

SUMMARY
Multimeric adaptors are broadly involved in vesicle-mediated membrane trafficking. AP2 adaptor, in particular, plays a central role in clathrin-mediated endocytosis (CME) by recruiting cargo and clathrin to endocytic sites. It is generally thought that trafficking adaptors such as AP2 adaptor assemble spontaneously. In this work, however, we discovered that AP2 adaptor assembly is an ordered process controlled by alpha and gamma adaptin binding protein (AAGAB), an uncharacterized factor identified in our genome-wide genetic screen of CME. AAGAB guides the sequential association of AP2 subunits and stabilizes assembly intermediates. Without the assistance of AAGAB, AP2 subunits fail to form the adaptor complex, leading to their degradation. The function of AAGAB is abrogated by a mutation that causes punctate palmoplantar keratoderma type 1 (PPKP1), a human skin disease. Since other multimeric trafficking adaptors operate in an analogous manner to AP2 adaptor, their assembly likely involves a similar regulatory mechanism.
Keywords: membrane trafficking, adaptor complex, clathrin-mediated endocytosis, AP2 adaptor, AP1 adaptor
INTRODUCTION
Clathrin-mediated endocytosis (CME) is a major route for internalization of cell surface and extracellular molecules (Fotin et al., 2004; Kaksonen et al., 2006; Kirchhausen et al., 2014; McMahon and Boucrot, 2011; Mettlen et al., 2018; Traub and Bonifacino, 2013). Since the coat protein clathrin does not bind directly to cargo proteins, it relies on adaptors to recruit cargo proteins to endocytic sites (Kaksonen and Roux, 2018; Mettlen et al., 2018). The predominant clathrin adaptor in CME is the heterotetrameric AP2 adaptor complex, which is comprised of two large subunits (α and β2), one medium subunit (μ2), and one small subunit (σ2) (Collins et al., 2002; Conner and Schmid, 2003; Pearse and Robinson, 1984). The AP2 adaptor complex assembles in the cytosol, initially adopting a “locked” conformation in which the cargo- and clathrin-binding sites are masked (Jackson et al., 2010). Upon recruitment to the cell surface through binding to phosphatidylinositol 4,5-bisphosphate (PIP2), AP2 adaptor undergoes conformational changes to expose its cargo-binding sites, which then recognize the sorting signals of cargo proteins (Di Paolo and De Camilli, 2006; Jackson et al., 2010).
AP2 adaptor recognizes two types of sorting signals – the acidic dileucine motif [DE]xxxL[LI] and the tyrosine-based motif Yxx∅ (x: any residue; ∅: hydrophobic residue) (Boll et al., 1996; Jackson et al., 2010; Kelly et al., 2008). The acidic dileucine motif binds to the α and σ2 subunits of AP2 adaptor, whereas the Yxx∅ motif is recognized by the μ2 subunit (Jackson et al., 2010; Kelly et al., 2008; Ohno et al., 1995; Yu et al., 2010). Upon binding to sorting signals, AP2 adaptor undergoes further conformational changes to expose its clathrin-binding domain, which then recruits clathrin to the endocytic sites to form clathrin-coated pits (CCPs) (Kelly et al., 2014). Subsequently, CCPs bud toward the cytosol to generate clathrin-coated vesicles (Brodsky, 2012; Fotin et al., 2004; Kirchhausen et al., 2014; McMahon and Boucrot, 2011; Ramanan et al., 2011).
The function of AP2 adaptor relies on the proper assembly of the four AP2 subunits into the heterotetrameric adaptor complex, a process still poorly understood at the molecular level. In this work, we identified AAGAB (also known as AAGAB p34) as a crucial regulator of AP2 adaptor assembly. We discovered that AAGAB guides the ordered assembly of AP2 subunits and stabilizes assembly intermediates. Without the assistance of AAGAB, AP2 subunits fail to form the adaptor complex, resulting in their degradation and CME defects. The function of AAGAB is abolished by a mutation that causes PPKP1, a human skin disease characterized by pathological thickening of palm and sole skin. Together, these findings demonstrated that the AP2 adaptor complex does not assemble spontaneously as previously assumed. Instead, AP2 adaptor assembly is a highly orchestrated process involving ordered molecular interactions controlled by AAGAB. We suggest that the assembly of other multimeric trafficking adaptors likely involves a similar regulatory mechanism.
RESULTS
Identification of CME regulators in a genome-wide genetic screen
To identify new components of the CME pathway, we performed an unbiased genome-wide CRISPR genetic screen using the CME cargo reporter HA-GLUT4-GFP, in which the glucose transporter GLUT4 was tagged with a cytosolic green fluorescent protein (GFP) and an exoplasmic hemagglutinin (HA) epitope (Figure 1A). Like native GLUT4 proteins, the reporter is continuously internalized through clathrin-mediated endocytosis (Antonescu et al., 2014; Bogan and Kandror, 2010; Jewell et al., 2010; Rowland et al., 2011). The relative surface levels of the reporter were calculated based on GFP fluorescence and HA staining measured by fluorescence-activated cell sorting (FACS).
Figure 1. Dissection of CME using a genome-wide CRISPR genetic screen.

(A) Diagram of the CME cargo reporter in which the transmembrane protein GLUT4 was tagged with a cytosolic green fluorescent protein (GFP) and an exoplasmic hemagglutinin (HA) epitope (Gulbranson et al., 2017; Muretta et al., 2008). Since this GLUT4-based reporter is constantly removed from the cell surface by CME (Antonescu et al., 2014), genetic ablation of CME is expected to elevate the surface levels of the cargo reporter. (B) Illustration of the genome-wide genetic screen to dissect CME in HeLa cells. (C) Flow cytometry analysis of the starting mutant population and the final sorted population (after three rounds of sorting) in the genome-wide CRISPR screen (top) and the secondary screen (bottom). CME cargo reporters on the cell surface were labeled using anti-HA antibodies and APC-conjugated secondary antibodies. APC and GFP fluorescence of the cells was measured by flow cytometry and used to calculate the relative surface levels of the reporter. (D) Ranking of genes in the CRISPR screen based on P values obtained from MAGeCK algorithm (Li et al., 2014). Each dot represents a gene. Genes above the horizontal cutoff line were tested in the pooled secondary screen. A gene is shown as a large grey dot if it was validated in the secondary screen while other genes are shown as small grey dots. Selected genes are labeled in red. AP2S1 and AP2M1 encode the σ2 and μ2 subunits, respectively, of the AP2 adaptor whereas FCHO2 encodes a key regulator in the early stage of CME (McMahon and Boucrot, 2011). See also Figure S1.
HeLa cells expressing the reporter were mutagenized by a pooled lentiviral CRISPR library containing 123,411 independent single guide RNAs (sgRNAs) targeting 19,050 proteincoding genes and 1,864 miRNAs (Figure 1B) (Sanjana et al., 2014). The mutagenized cells were consecutively sorted for three rounds to enrich the mutant cell population exhibiting elevated surface levels of the CME reporter (Figure 1B–C). Subsequently, sgRNAs were recovered from the sorted cells and analyzed by deep sequencing. The abundance of many sgRNAs (e.g., sgRNAs targeting AP2S1) in the sorted populations were substantially increased compared to an unsorted control population (Figure S1 and Table S1). By contrast, non-targeting control sgRNAs exhibited little enrichment (Figure S1). Genes were ranked based on the enrichment of their targeting sgRNAs using the MAGeCK algorithm (Table S2) (Gulbranson et al., 2017; Li et al., 2014). The majority of the hits from the genome-wide screen were validated in a pooled secondary screen (Figure 1D, Tables S3–4). The validated hits include genes encoding known CME regulators such as AP2S1, AP2M1 and FCHO2 (Figure 1D). Recovery of these known genes suggests that the screens were sensitive and specific. Many of the identified genes, however, were not previously linked to CME.
AAGAB is essential to CME
A top-ranking hit from the CRISPR genetic screen is AAGAB, which encodes a ubiquitously expressed soluble factor of 34 kDa in size (Figure 1D). Heterozygous loss-of-function mutations in AAGAB are frequently observed in PPKP1, a human skin disease characterized by pathological thickening of palm and sole skin (Nomura et al., 2015; Pohler et al., 2012). The biological function and molecular mechanism of AAGAB, however, remained unknown. Knockout (KO) of AAGAB strongly increased the surface level of the CME cargo reporter in HeLa cells (Figure 2A), confirming the results of the CRISPR screen. Next we directly examined the functional role of AAGAB in CME. Using a CME-mediated internalization assay, we observed that the uptake of the cargo reporter was abolished in AAGAB KO cells or cells deficient in AP2S1, which encodes the σ2 subunit of AP2 adaptor (Figure 2B). These results established that AAGAB mutation leads to CME defects. We reasoned that, if AAGAB is a general regulator of CME, its function should not be restricted to a specific cell type or cargo protein. Indeed, the surface levels of the CME cargo reporter were also strongly increased in AAGAB KO adipocytes (Figure 2C), similar to the observations in HeLa cells (Figure 2A). Surface levels of transferrin receptor (TfR), another known CME cargo (Conner and Schmid, 2003), were also strongly elevated in AAGAB KO cells (Figure 2D). By contrast, the surface levels of FAS and PVR, which are not CME cargo proteins (Figure 2E), remained unchanged in AAGAB KO cells (Figure 2E). Together, these results demonstrated that, like AP2 adaptor, AAGAB is a general regulator of CME.
Figure 2. AAGAB is required for CME.
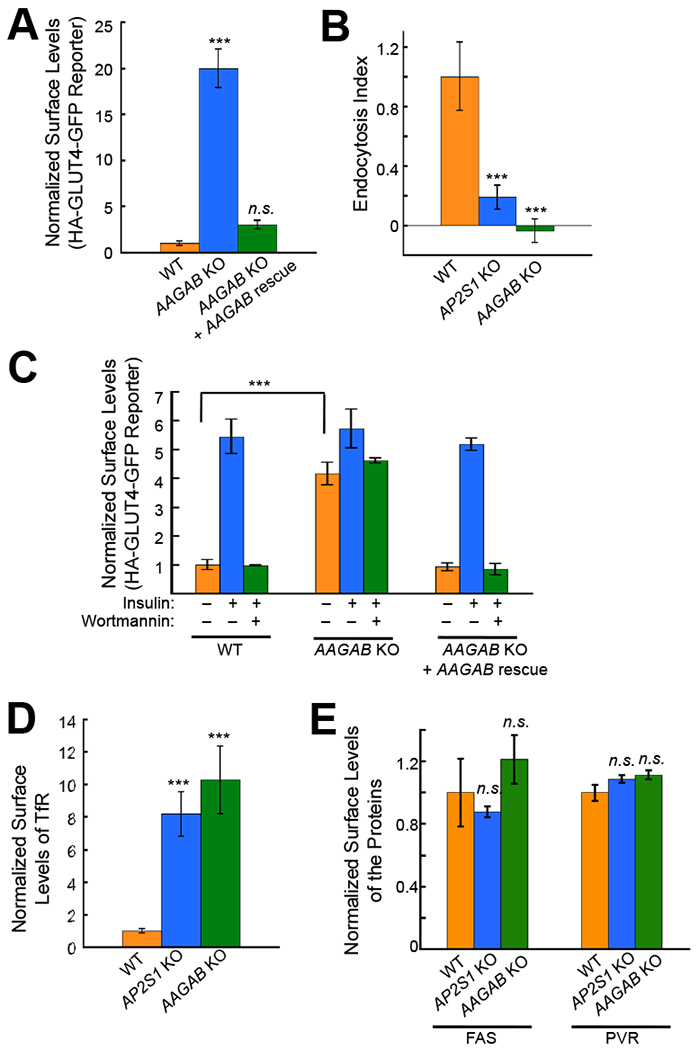
(A) Surface levels of the CME cargo reporter HA-GLUT4-GFP in HeLa cells were measured by flow cytometry. In AAGAB KO rescue cells, the WT AAGAB gene was stably expressed in AAGAB KO cells using lentiviral infection. The cells were labeled with anti-HA antibodies and APC-conjugated secondary antibodies before the APC and GFP fluorescence was measured by flow cytometry. The mean APC and GFP fluorescence of each sample was derived from >5,000 cells and the mean APC/GFP ratio was normalized to that of untreated WT cells. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the WT sample using six samples/group obtained from two independent experiments. Error bars indicate standard deviation. *** P<0.001. n.s., P>0.05. (B) Endocytosis rates of the CME cargo reporter HA-GLUT4-GFP in WT or mutant HeLa cells were quantified by flow cytometry. Live cells were stained at 37 °C in the presence or absence of Dynasore. The endocytosis Index was calculated using the following formula: (α – δ)/δ, where α is the mean fluorescence of the cells without dynasore treatment and δ is the mean fluorescence of dynasore-treated cells. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the WT sample. Each sample is based on the mean fluorescence from >5,000 cells and statistics was calculated using six samples/group obtained from two independent experiments. Error bars indicate standard deviation. *** P<0.001. (C) Experiments were performed as in A except that adipocytes were used. To measure insulin responses, cells were treated with 100 nM insulin for 30 minutes. To inhibit insulin signaling, 100 nM wortmannin was added 10 minutes prior to insulin stimulation. A Student’s t-test was used to compare samples from WT and mutant adipocytes using six samples/group obtained from two independent experiments. Error bars indicate standard deviation. *** P<0.001. (D) Flow cytometry measurements showing normalized surface levels of TfR in WT or mutant HeLa cells. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the WT sample. Each sample is based on the mean fluorescence from >5,000 cells and statistics was calculated using six samples/group obtained from two independent experiments. Error bars indicate standard deviation. *** P<0.001. (E) Surface levels of FAS and PVR in WT or mutant HeLa cells were measured by flow cytometry. The bar graphs depict mean values obtained from six samples/group from two independent experiments where each sample was a mean fluorescence obtained from >5,000 cells. Data of the mutant cells were normalized to those of WT cells. Error bars indicate standard deviation. n.s., P>0.05.
Loss of AP2 adaptor in AAGAB KO cells
Next we sought to unravel the molecular mechanism by which AAGAB regulates CME. AAGAB was found to associate with the α subunit (α-adaptin) of AP2 adaptor in a yeast two-hybrid assay (Page et al., 1999), but it was unclear whether the interaction was direct or biologically significant. To examine the binding of AAGAB to the α subunit, we co-expressed AAGAB and the large trunk domain of the α subunit in E. coli cells. Using an in vitro GST pull-down assay, we observed that AAGAB stoichiometrically bound to the α subunit monomer (Figure 3A–B). Thus, AAGAB directly interacts with the α subunit of AP2 adaptor.
Figure 3. Loss of AP2 adaptor in AAGAB KO cells.

(A) Diagram of the GST pull-down assay measuring AAGAB binding to the α subunit (trunk domain, a.a. 1-621) of AP2 adaptor. (B) Left: Coomassie blue-stained gel showing the binding of His6-SUMO-AAGAB to GST-tagged α subunit from GST pull-down assays. Right: Coomassie blue-stained gel showing His6-SUMO-AAGAB proteins precipitated by nickel beads. AAGAB expression levels were much higher than GST-α-subunit such that the latter was often not evident in nickel bead pull-down samples. (C) Representative confocal microscopy images showing the α subunit of AP2 adaptor in WT or mutant HeLa cells. Bar: 10 μm. (D) Representative TIRF microscopy images showing the α subunit of AP2 adaptor in WT or mutant HeLa cells. Bar: 10 μm. (E) Immunoblots showing the expression levels of the indicated proteins in WT or AAGAB KO HeLa cells.
The discovery of a direct interaction between AAGAB and the α subunit of AP2 adaptor prompted us to examine how AAGAB regulates the function of AP2 adaptor in the cell. Confocal imaging revealed abundant AP2 puncta on the surface of WT cells (Figure 3C). Strikingly, the AP2 puncta disappeared in AAGAB KO cells (Figure 3C). Total internal reflection fluorescence (TIRF) microscopy, which selectively visualizes events near the plasma membrane, confirmed the absence of AP2 puncta on the surface of AAGAB KO cells (Figure 3D). The loss of surface AP2 puncta in AAGAB KO cells could be due to defects in the recruitment of AP2 adaptors to the cell surface. Alternatively, the expression of AP2 adaptors could be diminished in the absence of AAGAB. To distinguish between these possibilities, we examined the expression levels of AP2 subunits in AAGAB KO cells. We observed that the expression of the α subunit was largely abolished in AAGAB KO cells (Figure 3E). Likewise, the expression of other AP2 subunits was also strongly reduced in AAGAB KO cells (Figure 3E). These results demonstrated that deletion of AAGAB leads to the loss of AP2 adaptor, correlating with CME defects in the mutant cells.
Stabilization of α subunit monomer by AAGAB
Since the stability of multimeric complexes is usually dependent on proper assembly (Kim et al., 2013), we postulated that AAGAB may be required for the assembly of the AP2 adaptor complex in the cytosol. Consistent with this model, AAGAB is a cytosolic protein (Figure 4A) (Pohler et al., 2012), exhibiting no co-localization with mature AP2 adaptor complexes on the cell surface (Figure 4A). We reasoned that, if AAGAB regulates AP2 adaptor assembly in the cytosol, it should remain active in CME when tethered to the cytosolic face of membrane-bound organelles. To test this possibility, we fused AAGAB to the cytosolic N-terminus of ATF6, an endoplasmic reticulum (ER)-resident membrane protein (Figure 4B) (Shen et al., 2002). Like ATF6, the AAGAB-ATF6 chimera exhibited a perinuclear network-like localization pattern characteristic of ER membrane proteins (Figure S2). We observed that TfR endocytosis was restored when either WT AAGAB or the AAGAB-ATF6 chimera was expressed in AAGAB KO cells (Figure 4C–D). By contrast, ATF6 itself failed to rescue TfR endocytosis (Figure 4C–D). Thus, the ER-anchored AAGAB protein is still functional in CME, indicating that it does not need to associate with the cell surface to regulate CME. Together, these findings further support the notion that AAGAB is involved in the assembly of the AP2 adaptor complex in the cytosol, prior to the trafficking of the adaptor complex to the cell surface.
Figure 4. AAGAB stabilizes the α subunit.
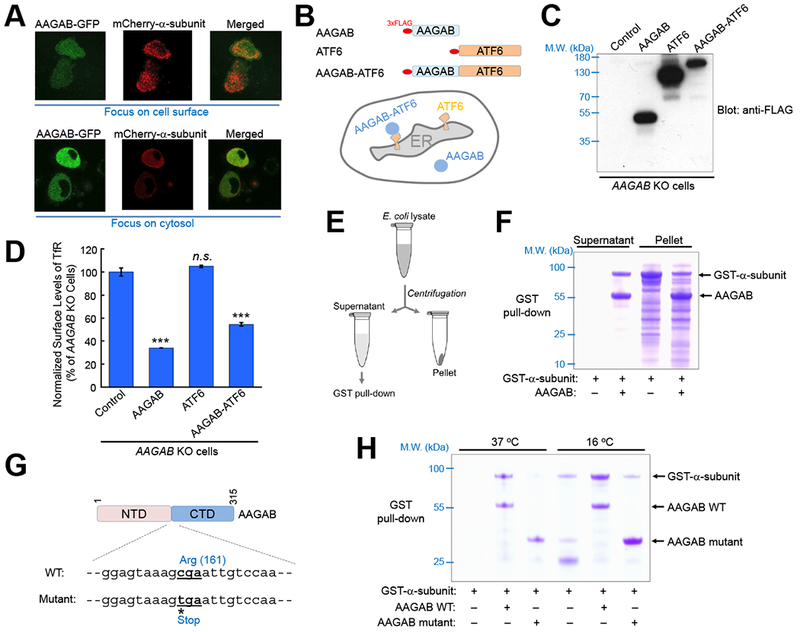
(A) Disparate subcellular localization of AAGAB and AP2 adaptor. GFP-tagged AAGAB and mCherry-tagged α subunit were transiently expressed in AAGAB KO HeLa cells. Their subcellular localization was visualized in live cells on an N-SIM microscope with the focus either at the cell surface (top) or the cytosol (bottom). (B) Diagrams of AAGAB, ATF6, and AAGAB-ATF6 chimera, which were N-terminally tagged with three tandem copies of the FLAG epitope (3×FLAG). (C) Immunoblot showing the expression of the 3×FLAG-tagged proteins in AAGAB KO HeLa cells. Control: an empty vector was transfected into AAGAB KO HeLa cells. (D) Flow cytometry measurements of the surface levels of TfR in AAGAB KO HeLa cells expressing the indicated proteins. TfR levels were normalized to the control sample in which an empty vector was transfected into AAGAB KO cells. Error bars indicate standard deviation. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the control sample. Each sample is based on the mean fluorescence from >5,000 cells and statistical analyses were carried out based on at least six samples/group obtained from at least two independent experiments. *** P<0.001. n.s. P>0.05. (E) Diagram of the assay measuring the solubility of α subunit (trunk domain) in E. coli with or without co-expression with His6-SUMO-AAGAB. Unless indicated otherwise, E. coli cells were grown at 37 °C. (F) Coomassie blue-stained gel showing the presence of GST-α-subunit (trunk domain) in the supernatant and pellet fractions of E. coli lysates. (G) Illustration of a PPKP1-causing mutation in which cytosine 481 was substituted with a thymine (Giehl et al., 2012; Pohler et al., 2012). The mutation resulted in a truncated AAGAB protein (a.a. 1-160). (H) Coomassie blue-stained gel showing proteins from GST pull-down assays. GST-α-subunit (trunk domain) was expressed in E. coli at either 16 °C or 37 °C with or without His6-SUMO-AAGAB (WT or the mutant shown in G). Note: the AAGAB mutant was prone to precipitate with glutathione beads but the experiment was performed using fully soluble proteins prior to GST pull-down. See also Figures S2–4.
Next we directly examined how AAGAB regulates AP2 adaptor assembly. Since AAGAB physically interacts with the α subunit and is required for the latter’s expression (Figure 3), AAGAB may stabilize the α subunit monomer. In support of this notion, the mRNA levels of the α subunit remained intact in AAGAB KO cells (Figure S3A). We then reconstituted AAGAB and the α subunit into E. coli cells, which lack both proteins. We observed that recombinant α subunit was readily expressed in E. coli with or without AAGAB co-expression (Figure 4E–F). However, soluble α subunit was obtained only from E. coli cells co-expressing AAGAB (Figure 4F). Without AAGAB co-expression, α subunit proteins were found exclusively in the insoluble fraction (Figure 4F). Thus, the α subunit monomer is an intrinsically unstable protein prone to misfolding and must be stabilized by AAGAB. Together, these data demonstrated that AAGAB acts as a specific chaperone for the α subunit monomer.
AAGAB mutations in PPKP1 usually cause premature translation termination, resulting in C-terminally truncated proteins (Nomura et al., 2015; Pohler et al., 2012). Next we examined the activity of one such truncated AAGAB mutant (Figure 4G). We observed that, when reconstituted in E. coli, the AAGAB mutant failed to stabilize the α subunit monomer such that the α subunit was absent in the soluble fraction (Figures 4H and S4). We then lowered the E. coli culture temperature from 37 °C to 16 °C to thermodynamically enhance protein folding and stability. We found that small amounts of soluble α subunit were obtained from the low temperature culture even without AAGAB co-expression (Figure 4H), in agreement with the chaperone function of AAGAB. The level of soluble α subunit in the 16 °C culture was strongly increased by co-expression with WT AAGAB but not the disease-mimicking AAGAB mutant (Figure 4H). Thus, the chaperone function of AAGAB is abolished by the presence of the PPKP1-causing mutation, correlating with the inability of the AAGAB mutant to rescue TfR endocytosis or AP2 expression (Figure S3B–C).
Together, these results demonstrated that AAGAB acts as a molecular chaperone to stabilize the α subunit monomer. Without AAGAB, the intrinsically unstable α subunit monomer is degraded. Since the stability of AP2 subunits is interdependent (Motley et al., 2003), loss of the α subunit leads to degradation of other AP2 subunits.
Sequential assembly of AP2 subunits controlled by AAGAB
The ability of AAGAB to bind and stabilize the α subunit monomer suggests that the assembly of the AP2 adaptor complex begins with the AAGAB-α-subunit dimer. Next we sought to define how the AAGAB-α-subunit dimer initiates AP2 adaptor assembly. To simultaneously monitor all AP2 subunits in co-immunoprecipitation (co-IP) assays, we developed an HA-tagging system in which each AP2 subunit was C-terminally tagged with an HA epitope (Figure 5A). As expected, AAGAB bound to the α subunit in co-IP (Figure 5B–C). Interestingly, while the σ2 subunit itself did not associate with AAGAB in co-IP, it interacted with AAGAB when the α subunit was co-expressed (Figure 5B–C). By contrast, the μ2 subunit did not associate with AAGAB or AAGAB-containing complexes in co-IP (Figure 5B–C). To distinguish the α and β2 subunits, which co-migrate on SDS-PAGE, we substituted the HA-tagged α subunit with an untagged version of the protein (Figure 5D). Co-IP assays using this tagging system showed that the β2 subunit did not bind AAGAB or AAGAB-containing complexes (Figure 5E–F). These co-IP assays also confirmed that the σ2 subunit associated with AAGAB only in the presence of the α subunit while the μ2 subunit did not bind AAGAB regardless of the presence of other subunits (Figure 5E–F).
Figure 5. AAGAB regulates the sequential assembly of AP2 subunits.
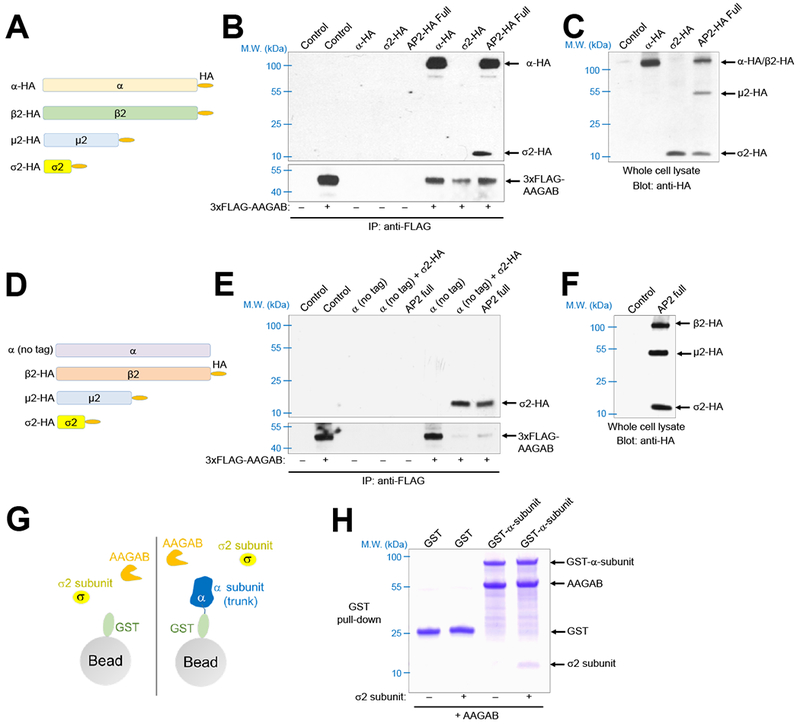
(A) Diagrams of HA-tagged full-length (FL) AP2 subunits used in co-IP. (B) Immunoblots showing the interaction of 3×FLAG-AAGAB with HA-tagged AP2 subunits. The 3×FLAG-AAGAB protein was transiently expressed in AAGAB KO HeLa cells with an empty vector (control) or vectors encoding the indicated HA-tagged AP2 subunits. The 3×FLAG-AAGAB protein was immunoprecipitated from the cell lysates using anti-FLAG antibodies and the presence of 3×FLAG-AAGAB (bottom) and HA-tagged AP2 subunits (top) in the immunoprecipitates was detected using anti-FLAG and anti-HA antibodies, respectively. AP2 full: co-expression of all four AP2 subunits depicted in A. (C) Immunoblot showing the expression of HA-tagged AP2 subunits in HeLa whole cell lysates. Note: α and β2 subunits co-migrated on SDS-PAGE. (D) Diagrams of FL AP2 subunits used in co-IP. AP2 subunits were C-terminally tagged with an HA epitope except the α subunit. (E) Immunoblots showing the interaction of 3×FLAG-AAGAB (bottom blot) with AP2 subunits (top blot). Co-IP and immunoblotting were carried out as described in B except that untagged α subunit was used. AP2 full: co-expression of all four AP2 subunits depicted in D. (F) Immunoblot showing the expression of AP2 subunits in HeLa cells. Note: α subunit was not detected due to a lack of an HA epitope. (G) Diagram of the GST pull-down assay measuring the interactions of AAGAB with the α and σ2 subunits. GST and GST-α-subunit were co-expressed with AAGAB in the absence or presence of the σ2 subunit. (H) Coomassie blue-stained gel showing the binding of His6-SUMO-AAGAB to GST-tagged α-subunit (trunk domain) and untagged σ2 subunit.
To further examine the binding of the σ2 subunit to AAGAB and the α subunit, we generated recombinant proteins in E. coli and examined their interactions using in vitro GST pull-down assays (Figure 5G). We observed that the σ2 subunit interacted stoichiometrically with AAGAB and the α subunit (Figure 5H), consistent with the co-IP data (Figure 5A–F). These results demonstrate that the AAGAB-α-subunit dimer recruits the σ2 subunit to form an AAGAB-α-σ2 trimer, in which the α-σ2 intermediate is stabilized by AAGAB. The inability of AAGAB to bind the β2 and μ2 subunits suggests that AAGAB dissociates from the α and σ2 subunits prior to the full assembly of AP2 adaptor. These molecular events occur in solution without requiring the membrane bilayer, in agreement with the cytosolic localization of AAGAB (Figure 4A). Thus, AAGAB regulates the assembly of the AP2 adaptor complex in the cytosol but does not follow the latter to the cell surface. These results also indicate that AAGAB not only acts as a chaperone for the α subunit monomer but also regulates the sequential assembly of AP2 subunits.
AAGAB-AP2 association requires the cargo-binding sites of AP2 subunits
The acidic dileucine motif of CME cargo proteins is recognized by the α and σ2 subunits of AP2 adaptor (Kelly et al., 2008). The acidic dileucine motif binds to the arginine 21 (R21) residue of the α subunit and the valine 88 (V88) and leucine 103 (L103) residues of the σ2 subunit (Figure 6A) (Kelly et al., 2008). Mutations of these residues abrogate the binding of AP2 adaptor to the acidic dileucine motif without affecting the folding of the proteins (Kelly et al., 2008). Using recombinant proteins co-expressed in E. coli (Figure 6B–C), we observed the binding of the σ2 subunit to the AAGAB-α-subunit complex was markedly inhibited by a V88D or L103S mutation in the σ2 subunit (Figure 6D), while the AAGAB-α-subunit complex itself remained intact (Figures 6E–F and S5A–B). Thus, these mutations interfere with the binding of the σ2 subunit to the AAGAB-α-subunit complex. Similarly, a R21E mutation in the α subunit markedly reduced the expression level of soluble α-subunit (Figures 6E and S5A–B), suggesting that the chaperone function of AAGAB involves the R21 residue of the α subunit. The total expression levels of the AAGAB protein in E. coli were not influenced by the mutations (Figures 6G and S5B). Consistent with these biochemical data, we observed that the R21E and L103S mutations diminished the expression of the α and σ2 subunits in WT HeLa cells (Figure S5C–D). Together, these data suggest that the cargo-binding residues of the α and σ2 subunits are required for AAGAB-AP2 interaction.
Figure 6. AAGAB recognizes the CME cargo-binding sites of AP2 subunits.
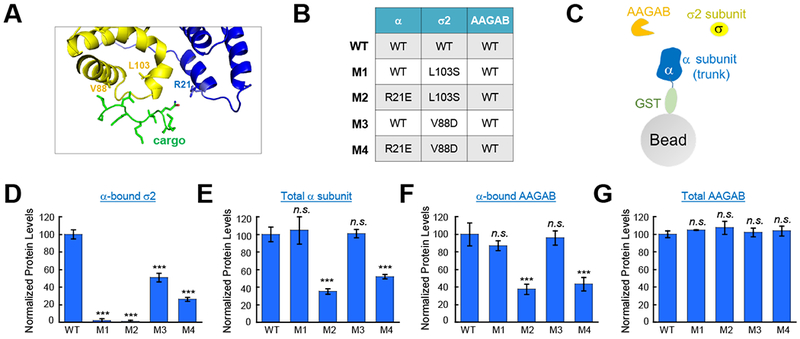
(A) Crystal structure showing the binding of a CME cargo peptide (green) to the α (blue) and σ2 (yellow) subunits of AP2 adaptor (PDB: 2jkr) (Kelly et al., 2008). Interacting side chains are shown. The R21 residue of the α subunit and the V88 and L103 residues of the σ2 subunit contact the acidic dileucine peptide. (B) Co-expression of WT His6-SUMO-AAGAB, GST-α-subunit (trunk domain, WT or R21E mutant), and untagged σ2 subunit (WT, V88D mutant, or L103S mutant) in E. coli. (C) Diagram showing the GST pull-down assay used to precipitate proteins from the E. coli lysates described in B. GST-α-subunit and associated proteins were precipitated from the E. coli lysates using glutathione beads and resolved on SDS-PAGE. Intensity of Coomassie blue-stained bands was quantified using Image J. The levels of the σ2 subunit (D), GST-tagged α subunit (E), and His6-SUMO-AAGAB (F) in the precipitates were normalized to their WT samples. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the WT sample based on at least three datasets from two independent experiments. Error bars indicate standard deviation. ***P<0.001. n.s., P>0.05. A representative Coomassie blue-stained gel is shown in Figure S6A. (G) Normalized expression levels of AAGAB in E. coli samples described in B. His6-SUMO-AAGAB was precipitated from E. coli lysates using nickel beads and resolved on SDS-PAGE. Note: the expression of AAGAB was in great excess to AP2 subunits. The protein levels were quantified as described in D-F and normalized to the WT sample. A representative Coomassie blue-stained gel is shown in Figure S6B. One-way ANOVA with Holm-Sidak correction for multiple testing was performed by comparing each dataset to the WT sample based on three samples from two independent experiments. Error bars indicate standard deviation. n.s., P>0.05. See also Figures S5–6.
In a yeast two-hybrid assay, AAGAB also associated with the γ subunit (γ-adaptin) of AP1 adaptor, which is involved in clathrin-mediated trafficking from the trans-Golgi to the endosome (Gorynia et al., 2012; Page et al., 1999). The biological significance of this interaction, however, was unclear. We observed that the expression of the γ subunit was strongly reduced in AAGAB KO cells whereas the δ subunit (δ-adaptin) of AP3 adaptor remained intact (Figure S6A). We next reconstituted AAGAB and the γ subunit into E. coli cells. We observed that soluble γ subunit proteins were obtained only when AAGAB was co-expressed (Figure S6B). In the absence of AAGAB, γ subunit was found in the insoluble fraction (Figure S6B). Thus, AAGAB binds and stabilizes the γ subunit, suggesting that it is also involved in the assembly of the AP1 adaptor complex.
DISCUSSION
In this work, we discovered that the AP2 adaptor complex does not assemble spontaneously as previously thought. Instead, it is a highly orchestrated process involving ordered molecular interactions controlled by AAGAB (Figure 7). AP2 adaptor assembly begins with the AAGAB-α-subunit dimer, which serves as a “seed” for subsequent association with other AP2 subunits. The AAGAB-α-subunit dimer recruits the σ2 subunit to form an AAGAB-α-σ2 trimer. In these complexes, AAGAB stabilizes the α subunit monomer and the α-σ2 intermediate, maintaining them at assembly-competent states. Subsequently, the β2 and μ2 subunits displace AAGAB, leading to the formation of the tetrameric AP2 adaptor complex (Figure 7). The β2 and μ2 subunits may transiently interact with the AAGAB-α-σ2 complex and induce conformation changes in the latter to trigger AAGAB release. Without the assistance of AAGAB, the intrinsically unstable AP2 subunits are prone to forming kinetically trapped aggregates and folding/assembly intermediates susceptible to degradation. Given the overall structural and functional similarity between AP1 and AP2 adaptors (Doray et al., 2007; Ren et al., 2013; Traub and Bonifacino, 2013), their assembly is likely controlled by AAGAB in a similar manner (Figure 7). Since AAGAB does not regulate AP3 adaptor and is not known to recognize other substrates, we posit that AAGAB specifically regulates AP1 and AP2 adaptors.
Figure 7. Model depicting the role of AAGAB in AP2 adaptor assembly.
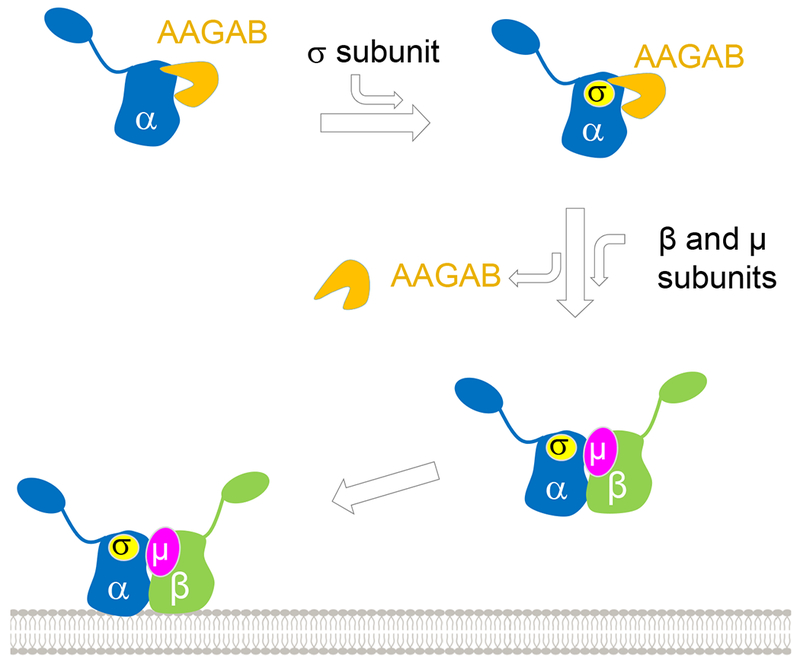
See also Figure S7.
AAGAB is not homologous to any known chaperone. Its N-terminal portion appears to adopt a GTPase-like structure but does not possess a canonical GTPase catalytic site (Gorynia et al., 2012; Pohler et al., 2012). The C-terminal half of AAGAB does not resemble any known protein or domain. AAGAB does not appear to act in a chaperone complex because our biochemical experiments showed that AAGAB itself is sufficient to stabilize the α subunit of AP2 adaptor and the γ subunit of AP1 adaptor. While further studies are needed to elucidate the pathogenesis of AAGAB-linked PPKP1, our findings suggest that the disease is caused by deficiencies in clathrin adaptors. Thus, compounds that promote the folding/stability of clathrin adaptors (e.g., chemical chaperones) may offer therapeutic benefits to PPKP1 patients.
The sorting signals of CME cargo proteins are short stretches of residues also found in many non-cargo proteins including cytosolic proteins and the cytosolic domains of membrane proteins anchored to intracellular organelles. Since the AP2 adaptor complex assembles in the cytosol, its binding to these non-cargo cytosolic proteins must be prevented. When the AP2 adaptor complex is fully assembled in the cytosol, the β2 subunit masks the cargo-binding sites until the adaptor complex is recruited to the plasma membrane (Jackson et al., 2010). However, prior to full assembly of the AP2 adaptor, another mechanism is required to shield the α and σ2 subunits from promiscuous interactions with cytosolic proteins. Interestingly, the cargo-binding sites of the α and σ2 subunits are required for AAGAB-AP2 association, suggesting that AAGAB may directly interact with the cargo-binding sites and prevent the latter from binding to acidic dileucine motifs on cytosolic proteins. Subsequent displacement of AAGAB is accompanied by the joining of the β2 subunit to bury the cargo-binding sites on the α and σ2 subunits. This AAGAB-dependent mechanism may also prevent premature binding of AP2 assembly intermediates to CME cargo proteins prior to full assembly of the adaptor complex. In addition to the cargo-binding residues of the α and σ2 subunits, AAGAB-AP2 association likely also involves other motifs of AP2 subunits. When artificially expressed in the absence of AAGAB, the α and σ2 subunits could form a AAGAB-free complex (Figure S7), suggesting that AAGAB stabilizes AP2 subunits and their assembly intermediates but is not required for α-σ2 association. However, it is also possible that the AAGAB-free α-σ2 complex represents a non-physiological, off-pathway assembly. Further structural and biochemical studies will be needed to determine how AAGAB sequentially interacts with AP2 subunits and whether it helps improve cargo-binding fidelity.
Multimeric adaptors are broadly involved in vesicle-mediated membrane trafficking (Bonifacino and Glick, 2004). These trafficking adaptors assemble in the cytosol before being recruited to membrane-bound organelles (Bonifacino and Glick, 2004; Rothman, 1994; Schekman and Novick, 2004). Since other adaptor complexes face the same challenges in their assembly as AP1 and AP2 adaptors, they likely also require AAGAB-like factors to control their assembly.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jingshi Shen (jingshi.shen@colorado.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa cells, 293T cells, and mouse preadipocytes (derived from inguinal white adipose tissues) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with penicillin/streptomycin and 10% FB Essence.
METHOD DETAILS
Plasmids
To express recombinant proteins in E. coli, we subcloned the human AAGAB gene into the pET-SUMO expression vector (Rathore et al., 2010), the mouse AP2A2 gene encoding the trunk domain of α subunit (a.a. 1-621) into the pGEX4T-3 vector, and the rat AP2S1 gene into the pACYC-Duet vector. For transient expression in mammalian cells, the human AAGAB gene was subcloned into the p3×FLAG 7.1 vector. The mouse Ap2a2 gene, the rat Ap2s1 gene, the human AP2B1 gene, and the mouse Ap2m1 gene were individually subcloned into the pcDNA3.1 vector with or without a C-terminal HA epitope tag. The rat Ap2s1 gene was subcloned into the p3XFLAG-CMV™-14 vector. To express the AAGAB-GFP fusion protein, the human AAGAB gene was subcloned into a modified pEGFP-N1 vector that encodes a monomeric eGFP bearing the A207K mutation. For lentiviral expression, the human AAGAB gene was subcloned into the pCSII vector. Point mutations were introduced using a site-directed mutagenesis kit. Oligonucleotides used in CRISPR KO are listed in Table S5.
Cell culture
All mammalian cells were grown at 37 °C with the CO2 level set to 5%. HeLa and preadipocyte cells stably expressing the HA-GLUT4-GFP reporter were generated using lentiviral infection (Gulbranson et al., 2017). Lentiviruses were produced by transfecting 293T cells with a mixture of plasmids including HA-GLUT4-GFP (Muretta et al., 2008), pAdVAntage, pCMV-VSVG, and psPax2. Lentiviral particles were collected 40 hours after transfection and every 24 hours thereafter for a total of four collections. Lentiviruses were pooled and concentrated by centrifugation in a Beckman SW28 rotor at 25,000 rpm for 1.5 hours. The viral pellets were resuspended in PBS and used to transduce HeLa cells and preadipocytes. A subclone of HeLa cells stably expressing HA-GLUT4-GFP was established and used for subsequent experiments. Immortalized mouse preadipocytes stably expressing HA-GLUT4-GFP were used without further subcloning. To differentiate into adipocytes, preadipocytes were cultured to ~95% confluence before a differentiation mixture was added at the following concentrations: 5 μg/mL insulin, 1 nM triiodo-l-thyronine, 125 mM indomethacin, 5μM dexamethasone, and 0.5 mM isobutylmethylxanthine. After 2 d, the cells were switched to DMEM supplemented with 10% FBS, 5 μg/mL insulin, and 1 nM triiodo-l-thyronine. After another 2 days, fresh medium of the same composition was supplied. Differentiated adipocytes were usually analyzed 6 days after addition of the differentiation mixture. For transient transfections, cells were plated at ~50% confluence, transfected with FuGENE HD following the manufacture’s protocol, and analyzed 24-48 hours after transfection.
Genome-wide CRISPR genetic screen
In the genome-wide CRISPR screen, ~40 million mutagenized HeLa reporter cells were seeded at ~1.2 million cells per 10-cm dish. On the following day, the cells were incubated in the KRH buffer (12 mM HEPES [pH7.0], 121 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, and 0.33 mM CaCl2) for two hours. The dishes were subsequently chilled on an ice bath and the cells were labeled with anti-HA antibodies and APC-conjugated secondary antibodies. After dissociation from the plates by Accutase, the cells were concentrated by centrifugation and the top 1.2% of the cells with the highest fluorescence were harvested in the first round of the sorting by FACS. The collected cells were expanded and sorted for another two rounds using the same fluorescence gating.
Genomic DNA was isolated from the final sorted population using a genomic DNA isolation kit. The unsorted control population contained ~50 million cells whereas the sorted populations contained about five million cells. The isolated genomic DNA was used as template to amplify guide sequences. In the first round of PCR, each reaction was performed in a total volume of 100 μL containing 10 μg genomic DNA and SequencingAdaptors (F and R) (Table S5).
The second round of PCR reactions was performed in a total volume of 50 μL using 5 μL of the PCR products from the first round as the template. Of the 12 forward barcoded primers, six were used for the sorted populations and six were used for the unsorted control population. The barcoded forward primers (F01-12) and the reverse primer (R01) are listed in Table S5. Stagger sequences are shown to the 5’ of the barcode in lower case, while the priming sites are shown to the 3’ of the barcode in lower case. PCR products were pooled, purified using a gel purification kit, and sequenced on an Illumina HiSeq2000 using 1×125 v4 Chemistry. Sequencing reads were demultiplexed and processed to contain only the 20-bp unique guide sequences using the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Readcount tables and gene enrichment analysis were performed using the MAGeCK algorithm (http://sourceforge.net/proiects/mageck/).
Pooled secondary CRISPR screen
A pooled secondary CRISPR library was built based on the guide sequences of an activity-optimized library (Wang et al., 2015). Ten sgRNAs were selected for each of the top 572 genes from the genome-wide genetic screen. The secondary library also contained 1,000 non-targeting control sgRNAs and sgRNAs for 598 unrelated genes (ten sgRNAs for each gene). Oligonucleotides containing the guide sequences were synthesized by CustomArray and amplified by PCR using the Secondary Library Amplification F and R primers (Table S5). PCR products were ligated into the pLenti-CRISPR vector using a Gibson assembly kit. The pLenti-CRISPR vector was digested using BsmBI followed by alkaline phosphatase treatment and gel purification. Each Gibson assembly reaction contained 100 ng vector and 40 ng PCR products in triplicates. The reactions were subsequently dialyzed against deionized water and transformed into electrocompetent E. coli cells.
CRISPR-Cas9 editing of candidate genes
A candidate gene was edited using two independent guide sequences targeting an early constitutive exon(s) of the gene. One of the guide sequences was subcloned into the pLenti-CRISPR-V2 vector (Gulbranson et al., 2017). The other guide sequence was subcloned into a modified version of the pLenti-Guide-Puro vector in which the puromycin selection marker was replaced with a hygromycin selection marker (pLenti-Guide-Hygro). Lentiviruses generated from the CRISPR plasmids were used to infect target cells. The infected cells were consecutively selected using 1 μg/mL puromycin and 500 μg/mL hygromycin B.
Flow cytometry
To measure the relative surface levels of the HA-GLUT4-GFP reporter, cells were washed three times with the KRH buffer (12 mM HEPES [pH to 7.4], 121 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, and 0.33 mM CaCl2) (Menasche et al., 2018). The cells were then starved for two hours at 37 °C in KRH buffer before being rapidly chilled on an ice bath. The surface reporters were labeled using anti-HA antibodies and APC-conjugated secondary antibodies. The cells were dissociated from the plates by Accutase before their APC and GFP fluorescence was measured on a CyAN ADP analyzer. To measure the surface levels of other proteins, cells were washed three times with the KRH buffer and then rapidly chilled on an ice bath. The cells were then labeled using primary antibodies and APC-conjugated secondary antibodies. After dissociation from the plates by Accutase, the APC fluorescence of the cells was measured on a CyAN ADP analyzer. Data from populations of ~5,000 cells were analyzed using FlowJo.
Endocytosis assay
WT or mutant HeLa cells were incubated with the KRH buffer for two hours followed by treatment with 100 nM insulin for 30 minutes. The plates were then transferred to a 37 °C water bath and, where appropriate, treated with 200 μM Dynasore for five minutes. After addition of 5 μg/mL anti-HA antibodies, the cells were incubated at 37 °C for another five minutes. The cells were then washed with a wash buffer (KRH buffer supplemented with 5% FBS) and dissociated from the plate by Accutase. The cells were then fixed by 2% paraformaldehyde (PFA) for 15 minutes at room temperature. After washing, the cells were incubated with APC-conjugated anti-mouse secondary antibodies at room temperature for one hour in the KRH buffer supplemented with 2% FBS and 0.2 % saponin. Subsequently the cells were washed and analyzed on a Cyan ADP Analyzer. Endocytosis Index was calculated using the following formula: (α – δ)/δ, where α is the mean fluorescence of the cells without dynasore treatment whereas δ is the mean fluorescence of dynasore-treated cells.
IP and immunoblotting
HeLa cells were transiently transfected with the indicated plasmids using FuGENE HD and harvested 24-48 hours after transfection for analysis. In IP experiments, cells were lysed in an IP buffer (25 mM HEPES [pH 7.4], 138 mM NaCl, 10 mM Na3PO4, 2.7 mM KCL, 0.5% Triton X-100, and a protease inhibitor cocktail). Proteins were precipitated from cell lysates using anti-FLAG M2 antibodies and protein A agarose beads. Immunoprecipitates and whole cells were lysed in 1× SDS protein sample buffer and resolved on 8% Bis-Tris SDS-PAGE. Proteins were detected using primary antibodies and HRP-conjugated secondary antibodies. HA-tagged proteins were detected directly using HRP-conjugated anti-HA antibodies.
Immunostaining and imaging
HeLa cells were either untransfected or transfected with the indicated plasmids using FuGENE HD. Cells were harvested 48 hours after transfection for analysis. Cells grown on coverslips were fixed using 2% PFA and permeabilized in PBS supplemented with 5% FBS and 0.2% saponin. The α subunit was labeled using anti-μ-adaptin antibodies and Alexa Fluor 568-conjugated anti-mouse secondary antibodies. FLAG-tagged proteins were labeled using anti-FLAG M2 antibodies and Alexa Fluor 488-conjugated anti-mouse secondary antibodies. After mounting on glass slides using the ProLong Antifade mountant with DAPI, the cells were visualized on a Carl Zeiss LSM780 confocal microscope or a Nikon Ti-E inverted microscope. To visualize cells using TIRF microscopy, cells grown on Delta T dishes were fixed and stained in a similar way as in confocal microscopy. The cells were submerged in the PBS-based Citifluor AF3 anti-fade solution and visualized on a Carl Zeiss Observer Z1 microscope equipped with a Stable Z heating system (Bioptechs). The TIRF angle was set at 66° for Alexa Fluor 568 fluorescence. Cell images were processed using Image J.
Live cell microscopy
HeLa cells were seeded in CellView cell culture dishes and transfected with plasmids encoding AAGAB-GFP and mCherry-α-adaptin using FuGENE HD. After 48 hours, the cells were switched to PBS supplemented with 5% FBE and visualized on a Nikon Inverted Structured Illumination (N-SIM)/A1 microscope.
qPCR assay
Total RNA was isolated using TRIzol reagent and chloroform extraction. Total RNA was purified using Zymo RNA clean and concentrator columns. Total RNA (500 nanograms) was used to synthesize cDNAs with the Superscript IV first strand synthesis system using random hexamers according to the manufacturer’s instructions. Gene expression was determined by RT-qPCR on an Applied Biosystems 7500 Fast Real-time PCR machine using SsoAdvanced Universal SYBER Green Supermix with gene-specific primer sets (Table S5). The cycle threshold values of a candidate gene was normalized to those of GAPDH, a reference gene, and the Δcycle threshold values were calculated. The results were plotted as fold changes relative to the control sample.
Recombinant protein expression and purification
Recombinant proteins were expressed and purified using our established procedures (Yu et al., 2019; Yu et al., 2015). GST-tagged α subunit (a.a. 1-621) was expressed in BL21 E. coli cells with or without His6-SUMO-AAGAB and untagged σ2 subunit. In a standard experiment, 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to induce protein expression when the O.D. 600 of the E. coli culture in LB media reached ~0.6. The cells were incubated at 37 °C for three hours before the cells were harvested and lysed. After centrifugation, proteins in the supernatant were precipitated using glutathione beads or nickel beads, resolved on SDS-PAGE, and stained with Coomassie blue. In GST control experiments, the empty pGEX4T-3 vector was used to express the GST protein. To express proteins at 16 °C, E. coli cells were induced with 0.2 mM IPTG for 16 hours at 16 °C. GST-tagged AP1 γ subunit (a.a. 1-595) was expressed with or without His6-SUMO-AAGAB at 37 °C in a similar manner as GST-tagged AP2 α subunit.
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance for the genome-wide screen was calculated with MAGeCK algorithm. For flow cytometry experiments, quantification was based on the mean fluorescence from >5,000 cells in each sample. Statistical significance was calculated using datasets from multiple independent experiments. Detailed procedures and results of statistical analyses are shown in the Figure Legends of the corresponding experiments.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA | BioLegend | Cat# 901501, RRID:AB_2565006 |
| Allophycocyanin (APC)-conjugated secondary antibodies | eBioscience | Cat# 17-4015-82 |
| TfR | DSHB | Cat# G1/221/12, RRID:AB_2201506 |
| FAS | eBioscience | Cat# 14-0959-80, RRID:AB_467396 |
| PVR | BioLegend | Cat# 337602, RRID:AB_2300508 |
| FLAG | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| α-adaptin (α subunit of AP2 adaptor) | BD Biosciences | Cat# 610502, RRID:AB_397868 |
| α-adaptin | Santa Cruz Biotechnology | Cat# SC-17771, RRID:AB_2274034 |
| α-tubulin | eBioscience | Cat# 14-4502-82, RRID:AB_1210456 |
| AAGAB | Bethyl | Cat# A305-593A |
| β2 subunit | Bethyl | Cat# A304-719A, RRID:AB_2620914 |
| μ2 subunit | BD Biosciences | Cat# 611350, RRID:AB_398872 |
| γ-adaptin | Sigma-Aldrich | Cat# A4200, RRID:AB_476720 |
| δ-adaptin | DSHB | Cat# anti-delta SA4, RRID:AB_2056641 |
| Horseradish peroxidase (HRP)-conjugated anti-HA | Roche | Cat# 12013819001, RRID:AB_390917 |
| HRP-conjugated anti-rabbit IgG | Sigma-Aldrich | Cat# A6154, RRID:AB_258284 |
| HRP-conjugated anti-mouse IgG | Sigma-Aldrich | Cat# A6782, RRID:AB_258315 |
| Alexa Flour 488-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat# A11008, RRID:AB_142165 |
| Alexa Flour 568-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat# A11004, RRID:AB_2534072 |
| Bacterial and Virus Strains | ||
| Electrocompetent E. coli | Lucigen | Cat# 60242 |
| BL21 Gold DE3 competent E. coli | Stratagene | Cat# 230132 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase | Innovative Cell Technologies | Cat# AT 104 |
| Insulin | Sigma-Aldrich | Cat# I0516 |
| Citifluor AF3 anti-fade solution | Electron Microscopy Sciences | Cat# 17972-25 |
| Dynasore | Sigma-Aldrich | Cat# D7693 |
| FB Essence | VWR | Cat# 10803-034 |
| 3,3’,5-Triiodo-L-thyronine | Sigma-Aldrich | Cat# T2877 |
| Fetal bovine serum (FBS) | Sigma-Aldrich | Cat# F2442 |
| Indomethacin | Sigma-Aldrich | Cat# I7378 |
| ProLong Antifade mountant with DAPI | Thermo Fisher Scientific | Cat# P36931 |
| Protease inhibitor cocktail | Roche | Cat# 05056489001 |
| Dexamethasone | Sigma-Aldrich | Cat# D1756 |
| Isobutylmethylxanthine | Sigma-Aldrich | Cat# I5879 |
| Gibson assembly kit | Synthetic Genomics | Cat# GA1200 |
| Saponin | Sigma-Aldrich | Cat# 47036 |
| FuGENE HD | Promega | Cat# E2311 |
| TRIzol reagent | Thermo Fisher Scientific | Cat# 15596018 |
| Critical Commercial Assays | ||
| Genomic DNA isolation kit | Thermo Fisher Scientific | Cat# K0721 |
| Site-directed mutagenesis kit | Agilent | Cat# 210518 |
| Zymo RNA Clean and Concentrator columns | Zymo Research | Cat# R1018 |
| Superscript IV first strand synthesis system | Thermo Fisher Scientific | Cat# 18091050 |
| Sso Advanced SYBER Green Supermix | Bio-Rad | Cat# 1725270 |
| Experimental Models: Cell Lines | ||
| HeLa cells | ATCC | CCL-2 |
| 293T cells | ATCC | CRL-3216 |
| Mouse preadipocytes: inguinal white adipose tissues | Dr. Shingo Kajimura | N/A |
| Oligonucleotides | ||
| Sequencing adaptors, see table S5. | Sanjana et al. (2014) | N/A |
| Sequencing primers, see table S5. | Sanjana et al. (2014) | N/A |
| Secondary library amplification primers, see table S5. | Sanjana et al. (2014) | N/A |
| Secondary library oligos, see table S3. | This paper | N/A |
| CRISPR guide sequence primers, see table S5. | This paper | N/A |
| AP2A2 qPCR primers, see table S5. | This paper | N/A |
| GAPDH qPCR primers, see table S5. | Dr. Roy Parker | N/A |
| Recombinant DNA | ||
| Plasmid: pET-SUMO-humanAAGAB | This paper | N/A |
| Plasmid: pGEX4T-3-mouseAP2A2 | This paper | N/A |
| Plasmid: pACYC-Duet-ratAP2S1 | This paper | N/A |
| Plasmid: p3×FLAG-humanAAGAB | This paper | N/A |
| Plasmid: pcDNA3.1-mouseAP2A2-HA | This paper | N/A |
| Plasmid: pcDNA3.1-ratAP2S1-HA | This paper | N/A |
| Plasmid: pcDNA3.1-humanAP2B1-HA | This paper | N/A |
| Plasmid: pcDNA3.1-mouseAP2M1-HA | This paper | N/A |
| Plasmid: pcDNA3.1-mouseAP2A2 | This paper | N/A |
| Plasmid: pCMV-ratAP2S1-3×FLAG | This paper | N/A |
| Plasmid: pEGFP-N1-humanAAGAB | This paper | N/A |
| Plasmid: pCSII-humanAAGAB | This paper | N/A |
| Plasmid: p3×FLAG-CMV™-14 | Sigma-Aldrich | Cat# E7908 |
| Plasmid: p3×FLAG-CMV™-7.1 | Sigma-Aldrich | Cat# E7533 |
| Plasmid: pcDNA™3.1 (+) | Thermo Fisher Scientific | Cat# V79020 |
| Plasmid: pLenti-HA-GLUT4-GFP | (Muretta et al., 2008) | N/A |
| Plasmid: pLentiCRISPRv2 | (Sanjana et al., 2014) | Addgene: 52961 |
| Plasmid: pLenti-Guide-Hygro | (Gulbranson et al., 2017) | N/A |
| Plasmid: pLenti-CRISPR | (Shalem et al., 2014) | Addgene: 49535 |
| Plasmid: pAdVAntage | Promega | Cat# E1711 |
| Plasmid: pCMV-VSVG | (Stewart et al., 2003) | Addgene: 8454 |
| Plasmid: psPAX2 | Dr. Diderio Trono | Addgene: 12260 |
| Plasmid: pLenti-Guide-Puro | (Sanjana et al., 2014) | Addgene: 52963 |
| Plasmid: pAP1G1 (1-595)-GST | (Ren et al., 2013) | |
| Plasmid: pmCherry-α-adaptin | Dr. Juan Bonifacino | |
| Software and Algorithms | ||
| FASTX-toolkit | Hannon lab website | http://hannonlab.cshl.edu/fastx_toolkit/ |
| MAGeCK | (Li et al., 2014) | https://sourceforge.net/p/mageck/wiki/Home/ |
| EXCEL | Microsoft | N/A |
| KaleidaGraph | Synergy Software | http://www.synergy.com/wordpress_650164087/ |
| Prism (version 7) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R studio | R-project | N/A |
| FlowJo (version 8 and 9) | FlowJo | N/A |
AAGAB binds to the AP2 complex and is essential for clathrin-mediated endocytosis
AAGAB controls the sequential assembly of the AP2 adaptor complex
AAGAB stabilizes AP2 complex intermediates
A skin disease-causing mutation impairs the function of AAGAB
The AP2 complex is required for clathrin-mediated endocytosis. Gulbranson et al. identifies AAGAB as a regulator of AP2 adaptor assembly, which guides the sequential association of AP2 subunits and stabilizes assembly intermediates. The function of AAGAB is disrupted by a mutation that causes punctate palmoplantar keratoderma type 1.
ACKNOWLEDGEMENTS
We thank Drs. Shingo Kajimura, Frances Brodsky, Juan Bonifacino, David James, Yuan Tian, Qian Yin, David Gershlick, Joaquin Espinosa, James Hurley, and Nausica Arnoult for reagents. We thank Drs. Michael Stowell, Margaret Robinson, and Soyeon Park for helpful discussions. We are grateful to Dr. James Orth, Yuming Han, Katrina Diener, Jeff Reece, Benjamin Dodd, and Molishree Joshi for technical assistance. This work was supported by NSFC grants 31871425 and 91854117 (both to H.Y.), NIH grants DK095367 and GM126960 (both to J.S.), a University of Colorado seed grant (J.S.), an American Heart Association Predoctoral Fellowship (D.R.G.), and an NIH Predoctoral Training Grant GM088759 (L.C. and B.L.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
See supplementary tables 1–5.
Supplementary Table 1. Summary of deep sequencing reads in genome-wide primary screen, related to Figure 1.
Supplementary Table 2. Ranking of genes from the genome-wide CRISPR screen, related to Figure 1.
Supplementary Table 3. List of sgRNAs in the secondary CRISPR library, related to Figure 1.
Supplementary Table 4. Summary of deep sequencing reads in secondary screen, related to Figure 1.
Supplementary Table 5. Oligonucleotides, related to STAR Methods.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Antonescu CN, McGraw TE, and Klip A (2014). Reciprocal regulation of endocytosis and metabolism. Cold Spring Harbor perspectives in biology 6, a016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS, and Kandror KV (2010). Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Current opinion in cell biology 22, 506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boll W, Ohno H, Songyang Z, Rapoport I, Cantley LC, Bonifacino JS, and Kirchhausen T (1996). Sequence requirements for the recognition of tyrosine-based endocytic signals by clathrin AP-2 complexes. EMBO J 15, 5789–5795. [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, and Glick BS (2004). The mechanisms of vesicle budding and fusion. Cell 116, 153–166. [DOI] [PubMed] [Google Scholar]
- Brodsky FM (2012). Diversity of clathrin function: new tricks for an old protein. Annu Rev Cell Dev Biol 28, 309–336. [DOI] [PubMed] [Google Scholar]
- Collins BM, McCoy AJ, Kent HM, Evans PR, and Owen DJ (2002). Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535. [DOI] [PubMed] [Google Scholar]
- Conner SD, and Schmid SL (2003). Differential requirements for AP-2 in clathrin-mediated endocytosis. The Journal of cell biology 162, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, and De Camilli P (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Doray B, Lee I, Knisely J, Bu G, and Kornfeld S (2007). The gamma/sigma1 and alpha/sigma2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Molecular biology of the cell 18, 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, and Walz T (2004). Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature 432, 573–579. [DOI] [PubMed] [Google Scholar]
- Giehl KA, Eckstein GN, Pasternack SM, Praetzel-Wunder S, Ruzicka T, Lichtner P, Seidl K, Rogers M, Graf E, Langbein L, et al. (2012). Nonsense Mutations in AAGAB Cause Punctate Palmoplantar Keratoderma Type Buschke-Fischer-Brauer. American Journal of Human Genetics 91, 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorynia S, Lorenz TC, Costaguta G, Daboussi L, Cascio D, and Payne GS (2012). Yeast Irc6p is a novel type of conserved clathrin coat accessory factor related to small G proteins. Molecular biology of the cell 23, 4416–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbranson DR, Davis EM, Demmitt BA, Ouyang Y, Ye Y, Yu H, and Shen J (2017). RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc Natl Acad Sci U S A 114(39), E8224–E8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, Collins BM, Honing S, Evans PR, and Owen DJ (2010). A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Oh E, and Thurmond DC (2010). Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol 298, R517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M, and Roux A (2018). Mechanisms of clathrin-mediated endocytosis. Nature reviews Molecular cell biology 19, 313–326. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, and Drubin DG (2006). Harnessing actin dynamics for clathrin-mediated endocytosis. Nature reviews Molecular cell biology 7, 404–414. [DOI] [PubMed] [Google Scholar]
- Kelly BT, Graham SC, Liska N, Dannhauser PN, Honing S, Ungewickell EJ, and Owen DJ (2014). Clathrin adaptors. AP2 controls clathrin polymerization with a membrane-activated switch. Science (New York, NY) 345, 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BT, McCoy AJ, Spate K, Miller SE, Evans PR, Honing S, and Owen DJ (2008). A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 456, 976–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, and Hartl FU (2013). Molecular chaperone functions in protein folding and proteostasis. Annual review of biochemistry 82, 323–355. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Owen D, and Harrison SC (2014). Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harbor perspectives in biology 6, a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome biology 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, and Boucrot E (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nature reviews Molecular cell biology 12, 517–533. [DOI] [PubMed] [Google Scholar]
- Menasche BL, Crisman L, Gulbranson DR, Davis EM, Yu H, and Shen J (2018). Fluorescence Activated Cell Sorting (FACS) in Genome-Wide Genetic Screening of Membrane Trafficking. Curr Protoc Cell Biol, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M, Chen PH, Srinivasan S, Danuser G, and Schmid SL (2018). Regulation of Clathrin-Mediated Endocytosis. Annu Rev Biochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, and Robinson MS (2003). Clathrin-mediated endocytosis in AP-2-depleted cells. The Journal of cell biology 162, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muretta JM, Romenskaia I, and Mastick CC (2008). Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J Biol Chem 283, 311–323. [DOI] [PubMed] [Google Scholar]
- Nomura T, Yoneta A, Pohler E, Suzuki S, Osawa R, Mizuno O, Ohguchi Y, Nomura Y, Yamashita T, McLean WHI, et al. (2015). Punctate Palmoplantar Keratoderma Type 1: A Novel AAGAB Mutation and Efficacy of Etretinate. Acta Derm-Venereol 95, 110–111. [DOI] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, and Bonifacino JS (1995). Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science (New York, NY) 269, 1872–1875. [DOI] [PubMed] [Google Scholar]
- Page LJ, Sowerby PJ, Lui WW, and Robinson MS (1999). Gamma-synergin: an EH domain-containing protein that interacts with gamma-adaptin. The Journal of cell biology 146, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse BM, and Robinson MS (1984). Purification and properties of 100-kd proteins from coated vesicles and their reconstitution with clathrin. EMBO J 3, 1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohler E, Mamai O, Hirst J, Zamiri M, Horn H, Nomura T, Irvine AD, Moran B, Wilson NJ, Smith FJ, et al. (2012). Haploinsufficiency for AAGAB causes clinically heterogeneous forms of punctate palmoplantar keratoderma. Nat Genet 44, 1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan V, Agrawal NJ, Liu J, Engles S, Toy R, and Radhakrishnan R (2011). Systems biology and physical biology of clathrin-mediated endocytosis. Integr Biol (Camb) 3, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, and Shen J (2010). Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci U S A 107, 22399–22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Farias GG, Canagarajah BJ, Bonifacino JS, and Hurley JH (2013). Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE (1994). Mechanisms of intracellular protein transport. Nature 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Rowland AF, Fazakerley DJ, and James DE (2011). Mapping Insulin/GLUT4 Circuitry. Traffic 12, 672–681. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman R, and Novick P (2004). 23 genes, 23 years later. Cell 116, S13–15, 11 p following S19. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (New York, NY) 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, and Prywes R (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Developmental cell 3, 99–111. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM, and Bonifacino JS (2013). Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harbor perspectives in biology 5, a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science (New York, NY) 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Xing Y, Harrison SC, and Kirchhausen T (2010). Structural analysis of the interaction between Dishevelled2 and clathrin AP-2 adaptor, a critical step in noncanonical Wnt signaling. Structure 18, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Crisman L, Stowell MHBand Shen J (2019). Functional Reconstitution of Intracellular Vesicle Fusion Using Purified SNAREs and Sec1/Munc18 (SM) Proteins. Methods Mol Biol I860, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Rathore SS, Shen C, Liu Y, Ouyang Y, Stowell MH, and Shen J (2015). Reconstituting Intracellular Vesicle Fusion Reactions: The Essential Role of Macromolecular Crowding. J Am Chem Soc 137, 12873–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.