

SUMMARY
During autophagy, phagophores grow into doublemembrane vesicles called autophagosomes, but the underlying mechanism remains unclear. Here, we show a critical role of Atg2A in phagophore expansion. Atg2A translocates to the phagophore at the mitochondria-associated ER membrane (MAM) through a C-terminal 45-amino acid domain that we have termed the MAM localization domain (MLD). Proteomic analysis identifies the outer mitochondrial membrane protein TOM40 as a MLD-interacting partner. The Atg2A-TOM40 interaction is responsible for MAM localization of Atg2A and requires the TOM receptor protein TOM70. In addition, Atg2A interacts with Atg9A by a region within its N terminus. Inhibition of either Atg2A-TOM40 or Atg2A-Atg9A interactions impairs phagophore expansion and accumulates Atg9A-vesicles in the vicinity of autophagic structures. Collectively, we propose a model that the TOM70-TOM40 complex recruits Atg2A to the MAM for vesicular and/or nonvesicular lipid transport into the expanding phagophore to grow the size of autophagosomes for efficient autophagic flux.
In Brief
Tang et al. show that human Atg2 is a key regulator for phagophore expansion. TOM40/70 directs Atg2A to MAM to mediate phagophore expansion. On the MAM, Atg2A facilitates Atg9-vesicle delivery and retrograde trafficking to promote phagophore expansion and efficient autophagic flux.
Graphical Abstract
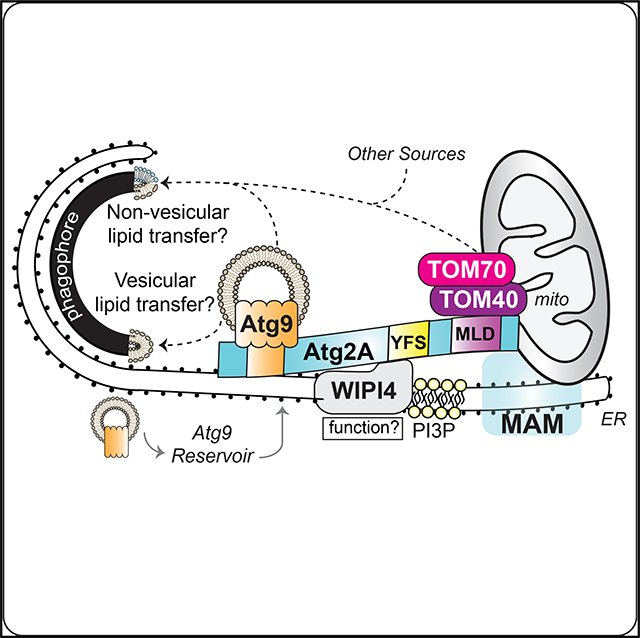
INTRODUCTION
Macroautophagy (hereafter autophagy) is an evolutionarily conserved intracellular degradation process that plays a vital role in the maintenance of cellular and tissue homeostasis (Mizushima et al., 2008). Dysregulation of autophagy has been implicated in the pathogenesis of various human diseases, including cancer, neurodegenerative disease, and vascular disease (Choi et al., 2013). Moreover, at the cellular level, autophagosomal membranes have been reported to function as signaling platforms for cell death and survival, including apoptosis, necroptosis, and mitogen-activated protein (MAP)-kinase pathways (Goodall et al., 2016; Martinez-Lopez et al., 2013; Tang et al., 2017). Thus, understanding the molecular mechanism of autophagosome biogenesis is pivotal for the development of future autophagy-targeting therapeutics.
The process of autophagy begins with the formation of the phagophore, which undergoes membrane expansion and eventually seals to form a double-membrane vesicle, known as the autophagosome (Mizushima et al., 2011). The autophagosome is then fused with endosomes and lysosomes to deliver the sequestered materials for hydrolytic degradation and nutrient recycling. Although the molecular mechanism of autophagosome biogenesis remains far from clear, yeast genetic screens have identified over 30 autophagy-related (ATG) genes that are important for autophagy (Klionsky, 2004; Nakatogawa et al., 2009). Several upstream ATG proteins, including components of the class III phosphatidylinositol 3-kinase complex, translocate to the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM) for autophagosome formation (Hamasaki et al., 2013). Among these ATG proteins, the Atg8/LC3 family of proteins is one of the most studied molecules involved in autophagosome formation. During autophagy, cytosolic LC3-I is conjugated to the lipid phosphatidylethanolamine (PE) to generate LC3-II on the phagophore. The LC3-PE conjugation occurs during phagophore expansion and serves to recruit ubiquitinated cargo through direct interaction with cargo adaptors, such as p62/sequestosome-1. Upon phagophore closure, LC3-II on the outer autophagosomal membrane (OAM) is delipidated and recycled to the cytosol prior to lysosomal fusion, while LC3-II on the inner autophagosomal membrane (IAM) is degraded upon autophagosome-lysosome fusion (Kabeya et al., 2000; Mizushima et al., 2001; Tanida et al., 2005). Therefore, LC3-II serves as a widely used marker for autophagy (Klionsky et al., 2016).
Although the lipidation of LC3 is associated with phagophore expansion, the mechanism and membrane source for such process remains unknown. Atg9 is the sole transmembrane ATG protein that is required for autophagosome formation (Webber et al., 2007). Although Atg9 basally resides in the trans-Golgi network (TGN) and recycling endosomes known as the Atg9 reservoir, vesicles containing Atg9 bud off from the reservoir in response to autophagic stimuli and traffic to the autophagosome formation site (Orsi et al., 2012; Takahashi et al., 2011; Takahashi et al., 2016; Yamamoto et al., 2012). Here, Atg9 only transiently associates with autophagosomal membranes and is rapidly recycled back to the reservoir for reuse (Orsi et al., 2012; Webber et al., 2007). As Atg9 shuttling between the reservoir and phagophore is important for controlling the number and size of autophagosomes (Imai et al., 2016; Yamamoto et al., 2012), Atg9 (legend continued on next page) vesicles are proposed to deliver membrane and other components for phagophore expansion.
In yeast, Atg2 interacts with Atg9 to promote autophagy and regulate Atg9 retrieval from the pre-autophagosomal structure (PAS) (Feng et al., 2014; Gó mez-Sánchez et al., 2018). Atg2 is one of the least understood ATG proteins required for autophagy (Gómez-Sánchez et al., 2018; Suzuki et al., 2013; Velikkakath et al., 2012). Previous studies in yeast have shown that Atg2 can be recruited to PAS through the interaction with the phosphatidylinositol 3-phosphate (PI3P)-binding protein Atg18 (Obara et al., 2008; Suzuki et al., 2007). In mammalian cells, two Atg2 orthologs (Atg2A/B) and four Atg18 orthologs (WIPI1–4) have been identified (Proikas-Cezanne et al., 2015; Velikkakath et al., 2012). Although Atg2A and Atg2B are functionally redundant to promote autophagic degradation (Velikkakath et al., 2012), WIPI proteins appear to be functionally non-redundant (Proikas-Cezanne et al., 2015). Among WIPIs, WIPI4 exhibits the strongest physical interaction with mammalian Atg2 and has been reported to be involved in omegasome maturation and autophagosome formation (Lu et al., 2011; Zheng et al., 2017). Although the physiological significance of mammalian Atg2-WIPI4 interaction remains unknown, the loss of Atg2 accumulates cytoplasmic punctate structures containing Atg9/ Atg9A, Atg1/ULK1, Atg14, and Atg16/Atg16L1 in both yeast and human cells(Tang et al., 2017; Velikkakath et al., 2012), suggesting the importance of Atg2 proteins in autophagosome biogenesis.
Here, we demonstrate that the loss of mammalian Atg2A/B significantly impairs autophagy at the phagophore expansion step. During autophagy, Atg2A translocates to the autophagosome formation site at the ER-mitochondria contact site by a C-terminal MAM localization domain (MLD). Although inhibition of Atg2A recruitment to the MAM impairs phagophore expansion and autophagic flux, the MAM targeting of Atg2A occurs independent of its binding to WIPI4 but requires the TOM70-TOM40 complex. Furthermore, we demonstrate that Atg2A directly interacts with Atg9A at the MAM to promote phagophore growth. These results implicate mammalian Atg2A as a critical regulator of Atg9A vesicle delivery and phagophore expansion during autophagosome biogenesis at the MAM.
RESULTS
Atg2 Is Required for Autophagosomal Membrane Expansion
Mammalian Atg2A/B proteins are functionally redundant and have been reported to be indispensable for basal and starvation-induced autophagic flux in THP-1 and HeLa cells (Tang et al., 2017; Velikkakath et al., 2012). Consistently, we found that the loss of Atg2A/B impaired lysosomal turnover of LC3-II and p62 in U-2 OS cells under both basal and starved conditions (Figures S1A and S1B). To clarify the role of Atg2A/B in autophagosome biogenesis, we next took the advantage of the HaloTagLC3 (HT-LC3) autophagosome completion assay. Briefly, this assay is able to distinguish expanding phagophores from nascent autophagosomes and mature autophagosomes or autolysosomes by sequentially labeling cytosol-accessible and autophagosome-sequestered HT-LC3 by using fluorescently tagged membrane-impermeable (MIL) and membrane-permeable (MPL) HaloTag ligands, respectively (Takahashi et al., 2018). As expected, phagophores (MIL+MPL−), autophagosomes (MIL+MPL+), and mature autophagic structures (MILMPL+) appeared in the cytoplasmic region of starved wild-type (WT) cells (Figures 1A a–c). Interestingly, all three autophagic structures (MIL+MPL−, MIL+MPL+, and MIL−MPL+ HT-LC3 foci) were also detected in Atg2A/B double knockout (CrAtg2A/B) cells (Figure 1A, d–j). Moreover, the addition of the lysosomal V-ATPase inhibitor Bafilomycin A1 (Baf-A1) slightly but significantly increased MPL signals in CrAtg2A/B cells (Figure 1C), indicating that autophagosomes still can form and mature into autolysosomes in the absence of Atg2A/B. Strikingly, however, we found that the MIL/MPL ratio in the presence of Baf-A1 in CrAtg2A/B cells was significantly higher than that in WT cells to indicate the accumulation of phagophores and/or autophagosomes (Figure 1D). Moreover, MIL signals were significantly increased in CrAtg2A/B cells even under non-starved conditions (Figures 1B), suggesting that Atg2A/B loss delays autophagosome completion under both basal and induced conditions.
Figure 1. Atg2A/B Is Required for Autophagosomal Membrane Expansion.

(A) HaloTag-LC3 (HT-LC3)-expressing wild-type (WT) and Atg2A/B double knockout (CrAtg2A/B) U-2 OS cells were incubated in complete media (CM) or starvation media (SM) in the presence or absence of 100 nM Baf-A1 for 2 h and subjected to the HT-LC3 autophagosome completion assay followed by confocal microscopy. Magnified images of the boxed (i–iii) and arrow-indicated areas (a–j) are shown in the middle and right panels, respectively. Schematic illustration of phagophore (MIL+MPL−), nascent autophagosome (MIL+MPL+), and mature autophagosome (MIL−MPL+) are shown on the upper right panel. Scale bars represent 20 μm and 1 μm in the magnified images.
(B and C) The cytoplasmic fluorescence intensities of MIL (B) and MPL (C) in each cell under indicated conditions were quantified and normalized to the respective mean fluorescence intensities of WT cells starved in the presence of Baf-A1 (n = 50).
(D) MIL/MPL ratio for each cell incubated in CM or SM in the presence of Baf-A1 was calculated and normalized to the mean of fluorescence intensities of WT cells (n = 50).
(E) The diameters of HT-LC3 foci randomly selected from the starved WT and CrAtg2A/B were quantified using the Velocity software (n = 100).
(F) WT and CrAtg2A/B U-2 OS cells were starved for 2 h and subjected for TEM. Representative autophagosome-like and autolysosome-like structures are shown on the upper and lower panels, respectively. Scale bars represent 200 nm.
(G) The diameter of each autophagic structure randomly selected from the electron micrographs was quantified (n = 20).
(H) HT-LC3-expressing CrAtg2A/B U-2 OS cells were starved for 2 h, stained for with MIL and MPL, and subjected to CLEM. Magnified images of the boxed areas are shown on the right panels.
Arrows and arrowheads indicate autophagosome-like and autolysosome-like structures, respectively. Scale bars represent 10 μm and 1 μm in the magnified image. Statistical significance was determined by one-way ANOVA followed by Dunn’s multiple comparisons test (B and C) or t test (D, E, and G). All values are mean ± SD; n.s., not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Notably, the sizes of HT-LC3 puncta in CrAtg2A/B U-2 OS cells were significantly smaller than those in WT cells (Figure 1E). To verify this phenotype, we performed transmission electron microscopy (TEM). Consistently, although autophagosomeand autolysosome-like structures were observed in both WT and CrAtg2A/B cells (Figure 1F), these autophagic structures were significantly smaller in Atg2A/B-deficient cells (Figure 1G). Taken together, these results indicate that mammalian Atg2A/B functions at the membrane expansion step during autophagosome biogenesis.
It has recently been reported that Atg2A/B deficiency in HeLa cells fails to form typical phagophores and autophagosomes and results in the accumulation of LC3 signals on small, single-membrane vesicle-like structures (Tamura et al., 2017). We, thus, next performed correlative light and electron microscopy (CLEM) and confirmed that HT-LC3-positive foci in CrAtg2A/B U-2 OS cells indeed represented autophagosome- and autolysosome-like structures (Figure 1H). To determine if the discrepancy is due to the difference in cell types, we generated Atg2A/B-deficient HeLa and HEK293T cells (Figures S1C and S1G) and analyzed autophagic structures by the HT-LC3 assay and electron microscopy. Consistent with the phenotype observed in U-2 OS cells, small autophagic structures were detected in Atg2A/B-deficient HEK293T and HeLa cells (Figures S2C–S2I), further supporting the role of mammalian Atg2A/B in phagophore expansion rather than formation.
Atg2A Localizes to ER-Mitochondria Contact Sites
To understand the mechanism behind the regulation of phagophore expansion by Atg2, we first determined the precise intracellular location of Atg2 by immunoelectron microscopy by using CrAtg2A/B cells stably expressing single guide RNA (sgRNA)-resistant EGFP-Atg2A. Interestingly, upon nutrient starvation, EGFP-Atg2A signals were detected at ER-mitochondria contact sites, also known as the MAM (Figure 2A). The localization of Atg2A was further determined by confocal microscopy. Consistent with a previous report (Velikkakath et al., 2012), EGFP-Atg2A signals were detected throughout the cytoplasmic region as well as in small foci structures under fed conditions (Figure 2B). The fluorescence intensity as well as the number of EGFPAtg2A puncta was significantly increased by starvation (Figures 2B–2D), suggesting the translocation of Atg2A during autophagy. Notably, although nearly all Atg2A puncta were found to be associated with the ER regardless of the nutrient status, a portion of the ER-associated Atg2A signals was also positive for a mitochondrial marker under fed conditiosn (Figures 2B and 2E). However, the percentage of Atg2A puncta associated with both ER and mitochondrial markers was significantly increased by starvation (Figures 2B and 2E), indicating the accumulation of Atg2A at MAM during autophagy.
Figure 2. Atg2A Localizes to MAM upon Autophagy Induction.

(A) CrAtg2A/B U-2 OS cells were transduced with lentiviruses encoding sgRNA-resistant EGFP-Atg2A, starved for 2 h and subjected to immunoelectron microscopy using anti-GFP antibody. Scale bars represent 200 nm.
(B) EGFP-Atg2A- and dsRed-ER-expressing CrAtg2A/B U-2 OS cells were incubated in CM or SM for 2 h, stained for TOM20, and analyzed by confocal microscopy. Magnified images of the boxed areas are shown in the lower panels. Scale bars represent 10 μm and 2.5 μm in the magnified images.
(C) The fluorescence intensity of EGFP-Atg2A in each cell under indicated conditions was quantified and normalized to the respective mean fluorescence intensity of cells incubated in CM (n = 40).
(D and E) Total EGFP-Atg2A puncta number per cell (D) and the percentage of EGFP-Atg2A puncta localize to cytosol (ER−MITO−), ER (ER+MITO−), mitochondria (ER−MITO−), or MAM (ER+MITO+) under CM or SM were quantified (n = 30) (E).
Statistical significance was determined by t test (C and D) or one-way ANOVA followed by Dunn’s multiple comparisons test (E). All values are mean ± SD. *p ≤ 0.05; ****p ≤ 0.0001.
Most recently, the membrane-tethering function of Atg2 has been demonstrated in vitro using liposomes (Neubert et al., 2019; Osawa et al., 2019; Valverde et al., 2019). Because Atg2A translocates to the MAM during starvation, we next examined if Atg2A/B is important for the formation of ER-mitochondrial contact sites. Electron microscopy showed that the number of MAM in CrAtg2A/B cells was comparable to that in WT cells (Figures S2A and S2B), indicating that the reduction of autophagosomal membrane size by Atg2A/B depletion is not due to the impairment of MAM formation.
Atg2A-WIPI4 Interaction Is Dispensable for Atg2A Function in Phagophore Expansion
It has been reported that knock down of Atg2A/B accumulates GFP-WIPI1-positive LC3 foci (Velikkakath et al., 2012). Consistently, we observed an accumulation of the PI3P marker mRFP-FYVE on GFP-ULK1-positive early autophagic structures in CrAtg2A/B cells in a manner that is dependent on the activation of the PI3-kinase (Figures S2C–S2E). Because the PI3Peffector Atg18 is required for the PAS recruitment of Atg2 in yeast (Obara et al., 2008), we next examined if WIPI4, a mammalian Atg18 ortholog, is crucial for Atg2A MAM localization and the function of Atg2A in phagophore expansion. Instead of WIPI4 knockout, which will disrupt multiple cellular functions of WIPI4, we used an Atg2A(YFS-AAA) mutant that abolishes Atg2A-WIPI4 interaction (Figures S3A and S3B), as previously reported (Zheng et al., 2017). To our surprise, the mutations in the YFS motif did not block the starvation-induced colocalization of EGFP-Atg2A with dsRed-ER and TOM20 (Figure S3C). Moreover, similar to EGFP-Atg2A(WT), EGFP-Atg2A(YFS-AAA) rescued the defects in phagophore expansion and autophagic degradation (Figures S3D–S3G) in CrAtg2A/B cells. Collectively, these results indicate that the Atg2A-WIPI4 interaction is dispensable for Atg2A translocation to the MAM and the phagophore expansion function of Atg2A.
TOM40 Is an Interacting Partner of Atg2A That Binds to the MLD
To determine the minimal region of Atg2A that is required for MAM localization, we next generated a series of Atg2 deletion mutants (Figure S4A). We found that a 45-amino acid sequence near the C-terminal end of Atg2A (amino acid 1776–1820) and located within the previously characterized autophagosome and lipid droplet localization region (Velikkakath et al., 2012) is sufficient for Atg2A localization to ER-mitochondria contacting sites and, thus, named this region the MLD. Notably, expression of EGFP-Atg2A(1776–1820) (EGFP-MLD) in CrAtg2A/B cells results in MAM localization regardless of nutrient status (Figures S4B and S4C).
To understand the mechanism underlying the MAM localization of Atg2A, we next took a proteomic approach to identify proteins that associate with the MLD. As Atg2A was previously suggested to only transiently associate with LC3-positive foci (Velikkakath et al., 2012), we took the advantage of photo-amino acid UV-crosslinking coupled with affinity purification and proteomics. To minimize the contamination of non-labeled bait proteins and the induction of apoptosis that is observed upon prolonged overexpression of EGFP-MLD (Figures S4D and S4E), we generated doxycycline-inducible EGFP-empty or EGFP-MLD-expressing CrAtg2A/B cells. The resultant cells were incubated with doxycycline together with photo-leucine and photo-methionine followed by UV-crosslinking, GFPTrap, and mass spectrometry. We recovered a total of 750 preys, and by employing EGFP-empty as a normalization control and a 5-fold increase threshold, 75 potential MLD interacting partners were identified and categorized according to their subcellular localization (Table S1). Consistent with the MAM localization of the MLD, over 55% of the potential interacting partners are either mitochondria- or ER-related proteins (Table S1), and these were selected for further analyses.
After validation by co-immunoprecipitation in the absence of photo-amino acid labeling and UV photo-crosslinking, we identified TOM40, the central component of the translocase of the outer mitochondrial membrane (TOM) complex (Dekker et al., 1998), as a MLD-binding protein (Figures 3A and 3B). The interaction of TOM40 with full-length Atg2A was confirmed by co-immunoprecipitation using anti-TOM40 antibodies or ectopic expression of EGFP-Atg2A followed by GFP-Trap (Figures S5A and 3C). To confirm that MLD mediates the interaction between full-length Atg2A and TOM40, we generated an EGFP-Atg2A mutant lacking the MLD (Figure 3A) and examined its interaction with TOM40. As expected, TOM40 interaction was abolished in the absence of MLD (Figure 3D), indicating that Atg2A interacts with TOM40 through the MLD. Importantly, the interaction of Atg2A with TOM40 was enhanced by starvation (Figures S4A and S4B), suggesting a role of TOM40 in the MAM translocation of Atg2A during autophagy. In contrast, the depletion of upstream ATGs including Atg13 and Atg9A had little effect on the interaction of Atg2A with TOM40 (Figures S5C and S5D). Collectively, these results identify TOM40 as an interacting partner of Atg2A that binds by its N-terminal MLD.
Figure 3. The MLD of Atg2A Is Responsible for the Interaction with TOM40.

(A) Schematic illustration of EGFP-Atg2A variants.
(B–D, G, and H) CrAtg2A/B HEK293T cells were transfected with the indicated EGFP-Atg2A variants for 24 h and subjected to immunoprecipitation with GFPTrap beads followed by immunoblotting with the indicated antibodies. EV and WCL represent EGFP-empty vector and whole-cell lysate, respectively.
(E) Amino acid sequences of the MLD and its variants used in the study are shown. Red underlined characters indicate the point mutations introduced into the sequences.
(F) Helical wheels of N-terminal (left) and C-terminal (right) α helices of MLD were plotted using HeliQuest. Red asterisks indicate amino acid residues substituted to alanine.
MLD Harbors Two Putative α Helices That Are Essential for TOM40 Interaction
To determine the residues of the MLD that are responsible for binding to TOM40, we first aligned the human Atg2A MLD protein sequence with its orthologs. We found that the MLD region and especially several positively charged and hydrophobic residues within this domain are highly conserved among all higher eukaryotes analyzed (Figure S6A). We next predicted the secondary and tertiary structure of the human MLD of Atg2A (Figures S6B and S6C) (Xu and Zhang, 2012, 2013). We found that the 45 amino acids of MLD comprise two α helices that are linked by a short coil (Figure S6C). Notably, three highly conserved arginine residues in the N-terminal helix of MLD are oriented on the same side (Figures 3E, 3F, and S6C), suggesting that these residues may mediate membrane binding (Li et al., 2013; Schwieger and Blume, 2009). Indeed, we observed that the arginine substitution to alanine (Figure 3F) reduced the foci formation of the MLD (Figure S6D). We next determined if the arginine residues are also important for the interaction with TOM40. We found that the MLD and TOM40 interaction was greatly suppressed by the arginine to alanine mutations (Figure 3G), suggesting that the arginine residues on the N-terminal helix of MLD are important not only for membrane recognition but also for the interaction of Atg2A with TOM40.
Photo-leucine and photo-methionine are used for the photoamino acid UV-crosslinking assay because protein-protein interactions frequently require the adaptation of hydrophobic residues into hydrophobic pockets of its interacting partner, such as p21-PCNA and BH3-mediated interactions of Bcl-2 family proteins (Gulbis et al., 1996; Shamas-Din et al., 2013). To determine if the conserved hydrophobic leucine residues on the C-terminal helix of MLD (Figure S6A) are required for interaction with TOM40, we generated a MLD mutant harboring leucine to alanine mutations (Figures 3E and 3F). Notably, both foci formation (Figure S6D) and TOM40 interaction (Figure 3G) were abolished by the leucine to alanine substitutions in the MLD. To confirm the importance of both helical regions of the MLD in the interaction of Atg2A with TOM40, we generated a full-length Atg2A mutant harboring both arginine and leucine to alanine substitutions in the MLD. Consistently, mutation of the conserved residues abolished the interaction of Atg2A with TOM40 (Figure 3H). Notably, Atg2A(L/R-A) maintains its interaction with WIPI4, whereas the WIPI4 interaction-defective Atg2A(YFS-AAA) mutant interacts with TOM40, indicating that the WIPI4 and TOM40 interactions of Atg2A are achieved by two independent domains (Figure S6E). Taken together, these data indicate that the MLD of Atg2A harbors two putative α helices that are essential for TOM40 interaction.
The MAM Localization of Atg2A Is Required for Phagophore Expansion and Autophagic Flux
To investigate whether TOM40 interaction of Atg2A is required for its MAM localization, we first analyzed the intracellular localization of the TOM40 interaction-defective Atg2A(MLD) and Atg2A(L/R-A) mutants. We found that neither EGFPAtg2A(ΔDMLD) nor EGFP-Atg2A(L/R-A) formed puncta or localized to MAM in response to nutrient starvation (Figures 4A and 4B), suggesting the importance of the Atg2A-TOM40 interaction in the membrane localization of Atg2A. We next investigated whether the MAM localization of Atg2A is crucial for the expansion of phagophores. In contrast to EGFP-Atg2A(WT), neither EGFP-Atg2A(L/R-A) nor EGFPAtg2A(ΔMLD) rescued the phagophore expansion defect in Atg2A/B-deficient cells (Figures 4C and 4D), indicating that MAM localization is a prerequisite for Atg2A to exert its biological function for proper membrane expansion. Furthermore, only EGFP-Atg2A(WT), but not EGFP-Atg2A(L/R-A) nor EGFP-Atg2A(ΔMLD), restored the impaired autophagic flux in CrAtg2A/B cells (Figures 4E–4J). Taken together, these data indicate that Atg2A exerts pro-autophagic function at the ER-mitochondria contacting sites by promoting phagophore expansion for efficient autophagic degradation.
Figure 4. MAM Localization of Atg2A Is Required for Its Biological Function in Autophagy.

(A) CrAtg2A/B U-2 OS cells stably expressing the indicated EGFP-Atg2A variants and dsRed-ER were starved for 2 h, stained for TOM20, and analyzed by confocal microscopy. Magnified images in the boxed areas are shown in the lower panels. Scale bars represent 20 μm and 5 μm in the magnified images.
(B) The fluorescence intensity in each cell expressing the indicated Atg2A variant was quantified and normalized to the respective mean fluorescence intensity of EGFP-Atg2A(WT) cells (n = 38).
(C and D) U-2 OS cells expressing the indicated EGFP-Atg2A variants were starved for 2 h, stained for LC3, and analyzed by confocal microscopy. In (C), LC3 is pseudo-colored green for visualization, and magnified images in the boxed areas are shown in the insets. Scale bars represent 20 μm and 5 μm in the insets. EV represents EGFP-empty vector. In (D), the diameters of LC3 foci were randomly selected and quantified using the Velocity software (n = 50).
(E–J) The indicated U-2 OS stable transductants were incubated in CM or SM for 2 h in the presence or absence of 100 nM Baf-A1 and subjected to immunoblotting with the indicated antibodies (E, G, and I). Basal and starvation-induced autophagic flux was quantified (F, H, and J; n = 3) as described in the STAR Methods. Statistical significance was determined by one-way ANOVA followed by Dunn’s multiple comparisons test. All values are mean ± SD; n.s., not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
TOM70 Mediates the Atg2A-TOM40 Interaction for Phagophore Expansion
Several additional TOM components, including TOM70, were detected in our proteomics analysis but failed to reach the five-fold enrichment threshold (Figure 5A). TOM70 and TOM20 are two of the major TOM receptors that transiently associate with the core TOM complex to deliver targeted proteins to TOM40 (Rapaport, 2002). Interestingly, a recent study has shown that TOM70, but not TOM20, clusters at MAM to recruit inositol trisphosphate receptor calcium transfer (Filadi et al., 2018). To determine if the TOM70-dependent delivery process is required for the Atg2A-TOM40 complex formation, TOM70-deficient U-2 OS cells were generated (Figure 5B) and subjected to co-immunoprecipitation analysis. Our data showed that the interaction of Atg2A with TOM40 was impaired by the loss of TOM70 (Figure 5C), suggesting a role of the TOM70 pathway in the recruitment of Atg2A to MAM during autophagy. To examine the importance of TOM70-dependent recruitment of Atg2A in phagophore expansion, we used the rapamycininduced FKBP (FK506-binding protein)/FRB (FKBP-rapamycin binding domain) dimerization system (Inobe and Nukina, 2016). To artificially induce the recruitment of Atg2A to MAM and the outer mitochondrial membrane (OMM) or the OMM alone, FRB-GFP-Atg2A(ΔMLD) was co-transduced to CrAtg2A/B cells with TOM70-mRFP-FKBP or TOM20-mRFP-FKBP, respectively. Rapamycin-induced dimerization of FRB-GFP-Atg2A (ΔMLD) and TOM70-mRFP-FKBP, but not TOM20-mRFPFKBP, restored the size of LC3 puncta in CrAtg2A/B cells (Figures 5D and 5E), indicating that TOM70-dependent recruitment of Atg2A to MAM is sufficient to induce phagophore growth upon the induction of autophagy. Notably, however, dimerization of FRB-GFP-Atg2A(ΔMLD) and TOM70-mRFP-FKBP occasionally generated abnormally enlarged LC3-positive structures (Figures 5E), and autophagic flux remained impaired in these cells (Figure 5F). These observations suggest that the dissociation of Atg2A from MAM may be required for functional autolysosome formation.
Figure 5. TOM70 Mediates the Atg2A-TOM40 Interaction and Atg2A-Dependent Phagophore Growth.

(A) TOM components and their corresponding peptide numbers identified in the proteomic analysis using EGFP-MLD are listed.
(B) HEK293T cells were transduced with lentiviruses encoding Cas9 and sgRNAs targeting TOM70 (CrTOM70) and subjected to immunoblotting with the indicated antibodies.
(C) WT or CrTOM70 HEK293T cells were transfected with the EGFP (EV) or EGFP-Atg2A (Atg2A) for 24 h and subjected to immunoprecipitation with GFP-Trap beads followed by immunoblotting with the indicated antibodies.
(D–F) The indicated CrAtg2A/B U-2 OS stable transductants were treated with DMSO or 150 nM rapamycin in the presence or absence of 100 nM Bafilomycin A1 for 8 h and subjected to LC3 immunostaining and confocal microscopy (D) or immunoblotting with the indicated antibodies (F). In (E), the diameters of LC3 foci in (D) were randomly selected and quantified using the Velocity software (n = 60).
Statistical significance was determined by one-way ANOVA followed by Dunn’s multiple comparisons test. All values are mean ± SD; n.s., not significant; ****p ≤ 0.0001.
Atg2A Interacts with Atg9A and Promotes Atg9A Vesicle Delivery for Phagophore Expansion
In yeast, Atg2 has been shown to interact with Atg9 to promote autophagy (Gómez-Sánchez et al., 2018). Consistently, we observed the interaction between Atg2A and Atg9A in mammalian cells (Figures 6A and S7B). Because Atg9 vesicles have been suggested to be a membrane source for phagophore growth (Webber et al., 2007), we next examined whether mammalian Atg9A is required for proper phagophore expansion. Similar to the loss of Atg2A/B, autophagic flux is impaired and LC3B puncta diameter is significantly reduced in Atg9A-deficient cells (Figures 6B–6D), supporting the importance of Atg9 vesicles in phagophore expansion. We next determined if mammalian Atg2 proteins are involved in Atg9A trafficking during autophagy. Consistent with previous studies (Orsi et al., 2012; Takahashi et al., 2011), Atg9A signals in starved WT cells were dispersed throughout the cytoplasmic region with only a small portion of the signals colocalizing with LC3 (Figures 6E and 6F). In contrast, the loss of Atg2A/B resulted in the accumulation of Atg9A signals on LC3-positive structures (Figure 6E). Using CLEM, we confirmed that the LC3 and Atg9A-positive structures in CrAtg2A/B cells represent autophagic structures (Figure 6G). Interestingly, the autophagic structures in CrAtg2A/B cells were surrounded by 30- to 50-nm Atg9A-GFP-positive vesicles (Figure 6G). Collectively, these results suggest that mammalian Atg2 proteins are dispensable for the anterograde trafficking of Atg9A but may mediate Atg9A vesicle delivery for phagophore expansion.
Figure 6. Atg2A/B Deficiency Accumulates Atg9 Vesicles at the Autophagosome Formation Sites in the Close Proximity of the MAM.

(A) GFP- or Atg9A-GFP-expressing CrAtg2A/B HEK293T cells were transfected with myc-Atg2A for 24 h and subjected to immunoprecipitation with GFP-Trap beads followed by immunoblotting with the indicated antibodies.
(B) WT and CrAtg9A U-2 OS cells were incubated in CM or SM in the presence or absence of 100 nM Baf-A1 and subjected to immunoblotting with the indicated antibodies.
(C) The indicated U-2 OS cells were starved for 2 h, stained for LC3, and analyzed by confocal microscopy. Magnified images in the boxed areas are shown in the insets. Scale bars represent 20 μm and 5 μm in the magnified images.
(D) The diameters of LC3 foci were randomly selected and quantified using the Velocity software (n = 100).
(E) HT-LC3-expressing WT and CrAtg2A/B U-2 OS cells were starved for 2 h, stained for Atg9A and MPL (TMR), and analyzed by confocal microscopy. Magnified images of the boxed areas are shown in the right panels (i and ii) and insets (iii). Scale bars represent 20 μm, 2 μm in the magnified images, and 0.5 μm in the insets.
(F) Colocalization coefficient of HT-LC3 with Atg9A per cell was quantified using the Velocity software and shown (n = 40).
(G) CrAtg2A/B U-2 OS cells stably expressing HT-LC3 and Atg9A-GFP were starved for 2 h, stained for MPL (TMR), and subjected to CLEM. Scale bars represent 10 μm and 0.5 μm in the magnified images. Magnified images of the boxed areas are shown in the middle (i) and right panels (a and b). Red and blue arrowheads indicate Atg9A-GFP-positive vesicles and HT-LC3-positive autophagic structures, respectively.
To better understand the function of mammalian Atg2 proteins at MAM, we next examined if the Atg2-Atg9 interaction is crucial for phagophore expansion and autophagic degradation. Using a series of deletion mutants (Figure S7A), we identified that amino acids 237–431 in the N terminus of Atg2A are essential for the interaction of Atg2A with Atg9A (Figures 7A, 7B, and S7B). As both EGFP-Atg2A and EGFP-Atg2A(Δ237–431) harbor intact MLD, similar TOM40 interactions were detected (Figure S7B), suggesting that the Atg2A-Atg9A interaction is achieved independently from the MLD-mediated TOM40 interaction. Consistently, the starvation-induced MAM localization of EGFP-Atg2A(Δ237–431) remains unaffected (Figure S7C). Taken together, these data indicate that EGFP-Atg2A(Δ237–431) localizes to MAM through interaction with TOM40 in response to autophagic stimuli; however, its interaction with Atg9A is abolished.
Figure 7. The Interaction of Atg2A with Atg9 Is Indispensable for Phagophore Expansion.

(A) Schematic diagrams of EGFP-tagged WT and Atg9A binding-defective Atg2A variant.
(B) GFP- or Atg9A-GFP-expressing CrAtg2A/B HEK293T cells were transfected with myc-Atg2A variants for 24 h and subjected to immunoprecipitation with GFP-Trap beads followed by immunoblotting with the indicated antibodies.
(C) U-2 OS cells expressing the indicated EGFP-Atg2A variants were starved for 2 h, stained for LC3, and analyzed by confocal microscopy. LC3 is pseudocolored green for visualization, and magnified images in the boxed areas are shown in the insets. Scale bars represent 20 μm and 5 μm in the magnified images. EV represents EGFP-empty vector.
(D) The diameters of LC3 foci were randomly selected and quantified using the Velocity software (n = 120). EV, WT, and Δ represent EGFP-empty vector, EGFP-Atg2A(WT), and EGFP-Atg2A(Δ237–431), respectively.
(E and F) The indicated U-2 OS cells were incubated in CM or SM for 2 h in the presence or absence of 100 nM Baf-A1 and subjected to immunoblotting with the indicated antibodies (E). Basal and starvation-induced autophagic flux in the indicated U-2 OS stable transductants were quantified as described in the STAR Methods (n = 3) (F).
Statistical significance was determined by one-way ANOVA followed by Dunn’s multiple comparisons test. All values are mean ± SD; n.s., not significant; **p ≤ 0.01; ****p ≤ 0.0001.
We next examined whether the Atg2A-Atg9A interaction at the MAM is required for the phagophore expansion functions of Atg2A. Although both Atg2A(WT) and Atg2A(Δ237–431) localize to MAM upon nutrient starvation (Figure S7C), the defects in phagophore expansion (Figures 7C and 7D) and autophagic degradation (Figures 7E and 7F) in CrAtg2A/B cells were only rescued by the restoration of EGFP-Atg2A(WT) but not the EGFP-Atg2A(Δ237–431) mutant. Moreover, unlike full-length Atg2A, which restored the defective Atg9A trafficking in CrAtg2A/B cells, the accumulation of Atg9A puncta adjacent to LC3-positive foci remained upon the expression of EGFPAtg2A(Δ237–431) (Figure S7D), indicating the importance of the Atg2A-Atg9A interaction in Atg9A vesicle delivery and subsequent retrograde trafficking. Collectively, these data indicate that Atg2A recruited to the MAM by a C-terminal MLD domain interacts with Atg9A at its N terminus to regulate phagophore expansion.
DISCUSSION
It has been proposed that the phagophore forms and grows at ER-mitochondria contact sites (Hamasaki et al., 2013), but the underlying molecular mechanisms have not been fully elucidated. In this study, we demonstrate the importance of the MAM localization of Atg2A in phagophore expansion. We found that the starvation-induced MAM localization of Atg2A is mediated through the interaction of OMM protein TOM70 and TOM40 and that Atg2A-MAM localization is required for proper Atg9A trafficking and autophagosomal membrane expansion. These results provide a mechanistic insight into phagophore growth and further support the importance of MAM in autophagosome biogenesis.
The observation that Atg2A/B depletion accumulates small immature autophagosomal membranes is consistent with previous reports (Tang et al., 2017; Velikkakath et al., 2012). However, although these studies initially suggested a role of Atg2A/B in phagophore closure, our data show that small phagophores in CrAtg2A/B cells still can undergo membrane closure to form autophagosomes that mature into autolysosomes. Because the size of autophagic structures in CrAtg2A/B cells is much smaller than that in WT cells, we propose that the failure of membrane expansion limits efficient membrane closure to accumulate immature autophagosomal membranes. Importantly, the observations that Atg2A/B deficiency significantly reduces the lysosomal turnover of LC3-II and impairs p62 degradation are consistent with the conclusion that Atg2 is required for efficient autophagic flux. Most recently, the involvement of endosomal sorting complex required for transport-III (ESCRT-III)-mediated membrane fission in phagophore closure has been demonstrated (Takahashi et al., 2018). Interestingly, although the sizes of phagophores were minimally affected by the depletion of ESCRT-III CHMP2A, Atg2A accumulated on p62- and Atg9-positive immature autophagic structures in CHMP2A-depleted cells (Takahashi et al., 2018). As the mechanism leading to ESCRT-III recruitment to phagophores is unknown, it will be of interest to determine whether Atg2A-mediated membrane expansion is a prerequisite for efficient recruitment of the ESCRT machinery.
During autophagosome biogenesis, many ATG proteins, including ATG14, DFCP1, ATG16, Beclin1, and VPS34, translocate to the autophagosome formation sites at MAMs (Hamasaki et al., 2013). In this study, we find that Atg2 also accumulates at MAMs during starvation. Interestingly, the MAM localization of Atg2 occurs independent of its ability to bind WIPI4, as the WIPI4 interaction-defective Atg2(YFS-AAA) mutant localizes to the MAM and rescues the defects in phagophore expansion and autophagic degradation in CrAtg2A/B cells. These results are unexpected, as yeast Atg18 has been suggested to dictate the PAS localization of Atg2 through the binding of PI3P (Kobayashi et al., 2012; Obara et al., 2008). Instead, we find that Atg2A localizes to MAM through a 45-amino acid domain located within the previously characterized autophagosome and lipid droplet localization region at the C terminus (Velikkakath et al., 2012). Notably, this domain is highly conserved and contains several essential arginine and leucine residues. As it is the minimal domain required for MAM localization and localizes to the MAM regardless of nutrient status, we named this the MLD.
Our proteomic analysis identifies the TOM component TOM40 as a MLD-interacting protein, and we have shown that TOM40 is responsible for the MAM localization of Atg2A through the TOM receptor TOM70. Despite restored phagophore expansion in CrAtg2A/B cells upon the artificial targeting of Atg2A(ΔMLD) to MAM by rapamycin-induced dimerization with TOM70, abnormally enlarged autophagic membranes can be observed and the defect in autophagic flux persists. As immature autophagic structures are reported to be detached from the ER in Atg2A/ B-depleted cells (Kishi-Itakura et al., 2014), the regulated assembly and disassembly of Atg2A-TOM40 interactions may be responsible for the establishment of phagophore-MAM contacting sites during phagophore expansion and the proper release of autophagosomes, but this remains to be studied. It also remains to be characterized how the MAM localization of Atg2 is controlled by nutrient status. Unlike full-length Atg2A, EGFPMLD strongly accumulates at MAM even under nutrient-rich culture conditions, suggesting that Atg2A may need to undergo conformational change or interact with an unknown co-factor in order to achieve MAM localization in response to autophagy induction.
During the revision of this manuscript, in vitro membrane tethering and lipid transfer activities have been demonstrated to reside within the N-terminal region of Atg2 (Neubert et al., 2019; Osawa et al., 2019; Valverde et al., 2019). As mitochondria have been shown to supply lipids including PE for the biogenesis of autophagosomes (Hailey et al., 2010), it is of future interest to investigate if Atg2A also mediates non-vesicular lipid transport at the MAM for phagophore expansion. Furthermore, the membrane source for the formation and/or expansion of the phagophore may also be supplied by vesicular trafficking (Longatti and Tooze, 2009). In this study, we observed that Atg2A/B deficiency accumulates Atg9A signals around LC3-positive autophagic structures. Although this observation is consistent with the notion that Atg2 regulates Atg9 retrieval from the PAS in yeast (Feng et al., 2014), we find that Atg9A vesicle-like structures are accumulated around autophagic structures at MAM in CrAtg2A/B cells to suggest that the membranes fail to be delivered to the expanding phagophore. Moreover, the Atg9-bindingdefective Atg2A(Δ237–431) mutant is capable of localizing to the MAM upon nutrient starvation but cannot restore phagophore expansion in CrAtg2A/B cells; thus, Atg2 at the MAM may function to tether or properly orient Atg9-containing vesicles for lipid delivery during phagophore expansion.
Collectively, we identify a minimal 45-amino acid MLD domain within the C terminus of Atg2A that is required for the TOM70/ TOM40-mediated translocation of Atg2A to the autophagosome formation site at the MAM for phagophore expansion. Although we show that an N-terminal domain of Atg2A supports Atg9mediated vesicular lipid transport for efficient autophagic flux, additional membrane tethering functions and/or lipid transfer activities of Atg2A at the MAM is an intriguing area for future study.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, HongGang Wang (hwang3@pennstatehealth.psu.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
U-2 OS (female), HeLa (female), and HEK293T (female) cells and indicated transfectants/transductants were cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM) supplemented with 10% FBS (VWR, 97068–091) and 1 × Antibiotic-Antimycotic solution (Corning, 30–004-CI). Cells were incubated at 37°C and 5% CO2.
METHOD DETAILS
Transfection and transduction
HEK293T cells were transfected using Calcium Phosphate method. Briefly, for 10 cm culture dish HEK293T cells, plasmid DNA (up to 15 μg) was diluted in 500 μL of 250 mM CaCl2 solution. 500 μL of 2 × HEPES-buffered saline (140 mM NaCl, 1.5mM Na2HPO4-2H2O, 50mM HEPES, pH 7.05) was added to DNA-CaCl2 mixture dropwise while vortex and incubated for 10 minutes at room temperature before adding to cells. Plasmid DNA and siRNA transfection in U-2 OS and HEK293T cells were performed using Nucleofector Kit V (Lonza, VCA-1003), jetPRIME (Polypus, 114–01) or FuGENE HD Transfection Reagent (Promega, E2311) per manufacturer’s instructions. Lentivirus production and transduction were performed as described previously (Young et al., 2012).
CRISPR/Cas9 genomic editing
CRISPR/Cas9-mediated knockout was performed as described previously (Tang et al., 2017). Briefly, sgRNAs targeting human TOM70, Atg2A, and Atg2B were sub-cloned into Lenti-sgRNA-EFS-GFP (TOM70), pLenti-CRISPR-V2 (Atg2A), and pLX-sgRNA (Atg2B) vectors, respectively. CrAtg2A/B U-2 OS, HEK293T, and HeLa cells were generated by infecting cells with pLenti-CRISPR-V2-sghAtg2A lentivirus followed by 7 days of puromycin selection. Cells were then subjected to transduction with pLXsgAtg2B lentivirus and blasticidin selection for 10 days. CrTOM70 HEK293T cells were generated by infecting cells with lentiCas9-Blast virus followed by 10 days of blasticidin selection. Cells were then subjected to transduction with Lenti-sgTOM70-EFS-GFP for 2 days and followed by GFP-positive FACS sorting. To generate Atg9A knockout cells, U-2 OS cells were transiently transfected with sgAtg9A-Cas9-P2A-GFP plasmid (Santa Cruz sc-408011) and followed by GFP-positive FACS sorting. Single clones were isolated, expanded, and screened for complete knockout by immunoblotting.
Autophagic flux assay
Cells were treated as described in the text prior to immunoblotting. The intensity of p62 and loading control was quantified using LI-COR Biosciences Image Studio software, and p62 levels were normalized to loading control. Basal autophagic flux (1) and starvation-induced autophagic flux (2) were calculated using the following formulas (Tooze et al., 2015): (1) [(complete media with BafA1 – complete media)/(complete media with BafA1)] × 100; (2) [(starvation media with BafA1 – starvation media)/(starvation media with BafA1)] × 100. All data were normalized to WT.
FACS cell death analysis
Cells were treated as described in the text. Cells were harvested by trypsinization and washed once in PBS prior to 5% Annexin-V and 7-AAD staining for 15 mins on ice. Cells were then washed once in Annexin-V binding buffer prior to FACS analysis.
HaloTag-LC3 autophagosome completion assay
The assay was performed as previously described (Takahashi et al., 2018). Briefly, cells were incubated at 37°C with AlexaFluor488-MIL in permeabilization buffer (3 nM XF-PMP in 1 × MAS buffer (220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA)) for 15 minutes. Equal volume of 2 × fixative (8% PFA and 400 mM sucrose in 1 × PBS) was added to cells and incubated at room temperature for 7 minutes. Cells were then washed with PBS and incubated with tetramethylrhodamine (TMR)-MPL in PBS at room temperature for 30 minutes. Finally, cells were washed with PSB before confocal microscopy.
Immunoblotting
Cells were collected on ice by scraping and washed with ice-cold PBS and then resuspended and incubated in RIPA buffer (25 mM Tris-HCl pH 8.0, 150mM NaCl, 1mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail) for 30 minutes on ice. Cell lysate supernatant was collected after 15 minutes 20,000 × g centrifugation. Protein concentrations were determined using BCA assay. Equal amount protein from each sample was denatured using 2 × laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris- HCL pH 6.8, 0.02% bromophenol blue, 10% 2-mercaptoethanol). Samples were then heated (100°C, except 70°C for Atg9A) for 10 minutes before electrophoresis. Proteins were then transferred to PVDF membrane overnight followed by blocking and antibody incubation. Membranes were imaged using Li-Cor Odyssey CLx Infrared Imaging System.
Co-immunoprecipitation
Co-IP using transfected HEK293T cells was performed as described previously with minor optimization (Tang and Takahashi, 2018). For TOM40 co-IP, cells were lysed in NP-40 lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40). For Atg9A-GFP co-IP, cells were lysed in TX-100 lysis buffer (50 mM Tris-HCl pH 7.4, 75 mM NaCl, 0.8% TX-100). For Atg9A co-IP, cells were homogenized in PBS by going through 27.5-gauge needle 20 times. Triton X-100 was then added to cell homogenate (0.01% final concentration). For WIPI4 co-IP, lysis buffer recipe was obtained from previous publication (Zheng et al., 2017). Cells were lysed for 30 minutes at 4°C while rotating. Cell lysates were then centrifuged for 15 minutes (20,000 × g, except 4000 × g for Atg9A) and supernatants were collected and subjected to BCA assay. 500 μg protein lysate from each sample was incubated with 25 μL post-washing GFP-Trap_MA beads for 3 hours at 4C while rotating. Beads were then washed 3 times with washing buffer (lysis buffer with 10% original detergent concentration) before immunoblotting analysis. For TOM40-Atg2A endogenous co-IP, magnetic protein-G beads (Bio-Rad, 1614021) were incubated with 10 μg TOM40 antibody (Santa Cruz) or normal mouse IgG in diluted (50% original detergent concentration) NP-40 lysis buffer for 2 hours at room temperature. Antibody-protein-G bead conjugates were washed 3 times in washing buffer prior to incubation with cell lysates (NP-40 lysis buffer) for 2 hours at room temperature. Beads were then washed 3 times with washing buffer (lysis buffer with 10% original detergent concentration) before immunoblotting analysis.
Fluorescence microscopy
Cells were seeded on 8-well chamber slides (Fisher Scientific, 12–565-1) overnight and subjected to indicated treatments. For TOM20 and Atg9A immunostaining, cells were fixed in 4% paraformaldehyde-PBS for 20 minutes in dark at room temperature and then subjected to 0.15% Triton X-100-PBS for 20 minutes or 100 μg/ml digitonin-PBS for 10 minutes at room temperature. Cells were rinsed with PBS 3 times and then incubated with 10% normal goat serum-PBS for 1h at room temperature before overnight incubation of indicated primary antibodies in 1% normal goat serum-PBS at 4°C. Cells were washed 3 time with PBS and then incubated with indicated secondary antibodies for 1 hour in dark at room temperature. Cells were rinsed 3 time with PBS before confocal microscopy. For endogenous LC3B immunostaining, cells were fixed in 4% paraformaldehyde-PBS briefly at room temperature (approximately 15–30 s) and then incubated with ice-cold methanol for 10 mins at −20°C. Cells were rinsed with PBS 3 times before 1.5 hours incubation with anti-LC3B antibody in 1% BSA-0.1% TWEEN-20-PBS. Cells were then rinsed with 0.1% TWEEN-20-PBS 3 times and subjected to incubation with indicated secondary antibodies and confocal microscopy. For HaloTag-LC3 MPL staining, cells were incubated with MPL in PBS for 1 hour and then washed 3 times with PBS. Leica AOBS SP8 with 63 × water-immersion lens or 63 × oil-immersion lens was used to analyze samples. Fluorescent images were then deconvolved, processed, and quantified using Huygens (Scientific Volume Imaging), IMARIS (Oxford Instruments), and Volocity (PerkinElmer) software, respectively.
Transmission electron microscopy, immuno-gold electron microscopy, and correlative light electron microscopy
TEM, immuno-TEM, and CLEM were performed as described previously (Takahashi et al., 2018). Briefly, for TEM, cells were seeded on plastic coverslip (Thermo Scientific 174950) in 24-well plate overnight. Cells were then treated with indicated conditions followed by 1 hour fixation in dark at room temperature (2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.3). Samples were then incubated with osmium fixation buffer (1% osmium tetroxide with or without 1.5% potassium ferrocyanide in 0.1 M sodium cacodylate, pH 7.3) for 1 hour followed by serial ethanol dehydration and resin embedding (Electron Microscopy Sciences, 14120). 70 nm sample sections were mounted on mesh copper grids followed by uranyl acetate and lead citrate staining before analyzed using JEOL JEM 1400 transmission electron microscope. For immuno-TEM, cells were seeded on plastic coverslip in 24-well plate overnight and treated with indicated conditions followed by 2 hours fixation (4% paraformaldehyde in phosphate buffer (PB), pH 7.4) in dark at room temperature. Samples were incubated in permeabilization buffer (0.25% saponin in PB) for 30 minutes followed by 1 hour incubation with blocking buffer (10% BSA, 10% normal goat serum, 0.1% cold water fish gelatin, 0.1% saponin in PB). Samples were incubated with the primary antibody at 4°C overnight and incubated with the gold-conjugated secondary antibody at room temperature for 1 hour followed by 10 minutes fixation (1% glutaraldehyde in PB), 15 minutes washing (50 mM glycine in PBS), and signal intensification using GoldEnhance EM for 5 minutes. Samples were then processed as described above. For CLEM, cells were seeded on Gridded Glass Bottom Dish (MatTek, P35G-1/5–14-C-GRID) overnight. Cells were then treated with indicated conditions and subjected to HaloTag-LC3 autophagosome completion assay or solely MPL(TMR) staining. Samples were analyzed by confocal microscopy and cells of interest were then processed for TEM, identified by grid number.
Photo-amino acid, UV crosslinking, and proteomic analysis
HEK293T cells were transduced with pCDH1-tet-on-EGFP or pCDH1-tet-on-EGFP-MLD lentivirus. Cells were then selected with 2 μg/ml puromycin for 4 days. 9 × 106 cells were seeded on 10 cm dish overnight and medium was replaced with 10 mL photoleucine and photo-methionine containing medium in the presence of 2 μg/ml doxycycline hyclate for 24 hours. Cells were then placed 2 cm under a 20 W 365 nm UV bench lamp (Blak-Ray XX-20BLB) and irradiated for 20 minutes. Cells were harvested and lysed on ice using NP-40 co-IP lysis buffer. Protein concentrations were determined using BCA assay. 500 μg lysate from each sample was incubated with 25 μL of post-washed GFP-Trap_MA beads for 3 hours at 4°C while rotating. Beads were then washed for 10 times and proteins were eluted with 25 μL 2 × laemmli buffer and 100°C heating for 10 minutes. Samples were then submitted for proteomic analysis at the Rutgers University Biological Mass Spectrometry Facility.
Rapamycin-induced oligomer formation system of FRB-FKBP fusion proteins
U-2 OS cells were transduced with lentivirus encoding FRB-EGFP-Atg2A(ΔMLD) followed by TOM20-mRFP-FKBP or TOM70-smRFP-FKBP prior to EGFP and mRFP double positive FACS sorting. Cells were treated with DMSO or 150 nM rapamycin for 8 hours prior to immunostaining and confocal microscopy.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence microscopy quantifications were performed using Velocity and IMARIS software. Immunoblotting quantifications were performed using Image Studio software.
Statistical significance was determined using Graph Pad Prism 7.0. Statistical computation, threshold for significance, and n numbers were indicated on each figure.
DATA AND CODE AVAILABILITY
Original proteomics data for Figure 5A and Table S1 in the paper is available at Mendeley Data https://doi.org/10.17632/tn4p7g48hm.2.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal anti-Atg2A | MBL International | Cat# PD041; RRID: AB_2810871 |
| rabbit polyclonal anti-Atg2B | Proteintech Group | Cat# 25155–1-AP; RRID: AB_2810874 |
| rabbit monoclonal anti-Atg9A | Cell Signaling Technology | Cat# 13509; RRID: AB_2798241 |
| mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| mouse monoclonal β-actin | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| rabbit polyclonal anti-GFP | Abcam | Cat# ab6556; RRID: AB_305564 |
| mouse monoclonal anti-GST | Santa Cruz Biotechnology | Cat# sc-138; RRID: AB_627677 |
| rabbit monoclonal anti-HSP90 | Cell Signaling Technology | Cat# 4877; RRID: AB_2233307 |
| rabbit monoclonal anti-LC3B | Cell Signaling Technology | Cat# 3868; RRID: AB_2137707 |
| rabbit monoclonal anti-Atg13 | Cell Signaling Technology | Cat# 13273; RRID: AB_2798169 |
| rabbit polyclonal anti-LC3 | Novus Biologicals | Cat# NB100–2220; RRID: AB_10003146 |
| mouse monoclonal anti-mCherry | Abcam | Cat# ab125096; RRID: AB_11133266 |
| mouse monoclonal anti-Myc-Tag | Cell Signaling Technology | Cat# 2276; RRID: AB_331783 |
| normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025; RRID: AB_737182 |
| mouse monoclonal anti-TOM70 | Santa Cruz Biotechnology | Cat# sc-390545; RRID: AB_2714192 |
| guinea pig polyclonal anti-p62 | American Research Product | Cat# 03-GP62-C; RRID: AB_1542690 |
| mouse monoclonal anti-TOM20 | Santa Cruz Biotechnology | Cat# sc-17764; RRID: AB_628381 |
| mouse monoclonal anti-TOM40 | Santa Cruz Biotechnology | Cat# sc-365467; RRID: AB_10847086 |
| donkey anti-rabbit IgG IRDye 800CW | LiCOr | Cat# 925–32213; RRID: AB_2715510 |
| donkey anti-rabbit IgG IRDye 680RD | LiCOr | Cat# 925–68073; RRID: AB_2716687 |
| donkey anti-mouse IgG IRDye 800CW | LiCOr | Cat# 925–32212; RRID: AB_2716622 |
| donkey anti-mouse IgG IRDye 680RD | LiCOr | Cat# 925–68072; RRID: AB_10953628 |
| donkey anti-guinea pig IgG IRDye 680RD | LiCOr | Cat# 925–68077; RRID: AB_10956079 |
| Nanogold-Fab goat anti-rabbit IgG | Nanoprobes | Cat# 2004; RRID: AB_2631182 |
| goat anti-rabbit IgG Alexa Fluor 488 | Life Technologies | Cat# A-11008; RRID: AB_143165 |
| goat anti-rabbit IgG Alexa Fluor 568 | Life Technologies | Cat# A-11031; RRID: AB_144696 |
| goat anti-mouse IgG Alexa Fluor 647 | Life Technologies | Cat# A-21238; RRID: AB_2535807 |
| GFP-Trap | ChromoTek | Cat# gtma-20; RRID: AB_2631406 |
| myc-Trap | ChromoTek | Cat# ytma-20; RRID: AB_2631370 |
| Protein G Magnetic Beads | Bio-Rad | Cat# 1614021; RRID: AB_2021282 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bafilomycin A1 | LC Laboratories | Cat# B-1080 |
| 7-AAD | BioLegend | Cat# 420404 |
| APC Annexin V | BioLegend | Cat# 640941 |
| Rapamycin | LKT Laboratories | Cat# R0161 |
| 3-Methyladenine | Sigma-Aldrich | Cat# M9281 |
| XF Plasma Membrane Permeabilizer | Seahorse Bioscience | Cat# 102504–100 |
| HaloTag Alexa Fluor 488 Ligand | Promega | Cat# G1001 |
| HaloTag TMR Ligand | VWR | Cat# PAG8251 |
| 16% Paraformaldehyde Aqueous Solution | Electron Microscopy Sciences | Cat# 15710 |
| GoldEnhance EM (original) | Nanoprobes | Cat# 2113 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| L-Photo-Methionine | Thermo Fisher Scientific | Cat# 22615 |
| L-Photo-Leucine | Thermo Fisher Scientific | Cat# 22610 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8340–5ML |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23252 |
| Deposited Data | ||
| Original proteomics dataset | Mendeley | https://data.mendeley.com/datasets/tn4p7g48hm/draft?a=b57fc569–4c2b-4012–9fee-f989309ebed2 |
| Experimental Models: Cell Lines | ||
| U-2 OS | ATCC | HTB-96 |
| HEK293T | ATCC | CRL-3216 |
| HeLa | ATCC | CCL-2 |
| Oligonucleotides | ||
| Atg13 ON-TARGETplus SMARTpool - Human | Dharmacon | Cat# L-020765–01-0005 |
| TOM40 ON-TARGETplus SMARTpool - Human | Dharmacon | Cat# L-012732–00-0005 |
| sgAtg2A 5′-CGCTGCCCTTGTACAGATCG-3′ | Tang et al., 2017 | N/A |
| sgAtg2B 5′-ATGGACTCCGAAAACGGCCA-3′ | Tang et al., 2017 | N/A |
| sgAtg9A | Santa Cruz Biotechnology | Cat# sc-408011 |
| sgTOM70 5′-GGCGCGTATACAGCGGGCTA-35′-TAGCCCGCTGTATACGCGCC-3 | This paper | N/A |
| Recombinant DNA | ||
| pCDH1-HTLC3 | Takahashi et al., 2018 | N/A |
| pCDH1-EGFP-Atg2A(WT) | Tang et al., 2017 | N/A |
| pEGFP-Atg2A(WT) | This paper | N/A |
| pEGFP-MLD | This paper | N/A |
| pCDH1-dsRed-ER | This paper | N/A |
| pCDH1-EGFP-Atg2A(ΔMLD) | This paper | N/A |
| pEGFP-Atg2A(ΔMLD) | This paper | N/A |
| pEGFP-MLD(R-A) | This paper | N/A |
| pEGFP-MLD(L-A) | This paper | N/A |
| pCDH1-EGFP-Atg2A(L/R-A) | This paper | N/A |
| pEGFP-Atg2A(L/R-A) | This paper | N/A |
| pCDH1-EGFP-Atg2A(YFS-AAA) | This paper | N/A |
| pEGFP-Atg2A(YFS-AAA) | This paper | N/A |
| pCDH1-FRB-EGFP-Atg2A(ΔMLD) | This paper | N/A |
| pCDH1-TOM20-mRFP-FKBP | This paper | N/A |
| pCDH1-TOM70-mRFP-FKBP | This paper | N/A |
| pCAG-GST-WIPI4 | This paper | N/A |
| pCDH1-Atg9A-GFP | This paper | N/A |
| pBW-myc-Atg2A(WT) | This paper | N/A |
| pCDH1-EGFP-Atg2A(Δ237–431) | This paper | N/A |
| pEGFP-Atg2A(Δ237–431) | This paper | N/A |
| pBW-myc-Atg2A(Δ237–431) | This paper | N/A |
| pCDH1-tet-on-EGFP-EF1-Puro-hUbC-rtTA | This paper | N/A |
| pCDH1-tet-on-EGFP-Atg2A(MLD)-EF1-PurohUbC-rtTA | This paper | N/A |
| pLenti-CRISPR-V2-sghAtg2A | Tang et al., 2017 | N/A |
| pLX-sghAtg2B | Tang et al., 2017 | N/A |
| LRG-sgTOM70 | This paper | N/A |
| Software and Algorithms | ||
| Prism 7 | GraphPad | https://www.graphpad.com |
| IMARIS | Oxford Instruments | https://imaris.oxinst.com |
| Velocity | PerkinElmer | https://www.perkinelmer.com/category/imageanalysis-software |
| Huygens Professional | Scientific Volume Imaging | https://svi.nl/Huygens-Professional |
| Image Studio Lite Ver 5.2 | Li-Cor | https://www.licor.com/bio/image-studio-lite/ |
Highlights.
Human Atg2 functions at the phagophore expansion step
Atg2A translocates to MAM during autophagy
TOM40/70 complex recruits Atg2A to MAM
Atg2A regulates Atg9-vesicle delivery for phagophore expansion at MAM
ACKNOWLEDGMENTS
This work was supported by the NIH grants GM127954 and CA222349, the Hyundai Hope On Wheels, the Four Diamonds, and the Lois High Berstler Research Endowment Fund. Authors would like to thank Nate Sheaffer, Joseph Bednarczyk, and Jade Vogel from the Penn State College of Medicine Flow Cytometry Core for assistance with the flow cytometry; Thomas Abraham and Wade Edris of the Penn State College of Medicine Imaging Core for assistance with the confocal microscopy; and Haiyan Zheng of the Rutgers University Biological Mass Spectrometry Facility for assistance with the proteomic analysis.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.036.
REFERENCES
- Choi AM, Ryter SW, and Levine B (2013). Autophagy in human health and disease. N. Engl. J. Med 368, 651–662. [DOI] [PubMed] [Google Scholar]
- Dekker PJ, Ryan MT, Brix J, Müller H, Hönlinger A, and Pfanner N (1998). Preprotein translocase of the outer mitochondrial membrane: molecular dissection and assembly of the general import pore complex. Mol. Cell. Biol 18, 6515–6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, and Klionsky DJ (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Leal NS, Schreiner B, Rossi A, Dentoni G, Pinho CM, Wiehager B, Cieri D, Calì T, Pizzo P, and Ankarcrona M (2018). TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol 28, 369–382.e6. [DOI] [PubMed] [Google Scholar]
- Gómez-Sánchez R, Rose J, Guimarães R, Mari M, Papinski D, Rieter E, Geerts WJ, Hardenberg R, Kraft C, Ungermann C, and Reggiori F (2018). Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol 217, 2743–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, and Thorburn A (2016). The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 37, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, and Kuriyan J (1996). Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 87, 297–306. [DOI] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, and Lippincott-Schwartz J (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oo-mori H, Noda T, Haraguchi T, Hiraoka Y, et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Imai K, Hao F, Fujita N, Tsuji Y, Oe Y, Araki Y, Hamasaki M, Noda T, and Yoshimori T (2016). Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci 129, 3781–3791. [DOI] [PubMed] [Google Scholar]
- Inobe T, and Nukina N (2016). Rapamycin-induced oligomer formation system of FRB-FKBP fusion proteins. J. Biosci. Bioeng 122, 40–46. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, and Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi-Itakura C, Koyama-Honda I, Itakura E, and Mizushima N (2014). Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci 127, 4089–4102. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ (2004). Cell biology: regulated self-cannibalism. Nature 431, 31–32. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Suzuki K, and Ohsumi Y (2012). Autophagosome formation can be achieved in the absence of Atg18 by expressing engineered PAS-targeted Atg2. FEBS Lett. 586, 2473–2478. [DOI] [PubMed] [Google Scholar]
- Li L, Vorobyov I, and Allen TW (2013). The different interactions of lysine and arginine side chains with lipid membranes. J. Phys. Chem. B 117, 11906–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longatti A, and Tooze SA (2009). Vesicular trafficking and autophagosome formation. Cell Death Differ. 16, 956–965. [DOI] [PubMed] [Google Scholar]
- Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, Lin L, Kovács AL, Yu L, and Zhang H (2011). The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell 21, 343–357. [DOI] [PubMed] [Google Scholar]
- Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, and Singh R (2013). Autophagy proteins regulate ERK phosphorylation. Nat. Commun 4, 2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, and Yoshimori T (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol 152, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, and Ohsumi Y (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol 10, 458–467. [DOI] [PubMed] [Google Scholar]
- Neubert P, Weichselbaum A, Reitinger C, Schatz V, Schröder A, Ferdinand JR, Simon M, Bär AL, Brochhausen C, Gerlach RG, et al. (2019). HIF1A and NFAT5 coordinate Na+-boosted antibacterial defense via enhanced autophagy and autolysosomal targeting. Autophagy 14, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara K, Sekito T, Niimi K, and Ohsumi Y (2008). The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem 283, 23972–23980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, and Tooze SA (2012). Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 23, 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, Ohsumi Y, and Noda NN (2019). Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol 26, 281–288. [DOI] [PubMed] [Google Scholar]
- Proikas-Cezanne T, Takacs Z, Dönnes P, and Kohlbacher O (2015). WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci 128, 207–217. [DOI] [PubMed] [Google Scholar]
- Rapaport D (2002). Biogenesis of the mitochondrial TOM complex. Trends Biochem. Sci 27, 191–197. [DOI] [PubMed] [Google Scholar]
- Schwieger C, and Blume A (2009). Interaction of poly(L-arginine) with negatively charged DPPG membranes: calorimetric and monolayer studies. Biomacromolecules 10, 2152–2161. [DOI] [PubMed] [Google Scholar]
- Shamas-Din A, Kale J, Leber B, and Andrews DW (2013). Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol 5, a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kubota Y, Sekito T, and Ohsumi Y (2007). Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Akioka M, Kondo-Kakuta C, Yamamoto H, and Ohsumi Y (2013). Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci 126, 2534–2544. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Meyerkord CL, Hori T, Runkle K, Fox TE, Kester M, Loughran TP, and Wang HG (2011). Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Tsotakos N, Liu Y, Young MM, Serfass J, Tang Z, Abraham T, and Wang HG (2016). The Bif-1-Dynamin 2 membrane fission machinery regulates Atg9-containing vesicle generation at the Rab11-positive reservoirss. Oncotarget 7, 20855–20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, Serfass JM, Chen L, Gebru M, Chen C, et al. (2018). An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun 9, 2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Nishimura T, Sakamaki Y, Koyama-Honda I, Yamamoto H, and Mizushima N (2017). Differential requirement for ATG2A domains for localization to autophagic membranes and lipid droplets. FEBS Lett. 591, 3819–3830. [DOI] [PubMed] [Google Scholar]
- Tang Z, and Takahashi Y (2018). Analysis of Protein-Protein Interaction by Co-IP in Human Cells. Methods Mol. Biol 1794, 289–296. [DOI] [PubMed] [Google Scholar]
- Tang Z, Takahashi Y, Chen C, Liu Y, He H, Tsotakos N, Serfass JM, Gebru MT, Chen H, Young MM, and Wang HG (2017). Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 24, 2127–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Minematsu-Ikeguchi N, Ueno T, and Kominami E (2005). Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1, 84–91. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Dooley HC, Jefferies HB, Joachim J, Judith D, Lamb CA, Razi M, and Wirth M (2015). Assessing mammalian autophagy. Methods Mol. Biol 1270, 155–165. [DOI] [PubMed] [Google Scholar]
- Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, Reinisch KM, and Melia TJ (2019). ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol 218, 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, and Mizushima N (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber JL, Young AR, and Tooze SA (2007). Atg9 trafficking in Mammalian cells. Autophagy 3, 54–56. [DOI] [PubMed] [Google Scholar]
- Xu D, and Zhang Y (2012). Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, and Zhang Y (2013). Toward optimal fragment generations for ab initio protein structure assembly. Proteins 81, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, and Ohsumi Y (2012). Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol 198, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al. (2012). Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem 287, 12455–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JX, Li Y, Ding YH, Liu JJ, Zhang MJ, Dong MQ, Wang HW, and Yu L (2017). Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy 13, 1870–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original proteomics data for Figure 5A and Table S1 in the paper is available at Mendeley Data https://doi.org/10.17632/tn4p7g48hm.2.