

SUMMARY
CD4+ Foxp3+ T regulatory (Treg) cells are key players in preventing lethal autoimmunity. Tregs undertake differentiation processes and acquire diverse functional properties. However, how Treg’s differentiation and functional specification are regulated remains incompletely understood. Here, we report that gradient expression of TCF1 and LEF1 distinguishes Tregs into three distinct subpopulations, particularly highlighting a subset of activated Treg (aTreg) cells. Treg-specific ablation of TCF1 and LEF1 renders the mice susceptible to systemic autoimmunity. TCF1 and LEF1 are dispensable for Treg’s suppressive capacity but essential for maintaining a normal aTreg pool and promoting Treg’s competitive survival. As a consequence, the development of T follicular regulatory (Tfr) cells, which are a subset of aTreg, is abolished in TCF1/LEF1-conditional knockout mice, leading to unrestrained T follicular helper (Tfh) and germinal center B cell responses. Thus, TCF1 and LEF1 act redundantly to control the maintenance and functional specification of Treg subsets to prevent autoimmunity.
Graphical Abstract
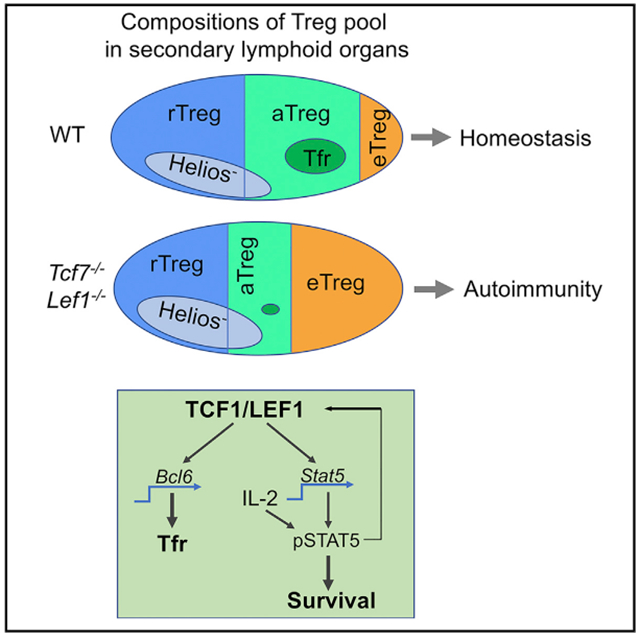
In Brief
Transcriptional regulation of Treg differentiation and function remains incompletely understood. Yang et al. report that two TCF family transcription factors regulate the survival and functional specification of a subset of Treg cells to prevent autoimmunity.
INTRODUCTION
CD4+ Foxp3+ regulatory T cells (Treg) play a pivotal role in immune tolerance and tissue homeostasis (Josefowicz et al., 2012; Sakaguchi et al., 2008). The deficit of Treg number or function causes lymphoproliferative and multi-organ autoimmune disorders (Allan et al., 2008; Salomon et al., 2000; Tang et al., 2008). Conversely, excessive Treg accumulation promotes persistent infection and cancer (Curiel et al., 2004; Enarsson et al., 2006; Liyanage et al., 2006; Mendez et al., 2004; Xu et al., 2006). Therefore, homeostatic regulation of the differentiation and function of Tregs are essential for these cells to exert their physiological roles (Campbell, 2015; Liston and Gray, 2014; Smigiel et al., 2014b). Several mechanisms have been proposed to maintain Treg homeostasis, including the balance between proliferation and apoptosis (Pierson et al., 2013), the dependence and negative feedback of Treg on paracrine interleukin-2 (IL-2) (Liston and Gray, 2014), and metabolic regulations (He et al., 2017; Yang et al., 2017).
TCF1 (encoded by Tcf7) plays a critical role in thymic T cell development and lineage commitment (Germar et al., 2011; Ioannidis et al., 2001; Steinke et al., 2014; Weber et al., 2011; Yu et al., 2012). In the periphery, TCF1 is required for self-renewal of memory CD8+ T cells and the repression of effector CD8+T cells (Gattinoni et al., 2009; Jeannet et al., 2010; Tiemessen et al., 2014; Zhao et al., 2010; Zhou and Xue, 2012). In peripheral CD4+ T cell lineage, TCF1 and LEF1 have been shown to promote the development and function of T follicular helper (Tfh) cells (Choi et al., 2015; Wu et al., 2015; Xu et al., 2015). As for Tregs, our earlier studies have found that TCF1 and LEF1 were among a list of transcription factors (TFs) that could cooperate with Foxp3 to “lock in” a Treg transcriptional program (Fu et al., 2012). However, which modules of Treg activities are specifically controlled by TCF1 or LEF1 remains unclear. Previous studies have shown that the activation of β-catenin negatively impacted thymic Treg development (Barra et al., 2015), or Treg suppressive function (van Loosdregt et al., 2013). In those studies, TCF1 was implicated to act as a downstream regulator to dampen Treg thymic generation or Foxp3 transcriptional activity. However, Ding et al. reported that β-catenin activation did not alter Treg function but instead improved their survival, so that fewer β-catenin+ Tregs were required to suppress CD45RBhi cell-induced colitis (Ding et al., 2008). Paradoxically, another study reported that sustained activation of β-catenin in Tregs contributed to the development of colitis and cancer (Keerthivasan et al., 2014). Thus, how exactly TCF1 and LEF1 impact Treg function remains to be defined.
In this study, we directly assessed the role of TCF1 and LEF1 in Treg physiology. Based on the expression of TCF1 and LEF1, we proposed a revised model of peripheral Treg differentiation, in which a subset of TCF1+ CD44hi “aTregs” was identified. Importantly, Treg-specific deletion of TCF1 and LEF1 led to systemic autoimmunity. To determine the underlying mechanisms, we used both gain- and loss-of-function approaches to modulate TCF1 and LEF1 and examined the differentiation, survival, functional specification, and suppressive capacity of Tregs. Our data revealed critical roles of TCF1 and LEF1 in regulating the competitive survival of pSTAT5+ aTreg subset and the development of Tfr to prevent autoimmune diseases.
RESULTS
Gradient Expression of TCF1 and LEF1 Distinguishes Peripheral Tregs into Distinct Subsets
We examined the expression of TCF1 and LEF1 on protein level in Tregs. We found that all CD62L+ CD44lo cells were TCF1+ (Figure 1A, middle panel, R1 gating). In contrast, CD62L− CD44hi Tregs can be divided into two distinct subsets: TCF1+ (R2) and TCF1− (R3) (Figures 1A, S1A, and S1B). The complete “tuning off” of TCF1 expression in the R3 subset was confirmed by analyzing the expression of TCF1 in Tcf7gfp/gfp mice (Tcf7 encodes TCF1) and Foxp3CreTcf7fl/fl mice (Figure S1C). Back-gating of the three subsets onto the CD44/CD62L plot showed that the expression of CD44 in R3 was slightly but significantly higher than that in R2 (Figures 1A and 1B). The compositions of these three Treg subsets were similar across different peripheral lymphoid organs at steady state (B6 mice, 7–9 weeks of age), with R1 the most, and R3 the least, abundant (Figure 1C). However, the proportions of R3 dramatically increased in non-lymphoid tissues, such as pancreas and intestinal lamina propria (Figure S1D). Aging is another factor impacting the compositions of Treg pool, characterized by significantly increased percentage of TCF1− R3 subset (Figures 1D and S1E). Different from the clear ON/OFF switch of TCF1 among these newly defined Treg subsets, the expression of LEF1 exhibited a gradient reduction and was negative (based on isotype control) in the R3 subset (Figures 1E, S1F, and S1G). Interestingly, immunostaining of spleen sections showed that TCF1+ Tregs were enriched in the T cell zone, whereas TCF1− Tregs formed clusters in the red pulp or marginal zone (Figure S1H). Thus, gradient expression of TCF1 (and LEF1 to a less extent) distinguishes peripheral Tregs into three subpopulations.
Figure 1. Gradient Expression of TCF1 and LEF1 Distinguishes Peripheral Treg into Distinct Subsets.

(A) Flow cytometric analysis of the expression of CD44, CD62L, and TCF1 in Tregs from the spleens of 6- to 8-week old B6 mice.
(B) Histograms summarize the MFIs of TCF1, CD62L, and CD44 in the R1 (blue), R2 (green), and R3 (red) subsets, as depicted in (A).
(C) The proportions of the R1, R2, and R3 Treg subsets in the spleen, pLNs, and mLNs.
(D) Flow cytometric analysis of TCF1 expression in splenic Tregs of adult (11 weeks old) and aged (55 weeks old) mice.
(E) Flow cytometric analysis of the expression of LEF1 and CD62L in cells prepared as in (A).
The data are representative of n = 3 independent experiments with n = 3–4 mice in each group. Shown are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; n.s., non-significant (two-tailed unpaired Student’s t test). wk, week; Spl, spleen; pLN, peripheral (cervical, axillary, brachial, and inguinal) lymph nodes; mLN, mesenteric lymph nodes.
See also Figure S1.
TCF1+ and TCF1− Tregs Have Distinct Transcriptomes
To further characterize the differences among these Treg subsets, we sorted each of them from pooled spleens and lymph nodes (LNs) of Tcf7gfp reporter mice (Figures S2A and S2B) and performed RNA sequencing (RNA-seq) of their transcriptomes. For data analysis, we first used the principal-component analysis (PCA) to parse out the similarities and differences among their transcriptomes. PCA showed that the replicates within the same subset were grouped together, whereas the distinctions between different subsets were clearly revealed. Interestingly, even though both the R2 and R3 subsets were CD62Llo CD44hi, the divergence of their transcriptomes between R2 and R3 was bigger than that between R2 and R1, based on the principal component 1 (which explained 94% of the variances) (Figure 2A).
Figure 2. TCF1+and TCF1−Tregs Have Distinct Transcriptomes.

(A) Principle-component analysis (PCA) of the transcriptomes of the R1, R2, and R3 Treg subsets as defined in Figure 1.
(B) Volcano plots showing the comparisons among the three Treg subsets. The T cell activation gene signature (red open circles) was superimposed onto each plot. The numbers of T cell activation signature genes that were over- or under-represented in each comparison and the p values (by χ2 test) were shown.
(C and D) Heatmaps (row normalized) depicting the expression of the selected transcription factor (C) and key functional molecules (D) in the R1, R2, and R3 Treg subsets. Genes were ranked and clustered according to their correlation to Tcf7 (C) or Icos (D). r2, correlation coefficient; min, lowest expression in each row; max, highest expression in each row.
(E and F) Flow cytometric analysis of surface molecule: ICOS, CD103, and KLRG1 (E) and transcription factors: IRF4 and Blimp1 (F) in the R1, R2, and R3 Treg subsets.
(G) Graphic summary illustrating the revised model of the Treg pool in peripheral lymphoid organs.
The data are representative of one experiment with n = 2 replicates for each Treg subsets (A–D) or n = 3 independent experiments (E and F). Shown are mean ± SEM (E and F). p values in (B) were calculated using the chi-square (χ2) test. p values in (E) and (F) were calculated using the two-tailed unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s., non-significant.
To more accurately assess the activation status in each of the three subsets, we superimposed T cell activation gene signature (Hill et al., 2007) onto our RNA-seq datasets. Using the volcano plots, we can determine to what extent a T cell activation signature was over- or under-represented. Compared to the R1 subset, both the R2 and R3 subsets exhibited a clear profile of T cell activation, depicted by the numbers of upregulated T cell activation signature genes (Figure 2B). The comparison between the R2 and R3 subsets revealed that the latter (TCF1−) gained a further elevated degree of activation.
To parse out the links between differentially expressed (DE) genes and Treg phenotype and function, we examined the expression patterns of two panels of genes across the three Treg subsets. The first panel (listed in Figure 2C) was composed of TFs that have been reported to participate in certain aspects of Treg differentiation and/or function. Using row-normalization algorithm, these 22 TFs fell into three clusters. In cluster 1 (including Tcf7, Gata1, Bach2, Ikzf1, Lef1, Myb, and Satb1), all TFs exhibited a stepwise downregulation and were noticeably under-represented in the R3 (TCF1−) subset. In contrast, the TFs in cluster 3 (including Irf4, Ikzf2, Gata3, Prdm1, and Tbx21) were significantly over-represented in the R3 subset. The rest of TFs in the list (cluster 2, including Jun, Fos, and Ikzf4) in this analysis did not show a clear pattern. However, interestingly, the expression of Bcl6 (a TF associated with Tfh and Tfr development) (Choi et al., 2015; Chung et al., 2011; Linterman et al., 2011; Wu et al., 2015; Xu et al., 2015) was relatively higher in R2 (TCF1 + CD44hi) subset, compared to R1 or R3. The expression of Ikzf2 showed a gradient increase from R1 to R3 subset (Figure S2C). We then analyzed the expression of Helios (encoded by Ikzf2) on protein level. Interestingly, we found that Helios+ Tregs comprised TCF1 + and TCF1− subsets, whereas the majority of Helios− Tregs were TCF1+ (Figure S2D). Further analysis of Helios expression in pre-gated R1, R2, and R3 showed that Helios− Tregs were predominantly enriched in the R1 pool (Figure S2E).
Thus, the over- or under-representation of these TFs occurs in a Treg subset-specific manner and the ON/OFF switch of TCF1 and LEF1, not that of CD44/CD62L, explains a greater degree of the distinctions among these Treg subsets. This notion was further supported by the expression of a list of genes encoding Treg-associated surface markers and secreted effector proteins (listed in Figure 2D). These genes fell into two clearly distinct clusters. Those Treg function-associated molecules (such as Icos, Ctla4, Lag3, Il10, and Tigit) were significantly upregulated in the R3 subset, whereas the genes in cluster 2 (including S1pr1, Ly6c, Sell, and Ccr7) exhibited an opposite trend of regulation.
We next evaluated the expression of Treg effector proteins and two key TFs on protein level using flow cytometry. About 80% of the R3 subset were ICOS+ or CD103+, whereas the expression levels of ICOS and CD103 were significantly lower in the R2 subset. Likewise, the expression of KLRG1, another surface marker associated with Treg terminal differentiation (Feuerer et al., 2010), was only detected in a fraction of R3 (Figure 2E). Likewise, the expression of IRF4 and Blimp1 showed a stepwise increase across the three subsets (Figure 2F), indicative of an increased need of these two TFs in Treg differentiation as previously reported (Cretney et al., 2011; Levine et al., 2014). Thus, based on the RNA-seq and flow cytometric analyses, we proposed a revised model of the compositions of Treg pool in peripheral lymphoid organs. In this model, three Treg subpopulations were identified and termed resting (rTreg), activated (aTreg), and effector (eTreg), reflecting their status of activation and differentiation (Figure 2G).
Treg-Specific Ablation of TCF1 and LEF1 Provokes the Spontaneous Onset of Systemic Autoimmunity
To determine whether TCF1 and LEF1 affect Treg’s function in immune tolerance, we took a loss-of-function approach to conditionally deplete Tcf7 and Lef1 in Tregs. In this regard, we crossed the mice bearing loxP-flanked Tcf7 or Lef1 alleles (Choi et al., 2015; Steinke et al., 2014; Yu et al., 2012) with Foxp3YFP-Cre mice (Rubtsov et al., 2008) (all on the B6 background) to generate Treg-specific ablation of Tcf7, or Lef1, or both (termed Tcf7KO, Lef1KO, or dKO, respectively). Of note, dKO mice began to develop immuno-pathologies from young ages (as early as 6–7 weeks old). There were several abnormalities. First, the sizes and cellularities of peripheral LNs (pLNs), not spleen, were significantly increased in dKO mice, compared to littermate controls (Figure 3A). Second, histological analysis showed that multiple organs (including thyroid and salivary glands, lung, and small and large intestines) succumbed to immune infiltrations (Figure 3B). These tissue pathologies were unlikely caused by the defects of Treg accumulation at tissue sites because the percentages and numbers of Tregs and the mean fluorescence intensities (MFIs) of Foxp3 protein were comparable, or even slightly increased, in colon lamina propria of dKO mice, compared to wild-type (WT) littermate controls (Figure S3A). Third, we detected an increased production of interferon (IFN)-γ, but not IL-17A, in CD4+ conventional T (Tconv) cells in pLNs (Figures 3C and 3D) and spleen (Figures S3B and S3C) of dKO mice. Meanwhile, the humoral immune responses were also significantly elevated in dKO mice, reflected by increased productions of immunoglobulin E (IgE) and IgG in the sera of these mice (Figure 3E). Among the IgG subclasses, the IgG2b production was particularly increased (Figure S3D). Importantly, we found that the sera of dKO mice contained higher concentrations of autoantibodies, reacting to self-antigens from multi-organs including kidney, pancreas, liver, stomach, and salivary and thyroid glands (Figure 3F), indicative of the manifestation of bona fide autoimmune diseases in dKO mice.
Figure 3. Treg-Specific Ablation of TCF1 and LEF1 Leads to Spontaneous Development of Systemic Autoimmunity.

(A) Representative images of spleen and pLNs from the mice with indicated genotypes. (right) The total numbers of live cells in the pLNs and spleen from the indicated mice.
(B) H&E staining of thyroid glands, salivary glands, lung, small intestine, and colon from Tcf7wt/wtLef1wt/wt;Foxp3Cre (WT) and Tcf7fl/flLef1fl/fl;Foxp3Cre (dKO) mice. Green arrows are pointing to the immune infiltrations.
(C) Flow cytometric analysis of the percentages and numbers of IFN-γ-producing CD4+ cells from pLNs of mice as indicated.
(D) Flow cytometric analysis of the percentages and numbers of IL-17A-producing CD4+ cells from the pLNs of mice as indicated.
(E) ELISA of the IgE (left) and IgG (right) production in the serum of mice with indicated genotypes.
(F) Western blot of homogenized tissue digestions (kidney, pancreas, liver, stomach, thyroid glands, and salivary glands) from female Rag1−/− mice with the sera from WT or dKO mice.
The data are representative of n = 3 independent experiments with n = 3–4 mice in each group. Shown are mean ± SEM. *p < 0.05, **p < 0.01, n.s., non-significant (two-tailed unpaired Student’s t test). Scale bar, 200 μm.
See also Figure S3.
Thus, Treg-specific ablation of Tcf7 and Lef1 renders the mice susceptible for early onset of systemic autoimmunity, caused by unrestrained cellular and humoral immune responses.
The Deficiency of Tcf7 and Lef1 Does Not Impair Treg’s Suppressive Capacity
Given the aberrant autoimmune phenotype in dKO mice, we asked whether Treg’s suppressive capacity was affected by conditional knockouts of Tcf7 and Lef1. We used an established approach of in vitro coculture of dendritic cells (DCs), responder T (Tresp), and Tregs (Onishi et al., 2008; Wing et al., 2008). As illustrated in Figure 4A, DCs and Tresp cells were sorted and co-cultured with Tregs isolated from either WT or dKO mice (Figure S4), in the presence of anti-CD3 mAb. On day 4, co-cultured cells were collected and analyzed for Tresp proliferation and DC maturation. Both WT and dKO Tregs suppressed Tresp proliferation. Of note, dKO Tregs were slightly more suppressive than WT Tregs when the ratios between Treg and Tresp were relatively higher (1:2 and 1:1) (Figure 4B).
Figure 4. TCF1 and LEF1 Are Dispensable for Treg’s Suppressive Capacity.

(A) Schematic diagram of experimental settings.
(B) Representative histograms of cell trace violet (CTV) dilution in indicated groups (left) and statistical data of Treg-mediated suppression on Tresp cells (right).
(C) Representative histograms of CD80 expression in DCs in indicated groups (top) and statistical data of Treg-mediated suppression on CD80 expression at indicated ratios (bottom).
(D) Representative histograms of CD86 expression in DCs in indicated groups (top) and statistical data of Treg-mediated suppression on CD86 expression at indicated ratios (bottom).
(E) Representative histograms of CTLA4 expression in DCs in WT (left) and dKO Tregs and statistical data (right).
The data are representative of n = 2 independent experiments with pooled n = 3 mice in each group. Shown are mean ± SEM. *p < 0.05; **p < 0.01; n.s., non-significant (two-tailed unpaired Student’s t test).
See also Figure S4.
Previous studies have reported that the downregulation of CD80 and CD86 on DCs is one key mechanism by which Tregs suppress effector T cells (Onishi et al., 2008; Wing et al., 2008). We examined the expression of CD80 and CD86 on DCs after the coculture. In line with their suppressive capacity on Tresp cells, both WT and dKO Tregs downregulated the expression of CD80 and CD86 on DCs. Again, dKO Tregs were slightly more efficient to downregulate CD80 and CD86 (Figures 4C and 4D).
CTLA4 is a critical functional molecule for Treg’s suppressive function via inhibiting DC maturation (Wing et al., 2008). We found that the expression of CTLA4 was comparable between dKO and WT Tregs (Figure 4E).
Together, these results suggest that the ablation of Tcf7 and Lef1 does not directly impair Treg’s suppressive capacity.
TCF1 and LEF1 Preferentially Maintain the aTreg Pool
We next asked whether TCF1 and LEF1 regulated Treg differentiation. We found that thymic Treg development was not affected by Foxp3Cre-mediated depletion of Tcf7, Lef1, or both (Figure S5A). In the periphery, the deficiency of Tcf7 and Lef1 did not alter the total numbers of bulk Tregs in secondary lymphoid organs, although the percentages of Tregs in mesenteric LNs (mLNs) and Peyer’s patch (PP) of dKO mice were slightly reduced (Figure S5B). The expression level of Foxp3 protein was not affected (or even slightly increased in spleen and mLNs) in dKO mice (Figure S5C).
Because the overt systemic inflammation in dKO mice may influence Treg phenotype and function, we attempted to examine the impacts of TCF1 and LEF1 on Tregs under non-inflammatory conditions. To do this, we used heterozygous female Foxp3YFP-Cre/wt mice. We sorted YFP+ dKO and WT Tregs and did RNA-seq analysis. Overall, the transcriptional profiles of dKO and WT Tregs were tightly linear correlated (Figure 5A). To parse out specific pathways that were influenced by TCF1 and LEF1, we did gene set enrichment analysis (GSEA) with a focus on the “hallmark gene sets” from the MSigDB (Liberzon et al., 2015; Subramanian et al., 2005). Interestingly, the top enriched pathways in dKO compared to WT Tregs were all linked to active cell cycle (E2F targets, G2M checkpoint, and Myc targets) (Figure 5B). Indeed, dKO Tregs exhibited an over-representation of T cell activation gene signature (Figure 5C) and increased rate of proliferation (Figure 5D), compared to WT Tregs. At the same time, dKO Tregs also showed increased expression of pro-apoptotic protein Bim but not the anti-apoptotic Bcl2 (Figure S5D). On the other hand, as expected, Wnt-β-catenin signaling was more skewed toward WT Tregs. Interestingly, IL-2/STAT5 signaling was also significantly under-represented in dKO Tregs (Figure 5B).
Figure 5. The Deficiency of TCF1 and LEF1 Leads to a Severe Reduction of aTregs.

(A) Expression-expression plot of transcriptomes of WT Tregs isolated from Foxp3YFP-Cre/wtTcf7wt/wtLef1wt/wt mice and dKO Tregs from Foxp3YFP-Cre/wtTcf7fl/flLef1fl/fl mice. Superimposed is the bulk Treg gene signature. Numbers depict the Treg signature genes over- or under-represented in dKO versus WT samples.
(B) GSEA analysis of the “hallmark” gene sets deposited in MSigDB. Both p values and FDR q values were shown. NES, normalized enrichment score.
(C) Volcano plots showing the comparisons between WT and dKO Tregs. The T cell activation gene signature (red) was superimposed. The numbers of T cell activation signature genes that were over- or under-represented in each comparison and the p value (by χ2 test) were shown.
(D) The proliferation of WT and dKO Tregs in Spleen, pLNs, and mLNs measured by Ki67 staining.
(E) Row-normalized heatmap of the expression of bulk Treg signature genes across r-, a-, and e-Treg subtypes (left). Superimposing each sub-cluster defined on the left to the FC plot comparing dKO versus WT Tregs (right).
(F) The percentages of Treg subsets in dKO and WT littermate controls using the gating strategy defined in Figure S5E.
(G) Representative fluorescence-activated cell sorting (FACS) plots showing the gating strategy and the compositions of Treg pool based on the expression of Helios, ICOS, and CD62L in WT and dKO mice (top). The percentages and numbers of each Treg subsets in Helios+ Treg compartment (bottom).
(H) The effect of retroviral transduction of Tcf7 on Treg differentiation in vitro. Experimental settings (top) and statistical data of Treg subsets in control (pMIG) and Tcf7-transduction groups (bottom).
The data are representative of one experiment with n = 2 replicates for each Treg subsets (A–C and E) or n = 2 (F–H), n = 3 (D) independent experiments. n = 3 mice in each group. Shown are mean ± SEM. p values in (C) were calculated using the chi-square (χ2) test. p values in (D) and (F)–(H) were calculated using the two-tailed unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s., non-significant.
See also Figure S5.
We next asked to what extent Treg gene signature was affected by the Tcf7/Lef1 depletion. In this regard, we superimposed Treg signature genes (Fu et al., 2012) onto the expression-expression plot shown in Figure 5A. Two-thirds of Treg signature genes were under-represented, while the remaining one-third was over-represented, in dKO Tregs, suggesting that different components of Treg transcriptional program were differentially influenced by TCF1 and LEF1. Based on their differential expression in our defined three subsets, the bulk Treg gene signature shown in Figure 5A can be further divided into three sub-clusters, reflecting their preferential expression in r-, a-, or e-Tregs (Figure 5E, left panel). We then examined how the expression of the genes in each sub-cluster was altered by Tcf7/Lef1 depletion (Figure 5E, right). Strikingly, almost all “aTreg-favorable” genes were under-represented in dKO samples, compared to WT controls, suggesting that TCF1 and LEF1 may have a preferential effect on aTreg differentiation.
To assess this possibility, we examined the relative abundance of each Treg subset in dKO mice. Because we could not use the TCF1/CD62L plot to distinguish these Treg subsets due to the lack of TCF1 expression in dKO Tregs, we found that ICOS can be used as a surrogate, in combination with CD62L, to gate on each subset (with >80% overlap with the TCF1/CD62L plot) (illustrated in Figure S5E). Indeed, we found that the fraction of aTreg was significantly decreased (~2-fold), with a simultaneously increased eTreg proportion, in dKO mice (Figure 5F). However, unexpectedly, the percentage of rTregs was also increased. Because the majority of Helios− Tregs (likely converted from Tconv cells in the periphery [Thornton et al., 2010]) are in the rTreg pool (Figure S2E), in repeated experiments, we first separated Tregs into Helios+ and Helios−. When we gated on Helios+ fractions (Figure 5G), again the deficiency of Tcf7 and Lef1 resulted in a severely diminished aTreg pool. Now the size of Helios+ rTreg pool was also reduced, though to a less extent. Again, we found increased percentages and numbers of eTregs in dKO mice (Figure 5G). In contrast, the numbers and compositions of Helios− Tregs were not affected by Tcf7 and Lef1 depletion (Figure S5F). To further determine the effect of TCF1 and LEF1 on the compositions of Treg pool, we took a gain-of-function approach by retrovirally transducing Tcf7 (encoding TCF1) into Tregs and found that compared to control transduction, ectopic expression of Tcf7 significantly increased aTreg proportion (Figure 5H).
In summary, TCF1 and LEF1 have a modest effect on bulk Treg homeostasis but are essential for maintaining a normal aTreg pool.
TCF1 and LEF1 Promote Treg’s Competitive Fitness by Regulating STAT5 Expression
We found that the percentage of CD25+ was significantly higher in TCF1+ than TCF1− Tregs (Figure 6A). In line with this, TCF1+ Tregs exhibited more potent responses to IL-2 in an in vitro STAT5 phosphorylation assay (Figure 6B). In addition, the expression of both Stat5a and Stat5b was also relatively higher in TCF1+ Tregs, compared to TCF1− counterparts (Figure S6A). Together, these findings suggest a positive correlation between TCF1 and STAT5 in Tregs.
Figure 6. TCF1 and LEF1 Are Required for Treg’s Competitive Fitness by Regulating STAT5 Expression and Activation.

(A) The percentages of CD25 expression in TCF1+ and TCF1− Tregs.
(B) In vitro pSTAT5 assays in TCF1 + and TCF1− Tregs, stimulated with IL-2 at various concentrations.
(C) GSEA analysis revealing that IL-2/STAT5 signaling was significantly under-represented in dKO Tregs. NES, normalized enrichment score.
(D) In vitro pSTAT5 assays in dKO and WT Tregs, stimulated with IL-2 at various concentrations. (left) The percentages of pSTAT5; (right) the MFI of pSTAT5 (gated on pSTAT5+).
(E) Ex vivo analysis of total STAT5 protein and pSTAT5 in dKO and WT Tregs. Representative FACS plots (top). Percentages of total STAT5 protein and pSTAT5, and the ratios of pSTAT5/total STAT5, respectively (bottom).
(F) ChIP-PCR analysis showing enriched binding of TCF1 to Stat5b promoter.
(G) In vivo Treg survival assays. Splenic CD4+ T cells were isolated from WT (Foxp3Cre) or dKO mice, i.p. transferred into Tcrα−/− mice and analyzed 5 weeks later. FACS plots and statistical data show the percentages of Tregs reconstituted from transferred donor cells.
(H) In vivo Treg survival assays under competitive conditions. Splenic CD4+ T cells were isolated from CD45.1 WT (Foxp3Cre), CD45.2 WT (Foxp3Cre), or CD45.2 dKO mice, mixed at a ratio of 3:1 between CD45.1 versus CD45.2 donor cells, i.p. transferred into Tcrα−/− mice and analyzed 5 weeks later. FACS plots and statistical data show the percentages of Tregs reconstituted from each type of donor cells in spleen, pLNs, and mLNs.
(I) Schematic diagram showing that splenic CD4+ T cells from WT or dKO mice were MACS enriched and transduced with retroviruses expressing Tcf7, or caSTAT5, i.p. transferred into Tcrα−/− mice and analyzed 3 weeks later (top). Representative FACS plots and statistical data showing the reconstitution of Tregs from different inputs as described in the inset legend (middle). The MFIs of TCF1 and LEF1 in Tregs from the conditions as described in the inset legend (bottom). The data are representative of n = 3 (A, B, D, and E) and n = 2 (F, G, and I) independent experiments and one experiment with n = 2 replicates for each Treg subsets (C) or one experiment with n = 3 mice in each group (H). n = 4–6 mice in each group. Shown are mean ± SEM. The p values were calculated using two-tailed unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. RV, retrovirus; Ctl, control; wk, week.
See also Figure S6.
Next, we asked whether the modulation of TCF1 and LEF1 in Tregs would affect their responsiveness to IL-2 and STAT5 activation. GSEA analysis showed that IL-2/STAT5 signaling was significantly under-represented in dKO Tregs (Figure 6C). Consistent with this, IL-2-stimulated STAT5 phosphorylation was less efficient in dKO Tregs (Figure 6D, left). Importantly, the intensity of pSTAT5 in dKO, not WT Tregs, fell to the same level as that in activated CD4+ non-Tregs (Figures 6D, right, and S6B), suggesting that compared to WT Tregs, dKO Tregs lost the competitive advantage for IL-2. To understand what caused the defective IL-2/STAT5 signaling in dKO Tregs, we first examined CD25 expression and found no difference between dKO and WT Tregs (Figure S6C). Next, in an ex vivo assay, we found that about 30% of WT Tregs were readily positive for total STAT5 protein (Figure 6E), and the same STAT5hi population was also positive for pSTAT5. Ex vivo TCF1+ Tregs exhibited a higher degree of STAT5 expression and activation than that in TCF1− Tregs (Figure S6D). In dKO Tregs, both total STAT5 and pSTAT5 were significantly reduced (Figure 6E). Thus, the impaired responsiveness to IL-2 in dKO Tregs was more likely due to the reduced expression of total STAT5 protein and reduced pSTAT5 as a consequence. Using chromatin immunoprecipitation (ChIP)-PCR, we found that TCF1 can also bind to Stat5b promoter in Tregs (Figure 6F). Thus, in Tregs, TCF1 acts upstream of STAT5 to regulate its expression.
We next assessed whether this compromised responses to IL-2 in dKO Tregs would affect their survival in vivo. In this regard, we isolated splenic CD4+ T cells from WT or dKO mice, transferred them into Tcrα −/− mice, and analyzed the reconstitution of donor cells 5 weeks later. Treg compartment was reconstituted in secondary lymphoid organs in recipient mice that have been transferred with WT CD4+ T cells. However, Treg reconstitution was significantly diminished in recipient mice received CD4+ T cells derived from the dKO donors (Figure 6G), indicative of an impaired survival of dKO Tregs in vivo under lymphopenic conditions. To more directly assess the competitive fitness between WT and dKO Tregs, we did co-transfer experiments by mixing splenic CD4+ T cells from WT (CD45.1+) and dKO (CD45.2+) mice at a ratio of 3:1 before transferring them into Tcrα −/− mice. In parallel, as controls, a mixture of CD45.1 and CD45.2 splenic CD4+ T cells (both were WT cells from Foxp3Cre mice) at a ratio of 3:1 was transferred into the Tcrα −/− mice. After 5 weeks, we found that WT Tregs were comparably reconstituted from both CD45.1 and CD45.2 donors. By contrast, the reconstitution of dKO Tregs was significantly diminished (Figure 6H). Consistently, the degree of STAT5 activation in residual dKO Treg was significantly lower, compared to that in co-transferred WT Tregs (Figure 6H). Together, these data showed that Tcf7/Lef1-deficient Tregs failed to compete with WT Tregs, or activated Tconv cells, for survival.
The MFIs of Foxp3 on a per cell basis were comparable in re-constituted Tregs from either dKO or WT mice (Figure S6E). The production of inflammatory cytokines (such as IFN-γ and IL-17A) was slightly (not significantly) increased in Tregs deficient for Tcf7, Lef1, or both (Figure S6F). It has been reported that Tregs lose Foxp3 expression when transferred into lymphopenic recipients (Duarte et al., 2009; Gavin et al., 2007). We asked whether dKO Tregs exhibited a higher degree of instability under such conditions. In this regard, we transferred highly purified congenically labeled WT and dKO Tregs into Tcrα−/− mice and found that the percentages of Foxp3− “ex-Treg” cells were comparable between WT and dKO donors, suggesting that the depletion of Tcf7/Lef1 did not exacerbate Treg’s instability under lymphopenic conditions (Figure S6G).
Next, we assessed whether the defect of dKO Treg survival can be corrected by ectopic expression of Tcf7 or constitutively activated STAT5 (caSTAT5). In this regard, we isolated CD4+ T cells from pooled spleen and pLNs of dKO mice, transduced these cells with Tcf7, caSTAT5 (Johnston et al., 2012; Onishi et al., 1998), or control retroviruses and transferred them into Tcrα−/− mice and analyzed them 3 or 5 weeks later. We found that ectopic expression of caSTAT5 was able to rescue the survival of dKO Tregs in lymphopenic hosts (Figure S6H). Similarly, transduction of dKO Tregs with Tcf7-expressing retroviruses also significantly improved the in vivo survival of these cells (Figure S6H). Next, we did competition assays by mixing congenically labeled splenic CD4+ T cells from dKO and WT mice at a 1:1 ratio and transduced them with either caSTAT5, Tcf7, or control retroviruses and then transferred them into Tcrα −/− mice. After 3 weeks, we found that again the transduction of caSTAT5 into dKO Tregs significantly improved the reconstitution of these cells. Importantly, caSTAT5 had a more profound effect on dKO Tregs (31-fold increase), compared to WT Tregs (1.9-fold increase) (Figure 6I), further supporting the notion that STAT5 acts downstream of TCF1 to promote Treg’s competitive survival. Interestingly, under these competition conditions, transduction of Tcf7 into dKO Tregs was less efficient in rescuing their survival, compared to that in WT Tregs (Figures 6I and S6I, filled versus open yellow dots) or non-competitive settings (Figure S6H). This was due to the different amounts of TCF1 protein after retroviral transduction, because, while the expression of TCF1 in dKO Tregs was markedly increased after Tcf7-RV transduction, it did not reach the level of that in Tcf7-RV-transduced WT Tregs (Figure 6I, bottom left). More interestingly, we found that caSTAT5 transduction significantly enhanced the expression of LEF1 (not TCF1) (Figure 6I), indicative of a feedforward loop of TCF1-STAT5-LEF1 in promoting Treg competitive fitness. Last, using the gating strategy illustrated in Figure S5E, we found that aTreg subset was preferentially reconstituted by retroviral transduction of either Tcf7 or caSTAT5 (Figure S6J).
In summary, TCF1 regulates the expression of STAT5 in Tregs. Subsequently activated STAT5 promotes the expression of LEF1, thus forming a positive feedback loop in TCF1+ LEF1+ Tregs to promote their competitive fitness.
Tfr Cells Are a Subset of TCF1+ aTregs
Our RNA-seq analysis of fractionated Treg subsets revealed that the expression of Bcl6 and Cxcr5 was relatively higher in aTregs than the other two subsets (Figure 7A), suggesting a possible link between TCF1 and Tfr development. In line with this, the expression of Bcl6 gene signature (a set of genes targeted by Bcl6 in Tregs [Liu et al., 2016]) was over-represented in aTreg, not eTregs (Figure 7B), suggesting that Tfr differentiation is more permissive at the TCF1+ aTreg stage. To further evaluate this hypothesis, we did k-means clustering and gene ontology (GO) analyses of the transcriptomes of the three Treg subsets, aiming to parse out Treg differentiation stage-associated over- or under-represented genes (Figure S7A) and their functional annotations (Table S5). Interestingly, “regulation of B cell-mediated immunity” was among the top enriched GO terms in the genes over-represented in TCF1+ aTregs (cluster 5) (Figure 7C).
Figure 7. TCF1 and LEF1 Redundantly Control Tfr Generation and Germinal Center B Cell Responses.

(A) The expression of Bcl6 (left) and Cxcr5 (right) detected by RNA-seq in the three Tregs.
(B) GSEA analysis revealing that Bcl6 gene signature was enriched in TCF1+ aTregs not TCF1− eTregs. NES, normalized enrichment score.
(C) Gene ontology (GO) annotations of the over-represented genes in TCF1+ aTregs identified by k-means clustering analysis (see Figure S7A for detail).
(D) Flow cytometric analysis of Bcl6 and CXCR5 in the three Treg subsets.
(E) Superimposing Tfr cells onto the bulk Treg plot (TCF1 versus CD62L). Inset numbers show the percentages of Tfr cells fell into each quadrant.
(F) Flow cytometric analysis of Foxp3 expression in CD4+ Bcl6+ CXCR5+ T cells from spleen of indicated mice (left). The percentages and numbers of Tfr and Tfh cells (right).
(G) Flow cytometric analysis of the expression of Bcl6 and FAS expression in CD4− B220+ cells in the spleen of indicated mice.
(H) Immunofluorescence staining of splenic frozen sections from Tcf7wt/wtLef1wt/wt; Foxp3Cre (WT) and Tcf7fl/flLef1fl/fl;Foxp3Cre (dKO) mice. Statistical data show the intensities of GL7 staining in each individual B220+ follicles normalized by the area of B cell follicles.
(I) Immunofluorescence staining of samples as in (H) with Foxp3, GL7, and CD4. Arrows in the top panel (WT) depicting Tfr cells. The numbers of Foxp3+ T cells within each individual GCs was counted and then normalized by the size of the GC area.
(J) ChIP-PCR analysis showing enriched binding of TCF1 to Bcl6 (upstream −500) in WT Tregs. Splenic CD4+ T cells from Tcf7fl/flCD4Cre mice were used as negative control to show the background of TCF1 binding.
The data are representative of n = 2 replicates for each Treg subsets (A–C); n = 3 independent experiments (D, E, and J) or n = 2 experiments with n = 3–4 mice in each group (F–I). The statistic calculation in (B) was false discovery rate (FDR) q value. Shown are mean ± SEM. The p values in (A, D, and F–I) were calculated using the two-tailed unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bars, 100 μm (H) and 50 μm (I).
Using Bcl6 and CXCR5 to gate Tfr cells within the Treg pool (Sage et al., 2013), we analyzed the abundance of Tfr in each fractioned Treg sub-compartment from peripheral lymphoid organs at steady state. Notably, Tfr cells can only be detected in the TCF1+ Treg pool (Figure 7D). About 6% of the TCF1+ aTregs exhibited a Tfr phenotype. In other words, ~95% of the Tfr cells were found within the TCF1+ aTreg pool, and none of the TCF1− eTregs expressed phenotypic markers of Tfr cells (Figure 7E).
Thus, Tfr cells are a subset of TCF1+ aTregs. These data also suggest that the development of Tfr cells occurs at a distinct stage of Treg differentiation, prior to “turning off” TCF1 expression.
TCF1 and LEF1 Are Indispensable for Tfr Generation
Next, we asked whether TCF1 and LEF1 played a role in Tfr differentiation. RNA-seq analysis showed that the expression of both Bcl6 and Cxcr5 was reduced in dKO versus WT Tregs (Figure S7B). Using flow cytometry, we analyzed the abundance of Tfr cells in mice bearing Treg-specific ablation of Tcf7, Lef1, or both. Using a gating strategy to show both Tfr and Tfh in the same plot (illustrated in Figure S7C), we found that Tfr cells were almost completely abolished in peripheral lymphoid organs of dKO mice (Figures 7F, S7D, and S7E). The percentages of Tfr cells within the pool of Bcl6hi CXCR5hi T cells were reduced about 10-fold. Likewise, the total numbers of Tfr cells were also significantly reduced in dKO mice. On the other hand, the total numbers of Tfh cells were significantly increased (Figure 7F), suggesting that Treg-specific ablation of Tcf7 and Lef1 altered the balance between Tfr and Tfh. Either Tcf7 or Lef1 single knockout exerted partial effects on the reduction of Tfr cells, supporting a redundancy between these two TFs.
We next asked whether the imbalanced Tfr-Tfh ratio in dKO mice led to altered germinal center (GC) B cell responses. Indeed, in dKO mice even at a relatively young age (7 weeks old), we already observed significantly increased GC B cells (defined as Bcl6+ FAS+), reflected by both their percentages within the B220+TCRβ− pool and the total numbers (Figures 7G, S7F, and S7G). To further verify the link between Treg-specific Tcf7/Lef1 ablation and GC B cell responses, we performed immunostainings of splenic frozen sections of 7-week-old dKO and littermate control mice. At this age, the GCs in the spleen of control mice were small but detectable. In contrast, in dKO mice we found significantly increased GC areas (Figures 7H and S7H). As expected, Tfr cells can be readily detected in most GCs in control mice (due to the small sizes of GCs at a young age in control mice, the number of Tfr cells per GC was relatively low). In contrast, the abundance of Tfr cells in each GC of dKO mice was significantly reduced, or not detected (Figure 7I), which is consistent with flow cytometric assays showing a dearth of Tfr cells in the dKO mice. Using ChIP-PCR, we found that TCF1 bound to the promotor of Bcl6 in WT Tregs, not Tcf7-deficicent T cells (Figure 7J), consistent with the results from a recent study (Xu et al., 2017). Thus, TCF1 and LEF1 act redundantly to control Tfr development by regulating Bcl6 expression.
DISCUSSION
The phenotypic and functional heterogeneity of Tregs has been increasingly reported (Liston and Gray, 2014; Panduro et al., 2016). Different regulatory mechanisms, mediated by specific TFs, TCR repertoires, or cytokines, are required for the differentiation, maintenance, and functional specification of each Treg subset (Campbell, 2015; Cretney et al., 2013). In this study, we report regulatory modules in Tregs that are essential for these cells to maintain immune tolerance. These modules are controlled by TCF1 and LEF1, two TCF family TFs. First of all, TCF1 and LEF1 are required for homeostatic maintenance of CD44hi TCF1+ aTreg, a newly defined subset. In the absence of TCF1 and LEF1, the expression and subsequent activation of STAT5 is diminished, causing Tregs unable to compete with activated Tconvs for IL-2. In parallel, the lack of TCF1 and LEF1 also abolishes Tfr generation, leading to unrestrained Tfh and GC B responses, which contributes to the onset of systemic autoimmunity.
The pool of Tregs in peripheral lymphoid organs is composed of CD62L+CD44lo and CD62L−CD44hi populations, reflecting their state of activation and effector maturation (Liston and Gray, 2014; Smigiel et al., 2014b). Our characterizations of TCF1 and LEF1 expression in Tregs revealed that CD62L−CD44hi cells can be further divided into two subsets (termed aTreg and eTreg, respectively). aTregs express no or low level of effector Treg-associated TFs (such as IRF4, Blimp1, T-bet, or Gata3) or functional molecules (i.e., ICOS, CTLA4, or IL-10). However, aTregs are not merely an immature or transient subset, because these cells preferentially express both Bcl6 and CXCR5, key genes for Tfr differentiation. This is confirmed by our further analyses including GSEA of DE genes and flow cytometry of fractionated Treg subsets. More interestingly, we find that Tfr cells are almost exclusively derived from the aTreg pool. This is important because it shows that the differentiation of Tfr, a specialized functional subset, occurs at a distinct (TCF1+) stage of Treg differentiation, compared to T-bet+, STAT3+, or other eTreg subsets (which are TCF1−). TCF1 acts upstream of Bcl6 in Tfr generation, consistent with a previous report (Xu et al., 2017). Furthermore, our data show that LEF1 also participates in the Tfr generation because Foxp3-Cre-mediated double knockouts of both Tcf7 and Lef1 led to almost completely abolished Tfr generation, an effect more profoundly than that by Tcf7 or Lef1 single knockout. TCF1 and LEF1 have been reported to promote early differentiation and effector function of Tfh cells, via promoting Bcl6 expression (Choi et al., 2015; Wu et al., 2015; Xu et al., 2015). Therefore, Tregs acquire a similar TF circuit to differentiate into Tfr, as that by activated Tconvs to undertake Tfh differentiation.
At steady state, TCF1− LEF1− eTregs comprise about 10% of total Treg pool in secondary lymphoid organs. In contrast, in non-lymphoid tissues, the majority of Tregs are TCF1− LEF1−. Moreover, tissue Treg gene signatures are largely recapitulated in TCF1− LEF1−, but not TCF1+ LEF1+ Tregs in lymphoid organs (B.-H.Y. and W.F., unpublished data). We thus propose that the small fraction of TCF1− LEF1− Tregs found in lymphoid organs (such as spleen and LNs) could be the precursors of those Tregs accumulated at non-lymphoid tissues. Further studies are needed to determine whether the primary function of these TCF1−LEF1− Tregs is to populate tissue Treg pools and regulate tissue homeostasis through participating in immunological or non-immunological processes.
Tcf7/Lef1 dKO mice spontaneously develop systemic autoimmunity. However, we found that the total numbers and percentages of bulk Tregs in secondary lymphoid organs were not affected by the ablation of Tcf7 and Lef1. In addition, Tregs can be detected in non-lymphoid tissues of dKO mice with comparable abundance to that in WT mice. Furthermore, dKO Tregs were equally (or even slightly more) suppressive for the proliferation of effector T cells and DC maturation, indicating that TCF1 and LEF1 are dispensable for Treg suppressive capacity. Then, what causes the onset of autoimmunity in dKO mice? We propose that the deficit of aTreg pool and the lack of Tfr cells in these mice are two plausible causes. We show that TCF1 and LEF1 are essential for maintaining a normal aTreg pool via regulating the expression and subsequent activation of STAT5. TCF1 directly binds to State promotor and regulates its expression. In line with this, TCF1+ Tregs express higher levels of Stat5a and Stat5b and more potently respond to IL-2. Conversely, the deficiency of Tcf7 and Lef1 remarkably reduces STAT5 expression in Tregs. The expression level of total STAT5 protein in Tregs is correlated with its activation (pSTAT5). However, the ratio of pSTAT5/STAT5 was not affected by Tcf7/Lef1 depletion, suggesting that TCF1 primarily influences the expression, not the activation of STAT5. Interestingly, a constitutively activated form of STAT5 promotes the expression of LEF1 in Tregs but not Tconv cells. Thus, our data support a positive feedback loop of TCF1-STAT5-LEF1 in Treg homeostatic maintenance. Of note, Liu et al. reported that pSTAT5+ Tregs formed clusters with self-antigen activated IL-2-producing T cells and were essential for suppressing incipient autoimmunity (Liu et al., 2015). We predict that the population of pSTAT5+ cells described by Liu et al. is overlapped with TCF1+ aTregs. Indeed, we found that TCF1+ Tregs were enriched in the T cell zones, which is also in line with another study showing that central (resting) Tregs gain more access to paracrine IL-2 than effector Tregs (Smigiel et al., 2014a). Together, our data reinforce the role of pSTAT5+ Tregs in keeping autoimmunity in check and highlight the importance of TCF1 and LEF1 as two upstream regulators.
It has been reported that thymic Tregs are derived from CD25+ Foxp3− precursors in a STAT5-dependent manner (Burchill et al., 2008; Lio and Hsieh, 2008). However, we found that thymic Treg profile was comparable between dKO and WT mice, suggesting that the deficiency of Tcf7 and Lef1 does not alter Treg development in the thymus. Interestingly, during the revision of this manuscript, Owen et al. reported that a second developmental program involving CD25− Foxp3lo Treg precursors also contributed to the generation of thymic Tregs with high efficiency (Owen et al., 2019). Indeed, we found that the expression of TCF1 in CD25− Foxp3lo Treg precursors was lower than that in CD25+ Foxp3− precursors, suggesting that CD25− Foxp3lo precursor-mediated thymic Treg development is less dependent on TCF1 (Figure S5A, right). We propose that CD25− Foxp3lo precursors may compensate for the defect of thymic Tregs caused by Tcf7/Lef1 deficiency. In addition, it would be also interesting to determine whether CD25− Foxp3lo thymic Treg precursors can directly populate the eTreg pool in peripheral organs. Though continuous thymic output and increased proliferation rate may partially rescue bulk Treg homeostasis in dKO mice in the periphery, the aTreg pool cannot be restored. A more deleterious consequence of aTreg defect is the lack of Tfr generation, causing unrestrained Tfh and GC B responses. Of note, while the size of eTreg pool is significantly increased and their suppressive capacity remains intact in dKO mice, the early onset of autoimmune diseases in these mice suggests that eTregs alone are not sufficient to keep autoimmunity fully in check. In other words, our data support the importance of the “wholeness” of Treg’s diverse subsets and functions to prevent the onset of autoimmune diseases.
We show that Tfr cells are almost exclusively derived from the TCF1+ aTreg pool, which relies on IL-2/STAT5 signaling for their maintenance. It has been reported that Tfr cells downregulate CD25 (Botta et al., 2017; Ritvo et al., 2017). Further studies by Wing et al. revealed that Tfr cells can be divided into CD25+ and CD25− subsets (Wing et al., 2017), and the authors proposed a “two-step” model for Tfr development (Wing et al., 2018). Thus, our data suggest that the axis of TCF1-STAT5 plays a more critical role in the initiation, not the maturation, of Tfr lineage. TCF1 and LEF1 promote the development and function of Tfh cells (Choi et al., 2015; Wu et al., 2015; Xu et al., 2015).
Our findings are in line with a previous study showing that β-catenin activation did not alter Treg function but instead improved their survival (Ding et al., 2008). Conversely, the activation of β-catenin has also been reported to negatively impact thymic Treg development (Barra et al., 2015) or Treg suppressive function (van Loosdregt et al., 2013). In those studies, TCF1 was implicated as a downstream regulator. During the revision of this manuscript, another study was published showing that Treg-specific β-catenin stabilization led to lethal autoimmunity in mice with dysfunctional Treg phenotype (Sumida et al., 2018). Interestingly, they found that β-catenin stabilization was associated with human IFN-γ-secreting Tregs, which also expressed a relatively higher level of TCF7. While these discrepancies could be due to the differences of experimental settings or methods used to examine the functions of Tregs, it is also possible that excessive activation of β-catenin and downstream TCF1 disturbs Treg homeostasis, likely causing abnormalities within the eTreg compartment, as our data suggest that the downregulation of TCF1 and LEF1 is a prerequisite for eTreg differentiation. Indeed, we find that the retroviral transduction of Tcf7 in dKO Treg can reconstitute the aTreg pool but fails to rescue eTregs, owing to the constitutive expression of TCF1 in transduced cells.
Genome-wide association studies (GWASs) have found that TCF7 polymorphisms are associated with the risk of human autoimmune diseases, T1D (Cooper et al., 2007; Erlich et al., 2009; Julier et al., 2009), and systemic lupus erythematosus (SLE) (Odhams et al., 2017; Sun et al., 2016). Altered Treg homeostasis and function caused by mutant TCF7 or LEF1 may contribute to the development of these autoimmune disorders. As demonstrated in animal models in this study, compromised Treg competitive fitness and defects of aTreg maintenance and associated Tfr generation could be the underlying mechanisms.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wenxian Fu (w3fu@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J (B6), Foxp3YFP-Cre (Rubtsov et al., 2008), B6/Rag1−/−, B6/Tcrα−/− and B6/CD45.1 mice were purchased from the Jackson Laboratory. Foxp3Thy1.1 (Liston et al., 2008), Tcf7fl/fl and Lef1fl/fl (Steinke et al., 2014; Yu et al., 2012) and Tcf7GFP(Choi et al., 2015; Yang et al., 2015) mice have been described. Tcf7fl/fl and Lef1fl/fl mice were crossed with Foxp3YFP-Cre to generate Foxp3CreTcf7fl/fl (termed Tcf7KO), Foxp3CreLef1fl/fl (Lef1KO), Foxp3CreTcf7fl/flLef1fl/fl (dKO), Foxp3Cre/wtTcf7fl/flLef1fl/fl, and littermate control mice, respectively. Tcf7GFP mice were crossed with Foxp3Thy1.1 to generate Foxp3Thy1.1Tcf7GFP/wt.
Both female and male mice were used. There was no sex-bias in the profile of peripheral Tregs. However, in one set of experiments, we use female heterozygous mice (i.e., Foxp3Cre/wtTcf.7fl/fl Lef1fl/fl and Foxp3Cre/wtTcf7wt/wt Lef1wt/wt) to examine Treg phenotype under non-inflammatory conditions. 6-9-week-old mice were used unless specified in the text or figure legends.
Study Approval
All mice were housed under the specific pathogen free (SPF) conditions in our animal facility at University of California, San Diego, in accordance with the ethical guidelines of the Institutional Animal Care and Use Committee.
METHOD DETAILS
Immune cell isolation
Single-cell suspensions of lymphoid organs were prepared by mechanic disruption through 100-μm strainers (Thermo Fisher Scientific). Red blood cells were lysed using Ammonium-Chloride-Potassium (ACK) buffer (Lonza, Walkersville, MD). Small intestinal and colonic lamina propria cells were prepared as previously described (Yuan et al., 2017). Briefly, the intestine was physically emptied, opened longitudinally and sliced into 0.2–0.5 cm pieces, followed by incubating in 20 mL PBS containing 5 mM EDTA at 37°C on a shaker (200 x rpm) for 15 min. The intestine fragments were further digested in 20 mL RPMI 1640 containing 2% FBS, Collagenase D (1 mg/ml) (Roche) and DNase I (0.1 mg/ml) (Sigma-Aldrich) at 37°C on a shaker (200 x rpm) for 45 min. Samples were collected by passing through 70 μm cell strainers. The cells were then purified using a 40/80% Percoll (GE Healthcare Life Science) gradient by centrifugation (900 x g, 20 min, no break at room temperature). The interphase was harvested and washed. The cells were resuspended in PBS containing 0.2% bovine serum albumin or medium containing fetal calf serum before extra/intracellular staining.
Flow cytometry
The antibodies used in this study were listed in the Key Resources Table. All stainings began by an incubation with anti-CD16/32 (2.4G2, Tonbo). Dead cells were excluded from analyses by using LIVE/DEAD fixable Aqua dead cell stain kit (Invitrogen) according to the manufacturer’s instructions. Surface staining was performed in PBS containing 0.2% BSA at 4°C. Intracellular staining was performed using Foxp3/transcription factor staining buffer set (eBioscience) according to the manufacturer’s instruction. For analyzing retrovirus transduced GFP+ cells, the cells were fixed with 0.8% PFA for 10 mins at 37°C and followed by intracellular staining using Foxp3/transcription factor staining buffer set. For intracellular IFNγ and IL-17A detection, the cells were cultured in complete medium in the presence of phorbol myristate acetate (PMA), Ionomycin (Signa-Aldrich) at 37°C for 4 hr. Brefeldin A (BioLegend) was added 2 hr prior to cell harvest. Samples were acquired with a BD LSRFortessa or LSRFortessa X20 (BD) and analyzed with FlowJo software (FlowJo, LLC).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD4 (GK1.5) FITC | BioLegend | Cat#100406; RRID:AB_312691 |
| Rat monoclonal anti-CD4 (GK1.5) PE | BioLegend | Cat#100408; RRID:AB_312693 |
| Rat monoclonal anti-CD4 (GK1.5) PE/Cy7 | TONBO | Cat#60-0041-U100; RRID:AB_2621828 |
| Rat monoclonal anti-CD4 (RM4-5) APC | BioLegend | Cat#100516; RRID:AB_312718 |
| Rat monoclonal anti-CD4 (RM4-5) BV605 | BioLegend | Cat#100548; RRID:AB_2563054 |
| Rat monoclonal anti-CD8a (53-6.7) APC | TONBO | Cat#20-0081-U100; RRID:AB_2621550 |
| Rat monoclonal anti-CD8a (53-6.7) PE | TONBO | Cat#50-0081-U500; RRID:AB_2621741 |
| Rat monoclonal anti-CD8a (53-6.7) PE/Cy7 | eBioscience | Cat#25-0081-81; RRID:AB_469583 |
| Rat monoclonal anti-CD8a (53-6.7) Biotin | BioLegend | Cat#100704; RRID:AB_312743 |
| Rat monoclonal anti-CD8a (53-6.7) APC/Cy7 | BioLegend | Cat#100714; RRID:AB_312753 |
| Rat monoclonal anti-CD11b (M1/70) BV605 | BioLegend | Cat#101237; RRID:AB_11126744 |
| Rat monoclonal anti-CD11b (M1/70) PE | BioLegend | Cat#101208; RRID:AB_312791 |
| Rat monoclonal anti-CD11b (M1/70) Biotin | BioLegend | Cat#101204; RRID:AB_312787 |
| American Hamster monoclonal anti-CD11c (N418) Biotin | BioLegend | Cat#117304; RRID:AB_313773 |
| American Hamster monoclonal anti-CD11c (N418) PE | BioLegend | Cat#117308; RRID:AB_313777 |
| Rat monoclonal anti-CD19 (6D5) Biotin | BioLegend | Cat#115504; RRID:AB_313639 |
| Rat monoclonal anti-CD19 (6D5) PE | BioLegend | Cat#115508; RRID:AB_313643 |
| Rat monoclonal anti-CD25 (PC61) Biotin | BioLegend | Cat#102004; RRID:AB_312853 |
| Rat monoclonal anti-CD25 (PC61) PE | BioLegend | Cat#102008; RRID:AB_312857 |
| Rat monoclonal anti-CD25 (PC61) PE/Cy7 | BioLegend | Cat#102016; RRID:AB_312865 |
| Rat monoclonal anti-CD25 (PC61) PerCP-Cy5.5 | BioLegend | Cat#102030; RRID:AB_312865 |
| Rat monoclonal anti-CD44 (IM7) APC | BioLegend | Cat#103012; RRID:AB_312963 |
| Rat monoclonal anti-CD44 (IM7) FITC | BioLegend | Cat#103006; RRID:AB_312957 |
| Rat monoclonal anti-CD44 (IM7) PE | BioLegend | Cat#103008; RRID:AB_312959 |
| Mouse monoclonal anti-CD45.1 (A20) PerCP/Cy5.5 | BioLegend | Cat#110728; RRID:AB_893346 |
| Mouse monoclonal anti-CD45.2 (104) APC/Cy7 | BioLegend | Cat#109824; RRID:AB_830789 |
| Rat monoclonal anti-CD45R (RA3-6B2) APC | TONBO | Cat#20-0452-U025; RRID:AB_2621574 |
| Rat monoclonal anti-CD45R (RA3-6B2) PerCP/Cy5.5 | BioLegend | Cat#103236; RRID:AB_893354 |
| Rat monoclonal anti-CD45R (RA3-6B2) Biotin | BioLegend | Cat#103204; RRID:AB_312989 |
| Rat monoclonal anti-CD45RB (C363-16A) PerCP/Cy5.5 | BioLegend | Cat#103314; RRID:AB_2284707 |
| Rat monoclonal anti-CD49b (DX5) Biotin | BioLegend | Cat#108904; RRID:AB_313411 |
| Rat monoclonal anti-CD62L (MEL-14) APC | BioLegend | Cat#104412; RRID:AB_313099 |
| Rat monoclonal anti-CD62L (MEL-14) PE | BioLegend | Cat#104407; RRID:AB_313094 |
| Rat monoclonal anti-CD62L (MEL-14) PE/Cy7 | BioLegend | Cat#104418; RRID:AB_313103 |
| Armenian Hamster monoclonal anti-CD69 (H1.2F3) PerCP/Cy5.5 | BioLegend | Cat#104522; RRID:AB_2260065 |
| Rat monoclonal anti-CD73 (TY/11.8) Biotin | BioLegend | Cat#127203; RRID:AB_1089063 |
| Armenian Hamster monoclonal anti-CD80 (16-10A1) Al488 | BioLegend | Cat#104716; RRID:AB_492822 |
| Rat monoclonal anti-CD86 (GL-1) APC/Cy7 | BioLegend | Cat#105029; RRID:AB_2074993 |
| Mouse monoclonal anti-CD90.1 (OX-7) Biotin | BioLegend | Cat#202510; RRID:AB_2201417 |
| Mouse monoclonal anti-CD90.1 (OX-7) PE | BioLegend | Cat#202524; RRID:AB_1595524 |
| Mouse monoclonal anti-CD90.1 (OX-7) PerCP/Cy5.5 | BioLegend | Cat#202516; RRID:AB_961437 |
| Mouse monoclonal anti-CD95 (SA367H8) FITC | BioLegend | Cat#152606; RRID:AB_2632901 |
| Armenian Hamster monoclonal anti-CD103 (2E7) AI488 | BioLegend | Cat#121408; RRID:AB_535950 |
| Armenian Hamster monoclonal anti-CD103 (2E7) Biotin | BioLegend | Cat#121403; RRID:AB_535946 |
| Rat monoclonal anti-CD138 (281-2) BV605 | BioLegend | Cat#142531; RRID:AB_2715767 |
| Armenian Hamster monoclonal anti-CD152 (CTLA4) (UC10-4F10-11) PE/Cy7 | TONBO | Cat#60-1522-U025; RRID:AB_2621861 |
| Mouse monoclonal anti-CD161 (NK1.1) (PK136) PE | TONBO | Cat#50-5941-U 100; RRID:AB_2621804 |
| Rat monoclonal anti-CD185 (CXCR5) (L138D7) Biotin | BioLegend | Cat#145510; RRID:AB_2562126 |
| Rat monoclonal anti-CD278 (ICOS) (7E.17G9) PE | BioLegend | Cat#117405; RRID:AB_961244 |
| Rat monoclonal anti-CD278 (ICOS) (7E.17G9) PE/Cy7 | eBioscience | Cat#25-9942-82; RRID:AB_2573564 |
| Rat monoclonal anti-CD279 (PD-1) (RMP1-30) APC | eBioscience | Cat#17-9981-80; RRID:AB_10853186 |
| Rat monoclonal anti-CD304 (Neuropilin-1) (3E 12) APC | BioLegend | Cat#145205; RRID:AB_2562031 |
| Rat monoclonal anti-CD366 (Tim-3) (RMT3-23) PE/Cy7 | BioLegend | Cat#119715; RRID:AB_2571932 |
| Rat monoclonal anti-Lag3 (C9B7W) PE | BioLegend | Cat#125207; RRID:AB_2133344 |
| Rat monoclonal anti-GL7 (GL7) PE | BioLegend | Cat#144608; RRID:AB_2562926 |
| Syrian hamster monoclonal anti-KLRG1 (MAFA) (2F1/KLRG1) Biotin | BioLegend | Cat#138406; RRID:AB_10575641 |
| Armenian Hamster monoclonal anti-TCR β (H57-597) Al488 | BioLegend | Cat#109215; RRID:AB_493344 |
| Armenian Hamster monoclonal anti-TCR β (H57-597) APC/Cy7 | BioLegend | Cat#109220; RRID:AB_893624 |
| Armenian Hamster monoclonal anti-TCR β (H57-597) PE-Cy7 | BioLegend | Cat#109222; RRID:AB_893625 |
| Armenian Hamster monoclonal anti-TCR γ/δ (GL3) PE | BioLegend | Cat#118108; RRID:AB_313832 |
| Armenian Hamster monoclonal anti-TCR γ/δ (eBioGL3) Biotin | eBioscience | Cat#13-5711-85; RRID:AB_466669 |
| Rat monoclonal anti-TER-119/Erythroid Cells (TER-119) Biotin | BioLegend | Cat#116204; RRID:AB_313705 |
| Mouse monoclonal anti-BCL-6 (K112-91) Al647 | BD PharMingen | Cat#561525; RRID:AB_10898007 |
| Rabbit monoclonal anti-Bim (C34C5) Al488 | Cell Signaling | Cat#94805S; RRID not available |
| Rabbit monoclonal anti-Bim (C34C5) Al647 | Cell Signaling | Cat#10408S; RRID not available |
| Rat monoclonal anti-Blimp1 (5E7) PE | BD PharMingen | Cat#564268; RRID:AB_2738718 |
| Rabbit monoclonal anti-active Caspase-3 (C92-605) PE | BD Bioscience | Cat#561011; RRID:AB_2033931 |
| Rat monoclonal anti-Foxp3 (FJK-16 s) APC | eBioscience | Cat#17-5773-82; RRID:AB_469457 |
| Rat monoclonal anti-Foxp3 (FJK-16 s) eF450 | eBioscience | Cat#48-5773-82; RRID:AB_1518812 |
| Rat monoclonal anti-GFP/YFP (5F12.4) PerCP-eFluor 710 | eBioscience | Cat#46-6498-80; RRID:AB_11042700 |
| Armenian Hamster monoclonal anti-Helios (22F6) PE | BioLegend | Cat#137206; RRID:AB_315400 |
| Rat monoclonal anti-IFNγ (XMG1.2) FITC | BioLegend | Cat#505806; RRID:AB_315402 |
| Rat monoclonal anti-IgD (11-26c.2a) Pacific Blue | BioLegend | Cat#405711; RRID:AB_10899576 |
| Rat monoclonal anti-IL-17A (TC11-18H10.1) APC | BioLegend | Cat#506916; RRID:AB_961384 |
| Rat monoclonal anti-IRF4 (IRF4.3E4) Al488 | BioLegend | Cat#646406; RRID:AB_2563004 |
| Rat monoclonal anti-IRF4 (IRF4.3E4) PE | BioLegend | Cat#646403; RRID:AB_2266296 |
| Mouse monoclonal anti-KI-67 (B56) PE | BD PharMingen | Cat#556027; RRID:AB_10611574 |
| Mouse monoclonal anti-KI-67 (B56) PerCP/Cy5.5 | BD PharMingen | Cat#561284; RRID:AB_10949502 |
| Rabbit monoclonal anti-LEF1 (C12A5) Al488 | Cell Signaling | Cat#8490S; RRID:AB_10949503 |
| Rabbit monoclonal anti-LEF1 (C12A5) PE | Cell Signaling | Cat#14440S; RRID:AB_1257210 |
| Mouse monoclonal anti-Nur77 (12.14) PE | eBioscience | Cat#12-5965-80; RRID:AB_2573310 |
| Mouse monoclonal anti-Stat5a + Stat5b (A-9) Al647 | Santa Cruz Biotechnology | Cat#sc-74442 AF647; RRID not available |
| Mouse monoclonal anti-Phospho-Stat5(pY694) (47) PE | BD Phosflow | Cat#612567; RRID:AB_2572664 |
| Rat monoclonal anti-RORgt (B2D) PerCP-eFluor710 | eBioscience | Cat#46-6981-82; RRID:AB_1595466 |
| Rabbit monoclonal anti-TCF1 (C63D9) Al647 | Cell Signaling | Cat#6709S; RRID not available |
| Armenian Hamster monoclonal anti-CD3ε (145-2C11) | BioLegend | Cat#100340; RRID:AB_11149115 |
| Rat monoclonal anti-CD16/CD32 (2.4G2) | TONBO | Cat#70-0161-U500; RRID:AB_2621487 |
| Syrian Hamster monoclonal anti-CD28 (37.51) | BioLegend | Cat#102112; RRID:AB_312877 |
| SA PerCP/Cy5.5 | BioLegend | Cat#405214; RRID:AB_2716577 |
| SA Al488 | BioLegend | Cat#405235; RRID not available |
| SA PECy7 | BioLegend | Cat#405206; RRID not available |
| Bacterial and Virus Strains | ||
| pCL-Eco | Addgene; Plasmid #12371 | |
| pMIGRI | Cho et al. (2017) | N/A |
| pMIGRI-Tcf7-long | Cho et al. (2017) | N/A |
| pMIGRI-caSTAT5b | Johnston et al. (2012) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IL-2 | PeproTech | Cat#AF-200-02 |
| Critical Commercial Assays | ||
| LIVE/DEAD fixable Aqua dead cell stain kit | Thermo Fisher Scientific | Cat#L34966 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat#00-5523-00 |
| TruSeq® Stranded mRNA LT - Set A | Illumina | Cat#RS-122-2101 |
| truChIP Chromatin Shearing Kit with Formaldehyde | Covaris | Cat#520154 |
| BD Cytofix/Cytoperm solution | BD | Cat#554722 |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | Cat# C34557 |
| Mouse Anti-dsDNA Ig’s (Total A+G+M) ELISA Kit, 96 tests, Quantitative | Alpha Diagnostic | Cat#5110 |
| Mouse Ig Isotyping ELISA Ready-Set-Go!® 10 × 96 tests | eBioscience | Cat#88-50630-88 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE117726 |
| Experimental Models: Cell Lines | ||
| 293T cells | Cho et al. (2017) | RRID:CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Foxp3YFP-Cre: B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J | The Jackson Laboratory | RRID:IMSR_JAX:016959 |
| Mouse: B6/CD45.1: B6.SJL-Ptprca PepCb/BoyJ | The Jackson Laboratory | RRID:IMSR_JAX:002014 |
| Mouse: B6/Tcrα−/−: B6.129S2-Tcratm1Mom/J | The Jackson Laboratory | RRID:IMSR_JAX:002116 |
| Mouse: B6/Rag1−/−: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | RRID:IMSR_JAX:002216 |
| Mouse: Foxp3Thy1.1 | Liston et al. (2008) | N/A |
| Mouse: Tcf7GFP | Choi et al. (2015) | N/A |
| Mouse: Tcf7fl/fl | Yu et al. (2012) | N/A |
| Mouse: Lef1f1/f1 | Steinke et al. (2014) | N/A |
| Oligonucleotides | ||
| ChIP-PCR Primer: Bcl6 –0.5K_F GGGTCTGGGGCTAATTCTTC | Xu et al. (2017) | N/A |
| ChIP-PCR Primer: Bcl6 –0.5K_R TAGCTGGAAGGAGCTGTGGT | Xu et al. (2017) | N/A |
| ChIP-PCR Primer: Stat5b_ F TCAGCTCCGTGTGGTTTCTAAC | This paper | N/A |
| ChIP-PCR Primer: Stat5b_ R TTCATCTCGAGCTGGGTGTTC | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| STAR | Dobin et al. (2013) | https://code.google.com/archive/p/rna-star/ |
| Cuffdiff | Trapnell et al. (2013) | https://software.broadinstitute.org/cancer/software/genepattern/modules/docs/Cuffdiff/7 |
| Extraction of Differential Gene Expression (EDGE) | Leek et al. (2006) | https://github.com/StoreyLab/edge |
| Gene Set Enrichment Assay (GSEA) | Subramanian et al. (2005) | http://software.broadinstitute.org/gsea/index.jsp |
| GenePattern software package | Reich et al. (2006) | http://software.broadinstitute.org/cancer/software/genepattern/use-genepattern |
| BISMA | Rohde et al. (2010) | http://services.ibc.uni-stuttgart.de/BDPC/BISMA/ RRID:SCR_000688 |
RNA-sequencing and data analysis
Tregs were first enriched by magnetic positive selection for Thy1.1 (MACS) from pooled spleens and LNs of Foxp3Thy1.1Tcf7GFP mice and then sorted with BD Aria2. For dKO and WT Treg isolation, CD4+ cells were first enriched by magnetic depletion (STEMCELL) from pooled spleens and LNs of Foxp3YFP-cre/wtTcf7fl/flLef1fl/fl or Foxp3YFP-cre/wtTcf7wt/wtLef1wt/wt mice, respectively. RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s instructions. Quality and integrity of total RNA were controlled on Tapestation (Agilent Technologies). cDNA library was prepared using TruSeq® Stranded mRNA LT - Set A (Illumina) according to the manufacturer’s instructions. Sequencing was conducted on Illumina Hiseq 4000 (SR50 or SR75) at the UCSD IGM Genomics Center. Fastq files from sequencing experiments were mapped to the mouse GRCm38 genome using default parameters for STAR (Dobin et al., 2013). Next, we performed gene differential expression analysis using Cuffdiff (Trapnell et al., 2013) and the R package Extraction of Differential Gene Expression (EDGE) (Storey and Tibshirani, 2003). Principle component analysis (PCA), Gene Set Enrichment Assay (GSEA) (Subramanian et al., 2005), and K-means clustering analysis were performed using the modules embedded in GenePattern software package (Reich et al., 2006).
In vitro Treg suppression assay
CD11c+ splenic DCs were enriched by MACS from spleens treated with Liberase TL (Roche). T responder cells (Tresp) and WT Tregs were FACS sorted from spleen and LNs of male Foxp3Y/creTcf7wt/wtLef1wt/wt mice. dKO Tregs were sorted from male Foxp3Y/creTcf7fl/flLef1fl/fl mice. Tresp cells were prelabeled with CTV (Molecular Probes, Invitrogen) and co-cultured with DCs and Tregs at the indicated ratio in the presence of 0.5 ug/ml soluble anti-CD3 antibody and analyzed on day 4 by flow cytometry for CTV dilution and DC maturation.
Cell line
293T cells were purchased from ATCC (ATCC® CRL-3216) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% Fetal Bovine Serum.
Retroviral transduction
Viruses were prepared by transfecting 293T cells with retroviral expression plasmids (pMIG, MSCV-IRES-GFP) encoding GFP only, Tcf7 (Cho et al., 2017), or caSTAT5 (Johnston et al., 2012). Retrovirus-containing supernatants were collected and concentrated by full speed centrifugation at 4°C overnight and resuspended in the desired volume of complete medium with 1.6 ug/ml polybrene (Sigma-Aldrich). CD4+ cells were enriched from spleens and lymph nodes of indicated mice by magnetic depletion with mAbs against CD8α, CD11b, CD11c, CD19, TCRγδ, CD119 and NK1.1. The purity of enriched CD4+T cells was > 95%. These cells were activated with plate-bound anti-CD3 (2.5 μg/ml), soluble anti-CD28 (2.5 μg/ml) and 200 U/ml of recombinant human IL-2 (PeproTech) in complete culture medium (RPMI 1640 supplemented with 10% fetal calf serum, 2 mM L-Glutamine, 100 U/ml penicillin/streptomycin, non-essential AA sup. and 50 μM 2-Mercaptoethanol). After 40 hr, activated T cells were spin-infected (400 x g, 32°C, 1 hr) with retrovirus supernatants prepared as above. For in vitro assay, transduced T cells were cultured for an additional 72 hr and analyzed by FACS. For in vivo transfer assays, transduced cells were washed twice with 1x PBS and transferred into Rag1−/− or Tcrα−/− mice.
Adoptive cell transfer
CD4+ T cells were enriched from the spleens of Foxp3Cre (WT) or Tcf7fl/flLef1fl/fl;Foxp3cre (dKO) mice by MACS (Miltenyi). 2 million cells were transferred i.p. into Tcrα−/− mice. For the co-transfer experiments, CD4+ T cells from CD45.1/Foxp3Cre and CD45.2/Foxp3Cre mice were mixed at 3:1 before transferring into Tcrα−/− mice; Similarly, CD4+ T cells from CD45.1/Foxp3Cre and CD45.2/Foxp3CreTcf7fl/flLef1fl/fl mice were mixed at 3:1 before transferring into Tcrα−/− mice.
Ex vivo assays of total STAT5 protein and pSTAT5 by flow cytometry
pLN and mLN were directly meshed into BD Cytofix/Cytoperm (BD), incubated for 1 hr at room temperature. Single-cell suspensions were then washed and permeabilized with 90% methanol on ice for 30 min. Surface and intracellular staining were performed using Foxp3/transcription factor staining buffer set (eBioscience) according to the manufacturer’s instruction.
In vitro STAT5 phosphorylation assays
Total splenocytes (1 × 106) first rested in complete RPMI1640 medium for 30 minutes and were then incubated with indicated human IL-2 concentration for 15 min at 37°C. After that, cells were fixed with paraformaldehyde (0.8%) for 10 minutes at 37°C. Fixed cells were permeabilized with ice-cold methanol for 30 minutes and were stained with anti-phospho-STAT5 (clone 47/Stat5[pY694]; BD Biosciences) and other surface or intracellular antibodies for 1 hour at room temperature.
Enzyme-linked immunosorbent assay (ELISA)
Sera IgG and IgE were quantitated using mouse uncoated ELISA Kits (Thermo Fisher) according to manufacturer’s instructions. The titters of Ig subclasses were measured using Ig isotyping mouse uncoated ELISA Kit (Thermo Fisher).
Chromatin immunoprecipitation
Tregs were isolated by magnetic separation from pooled spleens and LNs of Foxp3Thy1.1 mice and CD4+T cells from CD4Cre;Tcf7fl/fl mice. Collected cells were fixed with freshly prepared 1% methanol-free formaldehyde at room temperature for 5 min. Chromatin shearing was performed using truChIP Chromatin Shearing Kit with Formaldehyde (Covaris) on E220 (Covaris) according to the manufacturer’s manual. Chromatin immunoprecipitation was performed as previously described (Blecher-Gonen et al., 2013). Briefly, fragmented chromatin was incubated with protein A dynabeads (Invitrogen) pre-coupled with rabbit anti-TCF1 sera at 4°C overnight. Beads were then washed using ice-cold IP buffer and ice-cold TE buffer. 10% (w/v) Chelex 100 slurry was used to reverse crosslink at 100°C for 10 mins. The primers for PCR were listed in the Key Resources Table.
Immunostaining and histology
Tissue samples were snap-frozen in optimum cutting temperature (O.C.T., Fisher Scientific, Houston, TX). Cryo-sections of tissue sections (6 μm) were cut, fixed with pre-cold acetone for 20 min and blocked with normal rat serum. The following mAbs were used in different combinations as indicated in the figure legends: anti-B220 (RA3-6B2), anti-GL7 (GL7), anti-Foxp3 (FJK-16 s), anti-CD4 (RM4-5). Nuclei were stained with DAPI (4’,6-Diamidino-2–28 phenylindole dihydrochloride) (Invitrogen). Images were acquired on ZEISS AXIOSCAN Z1 SLIDE SCANNER (Zeiss), and were processed with Zen (Blue edition). For histology, the indicated organs were removed and fixed with 10% formalin solution. Fixed tissue blocks were transferred to 70% ethanol, paraffin-embedded, sectioned, stained with hematoxylin and eosin and scanned on ZEISS AXIOSCAN Z1 SLIDE SCANNER.
Western blot for autoantibody detection
Pancreas, salivary glands and kidney from female Rag1−/−mice were homogenized (VWR200 Homogenizer, VWR) on ice in a buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100,1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, and a protease inhibitor cocktail (Thermo Scientific). The homogenates were centrifuged at 12,000 x g per minute for 10 min, and the supernatants were mixed with a 4 x Laemmli Sample Buffer (Bio-Rad) and boiled for 5 min at 100°C. The extracts were separated by 10% SDS-PAGE gel and electrophoretically transferred onto a PVDF membrane (Bio-Rad). The membranes were blocked with PBS (VWR) containing 0.1% Tween-20 (Fisher Scientific) and 5% nonfat milk (Bio-Rad) for 1 h and assembled in a Mini-PROTEAN II Multiscreen Apparatus (Bio-Rad) and incubated overnight with 1:600 dilution of sera collected from dKO or control mice. After washing three times with PBST, the bound antibodies were reacted with Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (1:5,000, Jackson ImmunoResearch Laboratories) for 1 h, washed with PBST for three times, revealed with Clarity Western ECL Substrate (Bio-Rad) and autoradiography.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 6 software (GraphPad Software). Data are presented as mean ± s.e.m. Statistical significance was calculated by the Student’s t test with a 95% confidence interval. P values of 0.05 or less were considered statistically significant. P values for gene signature distribution were calculated using the Chi-square (χ2) test. Further detailed information of the number (n) of repeated experiments is indicated in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Data source: The accession number for RNA-seq reported in this paper is GEO: GSE117726.
Supplementary Material
Highlights.
The expression of TCF1 and LEF1 distinguishes Treg cells into distinct subsets
Treg-specific ablation of TCF1 and LEF1 provokes systemic autoimmunity
TCF1 and LEF1 are essential for Treg’s competitive fitness and survival
TCF1 and LEF1 are indispensable for the development of Tfr, a subset of aTregs
ACKNOWLEDGMENTS
We thank D. Mathis(Harvard), Y. Zheng (Salk), and C. Xiao (Scripps) for insightful discussions; Y. Zheng (Salk) for Rag1−/− mice; S. Crotty (La Jolla Institute for Allergy and Immunology) for the caSTAT5 retroviral plasmids; H. Kawamoto and K. Masuda (Kyoto University) for the anti-TCF1 sera; J. Olvera and C. Fine (UCSD) for cell sorting; Z. Mikulski and A. Denn (La Jolla Institute for Allergy and Immunology) for histology and microscopy; and W. Huang, Y. Qian, and Z. Wang for animal husbandry and genotyping. This work was supported by the National Institutes of Health, United States (AI108651, AI123782, and AI140095 to L.-F.L.; AI112579, AI121080, and AI139874 to H.-H.X.; and DK114427 and AI139753 to W.F.), the Juvenile Diabetes Research Foundation (2-SRA-2016-306-S-B to W.F.), the Veteran Affairs BLR&D Merit Review Program (BX002903A to H.-H.X.), and International Program for Ph.D. Candidates, Sun Yat-Sen University, Guangzhou, China (to S.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.061.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allan SE, Broady R, Gregori S, Himmel ME, Locke N, Roncarolo MG, Bacchetta R, and Levings MK (2008). CD4+ T-regulatory cells: toward therapy for human diseases. Immunol. Rev 223, 391–421. [DOI] [PubMed] [Google Scholar]
- Barra MM, Richards DM, Hansson J, Hofer AC, Delacher M, Hettinger J, Krijgsveld J, and Feuerer M (2015). Transcription Factor 7 Limits Regulatory T Cell Generation in the Thymus. J. Immunol 195, 3058–3070. [DOI] [PubMed] [Google Scholar]
- Blecher-Gonen R, Barnett-Itzhaki Z, Jaitin D, Amann-Zalcenstein D, Lara-Astiaso D, and Amit I (2013). High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc 8, 539–554. [DOI] [PubMed] [Google Scholar]
- Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, Zajac AJ, Randall TD, Lund FE, León B, and Ballesteros-Tato A (2017). Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat. Immunol 18, 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, and Farrar MA (2008). Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 28, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ (2015). Control of Regulatory T Cell Migration, Function, and Homeostasis. J. Immunol 195, 2507–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Wu CJ, Nguyen DT, Lin LL, Chen MC, Khan AA, Yang BH, Fu W, and Lu LF (2017). A Novel miR-24-TCF1 Axis in Modulating Effector T Cell Responses. J. Immunol 198, 3919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, Love PE, Peng W, Xue HH, and Crotty S (2015). LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol 16, 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, et al. (2011). Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med 17, 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Smyth DJ, Bailey R, Payne F, Downes K, Godfrey LM, Masters J, Zeitels LR, Vella A, Walker NM, and Todd JA (2007). The candidate genes TAF5L, TCF7, PDCD1, IL6 and ICAM1 cannot be excluded from having effects in type 1 diabetes. BMC Med. Genet. 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, Belz GT, Smyth GK, Busslinger M, Nutt SL, and Kallies A (2011). The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol 12, 304–311. [DOI] [PubMed] [Google Scholar]
- Cretney E, Kallies A, and Nutt SL (2013). Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 34, 74–80. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949. [DOI] [PubMed] [Google Scholar]
- Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, and Lafaille JJ (2008). Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat. Med 14, 162–169. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JH, Zelenay S, Bergman ML, Martins AC, and Demengeot J (2009). Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur. J. Immunol 39, 948–955. [DOI] [PubMed] [Google Scholar]
- Enarsson K, Lundgren A, Kindlund B, Hermansson M, Roncador G, Banham AH, Lundin BS, and Quiding-Jarbrink M (2006). Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin. Immunol 121, 358–368. [DOI] [PubMed] [Google Scholar]
- Erlich HA, Valdes AM, Julier C, Mirel D, and Noble JA; Type I Diabetes Genetics Consortium (2009). Evidence for association of the TCF7 locus with type I diabetes. Genes Immun. 10 (Suppl 1), S54–S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, and Benoist C (2010). Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. USA 107, 5919–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, Gazit R, Adoro S, Glimcher L, Chan S, et al. (2012). A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat. Immunol 13, 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. (2009). Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med 15, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, and Rudensky AY (2007). Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775. [DOI] [PubMed] [Google Scholar]
- Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, Arnett KL, Blacklow SC, Aifantis I, Aster JC, and Gounari F (2011). T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc. Natl. Acad. Sci. USA 108, 20060–20065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, Zheng Y, Downes M, and Evans RM (2017). Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl. Acad. Sci. USA 114, 12542–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, and Benoist C (2007). Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800. [DOI] [PubMed] [Google Scholar]
- Ioannidis V, Beermann F, Clevers H, and Held W (2001). The beta-catenin-TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat. Immunol 2, 691–697. [DOI] [PubMed] [Google Scholar]
- Jeannet G, Boudousquié C, Gardiol N, Kang J, Huelsken J, and Held W (2010). Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 107, 9777–9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Choi YS, Diamond JA, Yang JA, and Crotty S (2012). STAT5 is a potent negative regulator of TFH cell differentiation. J. Exp. Med 209, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30,531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julier C, Akolkar B, Concannon P, Morahan G, Nierras C, and Pugliese A; Type I Diabetes Genetics Consortium (2009). The Type I Diabetes Genetics Consortium ‘Rapid Response’ family-based candidate gene study: strategy, genes selection, and main outcome. Genes Immun. 10 (Suppl 1), S121–S127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, Weber C, Emmanuel AO, Sun T, Bentrem DJ, et al. (2014). β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med 6, 225ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Monsen E, Dabney AR, and Storey JD (2006). EDGE: extraction and analysis of differential gene expression. Bioinformatics 22, 507–508. [DOI] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, and Rudensky AY (2014). Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol 15, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, et al. (2011). Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med 17, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CW, and Hsieh CS (2008). A two-step process for thymic regulatory T cell development. Immunity 28, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, and Gray DH (2014). Homeostatic control of regulatory T cell diversity. Nat. Rev. Immunol 14, 154–165. [DOI] [PubMed] [Google Scholar]
- Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, and Rudensky AY (2008). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. USA 105, 11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, and Germain RN (2015). Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, Ichiyama K, Zhang X, Zhang L, Wen X, et al. (2016). Genome-wide Analysis Identifies Bcl6-Controlled Regulatory Networks during T Follicular Helper Cell Differentiation. Cell Rep. 14, 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage UK, Goedegebuure PS, Moore TT, Viehl CT, Moo-Young TA, Larson JW, Frey DM, Ehlers JP, Eberlein TJ, and Linehan DC (2006). Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J. Immunother 29, 416–424. [DOI] [PubMed] [Google Scholar]
- Mendez S, Reckling SK, Piccirillo CA, Sacks D, and Belkaid Y (2004). Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med 200, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odhams CA, Cortini A, Chen L, Roberts AL, Viñuela A, Buil A, Small KS, Dermitzakis ET, Morris DL, Vyse TJ, and Cunninghame Graham DS (2017). Mapping eQTLs with RNA-seq reveals novel susceptibility genes, non-coding RNAs and alternative-splicing events in systemic lupus erythematosus. Hum. Mol. Genet 26, 1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, and Kitamura T (1998). Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol. Cell. Biol 18, 3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi Y, Fehervari Z, Yamaguchi T, and Sakaguchi S (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 105, 10113–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, Ruscher R, Huang W, Proekt I, Miller CN, et al. (2019). Thymic regulatory T cells arise via two distinct developmental programs. Nat. Immunol 20, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D (2016). Tissue Tregs. Annu. Rev. Immunol 34,609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, Humblet-Baron S, Schönefeldt S, Herold MJ, Hildeman D, et al. (2013). Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat. Immunol 14, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, and Mesirov JP (2006). GenePattern 2.0. Nat. Genet 33, 500–501. [DOI] [PubMed] [Google Scholar]
- Ritvo PG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, Mariotti-Ferrandiz E, and Klatzmann D (2017). Tfr cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of Tfh cells. Sci. Immunol. 2, Published online September 8, 2017. 10.1126/sciimmunol.aan0368. [DOI] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, and Jeltsch A (2010). BISMA-fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 11, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 23, 546–558. [DOI] [PubMed] [Google Scholar]
- Sage PT, Francisco LM, Carman CV, and Sharpe AH (2013). The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat. Immunol 14, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, and Bluestone JA (2000). B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440. [DOI] [PubMed] [Google Scholar]
- Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ (2014a). CCR7 provides localized accessto IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med 211, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigiel KS, Srivastava S, Stolley JM, and Campbell DJ (2014b). Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol. Rev 253, 40–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, and Xue HH (2014). TCF-1 and LEF-1 act upstream of Th-POKto promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat. Immunol 15, 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102,15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida T, Lincoln MR, Ukeje CM, Rodriguez DM, Akazawa H, Noda T, Naito AT, Komuro I, Dominguez-Villar M, and Hafler DA (2018). Activated β-catenin in Foxp3+ regulatory T cells links inflammatory environments to autoimmunity. Nat. Immunol 19, 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, Ma J, Qi YY, Kim-Howard X, Motghare P, et al. (2016). High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet 43, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA (2008). Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 23, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, and Shevach EM (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol 134, 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemessen MM, Baert MR, Kok L, van Eggermond MC, van den Elsen PJ, Arens R, and Staal FJ (2014). T Cell factor 1 represses CD8+ effector T cell formation and function. J. Immunol 193, 5480–5487. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation attranscript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, Pals CE, Kurek D, Baert MR, Delemarre EM, et al. (2013). Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 39, 298–310. [DOI] [PubMed] [Google Scholar]
- Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, and Bhandoola A (2011).A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. [DOI] [PubMed] [Google Scholar]
- Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, Kidani Y, Matsuda K, Inoue T, Kurosaki T, et al. (2017). A distinct subpopulation of CD25− T-follicular regulatory cells localizes in the germinal centers. Proc. Natl. Acad. Sci. USA 114, E6400–E6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing JB,Tekgüç M, and Sakaguchi S (2018). Control of Germinal Center Responses by T-Follicular Regulatory Cells. Front. Immunol. 9, 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, Sen JM, Berg LJ, Gattinoni L, McGavern DB, and Schwartzberg PL (2015). TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell Rep. 12, 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, Zhao JM, Zhang B, Shi M, Ding X, et al. (2006). Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J. Immunol 177, 739–747. [DOI] [PubMed] [Google Scholar]
- Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, He R, Hao Y, Wang H, Zhao T, et al. (2015). The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat. Immunol 16, 991–999. [DOI] [PubMed] [Google Scholar]
- Xu L, Huang Q, Wang H, Hao Y, Bai Q, Hu J, Li Y, Wang P, Chen X, He R, et al. (2017). The Kinase mTORC1 Promotes the Generation and Suppressive Function of Follicular Regulatory T Cells. Immunity 47, 538–551 e5. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, Wang H, Wang X, Yu S, Zhou X, et al. (2015). TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol 16, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, Wu C, Shrestha S, Rankin S, Long L, et al. (2017). Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature 543, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, Jing X, Yokota Y, Meyerholz DK, Mullighan CG, et al. (2012). The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Yang BH, Dong Y, Yamamura A, and Fu W (2017). CRIg, a tissue-resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. eLife 6, e29540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, and Xue HH (2010). Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunol 134, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, and Xue HH (2012). Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J. Immunol 139, 2722–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data source: The accession number for RNA-seq reported in this paper is GEO: GSE117726.