

Abstract
Dihydroxyacetone phosphate (DHAP) is the endogenous by-product of fructose metabolism. Excess DHAP in cells can induce advanced glycation end products and oxidative stress. Dihydroxyacetone (DHA) is the triose precursor to DHAP. DHA is used as the active ingredient in sunless tanning products, including aerosolized spray tans, and is formed by the combustion of solvents found in electronic cigarettes. Human exposure to DHA has been increasing as the popularity of sunless tanning products and electronic cigarettes have grown. Topically applied DHA is absorbed through the viable layers of the skin and into the bloodstream. Exogenous exposure to DHA is cytotoxic in immortalized keratinocytes and melanoma cells with cell cycle arrest induced within 24 h and cell death occurring by apoptosis at consumer relevant concentrations of DHA within 72 h. Less is known about systemic exposures to DHA that occur following absorption through skin, and now through inhalation of the aerosolized DHA used in spray tanning. In the present study, HEK 293T cells were exposed to consumer-relevant concentrations of DHA to examine the cytotoxicity of systemic exposures. HEK 293T cells were sensitive to consumer-relevant doses of DHA with an IC50 value of 2.4 ± 0.3 mM. However, cell cycle arrest did not begin until 48 h after DHA exposure. DHA exposed cells showed altered metabolic activity with decreased mitochondrial function and decreased lactate and ATP production observed within 24 h of exposure. Autofluorescent imaging and NAD+ sensors also revealed an imbalance in the redox cofactors NAD+/NADH within 24 h of exposure. Cell death occurred by autophagy indicated by increases in LC3B and SIRT1. Despite DHA’s ability to be converted to DHAP and integrated into metabolic pathways, the metabolic dysfunction and starvation responses observed in the HEK 293T cells indicate that DHA does not readily contribute to the energetic pool in these cells.
Keywords: dihydroxyacetone, sunless tanning, mitochondria, cell cycle arrest, autophagy, NAD, redox, oxidative stress, GSH, glycolysis, Krebs, ATP
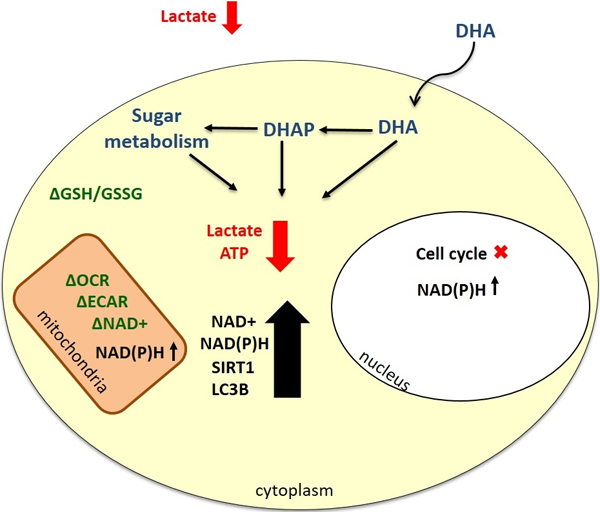
GRAPHICAL ABSTRACT
INTRODUCTION
A major alternative to ultraviolet (UV) tanning is the use of sunless tanning products (STPs), which contain dihydroxyacetone (DHA) as their active ingredient. DHA is the triose precursor to dihydroxyacetone phosphate (DHAP) formed along with glyceraldehyde-3-phosphate (GAP) during fructose metabolism. Used topically, DHA has been considered a safe tanning agent due to its simple carbohydrate structure and because it was thought to remain in the outer dead layers of skin. However, reports have established that up to 11% of the applied DHA (typically 5–15% w/v) penetrates into the viable layers of skin and 0.5% of the DHA applied enters the bloodstream. 1, 2 This amounts to high micromolar to low millimolar levels of DHA entering circulation.
Beyond topical application, DHA can also be inhaled through aerosolized applications of STPs or inhaled through electronic cigarette vapor. Characterization of the components in electronic cigarette vapor revealed that DHA is formed by the oxidation of glycerol via heat-initiated free radical chemistry. 3, 4 While inhalation measurements from spray tanning have not been conducted, electronic cigarettes release 0.5–2.33 µgDHA/puff ([DHA]puff =10 nm – 42.5 µM), which roughly translates to between 0.14 to 600 µM exposure per vaping session. 3, 5 Depending on the frequency of vaping, again high micromolar to low millimolar levels of DHA may enter the bloodstream.
We previously demonstrated that an exogenous exposure of DHA at 5 mM was cytotoxic to melanoma cells and a previous report demonstrated that DHA exposure at 5–25 mM induced DNA damage and was cytotoxic to immortalized keratinocytes. 6, 7 Interestingly, the sensitivity to DHA and mechanism of cell death differed between these two cell types with the melanoma cells becoming senescent before eventually becoming apoptotic, while the keratinocytes showed a rapid increase in DNA damage and a more rapid onset of apoptosis. 6, 7
Despite emerging evidence that DHA exposures are increasing and extending beyond the external-use-only ruling of the FDA, little is known about the systemic exposure effects of DHA. 8 Injection of hyperpolarized [2-13 C] DHA as a metabolic probe in mice has demonstrated that DHA is rapidly distributed by the bloodstream and metabolized by the liver and kidneys. 9, 10 These studies utilized high millimolar doses of DHA (80 mM), but only monitored organ behavior on the scale of minutes. 9 Given the cytotoxic response of low millimolar doses in skin models, here we examined low millimolar DHA exposure effects in the human embryonic kidney cell lines HEK 293T.
MATERIAL AND METHODS
Chemicals
Dihydroxyacetone (CAS 26776–70-5) was purchased from Sigma-Aldrich and used fresh.
Cell culture
HEK 293T human embryonic kidney cells were purchased from ATCC (Manassas, VA, USA) and grown at 37°C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Logan, UT, USA) supplemented with glutamine, 10% fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA, USA), and 1% sodium pyruvate (Gibco, Carlsbad, CA, USA). Cell passages were between 4 and 15 for all reported experiments. The cells were routinely tested for mycoplasma using Lonza MycoAlert kit (Walkersville, MD, USA) and found to be free of mycoplasma contamination.
Cytotoxicity studies
Cytotoxicity was determined by growth inhibition assays. HEK 293T were seeded at a density of 40,000 cells/well in six-well dishes. The following day, the cells were exposed to a range of DHA concentrations and incubated at 37 °C in a 5% CO2 incubator for 5 days. In our previous work with A375P melanoma cells, we found that 24 h exposure to 5 mM DHA and continuous exposure to 5 mM DHA gave the same IC50 value, so the cells here were dosed continuously. 7 After the treatment period, the medium was aspirated from the dish, and the cell monolayer was washed with phosphate buffered saline (PBS, VWR Life Sciences, Radnor, PA, US) and then exposed to 0.25% trypsin (Gibco, ThermoFisher, Waltham, MA, USA) for 5 secs to detach the cells. After aspirating trypsin, the cells were gently washed from the plate with PBS. The cells (triplicate wells for each DHA concentration) were counted using a Bio-Rad TC-20 cell counter, and the results were expressed as the number of cells in DHA-treated wells relative to cells in control wells (% control growth).
Flow cytometry
For cell cycle analysis, HEK 293T cells were seeded in 100-mm dishes at 0.1 × 106 per dish. The cells were allowed to attach overnight and were either left untreated or treated with 5 mM DHA for 24, 48, and 72 h. At the end of dosing, the cells were washed with PBS, detached with 0.05% trypsin (Gibco), and resuspended in PBS. The cells were pelleted, resuspended in 100 μl cold PBS, and fixed by slowly dropping into 70% ethanol, then stored overnight at 4°C. The cells were pelleted, washed in PBS, and pelleted again. The cell pellets were then resuspended in 250 μl PBS containing 50 µl of RNase A stock solution (10 mg/ml). After a 10 min incubation at 37°C, 20 μg/ml of propidium iodide (PI) was added, followed by 15 min at room temperature (~22°C) incubation. Samples were analyzed on a BD FACSCanto II (BD Biosciences) using FACSDiva software.
Western blotting
HEK 293T cells were seeded in 100 mm dishes at 0.5 × 106 allowed to attach overnight and then treated with 5 mM DHA for time periods indicated. At appropriate time points, the cells were washed twice with ice-cold PBS, scraped, and flash frozen. Pellets were stored overnight at −80°C, the lysed in two volumes of lysis buffer (25 mM β-glycerolphosphate, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2%Triton X-100, and 0.3% NP-40) plus Halt protease and phosphatase inhibitor (Pierce, Waltham, MA, USA) and incubated on ice for 30 minutes. Lysates were then centrifuged at 20,800 x g for 30 minutes at 4°C, and the supernatant fraction retained. Protein concentrations were determined by Bradford Quick Start protein assay (Bio-Rad, Hercules, CA, USA). 30 μg of protein was separated via 4–20% SDS-PAGE and transferred to PVDF membranes (Bio-Rad). Membranes were blocked in 5% skim milk in Tris-buffered saline (TBS, VWR Life Sciences) containing 0.1% Tween 20 (TBST) and then incubated with the following primary antibodies: anti-PARP1 (1:1000, #518639-GR) from BD Pharmingen (San Jose, CA, USA); LC3B (1:1000, PA1–46286) from ThermoFisher; SIRT1 (1:1000, #110304) from Abcam (Cambridge, MA, USA); Cyclin A2 (1:1000, #4656) and Cyclin B1 (1:1000, #4138) from Cell Signaling (Danvers, MA, USA); and β-Actin (1:5000, AM43002) ThermoFisher.
Mitochondrial membrane potential and morphology
HEK 293T cells were seeded at 1000 cells per well in black, 96-well, clear bottomed plates. Cells were allowed to adhere overnight then dosed with 5 mM DHA for 24, 48, and 72 h. Membrane potential was assayed using tetramethylrhodamine ethyl ester (TMRE) Mitochondrial Potential Assay Kit (Biovision, Milpitas, CA, USA). Carbon cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 20 μM, Biovision) was used as a control, incubated at 37°C for 20 minutes prior to dosing all samples with TMRE (200 nM) for 30 minutes at 37°C. Following incubation, the wells were washed and filled with assay buffer, and their fluorescence measured with a Tecan M1000 plate reader at an excitation wavelength of 549 nm and an emission wavelength of 575 nm.
To control for variance in cell number per well, the TMRE signal was normalized to nuclear content by staining with NucBlue live cell stain (Thermofisher) after TMRE fluorescence was recorded. Nuclear intensity was measured by exciting at 360 nm and recording fluorescence at 460 nm. Cell count normalized results were then expressed as relative to control. A minimum of three biological replicates were performed, and the values are expressed as relative fluorescence intensity ±SEM.
Mitochondrial morphology was evaluated by fluorescent staining with MitoTracker CMXRos (Thermofisher). HEK 293T cells were plated in 35 mm fluorodishes (World Precision) at densities of 0.1 X 106 cells per dish. The cells were adhered overnight before being dosed with fresh medium for untreated controls and 5 mM DHA for 72 h. At the end of the 72 h treatment, dishes were dosed with a 200 nM concentration of MitoTracker CMXRos and incubated for 30 minutes at 37°C prior. The cells were then fixed with 4% formaldehyde for 10 min at RT. After washing 3 times with PBS, the cells were mounted with ProLong Gold (ThermoFisher) and then imaged with a Nikon N-SIM super-resolution microscope.
ATP levels
HEK 293T cells were seeded at 5000 cells per well in white, 96-well clear-bottomed plates. The cells were allowed to adhere overnight, then either left untreated as a control or dosed with 5 mM DHA for 24 and 48 h. Prior to assessing ATP levels, the cells were counted on a Celigo Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA, USA). Then ATP was assayed using Cell Titer Glo Assay (Promega, Fitchburg, WI, USA). At the end of time points, 100 μl of Cell Titer Glo assay solution was added to each sample well of the 96 well plate. Plate contents were gently mixed, incubated for 10 minutes at room temperature, and then luminescence was recorded on a Tecan M1000 plate reader. Luminescence was normalized to cell count then expressed as relative to control, untreated cells. A minimum of three biological replicates were performed, and values are expressed as relative luminescence intensity ±SEM.
Intracellular Lactate levels
HEK 293T cells were seeded at 2000 cells per well for control, 24 h and 48 h time points; and at 4000 cells per well for the 72 h time point, in white, 96-well clear-bottomed plates. Cells were allowed to adhere overnight, then either left untreated as a control or dosed with 5 mM DHA for 24, 48, and 72 h. Following treatment, the cells were counted on a Celigo Imaging Cytometer. Intracellular lactate levels were then assayed using the Lactate Glo Assay (Promega, Fitchburg, WI, USA). Wells were washed once with PBS, and the manufacturer’s protocol for detection of intracellular lactate levels was followed precisely. After appropriate assay treatment and incubation times, the luminescence was read on a Tecan M1000 plate reader. Luminescence was normalized to cell count then expressed as relative to control, untreated cells. A minimum of three biological replicates were performed, and values are expressed as relative luminescence intensity ±SEM.
Extracellular Lactate and DHA levels
For NMR experiments, HEK 293T cells were seeded at 0.2 × 106 cells per well in 6-well dishes and allowed to adhere overnight. The cells were either left untreated or treated with 5 mM DHA for 24 and 48 h. Following treatment times, the cell culture medium was collected and centrifuged at 300 x g for 5 minutes. Following centrifugation, the supernatant was retained for 1H-NMR analysis. Each experimental sample was matched with control samples of appropriate time points: untreated cells, medium only, and cell-free medium with matched DHA dosage. NMR samples were prepared as follows: 450 μl of cell culture supernatant was mixed with 50 μl of D2O containing 10 mM acetone used as an internal standard (1H NMR δ 2.00ppm). The resulting mixture was vortexed three times. NMR spectral acquisition (ns=1024) was then performed using a Bruker Avance II NMR spectrometer equipped with 400 MHz magnet Ultrashield Plus, with temperature fixed to 300 K for all NMR measurements. TopSpin 3.2 (Bruker BioSpin) was used for all NMR spectral acquisition and pre-processing and the automation of sample submission was performed using ICON-NMR (Bruker BioSpin). The intensity of the lactate peaks at 1.35ppm (3Hs) and 4.10ppm (1H) and that of acetone at 2.00ppm were determined manually following FID processing. The relative changes in the intensity of the lactate peaks were normalized to the acetone’s peak intensity. The relative abundance of lactate in the supernatant was measured over three biological replicates and expressed as mean ± SEM.
DHA concentration in the supernatant was examined at 4.3 ppm (1H) for up to 48 h. The chemical shifts in the DHA peak are minimal even though the media’s pH changes over the course of the incubation period. However, a water suppression sequence must be applied in order to mask the HOD/HOH peak which comes at 4.6–4.8ppm in the 1H-NMR spectra. The DHA peak is too close to the water peak to remain undisturbed by the water suppression process and as a consequence, the intensity of the DHA peak cannot be reliably quantified, even though it can be detected. Over the 48 h period, the DHA peak is detectable in the supernatant consistent with sustained exposure to the chemical over the experimental period.
Extracellular flux analysis (Seahorse)
Extracellular Acidification Rate (ECAR), a measure of cellular acid efflux as an indicator of the amount of lactic acid produced, and Oxygen Consumption Rate (OCR) were measured by Seahorse XF24 Cellular Flux Analyzer (Agilent Technologies, Santa Clara, California, USA) according to manufacturer’s recommendations. Cells were plated in XF24 culture dishes at 5 X 103 cells per well. After adhering overnight, the cells were either left untreated or treated with 5 mM DHA for 24 and 48 h. Following treatment duration, wells were washed with Seahorse assay medium, then filled with 0.5 ml of fresh assay medium. Upon filling wells with fresh Seahorse assay medium, dishes were placed in Celigo cytometer for direct cell counting, then incubated in a CO2-free 37°C incubator for one hour prior to running plate in Seahorse machine. Values for OCR and ECAR were normalized to cell count in individual wells. A representative seahorse trace is presented, and the OCR and ECAR values were averaged over three biological replicates and expressed as mean ± SEM.
Intracellular NAD(P)H and NAD+ measurements
NAD(P)H levels were measured using the endogenous autofluorescence of these dinucleotides when they are excited with 355 nm laser as described in Blacker and Duchen 2016. 11 As above, the cells were plated in 8-well chambered coverglass at a density of 1 × 104 cells per chamber. Cells were treated with 5 mM DHA for 24, 48, and 72 h. After the indicated treatment time, the cells were imaged using a Nikon Arsi scanning confocal microscope modified to include a 355 nm laser (PicoQuant, Berlin, Germany) as previously described in Holton et al. 12. Experiments were performed using a UV passing 40× C-Apochromat (numerical aperture (NA) 1.2) oil immersion objective, and images were collected with an iXON III EMCCD camera (Andor Technology, Belfast, UK). A region of interest within a selected field of cells was scanned with the 355 nm laser beam while collecting a time-lapse series at the maximum frame rate of the camera (35 ms/frame) for four minutes. Emitted light was filtered using a UV-2E filter cube (Nikon Instruments). A composite image was generated via maximum intensity projection over the time-lapse series and then thresholded to generate a binary mask defining the NAD(P)H signal. Intensity values within the binary mask were measured, and statistical analysis was performed using one-way ANOVA followed by Dunnett’s multiple comparison test.
Free NAD+ was measured using fluorescence sensors expressed in HEK 293T cells as described in Cambronne at al. 13 Briefly, vectors containing circularly permuted Venus fluorescent protein (cpVenus) and the bipartite NAD+-binding domain targeted to the cytoplasm, mitochondria, or nucleus or a cpVenus control with no binding domain for each intracellular compartment were transfected into HEK 293T cells using jetPRIME (Polyplus, New York, NY, USA). Stable cell lines, Cyto-HEK293T, Mito-HEK293T, and Nuclear-HEK293T and the matched controls, CytoCtrl-HEK293T, MitoCtrl-HEK293T, and NuclearCtrl-HEK293T, were clonal selected and maintained in growth medium + puromycin.
Cyto-HEK293T, Mito-HEK293T, and Nuclear-HEK293T and their matched control cell lines were seeded at 5,000 cells per chamber in 8 chamber slides, one slide for each cell line. Cells were allowed to adhere overnight then a chamber was dosed with 5 mM DHA each day to have a 24, 48, and 72 h time points within a single chamber slide. Untreated, control medium was exchanged in an adjacent chamber when each 5 mM DHA dose was added to control for variations in NAD+ due to medium consumption by the cells over time. As a reference for increased NAD+ levels, a well within each chambered slide was dosed with 100 μM Nicotinamide riboside hydride (NRH) for 6 h with matching untreated medium control. 1 mM NRH has been demonstrated to boost intracellular NAD+ 4-fold in HEK293 cells. 14
After the appropriate dosing time, the cells were imaged using a Nikon A1rsi scanning confocal microscope with a 20x/0.75 objective. Excitation with 488 nm was used to monitor NAD+ binding and measurements at 405 nm, which are not affected by NAD+ binding, were used to normalize sensor expression levels as described in Cambronne et al. 13 The mean of the fluorescence ratio (488/405) was determined for control, 24, 48, and 72 h for each sensor Cyto-HEK293T, Mito-HEK293T, and Nuclear-HEK293T (F) and control CytoCtrl-HEK293T, MitoCtrl-HEK293T, and NuclearCtrl-HEK293T (F0), then normalized to the untreated mean value. The change in NAD+ concentration was then determined by F/F0 to normalize for variations in the fluorphore as described in Cambronne et al. 13 A minimum of 2000 cells were measured over at least two biological replicates for each experiment and the F/F0 mean ± SEM is reported.
Measuring GSH/GSSG
GSH (reduced glutathione) and GSSG (oxidized glutathione) were measured by use of GSH/GSSG Glo assay (Promega, Fitchburg, WI, US). The cells were seeded in white, 96-well plates at 4,000 cells per well. After adhering overnight, the cells were either left untreated or exposed to 5 mM DHA for 24 and 48 h. At the end of treatment time, cells were counted on a Celigo to normalize for variations in cell density, then assayed according to the manufacturer’s protocol. Following assay incubation, luminescence was recorded on Tecan M1000. Luminescence values were normalized to cell count and then expressed as a percentage of the untreated control. Results presented as the mean ± SEM of three replicates.
Statistical Analysis
Unless stated otherwise, data are represented as ± SEM of three biological replicates. Statistical tests were performed by one-way analysis of variance (ANOVA) with a posthoc test for multiple comparisons. Statistical significant *p < 0.05, **p < 0.01 and ***p < 0.001.
RESULTS
DHA induces cell cycle arrest and autophagy in HEK293T
To determine the cytotoxicity of DHA in HEK 293T cells, we evaluated inhibition of cell growth by sustained exposure to a range of DHA doses by direct cell counting. Cell proliferation was inhibited at doses greater than 1.5 mM, with IC50 and IC90 values of 2.4 mM and 4.6 mM DHA, respectively (Figure 1).
Figure 1. DHA is cytotoxic to HEK 293T cells.

HEK 293T cells were exposed to a range of DHA doses for 5 days, and then cell counting assays were performed, in triplicate (see Materials and Methods). Results are expressed as the average number of cells remaining to that of the control (% control growth) ± the standard error of the mean (SEM).
Previous studies in A375P and HaCaT cells observed G2/M cell cycle arrest within 24 h of exposure to IC90 doses of DHA. 6, 7 To observe cell cycle arrest, HEK 293T cells were dosed with 5 mM of DHA for 24, 48, and 72 h, followed by propidium iodide staining in order to observe cell cycle progression. No significant change in the cell cycle was observed 24 h after exposure to DHA (Figure 2A). However, by 48 h partial G2/M cell cycle arrest was observed with the percentage of cells in G1 phase decreased by nearly half and the portion in G2/M phase doubling (Figure 2B). At 72 h, the portion of cells in G2/M was unchanged, while very few cells remained in G1 (Figure 2B). Cell cycle arrest was also confirmed by increasing protein levels of Cyclin A2 and Cyclin B2, indicating an increase in cells in late S, G2, and early M phases (Figure 2C).
Figure 2. DHA exposure induced G2/M cell cycle arrest.

A. The cell cycle of HEK 293T cells treated with 5 mM DHA for 24, 48, and 72 h was determined using PI. B. Only minimal changes in the cell cycle were observed 24 h after exposure. By 48 h, the G2/M population doubled, while the S phase population continued to increase until 72 h, leaving a very small population of cells in G1. The mean percent cells in each cell cycle of three independent experiments are shown. C. Cell cycle arrest was confirmed by progressive increases in Cyclin B1 and Cyclin A2 from 48 to 72 h.
Apoptosis was the primary cell death mechanism for DHA-exposed A375P and HaCaT cells, yet up to 96 h after DHA exposure no cleavage of PARP1 or Caspase 3 was observed in HEK 293T cells (Figure 3A). An increase in the ratio of LC3B II to LC3B I was observed 48 h after exposure to 5 mM DHA, indicating autophagy as the mechanism of cell death. 15, 16 While a decrease in mTOR was not observed, expression of SIRT1, required for the deacetylation of LC3B and its export from the nucleus, also increases over the duration of cell exposure to DHA, supporting cell death by autophagy (Figure 3B). By 72 h, the cells are beginning to undergo cell death and degradation of LC3B by 96 h is consistent with the endpoint of autophagic flux (Figures 4 & 3).
Figure 3. DHA induced autophagy markers 48 h after exposure.

HEK 293T cells were treated with 5 mM DHA for 48, 72, and 96 h. A. The apoptotic marker, cleaved PARP-1, was not observed after DHA exposure. However, increased expression of the autophagy marker LC3B II was observed. B. Additional autophagy markers were examined, and only an increase in SIRT1 was observed after DHA exposure. β-actin was used as a loading control for immunoblotting, and representative immunoblots are shown.
Figure 4. Cell morphology changes induced by DHA exposure.

Differential interference contrast (DIC) images of HEK 293T cells exposed to 5 mM DHA. As a function of DHA exposure duration, the number of vacuoles increased in cells where they are most apparent 48 h after exposure. By 72 h, the cells show compromised plasma and nuclear membrane integrity. Representative images are shown with a scale bar of 50 µm.
DHA exposure alters the metabolic function of HEK 293T cells
Our previous study with A375P melanoma cells showed an increase in mitochondrial membrane potential after exposure to 5 mM DHA.7 Mitochondrial membrane potential was examined in the HEK 293T cells exposed to 5 mM DHA using tetramethylrhodamine, ethyl ester (TMRE). Fluorescence of TMRE was measured and normalized to cell number to better capture changes in membrane potential. Cells treated with 5 mM DHA showed a non-significant increase in membrane potential 24 h after exposure to DHA, but a steady decrease in potential thereafter. Reduced membrane potential was observed at 48 h, and 72 h exposures were consistent with 1 h Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), which uncouples the mitochondria resulting in a loss of membrane potential (Figure 5A). Super resolution imaging of mitochondria stained with MitoTracker CMXRos, a membrane sensitive dye that survives fixation, revealed that after 72 h of exposure to DHA, mitochondria are highly segmented, consistent with mitochondrial dysfunction and progressive cell death (Figure 5B).
Figure 5. DHA exposure alters mitochondrial membrane potential and morphology.

A. Mitochondrial membrane potential was assessed by measuring the mean fluorescence intensity of TMRE in untreated and 5 mM DHA treated cells. FCCP uncouples the membrane potential causing a loss in fluorescence signal and serves as a positive control. The graph shows the mean fluorescence intensity of three independent experiments normalized for cell number variations (see Material and Methods) ± the SEM. B. Mitochondrial morphology was examined by super resolution microscopy. MitoTracker CMXRos was used to stain mitochondria prior to fixation. Representative cells are shown.
Given the effects observed in mitochondria, we evaluated cellular oxygen consumption rates (OCR) and extracellular acidification rates (ECAR). After two hours after exposure to 5 mM DHA, the cells showed a significant reduction in OCR (Fig. 6A&B), but no significant changes in ECAR. This early reduction in OCR was recovered by 24 h but with substantial fluctuations in OCR observed over the three biological replicates out to 48 h (Figure 6C). At 24 h, ECAR began a steady decrease, continuing a significant downward trend out to 48 hours (Figure 6C).
Figure 6. DHA compromises mitochondrial function within 2 h of exposure.

A. Seahorse Biosciences XF24 extracellular flux analyzer was used to measure OCR (top) and ECAR (bottom). Top is a representative OCR trace of untreated (blue) and 5 mM DHA treated (red) HEK 293T cells after 2 h. Bottom is a representative ECAR trace of untreated (blue) and 5 mM DHA treated HEK 293T cells at 24 (red) and 48 h (green). B. OCR showed a significant decrease in oxygen consumption after 2 h exposure to 5 mM DHA. Though no change in ECAR was observed. C. OCR is highly variable 24 and 48 h after 5 mM DHA exposure. While ECAR showed a significant reduction in at both 24 and 48 h after 5 mM DHA exposure. Mean of three independent experiments ± SEM are shown. **p < 0.01 and ***p < 0.001
With the mitochondrial function changing over the exposure period, we evaluated intracellular ATP production using Cell Titer Glo. Cells were exposed to 5 mM DHA for 24 and 48 h and counted prior to lysis to normalize measurements. A significant reduction in total ATP was observed after DHA exposure at both 24 and 48 h (Figure 7).
Figure 7. ATP levels decrease after exposure to DHA.

Total intracellular ATP was assayed by CellTiter Glo in HEK 293T cells after treatment with 5 mM DHA for 24 and 48h. ATP levels decreased to 81.8 ± 5.5% at 24 h and further decreased to 71.5 ± 7.6% at 48 h. The graph shows the mean luminescence intensity of three independent experiments normalized for cell number variations and expressed relative to control ± the SEM. *p < 0.05
Lactate levels were assayed, in the supernatant, by NMR (Figure 8A); and intracellularly, by Lactate Glo assay (Figure 8B) over the same time period. Reduced lactate production was observed both in the supernatant and within the cell after DHA exposure, with the most significant decrease observed 48 h after exposure.
Figure 8. Extracellular and intracellular lactate are reduced after DHA exposure.

A. Nuclear Magnetic Resonance was used to assay levels of lactate in cell culture supernatant following exposure of HEK 293T cells to 5 mM DHA at 24 and 48 h. B. Lactate Glo was used to assess intracellular lactate levels at the same time points as the NMR samples. Mean of three independent experiments ± SEM are shown.
DHA exposure alters NAD(P)H levels
Conversion of DHA to DHAP by cells requires ATP and can shift the balance in metabolic pathways (Figure 9), as previously demonstrated by injection of hyperpolarized [2-13 C] DHA into mice. 9 This metabolic shift can also induce cofactor imbalance in NAD/NADH or NADP/NADPH (Figure 9A).
Figure 9. NAD(P)H utilization in DHA metabolism.

A. Glycolytic (Embden-Meyerhof) pathway showing DHA’s entry into the 3-carbon metabolic pathway. Utilization of NAD(P)H and ATP is highlighted in red. B. Autofluorescent images of endogenous NADH and NADPH in untreated and 5 mM DHA treated cells. C. Quantification of fluorescent signal for untreated and 5 mM DHA treated cells. Each time point has its own untreated control to account for changes in NAD(P)H due to medium consumption during the DHA exposure time. Representative images are shown in B and scale bar is 50 µm.
To examine cofactor imbalance, we exploited the intrinsic autofluorescence of NADH and NADPH when excited by 355 nm light. 11 DHA absorbance peaks at 271 nm, with no absorption observed in the 320 – 800 nm range. Within 24 h of exposure to DHA, HEK 293T cells showed a definite increase in NAD(P)H levels seen through an increase in the autofluorescent intensity (Figure 9B). The distribution of NAD(P)H in untreated cells appears to be mostly cytoplasmic and mitochondrial, though the mitochondrial staining is the most visible (Figure 9B, right). After DHA exposure, the NAD(P)H fluorescence is observed more intensely within the mitochondria, and the signal is now observed within the nuclear compartment. As the exposure duration increases, the fluorescence intensity continues to increase with 72 h showing the highest level of autofluorescence, consistent with loss in metabolic activity and cell death (Figure 9B, right).
Since the autofluorescence of NADH and NADPH are indistinguishable, we also employed NAD+ specific sensors developed by Cambronne et al. 13 These sensors target an NAD+ binding domain into the cytoplasm, mitochondria, or nucleus of the cells. Binding of NAD+ to the sensor causes a loss in cpVenus fluorescence reflected in the ratio change between the 488 nm excitation and the 405 nm excitation, which is unaffected by NAD+ binding. The ratio of the fluorescence change (F/F0) between the sensors which contain the NAD+ binding domain (F) and a control cpVenus vector without the binding domain (F0) reflect changes in the free NAD+ pool in each organelle. Using these sensors, we have measured the relative free NAD+ levels in each of these compartments as a function of DHA exposure time. NRH was used to boost intracellular levels of NAD+ in the HEK293T cells. A previous study noted a 4-fold increase in NAD+ after 6 h exposure to 1 mM NRH. Here, 100 µM NRH was exposed to HEK293T for 6 h. Cytoplasmic and nuclear NAD+ levels increased as observed by a ~ 15% and ~25% decrease in F/F0 for these sensors, respectively (Figure 10). 24 h exposure with 5 mM DHA induced a significant increase in free NAD+ in the cytoplasm (−20.5 ± 2.8% change in fluorescence compared to untreated, control) and a decrease in free NAD+ in the mitochondria (+19.5 ± 2.8%). NAD+ levels remained moderately elevated in the cytoplasm 48 h after exposure was initiated (−12.5 ± 5.2%), while the mitochondrial levels recovered to approximately untreated control levels (+7% ± 4.8%). No significant change in nuclear NAD+ levels was observed with DHA treatment.
Figure 10. NAD+ levels in the cytoplasm, mitochondria, and nucleus of DHA treated HEK 293T cells containing a cpVenus NAD+ binding sensor.

Quantification of the ratio of the normalized fluorescent signal from the organelle sensor (F) and the organelle control (F0) is shown for the cytoplasm (A), the mitochondria (B), and the nucleus (C) of stable HEK 293T cell lines expressing the organelle specific sensors and controls. A decrease in the ratio indicates an increase in free NAD+ levels within that organelle. 100 µM NRH is used as a control to spike the NAD+ levels. **p < 0.01
With the increase in total NAD(P)H observed by autofluorescence within the entire cell and the increase in NAD+ observed in the cytoplasm by the sensors, we assessed changes in reduced and oxidized glutathione (GSH/GSSG) to differentiate changes between NADH and NADPH after DHA treatment. Total intracellular GSH and GSSG were examined 24 and 48 h after exposure to 5 mM DHA (Figure 11). A reduction in GSSG (70% ± 5.5%) is observed 24 h after exposure to 5 mM DHA with no significant change in GSH observed. GSSG changes are reversed at 48 h with GSSG showing a significant increase compared to 24 h levels (104.6 ± 3.25%), and GSH reduced significantly compared to control and 24 h treatment (74.6 ± 6.5%) (Figure 11). Conversion of GSSG to GSH requires NADPH, so both NADPH and NADH are altered by DHA exposure, consistent with the autofluorescent measurements, but the contributions between the two species cannot be differentiated.
Figure 11. Reduced and oxidized glutathione levels change over DHA exposure period.

A. Reduced glutathione (GSH) levels in 5 mM DHA treated HEK 293T cells at 24 and 48 h. B. Oxidized glutathione (GSSH) in 5 mM DHA treated HEK 293T cells at 24 and 48 h. Mean of three independent experiments ± SEM are shown. *p < 0.05 between indicated comparison groups.
DISCUSSION
DHA is used as the active ingredient in sunless tanning products, including aerosolized products that allow spray tanning. DHA is also present in the inhaled vapors generated by the combustion of solvents found in electronic cigarettes. The increasing popularity of both spray tanning 17, 18 and electronic cigarettes 19 makes establishing whether DHA can promote cellular dysfunction once it is in systemic circulation critical. Once intracellularized, DHA is rapidly converted to DHAP, a metabolite of fructose metabolism. 9, 10, 20 Glucose and fructose cause endogenous increases in DHA and DHAP. This particular pathway and its physiological consequences have been well-studied in diabetes and in cases of triose-phosphate isomerase (Figure 9A; TPI) deficiency. There, increases in three carbon metabolism have been linked to increased protein damage through advanced glycation end products (AGE). 21–25 AGE protein modifications and their receptors (RAGE) have been implicated in the progressions of many intractable diseases, such as diabetes and atherosclerosis, and are also critical for pathologic changes in chronic degenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and alcoholic brain damage. 26 Methylglyoxal, a major contributor to AGE product formation, is generated directly from DHAP and its isomer glyceraldehyde 3-phosphate (GAP). In addition to being a precursor to methylglyoxal via DHAP, DHA, itself, can also act as a glycation agent. 27 While the importance of excess hexoses in promoting AGE product formation has been extensively explored, the effects of exogenous exposure to trioses such as DHA are far less well understood.
In this work, HEK 293T cells proved to have similar sensitivity to sustained exposure to DHA as previous reports with melanoma cell line A375P (IC50 of 2.4 mM vs. 1.9 mM). 7 However, unlike our previous work with A375P cells and the work with HaCaT cells, the HEK293T cells did not show a significant change in cell cycle 24 h after exposure to 5 mM DHA. 6, 7 The shift towards a partial G2/M cell cycle arrest was observed after 48 h of exposure with the percentage of cells in G1 phase decreased by nearly half and the portion in G2/M phase doubling over that period (Figure 2). The delay in cell cycle arrest also extended the cell death period with increases in LC3B, an autophagy marker, observed 48 and 72 h after exposure (Figure 3). Both A375P and HaCaT cells were observed to undergo apoptotic cell death with cleaved PARP-1 and caspase 3 observed. 6, 7 Here, no change in PARP-1 or caspase 3 was observed (Figure 3).
Autophagic cell death induced by DHA exposure is also supported by increased SIRT1. SIRT1 expression is often observed during glucose limiting conditions and when NAD+ increases in cells (Figure 10). 28–30 DHA exposure induced metabolic dysfunction accumulates over the exposure period and is consistent with the delayed onset of cell death (Figure 3 & 4). Measurements of the mitochondrial membrane potential show an increase in potential 24 h after exposure is initiated, followed by a significant decrease in potential that is consisted with the uncoupling observed by FCCP. Loss in mitochondrial function can be observed as early as 2 h after exposure to DHA, where a significant decrease in basal oxygen consumption is observed. However, the oxygen consumption rate appears to recover within 24 h, though the rates of consumption are highly variable and likely contribute to the altered membrane potential observed over time (Figure 6B & C).
In addition to OCR change, a reduced reserve respiratory capacity (RRC) is observed after DHA exposure, regardless of the length of the incubation period (Figure 6A). Mitochondrial Complex II has been associated with RRC, which is also regulated by pyruvate dehydrogenase kinase and AMPK in cardiac myocytes. 31 A compromised Krebs cycle would promote reduced RRC. The observed accumulation of NADH, and loss of NAD+, in mitochondria and the accumulation of NAD+ in the cytoplasm after DHA exposure, indicates imbalanced NADH consumption and compromised energy production (Figures 9 & 10). Alterations in NAD+/NADH were observed previously in starved rats after DHA exposure, but this work demonstrates changes can also occur in high nutrients states. 20
Accordingly, decreased ATP production is observed 24 and 48 h after DHA exposure (Figure 7). While decreased ATP production could result from a compromised Krebs cycle, the reduction in ECAR observed at 24 and 48 h and the reduction in intra- and extra-cellular lactate over the same period also indicate compromised glycolysis. An increased abundance of DHAP, from exogenous DHA, would result in an increase in GAP, which would be expected to yield an increased abundance of lactic acid compared to untreated cells. Yet, the lack of shifts in the production of lactic acid indicates that HEK 293T cells exposed to 5 mM DHA did not repurpose the DHA or glucose effectively to compensate for ATP loss. Additionally, the increase in total cellular NADH/NADPH and alterations in GSH/GSSG over the exposure time indicate the redox environment of the cells are compromised further supporting compromised glycolysis (Figure 10 & 11).
Altogether the data suggest that DHA slowly compromises metabolic function within the cell by altering both cytoplasmic and mitochondrial energy production. One possible explanation is that the normal electron transfer chain complexes present in the mitochondrial matrix are weakly compromised by DHA initially. Then, the increased abundance in three carbon sugars generates pyruvate that cannot be used efficiently by the cell to produce ATP. This may result from the production of AGEs and methylglyoxals that damage proteins involved in glycolysis, changes in the redox environment that limit enzymatic activity, or even by DHA itself inducing covalent modifications. More work is necessary to discern the fate of DHA within metabolic cycles at consumer relevant exposures.
In conclusion, DHA, a chemical used widely in sunless tanning products and e-cigarettes has been shown to alter the metabolic functions and survival capacity of HEK 293T cells, in a manner which differs from the two previous studies examining the cytotoxicity of DHA. Critically, in HEK 293T cells, the cytotoxic effect is delayed compared to the two other cell types, but the effect of 5 mM DHA on the mitochondrial function is fast acting and long lasting. These observations warrant further mechanistic investigations into the exposure effects of DHA exposure on nephrotic cells and more importantly, into other tissues likely to be exposed through DHA-containing product use.
ACKNOWLEDGMENTS
NRG and the Gassman lab are supported by start-up funding from the University of South Alabama Mitchell Cancer Institute and the National Institute of Health (ES028015). NRG, FH, and MEM are also supported by the National Institute of Health (AT009908). The authors would like to thank Steve McClellan and the Flow Cytometry and Imaging Core at the Mitchell Cancer Institute for assistance with flow experiments.
REFERENCES
- (1).Yourick JJ, Koenig ML, Yourick DL, and Bronaugh RL (2004) Fate of chemicals in skin after dermal application: does the in vitro skin reservoir affect the estimate of systemic absorption? Toxicology and applied pharmacology 195, 309–320. [DOI] [PubMed] [Google Scholar]
- (2).Goldman L, and Blaney DJ (1962) Dihydroxyacetone. Recent clinical investigative studies. Arch Dermatol 85, 730–734. [DOI] [PubMed] [Google Scholar]
- (3).Vreeke S, Korzun T, Luo W, Jensen RP, Peyton DH, and Strongin RM (2018) Dihydroxyacetone levels in electronic cigarettes: Wick temperature and toxin formation. Aerosol Science and Technology 52, 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Salamanca JC, Meehan-Atrash J, Vreeke S, Escobedo JO, Peyton DH, and Strongin RM (2018) E-cigarettes can emit formaldehyde at high levels under conditions that have been reported to be non-averse to users. Sci Rep 8, 7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lee YO, Nonnemaker JM, Bradfield B, Hensel EC, and Robinson RJ (2018) Examining Daily Electronic Cigarette Puff Topography Among Established and Nonestablished Cigarette Smokers in their Natural Environment. Nicotine Tob Res 20, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Petersen AB, Wulf HC, Gniadecki R, and Gajkowska B (2004) Dihydroxyacetone, the active browning ingredient in sunless tanning lotions, induces DNA damage, cell-cycle block and apoptosis in cultured HaCaT keratinocytes. Mutat Res 560, 173–186. [DOI] [PubMed] [Google Scholar]
- (7).Smith KR, Granberry M, Tan MCB, Daniel CL, and Gassman NR (2018) Dihydroxyacetone induces G2/M arrest and apoptotic cell death in A375P melanoma cells. Environ Toxicol 33, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).FDA. (2002) Code of Federal Regulations, Title 21, (Services, D. o. H. a. H., Ed.) p 1150, U.S. Food and Drug Administration, Washington, D.C. [Google Scholar]
- (9).Marco-Rius I, von Morze C, Sriram R, Cao P, Chang GY, Milshteyn E, Bok RA, Ohliger MA, Pearce D, Kurhanewicz J, Larson PE, Vigneron DB, and Merritt M (2017) Monitoring acute metabolic changes in the liver and kidneys induced by fructose and glucose using hyperpolarized [2–13 C]dihydroxyacetone. Magn Reson Med 77, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Moreno KX, Satapati S, DeBerardinis RJ, Burgess SC, Malloy CR, and Merritt ME (2014) Real-time detection of hepatic gluconeogenic and glycogenolytic states using hyperpolarized [2–13C]dihydroxyacetone. J Biol Chem 289, 35859–35867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Blacker TS, and Duchen MR (2016) Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med 100, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Holton NW, Andrews JF, and Gassman NR (2017) Application of Laser Micro-irradiation for Examination of Single and Double Strand Break Repair in Mammalian Cells. J Vis Exp, e56265. [DOI] [PMC free article] [PubMed]
- (13).Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, and Goodman RH (2016) Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yang Y, Mohammed FS, Zhang N, and Sauve AA (2019) Dihydronicotinamide riboside is a potent NAD(+) concentration enhancer in vitro and in vivo. J Biol Chem [DOI] [PMC free article] [PubMed]
- (15).Mizushima N (2004) Methods for monitoring autophagy. Int J Biochem Cell Biol 36, 2491–2502. [DOI] [PubMed] [Google Scholar]
- (16).Mizushima N, Yoshimori T, and Levine B (2010) Methods in mammalian autophagy research. Cell 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Daniel CL, and Gassman NR (2018) A truly safer alternative? Sunless tanning products and the unknown. Prev Med 112, 45–46. [DOI] [PubMed] [Google Scholar]
- (18).Daniel CL, Gassman NR, Fernandez AM, Bae S, and Tan MCB (2018) Intentional tanning behaviors among undergraduates on the United States’ Gulf Coast. BMC public health 18, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Murthy VH (2017) E-Cigarette Use Among Youth and Young Adults: A Major Public Health Concern. JAMA Pediatr 171, 209–210. [DOI] [PubMed] [Google Scholar]
- (20).Williamson DH, Veloso D, Ellington EV, and Krebs HA (1969) Changes in the concentrations of hepatic metabolites on administration of dihydroxyacetone or glycerol to starved rats and their relationship to the control of ketogenesis. Biochem J 114, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Aragno M, and Mastrocola R (2017) Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Beutler E, and Guinto E (1973) Dihydroxyacetone metabolism by human erythrocytes: demonstration of triokinase activity and its characterization. Blood 41, 559–568. [PubMed] [Google Scholar]
- (23).Rabbani N, Xue M, and Thornalley PJ (2016) Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci (Lond) 130, 1677–1696. [DOI] [PubMed] [Google Scholar]
- (24).Kitada M, Zhang Z, Mima A, and King GL (2010) Molecular mechanisms of diabetic vascular complications. J Diabetes Investig 1, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Orosz F, Olah J, and Ovadi J (2009) Triosephosphate isomerase deficiency: new insights into an enigmatic disease. Biochim Biophys Acta 1792, 1168–1174. [DOI] [PubMed] [Google Scholar]
- (26).Byun K, Yoo Y, Son M, Lee J, Jeong GB, Park YM, Salekdeh GH, and Lee B (2017) Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol Ther 177, 44–55. [DOI] [PubMed] [Google Scholar]
- (27).Seneviratne C, Dombi GW, Liu W, and Dain JA (2012) In vitro glycation of human serum albumin by dihydroxyacetone and dihydroxyacetone phosphate. Biochem Biophys Res Commun 417, 817–823. [DOI] [PubMed] [Google Scholar]
- (28).Canto C, and Auwerx J (2012) Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev 64, 166–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sathyanarayan A, Mashek MT, and Mashek DG (2017) ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell reports 19, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Qiu X, Brown KV, Moran Y, and Chen D (2010) Sirtuin regulation in calorie restriction. Biochim Biophys Acta 1804, 1576–1583. [DOI] [PubMed] [Google Scholar]
- (31).Pfleger J, He M, and Abdellatif M (2015) Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis 6, e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]