

Abstract
Cancer cells reprogram their metabolism to support growth and to mitigate cellular stressors. The serine synthesis pathway has been identified as a metabolic pathway frequently altered in cancers and there has been considerable interest in developing pharmacological agents to target this pathway. Here, we report a series of indole amides that inhibit human 3-phosphoglycerate dehydrogenase (PHGDH), the enzyme that catalyzes the first committed step of the serine synthesis pathway. Using X-ray crystallography, we show that the indole amides bind the NAD+ pocket of PHGDH. Through structure-based optimization we were able to develop compounds with low nanomolar affinities for PHGDH in an enzymatic IC50 assay. In cellular assays, the most potent compounds inhibited de novo serine synthesis with low micromolar to sub-micromolar activities and these compounds successfully abrogated the proliferation of cancer cells in serine free media. The indole amide series reported here represent an important improvement over previously published PHGDH inhibitors as they are markedly more potent and their mechanism of action is better defined.
Keywords: PHGDH, 3-phosphoglycerate dehydrogenase, Serine synthesis, Cancer metabolism, PHGDH inhibitor, inhibitor
Graphical Abstract
Serine serves an important role in cellular metabolism. Serine is required for protein synthesis and is incorporated into the head groups of certain lipids, such as sphingolipids and phosphatidylserine, the latter of which is an important constituent of cellular membranes. Furthermore, serine can be metabolized to glycine, which charges the folate pool with one carbon units. Glycine and folate one carbon units have numerous important roles including the production of nucleotides.1,2 Serine can be imported into a cell through amino acid transporters or can be produced de novo from glucose via the serine synthesis pathway (SSP).3,4 The SSP consists of three sequential enzymatic reactions. The first, and rate determining, step of SSP is the conversion of the glycolytic intermediate 3-phosphoglycerate (3-PG) to 3-phosphohydroxypyruvate (3-PHP). This reaction is catalyzed by the NAD+ dependent enzyme 3-phosphoglycerate dehydrogenase (PHGDH). Subsequently, 3-PHP is converted to 3-phosphoserine (3-PSER) by phosphoserine aminotransferase (PSAT) using glutamate as the nitrogen donor. Finally, phosphoserine phosphatase (PSPH) catalyzes the magnesium dependent hydrolysis of phosphoserine to yield serine.
Cancer cells are known to alter their metabolism not only in order to promote growth, but also to overcome a variety of cellular stresses.5-8 The SSP has been identified as one such pathway that cancer cells exploit for growth and stress resistance.6-13 In breast cancer and melanoma, amplifications of the gene encoding PHGDH that lead to overexpression of the enzyme have been identified.10,11 In non-small cell lung cancer, oncogenic mutations that activate NRF2 (nuclear factor erythroid-2-related factor) transcription factor signaling have been reported to induce PHGDH overexpression via ATF4.13 Proliferation of cancer cell lines that overexpress PHGDH is inhibited by shRNA mediated knockdown of PHGDH.10,11 In contrast, cancer cell lines with low PHGDH expression are insensitive to genetic inhibition of the pathway as they rely on extracellular serine import.10,11 Importantly, PHGDH promotes resistance to oxidative stress as the SSP maintains cellular NADPH levels.8,13 NADPH is the major source of cellular reducing power and fuels both the glutathione and thioredoxin systems.7 Increased PHGDH expression appears to increase the metastatic properties of cancer cells in animal and cell culture models perhaps by mitigating the oxidative stress associated with metastasis.6,8,9,12,13 Finally, in human patients, PHGDH expression correlates with shorter survival and more aggressive disease.9-13 Together these data suggest that targeting PHGDH may be an effective therapy for treating cancers with high SSP activity.
To date, no full-length crystal structure of human PHGDH has been reported. However, the structure of the truncated protein, amino acids 3-314, has been reported in the PDB (entry 2G76) with both a substrate analog, L-malate, and the NAD+ cofactor bound. The 2G76 structure shows that the truncated protein forms a dimeric species where the PHGDH catalytic sites (1 per monomer) form at the dimerization interface of two PHGDH monomers. Full-length PHGDH is thought to tetramerize by dimerization of two PHGDH dimers via interactions mediated by the C-terminal domain. A catalytically inactive partial digestion fragment of PHGDH composed of residues 93-298 that contains the NAD+ cofactor binding site missing a lid domain, which partially encloses the substrate binding site was also crystalized for the purpose of fragment based drug-screening by Unterlass et al.18
While a number of PHGDH inhibitors have been reported in the literature, identifying a potent and drug-like PHGDH inhibitor has proved challenging. Mullarky et al. identified CBR-5884 by screening a library of 800K compounds using an in vitro PHGDH enzyme assay.14,15 CBR-5884 is a weak but selective PHGDH inhibitor (IC50 = 33 ± 12 μM) that exhibits time dependent inhibition and disrupts the tetrameric state of PHGDH (Fig. 1). Pacold et al. screened a 400K-compound library using a similar PHGDH enzyme assay and discovered a thiourea class of inhibitors.16 A representative compound, NCT-503, showed moderate potency against PHGDH (IC50 = 2.5 ± 0.6 μM) (Fig. 1). Ravez et al. identified a class of α-ketothioamide derivatives of moderate potency, inhibition constant (Ki) of 27 ± 7 μM, again using a PHGDH enzyme assay to screen a fragment library of ~300 compounds (Fig. 1).17 While all three chemotypes showed encouraging anti-cancer activity in cellular assays, they all suffer from weak to moderate potency. Unterlass et al. conducted differential scanning fluorimetry (DSF) screening of a fragment library and discovered a dozen fragments that bind PHGDH competitively to NAD+ with millimolar affinities.18 Finally, AstraZeneca scientists reported inhibitor 1 (IC50 = 0.26 μM) that was identified via a fragment-based screen and was reported to bind the adenine region of the NAD+ pocket.19 The AstraZeneca disclosure was subsequently reviewed in Fuller et al.20 Interestingly, the fragments reported by Unterlass et al. also seemed to bind the adenine pocket.18
Fig. 1.

Selected small molecular PHGDH inhibitors published in the literature
Although only limited information was available for compound 1, we were interested in its sub-micromolar binding affinity and its mechanism of action (MOA), noncovalent binding to the NAD+ pocket. We decided to pursue 1 as a lead compound to develop more potent PHGDH inhibitors and study the biology of inhibiting PHGDH pharmacologically in cancer cells. Over the course of our studies, multiple patent applications from Raze Therapeutics and Boehringer Ingelheim were disclosed, all based on the indole amide series.21-25 Here, we report the structure-activity-relationship (SAR) of analogues of 1 and our biological results.
In order to test the activity of 1, we developed an in vitro enzyme assay for PHGDH similar to those previously reported with adaptations detailed in the supplementary materials.15,16 In brief, PHGDH produces NADH from NAD+ upon oxidation of 3-PG to 3-PHP. Using diaphorase as the coupling enzyme, NADH production was coupled to the reduction of resazurin to the fluorescent species resorufin. Thus, resorufin fluorescence was measured to monitor PHGDH activity. The downstream enzymes PSAT1 and PSPH were included in the reaction mixture to prevent product feedback inhibition of PHGDH by 3-PHP. We synthesized 1 and validated its PHGDH inhibitory activity in the enzymatic assay. Consistent with reports from surface plasmon resonance (SPR) experiments, we obtained an IC50 of 0.238 ± 0.035 μM.26 Next, we determined the Ki of 1 with respect to NAD+ (Fig. 2) in a manner analogous to that previously described.15,16 Compound 1 was a mixed mode inhibitor of PHGDH with Ki = 0.214 ± 0.026 μM, as evidenced by a decrease in Vmax and rightward shift in Km with increasing concentration of 1. These results were consistent with 1 binding the NAD+ pocket and the catalytic mechanism of PHGDH (ordered Bi Bi, NAD+ binds first).18,27 Because we were using an enzyme coupled assay, we were concerned that 1 was actually inhibiting another enzyme in the assay system and thus not an actual PHGDH inhibitor. However, given the agreement between the SPR data (obtained with only PHGDH immobilized enzyme immobilized on the assay chip) and our IC50 data, and, furthermore, that 1-mediated PHGDH inhibition was sensitive to increasing NAD+ concentration, we were confident that 1 was a bonafide PHGDH inhibitor. The tightness of structure guided SAR data provided below further suggest that 1 inhibits PHGDH directly rather than one of the coupling enzymes.
Fig. 2.

Compound 1 is a mixed mode inhibitor of PHGDH with respect to NAD+.
After confirming that compound 1 inhibited truncated PHGDH in the enzyme IC50 assay, we obtained an X-ray crystal structure of truncated PHGDH (residues 1-313) with compound 1 bound (protein data bank entry 6PLF; supplementary materials). Similar to the previously reported X-ray crystal structures (protein data bank entry 2G76 and 5N6C), truncated PHGDH formed a dimer.28 Compound 1 was found to bind in the adenine binding region of the NAD+ pocket (Fig. 3). One of the key interactions was a hydrogen bond between the amide bond N-H of 1 and the carboxylate of Asp175. The adjacent OH group also bound the carboxylate through a hydrogen bond. The indole ring was nestled into the adenine binding region of the NAD+ pocket surrounded by hydrophobic residues of P176, Y174, L151, L193, L216, T213, T207 and L210; thus, the lipophilicity of the indole ring would contribute to inhibitor binding affinity. The carboxylate of 1 extends to the pocket accommodating the diphosphate moiety of NAD+. This hydrophilic pocket is surrounded by two basic guanidine sidechains from Arg155 and Arg236, which could form charge-charge interactions with the carboxylate. The carboxylate also binds to Arg236 through a water bridge. There were several water molecules surrounding the ligand in the structure although none are directly bridged to the protein. One water molecule (HOH6 and HOH8 in the two chains) is buried underneath the ligand. An equivalent water molecule contacts the phosphate of NAD+ in the PDB structure 2G76.
Fig. 3.

X-ray co-crystal structure of compound 1 and PHGDH (PDB: 6PLF).
In order to interrogate the level of activity of the SSP in cells, we turned to uniformly carbon-13-labeled glucose (13C6-glucose) tracing using LC-MS in carney cells as described previously.15 In brief, we pretreated cells with inhibitor for 1 hour in the presence of 12C3-serine. After pretreatment, the cells were washed and labeled with 13C6-glucose media for 2 hours still in the presence of inhibitor. From the isotopic enrichment of serine, we could decouple newly synthesized serine from extracellular serine and serine that was synthesized prior to tracer addition.15 Assaying serine synthesis within three hours of drug treatment was preferred to longer term drug treatments to avoid false positives in which serine synthesis was decreased via an indirect effect, for example, off target cell death. Concentrations of compound 1 up to 40 μM were unable to inhibit serine synthesis as per the 13C6-glucose tracing assay (Fig. 5 A).
Fig. 5.

Inhibition of 13C3-serine synthesis form 13C6-glucose in Carney cancer cells.
Given the high polar surface area (PSA 150 Å2) of compound 1, the presence of five hydrogen-bond donors (HBD), and the low LogD (Table 1), we ascribed the lack of cellular activity to poor cell membrane permeability of 1. Thus, leveraging the crystallographic information, we sought to reduce the polarity of 1 while preserving key interactions necessary for inhibitor binding. Inspection of the co-crystal structure suggested the indole N-H of compound 1 was not necessary for binding affinity. In addition, the water network present near the oxazolidinone moiety was hypothesized to contribute little to the binding affinity of compound 1. Indeed, compound 2, made by removing the oxazolidinone and masking the indole N-H with a methyl group, had the same potency as 1, but was significantly less polar (PSA 101 Å2). Interestingly, removing the OH group (i.e. 3) had no effect on potency, but further lowered the PSA (81 Å2) and hydrogen-bond donor count. Thus, compound 3 had improved permeability as measured by the MDCK-MDR1 assay (A to B = 14 nm/s). Surprisingly, the attempt to introduce a charge-charge interaction with Asp175 by replacing the OH of 2 with an amino group as in 4 was strongly detrimental to potency. This finding could be rationalized by this amine forming a non-binding conformation with the adjacent amide carbonyl or the introduction of a formal charged center in the vicinity of the lipophilic surface formed by Ile177 and Ile178.
Table 1:
Structure-activity relationship of 1 and its analogues
| Compd. | Structure | IC50 (μM) |
HPLC LogD at pH7.4 |
PAMPA (nm s−1 at 10 μM, pH7.4) |
MDR1 Papp (AB, nm s−1) |
MDR1 Ratio (BA/AB) |
MW | PSA (Å2) |
|---|---|---|---|---|---|---|---|---|
| 1 | 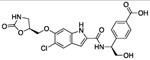 |
0.238 | −1.86 | ND | <2 | ND | 474 | 150 |
| 2 | 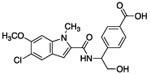 |
0.235 | 0.88 | <9 | 1 | ND | 403 | 101 |
| 3 | 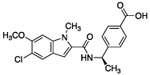 |
0.235 | 1.67 | ND | 14 | 1.3 | 387 | 80.6 |
| 4 | 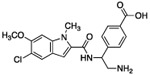 |
23.4 | – | – | – | – | 474 | 150 |
| 5 | 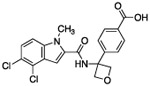 |
0.153 | – | 52 | 6 | 1.3 | 419 | 80.6 |
| 6 | 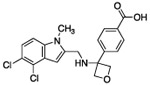 |
209 | – | – | 24 | 1.5 | 405 | 63.5 |
| 7 | 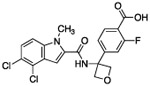 |
0.037a | 2.33 | ND 152 (pH 5.0) |
5 | 2.5 | 437 | 80.6 |
| 8 | 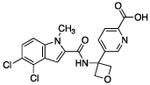 |
22 | 2.74 | 13 | 1 | 3 | 420 | 93.5 |
| 9 | 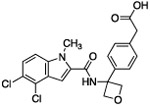 |
0.030a | 2.57 | <1 | <2 | ND | 433 | 80.6 |
| 10 | 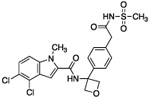 |
0.439a | 2.45 | <4 | <2 | ND | 510 | 115 |
| 11 | 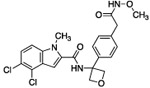 |
0.287 | 2.91 | 273 | 8 | 16 | 462 | 81.6 |
| 12 | 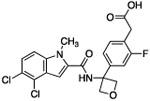 |
0.034a | 2.40 | 45 | ND | ND | 451 | 80.6 |
| 13 | 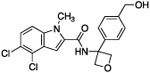 |
1.58 | 3.03 | 154 | 125 | 0.63 | 405 | 63.5 |
| 14 | 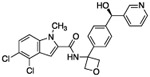 |
0.37 | 3.10 | 208 | 4.9 | 16 | 482 | 76.4 |
| 14-en | 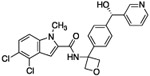 |
0.99 | 3.10 | 252 | 6.1 | 17 | 482 | 76.4 |
| 15 | 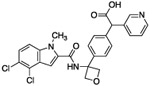 |
0.015a | 2.56 | <1 | <2 | ND | 510 | 93.5 |
| 16 | 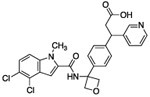 |
0.025a | 2.61 | 16 | – | – | 524 | 93.5 |
| 17 | 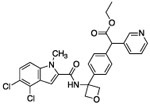 |
1.33 | – | – | – | – | 548 | 105 |
| 18 | 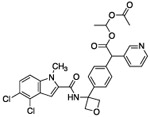 |
1.46 | 3.7 | 118 | 4.2 | 14 | 596 | 109 |
measured in enzyme assay with a low PHGDH concentration described in supplemental materials;
ND: not determined.
With a reduced PSA and HBD count, oxetane containing analog 5 showed moderate permeability in a parallel artificial membrane permeability assay (PAMPA). We also made analogues linked by other groups such as cyclobutane or furan. As predicted from X-ray structures, those compounds were almost as potent as the oxetane analogues (not shown). The furan-linked compounds are chiral (adding synthetic difficulty) and the cyclobutane-linked ones have higher LogD (poorer solubility) so oxetane 5 was preferred. In the 13C6-glucose tracing assay, compound 5 inhibited serine synthesis in a dose dependent manner, with approximately 40% inhibition at 10 μM and complete pathway inhibition at 40 μM (Fig. 5 B). We speculated that the dramatic decrease in potency observed in the 13C6-glucose tracing assay relative to the enzyme IC50 assay may derive from two factors. First, a kinetic effect where compound 5 crosses the cell membrane slowly compared to the time frame of the tracing experiment and/or that compound 5 was highly albumin bound (plasma protein binding >99%), greatly decreasing the free drug concentration. Experiments in which the drug pre-treatment period was increased from one to three hours prior to initiating 13C6-glucose labeling did not increase the extent of pathway inhibition (data not shown). Thus, the high plasma protein binding was likely a significant contributor to the shift in potency between the enzyme and 13C6-glucose tracing assays.
The hydrogen bond between the amide N-H and the carboxylate of Asp175 was critical; replacing the amide with an amine as in compound 6 resulted in a large loss of potency. Structural biology data suggested that charge-charge interactions between the carboxylate of the ligand and PHGDH arginine residues may be important. An ortho-substituting F- group next to the benzoic acid increased the potency of compound 7 by four fold (0.037 μM). It was reasoned that the increase in potency resulted from the decrease in the pKa of the benzoic acid. However, introducing a nitrogen heteroatom adjacent to the benzoic acid (compound 8) reduced potency. Homologation of the benzoic acid by one methylene unit was beneficial to potency, as demonstrated by compound 9. Unfortunately, replacing the carboxylic acid with bioisosteric groups, such as an acylsulfonamide (compound 10) or N-methoxyamide (compound 11), significantly decreased potency.29 Introduction of an ortho-substituting F-group (compound 12) failed to enhance potency, likely because the greater distance between the F- and the acid moiety decreases the electron-withdrawing effect.
As expected, replacing the carboxylic acid with a hydroxyl group decreased potency ten-fold (compound 13), but potency can be partially restored by the substitution with a 3-pyridyl group (compound 14). The hybrid of compound 9 and the pyridyl group gave a more potent compound, 15; the racemate of 15 had an IC50 of 015 μM. We assumed that, as with compounds 14 and 14-en, one of the two enantiomers of compound 15 was more active. However, the enantiomers of 15 were not separated due to concerns around the facile racemization of the chiral center. The increased enzymatic potency of 15 translated to greater potency in the 13C6-glucose tracing assay. Compound 15 achieved nearly 90% pathway inhibition at 10 μM (Fig. 5 C). Finally, compound 16 showed that the carboxylate could be extended by one methylene unit and retain potency.
We obtained a co-crystal structure of rac-15 in complex with truncated PHGDH (PDB entry 6PLG; supplementary materials). Although the resolution is moderate (3.1 Å), the structure suggested that the carboxylate may form a bidentate H-bond with two adjacent backbone N-Hs (Ile156 and Arg155) (Fig. 4). In this structure there are 8 monomers in the unit cell, all with compound 15 bound, and in some protein-ligand systems the pyridyl may form H-bonds with the Arg236 sidechain.
Fig. 4.

X-Ray co-crystal structure of compound 15 and PHGDH. (PDB entry 6PLG).
We profiled the selectivity of select PHGDH inhibitors (1, 4, 14, and 15) by performing biochemical IC50 experiments on a panel of three human NAD(P)+ dependent dehydrogenases (LDHA, IDH1, MDH1) related to PHGDH using commercially available proteins and assay kits (supplementary materials). No significant inhibitory activity of the PHGDH inhibitors towards other dehydrogenases was observed in the enzymatic IC50 assays at concentrations up to 10 μM (supplementary data). These data indicate that the PHGDH inhibitors bind to the NAD+ pocket of PHGDH specifically rather being pan dehydrogenase inhibitors.
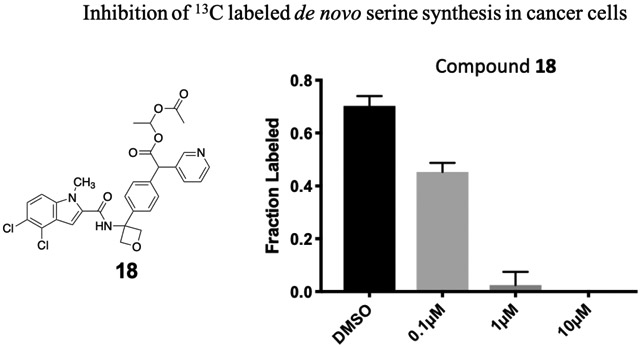
Genetic studies using RNAi suggested that greater than 75% pathway inhibition in the 13C6-glucose tracing assay was necessary in order to achieve robust anti-cancer efficacy in cell line proliferation assays (data not shown). To further improve potency in cell-based assays, we turned to a prodrug approach in an attempt to overcome the low membrane permeability liability of the carboxylates. We synthesized ester prodrugs 17 and 18 which mask the negatively charged carboxylic acid moiety of 15. A study suggested that the acetoxylethyl ester of tranexamic acid was more prone to hydrolysis in human plasma than its ethyl ester analogue (with a half-life time at 37 °C of 6-12 min compared to 240 min).30 The potency of compound 17 in the 13C6-glucose tracing assay was only marginally improved compared to that of the parent compound 15 (about 70% pathway inhibition at 10 μM, Fig. 5 D). We reasoned that perhaps the intracellular hydrolysis of the ethyl ester is slow compared to the time frame of the 13C6-glucose tracing assay and thus the improved membrane permeability of 17 was not detected in the assay as 17 did not efficiently bioconvert to its active form. Nevertheless, prodrug 18 successfully enhanced activity in the 13C6-glucose tracing assay. Compound 18 inhibited serine synthesis approximately 40% at 0.1 μM and achieved near complete pathway inhibition at 1 μM (Fig. 5 E). The 13Clabeling of 3-PG and pyruvate was not perturbed by drug treatment indicating that decreased serine labeling was due to on target PHGDH inhibition rather than changes in the availability of the PHGDH substrate, 3-PG (supplementary data).
Next, we examined whether the PHGDH inhibitors were able to inhibit the growth of a cancer cell line. To do so, we turned to Carney cells grown in serine depleted media, as described previously.15 Carney cells can grow in both serine replete and serine depleted medium, but removing serine from the media increases PHGDH protein expression and renders them dependent on the serine synthesis pathway. 15 Overall, the potency of the compounds in the 13C6-glucose tracing assay correlated with their ability to inhibit cell proliferation (Fig. 6). Prodrug 18 was significantly more potent than its parent compound 15 and almost completely abrogated Carney cell proliferation. Together these data establish compound 18 as a potent inhibitor of PHGDH able to abrogate proliferation of carney cancer cells in vitro.
Fig. 6.

Growth of Carney cancer cells in serine free media treated with PHGDH inhibitors.
In summary, we have developed an indole amide series of compounds that inhibit PHGDH with low nanomolar potencies in enzymatic assays. We were able to demonstrate that the compounds abrogated de novo serine synthesis in cells and that they inhibited the growth of carney cancer cells in serine free medium. These indole amide compounds represent an important improvement over previously published PHGDH inhibitors because they are markedly more potent and have a clear mechanism of action whereby they bind the adenine site of PHGDH in a reversible and non-covalent manner. We were able to greatly improve cellular potency by applying prodrug approaches and achieved proof-of-concept (POC) at the cellular level. However, these ester prodrugs are labile to hydrolysis in plasma and thus not suitable for in vivo studies. Further optimization will be focused on developing compounds suitable for in vivo POC.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge access to support that was generously provided by the Sanders Innovation & Education Initiative, as administered by the Tri-Institutional Therapeutics Discovery Institute. LCC has research reported in this publication that was supported by the National Cancer Institute of the National Institutes of Health under Award Number R35CA197588. LCC has research reported in this publication that was supported by a gift from The Roger and Susan Hertog Charitable Fund. Research reported in this publication was supported by The Daedalus Fund For Innovation Of Weill Cornell Medicine. We would like to thank members of the Cantley lab and the Tri-Institutional Therapeutics Discovery Institute for helpful discussions. We thank Ce Feng Liu for discussions and help with protein purification.
Footnotes
Note about conflicts of interest: LCC is a founder and member of the SAB of Agios Pharmaceuticals; he is also a co-founder, member of the SAB, and shareholder of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. LCC laboratory receives some funding support from Petra Pharmaceuticals. TDI personnel, as a condition of employment, assign all ownership and decision rights related to this research project to the originating institution WCM, MSK, or RU.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.de Koning TJ, Snell K, Duran M, et al. (2003) I-Serine in disease and development. Biochem J. 2003;371(3):653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Futerman AH, Riezman H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005;15(6):312–318. [DOI] [PubMed] [Google Scholar]
- 3.Palacín M, Estévez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 1998;78(4):969–1054. [DOI] [PubMed] [Google Scholar]
- 4.Barker G, Ellory J. The identification of neutral amino acid transport systems. Exp Physiol. 1990;75(1):3–26. [DOI] [PubMed] [Google Scholar]
- 5.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–73. [DOI] [PubMed] [Google Scholar]
- 6.Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullarky E, Cantley LC. Diverting glycolysis to combat oxidative stress In: Nakao K, Uemoto S, Minato N, eds. Innovative Medicine, Basic Research and Development. Springer Open; 2015;3–23. [PubMed] [Google Scholar]
- 8.Ye J, Fan J. Venneti S, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014;4(12):1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollari S, Käkönen SM, Edgren H, et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Tr. 2011;125(2):421–430. [DOI] [PubMed] [Google Scholar]
- 10.Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43(9):869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samanta D, Park Y, Andrabi SA, et al. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016;76(15):4430–4442. [DOI] [PubMed] [Google Scholar]
- 13.DeNicola GM, Chen P-H, Mullarky E, et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat Genet. 2015;47(12):1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullarky E, Lairson LL, Cantley LC, Lyssiotis CA. A novel small-molecule inhibitor of 3-phosphoglycerate dehydrogenase. Mol Cell Oncol. 2016;3(4):e1164280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullarky E, Lucki NC, Beheshti Zavareh R, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc National Acad Sci. 2016; 113(7): 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pacold ME, Brimacombe KR, Chan SH, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12(6):452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravez S, Corbet C, Spillier Q, et al. α-Ketothioamide derivatives: a promising tool to interrogate phosphoglycerate dehydrogenase (PHGDH). J Med Chem. 2017;60(4): 1591–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unterlass JE, Baslé A, Blackburn TJ, et al. Validating and enabling phosphoglycerate dehydrogenase (PHGDH) as a target for fragment-based drug discovery in PHGDH-amplified breast cancer. Oncotarget. 2018;9(17): 13139–13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The 5th RSC-BMCS fragment-based drug discovery meeting at Churchill College, Cambridge (UK), 2015 [Google Scholar]
- 20.Fuller N, Loredana S, Scott C, et al. An improved model for fragment-based lead generation at AstraZeneca. Drug Discov Today. 2016;21(8):1272–1283. [DOI] [PubMed] [Google Scholar]
- 21.Mainolfi N 2017, PCT Patent Appl. WO 2017156165A1.
- 22.Mainolfi N, Moyer MP. 2017, PCT Patent Appl. WO 2017156177A1.
- 23.Mainolfi N 2017, PCT Patent Appl. WO 2017156179A1.
- 24.Mainolfi N, Moyer MP, Saiah E. 2017, PCT Patent Appl. WO 2017156181A1.
- 25.Weinstabl H, Dahmann G, Treu M. et al. 2018, PCT Patent Appl. WO2018167019A1.
- 26.Prasad PN. Introduction to biophotonics. John Wiley & Sons, Inc., Hoboken, New Jersey, 2003 [Google Scholar]
- 27.Grant GA Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch Biochem Biophys. 2012;519(2): 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unterlass JE, Wood RJ, Baslé A, Tucker J, Cano C, et al. Structural insights into the enzymatic activity and potential substrate promiscuity of human 3-phosphoglycerate dehydrogenase (PHGDH). Oncotarget. 2017;8(61): 104478–104491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassalas P, Gay B, Lasfargeas C. et al. Structure property relationships of carboxylic acid isosteres. J Med Chem. 2016;59(7):3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svahn CM, Merenyi F, Karlson L. et al. Tranexamic acid derivatives with enhanced absorption. J Med Chem. 1986;29(4):448–453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.