

Abstract
In the last decade, we have seen increasing evidence of the importance of structural nuclear proteins such as lamins in nuclear architecture and compartmentalization of genome function and in the maintenance of mechanical stability and genome integrity. With over 400 mutations identified in the LMNA gene (encoding for A-type lamins) associated with more than ten distinct degenerative disorders, the role of lamins as genome caretakers and the contribution of lamins dysfunction to disease are unarguable. However, the molecular mechanisms whereby lamins mutations cause pathologies remain less understood. Here, we review pathways and mechanisms recently identified as playing a role in the pathophysiology of laminopathies, with special emphasis in Hutchinson Gilford Progeria Syndrome (HGPS). This devastating incurable accelerated aging disease is caused by a silent mutation in the LMNA gene that generates a truncated lamin A protein “progerin” that exerts profound cellular toxicity and organismal decline. Patients usually die in their teens due to cardiovascular complications such as myocardial infarction or stroke. To date, there are no efficient therapies that ameliorate disease progression, stressing the need to understand molecularly disease mechanisms that can be targeted therapeutically. We will summarize data supporting that replication stress is a major cause of genomic instability in laminopathies, which contributes to the activation of innate immune responses to self-DNA that in turn accelerate the aging process.
Keywords: Lamins, DNA damage, Genomic instability, Progeria, Replication stress, Innate immune response, cGAS-STING pathway
Introduction
Lamins are intermediate filament proteins that form a scaffold between the inner nuclear membrane and chromatin, which is known as the nuclear lamina. Two types of lamins form the nuclear lamina: A-type lamins (lamin A and C), encoded by the LMNA gene via alternative splicing, and B-type lamins (lamin B1, B2, and B3), encoded by LMNB1 and LMNB2 genes (Burke and Stewart 2013). Cryo-electron tomography shows that nuclear lamins assemble into tetrameric filaments that appear as globular-decorated fibers of 3.5-nm thickness (Turgay et al. 2017). The nuclear lamina is viewed as a center for the organization and distribution of genome function in cells, through its interaction with chromatin, transcription factors, nuclear envelope proteins, nuclear pore complexes, and the cytoskeleton (Guillin-Amarelle et al. 2018). Cells devoid of all lamins exhibit frequent, prolonged, and often nonhealing nuclear membrane ruptures, in addition to increased DNA damage (γH2AX foci) (Chen et al. 2018). Structurally, lamins consist of a central α-helical rod domain flanked by non-helical globular and tail domains (Burke and Stewart 2013). Lamin A is synthesized as a prelamin A precursor that undergoes extensive processing. In particular, its C-terminal –CAAX motif is farnesylated, the –AAX residues cleaved, and the terminal –Cys residue carboxyl methylated. Subsequently, the last 15 amino acids are cleaved by the Zmpste24/FACE1 metallopeptidase, producing mature lamin A (Schreiber and Kennedy 2013). In addition, lamin A/C are phosphorylated at multiple residues during interphase, which promotes filament disassembly and solubilization into the nucleoplasm (Kochin et al. 2014; Torvaldson et al. 2015). Alterations in the maturation of lamin A result in cellular damage and are associated with disease. Lamin-related diseases or laminopathies embody a range of tissue pathologies, including muscular dystrophies, peripheral neuropathies, lipodystrophies, dermopathies, leukodystrophies, and segmental progeroid syndromes such as Hutchinson Gilford Progeria Syndrome (HGPS), Atypical Werner Syndrome (AWS), and Restrictive Dermopathy (RD) (Worman et al. 2009; Gordon et al. 2014; Gonzalo and Kreienkamp 2015; Vidak and Foisner 2016; Ho and Hegele 2019). In addition to pathogenic mutations, altered expression levels of lamins are associated with cancer. In particular, reduced lamin A levels have been reported during the progression of a variety of cancers, including breast, ovarian, and cervical cancers, often associated with poor prognosis (Alhudiri et al. 2019; Wang et al. 2019).
Hutchinson Gilford Progeria Syndrome (HGPS) is caused by a single-base substitution in exon 11 of the LMNA gene that activates a cryptic splice site, leading to deletion of 50 residues near the C-terminus that include the Zmpste24 cleavage site De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003). The mutant protein, known as “progerin”, remains permanently farnesylated and carboxyl methylated and causes nuclear alterations similar to those of prelamin A-expressing cells. Progerin elicits, in a dose-dependent manner, nuclear deformation, loss of heterochromatin from the nuclear periphery, telomere shortening, deregulation of gene expression, mitochondria dysfunction, oxidative stress, DNA damage, and eventually premature entry into senescence (Goldman et al. 2004; Merideth et al. 2008; Pereira et al. 2008; Prokocimer et al. 2013; Gordon et al. 2014; Gonzalo and Kreienkamp 2015; Ullrich and Gordon 2015; Kreienkamp et al. 2016; Gonzalo et al. 2017). Despite the progress identifying cellular processes altered by progerin, we still lack a clear picture of the molecular mechanisms involved.
HGPS patients are normal at birth but exhibit a clear growth defect by 2 years of age, often not surpassing 4 ft of height and 30 kg of weight (Gordon et al. 2007; Kieran et al. 2007). Before their teenage years, patients show severe aging phenotypes, including almost complete alopecia, swollen veins, age spots, beak-shaped nose, shrunken chin, narrow chest, swollen and stiff joints, reduced fat and bone mineral density, and severe cardiovascular disease (CVD) (Merideth et al. 2008; Gordon, Gordon et al. 2011; Gordon et al. 2014; Ullrich and Gordon 2015). HGPS patients ultimately die at an average age of 14.6 years from cardiovascular complications such as stroke or myocardial infarction (Ullrich and Gordon 2015). The recent finding that progerin is expressed in the hearts of patients with dilated cardiomyopathy and correlates with left ventricular remodeling (Messner, Ghadge et al. Messner et al. 2018) supports a potential role of progerin in CVD during aging. Interestingly, HGPS pathology spares the central nervous system (CNS). Kids with progeria have normal brain function, with no evidence of cognitive or memory challenges usually associated with aging. Studies aiming to understand why cognitive functions in HGPS patients are preserved revealed that the LMNA gene in brain produces primarily lamin C and not lamin A or progerin. This is due to the expression of a micro RNA, miR-9, which specifically targets prelamin A/progerin transcripts in the brain (Jung et al. 2012; Jung, Tu et al. Jung et al. 2014). Importantly, expression of miR-9 in HGPS patient-derived fibroblasts alleviates cellular phenotypes of aging. Thus, miR-9 could be considered as a potential strategy to reduce progerin toxicity in different tissues, once all its targets are carefully examined.
Over the years, different mouse models of the disease have been generated, which have been instrumental for identifying mechanisms underlying premature aging and for testing therapies. A mouse model carrying the same mutation as in human patients in homozygosis (LmnaG609G/G609G) recapitulates more closely the phenotypes of HGPS patients, although with less severity (Osorio et al. 2011). Interestingly, our recent studies revealed that when these mice are fed high-caloric/high-fat diets, they experience a significant extension of lifespan, which is accompanied by increased disease severity (Kreienkamp et al. 2018a). These progeria mice on high-fat diet recapitulate more closely pathologies of HGPS patients, and thus might represent a better model to study the disease moving forward. Progeria mice and cells derived from HGPS patients are the focus of extensive research to identify molecular mechanisms underlying progerin-induced cellular toxicity and organismal decline, as well as to test therapies that ameliorate disease progression.
Here, we review new knowledge about how disruption of lamins function, and specially progerin expression, impacts cellular mechanisms that maintain genome integrity. In particular, recent studies show that lamins play a role in DNA replication and that the DNA damage that builds up in lamin-deficient cells is elicited in part by replication fork instability. In addition, we discuss the emerging model that DNA damage caused by exogenous or endogenous sources can be detected by cytosolic sensors of nucleic acids, which in turn activate innate immune responses at the crossroads of aging, cancer, and tumor immunity. Lastly, we bring awareness about the beneficial effects of the hormonal form of vitamin D (calcitriol) ameliorating phenotypes of genomic instability in cells with lamin dysfunction and replication stress.
Lamin dysfunction hinders DNA replication
A whole body of evidence supports that lamin dysfunction, either due to reduced expression or expression of mutants such as progerin, elicits DNA repair defects, telomere dysfunction, and overall genomic instability, reviewed elsewhere (Gonzalo and Kreienkamp 2015; Dobrzynska et al. 2016; Kubben et al. 2016b; Gonzalo et al. 2017; Burla et al. 2018). In recent years, interest in the effect of lamin dysfunction in DNA replication, a stage of special vulnerability for the genome, has been gaining momentum. Early studies in Xenopus egg extracts showed that disruption of lamin organization elicits a strong reduction in DNA replication (Spann et al. 1997; Moir et al. 2000). This defect was attributed to changes in the distribution of PCNA and the RFC complex, essential factors in the elongation phase, which formed aggregates with lamins. In mammalian cells, A-type lamins co-localize with PCNA in early sites of DNA replication (Kennedy et al. 2000; Dechat et al. 2008), and a direct interaction between PCNA and lamins via their C-terminus Ig-fold domain seems important for PCNA positioning on chromatin (Shumaker et al. 2008). In addition, lamins interact with DNA polymerases δ (lagging strand) and ε (leading strand) during S phase (Vaara et al. 2012), and lamin loss is associated with deficiencies in the resolution of stalled replication forks (Singh et al. 2013).
Prelamin A and progerin interact more robustly with PCNA than mature lamin A (Kubben et al. 2010) and are thought to sequester PCNA away from the replication fork, hindering replication fork progression (Wheaton et al. 2017). Consistently, prelamin A expression results in increased mono-ubiquitination of PCNA and induction of Pol η, markers of replication fork stalling (Cobb et al. 2016). In support of the PCNA sequestration model, progerin-expressing cells exhibit a marked reduction of PCNA at replicating DNA (Hilton et al. 2017; Wheaton et al. 2017), concomitant with markers of replication stress/DNA damage such as increased γH2AX, ATR activation, and RPASer33 phosphorylation (Wheaton et al. 2017). Progerin also causes aberrant accumulation of the nuclease XPA at stalled or collapsed forks (Hilton et al. 2017). Lowering the levels of progerin or XPA reinstates normal levels of PCNA at replication forks, with the consequent reduction of cell death. Moreover, RFC1 is degraded by a serine protease in HGPS cells, contributing to deficient loading of PCNA and Pol δ onto DNA (Tang et al. 2012). Interestingly, cells carrying other LMNA mutations (G232E and Q294P), associated with diseases such as Emery-Dreifuss muscular dystrophy (EDMD), overexpress a HEC-type E3 ubiquitin ligase, HECW2. It was recently shown that HECW2 interacts with PCNA and lamin B1 and mediates their ubiquitination and proteasomal degradation (Krishnamoorthy et al. 2018). HECW2 also interacts and ubiquitinates lamin A, but this interaction is reduced in presence of the mutants. Thus, the functional relationship between lamins, ubiquitinating enzymes, and replication factors should be further investigated as a potential mechanism contributing to genomic instability in laminopathies.
Altogether, these studies provide evidence for lamins playing a role in DNA replication, in part by modulating the association of key proteins such as PCNA to the replication fork, and for expression of prelamin A, progerin, and potentially other lamin mutants, causing replication stress (Fig. 1). In spite of these findings, our mechanistic understanding of lamins’ participation in DNA replication is limited. Similarly, the extent to which replication stress contributes to genomic instability in lamin-deficient cells and to the pathophysiology of laminopathies warrants further investigation. One could envision that different mutant lamins, with varying binding affinities for replication factors such as PCNA and DNA polymerases, could cause different DNA replication phenotypes. In addition, lamin dysfunction is associated with DNA repair deficiencies and accumulation of DNA damage, which poses a challenge for replication fork progression (Berti and Vindigni 2016; Bhat and Cortez 2018). Thus, lamin-deficient cells might need to deal with extra roadblocks that stall the replication fork. Given the increasing evidence that DNA repair proteins interact and protect the replication fork, while helping with the resolution of replication conflicts (Kolinjivadi et al. 2017), it is feasible that lamin deficiency hinders the recruitment/function of DNA repair factors at the replication fork. Some evidence along this line was obtained recently (Li, Chen et al. 2018). Li and colleagues demonstrated that lamin A binds the homologous recombinase RAD51, preventing its proteasomal degradation. They also identified a lamin-binding ligand (LBL1) that breaks lamin A-RAD51 interaction, rendering RAD51 available for proteasome-mediated degradation, which in turn inhibits DNA repair by HR (Li, Chen et al. 2018). Thus, it is tempting to speculate that loss of lamins or expression of mutant forms might inhibit RAD51 role in DNA replication, causing replication stress. New techniques such as genome-wide single-molecular replication assays (DNA fiber assays), in situ analysis of protein interactions at DNA replication forks (SIRF) (Roy et al. 2018), isolation of proteins on nascent DNA (iPOND) (Sirbu et al. 2011), and electron microscopy (Vindigni and Lopes 2017) are allowing us to identify specific mechanisms during replication that are regulated by lamins, and those that are altered in cells expressing different lamin mutations. Interestingly, there is evidence by iPOND that lamin A, but nor progerin, binds to nascent DNA (Wheaton et al. 2017).
Fig. 1.

Lamins preserve replication fork (RF) stability. Scheme depicts mechanisms of protection of RF integrity during instances of replication stress (RS) and models how lamin A and progerin impact these mechanisms. Left diagram shows that in lamin-proficient cells, lamin A binds to the RF, together with a broad range of DNA repair and remodeling factors (RPA, BRCA1/2, PALB2, RAD51), helping to ensure the stability of the fork by protecting it from nuclease-mediated degradation. These protective factors also mediate fork reversal and restart, ensuing genome stability. Right diagram shows a model whereby progerin sequesters PCNA, and potentially wild-type lamin A protein, away from the RF. This, in turn, could lead to RF stalling in addition to RF deprotection and nuclease (MRE11 and XPA)-mediated degradation of the stalled forks. As a consequence, progerin expression causes a robust replication fork instability phenotype
We recently used single-molecule replication assays to monitor the impact of progerin expression on DNA replication. We found increased frequency of replication fork stalling in progerin-expressing cells under normal conditions, but not in lamin A-depleted or -overexpressing cells (Kreienkamp et al. 2018b). Replication fork stalling was accompanied by fork deprotection and Mre11 nuclease-mediated degradation. Consistently, inhibition of Mre11 nuclease activity by the compound Mirin rescues progerin-induced replication defects. Replication fork instability was also observed in lamin-depleted cells, but only after treatment with fork-stalling compounds (unpublished results). These replication defects are associated with the increased DNA damage and the chromosomal aberrations characteristic of lamin-deficient cells. Ongoing studies are focused on understanding the molecular mechanisms whereby progerin expression or loss of lamins cause replication fork instability, with emphasis on potential deficiencies in DNA repair factors that stabilize stalled replication forks.
Activation of innate immune response to self-DNA in progeria
Recent work has established a strong relationship between DNA damage and immune signaling (Chatzinikolaou et al. 2014; Karakasilioti et al. 2013; Brzostek-Racine et al. 2011). DNA damage caused by exogenous sources (genotoxic agents, UV, ionizing radiation) or endogenous metabolic processes (DNA replication, telomere dysfunction, oxidative stress) is sensed in the nucleus by a repertoire of factors that trigger the DNA damage response (DDR) (Ciccia and Elledge 2010). The DDR, as well as other cellular programs such as the unfolded protein response, oxidative stress, senescence, or apoptosis, are coupled with the generation of signals that trigger systemic responses (Galluzzi et al. 2018). DNA damage for instance generates nucleic acid byproducts that can leak into the cytoplasm, where they are recognized by the machinery responsible for the detection of foreign nucleic acids (Kawai and Akira 2006; Ishikawa et al. 2009; Cai et al. 2014). One of the main sensors of cytoplasmic self-DNA and DNA from pathogens is cGAS (cyclic-GAMP synthase), an enzyme that upon binding to DNA synthesizes 2′,3′-cGAMP (cyclic-Guanosine MonoPhosphate-Adenosine MonoPhosphate Synthase) from ATP and GTP. cGAMP acts as a second messenger to activate the adaptor protein STING (stimulator of interferon genes), playing a crucial role in antimicrobial immune response (Li and Chen 2018). Upon cGAMP binding, STING is translocated from the ER to the ERGIC/Golgi compartments where it mediates IRF3 and NFκB activation and nuclear translocation, via kinases TBK1 and IKK, respectively. Nuclear IRF3 and NF-κB induce the expression of type I IFNs and other pro-inflammatory cytokines that boost the immune response. IFNs are then released into the extracellular milieu where in an autocrine or paracrine fashion bind IFN receptors (IFNAR), which through the JAK/STAT pathway activate hundreds of IFN-stimulated genes (ISGs) (Schneider et al. 2014). The role of the cGAS/STING/IFN pathway activating the innate immune system to clear pathogen-infected or damaged cells has been clearly demonstrated (Dou et al. 2017; Gluck et al. 2017; Harding et al. 2017; Yang et al. 2017; Li and Chen 2018). This pathway is also necessary for the establishment of senescence and has been shown to impact anti-tumor immunity and the response of tumors to immunotherapy (Brzostek-Racine, Gordon et al. 2011; Yu et al. 2015; Wang et al. 2017; Ng et al. 2018). Thus, the cGAS/STING/IFN cascade, which is activated in immune cells, tumor cells, and cells in the tumor microenvironment, is currently at the spotlight as a new target for immunotherapy.
The rapidly growing list of factors that trigger the cGAS/STING/IFN pathway includes carcinogens like DMBA, ionizing radiation, genotoxic agents such as cisplatin and etoposide, oncogenes, DNA repair/replication defects, telomere dysfunction, micronuclei, depletion of ssDNA-binding proteins RPA and RAD51, and loss of the cytoplasmic nuclease TREX-1 or the dNTPase/exonuclease SAMHD1 (West et al. 2015; Chen et al. 2016; Wolf, Rapp et al. Wolf et al. 2016; Bhattacharya, Srinivasan et al. Bhattacharya et al. 2017; Harding, Benci et al. Harding et al. 2017; Mackenzie et al. 2017; Parkes et al. 2017; Coquel et al. 2018; Li and Chen 2018; Ng, Marshall et al. Ng et al. 2018).
Our studies have shown that progerin expression activates the cGAS/STING/IFN pathway (Graziano et al. 2018; Kreienkamp et al. 2018b). Performing genome-wide RNAseq analysis of human skin fibroblasts derived from four HGPS patients and normal fibroblasts from three parents, we identified a robust signature of activation of genes in the IFN/antiviral/innate immunity category. Nearly 50 genes in this category were upregulated in HGPS fibroblasts of early and late passage in culture, compared to normal fibroblasts. These genes included pattern recognition receptors (PRR), proteins that normally survey the cytoplasmic space in search of nucleic acids from pathogens, but that also recognize self-nucleic acids that reach the cytoplasm (RIG-I, MDA5, OASs, PKR, TLR3). In addition, we found upregulation of PRR downstream signaling, including IRFs (IFN responsive factors), STAT1 (signal-transducing activators of transcription 1), and NFkB, as well as over 40 STAT1-regulated IFN-stimulated genes (ISGs). The results of our RNAseq analysis in HGPS fibroblasts were recapitulated upon expression of progerin, but not lamin A, in normal human-derived fibroblasts. In particular, induction of progerin expression rapidly leads to increased protein levels of cGAS and STING, global STAT1 and phosphorylated forms (Y701 and S727), IRF3, and ISG15 (interferon stimulated gene 15), a marker of IFN pathway activation (Graziano et al. 2018; Kreienkamp et al. 2018b). Intriguingly, IFNs themselves were not expressed in HGPS fibroblasts, indicating that this pathway represents a cell-intrinsic, IFN-independent activation of STAT1 downstream inflammatory signals. Activation of STAT1 and downstream ISGs in an IFN-independent fashion has been previously reported in other models (Dempoya, Matsumiya et al. Dempoya et al. 2012; Luu, Greenhill et al. Luu et al. 2014). Interestingly, a recent study showed that STAT1 mediates autoinflammation, lipoatrophy, and juvenile lethality in a mouse model that carries a gain-of-function mutation in the PDGFRB (platelet-derived growth factor receptor beta) gene (He et al. 2017). As such, crossing PDGRFB mutants with STAT1−/− mice rescues autoinflammation and improves lifespan. In contrast, deletion of IFNARs does not rescue wasting in these mice, indicating that STAT1 effect driving wasting in this context is independent of IFNs.
Our studies in progerin-expressing cells support a role for STAT1 linking DNA damage sensing with activation of innate immune responses. In particular, knockdown of STAT1 in progeria cells to levels observed in normal cells results in reduced expression of ISGs, in addition to improved cell proliferation and migration capabilities (Graziano et al. 2018; Kreienkamp et al. 2018b). These data suggest that STAT1 activation might contribute to cellular aging phenotypes. Further studies are needed to determine if STAT1 downregulation or pharmacological inhibition ameliorates organismal decline in mouse models of laminopathies, especially in progeria mice. This would be an important finding, given that STAT1 is associated with inflammation in immune and vascular cells during cardiovascular disease (Szelag, Piaszyk-Borychowska et al. Szelag et al. 2016), which underlies early death in HGPS patients (Prakash, Gordon et al. Prakash et al. 2018).
Another question that remains unanswered is the etiology of activation of the cGAS/STING pathway in HGPS. The robust replication fork instability phenotype observed in progerin-expressing cells suggests a model whereby replication stress causes DNA damage, leading to accumulation of nucleic acids in the cytosol that trigger the cGAS/STING pathway (Fig. 2). In support of this notion, immuno-FISH studies with lamin A antibody and a PNA-labeled telomere probe show that HGPS fibroblasts and MAFs from progeria mice accumulate telomeric DNA in the cytoplasm (Kreienkamp et al. 2018b). Given that telomeres are among the most difficult-to-replicate domains, it is likely that cytoplasmic telomeric DNA results from replication defects. However, this hypothesis remains to be formerly tested. Moreover, it is possible that the disruption of nuclear integrity by progerin exacerbates the leakage of DNA into the cytoplasm. Thus, the combination of replication stress/DNA damage and disruption of nuclear integrity might be responsible for the marked activation of the cGAS/STING pathway and the IFN-like response in progeria (Fig. 2).
Fig. 2.
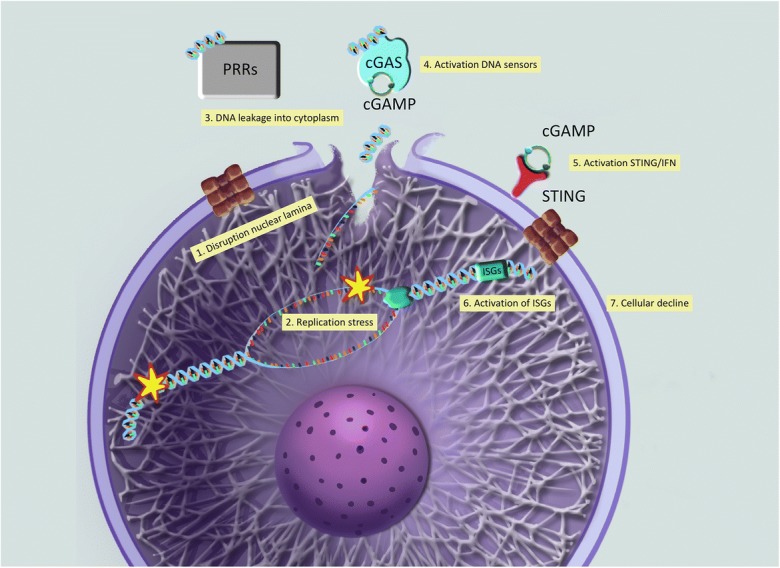
Model of how progerin activates innate immune responses to self-DNA. Our recent studies support a model whereby alterations in lamins function (1), such is the case of progerin expression, cause replication stress and genomic instability (2), in addition to disruption of nuclear integrity (1). These two events—increased DNA damage and nuclear fragility/leakage—contribute to the accumulation of DNA byproducts in the cytoplasm, where they are erroneously recognized as foreign by pattern recognition receptors or PRR (3). One of the key PRR is cGAS (cGAMP-synthase), which through the synthesis of cGAMP (4) activates STING (5) and a STAT1-mediated IFN response. Through transcriptional activation of hundreds of ISGs (6), the cells induce a robust innate immune response to self-DNA, which contributes to cellular and organismal aging (7). Graphic illustrations generated by Michael Andrus, BS, St Louis University
Vitamin D (calcitriol) reduces replication stress and represses innate immune response
Our work has revealed unique functions of the vitamin D/vitamin D receptor (VDR) axis maintaining genome stability. VDR loss in normal human fibroblasts immortalized with telomerase results in downregulation of homologous recombination proteins BRCA1 and RAD51, complete loss of recruitment of BRCA1 to DNA damage sites, accumulation of DNA double-strand breaks (DSBs), and premature entry into senescence (Graziano, Johnston et al. Graziano et al. 2016). Interestingly, cells undergoing oncogene (RasV12)-induced senescence exhibit downregulation of VDR concomitant with reduced expression of DNA repair factors BRCA1 and 53BP1. Treatment with calcitriol (hormonal active form of vitamin D) increases levels of VDR and DNA repair factors and improves DNA repair capabilities (Graziano, Johnston et al. Graziano et al. 2016). We also demonstrated that lamin-depleted and progerin-expressing cells display low VDR expression (Kreienkamp et al. 2016). Calcitriol increases VDR levels and ameliorates progerin-induced cellular phenotypes, including nuclear morphological abnormalities, DNA damage, and premature senescence. Interestingly, calcitriol rescues replication fork instability to the same extent as Mirin treatment in progerin-expressing cells and represses genes in the IFN/antiviral/innate immunity category that are upregulated in HGPS fibroblasts (Kreienkamp et al. 2018b). In particular, we observed reduced global levels of STAT1 and levels of phosphorylated STAT1 upon calcitriol treatment. As testament of the beneficial effects of calcitriol in progeria, treatment of HGPS fibroblasts with calcitriol for 10 days increased their efficiency of reprogramming into iPSCs (induced pluripotent stem cells) (Kreienkamp et al. 2018b). The effect of calcitriol rejuvenating HGPS patient-derived cells justifies preclinical studies to evaluate the effect of calcitriol in mouse models of progeria, and potentially other laminopathies.
The molecular mechanisms whereby calcitriol ameliorates HGPS cellular phenotypes await further investigation. It is possible that by rescuing replication fork instability, calcitriol reduces the DNA damage and accumulation of cytoplasmic DNA that activates the cGAS/STING pathway and the STAT1-mediated IFN-like response. However, calcitriol also improves nuclear morphological abnormalities, which could enhance the integrity of the nuclear envelope and reduce leakage of nucleic acids into the cytoplasm in HGPS cells. Unraveling how calcitriol rescues replication stress would be of critical importance for laminopathies, and might also provide a strategy to ameliorate replication stress in other contexts. Moreover, calcitriol has been shown to impact directly on STAT1, playing an important role in immune system function (Yang et al. 2013). Calcitriol decreases STAT1 and STAT3 phosphorylation/activation and inflammatory cytokine output in cancer cell lines and autoimmune disease mouse models (Muthian, Raikwar et al. Muthian et al. 2006; Wang, Li et al. Wang et al. 2013; Chen et al. 2015), as well as in large granular lymphocyte leukemia (LGLL) (Kulling et al. 2018; Osorio et al. 2011). Thus, it is possible that the beneficial effect of calcitriol in HGPS is the combination of many factors: (i) improved DNA repair/replication, (ii) reduced nuclear fragility, and (iii) direct downregulation of STAT1/IFN-like response. Studies in the next few years will hopefully shed light onto the relationship between vitamin D/VDR signaling, DNA damage, and immunogenicity of self-DNA.
Importantly, we demonstrated that different compounds known to ameliorate cellular decline in HGPS, also reduced the extent of replication stress and STAT1 activation (Kreienkamp et al. 2018b). These include all-trans-retinoic acid (ATRA), which represses progerin expression (Swift et al. 2013; Pellegrini, Columbaro et al. Pellegrini et al. 2015; Kubben et al. 2016a); Remodelin, an inhibitor of NAT10 (N-acetyltransferase-10) that rescues nuclear abnormalities via microtubule organization; and the combination of lonafarnib, farnesyltransferase inhibitor that inhibits progerin prenylation, and rapamycin, which increases autophagy-mediated progerin clearance (Cao et al. 2011; Gordon, Kleinman et al. Gordon et al. 2016). Overall, the data suggest that these phenotypes—replication stress and STAT1/IFN response—are drivers of the cellular decline in HGPS. Consistent with this idea, in aged normal human fibroblasts, activation of the STAT1/IFN pathway is concomitant with a decline in fibroblast functionality, including proliferation, differentiation, and migration (Midgley, Morris et al. Midgley et al. 2016). Importantly, progerin and prelamin A expression has been observed in different tissues from normal aged individuals (Dahl, Scaffidi et al. Dahl et al. 2006).
Concluding remarks
Many lines of evidence point to genomic instability driving cellular decline in lamin-related diseases. DNA replication, in particular, is at center stage as a process that requires an intact nuclear lamina. Although the specific role/s that nuclear lamins play in DNA replication are not fully understood, data suggest that the recruitment of factors critical for DNA replication and for protection of replication forks from degradation is hindered upon nuclear lamin dysfunction. Moreover, evidence of the implication of replication stress and DNA damage in the activation of antiviral/innate immune responses is growing strong. In the context of laminopathies, the characteristic nuclear fragility is likely to contribute to increase the robustness of the response. Importantly, this response has been placed at the crossroads of senescence/aging, cancer, immunosurveillance, and cancer immunotherapy, which illustrates the importance of understanding molecular mechanisms that allow its tunability. We envision that the near future will bring new knowledge also about pharmacological strategies that target this response. For instance, understanding how calcitriol, a hormone produced in our body, modulates genome stability and the innate immune response is of critical importance, and of potential immediate applicability to the clinic. Although still at the infancy of manipulating/tuning these pathways and mechanisms, new technologies and animal models will allow us to advance in ways that help patients with a broad range of diseases.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Alhudiri IM, Nolan CC, Ellis IO, Elzagheid A, Rakha EA, Green AR, Chapman CJ. Expression of Lamin a/C in early-stage breast cancer and its prognostic value. Breast Cancer Res Treat. 2019;174(3):661–668. doi: 10.1007/s10549-018-05092-w. [DOI] [PubMed] [Google Scholar]
- Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol. 2016;23(2):103–109. doi: 10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Cortez D. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol. 2018;25(6):446–453. doi: 10.1038/s41594-018-0075-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, Mishra R, Wakeland EK, Ghose S, Mukherjee S, Asaithamby A. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017;45(8):4590–4605. doi: 10.1093/nar/gkx126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187(10):5336–5345. doi: 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. 2013;14(1):13–24. doi: 10.1038/nrm3488. [DOI] [PubMed] [Google Scholar]
- Burla R, La Torre M, Merigliano C, Verni F, Saggio I. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus. 2018;9(1):368–379. doi: 10.1080/19491034.2018.1476793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54(2):289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3(89):89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- Chatzinikolaou, G., I. Karakasilioti and G. A. Garinis (2014). "DNA damage and innate immunity: links and trade-offs." Trends Immunol 35(9):429–435. [DOI] [PubMed]
- Chen PT, Hsieh CC, Wu CT, Yen TC, Lin PY, Chen WC, Chen MF. 1alpha,25-Dihydroxyvitamin D3 inhibits esophageal squamous cell carcinoma progression by reducing IL6 signaling. Mol Cancer Ther. 2015;14(6):1365–1375. doi: 10.1158/1535-7163.MCT-14-0952. [DOI] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- Chen NY, Kim P, Weston TA, Edillo L, Tu Y, Fong LG, Young SG. Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. Proc Natl Acad Sci U S A. 2018;115(40):10100–10105. doi: 10.1073/pnas.1812622115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb AM, Murray TV, Warren DT, Liu Y, Shanahan CM. Disruption of PCNA-lamins a/C interactions by prelamin a induces DNA replication fork stalling. Nucleus. 2016;7(5):498–511. doi: 10.1080/19491034.2016.1239685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, Promonet A, Cribier A, Sarrazin A, Niedzwiedz W, Lopez B, Costanzo V, Krejci L, Chabes A, Benkirane M, Lin YL, Pasero P. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. 2018;557(7703):57–61. doi: 10.1038/s41586-018-0050-1. [DOI] [PubMed] [Google Scholar]
- Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103(27):10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempoya J, Matsumiya T, Imaizumi T, Hayakari R, Xing F, Yoshida H, Okumura K, Satoh K. Double-stranded RNA induces biphasic STAT1 phosphorylation by both type I interferon (IFN)-dependent and type I IFN-independent pathways. J Virol. 2012;86(23):12760–12769. doi: 10.1128/JVI.01881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynska A, Gonzalo S, Shanahan C, Askjaer P. The nuclear Lamina in health and disease. Nucleus. 2016;7:233–248. doi: 10.1080/19491034.2016.1183848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, Capell BC, Xu C, Xu M, Kieckhaefer JE, Jiang T, Shoshkes-Carmel M, Tanim K, Barber GN, Seykora JT, Millar SE, Kaestner KH, Garcia BA, Adams PD, Berger SL. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in Lamin a cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19(11):731–745. doi: 10.1038/s41580-018-0068-0. [DOI] [PubMed] [Google Scholar]
- Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–1070. doi: 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant Lamin a causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford progeria syndrome. Curr Opin Cell Biol. 2015;34:75–83. doi: 10.1016/j.ceb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18–29. doi: 10.1016/j.arr.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120(4):824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW. Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26(7):1670–1679. doi: 10.1002/jbmr.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, Lopez-Otin C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156(3):400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Kleinman ME, Massaro J, D'Agostino RB, Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, Snyder BD, Ullrich NJ, Silvera VM, Liang MG, Quinn N, Miller DT, Huh SY, Dowton AA, Littlefield K, Greer MM, Kieran MW. Clinical trial of the protein Farnesylation inhibitors Lonafarnib, pravastatin, and Zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation. 2016;134(2):114–125. doi: 10.1161/CIRCULATIONAHA.116.022188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano S, Johnston R, Deng O, Zhang J, Gonzalo S. Vitamin D/vitamin D receptor axis regulates DNA repair during oncogene-induced senescence. Oncogene. 2016;35(41):5362–5376. doi: 10.1038/onc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano S, Kreienkamp R, Coll-Bonfill N, Gonzalo S. Causes and consequences of genomic instability in laminopathies: replication stress and interferon response. Nucleus. 2018;9(1):258–275. doi: 10.1080/19491034.2018.1454168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin-Amarelle C, Fernandez-Pombo A, Sanchez-Iglesias S, Araujo-Vilar D. Lipodystrophic laminopathies: diagnostic clues. Nucleus. 2018;9(1):249–260. doi: 10.1080/19491034.2018.1454167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Medley SC, Kim J, Sun C, Kwon HR, Sakashita H, Pincu Y, Yao L, Eppard D, Dai B, Berry WL, Griffin TM, Olson LE. STAT1 modulates tissue wasting or overgrowth downstream from PDGFRbeta. Genes Dev. 2017;31(16):1666–1678. doi: 10.1101/gad.300384.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton BA, Liu J, Cartwright BM, Liu Y, Breitman M, Wang Y, Jones R, Tang H, Rusinol A, Musich PR, Zou Y. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31(9):3882–3893. doi: 10.1096/fj.201700014R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho R, Hegele RA. Complex effects of laminopathy mutations on nuclear structure and function. Clin Genet. 2019;95(2):199–209. doi: 10.1111/cge.13455. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH, 2nd, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG. Regulation of prelamin a but not Lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 2012;109(7):E423–E431. doi: 10.1073/pnas.1111780109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Tu Y, Yang SH, Tatar A, Nobumori C, Wu D, Young SG, Fong LG. New Lmna knock-in mice provide a molecular mechanism for the 'segmental aging' in Hutchinson-Gilford progeria syndrome. Hum Mol Genet. 2014;23(6):1506–1515. doi: 10.1093/hmg/ddt537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasilioti, I., I. Kamileri, G. Chatzinikolaou, T. Kosteas, E. Vergadi, A. R. Robinson, I. Tsamardinos, T. A. Rozgaja, S. Siakouli, C. Tsatsanis, L. J. Niedernhofer and G. A. Garinis (2013). "DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria." Cell Metab 18(3): 403–415. [DOI] [PMC free article] [PubMed]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7(2):131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000;14(22):2855–2868. doi: 10.1101/gad.842600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran MW, Gordon L, Kleinman M. New approaches to progeria. Pediatrics. 2007;120(4):834–841. doi: 10.1542/peds.2007-1356. [DOI] [PubMed] [Google Scholar]
- Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack CG, Melo-Cardenas J, Imanishi SY, Goldman RD, Eriksson JE. Interphase phosphorylation of Lamin a. J Cell Sci. 2014;127(Pt 12):2683–2696. doi: 10.1242/jcs.141820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Techer H, Baldi G, Costanzo V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591(8):1083–1100. doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- Kreienkamp R, Croke M, Neumann MA, Bedia-Diaz G, Graziano S, Dusso A, Dorsett D, Carlberg C, Gonzalo S. Vitamin D receptor signaling improves Hutchinson-Gilford progeria syndrome cellular phenotypes. Oncotarget. 2016;7:30018–30031. doi: 10.18632/oncotarget.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp R, Billon C, Bedia-Diaz G, Albert CJ, Toth Z, Butler AA, McBride-Gagyi S, Ford DA, Baldan A, Burris TP, Gonzalo S. Doubled lifespan and patient-like pathologies in progeria mice fed high-fat diet. Aging Cell. 2018;18(1):e12852. doi: 10.1111/acel.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, Dorsett D, Kubben N, Batista LFZ, Gonzalo S. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by Progerin. Cell Rep. 2018;22(8):2006–2015. doi: 10.1016/j.celrep.2018.01.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy V, Khanna R, Parnaik VK. E3 ubiquitin ligase HECW2 targets PCNA and Lamin B1. Biochim Biophys Acta, Mol Cell Res. 2018;1865(8):1088–1104. doi: 10.1016/j.bbamcr.2018.05.008. [DOI] [PubMed] [Google Scholar]
- Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T. Identification of differential protein interactors of Lamin a and progerin. Nucleus. 2010;1(6):513–525. doi: 10.4161/nucl.1.6.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Brimacombe KR, Donegan M, Li Z, Misteli T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2016;96:46–58. doi: 10.1016/j.ymeth.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH, Misteli T. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016;165(6):1361–1374. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulling PM, Olson KC, Olson TL, Hamele CE, Carter KN, Feith DJ, Loughran TP., Jr Calcitriol-mediated reduction in IFN-gamma output in T cell large granular lymphocytic leukemia requires vitamin D receptor upregulation. J Steroid Biochem Mol Biol. 2018;177:140–148. doi: 10.1016/j.jsbmb.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BX, Chen J, Chao B, Zheng Y, Xiao X. A Lamin-binding ligand inhibits homologous recombination repair of DNA double-Strand breaks. ACS Cent Sci. 2018;4(9):1201–1210. doi: 10.1021/acscentsci.8b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92(9):761–769. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner M, Ghadge SK, Goetsch V, Wimmer A, Dorler J, Polzl G, Zaruba MM. Upregulation of the aging related LMNA splice variant progerin in dilated cardiomyopathy. PLoS One. 2018;13(4):e0196739. doi: 10.1371/journal.pone.0196739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley AC, Morris G, Phillips AO, Steadman R. 17beta-estradiol ameliorates age-associated loss of fibroblast function by attenuating IFN-gamma/STAT1-dependent miR-7 upregulation. Aging Cell. 2016;15(3):531–541. doi: 10.1111/acel.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Spann TP, Herrmann H, Goldman RD. Disruption of nuclear Lamin organization blocks the elongation phase of DNA replication. J Cell Biol. 2000;149(6):1179–1192. doi: 10.1083/jcb.149.6.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83(7):1299–1309. doi: 10.1002/jnr.20826. [DOI] [PubMed] [Google Scholar]
- Ng KW, Marshall EA, Bell JC, Lam WL. cGAS-STING and Cancer: dichotomous roles in tumor immunity and development. Trends Immunol. 2018;39(1):44–54. doi: 10.1016/j.it.2017.07.013. [DOI] [PubMed] [Google Scholar]
- Olson KC, Kulling Larkin PM, Signorelli R, Hamele CE, Olson TL, Conaway MR, Feith DJ, Loughran TP., Jr Vitamin D pathway activation selectively deactivates signal transducer and activator of transcription (STAT) proteins and inflammatory cytokine production in natural killer leukemic large granular lymphocytes. Cytokine. 2018;111:551–562. doi: 10.1016/j.cyto.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, Depetris D, de Carlos F, Cobo J, Andres V, De Sandre-Giovannoli A, Freije JM, Levy N, Lopez-Otin C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3(106):106ra107. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI, Salto-Tellez M, McQuaid S, Harte MT, Mullan PB, Harkin DP, Kennedy RD. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast Cancer. J Natl Cancer Inst. 2017;109(1):djw199. doi: 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini C, Columbaro M, Capanni C, D'Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6(30):29914–29928. doi: 10.18632/oncotarget.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira S, Bourgeois P, Navarro C, Esteves-Vieira V, Cau P, De Sandre-Giovannoli A, Levy N. HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Dev. 2008;129(7–8):449–459. doi: 10.1016/j.mad.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Prakash A, Gordon LB, Kleinman ME, Gurary EB, Massaro J, D'Agostino R, Sr, Kieran MW, Gerhard-Herman M, Smoot L. Cardiac abnormalities in patients with Hutchinson-Gilford progeria syndrome. JAMA Cardiol. 2018;3(4):326–334. doi: 10.1001/jamacardio.2017.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013;12(4):533–543. doi: 10.1111/acel.12070. [DOI] [PubMed] [Google Scholar]
- Roy S, Luzwick JW, Schlacher K. SIRF: quantitative in situ analysis of protein interactions at DNA replication forks. J Cell Biol. 2018;217(4):1521–1536. doi: 10.1083/jcb.201709121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber KH, Kennedy BK. When Lamins go bad: nuclear structure and disease. Cell. 2013;152(6):1365–1375. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker DK, Solimando L, Sengupta K, Shimi T, Adam SA, Grunwald A, Strelkov SV, Aebi U, Cardoso MC, Goldman RD. The highly conserved nuclear Lamin Ig-fold binds to PCNA: its role in DNA replication. J Cell Biol. 2008;181(2):269–280. doi: 10.1083/jcb.200708155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Hunt CR, Pandita RK, Kumar R, Yang CR, Horikoshi N, Bachoo R, Serag S, Story MD, Shay JW, Powell SN, Gupta A, Jeffery J, Pandita S, Chen BP, Deckbar D, Lobrich M, Yang Q, Khanna KK, Worman HJ, Pandita TK. Lamin a/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol Cell Biol. 2013;33(6):1210–1222. doi: 10.1128/MCB.01676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25(12):1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann TP, Moir RD, Goldman AE, Stick R, Goldman RD. Disruption of nuclear Lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J Cell Biol. 1997;136(6):1201–1212. doi: 10.1083/jcb.136.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE. Nuclear Lamin-a scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, Bluyssen HA. Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget. 2016;7(30):48788–48812. doi: 10.18632/oncotarget.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hilton B, Musich PR, Fang DZ, Zou Y. Replication factor C1, the large subunit of replication factor C, is proteolytically truncated in Hutchinson-Gilford progeria syndrome. Aging Cell. 2012;11(2):363–365. doi: 10.1111/j.1474-9726.2011.00779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torvaldson E, Kochin V, Eriksson JE. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus. 2015;6(3):166–171. doi: 10.1080/19491034.2015.1017167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K, Dubrovsky-Gaupp A, Sapra KT, Goldman RD, Medalia O. The molecular architecture of lamins in somatic cells. Nature. 2017;543(7644):261–264. doi: 10.1038/nature21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol. 2015;132:249–264. doi: 10.1016/B978-0-444-62702-5.00018-4. [DOI] [PubMed] [Google Scholar]
- Vaara M, Itkonen H, Hillukkala T, Liu Z, Nasheuer HP, Schaarschmidt D, Pospiech H, Syvaoja JE. Segregation of replicative DNA polymerases during S phase: DNA polymerase epsilon, but not DNA polymerases alpha/delta, are associated with lamins throughout S phase in human cells. J Biol Chem. 2012;287(40):33327–33338. doi: 10.1074/jbc.M112.357996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145(4):401–417. doi: 10.1007/s00418-016-1411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindigni A, Lopes M. Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys Chem. 2017;225:3–9. doi: 10.1016/j.bpc.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li H, Xie H, Fu M, Guo B, Ding Y, Li W, Yu H. 25-Hydroxyvitamin D3 attenuates experimental periodontitis through downregulation of TLR4 and JAK1/STAT3 signaling in diabetic mice. J Steroid Biochem Mol Biol. 2013;135:43–50. doi: 10.1016/j.jsbmb.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114(7):1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jiang J, He L, Gong G, Wu X. Effect of Lamin-a expression on migration and nuclear stability of ovarian cancer cells. Gynecol Oncol. 2019;152(1):166–176. doi: 10.1016/j.ygyno.2018.10.030. [DOI] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton K, Campuzano D, Ma W, Sheinis M, Ho B, Brown GW, Benchimol S. Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford progeria syndrome. Mol Cell Biol. 2017;37(14):e00659–e00616. doi: 10.1128/MCB.00659-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, Konig N, Kurth T, Wieczorek D, Kast K, Cardoso MC, Gunther C, Lee-Kirsch MA. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun. 2016;7:11752. doi: 10.1038/ncomms11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CY, Leung PS, Adamopoulos IE, Gershwin ME. The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2013;45(2):217–226. doi: 10.1007/s12016-013-8361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci: U S A; 2017. cGAS is essential for cellular senescence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, Lengner CJ, Greenberg RA, Johnson FB, Fuchs SY. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11(5):785–797. doi: 10.1016/j.celrep.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]