and other organs.
All the PDX studied (PDX1, 2 &3) were from newly diagnosed AML patients prior to treatment.
4. Drug Safety Assessment
Rats.
Three groups of rats (10 rats per group, 5 males and 5 females) were administered A51 (Di-PTSA [p-Toluenesulfonic acid] salt) once daily for up to seven consecutive days at dosages of 1 or 3 mg/kg/day (all within the mouse therapeutic dose range) or vehicle (DMSO 10%, Solutol 10% and 2-hydroxy prolyl-b-cyclodextrin (HP-b-CD, 10% w/v) in ultra-pure water) and evaluated for clinical signs of toxicity and body weight. All animals in the 0, 1, and 3 mg/kg/day groups survived until scheduled termination and were euthanized on Day 8 for collection of hematology and serum chemistry samples, and postmortem examination. Organs were weighed, and tissues preserved and retained in fixative without further processing or evaluation.
Dogs.
Four groups of four dogs (two per sex per group) were administered with vehicle or A51 (0.2 or 0.6 and 2.0 mg/kg/day, corresponding to the mouse therapeutic dose range and twofold beyond) once daily for up to seven consecutive days. Animals were evaluated for clinical signs of toxicity, body weight, and food consumption, and samples for evaluation of systemic exposure to A51 were collected from available animals on Days 1 and 7. On Day 8, surviving animals were euthanized and subjected to macroscopic examination, including organ weights and tissue collection/preservation.
In vivo and ex vivo treatments
For in vivo use (mouse oral treatment), A-series compounds (Di-HCL salts) were dissolved and administered in 1% methyl cellulose (sigma) in pure water, together with 0.01% tween 80 (sigma) and 0.02% PEG400 (sigma). For the PDX experiments, A51 Di-PTSA salt was dissolved and administered in DMSO 10%, Solutol 10% and 2-hydroxy prolyl-b-cyclodextrin (HP-b-CD, 10% w/v). For tissue culture experiments, compounds were dissolved at 1000X stocks in Dimethyl sulfoxide (DMSO) and diluted in growth medium.
PKC412 (midostaurin) 6% w/w in Gelucire 44/14 (Gattefosse, France) was stored at 4°C as a waxy-solid formulation. Prior to administration, the Gelucire/PKC412 waxy solid mixture was warmed in a 44°C water bath until liquid. The liquid mixture was then diluted with sterilized deionized water to produce the final PKC412 concentration. Dosing was performed 6d/week for 3 weeks by gavage at a concentration of 35mg/kg.
Crystallography studies
Crystals of CKIα (2.3Å), CKIδ (1.6Å) and P-TEFb (CDK9/Cyclin T1) (3.2Å) in complex with A86 were obtained using sitting drop vapor diffusion set-ups and datasets were collected at the ESRF synchrotron radiation source (Grenoble, FR, beamline ID30a1).
For CKIα Structure
Crystals of CSNK1A1 in complex with A86 were obtained using sitting drop vapor diffusion set-ups. CSNK1A1 at a concentration of5.8 mg/ml (50 mM Tris-HCl, 300 mM NaCl, 0.25 mM TCEP, pH 8.0) was pre-incubated with 0.7 mM (4.7-fold molar excess) of Mg2+-ATP (150 mM in DMSO) for 1 h. 0.1 mL of the protein solution was then mixed with 0.1 μL of reservoir solution (0.10 M MES/NaOH pH 6.8, 10.0%(v/v) 2-propanol, 26.0%(v/v) PEG 400) and equilibrated at 4°C over 0.06 mL of reservoir solution. Well diffracting crystals appeared within 4 days and grew to full size over 23 days (Figure 1). A 150 mM solution of A-86 in DMSO was diluted in 30% v/v PEG 400, 0.1 M MES/NaOH pH 6.8, 10% v/v 2-Propanol to 2 mM and 1 μL of the resulting solution was added to a 0.2 mL crystallization drop. Crystals were harvested after 90min and cryo-protected with Paratone-N. A complete 2.3Å dataset of a CSNK1A1/A-86 crystal was collected at the ESRF synchrotron radiation source (Grenoble, FR, beamline ID30a1). Molecular replacement was done using a published structure of CSNK1A1 (pdb accession code 5FQD) as starting model. Several rounds of alternating manual re-building and refinement with REFMAC5 resulted in the final model.
For CKIδ Structure
Wild-type CKIδ protein with the mutation R13N was used for crystallization. Crystals of CK1δ in complex with A86 were obtained using hanging drop vapor diffusion set-ups. CK1δ at a concentration of 13.6 mg/ml (50 mM HEPES, 266 mM NaCl, 1 mM EDTA, 1 mM DTT, 5 mM β-OG, pH 7.5) was pre-incubated with 2 mM (5.1-fold molar excess) of A-86 (150 mM in DMSO) for 1 hr. 1 μL of the protein solution was then mixed with 1 μL of reservoir solution (0.1 M Na3-Citrate, pH 4.9, 18%(w/v) PEG 3350) and equilibrated at 20° C over0.4 mL of reservoir solution. Well diffracting crystals appeared overnight. A complete 1.6Å dataset of a CK1δ/A-86 crystal was collected at the ESRF synchrotron radiation source (Grenoble, FR, beamline ID30a1). Molecular replacement was done using a published structure of CK1δ (pdb accession code 3UYT) as starting model. Several rounds of alternating manual re-building and refinement with REFMAC5 resulted in the final model.
For CDK9/Cyclin T1 Structure
Crystals of CDK9/Cyclin T1 in complex with A-86 were obtained using sitting drop vapor diffusion set-ups. CDK9/Cyclin T1 at a concentration of 4.5 mg/ml (20 mM Tris / HCl, 250 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.3) was pre-incubated with 0.6 mM (5.1-fold molar excess) of A-86 (150 mM in DMSO) and 4 mM TCEP for 1 hr. 0.1 μL of the protein solution was then mixed 0.1 μL of reservoir solution (0.01 M Ca-Chloride, 0.1 M MES/NaOH, pH 6.50, 1.2 M Na-Acetate) and equilibrated at 4°C over 0.06 mL of reservoir solution. Crystals were obtained using microseeding. Well diffracting crystals appeared within 4 days and grew to full size over 8 days. A complete 3.2Å dataset of a CDK9/A-86 crystal was collected at ESRF synchrotron radiation source (Grenoble, FR, beamline ID30a1). Molecular replacement was done using a published structure of CDK9/Cyclin T1 (pdb accession code 3BLH) as starting model. Several rounds of alternating manual re-building and refinement with REFMAC5 resulted in the final model.
All the structures were visualized using PyMOL (Version 1.8 Schrödinger, LLC). Multiple Sequence Analysis was performed with Clustal Omega, using the sequences for the human CKI and CKII gene family members.
Ex vivo assays
Mice with advanced leukemia (> 80% GFP+ in PB) were sacrificed and BM was flushed with PBS from the femur and tibia. Cells were treated with erythrocyte lysis buffer (NH4Cl 150 mM, KHCO3 10 mM, EDTA 0.1 mM) and centrifuged (200g, 5 min). Pellet was resuspended in PBS, white blood cells (WBCs) were counted with an automatic Abacus Junior Vet (Diatron) followed by centrifugation. Cells were strained (70 μm sterile strainer) and resuspended with StemPro-34 SFM media (GIBCO) supplemented with 10 ng/ml of IL3, IL6, thyroid peroxidase (TPO), stem cell factor (SCF); StemPro-34 Nutrient Supplement (25 μl/ml), PenSrep (Sigma, 1:100) and L-glutamine (Sigma, 1:100). Cells were diluted to 1.5*105 cells in 0.15 mL media and plated with or without the specific inhibitors in a U bottom 96 well plate with lid (Bar-Naor) for 16 – 18 hr. Inhibitors were dissolved in DMSO, and diluted 1:1000 to the final concentrations. Cells with DMSO only (diluted 1:1000) served as control. Incubation was done in a 5% CO2 TC incubator. For rapid-hit experiments, cells were centrifuged at the indicated time intervals, resuspended with fresh media without inhibitors and further incubated for a total of 18 hr. The following day, cells were spun (300g, 5 min), washed and stained with Annexin V-PE (Biolegend, 1:100) and 7AAD (TONBO biosciences) in Annexin buffer (150 mM NaCL, 4 mM KCl, 1.5 mM CaCl2, 10mM HEPES and 0.75 mM MgCl2). Cells were analyzed by Beckman coulter CytoFlex for GFP and PE.
CFU assay
Primary BM cells from WT and AML mice were harvested from the tibia and femur and treated as described above. WBCs were diluted in methylcellulose (MethoCult GF M3434; StemCell Technologies) supplemented with Gentamycin, Amphotericin B and PenStrep to a concentration of 2*104 cells/ml. Cells were plated in a 6 well TC dish, 1ml/well and incubated in a 5% CO2 TC incubator for 7–9 days with the indicated concentrations of the indicated inhibitor. CFUs were visualized and photographed with an Olympus SZX16.
Histology and immunohistochemistry (IHC)
5 μm sections were cut onto a poly lysine coated glass slide from 4% formaldehyde fixed and paraffin embedded tissue. Bones were fixed in 4% formaldehyde then decalcified before paraffin embedding in HCl/EDTA based solution (RDF D1210 Anapath, StatLab medical products) for four hr at room temperature. Hematoxylin and Eosin (H&E) staining were performed by the Pathology Department of Hadassah Ein-Karem hospital by an automated system.
Immunohistochemistry (IHC) for hCD45 and hc-Kit of PDX-3 samples was performed by automated BenchmarkUltra (Ventana Medical Systems) following standard procedures. Antibody staining was done with anti-human c-Kit (Cell MarQue #117R-16), anti-human CD45 (Dako #M0701) and at 1:200 and 1:250 dilutions, respectively. For cleaved caspase 3 staining sections were incubated with anti-cleaved caspase 3 (1:100; Cell Signaling Technology). ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit and 3,3′-Diaminobenzidine (DAB) chromogen (Lab Vision) were used for detection.
Blood Smear
Peripheral blood samples were smeared on a glass slide and were stained with Wright-Giemsa reagent by Hadassah Ein-Karem hospital Hematology Department.
Blood analysis
Peripheral venous blood was obtained from the mouse facial vein using standard techniques and analyzed using the auto hematology analyzer BC-2800 (Mindray) or an automatic Abacus Junior Vet (Diatron), following manufacturer’s instructions.
Flow cytometry
FACS analysis was performed with BECKMAN COULTER (CytoFLEX). Sorting was performed by FACS ARIA sorter. For immunostaining, cells were suspended in a 1% BSA/PBS buffer with 5μM EDTA. For viability staining, cells were suspended in Annexin buffer and stained for Annexin V (Biolegend) and 7AAD (TONBO biosciences). For human samples, blocking was done with 20% human serum.
FACS analysis for PDX-2 was done on live cells (propidium iodide (MBL #4700–200) negative or 7AAD negative) with the following antibodies: anti-hCD45-PB (eBioscience #304022), anti-hCD33-PE/Cy7 (Biolegend #366617), anti-hCD38-APC (Biolegend #356606) and anti-mCD45.1-FITC (Biolegend #110706). All antibodies used at 1:250 dilutions.
Antibodies used for HSC staining
Lineage cocktail- Biotin (Miltenyibiotec), c-Kit APC-eFluor780 (eBioscience), Sca-1 PE-Cy7 (eBioscience), Flt3 – APC (eBioscience), CD34 -eFluor450 (eBioscience), c-Kit–PE (Biolegend), strepavidin-percp/cy5.5 (eBioscience). The stem cell compartments were defined as following: LT-HSC, Lin−cKit+Sca1+CD34−FLT3−; ST-HSC, Lin−cKit+Sca1+CD34+FLT3−; and MPP, Lin−cKit+Sca1+ CD34+FLT3+.
Human shRNA sequences
ShCKIα (in Plko vector): 5′-CCGGGCAGAATTTGCGATGTACTTACTCGAGTAAGTACAT CGCAAATTCTGCTTTTT-3′
Western blot analysis
Proteins were extracted by whole-cell-extract protocols from cell pellets in RIPA buffer containing proteases and phosphatase inhibitors. Western blot analysis was performed by means of standard techniques. Blots were incubated with antibodies detecting β-catenin (1:2,500; BD Transduction Laboratories), phospho Ser45-β-catenin (1:750; Cell Signaling), phospho Ser33/Ser37/Thr41-β-catenin (1:750; Cell Signaling), phospho-Thr41/Ser45-β-catenin (1:750; Cell Signaling); CKIα (1:1000; Santa Cruz), Hsp90α (1:5000; Calbiochem), αTubulin (1:5000; Sigma), mouse p53 (1:1000; Cell Signaling), Cleaved Caspase 3 (1:750; Cell Signaling), c-Myc (1:750; Cell Signaling), MCL-1 (1:1000; Cell Signaling), human p53 (DO-1/1801 hybridoma mix; 1:20), MDM2 (2A9/4B2/4B11 hybridoma mix; 1:40), phospho-Histone H2A.X (Ser139; 1:1000; Millipore), PP2Ac (1:1000; rabbit serum kindly provided by David Virshup), Cyclin D1 (1:500; Lab Vision), Phospho-Rpb1 CTD (Ser 2/5; 1:1000; Cell Signaling), Phospho-Rpb1 CTD (Ser2; 1:1000; abcam), Phospho-Rpb1 CTD (Ser5; 1:1000; Merck). Secondary antibodies were HRP-linked goat anti-mouse, goat anti-rabbit and rabbit anti-goat (all 1:10,000; Jackson). Blots were developed using ECL (GE Healthcare).
RT-qPCR analysis
Total RNA was extracted from cell pellets using miRNeasy Kit (QIAGEN) and subjected to reverse transcription with qScript cDNA Synthesis Kit (QIAGEN). mRNA expression levels were measured by RT-qPCR using SYBR-Green (Invitrogen) in a QuantStudio 12 K Flex. Relative quantities of gene transcripts were analyzed in qBase software, and normalized to GAPDH transcripts. Sequences of RT-PCR primers are listed in the extended data information.
qPCR primers used in this study
| Gene name | Primer sequence (5′ to 3′) | |
|---|---|---|
| Mouse mRNA primers | Axin2 | GCAGTGTGAAGGCCAATGG |
| GCTTTCCAGCTCCAGTTTCAGT | ||
| Mcl1 | TTCCACAAAGGCATCCCAGC | |
| ATCCTGGGCAGCTTCAAGTC | ||
| Myc | TGAGCCCCTAGTGCTGCAT | |
| AGCCCGACTCCGACCTCTT | ||
| Mdm2 | TGTGTGAGCTGAGGGAGATG | |
| CACTTACGCCATCGTCAAGA | ||
| Gapdh | ACAACTTTGGCATTGTGGAA | |
| GATGCAGGGATGATGTTCTG | ||
| Ikzf2 | AGGAACACCCGCTGACAAAT | |
| CCCTTCGGGTGAAAGCTCAT | ||
| Erg | CTACGGCAGCTACATGGAGG | |
| GACATGGTCTGTGCTCCACA | ||
| Cdk6 | GAGTGCAGACCAGTGAGGAG | |
| TGACACTGTGCACACATCAAA | ||
| Cxcr4 | TGCAGCAGGTAGCAGTGAAA | |
| TGTATATACTCACACTGATCGGTTC | ||
| Myb | GGCTCTTGGCGGAGCC | |
| ATGGTCACACATCTCAATGTCTTCAT | ||
| Human mRNA primers | MYC | CTGGTGCTCCATGAGGAGA |
| TCCAGCAGAAGGTGATCCA | ||
| MDM2 | TGAATCTACAGGGACGCCATC | |
| TCACTTACACCAGCATCAAGATCC | ||
| AXIN2 | CACCCTTCTCCAATCCAAGC | |
| TCTTTGGCTCTTTGTGATCTTCTG | ||
| CYCLIN D1 | GGCGGAGGAGAACAAACAGA | |
| CTCCTCAGGTTCAGGCCTTG | ||
| UBC | ATTTGGGTCGCGGTTCTTG | |
| TGCCTTGACATTCTCGATGGT |
Chromatin Immunoprecipitation
WT or MLL-AF9-driven leukemic mice were sacrificed and BM was flushed with PBS from the femur and tibia. The cells were cultured ex vivo, treated with DMSO or A51 (1 μM) and harvested after 4 hr, (20 × 106 cells for each sample). The cells were then fixed and cross-linked in 1% (wt/vol) formaldehyde solution and neutralized with 0.125M glycine. Cells were resuspended and lysed in RIPA buffer containing 0.5% SDS. To solubilize and shear cross-linked DNAs, lysates were sonicated with Covaris M220 Sonicator at 75W with 20% duty factor, 200 cycles for 20 min. Immuno-beads were prepared by incubation of 80 μL Dynabeads Protein G magnetic beads (Life Technologies) for 6 hr with 2 mg of anti-H3K27Ac (Abcam, ab4729) antibodies, or with 4 mg of anti-RNA polymerase II CTD (phospho S2) antibodies (Abcam, ab5095). Sonicated lysates were centrifuged at 13,000 rpm for 5 mins and the chromatin was incubated overnight at 4°C with the anti-H3K27ac immune-beads, or with the anti-RNA polymerase II CTD (phospho S2) immune-beads. Following chromatin immunoprecipitation (ChIP), samples were washed, and bound complexes were eluted by Direct Elution Buffer (10 mM Tris-HCl pH-8, 0.3 M NaCl, 5 mM EDTA and 0.5% SDS), reverse cross-linked and treated with RNase A (ThermoFisher) and Proteinase K (Invitrogen). Chromatin was purified with QIAquick Gel Extraction Kit (QIAGEN) and processed for ChIP Seq. Immuno-precipitation performed with anti-IgG control yielded very low signal (not shown). Libraries for Illumina sequencing were prepared using TruSeq nano DNA sample prep kit (Illumina) using 50 ng of immunoprecipitated DNA as starting point. Libraries were prepared according to manufacturer’s instructions, quantified by Qubit (Invitrogen) and TapeStation (Agilent) and sequenced on an Illumina HiSeq2500 (50 SR lane V4 reagents).
ChIP-seq data processing
Sequence alignment and Peak identification
Following read duplication removal (picard-tools), all datasets were individually aligned to the mouse genome (GRCm38/mm10) using Bowtie2 (Langmead and Salzberg, 2012) with default parameters. Next, peaks were called on the aligned BAMs from each IP replicate using MACS2 with the relevant input sample (either AML or WT), a q-value threshold of 0.01, and MFOLD of 5 50. Pileup fragment profiles per million reads were generated by MACS2 using the SPMR parameter and were used for visualization of ChIP-seq data directly on the UCSC genome browser.
Super Enhancer identification
H3K27ac super-enhancers (SEs) and typical enhancers (TEs) were mapped using the ROSE software package (http://younglab.wi.mit.edu/super_enhancer_code.html) with a default stitching distance of 12.5Kb and a TSS exclusion zone of 2.5Kb. For Figure 4E, genes associated with super-enhancers were identified as those lying closest to a 50Kb window by ROSE geneMapper tool.
Quantifying Effects of A51 Treatment on Transcription Elongation
We quantified the effects of A51 treatment on transcription elongation of genes by measuring the change in pSer2 CTD density in whole gene body. The Log2 fold change in RNA Pol II density ± A51 treatment was calculated for all genes overlapping TE or SE by up to 5Kb.
Fold change and differential binding analysis
To determine the log2 fold change of H3K27ac or pSer2 binding levels in DMSO versus A51-treated samples, we used the Bio-conductor R package DiffBind (Stark and Brown, 2011). DiffBind was used on either typical enhancers or super-enhancers called by MACS and ROSE as explained above. The analysis was run wither in triplicates, for H3K27ac, or in duplicates, for pSer2. DiffBind was run using the DESeq2 method.
Chemical synthesis procedures
Preparation of (E)-1-cyclopropyl-4-(dimethylamino)-3-(2-(methylthio)pyrimidin-4-yl)but-3-en-2-one (Core A)
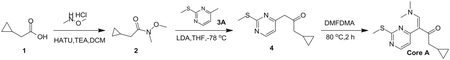
Step 1: N, O-Dimethylhydroxylamine hydrochloride (25.14 g, 257.69 mmol, 1.72 eq), HATU (56.97 g, 149.82 mmol, 1.00 eq) and TEA (45.48 g, 449.46 mmol, 3.00 eq) were added to a solution of 2-cyclopropylacetic acid (15.00 g, 149.82 mmol, 1.00 eq) in DCM (500 mL) at 0°C, and then the mixture was stirred at 30°C for 3 h. The resulting mixture was poured into water (500 mL). The aqueous washing phase was extracted with DCM (3*250 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (SiO2, petroleum ether(PE): ethyl acetate (EA) = 50:1 to 10:1) to give the desired compound 2 (13.20 g, 82.97 mmol, 55.4% yield) as colorless liquid.
1H NMR (CDCl3, 400 MHz) d 3.65 (s, 3H), 3.17 (s, 3H), 2.33 (d, J = 6.8 Hz, 2H), 1.09–1.06 (m, 1H), 0.54–0.50 (m, 2H), 0.16–0.14 (m, 2H).
Step 2: To a solution of 4-methyl-2-methylsulfanyl-pyrimidine (9.00 g, 64.19 mmol, 1.00 eq) in THF (500 mL) was added LDA (2 M, 48.46 mL, 1.51 eq) at − 78°C. After stirring for 1 h, a solution of compound 2 (13.79 g, 96.29 mmol, 1.50 eq) in THF (500 mL) was added drop wise at − 78°C and then the reaction mixture was stirred at 78°C for 4 h. Quenched with saturated aq. NH4Cl (100 mL), the aqueous phase was extracted with ethyl acetate (3 3 50 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was crystallized from petroleum ether/ethyl acetate to afford the desired compound 4 (13.60 g, 55.06 mmol, 85.8% yield) as a yellow solid.
LCMS: RT = 0.629 min, m/z 223.0 [M+H]+.
Step 3: A solution of compound 4 (13.60 g, 61.18 mmol, 1.00 eq) in DMF-DMA (51.42 g, 2.45 mol, 40 eq) was stirred at 90°C for 2 h. The solvent was removed in vacuum. The residue was purified by column chromatography on silica gel to give Core A (10.60 g, 36.30 mmol, 59.3% yield) as a yellow solid.
LCMS: RT = 0.634 min, m/z 278.2 [M+H]+
1H NMR (CDCl3, 400 MHz) d 8.38 (d, J = 6.8 Hz, 1H), 7.62 (s, 1H), 6.96 (s, 1H), 2.96–2.87 (m, 6H), 2.56 (s, 3H), 2.38 (d, J = 8.8 Hz, 2H),1.04–1.02 (m, 1H), 0.52–0.46 (m, 2H), 0.09–0.04 (m, 2H).
Preparation of 4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)-2-(methylsulfonyl)pyrimid ine (Core C)
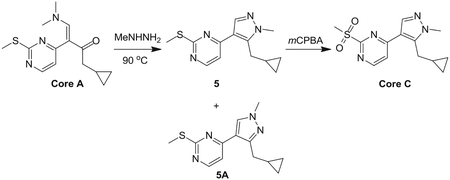
Step 1: A solution of Core A (6.20 g, 22.35 mmol, 1.00 eq) and methylhydrazine (8.00 g, 69.46 mmol, 3.11 eq) in ethanol (100 mL) was stirred at 90°C for 16 hr. The solvent was removed in vacuum. The residue was purified by pre-HPLC (basic condition) to afford compound 5 (1.80 g, 6.84 mmol, 30.6% yield) as a yellow solid and the isomer 5A (2.00 g, 7.30 mmol, 32.6% yield) as a yellow oil.
Compound 5:
LCMS: RT = 2.551 min, m/z 261.1 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 8.38 (d, J = 5.2 Hz, 1H), 7.90 (s, 1H), 7.11 (d, J = 5.2 Hz, 1H), 3.93 (s, 3H), 3.24 (d, J = 6.4 Hz, 2H), 2.62 (s, 3H), 1.12 – 1.09 (m, 1H), 0.54 – 0.49 (m, 2H), 0.32–0.28 (m, 2H).
Regioisomer 5A:
LCMS: RT = 2.486 min, m/z 261.1 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 8.41 (d, J = 5.2 Hz, 1H), 7.90 (s, 1H), 7.04 (d, J = 5.2 Hz, 1H), 3.92 (s, 3H), 2.96 (d, J = 6.4 Hz, 2H), 2.61 (s, 3H), 1.20 – 1.17 (m, 1H), 0.50 – 0.45 (m, 2H), 0.26 – 0.22 (m, 2H).
Step 2: To a solution of compound 1 (1.50 g, 5.76 mmol, 1.00 eq) in DCM (20 mL) was added m-CPBA (2.98 g, 17.28 mmol, 3.00 eq) at 0°C and stirred at 30°C for 2 hr. The resulting mixture was washed with NaHSO3 (2 3 100 mL), saturate aq. NaHCO3 (100 mL) and brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether: ethyl acetate = 10:1 to 1:1) to give the compound Core C (1.50 g, 5.08 mmol, 88.2% yield) as a yellow solid.
LCMS: RT = 1.891 min, m/z 293.0 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 8.74 (d, J = 5.6 Hz, 1H), 7.97 (s, 1H), 7.58 (d, J = 5.6 Hz, 1H), 3.94 (s, 3H), 3.37 (s, 3H), 3.25 (d, J = 6.8 Hz, 2H), 1.14 – 1.11 (m, 1H), 0.52 – 0.49 (m, 2H), 0.37 – 0.35 (m, 2H).
Preparation of (1R, 4R)-N1-(4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)pyrimidin-2-yl)cyclohexane-1,4-diamine (A-14)

The reaction mixture of amine n (1.20 eq) and Core C (1.00 eq) in dioxane (3 mL) was stirred at 130 C under microwave for 1.5 h. The reaction mixture was poured into H2O (20 mL). The aqueous layer was extracted EA (20 mL*3). The combined organic layers were washed with brine (20 mL*3), dried over Na2SO4, filtered and concentrated. The residue was purified by Pre-HPLC (basic condition) to give A-14. It was obtained as a yellow solid. Yield: 38.4%
LCMS: RT = 2.043 min, m/z 327.2 [M+H]+.
1H NMR (CDCl3, 400 MHz) δ 8.17 (d, J = 5.3 Hz, 1H), 7.83 (s, 1H), 6.70 (d, J = 5.3 Hz, 1H), 4.91 (s, 1H), 3.93 – 3.85 (m, 4H), 3.21 (d, J = 6.3 Hz, 2H), 2.74 (s, 1H), 2.16 (d, J = 4.0 Hz, 2H), 1.94 (d, J = 6.7 Hz, 2H), 1.30 – 1.25 (m, 4H), 1.11 – 1.10 (m, 1H), 0.50 – 0.46 (m, 1H), 0.27 – 0.24 (m, 1H)
Preparation of 4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)-N-((1R,4R)-4-(piperidin-1-yl)cyclohexyl)pyrimidin-2-amine (A-47)
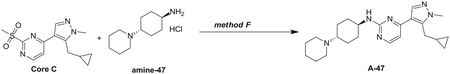
Compound A-47 was prepared according to the procedure described in method F with amine-47 and it was obtained as a white solid after purification by prep HPLC (HCl condition). Yield: 49.3%
LCMS: RT = 1.530 min, m/z 395.2 [M+H]+
1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 8.06 (d, J = 6.8 Hz, 1H), 7.24–7.21 (m, 1H), 4.10 (s, 1H), 3.88 (s, 3H), 3.49 (d, J = 11.2 Hz, 2H), 3.23 (s, 2H), 3.06–3.00 (m, 4H), 2.26 (d, J = 10 Hz, 4H), 1.96–1.57 (m, 10H), 1.15 (s, 1H), 0.50 (s, 2H), 0.31–0.30 (m, 2H).
Preparation of (1R, 4R)-N1-(5-chloro-4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)pyrimidin-2-yl)cyclohexane-1,4-diamine (A-51)
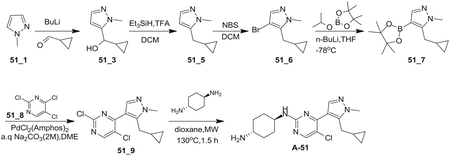
Step 1: To a solution of compound N-methylpyrazole (51_1, 8.00 g, 97.44 mmol, 1.00 eq) in THF (160 mL) was added drop-wise n-BuLi (2.5 M, 46.77 mL, 1.20 eq) at − 78°C. After the resulting mixture was stirred for 1hr at that temperature, the solution of cyclopropanecarbaldehyde (8.20 g, 116.93 mmol, 1.20 eq) in THF (80 mL) was added drop-wise. And then the reaction mixture was stirred at 20°C for 16 h. TLC (PE:EA = 2:1) showed reactant 1 (Rf = 0.3) was consumed and product (Rf = 0.05) was formed. The mixture was poured into aqueous NH4Cl (300 mL) and stirred for 10 min. The aqueous phase was extracted with ethyl acetate (100 mL*2). The combined organic phase was washed with brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by column chromatography (SiO2, PE:EA = 8:1 ~ 0:1) to afford compound 51_3 (12.00 g, 78.85 mmol, 80.9% yield, 100% purity) as a colorless oil.
LCMS: RT = 0.118 min, m/z 153.1 [M+H]+
Step 2: The reaction mixture of compound 51_3 (9.00 g, 59.14 mmol, 1.00 eq), TFA (40.46 g, 354.84 mmol, 26.27 mL, 6.00 eq) and Et3SiH (41.26 g, 354.84 mmol, 56.52 mL, 6.00 eq) in DCM (900 mL) was stirred at 40°C for 36h. The mixture was adjusted to pH = 8 with aqueous NaHCO3 and separated. The organic layer was concentrated and purified by prep HPLC (basic condition) to give compound 51_5 (2.10 g, 15.42 mmol, 26.1% yield) as a dark brown oil.
LCMS: RT = 0.565 min, m/z 137.1 [M+H]+
Step 3: To a solution of compound 51_5 (2.10 g, 15.42 mmol, 1.00 eq) in DCM (21 mL) was added NBS (3.02 g, 16.96 mmol, 1.10 eq) at 0°C. The mixture was stirred at 20°C for 2h. The mixture was concentrated and purified by column chromatography (SiO2, PE:EA = 20:1) to give compound 51_6 (3.00 g, 13.95 mmol, 90.5% yield) as a yellow oil.
LCMS: RT = 0.784 min, m/z 217.1 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 7.39 (s, 1 H), 3.87 (s, 3 H), 2.65–2.63 (d, J = 8.8 Hz, 2 H), 0.98–0.94 (m, 1H), 0.55–0.51 (m, 2H), 0.29–0.25 (m, 2H).
Step 4: To a solution of compound 51_6 (3.00 g, 13.95 mmol, 1.00 eq) in THF (60 mL) was added n-BuLi (2 M, 10.46 mL, 1.50 eq) drop-wise at − 78°C. After stirring for 0.5h at that temperature, a solution of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (5.19 g, 27.90 mmol, 2.00 eq) in THF (6 mL) was added. The resulting mixture was warmed to 20°C and stirred for 0.5h. TLC (PE:EA = 5:1) showed reactant (Rf = 0.6) was consumed and product (Rf = 0.5) was formed. The mixture was quenched with saturated NH4Cl (50 mL) and extracted with EA (100 mL). The organic layer was concentrated and purified by column chromatography (SiO2, PE:EA = 20:1 ~ 10:1) to give compound 51_ 7 (3.30 g, 11.40 mmol, 81.7% yield, 90.5% purity) as a colorless oil.
LCMS: RT = 0.801 min, m/z 263.2 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 7.67 (s, 1 H), 3.85 (s, 3 H), 2.82–2.81 (d, J = 6.8 Hz, 2 H),1.30 (s, 12H), 0.92–0.90 (m, 1H), 0.45–0.42 (m, 2H), 0.29–0.27 (m, 2H).
Step 5: To a solution of compound 51_ 7 (500.00 mg, 1.91 mmol, 1.00 eq) in DME (10 mL) were added 2, 4, 5-trichloropyrimidine (51_8, 420.40 mg, 2.29 mmol, 1.20 eq), Na2CO3 (2 M, 2.10 mL, 2.20 eq) and catalyst PdCl2(Amphos)2 (67.62 mg, 95.50 μmol, 0.05 eq) under nitrogen. The resulting mixture was stirred at 85°C for 2hr under nitrogen. TLC (PE:EA = 5:1) showed reactant (Rf = 0.4) was consumed and product (Rf = 0.5) was formed. The mixture was diluted with water (50 mL) and extracted with EA (50ml). The organic layer was concentrated and purified by silica gel column (PE:EA = 5:1) to afford compound 51_9 (350.0 mg, 1.05 mmol, 55% yield, 85% purity) as a yellow oil.
LCMS: RT = 0.819 min, m/z 283.1 [M+H]+
Step 6: The reaction mixture of compound 51_9 (300.00 mg, 1.06 mmol, 1.00 eq) and trans-cyclohexane-1, 4-diamine (484.17 mg, 4.24 mmol, 4.00 eq) in dioxane (4.5 mL) was stirred at 130°C for 2h under microwave. The mixture was filtered and concentrated. The crude was purified by prep-HPLC (HCl condition) to give A-51 (80.00 mg, 200.04 mmol, 18.9% yield, 99.3% purity, HCl) as a yellow solid.
LCMS: RT = 2.817 min, m/z 361.1 [M+H]+
1H NMR (DMSO-d6, 400 MHz) δ 8.37 (s, 1 H), 8.29 (s, 3 H), 8.08 (s, 1 H), 3.86 (s, 3 H), 3.70–3.68 (m, 2 H), 3.06–3.05 (d, J = 6.4 Hz, 2H),2.96 (s, 1H), 2.03–1.95 (m, 4H), 1.49–1.32 (m, 4H), 0.98 (s, 1H), 0.42–0.40 (m, 2H), 0.16 (m, 2H).
Preparation of N-((1R,4R)-4-(1H-pyrazol-1-yl)cyclohexyl)-5-chloro-4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)pyrimidin-2-amine (A64)
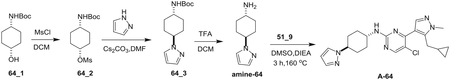
Step 1: To a solution of 64_1 (2.00 g, 9.29 mmol, 1.00 eq) in DCM (20 mL) were added TEA (1.88 g, 18.58 mmol, 2.58 mL, 2.00 eq) and methanesulfonyl chloride (1.06 g, 9.29 mmol, 719.03 μL, 1.00 eq) at 0°C. The mixture was stirred at 20°C for 2 hr. The mixture was diluted with H2O (30 mL) and extracted with DCM (20 mL*2). The combined organic layers were dried and concentrated to give 64_2(2.50 g, 8.52 mmol, 91.7% yield) as a white solid.
1H NMR (CDCl3, 400 MHz) δ 4.88 (s, 1 H), 4.48 (s, 1 H), 3.52 (s, 1 H), 3.01 (s, 3H), 2.07–2.03 (m, 2H), 1.83–1.82 (m, 2H), 1.73–1.70(m, 2H), 1.61–1.55 (m, 7H), 1.448 (s, 9H).
Step 2: To a solution of 1H-pyrazole (195.25 mg, 2.87 mmol, 1.20 eq) in MeCN (7.00 mL) were added Cs2CO3 (1.56 g, 4.78 mmol, 2.00 eq) and 64_2 (700.00 mg, 2.39 mmol, 1.00 eq). The mixture was stirred at 100°C for 2 hr. The mixture was diluted with H2O (20 mL) and extracted with EtOAc (10 mL*3). The combined organic layers were washed with brine (20 mL) and concentrated. The residue was purified by pre-HPLC (basic condition) to give 64_3(200.00 mg, 732.25 μmol, 30.6% yield, 97.1% purity) as a yellow solid.
MS: m/z RT = 0.888 min, 266.0 [M+H]+
Step 3: To a solution of 64_3 (180.00 mg, 678.35 μmol, 1.00 eq) in DCM (2 mL) was added TFA (400.00 mL) at 0°C. The mixture was stirred at 20°C for 1 hr. The mixture was diluted with H2O (30 mL) and extracted with DCM (10 mL*2). The aqueous layer was lyophilized to give amine-64 (180.00 mg, 633.91 μmol, 93. 5% yield, 98.3% purity, TFA) as colorless oil.
LCMS: RT = 0.272 min, m/z 166.2 [M+H]+
Step 4: To a solution of 51_9 (100.00 mg, 353.16 μmol, 1.00 eq) in DMSO (1.5 mL) were added amine-64 (95.48 mg, 529.74 μmol, 1.50 eq) and DIEA (182.57 mg, 1.41 mmol, 246.71 μL, 4.00 eq). The resulting mixture was stirred at 160°C for 3 hr, filtered and the mother liquid was concentrated. The residue was purified by pre-HPLC (basic condition) and then prep-HPLC (HCl condition) to give A-64 (10.00 mg, 23.42 μmol, 6.6% yield, 100% purity) as a yellow solid.
LCMS: RT = 1.981 m/z 412.2[M+H]+
1H NMR (MeOD, 400 MHz) δ 8.41–8.37 (m, 2 H), 7.83 (s, 1 H), 7.67 (s, 1 H),6.39 (s, 1H), 4.36 (s, 1H), 4.12–4.08 (m, 1H), 3.95 (s, 3H),3.25–3.23 (d, J = 6.4Hz, 2H), 2.26–2.23 (m, 4H), 2.05–2.02(m, 2H), 1.99–1.96 (m, 2H), 1.73–1.67(m, 2H), 1.09 (s, 1H), 0.55–0.50 (m, 2H),0.27–0.26 (m, 2H).
Preparation of (1R,4R)-N1-((1H-pyrazol-4-yl)methyl)-N4-(5-chloro-4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl) pyrimidin-2-yl)cyclohexane-1,4-diamine (A-75)
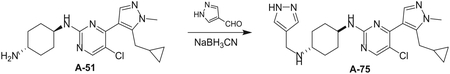
To a solution of A-51 (200.00 mg, 554.20 μmol, 1.00 eq) and 1H-pyrazole-4-carbaldehyde (53.25 mg, 554.20 μmol, 1.00 eq) were added AcOH (34.86 mL, 609.62 μmol, 1.10 eq) and NaBH3CN (69.65 mg, 1.11 mmol, 2.00 eq). The mixture was sitrred at 15°C for 16 hr. The mixture was quenched with aqueous NaHCO3 (1 mL) and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Gemini 150*25mm*10um; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 39%–39%,12min) to give A-75 (4.00 mg, 9.04 mmol, 1.6% yield, 99.7% purity) as a yellow solid.
LCMS: RT = 2.95 min, m/z 441.2 [M+H]+
1H NMR (CDCl3, 400 MHz) δ 8.21–8.12 (m, 2 H), 7.61 (s, 2 H), 4.90 (d, J = 8.4Hz, 1H), 3.91 (s, 3H), 3.85–3.80 (m, 3H), 3.06–3.05 (m, 2H), 2.70 (m, 1H), 2.19–2.07 (m, 4H), 1.42–1.23 (m, 4H), 1.09 (m, 1H), 0.50–0.45 (m, 2H), 0.18–0.14 (m, 2H).
Preparation of (1R,4R)-N1-(4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)-5-fluoropyrimidin-2-yl)cyclohexane-1,4-diamine (A-86)
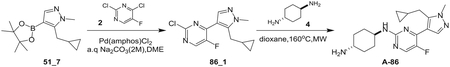
Step 1: To a solution of compound 51_7 (800 mg, 3.05 mmol, 1.00 eq) in DME (16 mL) were added compound 2 (509.54 mg, 3.05 mmol, 1.00 eq), Na2CO3 (2 M, 4.6 mL, 3.00 eq) and 4-ditert-butylphosphanyl-N,N-dimethyl-aniline;dichloropalladium (108.04 mg, 152.50 μmol, 108 uL, 0.05 eq) under nitrogen. The resulting mixture was stirred at 85°C for 2 hr under nitrogen. The mixture was diluted with H2O (50 mL) and extracted with EA (50 mL). The organic layer was concentrated to give compound 86_1(1.20 g, crude) as a yellow oil.
LCMS: RT = 1.819 min, m/z 267.1 [M+H]+
Step 2: The mixture of compound 86_1 (1.10 g, 4.12 mmol, 1.00 eq) and compound 4 (1.88 g, 16.48 mmol, 4.00 eq) in dioxane (16 mL) was stirred at 130°C for 8 hr under microwave. The mixture was filtered and concentrated. The residue was purified by pre-HPLC (HCl condition) to give A-86 (1.30 g, crude) as a yellow solid. A-86 (300 mg) was purified again by prep-HPLC (HCl condition) to give A-86 (20.00 mg, 52.51 μmol, 12.1% yield, 100% purity, HCl) as a yellow solid.
LCMS: RT = 2.321 min, m/z 345.2 [M+H]+
1H NMR (MeOD, 400 MHz) δ 8.37–8.36 (m, 1 H), 8.23–8.22 (m, 1 H), 4.07–4.03 (m, 1H), 3.96 (s, 3H), 3.21 (d, J = 6.4Hz, 2H), 2.24–2.18 (m, 4H), 1.68–1.57 (m, 2H), 1.19 (s, 1H), 0.57–0.55 (m, 2H), 0.38–0.34 (m, 2H).
Preparation of (S)-3-(4-methyl-1-oxoisoindolin-2-yl)azepane-2,7-dione (BTX161) Scheme.
Step 1
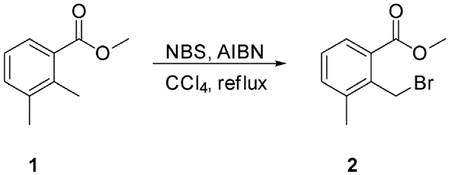
To a solution of methyl 2,3-dimethylbenzoate (1) (18.0 g, 109.6 mmol) in Carbon Tetrachloride (250 mL) at room temperature was added N-Bromosuccinimide (NBS) (23.4 g, 131.52 mmol). The reaction was heated to 85°C for 5 min. Then 2,2′-azobis(2-methylpropionitrile) (AIBN) (9.42 g, 54.8 mmol) was added and the reaction was stirred at 85°C for 16 hr. The reaction was cooled to room temperature and the solid was filtered off. The filtrate was concentrated to give the crude product, which was purified by silica-gel column chromatography eluting with Ethyl acetate−/−Petroleum ether from 0/1 to 1/50 to give crude methyl 2-(bromomethyl)-3-methylbenzoate (2) (23.4 g, yield: 87.8%) as a colorless oil. MS m/z 243.7 [M+].
Step 2
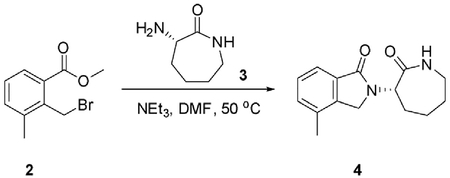
To a solution of methyl 2-(bromomethyl)-3-methylbenzoate (2) (23.4 g, 96.57 mmol) in N,N-dimethylformamide (300 mL) at room temperature was added (S)-3-aminoazepan-2-one (3) (12.36 g, 96.57 mmol). Then triethylamine (19.5 g, 193.14 mmol) was added, and the resulting mixture was heated to 50°C for 16 hr. The reaction was cooled to room temperature and the solvent was removed. The residue was diluted with water (100 mL). The suspension was stirred at room temperature for 20 min. The solid was filtered off and the solid was triturated with Ethyl acetate to give (S)-4-methyl-2-(2-oxoazepan-3-yl)isoindolin-1-one (4) (6.57 g, yield: 52.6%) as a white solid.
1H NMR (300 MHz, CDCl3) δ 7.81–7.78 (m, 1H), 7.52–7.49 (m, 1H), 7.41–7.38 (m, 2H), 4.91 (d, J = 11.4 Hz, 1H), 4.52 (dd, J = 17.4,58.5 Hz, 2H), 3.35–3.20 (m, 1H), 3.15–3.10 (m, 1H), 2.33 (s, 3H), 2.02–1.95 (m, 2H), 1.89–1.70 (m, 3H), 1.30–1.23 (m, 1H). MS m/z 259.1 [M+H]+.
Step 3
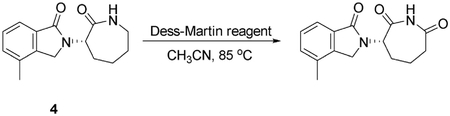
To a solution of (S)-4-methyl-2-(2-oxoazepan-3-yl)isoindolin-1-one (4) (5.8 g, 22.48 mmol) in Acetonitrile (300 mL) and wet DMSO (15 mL) at 0°C was added Dess-martin periodinane reagent (23.8 g, 56.2 mmol). The suspension was heated at 85°C for 16 hr. The reaction was cooled to room temperature and 100 mL of a saturated sodium thiosulfate solution was added followed by stirring for 5 min. The resulting mixture was extracted with Dichloromethane (30 mL × 2) and the combined solution was washed with 10% aq. Sodium thiosulfate / aq. Sodium bicarbonate (1:1 mixture) (50 mL) and brine. The organic layer was dried over Sodium sulfate, filtered and concentrated to afford the crude product, which was purified by silica-gel column chromatography eluting with Ethyl acetate / Petroleum ether from 1/2 to 1/1 to give the white solid. The solid was triturated with Methanol to give (S)-3-(4-methyl-1-oxoisoindolin-2-yl)azepane-2,7-dione (720 mg, yield: 11.8%) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.54 (dd, J = 1.6, 6.4 Hz, 1H), 7.44–7.41 (m, 2H), 5.26 (dd, J = 5.2, 12.4Hz, 1H), 4.48 (s, 2H), 3.12–3.04 (m, 1H), 2.61–2.55 (m, 1H), 2.34 (s, 3H), 2.32–2.30 (m, 1H), 2.13–2.02 (m, 2H), 1.87–1.78 (m, 1H). MS m/z 273.0 [M+H]+.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed by GraphPad Prism software. For survival studies the log-rank test was used to compare differences between the survival of two groups. For all in vitro, ex vivo, and in vivo experiments, unpaired two-tailed Student’s t test was used, except for Figure 1A where Mann-Whitney non-parametric test was used. Data are shown as mean ± SD as indicated in the figure legends. For bioinformatics analysis comparing pSer2 CTD ChIP-seq in SEs versus TEs (Figure 5E) we used Welch’s two-tailed t test as this is the standard test for this model. Statistical significance is as follows: ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. For each in vivo experiment number of mice (n) is indicated. For in vitro and ex vivo experiments number of technical or biological repeats is indicated in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The accession number for the ChIP-seq data reported in this paper is GEO: GSE117443. The accession numbers for the crystal structures of A86 in complex with CKIα, CKIδ, and CDK9/cyclinT1 reported in this paper are RCBS PDB: 6GZD, 6GZM, and 6GZH, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Lineage biotin ab cocktail | Miltenyibiotec | Cat#130-092-613 |
| anti-c-Kit APC | eBioscience | Cat#135107 |
| anti-Sca-1 Alexa Flour 700 | R&D SYSTEMS | Cat#FAB1226N |
| anti-Flt3 - PE | Biolegend | Cat#135306 |
| anti-Flt3 - APC | eBioscience | Cat#17-135-82 |
| anti-CD34 -FITC | BIO-RAD | Cat#MCA 1825FA |
| Anti-CD34-eFluor450 | eBioscience | Cat#48-0341-80 |
| anti-hCD45-PB | eBioscience | Cat#304022 |
| anti-hCD33-PE/Cy7 | Biolegend | Cat#366617 |
| anti-hCD38-APC | Biolegend | Cat#356606 |
| Anti-mCD45.1 | Biolegend | Cat#110706 |
| anti-Strepavidin-percp/cy5.5 | eBioscience | Cat#45-4317-82 |
| anti-β-catenin | BD Transduction Laboratories | Cat#610154 |
| anti-phospho Ser45-β-catenin | Cell Signaling | Cat#9564 |
| anti-phospho-Thr41/Ser45-β-catenin | Cell Signaling | Cat#9565 |
| anti-CKIα | Santa Cruz | Cat#C-19 |
| anti-Hsp90 | Calbiochem | Cat#ca1016 |
| anti-αTubulin | Sigma | Cat#t5168 |
| anti-p53 (mouse) | Cell Signaling | Cat#1C12 |
| anti-Cleaved Caspase 3 | Cell Signaling | Cat#9661s |
| anti-c-Myc | Cell Signaling | Cat#9402 |
| anti-MCL1 | Cell Signaling | Cat#5453 |
| anti-p53 (human) | Gift from Moshe Oren | Clone: DO-1/1801 |
| anti-human MDM2 | Gift from Moshe Oren | Clone: 2A9/4B2/4B1 |
| anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat#05-636 |
| Rabbit anti-PP2Ac | Gift from David Virshup | N/A |
| anti-Cyclin D1 | Lab Vision | Cat#SP4 |
| anti-Phospho -Rpb1 CTD (Ser 2/5) | Cell Signaling | Cat#4735 |
| anti-H3K27Ac | Abcam | Cat#ab4729 |
| anti-RNA polymerase II CTD (phospho Ser2) | Abcam | Cat#ab5095 |
| anti-RNA polymerase II CTD (phospho Ser5) | Mercury | Cat#04-1572 |
| c-Kit APC-eFlour780 | eBioscience | Cat#47-1171-82 |
| c-Kit-PE | Biolegend | Cat#105807 |
| Sca-1 PE-Cy7 | eBioscience | Cat#25-5981-82 |
| Peroxidase-conjugated Goat anti-Rabbit | Jackson | Cat#111-035-144 |
| Peroxidase-conjugated Rabbit anti-Goat | Jackson | Cat#705-035-003 |
| Peroxidase-conjugated Rabbit anti-Mouse | Jackson | Cat#315-035-003 |
| Donkey anti Mouse Alexa 488 | ThermoFisher | Cat#A21202 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EDTA (0.5 M), pH 8.0 | Ambion | Cat#AM9260G |
| RPMI 1640 Medium | GIBCO | Cat#11875119 |
| DMEM Medium | GIBCO | Cat#41965-039 |
| IMEM | Biological Industries, Israel | Cat#01-058-1A |
| L-Glutamine | Biological Industries, Israel | Cat#03-020 |
| Penicilin/Streptomycin | Biological Industries, Israel | Cat#03-031 |
| Recombinant murine stem cell factor (SCF) | PEPROTECH | Cat#250-03 |
| Recombinant murine IL-6 | PEPROTECH | Cat#216-16 |
| Recombinant murine IL-3 | PEPROTECH | Cat#213-13 |
| Recombinant murine G-CSF | PEPROTECH | Cat#250-05 |
| Recombinant murine TPO | PEPROTECH | Cat#315-14 |
| Recombinant human IL-6 | PEPROTECH | Cat#200-06 |
| Recombinant human IL-3 | PEPROTECH | Cat#200-03 |
| Recombinant human GM-CSF | PEPROTECH | Cat#300-03 |
| Recombinant human TPO | PEPROTECH | Cat#300-18 |
| MG132 | Calbiochem | Cat#133407-82 |
| Fetal Bovine Serum (FBS) | GIBCO | Cat#12657 |
| Triton X-100 | Alfa Aesar | Cat#A16046 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#D8418 |
| Methyl cellulose | Sigma-Aldrich | Cat#M7027-100G |
| Tween 80 | Sigma-Aldrich | Cat#P4780-500ML |
| PEG400 | Sigma-Aldrich | Cat#202398-500G |
| Dynabeads Protein G magnetic beads | Life Technologies | Cat#10003D |
| RNase A | ThermoFisher | Cat#EN0531 |
| Proteinase K | Invitrogen | Cat#100005393 |
| StemPro-34 SFM media | GIBCO | Cat#10639011 |
| SDS | Hy-labs | Cat#BP 494/100D |
| TRIzol | ThermoFisher | Cat#15596018 |
| EDTA (0.5 M), pH 8.0 | Ambion | Cat#AM9260G |
| DNase1 | Worthington | Cat#LS002006 |
| Triton X-100 | Alfa Aesar | Cat#A16046 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#D8418 |
| Gelucire 44/14 | Gattefosse, France | Cat#3051TPD |
| 2-hydroxy prolyl-β-cyclodextrin (HP-β-CD) | SIGMA | Cat#H-107 |
| StemPro-34 Nutrient Supplement | GIBCO | Cat#10640-019 |
| MethoCult™ GF M3434 | StemCell Technologies | Cat#03434 |
| Gentamycin | SIGMA | Cat#G1397-10ML |
| Amphotericin B | SIGMA | Cat#A2942-100ML |
| Formaldehyde | Electron Microscopy Sciences | Cat#15710 |
| Annexin V-PE | Biolegend | Cat#640908 |
| 7AAD | TONBO biosciences | Cat#13-6993-T500 |
| Propidium Iodide | MBL | Cat#4700-200 |
| A-series inhibitors (A14, A47, A51, A64, A75 and A86) | This paper | N/A |
| BTX161 | This paper | N/A |
| Lenalidomide | MedChem Express (MCE) | Cat#L328020 |
| PKC412 (midostaurin) | MedChem Express (MCE) | Cat#HY-10230 |
| Cytarabine | MedChem Express (MCE) | Cat#C998100 |
| THZ1 | MedChem Express (MCE) | Cat#T430000 |
| iCDK9 (NVP-2) | MedChem Express (MCE) | Cat#Y-12214A |
| Critical Commercial Assays | ||
| EasySep Mouse CD117 (cKIT) Positive Selection Kit | StemCell Technologies | Cat#18757 |
| miRNeasy Kit | QIAGEN | Cat#74104 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat#28704 |
| TruSeq nano DNA sample prep kit | Illumina | Cat#FC-121-4001 |
| qScript cDNA Synthesis Kit | QIAGEN | Cat#95047-025 |
| ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit | VECTOR | Cat#MP-7401 |
| 3,3′-Diaminobenzidine (DAB) chromogen | Thermo Scientific | Cat#TA-125-QHSX |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| Deposited Data | ||
| CKIα | This paper | PDB: 6GZD |
| CKIδ | This paper | PDB: 6GZM |
| CDK9/CyclinT1 | This paper | PDB: 6GZH |
| ChIP Seq data | This paper | GEO: GSE117443 |
| Experimental Models: Cell Lines | ||
| RKO | ATCC | CRL-2577 |
| MV4-11 | ATCC | CRL-9591 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | The Jackson laboratory | Stock No: 000664 |
| p53KI/KI | Christophorou et al., 2005 | N/A |
| NSG-SGM3 | The Jackson laboratory | Stock No: 013062 |
| NSG-hSCF | The Jackson laboratory | Stock No: 017830 |
| Csnk1a1fl/fl | The Jackson laboratory | Stock No: 025398 |
| Mx1-cre | The Jackson laboratory | Stock No: 005673 |
| CD45.1 C57BL/6 | The Jackson laboratory | Stock No: 002014 |
| Tet2−/−;Flt3ITD | Shih et al., 2015 | N/A |
| Oligonucleotides | ||
| ShCKIα (in Plko vector): 5′-CCGGGCAGAATTTGCGATGTACTTACTCGAGTAAGTACATCGCAAATTCTGCTTTTT-3′ | This paper | N/A |
| Scrambled control (in Plko vector): 5′ TCCTAAGGTTAAGTCGCCCTCG-3′ | This Paper | N/A |
| Primers used for RT-PCR, see Table in methods | This Paper | N/A |
| Recombinant DNA | ||
| MLL-AF9-IRES-GFP | Barabé et al., 2007 | N/A |
| Software and Algorithms | ||
| Picard-tools | The Broad Institute | http://broadinstitute.github.io/picard/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| ROSE software package | Loven et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| Bioconductor R package DiffBind | Stark and Brown, 2011 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com |
Highlights.
We developed orally active small molecule inhibitors co-targeting CKIα and CDK7/9
Crystallography studies provide structural basis for distinct kinase co-targeting
The inhibitors selectively kill AML progenitors and cure AML mice
The therapeutic mechanism entails p53 activation with super-enhancer shutdown
ACKNOWLEDGMENTS
We thank Amotz Frenkel for assistance with leukemia animal studies, Drs. Kevan Shokat (Cellular and Molecular Pharmacology, UCSF) and Kyle Chen (Bio-TheryX) for fruitful project discussions, Dr. Mark Minden (Ontario Cancer Institute- Princess Margaret Hospital, Toronto, Canada) for providing AML PDXs, Jeremy Hunt (DiscoverX, Fremont, California, USA) for the compound-kinome interaction analysis and data interpretation, and the Technion Genome Center, Technion-Israel Institute of Technology for sequencing. We are grateful to the Dr. Berta Strulovici, Dr. Noga Kozer and Dr. Haim Barr (The Nancy and Stephen Grand, Israel National Center for Personalized Medicine, The Weizmann Institute, Rehovot, Israel) for running the kinase library screens and Crelux-WuXi AppTec crystallography team (Martinsried, Germany) for assisting in solving the structures of inhibitor-bound CKIα, CKIδ and CDK9. We also thank Liming Xue at WuXi AppTec for the synthesis of compounds in Figure 1. This work was supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF) (to Y.B.-N., M.O., and E.P.), Israel Science Foundation (ISF)-Centers of Excellence (to Y.B.-N., M.O., and E.P.), the European Research Council within the FP-7 (294390 PICHO to Y.B.-N. and 281738 LIVERMICROENV to E.P.), Israel Cancer Research Fund Professorship (to Y.B.-N.), and Memorial Sloan Kettering Cancer Center Support Grant (NIH P30 CA008748 to R.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.cell.2018.07.045.
DECLARATION OF INTERESTS
P.E.E., R.W.S., L.F., and F.M. are BioTheryX Inc. employees. Y.B.-N. and J.V. are consultants to BioTheryX Inc. A patent for the new molecular entities detailed in the manuscript have been filed under PCT/IL2016/050852. Other authors declare no competing interests.
REFERENCES
- Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, et al. (2016). Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 534, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, and Alkalay I (2002). Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabé F, Kennedy JA, Hope KJ, and Dick JE (2007). Modeling the initiation and progression of human acute leukemia in mice. Science 316, 600–604. [DOI] [PubMed] [Google Scholar]
- Boddu P, Kantarjian HM, Garcia-Manero G, Ravandi F, Verstovsek S, Jabbour E, Borthakur G, Konopleva M, Bhalla KN, Daver N, et al. (2017). Treated secondary acute myeloid leukemia: a distinct high-risk subset of AML with adverse prognosis. Blood Adv. 1, 1312–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional addiction in cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Kuo CJ, Ito T, Su YY, Jiang ST, Chiu MH, Lin YH, Nist A, Mernberger M, Stiewe T, et al. (2017). CK1α ablation in keratinocytes induces p53-dependent, sunburn-protective skin hyperpigmentation. Proc. Natl. Acad. Sci. USA 114, E8035–E8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou MA, Martin-Zanca D, Soucek L, Lawlor ER, Brown-Swigart L, Verschuren EW, and Evan GI (2005). Temporal dissection of p53 function in vitro and in vivo. Nat. Genet 37, 718–726. [DOI] [PubMed] [Google Scholar]
- Coombs CC, Tallman MS, and Levine RL (2016). Molecular therapy for acute myeloid leukaemia. Nat. Rev. Clin. Oncol 13, 305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damalas A, Ben-Ze’ev A, Simcha I, Shtutman M, Leal JF, Zhurinsky J, Geiger B, and Oren M (1999). Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 18, 3054–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombret H, and Gardin C (2016). An update of current treatments for adult acute myeloid leukemia. Blood 127, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I, Haffner-Krausz R, Jung S, et al. (2011). CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 470, 409–413. [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. (2005). A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov 17, 353–377. [DOI] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järås M, Miller PG, Chu LP, Puram RV, Fink EC, Schneider RK, Al-Shahrour F, Peña P, Breyfogle LJ, Hartwell KA, et al. (2014). Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med 211, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, and Martins CP (2010). Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. (2017). The target landscape of clinical kinase drugs. Science 358, eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, and Shokat KM (2005). Features of selective kinase inhibitors. Chem. Biol 12, 621–637. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Krüger M, Richter J, Xu P, García-Reyes B, Peifer C, Halekotte J, Bakulev V, and Bischof J (2014). The CK1α family: contribution to cellular stress response and its role in carcinogenesis. Front. Oncol 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Ishizawa J, and Andreeff M (2016). Pharmacological activation of wild-type p53 in the therapy of leukemia. Exp. Hematol 44, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. [DOI] [PubMed] [Google Scholar]
- Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, and Oren M (2009). The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwenberg B, Downing JR, and Burnett A (1999). Acute myeloid leukemia. N. Engl. J. Med 341, 1051–1062. [DOI] [PubMed] [Google Scholar]
- Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, Faure M, Weisburd B, Ji X, Mercier A, et al. (2015). Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. eLife 4, e06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, and Zhou Q (2018). Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, and Kim HJ (2016). Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc. Natl. Acad. Sci. USA 113, 6248–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Abdel-Wahab O, and Aifantis I (2016). Emerging concepts of epigenetic dysregulation in hematological malignancies. Nat. Immunol 17, 1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016). Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med 374, 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gonen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, Taggart J, Lu Y, Deering RP, Hacohen N, et al. (2015). Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin. Invest 125, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, and Thomä NH (2016). Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130. [DOI] [PubMed] [Google Scholar]
- Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Bur-stain I, Morgenstern Y, Brachya G, Billauer H, et al. (2013). A senescenceinflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24, 242–256. [DOI] [PubMed] [Google Scholar]
- Salami J, and Crews CM (2017). Waste disposal-an attractive strategy for cancer therapy. Science 355, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Schneider RK, Ademà V, Heckl D, Järås M, Mallo M, Lord AM, Chu LP, McConkey ME, Kramann R, Mullally A, et al. (2014). Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer D, and Grant S (2016). Update on rational targeted therapy in AML. Blood Rev. 30, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et al. (2015). Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, and Cleary ML (2006). Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 10, 257–268. [DOI] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind: differential binding analysis of ChIP-Seq peak data. R package version 100. [Google Scholar]
- Vogelstein B, Lane D, and Levine AJ (2000). Surfing the p53 network. Nature 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Levêque D, Veal G, and Astier A (2014). Review of therapeutic drug monitoring of anticancer drugs part two–targeted therapies. Eur. J. Cancer 50, 2020–2036. [DOI] [PubMed] [Google Scholar]
- Wilson CS, and Medeiros LJ (2015). Extramedullary manifestations of myeloid neoplasms. Am. J. Clin. Pathol 144, 219–239. [DOI] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborowska J, Egloff S, and Murphy S (2016). The pol II CTD: new twists in the tail. Nat. Struct. Mol. Biol 23, 771–777. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the ChIP-seq data reported in this paper is GEO: GSE117443. The accession numbers for the crystal structures of A86 in complex with CKIα, CKIδ, and CDK9/cyclinT1 reported in this paper are RCBS PDB: 6GZD, 6GZM, and 6GZH, respectively.