

Summary
Centromeric nucleosomes are at the interface of the chromosome and the kinetochore that connects to spindle microtubules in mitosis. The core centromeric nucleosome complex (CCNC) harbors the histone H3 variant, CENP-A and its binding proteins, CENP-C (through its central domain; CD) and CENP-N (through its N-terminal domain; NT). CENP-C can engage nucleosomes through two domains: the CD and the CENP-C motif (CM). CENP-CCD is part of the CCNC by virtue of its high specificity for CENP-A nucleosomes and ability to stabilize CENP-A at the centromere. CENP-CCM is thought to engage a neighboring nucleosome, either one containing conventional H3 or CENP-A, and a crystal structure of a nucleosome complex containing two copies of CENP-CCM was reported. Recent structures containing a single copy of CENP-NNT bound to the CENP-A nucleosome in the absence of CENP-C were reported. Here, we find that one copy of CENP-N is lost for every two copies of CENP-C on centromeric chromatin just prior to kinetochore formation. We present the structures of symmetric and asymmetric forms of the CCNC that vary in CENP-N stoichiometry. Our structures explain how the central domain of CENP-C achieves its high specificity for CENP-A nucleosomes and how CENP-C and CENP-N sandwich the histone H4 tail. The natural centromeric DNA path in our structures corresponds to symmetric surfaces for CCNC assembly, deviating from what is observed in prior structures using artificial sequences. At mitosis, we propose that CCNC asymmetry accommodates its asymmetric connections at the chromosome/kinetochore interface.
Graphical Abstract
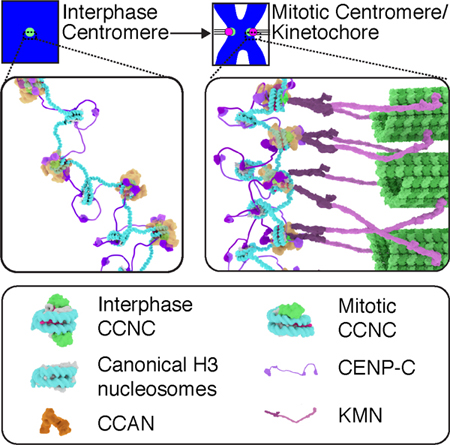
Allu et al. report the cryo-EM structures of the core centromeric nucleosome complex (CCNC). The structures reveal a distinct path of centromeric DNA that corresponds to robust CCNC assembly, and together with findings in cells, lead to a model for a CCNC structural transition at the onset of mitosis, immediately preceding kinetochore assembly.
Introduction
The faithful inheritance of the genome at cell division relies on specific connections of chromosomes to the microtubule-based spindle [1]. The connection point on the chromosome is the centromere, a locus that is not defined by a particular DNA sequence but rather by a specific form of chromatin. Chromatin with the CENP-A nucleosome at its foundation epigenetically specifies the location of the centromere throughout the cell cycle serving as the site for kinetochore formation at cell division [1,2]. The kinetochore, in turn, physically connects to spindle microtubules.
The relationship between DNA sequence and the function of the centromere is under heavy investigation. This is because of the paradoxical observations that the sequences typically found at centromeres (e.g. α-satellite DNA in humans and minor satellite DNA in mice) are dispensable for basic centromere function [3–7] but nonetheless play a role in formation of centromeres on naked DNA in human artificial chromosomes [8,9,10], critical when other centromere defects are imposed [11,12], and can strengthen centromeres in divergent strains of mouse when in competition in female meiosis I [13]. How DNA sequence impacts centromeric chromatin remains largely unanswered.
We recently reconstituted the core centromeric nucleosome complex (CCNC) consisting of the octameric CENP-A nucleosome wrapped with 147 bp of human α-satellite DNA bound by two copies each of the non-histone centromere proteins CENP-C (central domain [CD]; a.a. 426–537) and CENP-N (N-terminal domain [NT]; a.a. 1–240) [14]. The precise nucleosomal position of the CCNC within the monomer unit of repetitive α-satellite DNA matches the favored site at natural centromeres [5] and the preferred site of assembly in reconstituted CENP-A nucleosomes [15]. CENP-C and CENP-N represent the two subunits of the constitutive centromere-associated network (CCAN) of sixteen proteins that directly recognize the CENP-A nucleosome via binding sites on its surface [16–21].
In diverse eukaryotes, including mammals, multiple copies of the CCNC coalesce to form a discrete compartment on the surface of centromeric chromatin in mitosis that serves as the interface between the chromosome and the proteinaceous kinetochore [1]. CENP-C is thought to have an elongated structure that contains numerous binding modules for centromere and kinetochore components, while CENP-N is coupled to its partner CENP-L through its C-terminus and, in that context, recruits the ‘HIKM’ sub-complex of the CCAN [22,23]. Despite the centrality of connecting chromosomes to the microtubule-based spindle, how the CCNC organizes kinetochore formation remains unclear.
In this study, we find that the stoichiometry of the CCNC changes at the moment of mitotic onset, prior to kinetochore formation, and we report the cryo-EM structures of distinct forms of the CCNC: one harboring two copies each of CENP-C and CENP-N and one harboring two copies of CENP-C and one copy of CENP-N. These structures provide key insight into the fundamental unit of centromeric chromatin that guides chromosomal inheritance. Further, the natural DNA sequence used in our studies revealed a distinct path of nucleosomal DNA at the centromere. Indeed, the natural DNA sequence allows for robust CCNC assembly, compared to an artificial sequence used widely in nucleosome studies. Placing the CCNC structures in the context of the chromosome, we propose a model wherein CCNC symmetry in interphase confers stable centromere transmission, while asymmetry generated at the moment of mitotic onset orients connections to neighboring chromatin and the kinetochore on opposite CCNC surfaces.
Results
One copy of CENP-N is lost for every two copies of CENP-C upon mitotic entry
While the reconstituted CCNC [14]—and even higher-order complexes capable of binding to microtubules [22]—harbor two copies each of CENP-C and CENP-N bound to the CENP-A nucleosome, there is cell biological evidence that CENP-N levels at centromeres vary during the cell cycle [24,25]. Indeed, in one proposal, CENP-A nucleosome binding through CENP-NNT is somewhat destabilized in mitosis, while CENP-C binding through CENP-NCT and its binding partner CENP-L is stabilized [23]. In this light, we considered the possibility that CCNC stoichiometry could change as part of a particularly early step in kinetochore formation at the onset of mitosis. The onset of mitosis occurs over 5–10 minutes, and the centromere undergoes rapid, dramatic alterations during this window of time [1]. First, just prior to nuclear envelope breakdown (NEBD), sister centromeres separate from one resolvable locus to two loci separated by ~500 nm. Second, chromosomes condense as prophase initiates, coinciding with NEBD. Third, kinetochores are assembled capable of recruiting mitotic checkpoint components (e.g. MAD2) and the components that physically connect centromeres to spindle microtubules (e.g. the KMN network). Since CENP-A nucleosome levels are quantitatively retained throughout the cell cycle, including mitosis [26], we focused on the other subunits, CENP-C and CENP-N. We employed a human DLD-1 cell line [23] in which both alleles of the endogenous CENP-N gene have been edited with a tag including EGFP (Figure 1A). In this manner, we can focus on endogenous levels of CENP-N without necessitating antibody reagents that have been notoriously unreliable for this particular centromere component. In comparing a population of cells arrested in G2 just prior to mitotic onset to ones that have entered mitosis and progressed to prometaphase or metaphase, we found that CENP-N levels at centromeres drop to about one fourth their original levels per resolvable centromere locus (Figure 1B, C). This is in contrast to the expectation of a drop to one half the original level due to separation of sister centromeres. CENP-C, however, matches this expectation of dropping to about one half the original level per resolvable centromere locus (Figure 1B, D). Moreover, unlike CENP-C, the ratio of CENP-N to CENP-A nucleosomes drops in mitosis (Figure S1). Like CENP-N, its binding partner within the CCAN, CENP-L, drops moderately in abundance at centromeres in mitotic cells (Figure S1F).
Figure 1. CENP-C is quantitatively retained at centromeres upon mitotic entry, but CENP-N levels drop by half prior to kinetochore formation.
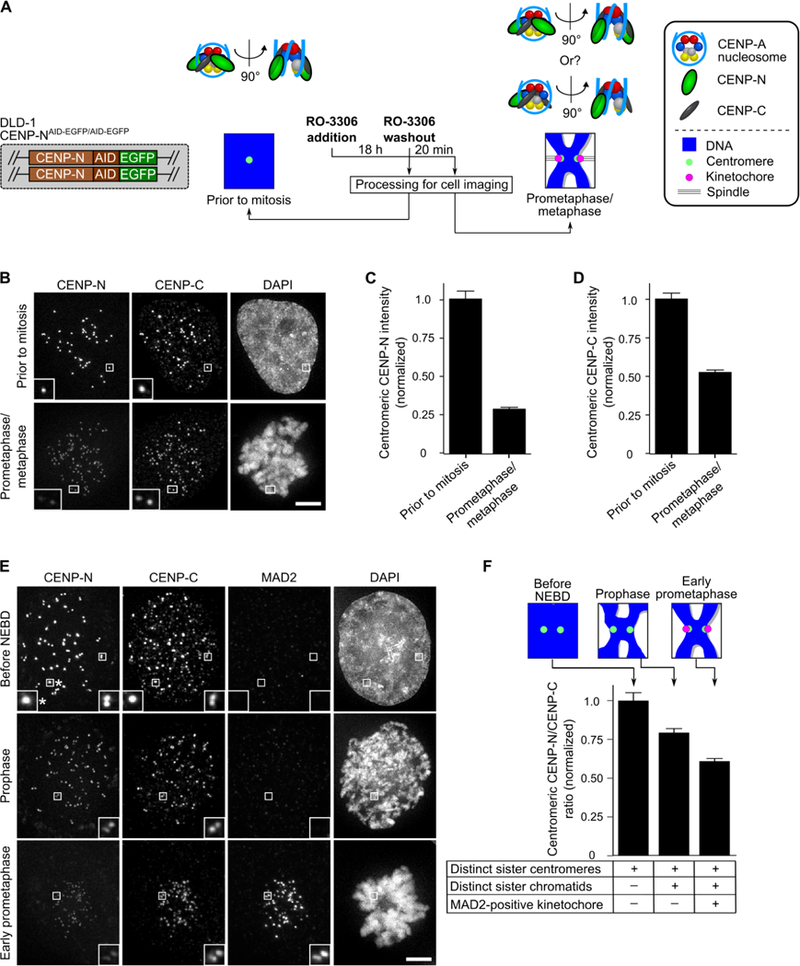
(A) Schematic representation for measuring CCNC components upon mitotic entry. (B) Representative images of cells treated as in panel A. Scale bar, 5 µm. (C,D) Quantification of experiment in panel B. Centromeric intensity of CENP-N (C) or CENP-C (D) were normalized to the values measured prior to mitosis and reported as the mean ± 95% confidence interval (n = 418 or 609 centromeres for the prior to mitosis and prometaphase/metaphase conditions, respectively). (See also Figure S1). (E) Representative images of cells imaged at various stages in the moments surrounding mitotic onset. Scale bar, 5 µm. Note that some, but not all, sister centromeres have separated to the point of being distinct pairs in the cells just prior to NEBD. The asterisk denotes an inset of sister centromeres not yet resolved into a pair. (F) Quantification of panel E, expressed as the mean ratio (± 95% confidence interval [n = 601, 889, or 1009 centromeres for the before NEBD, prophase, and early prometaphase conditions, respectively]) of centromeric CENP-N/centromeric CENP-C normalized to the measurement in cells prior to NEBD.
To determine if this change in CCNC stoichiometry occurs prior to formation of a functional kinetochore, we used sister centromere separation, chromosome condensation, and MAD2 recruitment to centromeres to stage each cell and thereby measure CENP-N/CENP-C ratios at the rapid steps leading into mitosis. By early prophase, before any functional kinetochores form (i.e. no detectable MAD2), CENP-N levels have dropped substantially relative to CENP-C (Figure 1E, F). By early prometaphase, when high levels of MAD2 are present at kinetochores because they have not yet formed stable interactions with spindle microtubules, the relative levels of CENP-N have dropped to nearly half relative to CENP-C (Figure 1E, F).
Structure of the CCNC
In performing multiple biochemical steps after its reconstitution to isolate a pure CCNC preparation at a concentration appropriate for cryo-EM studies, we found it existing as a mixture of nucleosome complexes that each contain two copies of CENP-CCD but with either one or two copies of CENP-NNT (Figure S2). Taking into account this known source of heterogeneity in our sample, we first employed a phase-plate [27,28] and collected small datasets from samples prepared under various conditions (see STAR Methods), one of which yielded ~200,000 particles sorted into 2D class averages, including several of which that clearly showed well-defined features of a nucleosome bound by either one or two copies of CENP-NNT (Figure S2 and Table S1). We then acquired many more micrographs from the optimized sample using accelerated data collection procedures (i.e. without the phase-plate), for a combined yield of >1.3 million particles successfully sorted into 2D class averages (Figure S3A). We undertook multiple rounds of 3D classification without imposing symmetry. After removing ~60% of particles that did not show well-ordered structure, the second round of 3D classification clearly revealed the two forms of the CCNC: one containing two copies each of CENP-CCD and CENP-NNT and the other containing two copies of CENP-CCD and one copy of CENP-NNT, accounting for 14% and 22% of the particles, respectively (Figure S3A). The latter was further classified into three maps, which chiefly differ from each other at the nucleosomal terminal DNA regions (Figure S3A). Thus, the two forms of the CCNC are present in our preparations both before (Figure S2A) and after final grid preparation, including the step of gradient separation/fixation (GraFix; see STAR Methods).
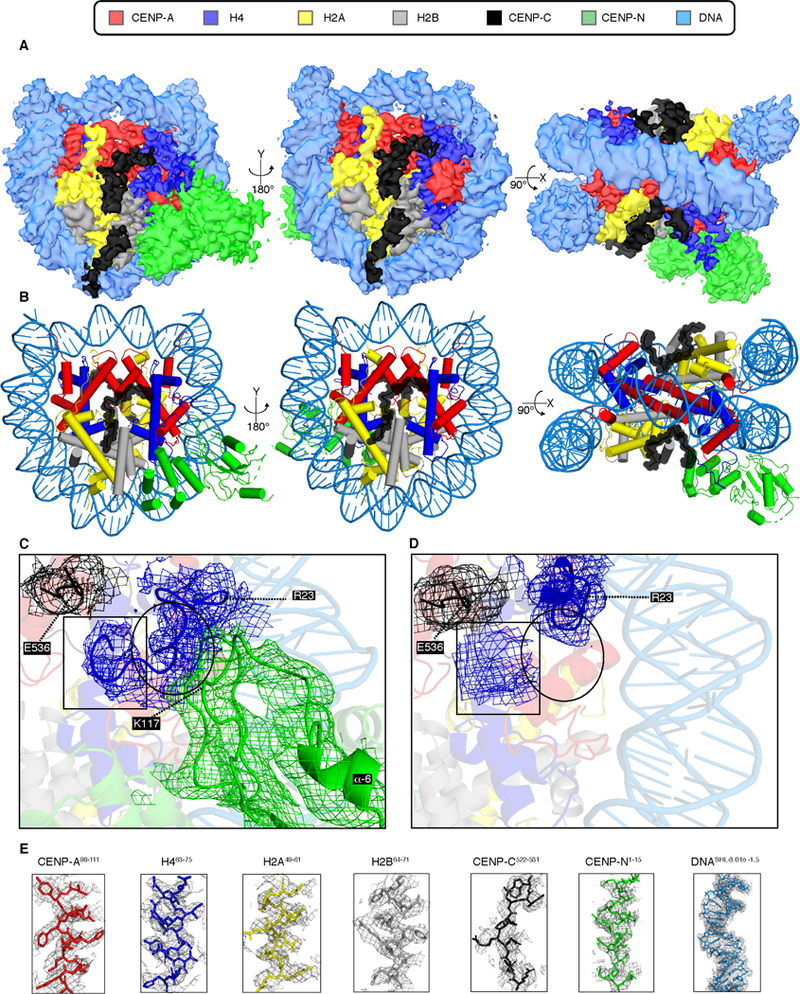
We first focused our attention on the most populated classes after three rounds of 3D classifications: those that contain the CCNC with two copies each of CENP-CCD and CENP-NNT. We obtained a reconstruction of it with an overall resolution of 3.5 Å, allowing us to preliminarily assign distinct regions of the map to each of its subunits (Figures 2A and S3B–E and Table S2). Coincident with direct interpretation of this map into an atomic model, we undertook studies utilizing crosslinking coupled to mass spectrometry (XL-MS). Using this approach, we identified many inter-subunit crosslinks involving each of the six protein subunits (Figures 2B and S4A–D and Table S3). Further, the calculated distances of our XL-MS data nicely fit the final atomic model (Figure 2C, D). Our atomic model shows the octameric CENP-A-containing histone core wrapped ~1.6 times by α-satellite DNA and bound symmetrically by two copies each of CENP-CCD and CENP-NNT (Figure 2E). Since the symmetric binding sites for CENP-CCD and CENP-NNT are both occupied, we conclude that this is the fully loaded, constitutive form of the CCNC present at interphase centromeres.
Figure 2. Structure of the centromeric nucleosome bound by two copies each of CENP-C and CENP-N.
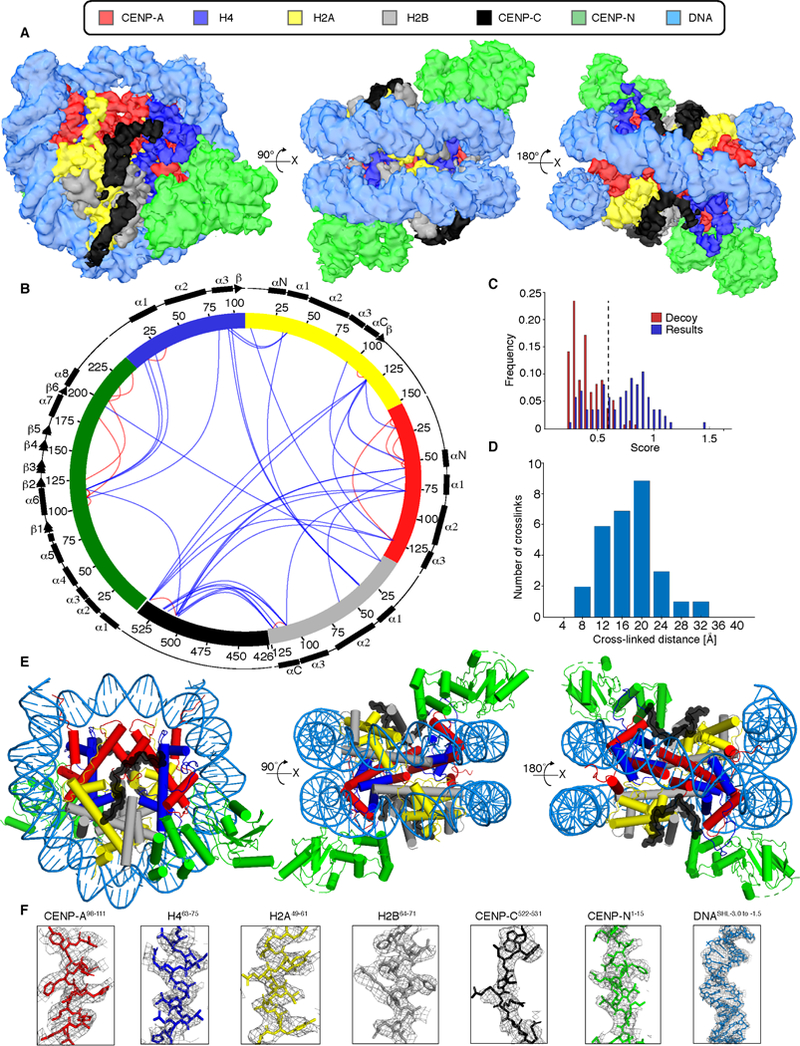
(A) Cryo-EM density map of the CCNC with colors assigned to regions of density corresponding to the DNA and each protein subunit (See also Figures S2, S3 and Tables S1, S2). (B) Crosslinking map of the CCNC with intramolecular (red) and intermolecular (blue) crosslinks between the indicated sites (See also Figure S4 and Table S3). (C) Only crosslinks with scores above 0.6 (dashed line) are reported. This threshold was determined based on decoy analysis (see STAR Methods) and set a false detection rate of ~2%. (D) Histogram of the Cα-Cα distances between crosslinked residues that could be mapped onto our atomic model. This is a very typical histogram for the BS3 crosslinker, suggesting a good fit between the XL-MS data and the model. (E) Structural model of the CCNC. (F) Model building and density features of CCNC bound with two copies of CENP-CCD and CENP-NNT. Representative regions of the EM density map to illustrate map quality and model fitting with CCNC components into the EM density with color assigned to each subunit.
Each histone-exposed face of the nucleosome is bound by a single copy of CENP-CCD and CENP-NNT. CENP-CCD is precisely positioned on the nucleosome surface, existing as a discontinuous polypeptide stretch lacking any secondary structure (Figure 2E, F). CENP-NNT exists as a folded, globular domain positioned at an adjacent site on the nucleosome with contacts to loop L1 of CENP-A and the adjacent nucleosomal DNA at sites 2 and 3 helical turns away from the nucleosomal dyad; a position consistent with previous reports of CENP-NNT bound to CENP-A nucleosomes in the absence of CENP-C [14,18,21]. Indeed, the CENP-NNT chains in the CCNC or when bound as a single copy to a CENP-A nucleosome (PDB# 6C0W) [21] align with an overall RMSD of 2.1 Å. CENP-CCD and CENP-NNT do not have an obvious site of direct contact with one another within the CCNC, thus inviting a closer examination of how they individually and collaboratively interact with CENP-A nucleosomes.
Contact points on the histone surface of the CENP-A nucleosome
The nucleosome contact surface of CENP-CCD (Figure 3A) includes a structured span from CENP-Ca.a.519−537 that was studied by prior solution-based biophysical approaches, nucleosome binding/mutagenesis, and homology modeling to a crystal structure containing a second, related nucleosome binding site, the CENP-C motif (CENP-CCM) [20]. CENP-CCM, unlike CENP-CCD, lacks high specificity for CENP-A nucleosomes relative to their canonical counterparts containing histone H3 [20]. Thus, the actual structure of CENP-CCD is of primary importance to understanding the connection between CENP-C and the CENP-A nucleosome. The region from CENP-Ca.a.519−537 in our cryo-EM map has clear side-chain densities, including key electrostatic interactions between CENP-CR521, R522, and R525 with the acidic patch of H2A (Figures 3A and S4E): CENP-CR521 with H2AE64, CENP-CR522 with H2AE61, D90 and E92, and CENP-CR525 with H2AE91 and H2BE105. This region of CENP-C also harbors neighboring hydrophobic residues (W530 and W531) that are packed with two hydrophobic residues on the C-terminal tail of CENP-A (L135 and L139), both absent in H3 and strongly implicated in its specific recognition of CENP-A [17,20], as well as the hydrophobic portion of the side-chain of CENP-AR131 (Figure 3A). In addition, there is another electrostatic interaction between CENP-CK534 and H4E52 (Figure 3A). Thus, remarkably, within a single stretch of 14 a.a. (CENP-Ca.a.521−534), CENP-C forms bonds with all four subunits of the histone surface of the nucleosome (Figures 3A and S4E–I).
Figure 3. Centromeric nucleosome interactions with non-histones, CENP-C and CENP-N.
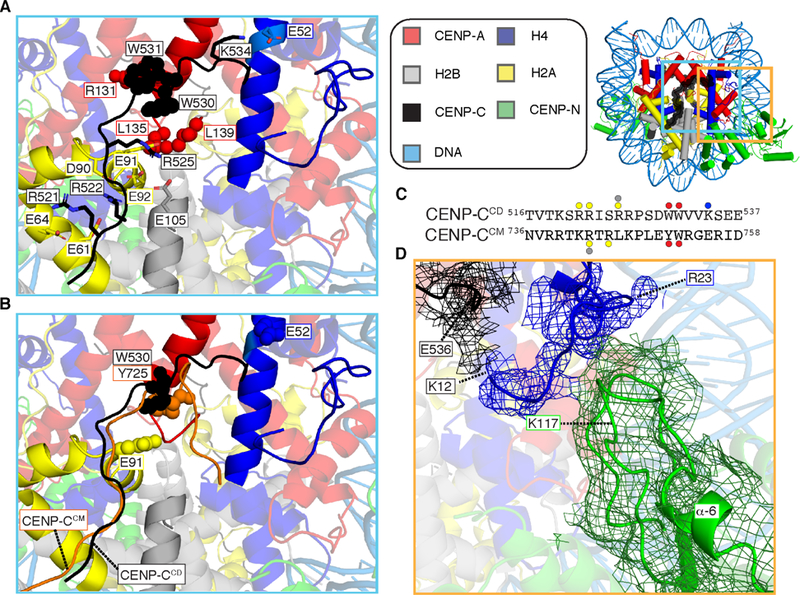
(A) CENP-Ca.a518−537 bonds with all four histone subunits of the centromeric nucleosome involve extensive electrostatic (sticks) and hydrophobic (spheres) interactions (See also Figure S4E) (B) CENP-CCM (orange) from PDB# 4X23 is aligned with the CCNC structure. The additional electrostatic interactions made by CENP-CCD with sites on the histone octamer (H2AE91 and H4E52) are shown in space fill, as are CENP-CW530 and CENP-CY725 at the site of contact with CENP-A or histone H3 C-termini, in CENP-CCD and CENP-CCM, respectively. (C) Protein sequence alignment of human CENP-CCD and CENP-CCM. The sites of contact with histone components are highlighted with circles color coded to match the histone subunits. (D) Cryo-EM density for the histone tail of H4 (mesh overlaying ribbon model) extends to H4K12, with contacts between H4a.a.12−20 with the C-terminal portion of CENP-CCD on one side and two nearby loops of CENP-NNT on the other. Density here was assigned to the H4 tail prior to B-factor correction, and H4R23 is labeled because its side-chain density in our map provided a landmark with which to orient main chain density N-terminal to it. Local refinement (see EMDB #9252 and see also Figure S5A) yielded the shown density map.
In comparison to CENP-CCM (Figure 3B), the N-terminal 12 a.a. of CENP-CCD follow a similar path across the nucleosome surface and share two electrostatic and two hydrophobic interaction sites (Figure 3B, C). The interaction between CENP-CR525 and H2AE91 is unique to CENP-CCD, and the more hydrophobic CENP-CW530 relative to CENP-CY725 is packed against the CENP-A-specific hydrophobic C-terminal leucines (Figure 3A–C). The C-terminal 7 a.a. of CENP-CCD strongly deviate from the path of CENP-CCM allowing the additional electrostatic contact with H4 (CENP-CK534 with H4E52), while CENP-CCM has no contact with H4 (Figure 3A–C). Indeed, we note that K534 is conserved in the CENP-CCD in diverse mammals [20], while the corresponding position in CENP-CCM has switched charge (E755). Thus, CENP-CCD has additional contacts that contribute to the stability and specificity of binding to the CENP-A nucleosome, explaining why it was initially identified as the CENP-A recognition domain [17] and has higher specificity for CENP-A nucleosomes relative to CENP-CCM [20].
While CENP-CCD and CENP-NNT do not have direct contact points with each other within the CCNC, we noted that clear density corresponding to the N-terminal tail of histone H4 was sandwiched between the C-terminal region of CENP-CCD and two adjacent loops extending from the globular CENP-NNT domain (Figure 3D). Indeed, we can assign the continuous path of the H4 tail from where it emerges from its histone fold domain all the way N-terminal to H4K12 (Figure 3D), in contrast to earlier structures of canonical nucleosomes [29] or centromeric nucleosomes [18,21,30] where less of the tail apparently populates a specific position as it emerges from the core of the nucleosome. CENP-CCD is nearest to the region near H4a.a.12−13 while CENP-NNT is nearest to H4a.a.18−22 (Figure 3D). Focused image classification of density corresponding to CENP-NNT and the H4 tail, performed by subtracting the remaining density of the CCNC, revealed the loop following the α6-helix of CENP-N as the point of contact with H4a.a.18−22 (Figures 3D and S5A–C). Notably, the orientation and length of this loop—longer at the cost of shortening the α6-helix—is one of only two clear deviations in CENP-NNT within the CCNC (Figure 3) relative to when it is bound to a CENP-A nucleosome in the absence of CENP-CCD [18,21] [the other deviation is a 2 Å shift of the α8 helix (Figure S5)]. This explains why this region of CENP-NNT is the only region we observed to undergo changes in backbone amide hydrogen exchange during CCNC assembly, where faster exchange—consistent with α6-helix distortions/unfolding—only occurs in the presence of CENP-CCD [14]. Strikingly, CENP-NK117 on a loop following the α6-helix that contacts histone H4 is the sole residue forming crosslinks with H4K8, K12, and K20 (Figures 2B and S5D). Together, CENP-CCD and CENP-NNT contacts guide the tail of H4 on a path that overrides the tendency in isolated CENP-A nucleosomes for it to exit the nucleosome in another direction altogether [31]. Removal of the H4 tail does not negatively impact CCNC assembly (Figure S5E–G), so the binding surface it provides does not appear to be key to nucleosome association of either subunit. Conversely, CENP-CCD and CENP-NNT drive the specific orientation of the tail of histone H4 along the surface of the nucleosome.
Centromeric DNA accommodates robust CCNC formation
Structural studies of canonical and centromeric nucleosome core particles and higher order assemblies have used a variety of DNA sequences, typically in the 145–147 bp range (Figure 4A). The CCNC DNA in our cryo-EM studies consists of 147 bp of cloned natural α-satellite [32] representing the primary position where CENP-A nucleosomes assemble at functional human centromeres, and an internal 145 bp sequence [15] was very recently used for structural analysis of CENP-A-containing nucleosomes [33]. Earlier crystallographic studies largely employed palindromic sequences, including those where the half site corresponds to cloned human α-satellite DNA (Figure 4A). The artifical, so-called ‘Widom 601’ nucleosome positioning sequence [34] has become the most common sequence used for structural studies, especially in recent cryo-EM studies, including of centromeric nucleosomes [18,21]. Comparison of the DNA path in the CCNC to that of other nucleosome structures containing CENP-A, as well as the classic crystal structure of the conventional nucleosome containing canonical histone H3, reveals a distinctive bulge four turns of DNA from the dyad axis that locally widens superhelical pitch by 3–4 Å (Figures 4B, C and S6A). This moves two regions of DNA at +35 and −35 bp relative to the dyad closer to CENP-N molecules by ~1.5–2.0 Å, compared to canonical nucleosome structures. Indeed, the superhelical pitch with the CCNC is an outlier compared to a broad survey of available high-resolution nucleosome structures [35]. Two of the structures (PDB# 6C0W and 6BUZ) [18,21] we assessed alongside the CCNC use the artificially super strong nucleosome positioning Widom 601 sequence. Two others use palindromic human α-satellite-derived sequences (PDB# 1AOI and 3AN2) [29,30]. Notably, the CENP-A nucleosome in isolation on natural α-satellite DNA has a small but clear deviation from typical nucleosome structures at the same position (Figure 4C; PDB# 6O1D), and binding of an antibody specific for nucleosomes to the surface essentially eliminates any bulge (Figure 4C; PDB# 6E0P) [33]. Thus, it appears that deviation in the path of DNA at this location is sensitive to the nature of the proteins that bind and assemble on the CENP-A nucleosome. The sequence-specific change to shape, most prominent in our structure (Figure 4A, B) is a strong indicator of the impact of sequence on nucleosomal structures. This likely relates to how DNA sequence impacts local and global changes to histone contacts [34], affecting DNA flexibility and contributing to important dynamic features in chromatin [36].
Figure 4. DNA sequence contributes to the structure and faithful assembly of the CCNC.
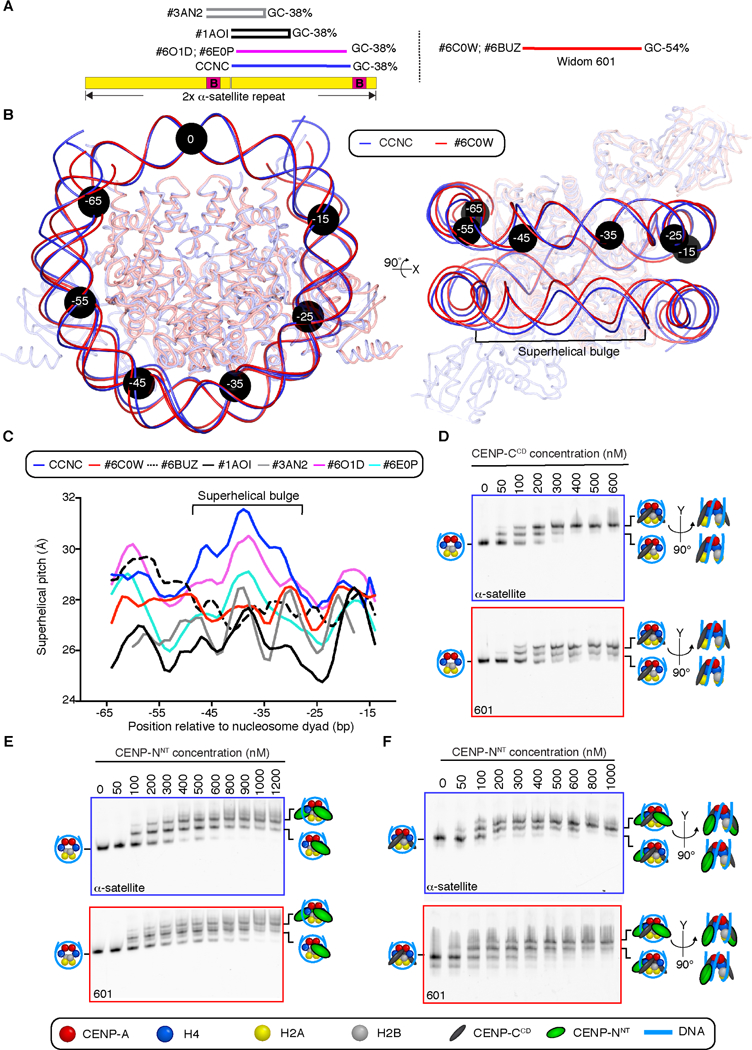
(A) Position of on a tandem copy of the 171 bp human α-satellite DNA sequence of palindromic sequences used in prior nucleosome structure studies [29,30], the natural sequence used in this study of the CCNC, and the unnatural “Widom 601” sequence that has emerged as the most common sequence for nucleosome structural studies. (B) Alignment using one CENP-A and histone H4 dimer of CCNC structure with PDB# 6C0W (CENP-A nucleosome wrapped with 601 DNA and bound by one copy of CENP-NNT; [21]. Black circles denote the indicated number of bp from the nucleosome dyad. Note that the DNA used for both structural studies is 147 bp of DNA, although the terminal 4 bp are not resolved in PDB# 6C0W (see also Figure S6). (C) Local superhelical pitch measurements comparing the interphase CCNC with the indicated related structures in the PDB. (D) Binding of CENP-CCD to CENP-A nucleosomes wrapped with either α-satellite or 601 DNA. (E) Binding of CENP-NNT to CENP-A nucleosomes wrapped with either α-satellite or 601 DNA. (F) Binding of CENP-NNT to CENP-A nucleosomes wrapped with either α-satellite or 601 DNA and bound by CENP-CCD. Binding assays in D-F are representative example of three independent experiments.
It is likely that the capacity to form the observed overall DNA 3–4 Å bulge locally accommodates the ~1.5 Å DNA shift favorable for CENP-NNT binding, enabling two CENP-N molecules to equivalently interact with both surfaces of the nucleosome. Consistent with this interpretation, although the symmetry was not imposed during data analysis, the CCNC exhibited a more symmetric nucleosome structure than a CENP-A nucleosome assembled with the 601 DNA and bound by one copy of CENP-NNT (PDB# 6C0W) [21]. Indeed, the CCNC harbors two nearly identical CENP-N binding sites on both surfaces of the nucleosome, whereas the 601 DNA-containing nucleosome exhibits an asymmetric DNA path on the second potential CENP-NNT-binding surface opposite the occupied site (Figure S6B, C). In particular, the asymmetric DNA path in the 601 DNA-containing nucleosome is in the region where the 3–4 Å DNA bulge is formed in the CCNC assembled with α-satellite DNA. We note that in one study [18], a low resolution (~11 Å) EM density map was obtained of a small population of 601 DNA-containing nucleosomes bound by two copies of CENP-NNT when an excess of it was introduced, but high-resolution maps were only obtained with a single copy of CENP-NNT bound [18,21]. We conclude that the unequal second binding site on 601 DNA-containing nucleosomes is likely to be unfavorable for binding the second copy of CENP-NNT.
To test this notion, we performed side-by-side comparisons of CCNC assembly steps on the natural, α-satellite sequence and the commonly used 601 artificial sequence. The first step of CCNC assembly in our approach is to add two copies of CENP-CCD. Strikingly, CENP-CCD is assembled on both faces of the nucleosome at lower concentrations when CENP-A nucleosomes are wrapped with α-satellite DNA, compared to 601 DNA (Figure 4D). It is possible, though, at high concentrations of CENP-CCD, to form populations of nucleosome complexes with a similar degree of saturation of CENP-CCD binding sites (i.e. 2 copies per nucleosome; Figure 4D). Two copies of CENP-NNT are readily assembled on CENP-A nucleosomes assembled on either type of sequence in the absence of CENP-CCD (Figure 4E), but CENP-NNT-bound CENP-A nucleosomes assembled on α-satellite DNA reliably yield uniform migration on native-PAGE gels, relative to the more heterogeneously migrating ones when 601 DNA is used (Figure 4E). Indeed, when CCNC formation is completed by the addition of CENP-NNT to CENP-A nucleosomes bound by CENP-CCD, α-satellite DNA-containing complexes have a relatively uniform migration behavior, indicating a stable conformation (Figure 4F). We further performed a densitometric analysis of the band representing the full CCNC to measure the extent to which α-satellite DNA-containing nucleosomes robustly assemble a discrete CCNC relative to those assembled with 601 DNA. The height of the intensity profile taken from a line scan intersecting the band was substantially higher for α-satellite than for 601 (2.1 +/− 0.6 vs. 1.1 +/− 0.1, respectively; arbitrary units normalized for area +/− s.d., n = 3) while the 601 profile was broader (3.4 +/− 0.3 vs. 4.4 +/− 0.1, for α-satellite and 601, respectively; arbitrary units normalized for area +/− s.d., n = 3). These measurements indicate that the CCNC complex assembled on α-satellite migrates as a more uniform species than the complex formed on 601. We reason that the CCNC components impact the path of the DNA, including the changes known to be imparted by CENP-CCD binding [15,37], and that the rigid 601 sequence (54% GC and several GC dinucleotide sequences at major groove contacts with the histone octamer known to lock it in place [38]) fails to readily accommodate the changes relative to the natural, flexible α-satellite sequence (38% GC and largely devoid of the GC dinucleotides at histone octamer contact points). We further reason that shape and physical properties conferred by natural centromere α-satellite DNA with more uniform CCNC assembly contributed to our success in achieving high-resolution cryo-EM density maps of the symmetric form of the CCNC (i.e. with two copies each CENP-CCD and CENP-NNT; Figures 2A, 4F), rather than differences in sample/grid preparation.
The structure of the CCNC harboring only a single copy of CENP-NNT
We next turned our attention to classes of particles containing only a single copy of CENP-NNT. Our 3D classification scheme (Figure S3A) proved to be a successful strategy to obtain a reconstruction of this form of the CCNC at an overall resolution of 3.6 Å (Figures 5A and S7). Our atomic model of the CCNC harboring a single copy of CENP-NNT (Figure 5B) is similar to the form harboring two copies (Figure 2E), even including the 3–4 Å bulge at the +36 to 38 bp position from the nucleosome dyad (Figure S7). One notable difference, though, is the reduced histone H4 tail density on the face lacking CENP-NNT relative to the face containing it (Figure 5C, D). This supports our notion (Figure 3D) that the H4 tail is stabilized by interactions with both CENP-CCD and CENP-NNT.
Figure 5. Structure of an asymmetric form of the CCNC that harbors only a single copy of CENP-NNT.
(A) Cryo-EM density map of this form of the CCNC with colors assigned to regions of density corresponding to the DNA and each subunit (see also Figure S7). (B) Structural model of the CCNC harboring a single copy of CENP-NNT. (C) EM density for the H4 tail (a.a. 9–23). Atomic model was built with Coot without side chains (with alanine at each position). H4 EM density extends to H4G9. H4a.a.9−13 is located near the C-terminus of CENP-CCD (indicated by box), whereas H4a.a.18−22 is in contact with the loop following the α6-helix of CENP-N (indicated by circle). (D) A small H4 tail EM density (indicated by box) near CENP-CCD was observed in the absence of CENP-NNT. The rest of the H4 tail was not observed (indicated by circle). (E) Model building and density features of core centromere nucleosome complex bound with two copies of CENP-CCD and CENP-NNT. Representative regions of the EM density map to illustrate map quality and model fitting with CCNC components into the EM density with color assigned to each subunit.
While the overall structure and resolution is similar in the two major forms of the CCNC (Figures 2A, E, F and 5A, B, E) the local resolution in our two structures substantially diverge at particular locations (Figure 6A). The CCNC with two copies of CENP-NNT has fairly even high-resolution (2.1–4.0 Å) throughout the core of the complex, with somewhat lower resolution in peripheral DNA and CENP-NNT (Figure 6A). In the CCNC with only a single copy of CENP-NNT, however, the core of the histone octamer has an overall higher resolution than that observed in the CCNC that harbors two copies of CENP-NNT, while the terminal DNA (i.e. the final 1.5 turns of DNA) on each side of the nucleosome and the single copy of CENP-NNT has a relatively poor resolution (>7 Å) (Figure 6A). The increased terminal DNA heterogeneity is present in the form of the CCNC harboring a single copy of CENP-NNT even after 3D classification was used to reduce it (Figure S3A). This implies that in this form of the CCNC the histone octamer strongly populates a fixed orientation but that constraints are relieved on CENP-NNT positioning, as well as the orientation of DNA as is enters/exits the nucleosome. Regarding the nucleosome termini, the most prominent path of DNA in both forms of the CCNC is nearly identical (Figure 6B), extending outwards relative to free CENP-A nucleosomes (i.e. in the absence of either CENP-CCD or CENP-NNT; Figure 6B [33]).
Figure 6. Nucleosome terminal DNA in the two forms of the CCNC.
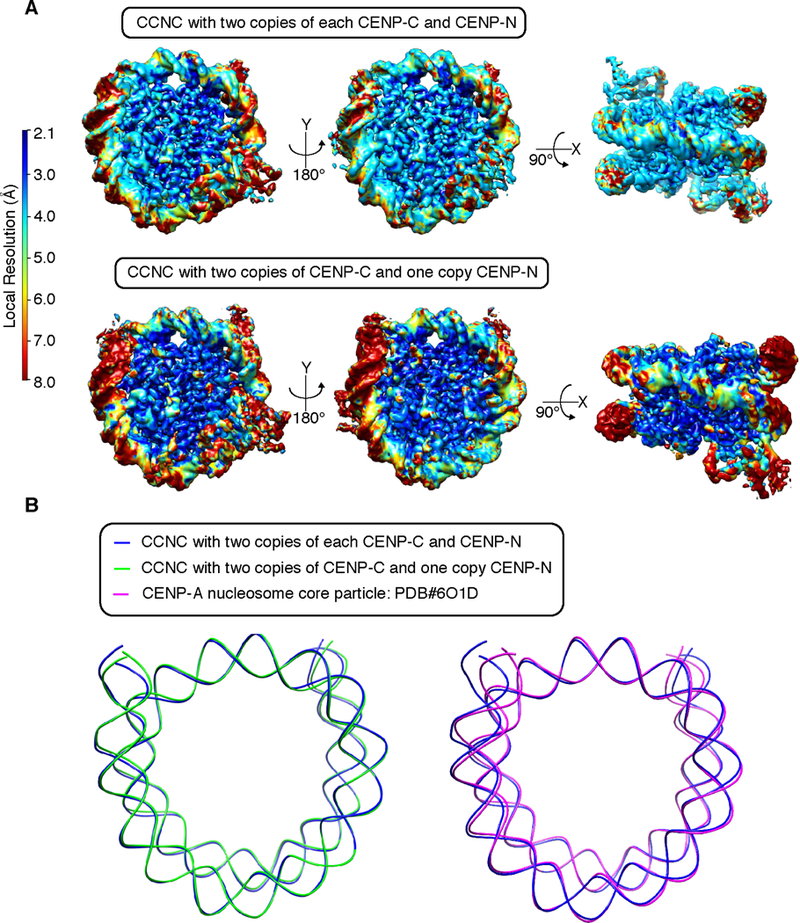
(A) Reconstructions of the two forms of the CCNC colored by local resolution. (B) Alignments of the forms of the CCNC and free CENP-A nucleosome core particles.
Discussion
For CENP-A nucleosomes to function at the centromere, they must faithfully form the complex that marks centromere location and the complex that forms the connection point between bulk chromatin and the kinetochore. We have solved the structures for the complexes that mark centromeres through the cell cycle and that form this vital connection. While it has been obvious since the earliest days of cell biology that the chromosome/spindle interface, itself, generates a stark asymmetry, our findings suggest that this asymmetry goes right down to the fundamental repeating unit of centromeric chromatin.
We took the approach of using the prominent position and sequence of human centromeric nucleosomal DNA [5] because we reasoned it would most accurately recapitulate what is found in nature. We were also acutely aware that it directly positions CENP-A nucleosomes, in vitro, and yields robust formation of CCNC components including the stabilizing features they impart to centromeric chromatin [14,15,23,37,39,40]. The surprising, stable superhelical 3–4 Å bulging, for which we were able to unambiguously trace phosphate residues in each DNA strand, in both forms of the CCNC is accommodated by α-satellite DNA sequence (Figure 4A, B). However, non-natural sequences, like Widom 601, appear to not accommodate such deviations from canonical nucleosome structure. Our structures also yielded clear density of the DNA at the nucleosome termini (i.e. where it enters/exits the nucleosome), with the CCNC harboring two copies of CENP-NNT containing clearer density than the form with only a single copy. Both forms of the CCNC have a slight bend in the terminal DNA away from the histone core relative to canonical nucleosomes, likely due to weaker DNA connections with the CENP-A αN-helix [30,41,42] and contributing to their nuclease sensitive nature at functional centromeres [5,13,43]. The notion that the non-histone components, CENP-C and CENP-N, could contribute to terminal DNA positioning [15,18,21,33] is supported because the second copy of CENP-NNT in the CCNC fixes the position of the terminal DNA in terms of the local resolution that we observed at that location from the same cryo-EM data set as particles lacking the second copy (Figure 6A). A prior crystal structure of the CENP-A nucleosome in isolation did not give clear electron density that could be assigned to the terminal 13 bp (i.e. +/− 60–73 bp from the dyad) on each side of the nucleosome [30]. This is likely to have resulted from the absence of CCNC components and the relative difficulty with crystallography to assign structure to flexible regions but it could also be explained because the nucleosome position and terminal DNA sequences used in that study [30] do not represent a favored position in mammalian centromeres [5,13].
Towards an emerging understanding of higher-order complex formation upon centromeric nucleosomes, our structure complements a very recent report of the budding yeast CCAN/Ctf19 cryo-EM structure (PDB# 6NUW) [19]. That structure contains most yeast orthologs of CCAN component but lacks CENP-CMif2 and the CENP-A nucleosome. We built composite models of CCAN/CCNC by aligning through the shared subunit, CENP-NNT, and slightly rotating CCAN subunits to avoid steric clashes with the nucleosome (Figure 7A). CENP-CCD, lacking its own secondary structure, fits tightly against the CENP-A nucleosome surface beneath the large globular CCAN complex. Further, we envision that it has flexible connectors extending to its other centromere scaffolding connection sites that lie distal to both N- and C-terminal directions [1].
Figure 7. Model of the transition of centromeric chromatin from interphase to mitosis.
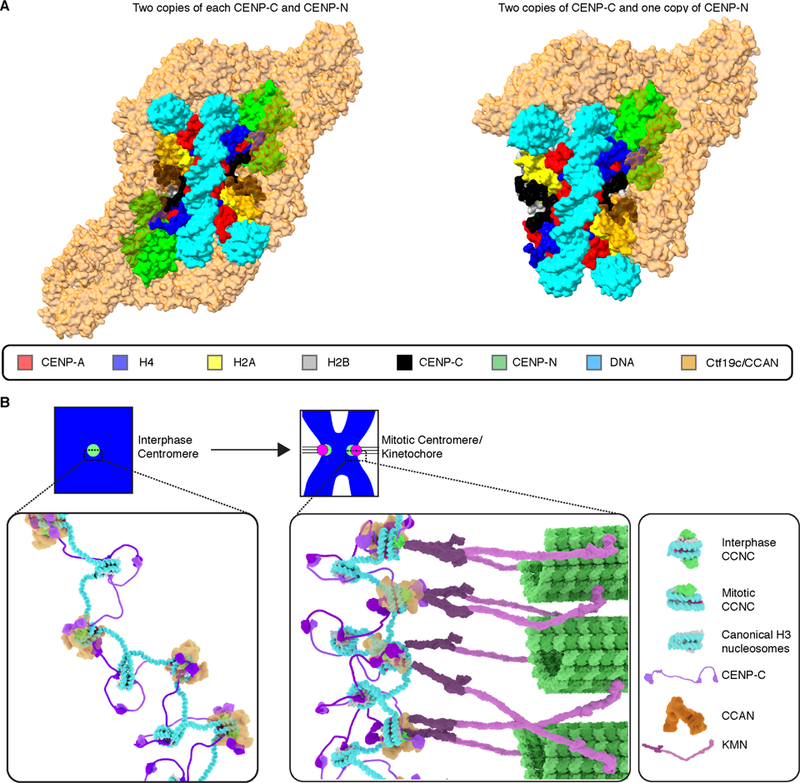
(A) Composite model of the two forms of the human CCNC with the yeast Ctf19c/CCAN complex (PDB# 6NUW; [19]) after first aligning N-terminal of CENP-NChl4 in Ctf19c to CENP-NNT in the CCNC and rigid body rotation of Ctf19c to remove steric clashes before performing global minimization in PHENIX. (B) Model of the CCNC transition from interphase to mitosis. See text for details and see also Video S1 for animation of the transition.
Regarding the function of the centromere in chromosome segregation, our structural and cell biological insights lead us to propose a model for the transition from an interphase to a mitotic CCNC (Figure 7B and Video S1). In interphase, two copies each of CENP-CCD and CENP-NNT bind symmetrically to each face of the nucleosome. We envision this is a very stable arrangement [14], and further culminates in surrounding the centromeric nucleosome with the CCAN. In addition, in our model, CENP-C would use its second nucleosome binding domain, CENP-CCM, to tether the CCNC to neighboring chromatin on either side of it. The loss of one copy of an essential foundational centromere component, CENP-N, at the very time in the cell cycle when this component is required for kinetochore assembly may appear paradoxical. In our model, however, the loss serves to orient the CCAN in order to properly orient the chromosome—kinetochore interface. As cells enter mitosis, perhaps due to the higher degree of chromatin condensation [24], one copy of CENP-NNT is lost. This asymmetry orients the face containing CENP-NNT towards the spindle, where CENP-N performs its essential role in recruiting multiple components of the CCAN that, in turn, recruit the KMN complex and the other constituents of the kinetochore. Having both CENP-C and CENP-N on the face where the kinetochore grows is key because both are essential for recruitment of downstream CCAN components, including CENP-T which makes critical direct contacts to the KMN complex [44–46]. The side of the CCNC lacking CENP-N would be strongly tethered to neighboring chromatin, in our model, with CENP-C on the chromatin-oriented face linked to a neighboring nucleosome. While reconstitutions of CCAN components yielded equal stoichiometry of individual components [19, 22], it is possible that the copy number of CCAN components besides CENP-N vary between interphase and mitosis. Measurements of subunit abundances in the CCAN at the transition from interphase to mitosis deserves future study. Our findings help to develop a model for the orientation of the CCAN on the CENP-A nucleosome, providing a framework upon which to understand the interphase to mitotic forms of the 16-subunit CCAN. In our model, the fixed core of the CENP-A nucleosome in the mitotic CCNC would resist disassembly under spindle forces, while the plasticity of the nucleosome terminal DNA would accommodate the forces and the geometrical constraints imposed by the fixed position of CENP-A between the spindle and the rest of the chromosome.
We envision that the change in stoichiometry from the interphase to mitotic form (i.e. reducing the number of CENP-N from two to one per CENP-A nucleosome) is coupled to chromosome condensation and the rearrangement of CENP-A containing nucleosomes on the surface of the mitotic chromosome. The CENP-A nucleosome, in any model of condensed mitotic chromatin, is predicted to be in close contact with neighboring nucleosomes. This contact is known to occlude the CENP-NNT binding site [24], thus CENP-N would be predicted to be rapidly evicted upon chromosome condensation, matching our measurements of loss of half of the CENP-N molecules at centromeres in the first minutes of mitosis (Figure 1). The other face, in our model, is free from nucleosome contacts. We favor this mode of the CCNC transition, in part, because CENP-A forms the foundation of the kinetochore, requiring it to have a surface accessible to the cytoplasm (i.e. not blocked by close packing with neighboring nucleosomes). Also, an earlier view that CENP-C and CENP-N occupy separate nucleosomes [24,47], would predict that removal of CENP-N occurs on a distinct set of CENP-A nucleosomes from those bound by CENP-C. Several emerging findings–complementary binding surfaces for CENP-CCM and CENP-NNT [14,18,20,21], their ready assembly on the same nucleosome [14], the strong disruption of the CCAN upon the rapid depletion of either subunit [23], and the well-ordered higher order structure of the CCAN [19]–are all more in line with our preferred model (Figure 7B), however.
In summary, our mammalian centromere model represents a mode by which centromeric nucleosomes generate a robust and organized interface between the chromosome and mitotic spindle.
STAR Methods:
LEAD CONTACT AND MATERIALS AVAILABLITY.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ben E. Black (blackbe@pennmedicine.upenn.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The DLD-1 CENP-NAID-EGFP/AID-EGFP cell line [23] was cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% tetracycline-free fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin and maintained at 37°C in a humidified incubator with 5% CO2. The DLD-1 Flp-In T-Rex cell line stably expressing Tir1 [48] was cultured as above.
METHOD DETAILS:
G2 arrest, release, and processing for cell imaging
Cells were treated with 9 µM RO-3306 overnight (~18 h) to inhibit Cdk1 and arrest cells in G2. Cells were then fixed either immediately or following RO-3306 washout. Cells were fixed in 4% formaldehyde in PBS for 10 min at room temperature, quenched with 100 mM Tris (pH 7.5) for 5 min, permeabilized in 0.5% Triton X-100 in PBS for 5 min, and washed three times in 0.1% Tween-20 in PBS for 5 min each. Coverslips were blocked in blocking buffer (PBS supplemented with 2% FBS, 2% bovine serum albumin, and 0.1% Tween-20) prior to antibody incubations in blocking buffer. The following primary antibodies were used: mouse monoclonal anti-CENP-A (1 µg/ml; Enzo Life Sciences ADI-KAM-CC006-E), rabbit anti-CENP-C (0.1 µg/ml; custom-made by Covance and affinity-purified in-house. [49]), and mouse monoclonal anti-MAD2 (0.4 µg/ml; Santa Cruz Biotechnology sc-65492). The following fluorophore-conjugated secondary antibodies from Jackson ImmunoResearch Laboratories were used at a dilution of 1:200: Dylight 649 anti-mouse (715–495-151), Cy3 anti-rabbit (111–165-144), Cy3 anti-mouse (715–165-151) and Cy5 anti-rabbit (711–175-152). For CENP-L immunofluorescence (Figure S1F) rabbit polyclonal anti-CENP-L (1:1000; [23]), cells were treated with 50 µM STLC for 2 h and pre-permeabilized in 0.1% Triton X-100 in PBS for 30 s prior to fixation. Cells were processed as above, except without permeabilization in 0.5% Triton X-100. PBS replaced 0.1% Tween-20 in PBS for the wash steps. DNA was stained with DAPI, and coverslips were mounted in VectaShield (Vector Laboratories). Asynchronous cells were fixed in 4% formaldehyde in PBS with no prior drug treatment and processed as above.
Cell image acquisition and quantitation
Images were captured at room temperature on an inverted fluorescence microscope (DMI6000 B; Leica) equipped with a charge-coupled device camera (ORCA AG; Hamamatsu Photonics) and a 100x, 1.4 NA oil immersion objective. Images were collected as 0.2 µm z-sections using identical acquisition conditions and as much of the dynamic range of the camera as possible without saturating any pixels. Nuclei were cropped, and z-series were deconvolved using LAS-AF software (Leica). Fluorescence intensity at centromeres was measured from deconvolved and maximum-projected images using an ImageJ macro [50], CRaQ v1.12 [51], under default parameters with slight modifications. Briefly, centromeres were identified in a reference channel (CENP-A for the experiment shown in Figures 1B and S1; CENP-C for the experiment shown in Figure 1E) and the integrated intensity within the 7 × 7-pixel box in each of the data channels (CENP-N, detected using EGFP fluorescence; CENP-A or CENP-C) was corrected for local background. A minimum of 350 centromeres were analyzed for each condition, and the mean ± 95% confidence interval is reported. For representative images, contrast in each channel has been adjusted linearly and identically across conditions. Solely for display purposes (i.e. not for quantitation), the representative images have been scaled to visibly display the large intensity range of CENP-N at resolved sister centromeres resulting in truncated intensity information at a small proportion of the brightest pixels within centromeres that have not resolved into two distinct sisters in cells staged prior to NEBD.
Recombinant protein and DNA production and purification
Human CENP-A and canonical histones were prepared, as described [52,53]. (CENP-A/H4(FL))2 or (CENP-A/H4(∆19))2 is expressed from a bicistronic construct as a soluble heterotetramer. (CENP-A/H4)2 is purified by hydroxyapatite column followed by cation exchange. Histones H2A and H2B are expressed as monomers in inclusion bodies. Histones H2A and H2B were purified under denaturing conditions and refolded as a soluble heterodimer. CENP-CCD is expressed as a GST-tagged protein, as described [15]. CENP-CCD is affinity purified by GST tag and GST is cleaved with PreScission protease followed by cation exchange. CENP-NNT is expressed and purified with a poly-His tag, as described [14]. CENP-NNT-His protein is affinity purified using a 1 ml HisTrap FF column (GE Healthcare).
The DNA used to reconstitute the CCNC corresponds to the α-satellite sequence from the human X chromosome [5,14,32,37]:
5’-ATCAAATATCCACCTGCAGATTCTACCAAAAGTGTATTTGGAAACTGCTCCATCA AAAGGCATGTTCAGCTCTGTGAGTGAAACTCCATCATCACAAAGAATATTCTGAGAA TGCTTCCGTTTGCCTTTTATATGAACTTCCTCGAT-3’. A plasmid containing six identical repeats of this 147 bp DNA sequence is harvested from bacteria, then purified, and subjected to EcoRV digestion. The plasmid is removed from the 147 bp monomer DNA by anion chromatography using Source 15Q resin (GE Healthcare).
Assembly and purification of the CCNC for cryo-EM studies
CENP-A nucleosome core particles were assembled with purified histones and DNA at equimolar ratio using gradual salt dialysis followed by thermal shifting for 2 h at 55°C [52]. To form the complex with CENP-CCD and CENP-NNT, 2.2 moles of CENP-CCD and 4 moles of CENP-NNT were added per mole of CENP-A nucleosome core particles in 10 mM HEPES (pH 7.5), 50 mM NaCl, 1 mM DTT, 1 mM EDTA and incubated on ice for 1 h. The CCNC was then purified by preparative electrophoresis (Prep Cell, BioRad) using 5% native PAGE. The peak fractions were then subjected to a modified GraFix [54] protocol wherein 0.05% glutaraldehyde is added to the sample, concentrated using centricon concentrators, and then applied to a 5–30% glycerol gradient containing 0.125% gluteraldehyde. Centrifugation was performed for 12 h at 4°C, 165,000 x g in a SW60 rotor (Beckman coulter). Gradient fractions from GraFix sample containing CCNC were selected for downstream EM analysis that had mixtures of complexes with either one or two copies of CENP-N, assessed by native PAGE analysis, in corresponding fractions of samples where no fixative was added.
Negative stain analysis and Cryo-EM sample preparation.
For EM grid preparation, the CCNC cryo-EM sample was then dialyzed in 10 mM HEPES, (pH 7.5), 50 mM NaCl, 1 mM EDTA and 1 mM DTT and was diluted with an appropriate volume of the dialysis buffer supplemented with or without NP-40 (0.004%). Negative stain analysis was performed to identify conditions that yielded potentially appropriate concentration, homogeneity, and homogenous distribution of particles for subsequent cryo-EM grid preparations. To do this, 3 µl (0.03 mg/ml) of dialyzed complex was loaded on glow-discharged 300 mesh Cu carbon grids (EMS) and incubated for 30 s before staining with 2% uranyl acetate. All samples were imaged on a FEI Tecnai 12 microscope operating at 120 kV, using a CCD camera (Gatan BM-Ultrascan) at a nominal magnification of 39,000x (Figure S2B). To prepare cryo-EM grids, 2 µl of the dialyzed CCNC (~0.06 and ~0.5 mg/mL without and with 0.004% NP-40) was applied to CF1.2/1.3 holey carbon grids that were glow discharged for 40 s before sample application, subsequently blotted for 8 s, and flash-frozen by plunging into liquid ethane with a Leica EM CPC manual plunger. EM grids were made in batches and freezing conditions were optimized using an FEI TF20 microscope operating at 200 kV equipped with a FEI Falcon II camera.
Cryo-EM data acquisition and processing
Cryo-EM specimens were imaged using a FEI Titan Krios transmission electron microscope (TEM) operating at 300 kV equipped with a K2 Summit direct electron detector with an energy quantum filter (Gatan) at a nominal magnification of 130,000x, resulting in image sampling at 0.55 Å per pixel in super resolution mode. Automated data acquisition was carried out using Serial EM software [55]. Image stacks of 40 frames were collected over 10 s with a total dose of 40 or 50 electrons Å−2 with Volta phase plate defocus 0.5 µm [28] and without the Volta phase plate defocus range 2–3 µm.
First, relatively small data sets containing ~200,000–300,000 CCNC particles without and with NP-40 were collected with the Volta phase plate (datasets 1 and 2, respectively). Micrographs in data set 1 exhibited preferred top views with partially or fully unwrapped DNA from the CCNC, whereas micrographs in data set 2 yielded fields of homogeneous nucleosomes wrapped with DNA viewed from varied angles (Figure S2C), indicating that the CCNC was stabilized by NP-40 during plunge freezing. Then, dose-fractioned frames of data set 2 were aligned with MotionCor2 [56], and CTF parameters were determined with Gctf (v1.06) [57]. 2D class averages obtained from ~10,000 manually picked particles were used as references for auto-picking in RELION [58]. From 2540 micrographs comprising data set 2 we obtained ~144,622 particles after particle sorting and 2D classification (Figure S2F). 3D classification was subsequently performed using PDB# 3AN2 (back-calculated and low-pass filtered at 60 Å resolution) as an initial model, yielding an EM map containing 55,124 particles showing clear nucleosome features and 2 copies of CENP-NNT (Figure S2G). We then refined and performed post-processing of this model to reach a resolution of 7.1 Å judged by FSC 0.143 criterion (Figure S2I). Lastly, accelerated data collection by image shift (~1 µm) with and without the use of the phase plate yielded many more micrographs (data sets 3 and 4, respectively) from the same batch as data set 2, followed by motion correction, CTF determination, and auto-picking using the best 2D class averages from data set 2 as references. Combined particles from data sets 2–4 were filtered by multiple rounds of 2D class averaging, yielding a total of 1,340,672 CCNC particles, which were used for iterative rounds of hierarchical 3D classification without symmetry (Figure S3A). After two rounds of 3D classification using a 60 Å low-pass filtered model from data set 2 reconstruction (3D classification-1 and 2 in Figure S3A), we obtained the two forms of the CCNC: one containing two copies each of CENP-CCD and CENP-NNT and the other containing two copies of CENP-CCD and one copy of CENP-NNT. Reconstruction of the CCNC containing two copies of CENP-NNT was obtained from ~188,995 particles, and further refined to a resolution of 3.5 Å by 3D auto-refinement, per-particle CTF refinement, and Bayesian polishing in RELION 3.0. Two EM maps with and without a B-factor correction (−168 Å2) have been deposited in a single entry in the Electron Microscopy Data Bank (accession #EMDB-9251). In a similar manner, reconstruction of the CCNC containing a single copy of CENP-NNT was obtained from ~113,830 particles with a resolution of 3.6 Å. Two EM maps with and without a B-factor correction (−204 Å2) have been deposited in a single entry in the Electron Microscopy Data Bank (accession #EMDB-9250). Local resolution estimates were determined using the Monores program [59].
Model building
B-factor uncorrected and B-factor corrected EM density maps were used for model building. PHENIX [60], Coot [61], and UCSF Chimera [62] programs were used for model fitting and refinement procedures. An initial model generated by replacing the DNA chains of PDB# 6C0W with those from PDB# 1KX5 DNA followed by global minimization in PHENIX was docked into EM density as a rigid body using Chimera [21,63]. The crystal structure of CENP-NNT (PDB# 6EQT; [21]) and a homology model (using PDB# 4X23 of CENP-CCM; [20] of CENP-CCD(a.a.518−537) were separately fitted as a rigid body into corresponding EM densities. The model of CENP-CCD(a.a.518−537) was manually adjusted into EM density by using model building tools and refined by regularization and real-space refinement in Coot. The N-terminal tail of histone H4 was extended without side chains (i.e. alanines were substituted at each position) by tracing EM density, and refined in a similar manner. Then all chains were combined into a single PDB model and they subsequently underwent real space refinement in Coot and PHENIX. The DNA sequence in the PDB model was then replaced with the α-satellite sequence used in this study and additional rounds of refinement were carried out with EM density maps.
Focused 3D classification and Modeling of CENP-N and H4 tail
For more detailed analysis of the interaction between CENP-NNT and H4 tail, ~528,000 CCNC particles after 3D classification-1 (Figure S3A) were subjected to focused 3D classification with subtraction of the residual densities corresponding to the nucleosome (see Figure S5A). Briefly, first, a mask (purple density in Figure S5A) was applied to ~528,000 particles, such that densities corresponding to the nucleosome were subtracted. Second, resulting particles were subjected to 3D classification into six different classes without alignment. A 3D class containing 54,139 particles, with predominant conformation for H4 and CENP-N were refined and post-processed to reach FSC 4.3 Å (accession #EMDB-9252). The local resolution estimation for the H4 tail is limited to ~6.5 Å in the EM map. The N-terminal tail of histone H4 and CENP-NNT were remodeled based on this EM map using Coot: the N-terminal tail of histone H4 was fine-tuned by tracing EM density without side-chains (i.e. using a polyalanine chain for H4a.a.12−22), whereas α6- and α8-helices of CENP-NNT were manually modified to fit into corresponding densities by using model building tools and refined by regularization and real-space refinement in Coot. Then the resulting model of CCNC was subjected to flexible fitting and real space refinement in Coot. Further real space refinement of this model was performed in PHENIX to optimize global geometry. Figure panels were generated using PyMol [64], UCSF Chimera, and UCSF ChimeraX [65].
XL-MS studies
CCNC was crosslinked with BS3 and digested to peptides as described previously [66]. 50 µg of purified CCNC was crosslinked in separate tubes with 1, 2, 3 mM BS3 on ice for 1 h, and then quenched with 30 mM ammonium bicarbonate for 15 min. After crosslinking, the sample was subsequently purified using a 5 to 30% sucrose gradient and centrifugation at 165,000 x g for 12 h at 4° C. This step eliminates higher-order contaminants. Peak fractions from the gradient centrifugation were concentrated and precipitated with acetone for 1 h at −80°C followed by the addition of 10 mM DTT and 50 mM iodoacetamide. Trypsin digestion was performed at 1:50 (trypsin:sample by mass) overnight at 37°C while shaking (600 rpm) and then quenched with 5% TFA. Peptides and crosslinked peptides were desalted on C18 stage-tips and eluted by 75% acetonitrile. The eluted peptides were dried in a SpeedVac, reconstituted in 0.1% formic acid, and measured immediately in the mass spectrometer. The peptides were analyzed by a 90 minute 0–40% gradient LC-MS on a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). The resulting data files were analyzed by Proteome Discoverer (Thermo Fisher Scientific) for protein identification. The crosslinks were identified using the FindXL software [67]. The search included fixed modification of cysteines by iodoacetamide, variable methionine oxidation (‘Mox’ symbol in the crosslink Excel table), and variable lysine modifications by mono-linked BS3 in either hydrolysed or non-hydrolysed form (‘K++’ and ‘K+’ symbols in Table S3). The data from the 1 mM, 2 mM, and 3 mM BS3 samples were pooled together into one (non-redundant) list of crosslinks. Crosslinks were included in the list only if they fulfilled three conditions: 1) the measured mass of the precursor ion of the two crosslinked peptides is within 6 ppm of the calculated mass; 2) at least four MS/MS fragments were identified on each peptide; 3) the score assigned to the crosslink (calculated as the number of identified MS/MS fragments divided by combined length of the two peptides) is 0.6 or higher. Estimation of the false-detection rate (FDR) in the final list was determined in the following way. The identification of crosslinks was repeated ten times with an erroneous crosslinker mass that differed from the true mass by 50.0, 55.0, 60.0, … 95.0 Da. This led to a distribution of scores that was much lower than the scores obtained with the correct crosslinker mass (decoy vs. results in Figure 2C). We therefore estimate the FDR to be 1 in 55 crosslinks or ~2%. The mass spectrometry proteomics data deposited to the ProteomeXchange Consortium via the PRIDE [68] repository with the dataset identifier PXD12605 and 10.6019/PXD12605.
Structural comparison and superhelical pitch calculations
Structures of a canonical nucleosome with palindromic ⍺-satellite DNA (PDB# 1AOI), CENP-A nucleosome on palindromic ⍺-satellite DNA (PDB# 3AN2), two structures of CENP-A nucleosome wrapped with 601 DNA and bound by one copy of CENP-NNT (PDB# 6C0W and 6BUZ) and the structures of the CCNC (PDB# 6MUO and 6MUP; this study) were aligned using the CENP-A/H4 dimer or the H3/H4 dimer in UCSF Chimera. RMSD analysis was conducted and figures were made in UCSF Chimera. Superhelical pitch of nucleosome per base-pair step was determined as a rise (Å) by 78 bp of DNA base pair midpoints calculated with Curves+ [69].
Binding assays
CENP-A nucleosomes were prepared with labelled Cy5-H2B on 147 bp α-satellite or 601 DNA by gradient dialysis [14]. Further, nucleosomes were purified by sucrose gradient centrifugation. CENP-CCD binding assay is performed by incubating 200 nM of nucleosomes with increasing concentration of CENP-CCD in buffer (20 mM Tris-Cl pH 7.5, 1 mM EDTA and 1 mM DTT). CENP-NNT binding assays were performed by incubating 200 nM of nucleosomes or 200 nM of nucleosomes saturated with two copies of CENP-CCD with increasing concentration of CENP-NNT protein in the same buffer. These samples were incubated on ice for 1 h before separating by 5% native PAGE and gels were analyzed in a Typhoon 9200 imager (GE Healthcare). Similar binding comparison experiments were performed with CENP-A nucleosomes containing either H4(FL) or H4(∆19). For densitometric analysis of the CCNC, intensity profile along the lane at 800 nM CENP-NNT from three independent experiments was generated using ImageJ. The height of the peak representing the fully assembled CCNC was calculated as the length of the vertical line from the top of the relevant peak to the baseline. The full width at half maximum (FWHM) value was calculated as the width of the peak at the half-maximal intensity. The peak height and FWHM were each normalized to the peak area for comparison between DNA sequences. The average of three experiments +/− S.D. is reported.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of the experiments can be found in the figure legends and the Methods Details section of the STAR methods. Statistical details include exact value of n, n represent the number of centromeres measured, definition of center, and dispersion and precision measures. Statistical tests were performed as described in the Method Details section using R [70].
DATA AND CODE AVAILABILITY
Data Resources
The accession numbers for the three cryo-EM structures reported in this paper are EMDB: #9250, #9251 and #9252 and for two coordinates PDB: #6MUO and #6MUP. The mass spectrometry proteomics data deposited to the PRIDE repository with the dataset identifier PXD12605 and 10.6019/PXD12605.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Dylight 649 anti-mouse | Jackson ImmunoResearch Laboratories | 715–495-151 |
| Cy3 anti-rabbit | Jackson ImmunoResearch Laboratories | 111–165-144 |
| Cy3 anti-mouse | Jackson ImmunoResearch Laboratories | 715–165-151 |
| Cy5 anti-rabbit | Jackson ImmunoResearch Laboratories | 711–175-152 |
| mouse monoclonal anti-CENP-A | Enzo Life Sciences | ADI-KAM-CC006-E |
| rabbit anti-CENP-C | [49] | N/A |
| mouse monoclonal anti-MAD2 | Santa Cruz Biotechnology | sc-65492 |
| rabbit anti-CENP-L | [23] | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CF1.2/1.3 holey carbon grids | EMS | CFT413–50 |
| Uranyl acetate | EMS | 22400 |
| STLC | Sigma | 164739 |
| VectaShield | Vector Laboratories | H-1000 |
| mineral oil | Sigma | M531 |
| RO-3306 | Enzo Life Sciences | ALX-270–463 |
| BS3 | Thermo Fisher Scientific | 21580 |
| Trypsin | Promega corporation | V5111 |
| Glutaraldehyde | Sigma | G5882–10x |
| Iodoacetamide | Sigma | I1149–5G |
| Deposited Data | ||
| CENP-A nucleosome bound by two copies of CENP-CCD and two copies CENP-NNT | This paper | EMDB:9251 |
| CENP-A nucleosome bound by two copies of CENP-CCD and one copy CENP-NNT | This paper | EMDB:9250 |
| CENP-A nucleosome bound by two copies of CENP-CCD and two copies CENP-NNT with local refinement | This paper | EMDB:92523 |
| Model of CENP-A nucleosome bound by two copies of CENP-CCD and two copies CENP-NNT | This paper | PDB:6MUP |
| Model of CENP-A nucleosome bound by two copies of CENP-CCD and two copies CENP-NNT | This paper | PDB:6MUO |
| CCNC BS3 crosslink Mass spectrometry proteomics data | This paper | PXD12605 and 10.6019/PXD12605 |
| Experimental Models: Cell Lines | ||
| DLD-1 Flp-In T-Rex | [48] | N/A |
| DLD-1 CENP-NAID-EGFP/AID-EGFP | [23] | N/A |
| Software and Algorithms | ||
| COOT | [61] | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| PHENIX | [60] | https://www.phenix-online.org |
| PyMOL | [64] | http://www.pymol.org |
| MotionCor2 | [56] | http://msg.ucsf.edu/em/software/motioncor2.html |
| Gctf | [57] | https://www.mrc-lmb.cam.ac.uk/kzhang/Gctf/ |
| Monores | [59] | https://www.ncbi.nlm.nih.gov/pubmed/29395788 |
| RELION | [58] | http://www2.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| UCSF Chimera | [62] | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | [65] | https://www.cgl.ucsf.edu/chimerax/ |
| Curves+ | [69] | https://bisi.ibcp.fr/tools/curves_plus/ |
| LAS-AF | Leica Microsystems | https://www.leica-microsystems.com |
| Fiji (Fiji Is Just ImageJ) | [50] | https://imagej.net/Fiji |
| CRaQ v1.12 | [51] | http://facilities.igc.gulbenkian.pt/microscopy/macros/CRaQ_v1.12.ijm |
| Serial EM | [55] | http://bio3d.colorado.edu/SerialEM/ |
| Find_XL | [66] | http://biolchem.huji.ac.il/nirka/software.html |
| RStudio | [70] | http://www.rstudio.com/. |
| Other | ||
| HisTrap FF | GE Healthcare | 11–0004-58 |
| Amicon Ultra concentrators | Millipore | UFC501024 |
| C18 stage-tips | Thermo Fisher Scientific | 89870 |
Highlights:
One copy of CENP-N is lost for every two copies of CENP-C at mitotic onset
Structures of core centromeric nucleosome complex (CCNC) with 1 or 2 copies CENP-N
Natural centromere nucleosomal DNA conformation corresponds to robust CCNC assembly
Emerging model of mitotic centromeric chromatin with global and local asymmetries
Acknowledgements
We thank our UPenn colleagues L. Guo, H. Zhang, and M. Lampson for helpful discussions and H-J. Kim for technical support. We thank A. Straight (Stanford), K. Luger (Colorado), and D. Cleveland (UCSD) for plasmids, I. Cheeseman (MIT) for DLD1 CENP-NAID-EGFP/AID-EGFP cells and CENP-L antibody, D. Williams (ASU) for technical assistance, and J. Iwasa and G. Hsu (Utah) at the Animation Lab for Video S1. We thank the Beckman Center for Cryo-EM (UPenn) for use of instrumentation. This work was supported by NIH research grants (GM130302, B.E.B.; GM123233, K.M.). M.B., M.S., and N.K. are funded by the Israel Science Foundation grant 15/1768.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare no conflict of interest.
Video S1. Model of the transition of centromeric chromatin from interphase to mitosis. Related to Figure 7.
References
- 1.Musacchio A, and Desai A (2017). A molecular view of kinetochore assembly and function. Biology 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black BE, and Cleveland DW (2011). Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Depinet TW, Zackowski JL, Earnshaw WC, Kaffe S, Sekhon GS, Stallard R, Sullivan BA, Vance GH, Van Dyke DL, Willard HF, et al. (1997). Characterization of neo-centromeres in marker chromosomes lacking detectable alpha-satellite DNA. Hum. Mol Genet 6, 1195–1204. [DOI] [PubMed] [Google Scholar]
- 4.Eichler EE (1999). Repetitive conundrums of centromere structure and function. Hum. Mol. Genet 8, 151–155. [DOI] [PubMed] [Google Scholar]
- 5.Hasson D, Panchenko T, Salimian KJ, Salman MU, Sekulic N, Alonso A, Warburton PE, and Black BE (2013). The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Struct. Mol. Biol 20, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.du Sart D, Cancilla MR, Earle E, Mao JI, Saffery R, Tainton KM, Kalitsis P, Martyn J, Barry AE, and Choo KH (1997). A functional neo-centromere formed through activation of a latent human centromere and consisting of non-alpha-satellite DNA. Nat. Genet 16, 144–153. [DOI] [PubMed] [Google Scholar]
- 7.Warburton PE, Cooke CA, Bourassa S, Vafa O, Sullivan BA, Stetten G, Gimelli G, Warburton D, Tyler-Smith C, Sullivan KF, et al. (1997). Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr. Biol 7, 901–904. [DOI] [PubMed] [Google Scholar]
- 8.Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, and Willard HF (1997). Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet 15, 345–355. [DOI] [PubMed] [Google Scholar]
- 9.Okada T, Ohzeki J, Nakano M, Yoda K, Brinkley WR, Larionov V, and Masumoto H (2007). CENP-B controls centromere formation depending on the chromatin context. Cell 131, 1287–1300. [DOI] [PubMed] [Google Scholar]
- 10.Logsdon GA, Gambogi CW, Liskovykh MA, Barrey EJ, Larionov V, Miga KH, Heun P, and Black BE (2019) Human artificial chromosomes that bypass centromeric DNA. Cell, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, and Cleveland DW (2015). DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 33, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, and Cleveland DW (2017). Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol 19, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwata-Otsubo A, Dawicki-McKenna JM, Akera T, Falk SJ, Chmátal L, Yang K, Sullivan BA, Schultz RM, Lampson MA, and Black BE (2017). Expanded satellite repeats amplify a discrete CENP-A nucleosome assembly site on chromosomes that drive in female meiosis. Curr. Biol 27, 2365–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo LY, Allu PK, Zandarashvili L, McKinley KL, Sekulic N, Dawicki-McKenna JM, Fachinetti D, Logsdon GA, Jamiolkowski RM, Cleveland DW, et al. (2017). Centromeres are maintained by fastening CENP-A to DNA and directing an arginine anchor-dependent nucleosome transition. Nat. Commun 8, 15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LET, Van Duyne GD, Vinogradov SA, et al. (2015). Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science 348, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll CW, Silva MCC, Godek KM, Jansen LET, and Straight AF (2009). Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nat. Cell Biol 11, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll CW, Milks KJ, and Straight AF (2010). Dual recognition of CENP-A nucleosomes is required for centromere assembly. J. Cell Biol 189, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chittori S, Hong J, Saunders H, Feng H, Ghirlando R, Kelly AE, Bai Y, and Subramaniam S (2018). Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science 359, 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinshaw SM, and Harrison SC (2019). The structure of the Ctf19c/CCAN from budding yeast. ELife 8, e44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato H, Jiang J, Zhou B-R, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, and Bai Y (2013). A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science 340, 1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pentakota S, Zhou K, Smith C, Maffini S, Petrovic A, Morgan GP, Weir JR, Vetter IR, Musacchio A, and Luger K (2017). Decoding the centromeric nucleosome through CENP-N. ELife 6, e33442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weir JR, Faesen AC, Klare K, Petrovic A, Basilico F, Fischböck J, Pentakota S, Keller J, Pesenti ME, Pan D, et al. (2016). Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537, 249–253. [DOI] [PubMed] [Google Scholar]
- 23.McKinley KL, Sekulic N, Guo LY, Tsinman T, Black BE, and Cheeseman IM (2015). The CENP-L-N complex forms a critical node in an integrated meshwork of interactions at the centromere-kinetochore interface. Mol. Cell 60, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang J, Liu Y, Wei Y, Deng W, Yu Z, Huang L, Teng Y, Yao T, You Q, Ruan H, et al. (2015). Structural transitions of centromeric chromatin regulate the cell cycle-dependent recruitment of CENP-N. Genes Dev 29, 1058–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClelland SE, Borusu S, Amaro AC, Winter JR, Belwal M, McAinsh AD, and Meraldi P (2007). The CENP-A NAC/CAD kinetochore complex controls chromosome congression and spindle bipolarity. EMBO J 26, 5033–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen LET, Black BE, Foltz DR, and Cleveland DW (2007). Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol 176, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chua EYD, Vogirala VK, Inian O, Wong ASW, Nordenskiöld L, Plitzko JM, Danev R, and Sandin S (2016). 3.9 Å structure of the nucleosome core particle determined by phase-plate cryo-EM. Nucleic Acids Res 44, 8013–8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danev R, Tegunov D, and Baumeister W (2017). Using the Volta phase plate with defocus for cryo-EM single particle analysis. Elife 6, e23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 30.Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi-Takanaka Y, Oda T, Sato M, Park S-Y, et al. (2011). Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 476, 232–235. [DOI] [PubMed] [Google Scholar]
- 31.Arimura Y, Tachiwana H, Takagi H, Hori T, Kimura H, Fukagawa T, and Kurumizaka H (2019). The CENP-A centromere targeting domain facilitates H4K20 monomethylation in the nucleosome by structural polymorphism. Nat. Commun 10, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang TP, Hansen SK, Oishi KK, Ryder OA, and Hamkalo BA (1982). Characterization of a cloned repetitive DNA sequence concentrated on the human X chromosome. Proc. Natl. Acad. Sci. U.S.A 79, 6593–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou B-R, Yadav KNS, Borgnia M, Hong J, Cao B, Olins AL, Olins DE, Bai Y, and Zhang P (2019). Atomic resolution cryo-EM structure of a native-like CENP-A nucleosome aided by an antibody fragment. Nat. Commun 10, 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42. [DOI] [PubMed] [Google Scholar]
- 35.Korolev N, Lyubartsev AP, and Nordenski.ld L (2018). A systematic analysis of nucleosome core particle and nucleosome-nucleosome stacking structure. Sci. Rep 8 1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngo TTM, Zhang Q, Zhou R, Yodh JG, and Ha T (2015). Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falk SJ, Lee J, Sekulic N, Sennett MA, Lee T-H, and Black BE (2016). CENP-C directs a structural transition of CENP-A nucleosomes mainly through sliding of DNA gyres. Nat. Struct. Mol. Biol 23, 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, Wang J-PZ, and Widom J (2006). A genomic code for nucleosome positioning. Nature 442, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nechemia-Arbely Y, Miga KH, Shoshani O, Aslanian A, McMahon MA, Lee AY, Fachinetti D, Yates JR, Ren B, and Cleveland DW (2019). DNA replication acts as an error correction mechanism to maintain centromere identity by restricting CENP-A to centromeres. Nat. Cell Biol 21, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitra S, Bodor DL, David AF, Mata JF, Neumann B, Reither S, Tischer C, and Jansen LET (2019). Genetic screening identifies a SUMO protease dynamically maintaining centromeric chromatin and the associated centromere complex. bioRxiv, 620088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conde N. e S., Black BE, Sivolob A, Filipski J, Cleveland DW, and Prunell A (2007). CENP-A-containing nucleosomes: easier disassembly versus exclusive centromeric localization. J. Mol. Biol 370, 555–573. [DOI] [PubMed] [Google Scholar]
- 42.Panchenko T, Sorensen TC, Woodcock CL, Kan Z, Wood S, Resch MG, Luger K, Englander SW, Hansen JC, and Black BE (2011). Replacement of histone H3 with CENP-A directs global nucleosome array condensation and loosening of nucleosome superhelical termini. Proc. Natl. Acad. Sci. U.S.A 108, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nechemia-Arbely Y, Fachinetti D, Miga KH, Sekulic N, Soni GV, Kim DH, Wong AK, Lee AY, Nguyen K, Dekker C, et al. (2017). Human centromeric CENP-A chromatin is a homotypic, octameric nucleosome at all cell cycle points. J. Cell Biol 216, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, and Cheeseman IM (2011). Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malvezzi F, Litos G, Schleiffer A, Heuck A, Mechtler K, Clausen T, and Westermann S (2013). A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J 32, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishino T, Rago F, Hori T, Tomii K, Cheeseman IM, and Fukagawa T (2013). CENP-T provides a structural platform for outer kinetochore assembly. EMBO J 32, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagpal H, Hori T, Furukawa A, Sugase K, Osakabe A, Kurumizaka H,and Fukagawa T (2015). Dynamic changes in CCAN organization through CENP-C during cell-cycle progression. Mol. Biol. Cell 26, 3768–3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holland AJ, Fachinetti D, Han JS, and Cleveland DW (2012). Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 109, E3350–E3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bassett EA, Wood S, Salimian KJ, Ajith S, Foltz DR, and Black BE (2010). Epigenetic centromere specification directs aurora B accumulation but is insufficient to efficiently correct mitotic errors. J. Cell Biol 190, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bodor DL, Rodríguez MG, Moreno N & Jansen LET (2012). Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol 55, 8.8.1–8.8.34. [DOI] [PubMed] [Google Scholar]
- 52.Sekulic NE & Black BE (2016). Preparation of recombinant centromeric nucleosomes and formation of complexes with nonhistone centromere proteins. Meth. Enzymol 573, 67–96. [DOI] [PubMed] [Google Scholar]
- 53.Sekulic N, Bassett EA, Rogers DJ, and Black BE (2010). The structure of (CENP-A–H4)2 reveals physical features that mark centromeres. Nature 467, 347–351.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stark H Chapter Five - GraFix: (2010). Stabilization of fragile macromolecular complexes for single particle Cryo-EM. Meth. Enzymol 481, 109–126. [DOI] [PubMed] [Google Scholar]
- 55.Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- 56.Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vilas JL, Gómez-Blanco J, Conesa P, Melero R, Rosa-Trevín JM de la Otón J, Cuenca J, Marabini R, Carazo JM, Vargas J, et al. (2018). MonoRes: Automatic and accurate estimation of local resolution for electron microscopy maps. Structure 26, 337–344. [DOI] [PubMed] [Google Scholar]
- 60.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python based system for macromolecular structure solution. Acta Cryst. D 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Cryst D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 63.Davey CA, Sargent DF, Luger K, Maeder AW, and Richmond TJ (2002). Solvent mediated interactions in the structure of the nucleosome core particle at 1.9Å resolution. J. Mol. Biol 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- 64.Schrödinger, LLC. (2015) The PyMOL molecular graphics system, v.2.0 Schrödinger. [Google Scholar]
- 65.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2017). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalisman N, Adams CM, and Levitt M (2012). Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A 109, 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slavin M, and Kalisman N (2018). Structural Analysis of Protein Complexes by Cross-Linking and Mass Spectrometry. Meth. Mol. Bio 1764, 173–183 [DOI] [PubMed] [Google Scholar]
- 68.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, and Zakrzewska K (2009). Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res 37, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.RStudio Team (2015). RStudio: Integrated Development for R RStudio, Inc, Boston, MA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Resources
The accession numbers for the three cryo-EM structures reported in this paper are EMDB: #9250, #9251 and #9252 and for two coordinates PDB: #6MUO and #6MUP. The mass spectrometry proteomics data deposited to the PRIDE repository with the dataset identifier PXD12605 and 10.6019/PXD12605.