

Abstract
In our previous studies of the molecular mechanisms of poly(ADP-ribose) polymerase 1 (PARP-1)-mediated transcriptional regulation we identified a novel class of PARP-1 inhibitors targeting the histone-dependent route of PARP-1 activation. Because histone-dependent activation is unique to PARP-1, non-NAD-like PARP-1 inhibitors have the potential to bypass the off-target effects of classical NAD-dependent PARP-1 inhibitors, such as olaparib, veliparib, and rucaparib. Furthermore, our recently published studies demonstrate that, compared to NAD-like PARP-1 inhibitors that are used clinically, the non-NAD-like PARP-1 inhibitor 5F02 exhibited superior antitumor activity in cell and animal models of human prostate cancer (PC). In this study, we further evaluated the antitumor activity of 5F02 and several of its novel analogues against PC cells. In contrast to NAD-like PARP-1 inhibitors, non-NAD-like PARP-1 inhibitors demonstrated efficacy against androgen-dependent and -independent routes of AR signaling activation. Our experiments reveal that methylation of the quaternary ammonium salt and the presence of esters were critical for the antitumor activity of 5F02 against PC cells. In addition, we examined the role of a related regulatory protein of PARP-1, called Poly(ADP-ribose) glycohydrolase (PARG), in prostate carcinogenesis. Our study reveals that PARG expression is severely disrupted in PC cells, which is associated with decreased integrity and localization of Cajal bodies (CB). Overall, the results of our study strengthen the justification for using non-NAD-like PARP-1 inhibitors as a novel therapeutic strategy for the treatment of advanced prostate cancer.
Keywords: PARP-1, PARG, poly(ADP-ribose), non-NAD-like PARP-1 inhibitors, prostate cancer, 5F02
Graphical Abstract
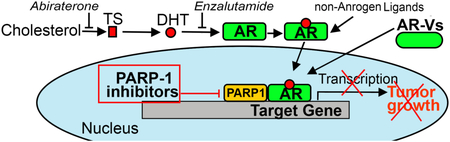
Therapeutic approaches targeting the androgen receptor (AR) signaling axis. PARP-1 inhibitors suppress AR transcriptional function and can therefore be effective against both androgen-dependent and -independent mechanisms of AR activation.
1. Introduction
PC is a significant public health concern, having an annual incidence of approximately 164,000 cases and 29,430 deaths in the United States [1]. Prostatic growth, both benign and malignant, is highly sensitive to changes in androgen production. At present, therapeutic agents such as abiraterone and enzalutamide (Fig.1A), have been shown to extend survival in patients with castration-resistant PC (CRPC). Nevertheless, nearly all patients ultimately develop a hormone-resistant form of PC.
Figure 1. Scientific rationale for targeting prostate cancer using non-NAD-like PARP-1 inhibitors.

A. Therapies targeting the AR signaling axis. Abiraterone, a potent and selective inhibitor of CYP17, blocks synthesis of testosterone (TS) and dihydrotestosterone (DHT). Enzalutamide inhibits binding of androgens to the AR. PARP-1 inhibitors reduce AR chromatin occupancy and thus diminish AR transcriptional output. B. Three ways of PARP-1 protein regulation: 1) competition with NAD for binding, 2) disruption of PARP-1 interaction with histones and 3) obstruction of binding with DNA.
Androgen receptor (AR) signaling is critical for both the initiation and maintenance of PC, even in castration resistant disease. For example, serum levels of prostate-specific antigen (PSA), a transcriptional target of the AR, rise in parallel with the emergence of castration-resistant disease, indicating that the AR is functionally active in CRPC [2–4]. These data suggest that AR-mediated functions are not completely abrogated by existing androgen deprivation therapies. Their therapeutic failure is often accompanied by various molecular alterations such as activation of the AR complex via molecular crosstalk with other signaling pathways, androgen-independent AR activation, and AR structural alterations, including expression of constitutively active AR variants that lack the ligand-binding domain [1,5,6].
Poly(ADP-ribose) polymerase 1 (PARP-1) is an abundant and ubiquitous nuclear enzyme. When active, it captures NAD+ to assemble long and branching molecules of poly(ADP-ribose) (pADPr), thereby modifying itself and surrounding nuclear proteins [7,8]. Previously we have demonstrated that PARP-1 binds the promoters of genes involved in the determination of cell self-identity and carcinogenesis [9]. The proposed function of adding pADPr to surrounding chromatin is for opening the chromatin structure, thereby allowing transcriptional machinery to access promoters and other gene elements necessary for gene expression. Current literature and recent clinical trials show that PARP-1 inhibitors are effective for the treatment of PC tumors that harbor mutations in DNA damage-repair genes [10,11]. In addition, recent studies have established that PARP-1 activity is required for AR function in PC cells, as well as generation and maintenance of castration resistance [12]. Given that both DNA repair and AR-mediated transcription depend on PARP-1 function, PC may be an especially susceptible target for PARP-1 inhibition.
PARP-1 activity has been shown to be regulated by three distinct mechanisms: 1) competitive binding with NAD; 2) disruption of PARP-1 interaction with histones; and 3) obstruction of PARP-1 binding with DNA [13] (Fig.1B). Most clinical PARP-1 inhibitors are designed to compete with NAD at the enzyme active site [14]. However, because NAD is ubiquitous in living cells and is utilized by several physiologically necessary enzymes, NAD competitors tend to have off-target effects. Multiple publications indicate that a vast majority of the currently available PARP-1 inhibitors do indeed have activities at distant unrelated targets, showing inter- family polypharmacology (promiscuous activities at targets of different families) and intra- family polypharmacology (non-selective activities at targets of the same family) as well as multi- signaling polypharmacology (multi-signaling activities mediated by the same target) [15–18]. Examples of off-target effects include inhibitory activities against mono-ADP-ribosyltransferases and sirtuins, which regulate essential metabolic processes [15,19–20]. In addition to the risk of unwanted side effects, the polypharmacologic effects of current NAD-like PARP-1 inhibitors may interfere with the interpretation of data obtained in basic biological research and clinical outcome studies.
To overcome the limitation of NAD-like PARP-1 inhibitors, we set out to identify molecules that inhibit PARP-1, but maintain structural independence from NAD [21]. By targeting the histone-dependent route of PARP-1 activation, a binding site that is unique to PARP-1, we have identified a novel class of small molecule inhibitors [22–25] that may be superior to the existing clinical therapies. Our new PARP-1 inhibitors have no obvious structural homologues among components of other enzymatic pathways, supporting the possibility that they may have greater inhibitory specificity and may minimize off-target effects observed in the current NAD targeting inhibitors such as olaparib and veliparib [24].
To corroborate the role of PARP-1 in PC, we examined the role of a PARP-1 regulating protein, called Poly(ADP-ribose) glycohydrolase (PARG) in PC survival. Thereafter we tested the antitumor activity of non-NAD-like PARP-1 inhibitors using an in vitro system composed of androgen-dependent and castration-resistant PC cell lines. We tested the antitumor activity of a lead group of seven non-NAD-like PARP-1 inhibitors using LNCaP, PC-3, and DU-145 cells, and the transcriptional activity of AR, pADPr expression, proliferation, and apoptosis were quantified. The effect of non-NAD-like PARP-1 inhibitors was further tested in the presence of the synthetic androgen agonist R1881, and their expected pharmacokinetic properties were assessed by physicochemical and ADME (Adsorption, Distribution, Metabolism and Excretion) analyses.
2. Materials and Methods
2.1. Reagents and Chemicals
Starting materials and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) in the highest purity available and used without further purification. Anhydrous solvents were purchased from Sigma-Aldrich in Sure/Seal™ bottles. All anhydrous solvents were > 99% pure. Other solvents were purchased from Fisher Scientific (Hampton, NH, USA) in Certified ACS grade and used without further purification.
2.2. Synthesis (Fig.2)
Figure 2. Synthesis of 5F02 analogues.

A. The structure of 5F02 is shown as compound 1. It contains both ester and N-methyl quaternary ammonium functionalities. It is possible that the N-methyl quaternary ammonium functionality served as a methyl donor to promote covalent methylation which could cause the observed biological activity. The ester functionality is likely to be metabolically labile upon in vivo administration by virtue of cleavage by esterases. Therefore, it was of interest to prepare and test analogs in which the quaternary methyl group is absent (2), the ester was converted to an amide (3), or both of these modifications being simultaneously (4). Compounds 2–4 were prepared starting from cyclododecanol (5) and cyclododecanamine (6), both reacting with 2-(1-piperidinyl)acetic acid to give ester 2 and amide 4. Compound 4 was taken on further in reaction with methyl iodide to give N-methyl quaternary ammonium amide 3. B. The structures of synthesized 5F02 analogs MC-270016, 270017, 270019 and 270021 are shown. The intention was to obtain some preliminary information on the SAR of the piperidine ring of 5F02. These compounds were prepared by reacting cyclododecanol (5) with chloroacetyl chloride to give the common intermediate (7). Compound (7) was then treated with the appropriate cyclic amine to provide the penultimate intermediates (8), which were methylated with methyl iodide to provide the final target molecules.
2.2.1. General methods
1H-NMR spectra were recorded on a Varian Mercury 300-MHz instrument or a Bruker Avance III 400 MHz spectrometer. Purity (%) and mass spectral data were determined with a Waters Alliance 2695 HPLC/MS (Waters Symmetry C18, 4.6 × 75 mm, 3.5 μm) with a 2996 diode array detector from 210–400 nm or an Agilent Technologies 1200 Series LC system (Zorbax SB-C18, 2.1 × 30 mm, 3.5 μm) with a diode array detector from 210 – 400 nM) coupled to a 6130 Quadrapole MS. Normal phase chromatographic purifications were performed on silica gel using a Teledyne ISCO Combiflash Rf system. LCMS/MS analyses were performed on a Waters TQ instrument (electrospray positive mode) coupled to a Waters Aquity® UPLC BEH column (C18, 1.7 μM, 2.1 × 50 cm). Detailed methods are provided in the appropriate sections below.
2.2.2. Synthetic procedures (Fig.2A)
Cyclododecyl 2-(piperidin-1-yl)acetate hydrochloride (4). Cyclododecanol (0.5 g, 2.7 mmol), 2-(piperidin-1-yl)acetic acid (0.38 g, 2.7 mmol), and 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (0.52 g, 2.7 mmol) were combined in dichloromethane (20 ml) and stirred overnight at room temperature. The reaction was washed with water (15 ml), aqueous sodium bicarbonate (10 ml), the organic layer separated, dried (potassium carbonate), filtered, and the solvent removed under vacuum to yield an oil that was dissolved in ether (20 ml) and treated with hydrochloric acid in dioxane (1 eq. of 1N HCl). Filtration provided a white solid, cyclododecyl 2-(piperidin-1-yl)acetate hydrochloride (535 mg, 64%). M+H+ = 310.5. 1H NMR (300 MHz, CDCl3) δ 5.11 (tt, J=4.69, 7.32 Hz, 1H), 3.75–3.85 (m, 3H), 3.29–3.44 (m, 4H), 1.63–1.78 (m, 4H), 1.29–1.45 (m, 24H). HRMS (ESI-TOF): calc’d for C19H36NO2 [M+H]+ 310.2741; found 310.2735.
N-cyclododecyl-2-(piperidin-1-yl)acetamide hydrochloride (5). Cyclododecanamine (0.5 g, 2.7 mmol), 2-(piperidin-1-yl)acetic acid (0.38 g, 2.7 mmol), and 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (0.52 g, 2.7 mmol) were combined in dichloromethane (20 ml) and stirred overnight at room temperature. The reaction was washed with water (15 ml) and the organic separated, dried (potassium carbonate), filtered, and the residue purified by reversed phase HPLC. The appropriate fractions were combined, 1N aqueous hydrochloric acid (5 ml) added, and the solution was frozen and lyophilized to yield a white solid: N-cyclododecyl-2-(piperidin-1-yl)acetamide hydrochloride (400 mg, 48%) LCMS: Rf = 4.21 min, (M+H)+ = 309.5. 1H NMR (300 MHz, CDCl3) δ 8.55–8.59 (m, 1H), 8.25 (s, 1H), 4.00–4.03 (m, 1H), 3.85 (s, 2H), 3.45–3.49 (m, 4H), 1.82–1.99 (m, 4H), 1.28–1.43 (m, 24H). HRMS (ESI-TOF): calc’d for C19H37N2O [M+H]+ 309.2900; found 309.2892.
1-[(Cyclododecylcarbamoyl)methyl]-1-methylpiperidin-1-ium iodide (6). N-Cyclododecyl-2-(piperidin-1-yl)acetamide (700 mg, 2.3 mmol) was dissolved in THF (10 ml), treated with methyl iodide (0.21 ml, 3.4 mmol), and strirred overnight at ambient temperature. The reaction was diluted with ethyl ether (10 ml) and the resulting solid was collected by filtration to afford a light yellow solid 1-[(cyclododecylcarbamoyl)methyl]-1-methylpiperidin-1-ium iodide (610 mgs, 86%). LCMS: Rf = min, (M+H)+ = .1H NMR (being added). HRMS (ESI-TOF): calc’d for C20H39N2O [M]+ 323.3057; found 323.3048.
2.2.3. Synthetic procedures (Fig.2B)
Cyclododecyl 2-chloroacetate (7). In a round bottom flask, cyclododecanol ((5), 20.8 g, 113 mmol.) was dissolved in benzene (225 ml), and then pyridine (11.4 ml, 141 mmol) was added. This mixture was cooled to 0°C and chloroacetyl chloride (12.7 g, 113 mmol) was added dropwise. The reaction was then heated at 78°C and allowed to stir for 3 hours. Upon completion, it was allowed to cool to room temperature. The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl and finally saturated sodium bicarbonate. The combined organic layers were dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue were purified by normal phase chromatography on silica gel (0–10% hexanes/ethyl acetate). The desired product was obtained as a white solid (19.1 g, 65%). LCMS: Rf = 4.41 min, (ESI+) (M+Na)+ = 282.3; 1H NMR (400 MHz, CDCl3) δ 5.11–5.04 (m, 1H), 4.00 (s, 2H), 1.78–1.67 (m, 2H), 1.57–1.46 (m, 2H), 1.44–1.26 (m, 18H).
General procedures for preparation of cyclododecyl acetyl amines (8). In a vial, cyclododecyl acetyl chloride ((7), 1 g, 4.1 mmol) was combined with the desired amine (5.33 mmol) and heated to 100°C for 48 hours. The resulting reaction mixtures were then cooled to room temperature, concentrated under reduced pressure and purified by normal phase column chromatography on silica gel (0–40% hexanes/ethyl acetate).
Cyclododecyl 2-(pyrrolidin-1-yl)acetate (8a). Obtained in 51% yield as an orange solid. LCMS: Rf = 2.68 min, (ESI+) (M+H)+ = 296.2; 1H NMR (400 MHz, CDCl3) 5.13–5.04 (m, 1H), 3.34 (s, 2H), 2.72–2.64 (m, 4H), 1.89–1.78 (m, 4H), 1.78–1.67 (m, 2H), 1.58–1.48 (m, 2H), 1.48–1.24 (m, 18H).
Cyclododecyl 2-(azepan-1-y)acetate (8b). Obtained in 44% yield as an orange solid. Rf = 2.98 min, (ESI+) (M+H)+ = 324.3; 1H NMR (400 MHz, CDCl3) δ 5.06–5.01 (m, 1H), 3.36 (s, 2H), 2.68–2.63 (m, 4H), 1.85–1.76 (m, 4H), 1.74–1.64 (m, 2H), 1.55–1.43 (m, 2H), 1.42–1.19 (m, 22H).
Cyclodedecyl 2-(4,4-dimethylpiperidin-1-yl)acetate (8c). Obtained in 58% yield as an orange solid. Rf = 2.88 min, (ESI+) (M+H)+ = 338.3; 1H NMR (400 MHz, CDCl3) δ 5.00–4.96 (m, 1H), 3.33 (s, 2H), 2.61–2.55 (m, 4H), 1.83–1.72 (m, 4H), 1.70–1.61 (m, 2H), 1.51–1.41 (m, 2H), 1.42–1.19 (m, 18H), 0.85 (s, 6H).
Cyclododecyl 2-thiomorpholinoacetate (8d). Obtained in 37% yield as a yellow solid. Rf = 2.71 min, (ESI+) (M+H)+ = 328.2; 1H NMR (400 MHz, CDCl3) δ 5.02–4.97 (m, 1H), 3.34 (s, 2H), 2.68–2.61 (m, 4H), 2.60–2.52 (m, 4H), 1.73–1.64 (m, 2H), 1.55–1.47 (m, 2H), 1.42–1.19 (m, 18H). Cyclododecyl 2-(azetidin-1-yl)acetate [MC-270008]. Obtained in 28% yield as a yellow solid. Rf = 2.53 min, (ESI+) (M+H)+ = 282.2; 1H NMR (400 MHz, CDCl3) δ 4.97–4.90 (m, 1H), 3.30 (s, 2H), 3.28–3.20 (m, 4H), 3.17–3.11 (m, 2H), 1.70–1.62 (m, 2H), 1.53–1.43 (m, 2H), 1.38–1.06 (m, 18H).
2.2.4. General procedures for preparation of 5F02 analogs (Fig.2B).
In a vial, the cyclododecylaminoacetate (8a-d), (1 mmol) was dissolved in methanol (2 mL), and then iodomethane was added (156 mg, 0.068 ml, 1.1 mmol). The vial was sealed and the mixture was heat to 60°C and allowed to stir overnight. The resulting reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting solid residue was triturated with diethyl ether, collected by vacuum filtration, washed with diethyl ether and dried in vacuo.
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-methylpyrrolidin-1-ium iodide [MC-270016]. Obtained in 85% yield as a yellow solid. Rf = 3.28 min, (ESI+) (M)+ = 310.2; 1H NMR (400 MHz, CDCl3) δ 5.10–5.03 (m, 1H), 4.85 (s, 2H), 4.08–4.01 (m, 4H), 3,41 (s, 3H), 2.39–2.26 (m, 2H), 2.26–2.12 (m, 2H), 1.76–1.65 (m, 2H), 1.54-.44 (s, 2H), 1.38–1.21 (m, 18H).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-methylazepan-1 ium iodide [MC-270017]. Obtained in 68% yield as an orange solid. Rf = 3.39 min, (ESI+) (M)+ = 338.3; 1H NMR (400 MHz, CDCl3) δ 5.10–5.03 (m, 1H), 4.71, (s, 2H), 4.07–3.98 (m, 2H), 3.92–3.83 (m, 2H), 3.59 (s, 3H), 2.06–1.86 (m, 4H), 1.83–1.62 (m, 6H), 1.55–1.43 (m, 2H), 1.41–1.18 (m, 18H).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1,4,4-trimethylpiperidin-1-ium iodide [MC-270019]. Obtained in 71% yield as a white solid. Rf = 3.39 min, (ESI+) (M)+ = 338.3; 1H NMR (400 MHz, CDCl3) δ 5.10–5.03 (m, 1H), 4.71, (s, 2H), 4.07–3.98 (m, 2H), 3.92–3.83 (m, 2H), 3.59 (s, 3H), 2.06–1.86 (m, 4H), 1.83–1.62 (m, 6H), 1.55–1.43 (m, 2H), 1.41–1.18 (m, 18H).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1,4,4-trimethylpiperidin-1-ium iodide [MC-270019]. Obtained in 71% yield as a white solid. Rf = 3.52 min, (ESI+) (M)+ = 352.3; 1H NMR (400 MHz, CDCl3) δ 5.11–5.02 (m, 1H), 4.51 (s, 2H), 3.63–3.51 (m, 2H), 3.51–3.40 (m, 2H), 3.21 (s, 3H), 1.80–1.46 (m, 8H), 1.42–1.22 (m, 18H), 1.02 (s, 3H), 1.01 (s, 3H).
4-(2-(Cyclododecyloxy)-2-oxoethyl)-4-methylthiomorpholin-4-ium iodide [MC-270021]. Obtained in 66% yield as a light yellow solid. Rf = 3.22 min, (ESI+) (M)+ = 342.3; 1H NMR (400 MHz, CDCl3) δ 5.17–5.09 (m, 1H), 4.51 (s, 2H), 4.35–4.14 (m, 4H), 3.99–3.79 (m, 4H), 3.56 (s, 3H), 1.81–1.67 (m, 2H), 1.59–1.47 (m, 2H), 1.43–1.23 (m, 18H).
2.3. Calculated in vitro physicochemical parameters
Predicted octanol/water partition coefficients (logP) and topological polar surface areas (TPSA) were calculated using the Marvin calculator plugin available in MarcinSketch from ChemAxon (Budapest, Hungary). The logP calculator employs a variation [26] on the atomic fragment method originally described by Viswanadhan et al. [27]. The TPSA calculator uses the fragment-based summation method described by Etrl et al. [28].
2.4. in vitro physicochemical/ADME assays
2.4.1. Kinetic aqueous solubility
Compounds were assessed for their aqueous solubility in phosphate buffered saline (PBS; 136.9 mM NaCl, 2.68 mM KCl, 8.1 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4) using the commercially available Millipore MultiScreen™ Solubility filter system (Millipore, Billerica, MA, USA). Liquid handling was performed using JANUS® Verispan and MTD workstations (PerkinElmer, Waltham, MA, USA). 4 μl of drug solutions (10 mM in DMSO) are added to 196 μl of PBS/well in a MultiScreen® HTS 96-wel plate to give a final DMSO concentration of 2% and a drug concentration of 200 μM (maximum solubility value of assay). Plates are gently shaken for 90 minutes and then subjected to vacuum on a manifold. Soluble drug is collected in a second 96 well plate. The filtrate is transferred to 96-well Griener UV Star® analysis plates (Sigma-Aldrich, St. Louis, MO) containing 40 μl of acetonitrile. The drug concentration in the filtrate is measured by UV absorbance on a Spectromax® Plus microplate reader (Molecular Devices, Sunnyvale, CA, USA) using Softmax Pro software V. 5.4.5. Absorbances at 5 wavelengths (280, 300, 320, 340 and 360 nm) were summed to generate the UV signal. Assays were performed in triplicate. Standard curves were generated by adding 4 μl of 50x of five concentrations of test compounds in DMSO to 40 μl of acetonitrile in UV Star® plates followed by 156 μl of PBS. Analysis and statistics were performed using GraphPad® Prism v. 5.04.
2.4.2. Microsomal stability
The clearance of test compounds in pooled mouse or human liver microsomes was determined at 37°C. Assays were conducted in 96-well deep well polypropylene plates. Microsomal protein content was adjusted to give accurate rates of substrate consumption [29]. Test compounds (1 μM) were incubated in 0.5 ml of 100 mM potassium phosphate buffer (pH 7.4) with 0.5 mg/ml polled liver microsomes from male CD-1 mice or pooled mixed-gender human liver microsomes (Life Technologies, Grand Island, NY, USA), 2 mM tetra-sodium NADPH and 3 mM magnesium chloride for 60 minutes at 37°C with gentle shaking. At five time points, 75 μl of reaction mixture was transferred to 96-well shallow well stop plates on ice containing 225 ml of acetonitrile with 0.1 μM propafenone. Control reactions (lacking NADPH) were performed in a similar manner to demonstrate NADPH dependency of compound loss. Standard curves for test compounds were generated using 5 concentrations in duplicate. Stop plates were centrifuged at 2,000g for 10 min and then 170 μl of the supernatants were transferred to a Waters Aquity® UPLC 700 μl 96-well sample plate with cap mat (Waters, Milford, MA, USA). The amount of compound remaining in the supernatant was quantified by LCMS/MS using a Waters TQ MS instrument (electrospray positive mode) coupled to a Waters Aquity® UPLC (BEH column, C18, 1.7 μM, 2.1 × 50 cm, 5% to 95% acetonitrile gradient with water with 0.1% formic acid). Propafenone was used as the internal standard. GraphPad Prism® v 5.04 was used for nonlinear fitting of time course data to generate t1.2 values.
2.4.3. Cytochrome P450 inhibition
Test compounds were assessed for their ability to inhibit the three major human cytochrome P450 enzymes, CYP3A4 (EC1.14.13.67, EC1.14.13.97, EC1.14.13.32), CYP2D6 (EC1.14.14.1) and CYP2C9 (1.14.13.80, EC1.14.13.48, EC1.14.13.49) as previously described [30]. Expressed enzymes were used to minimize non-specific binding and membrane partitioning issues [31]. The CYP3A4 assay used midazolam as a substrate and analysis was performed by LC/MS/MS on a Waters Xevo TQ MS instrument as described above. The CYP2D6 and CYP2C9 assays use fluorescent substrates and were analyzed on an Envision plate reader.
2.4.4. Mouse Plasma Stability
The assay was conducted at 37°C in single time point mode (60 min incubation) in triplicate. Test compounds or procaine were tested at a final concentration of 1 μM in either 2.5% DMSO / 97.5% mouse plasma (Bioreclamation MSEPLNAHP-C57-M; pooled filtered plasma from male C57BL/6 mice; sodium heparin as anticoagulant; pH adjusted to 7.4 with 2N HCl on day of use) or 2.5% DMSO / 97.5% PBS (pH = 7.4 PBS: 136.9 mM NaCl; 2.68 mM KCl; 8.1 mM Na2HPO4; 1.47 mM KH2PO4; 0.9 mM CaCl2; 0.49 mM MgCl2). Samples were analyzed with a Waters® Aquity UPLC (BEH C18 1.7 μm, 2.1 × 50 mm column) in tandem with Waters® TQ MS mass spectrometer. 5 μl of sample was injected with a 3 min linear 5% to 95% acetonitrile/water gradient in 0.1% formic acid with a 0.5 ml/min flow rate. Compound electrospray mode, capillary voltages, cone voltages, MRMs (one per compound) and quantization were optimized and determined using Waters® QuanOptimize software with propafenone as an internal standard. Standard curves (six concentrations from 0–1000 nM) were generated with room temperature PBS or plasma. In order to identify plasma-dependent stability effects, stability was expressed as % compound remaining (PBS). The reference compound procaine typically was 100% stable in PBS but was > 95% degraded in plasma.
2.4.5. Plasma protein binding
Equilibrium dialysis was performed as previously described [32] using 96-well equilibrium dialyzer plates with MW cutoff of 5K (Harvard Apparatus, Holliston, MA, USA) and a dual-plate rotator set to maximum speed (Harvard Aparatus, Holliston, MA, USA) located in a 37°C incubator with a 10% CO2 atmospheric environment. Test compounds (2.5 μM) were added to plasma in DMSO (2 μl, final DMSO concentration 0.4%) to give 10 μM. 200μl of plasma mixture (pH 7.4) and buffer (Dulbecco’s phosphate-buffered saline 1x without calcium and magnesium (Mediatech Inc., Herndon, VA, USA)) were placed in their respective sides, wells were capped and the plate was then placed in the rotator and allowed to dialyze for 22 hours. Following dialysis, 25μl of buffer and plasma mixture were removed and mixed with 25μl of the opposite matrix in 96-well deep plates. Concentrations of analytes from each side of the dialysis plate were determined by LC/MS/MS using a standard curve constructed in the same matrix. The fraction unbound (fu) was calculated by dividing the drug concentration in the buffer side of the dialysis plate by the drug concentration in the plasma side.
2.5. In vivo exposure in mice
The in vivo exposure of 5F02 was measured over a 24 h period in 6–8 week old male C57BL/6 mice in weight range 20–24 g (Harlan Labs, Indianapolis, IN, USA; n = 3 animals per time point) following intraperitoneal doses of 1.0 mg/kg, 2.5 mg/kg and 5.0 mg/kg. Animals were acclimatized for 72 h prior to any experimental procedures. They were housed 3–5 per cage on a 12 h light/12 h dark cycle and were given free access to food and water. Animals were observed at least once daily for any abnormalities or distress and for the development of infectious disease. Treatment of animals was in accordance with the regulations outlined in the USDA Animal Welfare Act (9 CFR Parts 1, 2 and 3) and the conditions specified in the Guide for the Care and Use of Laboratory Animals (ILAR publication, NEC, 2011, The National Academies Press). Test compounds were administered in a vehicle consisting of N-methylpyrrolidine (NMP) in sterile saline (10% NMP for 1.0 mg/kg; 25% NMP for 2.5 mg/kg; 50% NMP for 5.0mg/kg). The dosing volume was 0.2 ml/mouse. Animals were observed hourly for signs of distress or poor health. Time points (following drug administration) examined were 0, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h. Blood was drawn under CO2 anesthesia by cardiac puncture and collected in tubes containing EDTA. Plasma was isolated by centrifugation at 12,000 rpm for 7 min and stored at −20°C until analysis. At the end of the 24 h study all animals were euthanized by CO2 inhalation. Drug concentrations in plasma were quantified by LC/MS/MS. Assay acceptance criteria was 20% for all standards and 25% for the LLOQ. Statistics were analyzed using WinNonLin.
Ethics statement:
This study was carried out in strict accordance with the recommendations from the Guide for the Care and Use of Laboratory Animals, as provided by the American Association of Accreditation of Laboratory Animal Care (AAALAC).
2.6. Human cell cultures
Androgen-dependent LNCaP and androgen-independent PC-3 and DU-145 prostate cancer cells were obtained from ATCC (Rockville, MD, USA). Initial stocks were cryopreserved, and at every six-month interval a fresh aliquot of frozen cells was utilized for the experiments. Cells were cultured in RPMI 1640 (Bio-Whittaker, Walkersville, MD, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA), penicillin and streptomycin (10378–016, Thermo Fisher Scientific, USA), sodium pyruvate (11360–070, Thermo Fisher Scientific, USA) and nonessential amino acids (11140–050, Thermo Fisher Scientific, USA) according to the manufacturer protocol under conditions indicated in the figure legends. Normal prostate epithelium cells RWPE-1 were obtained from ATCC (Rockville, MD, USA). RWPE-1 cells were maintained in Keratinocyte-Serum Free medium (17005–042, Thermo Fisher Scientific, USA) supplemented with 5 ng/ml of human recombinant EGF (PHG0311, Thermo Fisher Scientific, USA) and 0.05 mg/ml of bovine pituitary extract (130280–14, Thermo Fisher Scientific, USA).
2.7. Luciferase reporter assay
Luciferase reporter assay was performed as described in our previous publication [33]. R1881 was obtained from Sigma-Aldrich (R0908, USA).
2.8. Confocal microscopy
Preparation and immunostaining of cells was performed using standard protocol as described [34]. The primary antibody used was anti-Coilin (1:2000; Ff-7, mouse sc-55594, Santa Cruz, USA)), anti-PARG (1:1000; rabbit, ab169639, Abcam, USA), and the secondary antibody used was goat anti-rabbit Alexa-563 (1:400, Molecular Probes, USA) and anti-mouse Alexa-488 (1:400, Molecular Probes, USA). Slides were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA) with Draq5 dye (424101, BioLegend, USA) at 0.05 mg/ml for DNA staining.
2.9. PARP-1 inhibitory assay in human cell culture
Different doses of new non-NAD-like PARP-1 inhibitors or classical PARP-1 inhibitors Olaparib (Astra Zeneca, USA), Veliparib (ALX-270–444, Enzo, USA) and Rucaparib (PZ0036, Sigma-Aldrich, USA) were added to the cells cultured in complete medium. After 24 or 48 h, cells were lysed, and protein samples were analyzed with SDS-PAGE and Western Blot using anti-pADPr antibody (1:500, mouse, 1020, Tulip Biolabs, USA).
2.10. Cell Proliferation Assay
Cell proliferation was determined by CellTiter-Blue assay (Promega) as described in our previous publication [35]. Cells were plated at a density of 103 cells/well (100 μl) in a 96-well plate, allowed to adhere overnight and treated with various agents as described in the figure legends.
2.11. Human PARG Plasmid cloning
To produce full length human PARG we used pCMV6-XL5-hPARG (PARG NM_003631 Untagged clone, SC117822, Origene; USA)) as a PCR template. Following PCR primers were used: T7 F: GTAATACGACTCACTATAGGGC and hPARG SalI R: TTATGTCGACTGGTCCCTGTCCTTTGCCCTG. The PCR fragment was digested with SalI HF (R3138, New England Biolab, USA) and SmaI (R0141, New England Biolab, USA), purified with Qiaquick PCR purification kit (28104, Qiagen, The Netherlands). The vector pEYFP-N3 (Clontech, USA) were cut with Hind III, then treated with Klenow (M0210L, New England Biolab, USA) according to product manual, purified with Qiaquick PCR purification kit, digested with SalI HF, purified with Qiaquick PCR purification kit again. The treated PCR fragment insert and vector were ligated with T4 DNA ligase (NEB M0202), pEYFP-N3-hPARG ligation was transformed to One shot Competent E.coli cells (C404010, Invitrogen, USA), colonies were cultured in LB broth (244620, BD Difco, USA), extracted with Qiaprep Spin miniprep kit (27106, Qiagen, The Netherlands), pEYFP-N3-hPARG plasmid was cut by KpnI (R3142, New England Biolab, USA) to check the digestion pattern and confirmed by sequencing.
2.12. Clonogenic Cell Survival Assay
Cells were transfected with pEYFP-N3 and pEYFP-N3-hPARG plasmids using Fugene 6 (E2691, Promega, USA) transfection reagent according to the manufacturer protocol. 48 h later, cells were trypsinized for Clonogenic Cell Survival Assay and Western blot. Cells were plated in Methylcellulose-based Media (HSC002, R&D Systems, USA), supplemented with L-glutamine and penicillin and streptomycin (10378–016, Life Technologies Corporation, USA), sodium pyruvate (11360–070, Life Technologies Corporation, USA) and nonessential amino acids (11140–050, Life Technologies Corporation, USA) according to the manufacturer protocol. All cells were seeded in 35 mm Petri dishes with different cell concentrations: PC-3 1,000 cells/dish and LNCaP 15,000cells/dish. Colonies were counted and documented on day 8–10. Data were fitted to exponential and logarithmic decay models using the nonlinear curve fitting module of Statistica 7.0 software. The best fitting models for each inhibitor are represented on the chart. Plating efficiencies (PE) were calculated as follows: PE = number of colonies / number of cells seeded. The surviving fraction (SF) was calculated as follows: SF = number of colonies / number of cells seeded × PE.
2.13. PARP-1 activity assay
1 μl of H4-histone (1μg/μl) or endonuclease-digested plasmid DNA (0.01 μg/μl) was mixed with 25 μl 200 μM NAD and 1 μl of inhibitor/water. This mixture was combined with 10x PARP-1 reaction buffer (500 mM Tris, pH8.0, 250 mM MgCl2, 1%Triton X-100) and 0.7 μl PARP-1 enzyme (0.1 μg/μl, 4668–02K-01, Trevigen, USA). All reactions were carried out for 30 min at room temperature. Samples were examined with SDS-PAGE and Western Blot using anti-pADPr antibody (1:500, mouse, 1020, Tulip Biolabs, USA).
2.14. Western blot
For semi-quantitative protein analysis, cells were lysed in 1xSDS sample buffer (25 mM Tris (pH 6.8), 2% 2-mercaptoethanol, 3% SDS, 0.1% bromophenol blue, and 5% glycerol) at 1 × 107 cells/ml and then boiled for 5 min. Proteins were resolved by SDS-PAGE and transferred to i-Blot (IB23001, Invitrogen, USA). The following antibodies were used for immunoblotting assays: anti-pADPr (1:4000, rabbit, 528815, Calbiochem, USA), anti-pADPr (1:500, mouse, 1020, Tulip Biolabs, USA), anti-hPARG (1:1000; rabbit, ab169639 Abcam, USA), anti PSA (1:1000, rabbit, 5153, Cell Signaling Technology, USA), anti-β-actin (1:5000, mouse, A5441, Sigma, USA), anti-GFP (1:1000, rabbit, TP401, Origene, USA), anti-GFP (1:5000, mouse, 632380, BD, USA), and either goat anti-rabbit or anti-mouse secondary antibody conjugated to horseradish peroxidase (Sigma, USA). Western blotting was done using the detection kit from Amersham/GE Healthcare (RPN2106, USA), according to the manufacturer’s instructions. Image digitizing and quantitative analysis were performed by Odyssey v1.2 software (LI-COR, Lincoln, NE, USA).
2.15. Statistics
All data are presented as mean ± SEM. Statistical analyses were done using 2-tailed Student’s t-test. A P value of 0.05 or less was considered significant.
3. Results
3.1. Poly(ADP-ribosyl)ating pathway plays an important role in prostate cancer malignancy
Cancer-derived cells sensitive to PARP-1 inhibitors often produce excessive amounts of pADPr [36]. The high level of endogenous pADPr appears to be the best predictor of tumor sensitivity to PARP-1 inhibition [21,36]. Excessive amounts of pADPr are known to accumulate in a variety of cancers and with respect to currently available therapies, pADPr accumulation is generally associated with a poor prognosis [36,37]. A significant elevation of pADPr, loss of PARG, and overproduction of the PARP-1 protein have previously been reported in PC cell lines [13,24], suggesting that PC cells may be an ideal model for studying the antitumor potential of our new PARP-1 inhibitors. Previously, we have reported that an over-accumulation of pADPr and loss of PARP-1 protein antagonist, Poly(ADP-ribose) glycohydrolase (PARG), is associated with the disruption of Cajal body (CB) functions and morphology [38]. CBs are nuclear sub-organelles, which are involved in the transcriptional splicing events, assembly, and regulation of nuclear proteins [39]. Upregulation of CBs correlates with global transcriptional changes [40]. Both, the malignant transformation and PARP-1/PARG dependent nuclear processes also correlate with genome-wide changes in transcription [8,9]. Therefore, we tested whether malignant PC cells showed any evidence of CBs dysregulation. We used a Coilin protein, a CB marker, to monitor CBs in malignant and normal prostate epithelial cells. Both, CB integrity and localization of PARG protein were severely disrupted in PC cells (Fig.3A). In contrast to one or two distinct CBs that were detected in normal prostate epithelial cells, diffuse patterns of small CBs were seen in LNCaP PC cells (Fig.3A). In LNCaP cells, PARG was not only downregulated but also had distinctly different subcellular localization than controls (Fig.3A). These data suggest a correlation between downregulation of PARG, CB alterations, and PC malignancy. To test the hypothesis that PARG regulates PC malignancy, we transiently transfected LNCaP PC cells with a plasmid constitutively expressing full-length human PARG or EYFP protein as a control. In these experiments, the effectivity of transfection was 60 – 70%. The level of EYFP and PARG proteins expression was stably maintained during cell culture amplification (Fig.3B). Our clonogenic assay showed that PC cells expressing PARG produce significantly fewer colonies than control EYFP-expressing PC cells (Fig.3C). Moreover, cells collected after clonogenic growth show no PARG expression at all (Fig.3D). These results confirm that the prostate malignancy is associated with PARG protein downregulation and can be suppressed by restoring PARG protein activity. These findings also suggest that PARP-1/PARG-dependent processes in PC involve a global regulation of transcription.
Figure 3. Non-NAD-like PARP-1 inhibitors suppress malignant phenotype of prostate cancer cells.

A. The morphology of Cajal bodies (CBs) in disrupted in PC cells. Normal prostate cells (RWPE1), prostate cancer cells (LNCaP), were fixed and immunostained with rabbit anti-PARG (Red) and Guinea Pig anti-Coilin antibody (green). Nuclear DNA were detected with the Draq5 dye (blue). The disruption of CBs in cancer-derived cells is clearly visible. B-D. LNCaP PC cells were transfected with plasmids expressing EYFP or the PARG proteins under control of CMV promoters. After cell amplification, we examined the levels of ectopic proteins expression, using Western blot analysis (B). We compared the malignant potential of EYFP and PARG expressing cells using Clonogenic Cell Survival Assay (C). After that, we retested the level of ectopic proteins expression in cells growing on plates (D). E-F. The effects of 5F02, olaparib, and enzalutamide on the AR transcriptional activity in LNCaP cells expressing both wild-type and constitutively active AR spliced variant AR-V7. LNCaP cells stably expressing AR-V7 and firefly luciferase driven by AR-dependent promoter were treated with the indicated agents for 8 hrs. Samples were assayed for firefly luciferase activity (E). LNCaP cells stably expressing AR-V7 were treated with 5F02, olaparib, or enzalutamide (all at 10μM) for 24 hrs. PSA and AR levels were examined by Western blot analysis (F). G. The effect of 5F02, olaparib, and enzalutamide on viability of LNCaP expressing both wild-type and constitutively active AR splice variant AR-V7. Cells were treated with escalating concentrations of each of the three inhibitors for 96 hours. Cellular viability was assessed using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software and presented in Table 1.
3.2. Non-NAD-like PARP-1 inhibitors suppress androgen-dependent and -independent activation of AR signaling
AR-mediated functions are not completely abrogated by existing androgen deprivation therapies and therapeutic failure is often accompanied by the expression of truncated constitutively active AR splice variants (AR-Vs) that lack the ligand-binding domain. AR-V7, which is found in approximately 70% of CRPC cases, is the most common and best characterized constitutively active AR variant [1–3]. Elevated AR-V7 expression is associated with resistance to androgen deprivation therapies [4] and poor clinical prognosis [2]. We examined the functional impact of non-NAD-like 5F02 PARP-1 inhibitor on AR transcriptional activity in LNCaP cells with stable expression of AR-V7. As expected, treatment with the NAD-competing inhibitors enzalutamide and olaparib produced a negligible inhibitory effect on AR transcriptional activity (Fig.3E) and PSA expression (Fig.3F). In contrast, treatment with 5F02 inhibited AR transcriptional activity (Fig.3E) and PSA expression (Fig.3F). A similar trend was observed in a cell viability assay (Fig.3G and Table 1). 5F02 exerted significantly higher antitumor efficacy against LNCaP cells expressing AR-V7 compared with olaparib and enzalutamide. Collectively our data demonstrate that unlike NAD-like PARP-1 inhibitors and antiandrogen agents that target androgen-mediated activation of AR, our novel non-NAD-like PARP-1 inhibitors suppress AR transcriptional function. These data suggest that non-NAD-like PARP-1 inhibitors may be effective against both androgen-dependent and -independent routes of AR activation.
Table 1.
The effect of 5F02, olaparib, and enzalutamide on the viability of LNCaP expressing both wild-type and constitutively active AR splice variant AR-V7. Cell viability was examined using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software.
| 5F02 | Olaparib | Enzalutamide | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM |
| 15.7 | 8.9 | 6.3 | 3.5 | 1835 | 228.7 | 61.9 | 7.3 | 4751.7 | 163.7 | 19.8 | 0.6 |
3.3. Structural alterations in 5F02 molecule changes its anti-PARP-1 activity
In our previous studies we performed analysis of 50,000 small compounds, selecting positive hits that reduced PARP-1 activity by at least 3-fold [22–24]. Based on the levels of anti-PARP-1 activity, structural analysis and ease of synthesis, we ultimately selected 5F02 as our lead compound for future studies (Fig.4A). As compared to the NAD-like PARP-1 inhibitor olaparib, 5F02 more effectively inhibits growth of tumor cells both in vitro and in animal models of human PC [5]. It is generally expected that quaternary ammonium salts would be more likely to spontaneously transfer a methyl group either metabolically or chemically, which presents the possibility of activity loss. Moreover, quaternary ammonium salts are predicted to have limited absorption upon oral administration due to their constitutive charge [41]. Therefore, with the intent of improving 5F02-mediated PARP-1 inhibition, we synthesized three analogues of 5F02 in which the ester and quaternary ammonium salt have been altered to amide and/or desmethyl modifications which we expected would increase the stability of 5F02. These 5F02 analogues were called FC-7218, FC-7219, FC-7220 (Fig.4B). To test which structural features were responsible for the biological activities of our lead compound, we also synthesized several additional derivatives of 5F02 (i.e. MC-270008, MC-270016, MC-270017, MC-270019, and MC-270021) (Fig.4B).
Figure 4. Structures and anti-PARP-1 activities of non-NAD-like PARP-1 inhibitors.

A. Molecular structures of 5F02 – a non-NAD-like PARP-1 inhibitor, veliparib, rucaparib, and olaparib - NAD-mimetic PARP-1 inhibitors, and enzalutamide – a clinically approved second-generation antiandrogen. Green and red circles indicate parts of 5F02 molecule that were chemically modified. B. 5F02 and its new derivatives. C. PARP-1 inhibition assay in cell free system using NAD-mimetic PARP-1 inhibitor olaparib, antiandrogen enzalutamide and new derivatives of non-NAD-like PARP-1 inhibitor 5F02. Reduction of pADPr (inhibition of PARP-1 activity) was detected by Western blot for inhibitor-treated reaction mixtures relative to DMSO-treated mixtures. All inhibitors were used in 10μM concentration. D. Quantification of PARP-1 activity in cell free system using colorimetric assay (22–24).
The PARP-1 inhibitory activity of each analogue or derivative was tested in a cell free in vitro system, as described [24]. Our results demonstrated that all three of the FC group compounds lost PARP-1 inhibitory potential (Fig.4C–D). One of the small molecules of the MC group (MC-270008) also demonstrated a diminished capacity to inhibit PARP-1, while the other four members of MC group inhibited PARP-1 to a similar degree as 5F02 (Fig.4C–D).
3.4. The methylation of the quaternary ammonium salt and the presence of ester are critical for the antitumor activity of 5F02
To further test of the FC compounds (Fig.5A), we added an androgen receptor agonist (R1881) and determined the ability of each inhibitor to reduce AR transcriptional activity (Fig.5A). Treatment of androgen-dependent LNCaP PC cells with our lead inhibitor 5F02 notably diminished R1881-mediated increase in AR transcriptional activity (Fig.5B). In contrast, treatment with FC-7218, FC-7219, and FC-7220 or with NAD-like PARP-1 inhibitors olaparib, veliparib, and rucaparib had only weak inhibitory effects on AR transcriptional activity (Fig.5B). We compared the effect of treatment with 5F02, FC-7218, FC-7219, FC-7220, and NAD-like PARP-1 inhibitors on cell viability in androgen-dependent LNCaP, or the castration-resistant PC cell lines, PC-3 and DU-145. Treatment with FC-7218 and FC-7219 had no significant effect on the viability of all three tested PC cell lines (Fig.5C–E). Given that the main difference between the derivatives is based on the loss of methylation of the quaternary ammonium salt, our data suggest that this moiety plays a critical role in the antitumor activity of 5F02. Replacement of the ester with an amide (FC-7220) also reduced the antitumor potential of 5F02 (Fig.5F), which coincided with reduced anti-PARP-1 activity (Fig.4D). Notably, 5F02 demonstrated a significantly higher antitumor efficacy against androgen-dependent LNCaP cells than the NAD-like PARP-1 inhibitors olaparib, veliparib, and rucaparib. 5F02 was also more effective in reducing the viability of castration-resistant PC-3 and DU-145 cells than NAD-like PARP-1 inhibitors (Fig.5G,H and Table 2).
Figure 5. Methylation of the quaternary ammonium salt and the presence of carboxyl group are critical for the antitumor activity of 5F02.

A. Chemical structures of 5F02 and its derivatives. Green circle indicates the area of the 5F02 molecule that was chemically modified. B. Treatment with 5F02 attenuates the transcriptional activity of the AR in LNCaP PC cells. LNCaP cells stably expressing firefly luciferase driven by AR-dependent promoter were treated with 5F02, 5F02 analogs (i.e. FC-7218, FC-7219, and FC-7220), or clinically relevant NAD-like PARP-1 inhibitors olaparib, veliparib, and rucaparib in the presence of a synthetic androgen agonist R1881 (2.5nM) for 8 hrs. Samples were assayed for firefly luciferase activity. C-E. Androgen-dependent LNCaP (C) and castration-resistant PC-3 (D) and castration-resistant DU-145 (E) PC cells were treated with escalating concentrations of 5F02, FC-7218, and FC-7219 for 96 hours. Cell viability was assessed using the CellTiter Blue assay. F-H. The effect of non-NAD-like and NAD-like PARP-1 inhibitors on the viability of androgen-dependent LNCaP (F), castration-resistant PC-3 (G), and castration-resistant DU-145 (H) PC cells. Cells were treated with escalating concentrations of different inhibitors for 96 hours. Cell viability was assessed using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software and presented in Table 2.
Table 2.
The effect of non-NAD-like and NAD-like PARP-1 inhibitors on viability of androgen-dependent LNCaP and castration-resistant PC-3 and DU-145 PC cells. Cell viability was examined using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software.
| LNCaP | PC-3 | DU-145 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM | |
| 5F02 | 20.4 | 9.4 | 5.8 | 2.6 | 38.6 | 12.5 | 6.1 | 1.9 | 29.1 | 11.3 | 6.2 | 2.3 |
| FC-7220 | 84.2 | 29.4 | 15.2 | 5.1 | 310.2 | 83.7 | 36.8 | 9.6 | 296.1 | 82.7 | 37.2 | 10.0 |
| Olaparib | 4451.1 | 162.9 | 20.4 | 0.7 | 383.6 | 39.0 | 9.3 | 0.9 | 125.8 | 22.0 | 7.4 | 1.2 |
| Veliparib | 768.3 | 153.1 | 55.6 | 10.6 | 1633.9 | 237.0 | 70.5 | 9.7 | 547.2 | 104.0 | 36.7 | 6.7 |
| Rucaparib | 154.7 | 52.6 | 26.7 | .8.8 | 157.0 | 23.2 | 7.0 | 1.0 | 82.7 | 14.8 | 5.0 | 0.9 |
3.5. Structural changes in the quaternary amine moiety of 5F02 analogs result in differences in cell line susceptibility
In order to obtain some preliminary structure-activity relationship (SAR) information we prepared a collection of 5F02 derivatives where the piperidinium moiety of 5F02 was replaced by other cyclic ammonium groups of varying length, volume and polarity (Fig.4B, see Materials and Methods). Our goal was to probe the length and width of any binding region on the PARP1 protein that was accommodating the piperidine ring of 5F02 by examining cyclic amine groups that possessed 1) less volume and length (azetidine ring of MC-270008; pyrrolidine ring of MC-270016); 2) greater length and equal volume (the dimethylpiperidine ring of MC-270019); 3) greater volume and length (the cycloheptyl ring of MC-270017); and, 4) the presence of a hydrogen bonding atom (the thiomorpholine ring of MC-270021). The analogs were prepared in a moderate yield in a three-step synthetic sequence that was amenable to parallel synthesis (see Materials and Methods). The non-quaternary free base of the azetidine derivative MC-270008 was prepared in modest yield, but methylation of the azetidine ring proved problematic, possibly due to the added constraint around the tertiary nitrogen caused by the 4-membered ring.
We then tested the novel 5F02 derivatives in order to elucidate the impact of altering the cyclic amine group on the biological activity of the 5F02 chemical scaffold (Fig.6A). Similarly to the non-quaternary analogs FC-7218 and FC-7219, MC-270008 had an insignificant effect on the PARP1 enzymatic activity in PC cells. Therefore, we excluded it and the other two non-quaternary analogs from further analysis. The AR transcriptional luciferase assay in LNCaP cells showed that the inhibitory effect of MC-270016, MC-270017 and MC-270019 were similar with that of 5F02 (Fig.6B). These results suggest that slight structural changes in this region of the 5F02 scaffold do not significantly disrupt their ability to inhibit PARP-1 pathways in the PC. Interestingly, while MC-270016, MC-270017 and MC-270019 were slightly more potent than 5F02 in reducing viability of androgen-dependent LNCaP cells (Fig.6C and Table 3), these agents were less effective than 5F02 in suppressing viability of castration-resistant AR-negative PC-3 and DU-145 cells (Fig.6D,E and Table 3). These findings suggest that the antitumor activity of MC-270016, MC-270017 and MC-270019 is mediated primarily via direct inhibition of AR or indirect inhibition of AR signaling, whereas 5F02 activity does not depend on the presence of AR. These experiments also largely revealed a correlation between anti-PARP-1 and anti-tumor activities of 5F02 and its derivatives (Fig.4D, Fig.6C,D,E and Table 3).
Figure 6. The effect of 5F02 and its analogs on AR transcriptional activity and viability in LNCaP PC cells.

A. Chemical structures of 5F02 and its derivatives. Red circle indicates the area of 5F02 molecule that was chemically modified. B. The effect of 5F02 and its analogs on the transcriptional activity of the AR in LNCaP PC cells. LNCaP cells stably expressing firefly luciferase driven by AR-dependent promoter were treated with 5F02 and its analogs in the presence of a synthetic androgen agonist R1881 (2.5nM) for 8 hrs. Samples were assayed for firefly luciferase activity. C-E. The effect of 5F02 and its derivatives on the viability of androgen-dependent LNCaP (C), castration-resistant PC-3 (D), and castration-resistant DU-145 (E) PC cells. Cells were treated with escalating concentrations each inhibitor for 96 hours. Cellular viability was assessed using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software presented in Table 3.
Table 3.
The effect of 5F02 and its derivatives on viability of androgen-dependent LNCaP and castration-resistant PC-3 and DU-145 PC cells. Cell viability was examined using the CellTiter Blue assay. The effective doses (ED) were calculated using XLfit software.
| LNCaP | PC-3 | DU-145 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM | ED90 μM | ED70 μM | ED50 μM | ED20 μM | |
| 5F02 | 20.4 | 9.4 | 5.8 | 2.6 | 38.6 | 12.5 | 6.1 | 1.9 | 29.1 | 11.3 | 6.2 | 2.3 |
| MC-270016 | 19.4 | 6.1 | 3.0 | 0.9 | 85.0 | 37.4 | 22.4 | 9.6 | 208.1 | 73.9 | 38.6 | 13.3 |
| MC-270017 | 10.1 | 4.1 | 2.3 | 0.9 | 40.8 | 24.2 | 17.5 | 10.2 | 61.1 | 31.6 | 20.9 | 10.6 |
| MC-270019 | 12.2 | 5.2 | 3.1 | 1.3 | 48.1 | 23.1 | 14.6 | 6.9 | 89.1 | 40.4 | 24.6 | 10.9 |
| MC-270021 | 52.1 | 20.3 | 11.2 | 4.2 | 3272.7 | 282.1 | 60.6 | 4.9 | N/A | N/A | N/A | N/A |
3.6. Characterization of physicochemical and ADME properties of 5F02 and its analogs
We analyzed in vitro physicochemical and ADME (Adsorption, Distribution, Metabolism and Excretion) properties of non-NAD-like PARP-1 inhibitors to provide the rationale for the observed differences in functional activities of 5F02 and its analogs. Such data are also needed to provide basic information about the properties of new probe and tool compounds prior to their entry into expensive and labor-intensive animal studies. We examined 5F02, the quaternary amide analog FC-7220 and the four quaternary 5F02 derivatives MC-270016, MC-270017, MC-270019, and MC-270021 in a battery of in vitro physicochemical and ADME assays. The results are presented in Tables 4–1 and 4–2.
Table 4–1.
In vitro assessment of ADME properties during lead selection and lead optimization. Maximum aqueous solubility for 5F02 and its analogs is moderate. The compounds are characterized by rapid clearance in mouse liver microsomes in the presence of NADPH, an indicator of lability to oxidative metabolism. In the absence of NADPH, the ester analogs (5F02, MC-270016, MC-270017, MC-270019 and MC-270021) demonstrated some instability in liver microsomes, suggesting some lability to hydrolytic metabolism. This result was confirmed in mouse plasma where the esters demonstrated moderate half-lives in the range of 35–120 minutes. The amide derivative FC-7820 appeared stable to hydrolysis in these systems.
| Compound | clogP | TPSA† | Solubility in PBS (pH 7.4) (μM) | Mouse Liver Microsome Stability | Mouse Plasma Stability T1/2 (min) | ||
|---|---|---|---|---|---|---|---|
| + NADPH | − NADPH | ||||||
| Clint (μL/min/mg) | % Remaining* | ||||||
| 5FO2 | 5.11 | 26.3 | 171 | 4.9 | 306 | 91% | 60 |
| FC-7220 | 4.54 | 29.5 | 191 | 7.1 | 196 | 101% | > 240 |
| MC270016 | 5.11 | 26.3 | 125 | 3.8 | 368 | 98% | 57.2 |
| MC270017 | 5.56 | 26.3 | 140 | 1.8 | 790 | 87% | 35.9 |
| MC270019 | 5.52 | 26.3 | 95.2 | 2.6 | 456 | 84% | 19.6 |
| MC270021 | 4.32 | 26.3 | 110 | 4 | 346 | 88% | 118 |
Topological Polar Surface Area;
Percent remaining after 60 minutes.
Table 4–2.
In vitro assessment of ADME properties during lead selection and lead optimization. As in mouse liver microsomes, 5F02 and its analogs were characterized by rapid metabolism in human liver microsomes (+ NADPH) and the ester derivatives (5F02, MC-270016, MC-270017, MC-270019 and MC-270021) showed a slight tendency toward being labile to enzymatic hydrolysis in that system (− NADPH). As in the mouse, the amide analog FC-7820 appeared stable to hydrolytic metabolism in human liver microsomes. All analogs tested showed no inhibitory activity against the human metabolic enzyme CYP2C9 at concentrations up to and including 10 uM but moderate inhibitory potency against human CYP3A4 and CYP2D6.
| Compound | Human Liver Microsome Stability | Inhibition hCYP3A4 IC50 (nM) | Inhibition hCYP2D6 IC50 (nM) | Inhibition hCYP2C9 IC50 (nM) | ||
|---|---|---|---|---|---|---|
| +NADPH | −NADPH | |||||
| Clint (μl/min/mg) | % Remaining* | |||||
| 5F02 | 3.4 | 404 | 88% | 3280 | 1580 | > 10000 |
| FC-7220 | 1.6 | 850 | 100% | 1930 | 2370 | > 10000 |
| MC270016 | 1.9 | 721 | 92% | 2980 | 8540 | > 10000 |
| MC270017 | 1.2 | 1200 | 95% | 1700 | 4400 | > 10000 |
| MC270019 | 1.2 | 1150 | 92% | 4670 | 9930 | > 10000 |
| MC270021 | 1.3 | 1070 | 85% | 3920 | 9250 | > 10000 |
Percent remaining after 60 minutes.
Maximum kinetic solubility in 2% DMSO/phosphate buffered saline for the analogs tested ranged from 95 μM to 195 μM. All of the compounds displayed very high clearance in mouse and human liver microsomes in the presence of NADPH, an indicator that the scaffold is susceptible to oxidative metabolism. The moderate half lives in mouse plasma and the indication of some small degree of disappearance in liver microsomes in the absence of NADPH suggests that the esters may possess some small degree of liability to enzymatic hydrolysis. As expected, the amide analog FC-7220 was more resistant to metabolic hydrolysis in our assay systems. The compounds displayed weak to moderate inhibitory potency against the two metabolizing cytochrome P450 enzymes CYP3A4 and CYP2D6 but no activity against CYP2C9 at concentrations up to and including 10 μM. For probe molecules such as these, the cytochrome P450 inhibition is not an issue. As further lead optimization continues, it would be beneficial to reduce the cytochrome P450 inhibition in order to avoid potential drug-drug interactions, although with drugs for life-threatening diseases such as cancer that will not be given long-term, some off-target effects can be tolerated because the benefit-to-risk ratio is high. All of the compounds have logP values that are surprisingly high for charged species. This is probably due to the high degree of lipophilicity associated with the cyclododecyl group and the likelihood that the positive charge is shielded from the solvent by the degree of lipophilic steric bulk surrounding it. These results, along with low topological polar surface areas (TPSA), suggest that the compounds should show reasonable permeability across cellular membranes. The fact that the logP and TPSA values for all of the tested analogs are very similar suggests that these physicochemical properties are not playing a significant role in the in vitro potency differences seen between analogs and between cell lines.
3.7. In vivo exposure of 5F02 in male C57Bl6 mice
To guide dosing for the in vivo efficacy model, the PK parameters of 5F02 were assessed. The results are summarized in Fig.7 and Table 5. 5F02 showed non-linear exposure across the dose range following intraperitoneal administration, with the peak plasma concentration increasing disproportionately to dose increases. Clearance was fast and half-life was relatively short, at the lower doses of 1.0 and 2.5 mg/kg. These results are consistent with the ADME data, which suggest that the molecule is labile to oxidative metabolism and to a lesser degree hydrolytic metabolism. At the higher dose of 5.0 mg/kg, the half-life was longer, possibly due to partial saturation of metabolism (e.g., the moderate inhibition of CTP3A4 and 2D6) or hepatic efflux transport. Based on the plasma protein binding of 59.1%, the peak plasma concentration at the 5.0 mg/kg dose is predicted to be approximately 0.36 μg/ml (1.11 μM).
Figure 7.

In vivo plasma levels of 5F02 following a single intraperitoneal dose of 1.0, 2.5 and 5.0 mg/kg. Plasma protein binding, as assessed by the equilibrium dialysis method, was 59.9%, giving a predicted free, unbound plasma concentration of 39.1%. The administration vehicle was N-methyl-pyrrolidine (NMP) in sterile saline (10% NMP for 1.0 mg/kg; 25% NMP for 2.5 mg/kg; 50% NMP for 5.0 mg/kg).
Table 5.
PK parameters of 5F02 in male C57Bl6 mice following a single intraperitoneal administration. The compound indicated non-linear kinetics, with the peak plasma concentration increasing non-proportionally with increasing dose. At the lower doses, the compound is characterized by rapid clearance and short half-life, which is expected given the in vitro ADME results. The unexpectedly low clearance and long half-life at the highest dose of 5.0 mg/kg may indicate that metabolism and/or hepatic efflux are becoming saturated.
| μg/ml | h | μg/ml-h | h | l/h | l | |
|---|---|---|---|---|---|---|
| 1 mg/kg ip | 0.0067 | 0.083 | 0.007 | 0.8 | 3.43 | 4.2 |
| 2.5 mg/kg ip | 0.2292 | 0.5 | 0.18 | 1.2 | 0.14 | 0.16 |
| 5.0 mg/kg ip | 0.6 | 2.0 | 1.87 | 4.6 | 0.07 | 0.3 |
4. Discussion
Historically, androgen ablation has been the most common treatment for advanced PC. Therapeutic failure of anti-androgen therapeutics is often accompanied by various molecular alterations resulting in androgen-independent activation of the AR signaling pathway. PARP-1 inhibitors have demonstrated efficacy as treatments for solid and hematological malignancies harboring mutations in DNA damage-repair genes. Apart from the role of PARP-1 in DNA repair, PARP-1 has been demonstrated to exert key roles in transcriptional regulation of protumorigenic genes in cancer cells of various origins [42,43]. Recent studies have established that PARP-1 plays a critical role in controlling androgen receptor function in PC cells [12]. Given that both DNA repair and AR-mediated transcription depend on PARP-1 function, PC may represent a disease state that is particularly susceptible to PARP-1 inhibition. Consistent with this prediction, our data demonstrate that several PC cell lines are susceptible to PARP-1 inhibition.
Our study of the molecular mechanisms of PARP-1-mediated transcriptional regulation has identified a novel class of PARP-1 inhibitors targeting the histone-dependent route of PARP-1 activation [22–24]. To date, there are no known examples of compounds capable of accessing this unusual pathway. Our recently published work demonstrates that our representative non-NAD-like PARP-1 inhibitor 5F02 has significant antitumor activity in androgen-dependent and castration-resistant cell and animal models of human PC [24]. As noted above, our novel, non-NAD-like PARP-1 inhibitors (e.g. 5F02) are superior to currently available NAD-like PARP-1 inhibitors, such as olaparib, veliparib, and rucaparib as well as conventional anti-androgen therapeutics such as abiraterone and enzalutamide. It has been previously demonstrated that the growth-suppressive effect of NAD-dependent PARP-1 inhibitors is largely confined to AR-positive PC cells [12]. Our findings demonstrate that unlike NAD-dependent PARP-1 inhibitors, the non-NAD-like PARP-1 inhibitor 5F02 is effective against both AR-positive PC cells and AR-negative PC cells, whereas the analogues of this compound are most effective against AR-positive PC. Overall, our data suggest 5F02 and related analogues are effective in inhibiting AR signaling, and 5F02 may have extended applications independent of the presence of AR. In the current study, we performed various chemical modifications of our lead PARP-1 inhibitor 5F02 and examined the potency and ADME properties of 5F02 and several other related analogues. The 5F02 analogues differed from the parent compound by changes in the ester and quaternary ammonium salt, which were altered to amide and/or desmethyl modifications respectively. These modifications were made to increase the stability of the analogues over the parent compound, however, replacement of ester with amide and desmethyl modifications reduced the antitumor potential of 5F02, suggesting that these alterations would not be feasible for the biological improvement of 5F02.
Within the each cell line, the aminoalkyl ester analogs of 5F02 (MC-270016, MC-270017 and MC-270019) displayed similar ED50 values on cellular viability (Table 3). The thiomorpholine derivative MC-270021 was 4–5 fold less potent in LNCaP and PC-3 cells compared to the aminoalkyl analogs. The divergence in potency for this molecule is not explained by physicochemical properties because the logP and TPSA values for MC-270021 are very similar to that seen for the more potent derivatives. Nor is this phenomenon explained by differences in aqueous solubility or by size (the thiomorpholine ring is similar in size to the cycloheptyl ring of MC-270017). The potency difference seen for MC-270021 could indicate that the binding pocket for this end of the scaffold on PARP-1 is composed exclusively of lipophilic residues (e.g., alkyl groups, aryl rings, etc) and that the more polar sulfur atom is encountering some unfavorable interactions with those highly lipophilic residues. Like MC-270021, the amide derivative FC-7820 is also slightly less potent in LNCaP cells compared to MC-270016, MC-270017 and MC-270019. This difference cannot be explained by lipophilicity or solubility differences. It is conceivable that the amide group of FC7820 must assume a somewhat higher energy conformation to bind to PARP1 compared to the more freely rotating ester analogs, but the current data cannot confirm or refute this hypothesis. A more detailed structural analysis of the binding of these analogs to PARP-1 will be required to fully understand these results.
Our group was the first to identify the agents that specifically target the histone-dependent route of PARP-1 activation, a mechanism of activation unique to PARP-1. The results described here suggest that our novel non-NAD-like PARP-1 inhibitors may be effective therapies for patients with castration-resistant prostate cancer. Importantly, non-NAD-like PARP-1 inhibitors appear to have the potential to be used as monotherapies, or in combination with existing strategies for the treatment of PC.
Acknowledgements
Drs. Othman Ghribi, Kimberly D. Hammer and Kate Pechenkina contributed valuable comments on the manuscript. We also thank Atreyi Ghatak for assistance with western-blot experiments. This work was supported in part by the National Institutes of Health Grants R03 CA212566 to A.V.T., R01 CA186238 to T.S. and W.E.C., RO3 CA167671 to V.M.K., RO3 CA216173 to P.M., and the Department of Defense grants PC160049 and KC170127 to A.V.T. and V.M.K.
Abbreviations:
- ADME
adsorption, distribution, metabolism and excretion
- AR
androgen receptor
- AR-Vs
androgen receptor splice variants
- ATCC
American Type Culture Collection
- BEH
ethylene bridged hybrid
- CB
Cajal bodies
- CD-1
cluster of differentiation 1
- CRPC
castration-resistant prostate cancer
- ED
effective dose
- EGF
epidermal growth factor
- ESI-TOF
electrospray ionisation time-of-flight mass spectrometry
- EYFP
enhanced yellow fluorescent protein
- FBS
fetal bovine serum
- fu
fraction unbound
- GFP
green fluorescent protein
- HPLC/MS
high-performance liquid chromatography–mass spectrometry
- HRMS
high resolution mass spectrometry
- LCMS/MS
liquid chromatography-tandem mass spectrometry
- LLOQ
lower limit of quantification
- MRMs
multiple reaction monitoring
- NMP
N-methylpyrrolidine
- pADPr
poly(ADP-ribose)
- PARG
poly(ADP-ribose) glycohydrolase
- PARP-1
poly(ADP-ribose) polymerase 1
- PC
prostate cancer
- PCR
polymerase chain reaction
- PE
plating efficiencies
- PSA
prostate-specific antigen
- RPMI
Roswell Park Memorial Institute medium
- SAR
structure-activity relationship
- SF
surviving fraction
- TPSA
topological polar surface areas
- UPLC
ultra performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA-Cancer J Clin 2019; 69(1): 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. (N. Y., NY, U. S.) 2004; 10: 33–9. [DOI] [PubMed] [Google Scholar]
- 3.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013; 32: 5501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsogiannou M, Ziouziou H, Karaki S, Andrieu C, Henry de Villeneuve M, et al. The hallmarks of castration-resistant prostate cancers. Cancer Treat. Rev 2015; 41: 588–97. [DOI] [PubMed] [Google Scholar]
- 5.Aragon-Ching JB. The evolution of prostate cancer therapy: targeting the androgen receptor. Front. Oncol 2014; 4: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsiades N A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res 2013; 73: 4599–605. [DOI] [PubMed] [Google Scholar]
- 7.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J 1999; 342(2): 249–68. [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol. Aspects Med 2013; 34: 1124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lodhi N, Kossenkov AV, Tulin AV. Bookmarking promoters in mitotic chromatin: poly(ADP-ribose)polymerase-1 as an epigenetic mark. Nucleic Acids Res 2014; 42: 7028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deshmukh D, Qiu Y. Role of PARP-1 in prostate cancer. Am. J. Clin. Exp. Urol 2015; 3: 1–12. [PMC free article] [PubMed] [Google Scholar]
- 11.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med 2015; 373: 1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discovery 2012; 2: 1134–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirsanov KI, Kotova E, Makhov P, Golovine K, Lesovaya EA, Kolenko VM, et al. Minor grove binding ligands disrupt PARP-1 activation pathways. Oncotarget 2014; 5: 428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012; 72: 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liscio P, Camaioni E, Carotti A, Pellicciari R, Macchiarulo A. From polypharmacology to target specificity: the case of PARP inhibitors. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2013; 13(23): 2939–54. [DOI] [PubMed] [Google Scholar]
- 16.Passeri D, Camaioni E, Liscio P, Sabbatini P, Ferri M, Carotti A, et al. Concepts and molecular aspects in the polypharmacology of PARP-1 inhibitors. ChemMedChem 2016; 11(12): 1219–26. [DOI] [PubMed] [Google Scholar]
- 17.Kirby IT, Cohen MS. Small-molecule inhibitors of PARPs: from tools for investigating ADP-ribosylation to therapeutics. Curr. Top. Microbiol. Immunol 2019; 420: 211–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekblad T, Camaioni E, Schüler H, Macchiarulo A. PARP inhibitors: polypharmacology versus selective inhibition. FEBS J 2013; 280(15): 3563–75. [DOI] [PubMed] [Google Scholar]
- 19.Mangerich A, Burkle A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer 2011; 128: 251–65. [DOI] [PubMed] [Google Scholar]
- 20.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotech 2012; 30: 283–8. [DOI] [PubMed] [Google Scholar]
- 21.Tulin A Re-evaluating PARP1 inhibitor in cancer. Nat. Biotech 2011; 29: 1078–9. [DOI] [PubMed] [Google Scholar]
- 22.Kotova E, Tulin AV. High-throughput colorimetric assay for identifying PARP-1 inhibitors using a large small-molecule collection. Methods Mol. Biol. (N. Y., NY, U. S.) 2017; 1608: 299–312. [DOI] [PubMed] [Google Scholar]
- 23.Kotova E, Pinnola AD, Tulin AV. Small-molecule collection and high-throughput colorimetric assay to identify PARP1 inhibitors. Methods Mol. Biol. (N. Y., NY, U. S.) 2011; 780: 491–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas C, Ji Y, Kotova E, Lodhi N, Pinnola AD, Golovine K, et al. New generation non-NAD-like PARP-1 inhibitors effectively eliminate drug resistant tumors in vivo. EBioMedicine 2016; 13: 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nieborowska-Skorska M, Maifrede S, Ye M, Toma M, Hewlett E, Gordon J, et al. Non-NAD-like PARP1 inhibitor enhanced synthetic lethal effect of NAD-like PARP inhibitors against BRCA1-deficient leukemia. Leuk. Lymphoma 2018; 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szegezdi JP, Schizmadia F. Prediction of distribution coefficient using microconstants. Poster presented at the 227th ACS National Meeting March 28-April 1, 2004, Anaheim, CA http://chemaxon.com/conf/
- 27.Viswanadhan VN, Ghose AK, Revankar GR, Robins RK. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certaion naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci 1989; 3: 163–72. [Google Scholar]
- 28.Etrl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem 2000; 43: 3714–17. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Jamei M, Yeo KR, Rostami-Hodjegan A, Tucker GT. Misuse of the well-stirred model of hepatic drug clearance. Drug Metab. Dispos 2007; 35: 501–2. [DOI] [PubMed] [Google Scholar]
- 30.Blass BB, Iyer P, Abou-Gharbia M, Childers WE, Gordon JC, Ramanjulu M, et al. Design and synthesis of functionalized piperazin-1yl-(E)-stilbenes as inhibitors of 17ahydroxylase-C17,20-lyase (Cyp17). Bioorg. Med. Chem. Lett 2018; 28: 2270–4. [DOI] [PubMed] [Google Scholar]
- 31.McMasters DR, Torres RA, Crathern SJ, Dooney D, Nachbar RB, Sheridan RP, et al. Inhibition of recombinant cytochrome P450 isoforms 2D6 and 2C9 by diverse drug-like molecules. J. Med. Chem 2007; 50: 3205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kochansky CJ, McMasters DR, Lu P, Koeplinger KA, Kerr HH, Shou M, et al. Impact of pH on plasma protein binding in equilibrium dialysis. Mol. Pharmacol 2008; 5: 438–48. [DOI] [PubMed] [Google Scholar]
- 33.Golovine K, Makhov P, Teper E, Kutikov A, Canter DJ, Uzzo RG, et al. Piperlongumine induces rapid depletion of the androgen receptor in human prostate cancer cells. Prostate (Hoboken, NJ, U. S.) 2013; 73(1): 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotova E, Jarnik M, Tulin AV. Uncoupling of the transactivation and transrepression functions of PARP1 protein. Proc. Natl. Acad. Sci. U. S. A 2010; 107: 6406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canter DJ, Kutikov A, Golovine K, Makhov P, Simhan J, Uzzo RG, et al. Are all multi-targeted tyrosine kinase inhibitors created equal? An in vitro study of sunitinib and pazopanib in renal cell carcinoma cell lines. Can. J. Urol 2011; 18(4): 5819–25. [PMC free article] [PubMed] [Google Scholar]
- 36.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res 2010; 70: 5389–98. [DOI] [PubMed] [Google Scholar]
- 37.Swindall AF, Stanley JA, Yang ES. PARP-1: friend or foe of DNA damage and repair in tumorigenesis? Cancers 2013; 5: 943–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotova E, Jarnik M, Tulin AV. Poly (ADP-ribose) Polymerase 1 is required for protein localization to Cajal body. PLoS Genet 2009; 5(2): e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinderman NB, LaVelle A. A nucleolus-associated coiled body. J. Neurocytol 1976; 5: 545–50. [DOI] [PubMed] [Google Scholar]
- 40.Gall JG. The centennial of the Cajal body. Nat. Rev. Mol. Cell Biol 2003; 4: 975–80. [DOI] [PubMed] [Google Scholar]
- 41.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 2011; 54(8): 2529–91. [DOI] [PubMed] [Google Scholar]
- 42.Schiewer MJ, Knudsen KE. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res 2014; 12: 1069–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraus WL, Lis JT. PARP goes transcription. Cell 2003; 113: 677–83. [DOI] [PubMed] [Google Scholar]