

Abstract
Cochlear implantation has become the standard‐of‐care for adults and children with severe to profound hearing loss. There is growing evidence that qualitative as well as quantitative deficits in the auditory nerve may affect cochlear implant (CI) outcomes. Auditory neuropathy spectrum disorder (ANSD) is characterized by dysfunctional transmission of sound from the cochlea to the brain due to defective synaptic function or neural conduction. In this review, we examine the precise mechanisms of genetic lesions causing ANSD and the effect of these lesions on CI outcomes. Reviewed data show that individuals with lesions that primarily affect the cochlear sensory system and the synapse, which are bypassed by the CI, have optimal CI outcomes. Individuals with lesions that affect the auditory nerve show poor performance with CIs, likely because neural transmission of the electrical signal from the CI is affected. We put forth a nuanced molecular classification of ANSD that has implications for preoperative counseling for patients with this disorder prior to cochlear implantation. We propose that description of ANSD patients should be based on the molecular site of lesion typically derived from genetic evaluation (synaptopathy vs. neuropathy) as this has implications for expected CI outcomes. Improvements in our understanding of genetic site of lesions and their effects on CI function should lead to better CI outcomes, not just for individuals with auditory neuropathy, but all individuals with hearing loss.
Keywords: Cochlear implants, genetics, auditory neuropathy spectrum disorder
INTRODUCTION
Cochlear implantation has revolutionized the care for children and adults with deafness. The first surgery for implantation of a single channel cochlear implant (CI) occurred in 1961, by House and Doyle, and was followed by the first multichannel CI in 1978.1
In the intervening six decades, the goals for CI recipients' sound perception have advanced and continue to be refined. Initially, the goal was perception of sound, then speech recognition, and serviceable hearing. Today, the goal is to improve quality of life through excellent speech recognition, functional hearing in noise, and the ability to appreciate nuanced sound with precisely coded temporal elements like music. In addition to including adults and children with severe to profound sensorineural hearing loss (SNHL) not improved with hearing aids, CI candidacy criteria has also changed considerably and now includes children as young as 12 months old (and younger under certain conditions) and adults with retained low frequency hearing via the hybrid CI which uses a combination of electrical and acoustic stimulation.2
CIs allow children with deafness to attain speech outcomes on par with their normal‐hearing peers.3 In addition, CIs improve quality of life for adults with hearing loss as well as children with hearing loss and their families.4, 5 While data show that the majority of CI users obtain excellent hearing and speech recognition in noise outcomes, a minority do not achieve expected hearing levels. This variability occurs for both children3 and adults.6 Since the early years of cochlear implantation there have been attempts to determine factors why some users experience better, or worse, outcomes than predicted preoperatively.7
Some of the strongest predictors of postoperative CI outcomes are clinical factors associated with hearing loss including duration of deafness, socioeconomic status, and preoperative audiometric thresholds.8 The largest studies of clinical factors predicting postoperative CI outcomes showed that two different statistical models that considered up to nine different clinical factors simultaneously could account for 10.5% or 22% of variance in postoperative speech outcomes.6, 9 A less complex and more encompassing measure to explain postoperative CI outcomes has so far been out of reach.
From a clinical perspective, there are few relative contraindications to cochlear implantation, but there is general agreement that outcomes are poor in patients with an absent cochlear nerve or severely narrowed internal auditory canal.10 The lack of a cochlear nerve would prevent the electrical stimulus from the CI from reaching the brainstem. For these patients, auditory brainstem implantation has led to improved speech outcomes.11
As opposed to a complete lack of an auditory nerve, a decreased number of spiral ganglion (SG) neurons has also been hypothesized to affect CI outcomes. Otte et al and Nadol et al evaluated spiral ganglion counts in temporal bones of individuals with hearing loss of various causes.12, 13 These authors identified wide variability in SG counts and predicted that this may affect CI outcomes. More recently, Seyyedi et al were the first to show that SG counts significantly predicts CI outcomes in six patients with matched temporal bones.14 Therefore, the binary presence or absence of auditory nerve fibers appears to play an important role in CI outcomes. A growing set of published data is revealing that differences in auditory nerve or spiral ganglion health may be just as important.
In this review, we will examine published data which show that qualitative, not solely quantitative, deficits in the auditory nerve affect CI outcomes. As opposed to a complete lack of an auditory nerve, it follows that individuals with a damaged, altered, or relatively nonfunctional auditory nerve or spiral ganglion could also have relatively poor CI outcomes. These individuals may have poor spiral ganglion or auditory nerve “health” which may affect outcomes as transmission of neural signal from the stimulating electrode of the CI to the brain. Although currently available data are limited by a small number of patients with inconsistent reporting of CI outcomes, a better understanding of lesions that affect the health of the auditory synapse or auditory nerve will lead to improved CI outcomes.
Auditory Neuropathy Spectrum Disorder
Auditory neuropathy is a term used to describe individuals with hearing loss and normally functioning outer hair cells. These individuals will have hearing loss characterized by normal otoacoustic emission (OAE) or cochlear microphonics (CM), indicating normal cochlear function, coupled with abnormal transmission of auditory signal from the synapse to the brain as evidenced by altered auditory brainstem responses (ABRs). The site of lesion causing auditory neuropathy may involve the presynaptic site of release of glutamate in the hair cell, the synapse, the postsynaptic site of neurotransmitter stimulation, the site of initiation of the excitatory postsynaptic potential (EPSP) at the terminal dendrite, or sites along the spiral ganglion that affect transmission of the neural signal along the auditory nerve to the brainstem. In cases of auditory neuropathy, the inner hair cells (IHC) and outer hair cells (OHC) function irrespective of transmission of neural signal, and therefore these lesions result in auditory dyssynchrony. Due to this dyssynchrony, individuals typically will have difficulty with temporal processing of sound resulting in impaired speech perception and sound localization. Because there is an inability to effectively transmit the neural signal to the brain, increasing the sound stimulus level, through hearing aids for instance, may have limited benefit for these patients.
Our understanding of auditory neuropathy continues to expand due to an improved understanding of the molecular physiology of hearing. There is a wide array of physiologic defects in the auditory system that can cause auditory neuropathy, which has led to the introduction of a more broad term for any form of hearing loss and aberrant transmission of neural signal to the brain: auditory neuropathy spectrum disorder (ANSD, reviewed in Moser and Starr15). This includes lesions affecting the auditory synapse (auditory synaptopathy) or nerve (auditory neuropathy). Today, ANSD is the preferred and more accurate term but is often used interchangeably with auditory neuropathy or auditory dyssynchrony.
ANSD is caused by both genetic and environmental factors. As will be discussed below, there are 13 known genes that cause ANSD. Environmental causes of ANSD primarily affect the newborn including hyperbilirubinemia, thiamine deficiency, and hypoxia although noise‐induced and age‐related auditory neuropathy affect adults.15 ANSD is a relatively common cause of hearing loss, affecting between 1.2% and 8.4% of those with hearing loss depending on the population.16, 17, 18
It has long been hypothesized that individuals with auditory neuropathy are poor CI candidates as transmission of the signal from electrical stimulation of the spiral ganglion provided by the CI could be affected.19 One large study of subjects with ANSD showed generally positive, though somewhat variable, CI outcomes.20 Recent studies have confirmed that a nuanced perspective of ANSD is needed to understanding CI outcomes: the exact site of lesion causing hearing loss becomes key in understanding the impact on CI function. Some individuals will have qualitative or quantitative deficits to the spiral ganglion or cochlear nerve and are hypothesized to have poor outcomes with CIs. However, some individuals with ANSD have a genetic lesion affecting the synapse itself, which would be bypassed by a CI and therefore would be expected to have outcomes on par with an individual with a genetic lesion affecting the inner hair cells. For example, one study specifically examined the difference between children with ANSD due to cochlear nerve hypoplasia compared to children with ANSD due to other causes.21 They found that children with ANSD and cochlear nerve hypoplasia had significantly worse postoperative outcomes. Other studies have shown that the effects of ANSD on the developing cortex may play an important role in CI outcomes.22 This sort of physiologic site‐of‐lesion analysis is required to improve preoperative counseling for patients with ANSD prior to cochlear implantation.
In this review, we will provide an overview of the sites of lesion caused by genetic insults that cause ANSD and how they are theorized to affect CI outcomes. We will first provide a brief overview of the molecular physiology of the auditory system, with a special attention to how a CI functions within this system. We will then review known genetic lesions that negatively affect spiral ganglion health and review postoperative CI outcome data, when available, for each molecular lesion to the peripheral auditory system. Our aim is to provide an overview of this rapidly advancing field with the goal of improving CI outcomes for all individuals by furthering research in this paradigm for understanding the human auditory system.
Division of the Peripheral Auditory System into Three Components
From the perspective of CI physiology, the human peripheral auditory system can be divided into three conceptual components or partitions: sensory, synaptic, and neural (Fig. 1). The sensory component is responsible for mechanoelectrical transduction of physical sound waves in to electrical signals through the action of the inner hair cells of the organ of Corti. Deflection of the stereocilia of the hair cells allows ion channels to open and transgression of potassium and calcium into the inner hair cell. The tectorial membrane functions, in concert with the outer hair cells which are fixed to its outer surface, to frequency match and fine tune pitch.
Figure 1.
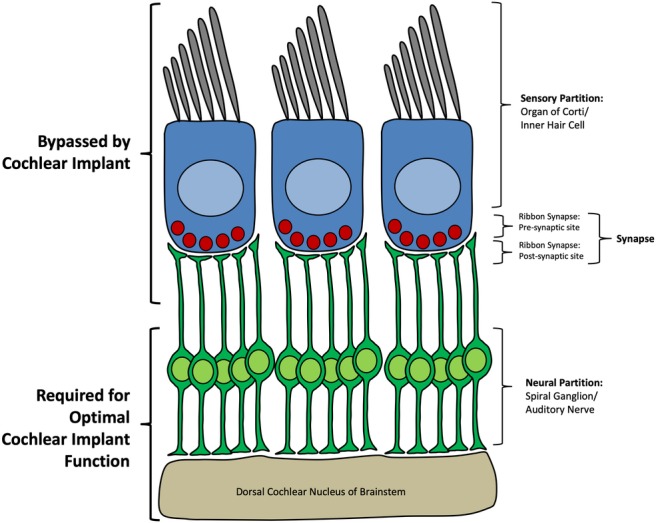
Overview of the peripheral auditory system with special attention to cochlear implant physiology. The inner hair cell and sensory partition as well as the presynaptic and postsynaptic portion of the synapse are bypassed by the cochlear implant whereas the auditory nerve, spiral ganglion, and brainstem are required for optimal cochlear implant function.
The synapse of the peripheral auditory system includes the presynaptic site (the base of the hair cell), the synapse itself, and the postsynaptic site (terminal dendrite of the spiral ganglion) (Fig. 1). The base of inner hair cells interface with terminal processes of spiral ganglion neurons. Presynaptic release of glutamate leads to transmission of sound signal to the spiral ganglion neurons and propagation of the signal centrally. The synapse between the inner hair cell and the spiral ganglion terminal process is composed of several unique features. First, each spiral ganglion neuron receives input from a single inner hair cell but each inner hair cell synapses with several spiral ganglion neurons. After deflection of inner hair cell stereocilia, the mechanoelectrical transduction channels located in the stereocilia allow cation influx. This triggers voltage‐dependent calcium channels near the synapse at the base of the inner hair cells to open. The influx of calcium leads to synaptic vesicle fusion and release of glutamate at the synapse between inner hair cell and spiral ganglion. This synapse is able to encode a high degree of temporal precision because of graded release of glutamatergic vesicles via the ribbon synapse. The ribbon synapse is composed of a long tether which holds synaptic vesicles close to the presynaptic release point in inner hair cells as it is anchored to the cell membrane at the presynaptic active zone in close approximation for release at the synaptic cleft. This presynaptic organization allows for transmission of a signal that is finely temporal encoded (up to 1 kHz) and indefatigable. The synaptic component also includes the postsynaptic glutamate receptors at the terminal dendrite of the spiral ganglion.
The neural partition of the human auditory system (Fig. 1) is comprised of the distal neurite of the spiral ganglion, the somata of the spiral ganglion which reside in Rosenthal's canal, and the central processes of the spiral ganglion which form the cochlear partition of the vestibulocochlear nerve (cranial nerve VIII) and branch to form synapses within the dorsal and ventral cochlear nucleus of the midbrain. Mechanistically the neural partition therefore encompasses spike initiation of the EPSP at the terminal dendrite. Lossless conduction of this signal occurs through the myelinated distal and central dendrites of the spiral ganglion. Central components of the human auditory system include the brainstem and auditory cortex of the temporal lobe which are together responsible for maintenance of the tonotopic organization of sound and processing of sound and speech, respectively.
The CI bypasses the sensory and synaptic partitions by directly stimulating the spiral ganglion somata leading to transmission of electrical signal to the midbrain (Fig. 1). Thus, it follows that the health of the organ of Corti or synapse will not affect outcomes of cochlear implantation. Conversely, the health of the spiral ganglion (or more generally the auditory nerve), the synapse between the spiral ganglion and the cochlear nucleus, midbrain, or auditory cortex may negatively affect the electrical transmission of sound provided by a CI and theoretically could lead to suboptimal CI outcomes.
EVALUATION OF SPIRAL GANGLION HEALTH
In 2002, Sininger and Trautwein wrote that “The exact nature of the lesions in patients with [auditory neuropathy] cannot be determined while they are alive.”23 Fortunately, due to advancements in our understanding of electrophysiology of the peripheral auditory system, this is no longer true. ABR and OAEs remain a mainstay of diagnosing ANSD. In some cases, electrocochleography (ECochG), which measures cochlear and early neural responses as acoustic signals are transmitted from the cochlea to the brain, can further confirm the site of lesion.24 ECochG can be performed via transcanal approach by placing a probe in the tympanic membrane, transtympanically through a probe placed near the round window, or during CI surgery at which time intraoperative intracochlear recordings can be made using electrical stimulus or auditory stimulus.25, 26, 27, 28 For individuals who have received a CI, the implant itself can be used as both the recording and stimulating electrode29 including the use of auditory stimuli in patients with residual hearing after cochlear implantation.30 ECochG measures used to determine the precise site of lesion in individuals with ANSD include cochlear microphonic, summating potential, auditory nerve neurophonic, and adaptation at low and high frequencies.15, 24
Assessment of spiral ganglion health using intraoperative ECochG measurements has become one of the best predictors of CI outcomes. One study showed that up to 47% of postoperative CI outcomes could be predicted from ECochG measures in adults.26 In pediatric CI recipients, up to 50% of the postoperative outcome variance could be predicted using intraoperative ECochG if clinical factors were also controlled for.27, 28 However, the primary disadvantage to intraoperative ECochG is that it is intraoperative: it does not allow for preoperative counseling.
A method for assessing SG health that can be done preoperatively with high reliability would provide valuable patient counseling information prior to implantation and could allow targeted programming and rehabilitation postoperatively for at‐risk individuals in particular. Precision CI programming could be especially important in children who are not able to verbalize satisfaction with hearing. This type of precision programming has already shown some progress for patients with ANSD.31, 32 And in the future, an improved understanding of CI genetics will lay the groundwork for tailor‐made CIs and implantable devices that provide molecular therapies.
MOLECULAR PHYSIOLOGY OF HEARING AND DEAFNESS, A CI PERSPECTIVE
Over the past 30 years, our understanding of genetics of deafness has greatly expanded our view of the molecular physiology of the auditory system. Deafness is extremely genetically heterogeneous. Unlike a disease like cystic fibrosis, where the vast majority of cases are due to variants affecting a single gene, there are more than 110 known deafness genes with more than 7,000 known deafness causing variations (http://deafnessvariationdatabase.org; http://hereditaryhearingloss.org). Genetic variants have been identified that cause deafness by affecting each component of hearing from sensory transduction to transmission of sound to the brain. This extreme genetic heterogeneity underscores the fragility, complexity, and importance of human hearing.
Approximately 80% of congenital hearing loss in the United States is due to a genetic cause and genetics are likely to contribute a significant partition of postlingual and adult onset hearing loss as well.33 Understanding these genes and how, when mutated, they cause deafness, provides a high resolution understanding of the molecular mechanisms of CI outcomes. We will briefly review CI outcomes for genes affecting the sensory partition of the peripheral auditory system and then focus individually on each of the genes known to cause ANSD by affecting the synapse or the spiral ganglion itself. Converging data supports the hypothesis that genetic mutations negatively affecting the spiral ganglion itself lead to worse CI outcomes, presumably through worsened spiral ganglion health.
GENETIC LESIONS TO THE SENSORY (COCHLEAR) PARTITION
Defects in any of the sensory components of the organ of Corti including inner hair cells, outer hair cells, the tectorial membrane, supporting cells, or cells of the stria vascularis can lead to deafness. Deafness due to a mutation in GJB2 was first described in 1997, and since that time has been shown to be one of the most frequent causes of congenital severe‐to‐profound hearing loss.34 The GJB2 gene encodes connexin 26, which forms a pore that allows the flow of K+ and maintains the ionic gradient of the scala media and allows the mechanotransduction of sound. Mutations in GJB2 have been shown in multiple studies to be associated with excellent CI outcomes.35, 36, 37, 38 Similarly, good CI outcomes have been reported in individuals with other genetic lesions affecting the sensory apparatus including mutations in genes that affect the cells of the stria vascularis (SLC26A4 38), the tip‐links of the stereocilia of the inner hair cells (CDH23 39, 40), the unconventional myosin motor proteins of the hair cells (MYO7A 40), and the stereocilia structure itself (LOXHD1 41), among others. One large study of 173 individuals who underwent cochlear implantation showed that any mutation in a deafness‐causing gene affecting the organ of Corti was associated with good CI outcomes.42 Our group performed a statistical analysis of 155 individuals who underwent cochlear implantation and found that those patients with deafness due to mutations in the sensory partition of the peripheral auditory system had postoperative CI outcomes significantly better than other groups.43 In summary, for patients with identifiable genetic site of lesion in the sensory partition of the peripheral auditory system, CI outcomes are expected to be excellent as the CI bypasses the defective partition of the auditory system.
GENETIC LESIONS TO THE SYNAPSE
Auditory synaptopathy is the term used for ANSD due to a defective or poorly functioning synapse. Persons with hearing loss caused by a genetic mutation affecting the synapse would be expected to have normal OAE response but an altered ABR, as in other forms of ANSD. The auditory synapse is bypassed by the CI and so we would hypothesize that individuals with a defect in the synapse would have good performance with a CI (Fig. 1).
Auditory synaptopathies can be distinguished from auditory neuropathies by ECochG: individuals with synaptopathy appear to have enhanced adaptation to frequency specific tones whereas those with neuropathy had abnormal low frequency adaptation.44 The inner hair cell ribbon synapse is unique to the auditory system. Therefore, genetic auditory synaptopathies typically only cause deafness; in contrast, genetic neuropathies frequently affect other peripheral neurons as well leading to syndromic phenotypes. Auditory synaptopathies are rare and so studying these genetic lesions is of utmost importance to improving our understanding the human auditory system and CI outcomes.
Calcium influx at the base of the inner hair cell near the ribbon synapse is mediated via the Cav1.3L‐type Ca2+ channel, encoded by the CACNA1D gene. Otoferlin, encoded by the gene OTOF, is one protein responsible for regulating exocytosis of glutamatergic vesicles at the presynaptic site. The vesicular glutamate transporter type 3 (VGLUT3 gene) is responsible for glutamate uptake at the postsynaptic site. Synaptopathies can therefore be classified specifically based on the site of lesion to the synapse: pre‐ or postsynaptic. This high resolution classification will assist with analysis of electrophysiologic data in these individuals. Specific genetic lesions that cause pre‐ or postsynaptic auditory synaptopathy will be described in detail below and are summarized in Table 1.
Table 1.
Genes identified to cause auditory neuropathy spectrum disorder including auditory synaptopathy and neuropathy.
| Functional Component | Site of Lesion | Gene | Protein | Function | Phenotype | Total Reported Deafness‐Causing Mutations | CI Outcomes | CI Outcomes References |
|---|---|---|---|---|---|---|---|---|
| Synapse | Presynaptic—inner hair cell | OTOF | Otoferlin | Calcium sensor involved in exocytosis of synaptic vesicles | Autosomal recessive NSHL and temperature‐sensitive deafness | 113 | Good | 42, 45, 46, 47, 48 |
| CACNA1D * | Calcium voltage‐gated channel subunit alpha1 D (Cav1.3) | Calcium influx at the presynaptic site | Sinoatrial node dysfunction and deafness syndrome (SANND) | 1 | — | — | ||
| CABP2 * | Calcium binding protein 2 | Interaction with calcium channels to alter voltage dependence at the presynaptic site | Autosomal recessive NSHL | 3 | — | — | ||
| SLC17A8 | VGLUT3: vesicular glutamate transporter 3 | Involved in presynaptic vesicular glutamate uptake and release | Autosomal dominant NSHL | 1 | Good | 43 | ||
| Postsynaptic—spiral ganglion | DIAPH3 | Diaphanous formin 3 | Involved in spiral ganglion terminal dendrite contact with inner hair cells | Autosomal dominant NSHL | 1 | Good | 49 | |
| OPA1 | Mitochondrial dynamin related GTPase protein | Involved in spiral ganglion terminal dendrite growth | DOA, or DOA and auditory neuropathy (DOA+) | 6 | Good | 24, 50 | ||
| ROR1 | Receptor tyrosine kinase‐like orphan receptor | Modulates spiral ganglion terminal dendrite growth | Common cavity malformation and auditory neuropathy, autosomal recessive | 1 | Good | 51 | ||
| ATP1A3 | Alpha‐3 catalytic subunit of the Na+/K+ ATPase transmembrane ion pump | Maintains resting membrane potentials in spiral ganglion terminal dendrites | NSHL and CAPOS syndrome | 1 | Good | 52 | ||
| Auditory nerve | Spiral ganglion cell bodies and proximal axons | TIMM8A | Translocase of inner mitochondrial membrane 8A | Defective mitochondrial transport leads to degeneration of auditory nerve | Mohr‐Tranebjaerg syndrome (DDON) | 6 | Poor | 53, 54 |
| AIFM1 | Apoptosis‐inducing factor 1 | Oxidative phosphorylation and respiratory chain | X‐linked auditory neuropathy and Cowchock syndrome | 11 | — | |||
| NARS2 | Mitochondrial asparagine‐tRNA ligase | Mitochondrial respiratory chain, oxidative phosphorylation | Autosomal recessive NSHL; Leigh syndrome (progressive neurodegenerative disease) | 2 | — | |||
| MPZ, PMP22 | — | — | Charcot‐Marie‐Tooth disease, hereditary sensory and motor neuropathy | — | Poor | 54 |
There is no current clinical evidence or data from animal models implicating this gene as a cause of ANSD, but in vitro analysis supports the hypothesis that this gene causes ANSD.
ANSD = auditory neuropathy spectrum disorder; CAPOS = cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss; CI = cochlear implant; DDON = deafness‐dystonia‐optic neuropathy; DOA = dominant optic atrophy; NSHL = non‐syndromic hearing loss.
OTOF—Presynaptic Synaptopathy
Mutations in the gene OTOF were first identified as a cause of autosomal recessive nonsyndromic deafness, DFNB9, nearly 20 years ago.55 DFNB9 deafness is severe‐to‐profound congenital or prelingual deafness characterized by present OAEs and abnormal ABR. OTOF was the first identified genetic cause of ANSD. In addition to nonsyndromic hearing loss (NSHL), mutations in OTOF can also cause temperature‐sensitive deafness whereby an elevation in body temperature can severely worsen hearing loss, in particular word understanding and tinnitus, but this then returns to close to normal hearing on body cooling.56 Similar mutations in OTOF may lead to disparate audiologic outcomes, as evidenced by one study of two siblings with ANSD and the same OTOF mutation but significantly different speech reception scores, electrically evoked potentials, and CI outcomes.57
The otoferlin protein acts as a calcium sensor, which is required for the final stages of exocytosis of the glutamatergic vesicles in inner hair cells at the ribbon synapse, but the exact mechanism by which it performs this function is unknown.58 Otof knockout mice are able to complete all stages of exocytosis aside from the final step, glutamate release. Detailed studies on humans with OTOF mutations show that these individuals have progressive auditory fatigue, which likely represents a failure of the normally indefatigable auditory synapse.44 These results show the mechanisms by which genetic alterations to OTOF can affect the temporal representation of auditory stimuli and cause auditory synaptopathy.
To date, a total of 113 deafness‐causing mutations in OTOF have been reported which encompass missense as well as loss of function changes (http://deafnessvariationdatabse.org). OTOF mutations are particularly prominent in the Spanish population where up to 8% of autosomal recessive nonsyndromic hearing loss is due to OTOF mutations likely due to a founder effect.45 In a large multiethnic population of 1,119 individuals with hearing loss, mutations in OTOF accounted for 2.4% of diagnoses, the 12th most common gene causing deafness in this cohort.34
Available data from multiple case‐series show that CI outcomes for individuals with OTOF are excellent and typically on par with individuals with genetic mutations affecting the sensory partition.42, 45, 46, 47, 48 A recent review identified 32 different patients from 11 published studies with different OTOF mutations, all of whom had good or excellent CI outcomes.59 The data from OTOF are the strongest to confirm that auditory synaptopathies are bypassed by a CI and therefore have good CI outcomes.
CACNA1D—Presynaptic Synaptopathy
CACNA1D encodes the voltage‐gated calcium channel subunit alpha1 D, also known as Cav1.3. Mutations in the gene CACNA1D cause a syndromic form of hearing loss called sinoatrial node dysfunction and deafness syndrome (SANND syndrome).60 The affected calcium channel, Cav1.3, is expressed in inner and outer hair cells, cardiomyocytes, neuroendocrine cells, and neurons resulting in the syndromic deafness when loss of function mutations occur. The gene encodes for part of the pore‐forming subunit of the calcium channel. Loss of function mutations of CACNA1D lead to impaired synaptic transmission at the ribbon synapse in knockout mice.61, 62
Only one mutation found in two Pakistani families has been identified to cause SANDD syndrome. Deafness caused by this mutation is congenital and severe to profound and accompanied by bradycardia. No evaluation of auditory neuropathy was provided but based on molecular characterization, these patients would be expected to display characteristics of ANSD. Further information on CI outcomes is pending but we would hypothesize that because this is a synaptopathy patients would have an excellent CI outcome.
CABP2—Presynaptic Synaptopathy
Calcium binding proteins interact with calcium channels to alter the voltage‐dependence of the voltage‐gated pore at the presynaptic site at the base of hair cells. Mutations in CAPB2 cause autosomal recessive nonsyndromic hearing loss at the DFNB93 locus.63 A lack of Ca2+ influx leads to impaired vesicular release of glutamate and an auditory synaptopathy. The original reported mutation causing deafness was identified in an Iraqi family with flat or cookie‐bite moderate to severe prelingual onset with Marfanoid features but no other syndromic features.63 A total of three deafness causing mutations in CABP2 have been published but there have been no reported CI outcomes.
SLC17A8—Presynaptic Synaptopathy
Mutations in the gene SLC17A8 cause autosomal dominant nonsyndromic hearing loss at the DFNA25 locus.64 Affected individuals have progressive high‐frequency sensorineural hearing loss. There are only three reported pathogenic mutations in SLC17A8 causing nonsyndromic hearing loss including the original report and two in Korean families.64, 65, 66 There are no reports of features of auditory neuropathy in these families but detailed electrophysiologic studies for these patients have not been published.
SLC17A8 encodes for VGLUT3, the vesicular glutamate transporter 3 protein. Mice with mutations in Slc17a8 have been shown to have auditory neuropathy.67 Hearing loss in Slc17a8 knockout mice is due to the lack of glutamate exocytosis by inner hair cells and therefore a lack of synaptic transmission at the IHC‐SG terminal dendrite synapse.64, 67 Importantly, viral mediated transfer of the Slc17a8 gene to IHCs rescues the hearing loss in these mice, representing an avenue for future gene therapy in humans.68
One study that included one patient with SLC17A8 deafness showed a relatively good CI outcome.43 This is consistent with the molecular physiology of this protein directly affecting the presynaptic site. Further studies are needed to confirm this finding.
DIAPH3—Postsynaptic Synaptopathy
Mutations in DIAPH3 cause nonsyndromic auditory neuropathy via a postsynaptic lesion. To date, there is only a single reported deafness‐causing mutation.69 Affected individuals have postlingual onset progressive hearing loss with initially retained OAEs that diminish over time.49 Electrophysiologic studies localized the lesion to the distal neurite, synapse, or inner hair cell.49 The DIAPH3 gene encodes the diaphanous formin 3 gene, overexpression of which appears to affect distal neurite contact with inner hair cells.69 Three individuals with DIAPH3 deafness have received CIs and had good outcomes, as could be hypothesized based on the site of lesion at the distal neurite.49
OPA1—Postsynaptic Synaptopathy
Mutations in the gene OPA1 cause dominant optic atrophy (DOA) or syndromic dominant optic atrophy (DOA+). DOA+ is characterized by optic atrophy and auditory neuropathy. Mutations that cause haploinsufficiency, or lack of a full component of the optic atrophy 1 protein, result in DOA whereas missense mutations that act via a dominant negative mechanism to inhibit the activity of the normal remaining protein cause DOA+.70 There are six reported DOA+ mutations, but the most commonly identified mutation is p.Arg445His.
The OPA1 gene encodes for the mitochondrial dynamin‐related GTPase protein which is crucial for mitochondrial functioning. It is hypothesized that mutations causing DOA+ result in degeneration of the terminal axons of spiral ganglion neurons and thus cause a postsynaptic auditory synaptopathy.70 Individuals with DOA+ have preserved OAEs but abnormal ABRs. The hearing loss is moderate to severe and variable in which frequencies are affected predominately, even within the same family.50, 70 Several individuals with DOA+ who received CIs were found to have good outcomes which is consistent with the hypothesized site of lesion at the peripheral axons of the spiral ganglion, preventing appropriate synaptic transmission, yet the neuronal somata are presumably preserved and responsive to electrical stimulation.70
ROR1—Postsynaptic Synaptopathy
ROR1 encodes for the receptor tyrosine kinase‐like orphan receptor, a protein which is important for neurite outgrowth. Two siblings from a Turkish family with cochlear common cavity malformation, moderate to severe downsloping SNHL, and normal OAE response, indicative of auditory neuropathy were found to have a mutation in the gene ROR1.51 This gene encodes for receptor tyrosine kinase‐like orphan receptor 1, a transmembrane protein involved in cell signaling via the Wnt pathway. Knockout mice showed multiple other defects including urogenital and skeletal abnormalities as well as growth retardation, but also showed common cavity malformation and deafness. Examination of the cochlea in knockout Ror1 mice showed a lack of innervation of the inner hair cells by peripheral axons of spiral ganglion neurons as well as aberrant growth of spiral ganglion neuron axons toward outer hair cells. Therefore, mutations in ROR1 cause an auditory synaptopathy affecting the postsynaptic site.51 There is only one reported deafness‐causing mutation in ROR1. One of the two affected individuals received a CI with good outcomes.51
ATP1A3—Postsynaptic Synaptopathy
Mutations in ATP1A3 cause CAPOS syndrome (cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss) as well as NSHL without any other neurologic features.52 Detailed audiometric analysis of two individuals with mutations in ATP1A3 showed auditory synaptopathy likely affecting the postsynaptic site.52 These two patients both showed excellent postoperative outcomes after cochlear implantation. ATP1A3 encodes the α3 catalytic subunit of Na+/K+ ATPase (adenosine triphosphotase), which is a membrane‐bound transporter that uses ATP to maintain a resting transmembrane potential in nerve dendritic terminals. This transporter was shown to be expressed abundantly in the peripheral axons of spiral ganglion neurons in rats.71 The exact mechanism(s) whereby mutations in ATP1A3 cause auditory synaptopathy remain unknown, but it is hypothesized that EPSPs are affected due to altered resting membrane potential.
GENETIC LESIONS AFFECTING THE SPIRAL GANGLION AND AUDITORY NERVE
Spiral ganglion neurons are bipolar, with cell bodies located in the modiolus of the cochlea, peripheral axons running through the spiral limbus toward the row of inner hair cells of the cochlea, and proximal axons synapsing at the midbrain in a tonotopical organization.72 Each spiral ganglion neuron is frequency‐tuned based, in part, on the inner hair cell from which it receives afferent information. There are multiple spiral ganglion peripheral fibers synapsing with each inner hair cell. The axons of spiral ganglion neurons are ensheathed by myelinating Schwann cells along their distal, peripheral axons projecting to the organ of Corti and the early partition of the central projections. As the proximal, central axons project to the brainstem, there is a transition from peripheral myelin by Schwann cells to central myelin by oligodendrocytes.73 In response to glutamatergic stimulation at the synapse, postsynaptic excitatory potentials at the peripheral axon lead to Na+ influx via a large number of channels in a graded fashion that allows for precise temporal coding of acoustic stimuli.
Genetic lesions that affect the signal transmission to the brain lead to a loss of temporal precision and dyssynchrony, or a degraded potential and altered perception and/or understanding of sound, either of which cause deafness through auditory neuropathy. There are several genes identified that cause auditory neuropathy and these are detailed below. A genetic lesion that specifically affects the auditory nerve would be not be bypassed by a CI and could theoretically affect CI outcomes. Genes that, when mutated, cause auditory neuropathy often lead to syndromic forms of hearing loss particularly hereditary neuropathies, as other peripheral neurons are often affected in addition to the spiral ganglion.
TIMM8A
Mohr‐Tranaebjaerg Syndrome, also known as deafness‐dystonia‐optic neuropathy (DDON) syndrome is a progressive neurodegenerative genetic disorder characterized by early childhood onset auditory neuropathy, dystonia and ataxia that present in the second decade, decreased visual acuity starting in the third decade, and dementia starting in the fifth decade.74 Each of these clinical findings is progressive. There are also psychiatric features such as paranoia. Mutations in TIMM8A cause DDON in an X‐linked recessive inheritance pattern.
Pathologic evaluation of human temporal bones in patients with DDON shows near total loss of cochlear neuronal cells and severe loss of vestibular neurons in four subjects examined. The disease process is therefore characterized as postnatal progressive degeneration of neurons, including cochlear, vestibular, and optic neurons.75 DDON therefore represents the prototypical auditory neuropathy. There are 12 reported pathologic mutations in the TIMM8A that cause DDON.
One report of a patient with DDON implanted with a CI at age 4 showed that after 2 years, he had only marginal performance with the CI even at high current levels.53 In patients for whom cochlear implantation has failed or for whom cochlear implantation is not feasible due to cochlear nerve aplasia or hypoplasia, auditory brainstem implantation is an option; one study of DDON patients showed reasonable outcome after removal of CIs and replacement with auditory brainstem implants.54
AIFM1
Mutations in the gene AIFM1 cause X‐linked auditory neuropathy as well as Cowchock syndrome, a progressive neuromuscular disorder associated with deafness and cognitive impairment. Individuals with isolated X‐linked auditory neuropathy also show a delayed onset peripheral sensory neuropathy presenting as extremity numbness, unsteadiness, and areflexia.76 To date, there are 11 reported deafness‐causing mutations.
AIFM1 encodes apoptosis‐inducing factor 1, a flavoprotein located in the mitochondrial intermembrane space and localized to inner and outer hair cells as well as spiral ganglion neurons. The AIFM1 protein plays a role in oxidative phosphorylation, redox control, and respiratory chain activity in healthy cells. Several patients with these mutations have been shown to have cochlear nerve hypoplasia which appears to be delayed in onset (not congenital).76 There are no reports of cochlear implantation in these subjects, but they would be expected to have poor outcomes, particularly if there is cochlear nerve deficiency.
NARS2
Mutations in NARS2 cause autosomal recessive nonsyndromic auditory neuropathy as well Leigh Syndrome, an early‐onset progressive neurodegenerative disorder affecting the central nervous system (CNS) with symptoms dependent on involved areas of the CNS.77 NARS2 encodes the mitochondrial asparagine‐tRNA ligase protein which is involved in energy metabolism including the respiratory chain complexes. In individuals with NARS2 mutations and Leigh syndrome, initial evaluation showed absent ABRs but present CM, however by 11 weeks OAEs were absent.77 There is only one family reported with isolated deafness and NARS2 mutations. This family had congenital severe to profound SNHL but detailed electrophysiologic information including ABR and OAE was not available.77
The NARS2 protein is expressed in the spiral ganglion as well as several cells of the organ of Corti. It is hypothesized that deafness is due to cellular damage that may affect spiral ganglion neurons first prior to affecting hair cells.77
Hereditary Demyelinating Neuropathies
Auditory neuropathy is a common finding in Charcot‐Marie‐Tooth disease (CMT). CMT is the most common inherited neurologic disorder, affecting 1 in 2,500 people and characterized by progressive motor and sensory neuropathy. There are several types of CMT which vary based on severity, involved nerves, and inheritance, and to date more than 80 genes have been identified to cause CMT.78 Two genes in particular, MPZ and PMP22, have been associated with CMT involving auditory neuropathy. Temporal bone histology of patients with autosomal dominant ANSD and demyelinating CMT showed normal cochlear hair cells but a marked degeneration of spiral ganglion neurons.79
One report of an individual with CMT who underwent cochlear implantation showed postoperative speech discrimination score of 54%, indicating a relatively poor result but that that some improvement in possible even in the face of likely auditory nerve degeneration.80 Other hereditary progressive motor and sensory neuropathies with auditory neuropathy such as Friedreich ataxia are likely to have similar lesions within the spiral ganglion.81 Cochlear implantation for these individuals will likely have less‐than ideal postoperative outcomes, but can still be offered as a treatment option for deafness.82
GENES WITH POSSIBLE EFFECTS ON THE AUDITORY SYNAPSE OR SPIRAL GANGLION
TMPRSS3
Mutations in the gene TMPRSS3 cause two different types of autosomal recessive NSHL: congenital severe‐to‐profound deafness (DFNB10), and postlingual progressive deafness (DFNB8).83, 84, 85 The type of deafness depends on the type of mutation, and there are clear genotype‐phenotype correlations between more damaging mutations with early onset and more severe hearing loss.86, 87 To date, there are 41 reported deafness‐causing mutations in TMPRSS3. The function of the protein encoded by TMPRSS3, transmembrane serine protease 3, is unknown. However, multiple studies have shown the protein to be expressed in inner and outer hair cells as well as the spiral ganglion, including in dissected human temporal bones.88, 89, 90, 91 A recent study showed that TMPRSS3 is expressed highly in type II spiral ganglion neurons in mice.92 TMPRSS3 function is required for hair cell survival in mice90and also spiral ganglion neuron survival in vitro.93
There are no reports that TMPRSS3 mutations cause ANSD. However, DFNB8 mutations in TMPRSS3 are the most common cause of genetic deafness in a large cohort of CI users with postlingual onset deafness.43 Therefore, understanding the role of TMPRSS3 in the peripheral auditory system is critical. CI outcomes with TMPRSS3 mutations have been variable, with some studies showing good outcomes86, 94 while others have shown below‐average outcomes.43, 95, 96 Understanding deafness due to TMPRSS3 mutations is complicated by multiple phenotypes which may indicate multiple molecular lesions. One hypothesis is that more severe TMPRSS3 mutations may predominantly affect the hair cells, while more mild mutations may primarily affect the spiral ganglion. Importantly a recent report on patients with TMPRSS3 mutations and hearing preservation CIs demonstrated disrupted neural responses and relatively well‐preserved cochlear microphonic responses.30 Further research is needed to determine the exact mechanisms by which mutations in TMPRSS3 cause deafness and how these mechanisms may affect CI outcomes.
TBC1D24
Mutations in the gene TBC1D24 cause nonsyndromic hearing loss, genetic epilepsy, and Deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS) syndrome (Table 2). Mutations in TBC1D24 have been shown to cause both congenital severe to profound autosomal recessive nonsyndromic SNHL97 as well as autosomal dominant progressive nonsyndromic SNHL.98 There are 25 reported mutations in TBC1D24 including nonsyndromic hearing loss, DOORS syndrome, and epilepsy. None of the affected individuals from these families had epilepsy.
Table 2.
Genes hypothesized to be involved in synaptic or spiral ganglion function but with unclear function.
| Gene | Diagnosis | Deafness Onset | Deafness Severity | Protein | Protein Expression | Protein Function | CI Outcomes |
|---|---|---|---|---|---|---|---|
| TMPRSS3 | Autosomal recessive NSHL | Prelingual (DFNB10) or postlingual (DFNB8) | Severe to profound (DFNB10) or progressive high frequency (DFNB8) | Transmembrane serine protease 3 | Inner hair cells, outer hair cells, type II spiral ganglion cells | Unknown | Variable |
| TBC1D24 | Autosomal recessive or dominant NSHL; epilepsy; DOORS | Congenital or postlingual | Severe to profound or progressive | TBC1D24 protein | Inner hair cells outer hair cells, spiral ganglion neurons | Involved in neural maturation in the cortex | Unknown |
| DFNB59 | Autosomal recessive NSHL | Congenital | Severe to profound | Pejvakin | Inner hair cells, outer hair cells, spiral ganglion cells | Functions in peroxisomes which protect cells from reactive oxygen species | Unknown* |
One case‐control study found that a polymorphism in DFNB59, p.G292R, was associated with worse cochlear implant outcomes.104
CI = cochlear implant; DOORS = deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures; NSHL = non‐syndromic hearing loss.
The TBC1D24 gene encodes the Tre2‐Bub2‐Cdc16 (TBC) domain‐containing RAB‐specific GTPase‐activating protein 24 which is expressed in inner and outer hair cells as well as in the spiral ganglion.97, 98 This protein has been shown to be crucial for distal neurite outgrowth and maturation of cortical neurons.99 Therefore, mutations in TBC1D24 would be hypothesized to cause a postsynaptic auditory synaptopathy.
Whether mutations in TBC1D24 cause ANSD is still unknown as detailed physiologic analysis of the affected families including OAE and ECochG are not available. The TBC1D24 gene is a good candidate for auditory neuropathy given its involvement in the CNS (epilepsy) and expression in the spiral ganglion, but further research is needed to determine the function of the TBC1D24 gene in the human auditory system.
DFNB59
Mutations in the gene DFNB59 were identified as the second genetic cause of auditory neuropathy (Table 2).100 Both affected individuals and knock‐in mice showed preserved OAEs and altered ABRs. However, a second study showed that affected individuals had no signs of auditory neuropathy with a lack of OAEs and CM,101 and a second Dfnb59 mutant mouse lacked OAEs.102 The protein encoded by DFNB59, pejvakin, plays a role in formation of peroxisomes which protect cells from damage during antioxidant response.103 Pejvakin is expressed in the inner and outer hair cells as well as spiral ganglion neurons.103 The exact role of DFNB59 in the peripheral auditory system is unclear. To date, there are 16 reported deafness‐causing mutations in this gene, two of which have been reported to cause auditory neuropathy.100 There are no reports of CI outcomes with individuals with deafness causing by DFNB59 mutations. However, one case‐control study showed that a polymorphism in the DFNB59 gene, p.G292R (rs79399438) which is present in 2.9% of individuals, is associated with worse CI outcomes.104 Further research is clearly needed to better understand the role of pejvakin in the auditory system including whether it truly causes an auditory neuropathy phenotype.
CONCLUSIONS
Aside from the binary absence or presence of a cochlear nerve, it is becoming apparent that all CI candidates will have a broad spectrum of auditory nerve functional ability. Assessment of the “health” of the spiral ganglion and auditory nerve has become a key focus of predicting CI outcomes.15, 24, 30
Many studies have attempted to determine if cochlear implantation will benefit individuals with auditory neuropathy. Initially, researchers felt that auditory neuropathy was a contraindication to implantation.19 However, there were some initial reports of success of cochlear implantation in these patients.105, 106 Subsequently, several studies have found worse CI outcomes in patients with ANSD; yet despite poorer outcomes compared to typical CI recipients, patients with ANSD often still benefit from cochlear implantation.21, 107 Other studies, including a systematic review, have shown more variable outcomes in patients with ANSD following cochlear implantation.108, 109 A primary flaw in studies of patients with ANSD and CIs is that the studies do not precisely define the site lesion involved in the individuals studied. The studied groups are composed of an unknown number of individuals with neuropathy or synaptopathy which, as described here, are expected to have different CI outcomes.
As described in this review, the precise site of lesion causing neuropathy can help to determine at least a portion of expected postoperative outcomes. Preferably, a molecular diagnosis along with physiologic data from ECochG to determine the exact site of lesion should be available for each individual to be studied. Only then we can accurately study postoperative CI outcomes in individuals with ANSD. An ideal study would compare large numbers of individuals with specific genetic forms of deafness, controlled for clinical variables including duration of deafness, time of implantation, socioeconomic status, and type of CI. Due to the extreme genetic heterogeneity of deafness, this will require a large number of patients to obtain groups large enough for comparison.
The data are currently limited to a small number of patients with variable tests used for diagnosis and evaluation of outcome. Accordingly, any study designed to report outcomes for patients with ANSD and cochlear implantation should include: 1) a detailed clinical history from patient including any family history of peripheral neuropathy, vision loss, or vestibular deficits; 2) audiologic data: degree of hearing, age of onset, speech recognition scores in quiet and in noise; 3) electrophysiologic data: OAE, ABR, ECochG including CM, summating potential, auditory nerve neurophonic, and adaptation; 4) CI device implanted; and 5) widely used postimplant measures including word recognition scores in quiet and in noise. In addition, genetic data should also be available for these patients. Such detailed information will aid in comparisons between studies for these relatively rare disorders in the future.
Another complicating factor in studying ANSD is that in approximately 20–30% of individuals with ANSD, the OAE response diminishes over time.15 This is certainly the case for the most studied gene, OTOF,59 but also appears to be the similar in other studied auditory synaptopathies and neuropathies.15, 59 This has implications for the evaluation of noncongenital cases and adults with deafness. In many instances, the physiologic determinates currently used to diagnose auditory neuropathy, namely OAE, may therefore not be available. The cochlear microphonic is another physiologic measure to evaluate for ANSD, but this test is currently not used for screening. This underscores the importance of molecular genetic testing for individuals, including newborns and children, as well as adults, who are undergoing cochlear implantation.
Further research is also needed on the effects of genetic lesions to the auditory cortex. Several known deafness genes have known effects on the midbrain and auditory cortex, including DFNB59, CACNA1D, and KCNQ4.110 Two genes known to be involved in the hair cell tip‐link, PCDH15 and CDH23 also are required for interneuron development in the auditory cortex in mice.111 Further research is required to determine if mutations in these genes affect CI outcomes due to lesions in central auditory circuits.
From a practical standpoint, the advances in our understanding of CIs and ANSD presented in this review will generally not alter patient care by CI surgeons today. However, in the near future, as more tailored CI devices and molecular therapies emerge an understanding of site of lesion to the auditory system will be crucial. Today, CI surgeons can consider the effect of genetic or other lesions to the auditory nerve when counseling patients about CI outcomes. However, at the current stage of research, even for patients with mutations affecting the auditory nerve, a CI would still be recommended given improved hearing outcomes in almost all patients with a CI. CI surgeons are encouraged to pursue genetic testing in evaluation of deafness, particularly children, as recommended by recent guidelines.112 To evaluate for mutations affecting the auditory nerve or synapse, a multigene comprehensive panel would be required as the genes discussed above are not typically included in single gene tests.113 Importantly, the CI surgeon should keep in mind that the site of lesion will not be the only determinate of CI outcome, as even two siblings with the same mutation causing ANSD can have markedly different audiologic measures and CI outcomes.57
A paradigm has emerged where the CI surgeon needs to consider not only clinical factors such as type and duration of deafness but molecular and physiology factors such as the health of the cochlea, synapse, spiral ganglion, and auditory nerve to provide the best outcomes for patients. This evaluation typically begins with molecular diagnosis of deafness and will often require multidisciplinary discussion with audiologists and geneticists. As our understanding of the physiology of hearing and CIs has improved, the importance of these factors becomes even more apparent. Such research lays the groundwork for targeted molecular therapies as well as custom CIs tailored to a specific site of lesion.
AUTHOR CONTRIBUTIONS
Both authors conceived of and wrote and edited the final manuscript.
Funding: This study was supported by National Institutes of Health grants R01 DC012578 (MRH) and UL1TR002537 (MRH).
Conflict of Interest: MRH is a cofounder and chief medical officer of iotaMotion, Inc. with equity interest. Otherwise, the authors declare no competing interests.
BIBLIOGRAPHY
- 1. Clark GM, Black R, Forster IC, Patrick JF, Tong YC. Design criteria of a multiple‐electrode cochlear implant hearing prosthesis [43.66.Ts, 43.66.Sr]. J Acoust Soc Am 1978;63(2):631–633. [DOI] [PubMed] [Google Scholar]
- 2. Roland JT, Gantz BJ, Waltzman SB, Parkinson AJ. United States multicenter clinical trial of the cochlear nucleus hybrid implant system. Laryngoscope 2016;126(1):175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Niparko JK, Tobey EA, Thal DJ, et al. Spoken language development in children following cochlear implantation. JAMA 2010;303(15):1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mo B, Lindbaek M, Harris S. Cochlear implants and quality of life: a prospective study. Ear Hear 2005;26(2):186–194. [DOI] [PubMed] [Google Scholar]
- 5. Loy B, Warner‐Czyz AD, Tong L, Tobey EA, Roland PS. The children speak: an examination of the quality of life of pediatric cochlear implant users. Otolaryngol Head Neck Surg 2010;142(2):247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lazard DS, Vincent C, Venail F, et al. Pre‐, per‐ and postoperative factors affecting performance of postlinguistically deaf adults using cochlear implants: a new conceptual model over time. PLoS One 2012;7(11):e48739–e48711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gantz BJ, Woodworth GG, Knutson JF. Multivariate predictors of audiological success with multichannel cochlear implants. Ann Otol Rhinol Laryngol 1993;102(12):909–916. [DOI] [PubMed] [Google Scholar]
- 8. Moberly AC, Bates C, Harris MS, Pisoni DB. The enigma of poor performance by adults with cochlear implants. Otol Neurotol 2016;37(10):1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blamey P, Artieres F, Baskent D, et al. Factors affecting auditory performance of postlinguistically deaf adults using cochlear implants: an update with 2251 patients. Audiol Neurootol 2013;18(1):36–47. [DOI] [PubMed] [Google Scholar]
- 10. Papsin BC. Cochlear implantation in children with anomalous cochleovestibular anatomy. Laryngoscope 2005;115(1 pt 2 suppl 106):1–26. [DOI] [PubMed] [Google Scholar]
- 11. Colletti V, Fiorino F, Sacchetto L, Miorelli V, Carner M. Hearing habilitation with auditory brainstem implantation in two children with cochlear nerve aplasia. Int J Pediatr Otorhinolaryngol 2001;60(2):99–111. [DOI] [PubMed] [Google Scholar]
- 12. Otte J, Schunknecht HF, Kerr AG. Ganglion cell populations in normal and pathological human cochleae. Implications for cochlear implantation. Laryngoscope 1978;88(8 pt 1):1231–1246. [DOI] [PubMed] [Google Scholar]
- 13. Nadol JB Jr, Young YS. Survival of spiral ganglion cells in profound sensorineural hearing loss: implications for cochlear implantation. Ann Otol Rhinol Laryngol 1989;98(6):411–416. [DOI] [PubMed] [Google Scholar]
- 14. Seyyedi M, Viana LM, Nadol JB Jr. Within‐subject comparison of word recognition and spiral ganglion cell count in bilateral cochlear implant recipients. Otol Neurotol: neurotology: official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology 2014;35(8):1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moser T, Starr A. Auditory neuropathy‐‐neural and synaptic mechanisms. Nat Rev Neurol 2016;12(3):135–149. [DOI] [PubMed] [Google Scholar]
- 16. Foerst A, Beutner D, Lang‐Roth R, Huttenbrink K‐B, von Wedel H, Walger M. Prevalence of auditory neuropathy/synaptopathy in a population of children with profound hearing loss. Int J Pediatr Otorhinolaryngol 2006;70(8):1415–1422. [DOI] [PubMed] [Google Scholar]
- 17. Penido RC, Isaac ML. Prevalence of auditory neuropathy spectrum disorder in an auditory health care service. Braz J Otorhinolaryngol 2013;79(4):429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vignesh SS, Jaya V, Muraleedharan A. Prevalence and audiological characteristics of auditory neuropathy spectrum disorder in pediatric population: a retrospective study. Indian J Otolaryngol Head Neck Surg 2014;68(2):196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain 1996;119(pt 3):741–753. [DOI] [PubMed] [Google Scholar]
- 20. Berlin CI, Hood LJ, Morlet T, et al. Multi‐site diagnosis and management of 260 patients with auditory neuropathy/dys‐synchrony (auditory neuropathy spectrum disorder). Int J Audiol 2010;49(1):30–43. [DOI] [PubMed] [Google Scholar]
- 21. Walton J, Gibson WPR, Sanli H, Prelog K. Predicting cochlear implant outcomes in children with auditory neuropathy. Otol Neurotol 2008;29(3):302–309. [DOI] [PubMed] [Google Scholar]
- 22. Sharma A, Cardon G, Henion K, Roland P. Cortical maturation and behavioral outcomes in children with auditory neuropathy spectrum disorder. Int J Audiol 2011;50(2):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sininger YS, Trautwein P. Electrical stimulation of the auditory nerve via cochlear implants in patients with auditory neuropathy. Ann Otol Rhinol Laryngol Suppl 2002;189:29–31. [DOI] [PubMed] [Google Scholar]
- 24. Santarelli R, del Castillo I, Cama E, Scimemi P, Starr A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear Res 2015;330(pt B):200–212. [DOI] [PubMed] [Google Scholar]
- 25. Abbas PJ, Brown CJ. Assessment of responses to cochlear implant stimulation at different levels of the auditory pathway. Hear Res 2015;322(pt C):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fitzpatrick DC, Campbell AT, Choudhury B, et al. Round window electrocochleography just before cochlear implantation. Otol Neurotol 2014;35(1):64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Formeister EJ, McClellan JH, Merwin WHI, et al. Intraoperative round window electrocochleography and speech perception outcomes in pediatric cochlear implant recipients. Ear Hear 2015;36(2):249–260. [DOI] [PubMed] [Google Scholar]
- 28. McClellan JH, Formeister EJ, Merwin WHI, et al. Round window electrocochleography and speech perception outcomes in adult cochlear implant subjects: comparison with audiometric and biographical information. Otol Neurotol 2014;35(9):e245–e252. [DOI] [PubMed] [Google Scholar]
- 29. Abbas PJ, Tejani VD, Scheperle RA, Brown CJ. Using neural response telemetry to monitor physiological responses to acoustic stimulation in hybrid cochlear implant users. Ear Hear 2017;38(4):409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shearer AE, Tejani VD, Brown CJ, et al. In vivo electrocochleography in hybrid cochlear implant users implicates TMPRSS3 in spiral ganglion function. Sci Rep 2018;8(1):14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelosi S, Rivas A, Haynes DS, et al. Stimulation rate reduction and auditory development in poorly performing cochlear implant users with auditory neuropathy. Otol Neurotol 2012;33(9):1502–1506. [DOI] [PubMed] [Google Scholar]
- 32. Peterson A, Shallop J, Driscoll C, et al. Outcomes of cochlear implantation in children with auditory neuropathy. J Am Acad Audiol 2003;14(4):188–201. [PubMed] [Google Scholar]
- 33. Korver AMH, Smith RJH, Van Camp G, et al. Congenital hearing loss. Nat Rev Dis Primers 2017;3:16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sloan‐Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016;135(4):441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Green GE, Scott DA, McDonald JM, et al. Performance of cochlear implant recipients with GJB2‐related deafness. Am J Med Genet 2002;109(3):167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bauer PW, Geers AE, Brenner C, Moog JS, Smith RJH. The effect of GJB2 allele variants on performance after cochlear implantation. Laryngoscope 2003;113(12):2135–2140. [DOI] [PubMed] [Google Scholar]
- 37. Abdurehim Y, Lehmann A, Zeitouni AG. Predictive value of GJB2 mutation status for hearing outcomes of pediatric cochlear implantation. Otolaryngol Head Neck Surg 2017;157(1):16–24. [DOI] [PubMed] [Google Scholar]
- 38. Wu C‐M, Ko H‐C, Tsou Y‐T, et al. Long‐term cochlear implant outcomes in children with GJB2 and SLC26A4 mutations. PLoS One 2015;10(9):e0138575–e0138513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu XZ, Angeli SI, Rajput K, et al. Cochlear implantation in individuals with Usher type 1 syndrome. Int J Pediatr Otorhinolaryngol 2008;72(6):841–847. [DOI] [PubMed] [Google Scholar]
- 40. Pennings RJ, Damen GW, Snik AF, Hoefsloot L, Cremers CW, Mylanus EA. Audiologic performance and benefit of cochlear implantation in Usher syndrome type I. Laryngoscope 2006;116(5):717–722. [DOI] [PubMed] [Google Scholar]
- 41. Edvardson S, Jalas C, Shaag A, et al. A deleterious mutation in the LOXHD1 gene causes autosomal recessive hearing loss in Ashkenazi Jews. Am J Med Genet A 2011;155A(5):1170–1172. [DOI] [PubMed] [Google Scholar]
- 42. Miyagawa M, Nishio SY, Usami SI. A comprehensive study on the etiology of patients receiving cochlear implantation with special emphasis on genetic epidemiology. Otol Neurotol 2016;37(2):e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shearer AE, Eppsteiner RW, Frees K, et al. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear Res 2017;348:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wynne DP, Zeng F‐G, Bhatt S, Michalewski HJ, Dimitrijevic A, Starr A. Loudness adaptation accompanying ribbon synapse and auditory nerve disorders. Brain 2013;136(pt 5):1626–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rodríguez‐Ballesteros M, Reynoso R, Olarte M, et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat 2008;29(6):823–831. [DOI] [PubMed] [Google Scholar]
- 46. Rouillon I, Marcolla A, Roux I, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol 2006;70(4):689–696. [DOI] [PubMed] [Google Scholar]
- 47. Wu C‐C, Liu T‐C, Wang S‐H, Hsu C‐J, Wu C‐M. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 2011;121(6):1287–1293. [DOI] [PubMed] [Google Scholar]
- 48. Loundon N, Marcolla A, Roux I, et al. Auditory neuropathy or endocochlear hearing loss? Otol Neurotol 2005;26(4):748–754. [DOI] [PubMed] [Google Scholar]
- 49. Starr A, Isaacson B, Michalewski HJ, et al. A dominantly inherited progressive deafness affecting distal auditory nerve and hair cells. J Assoc Res Otolaryngol 2004;5(4):411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang T, Santarelli R, Starr A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res 2009;1300:97–104. [DOI] [PubMed] [Google Scholar]
- 51. Diaz‐Horta O, Abad C, Sennaroglu L, et al. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc Natl Acad Sci U S A 2016;113(21):5993–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han K‐H, Oh D‐Y, Lee S, et al. ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Sci Rep 2017;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brookes JT, Kanis AB, Tan LY, Tranebjærg L, Vore A, Smith RJH. Cochlear implantation in deafness‐dystonia‐optic neuronopathy (DDON) syndrome. Int J Pediatr Otorhinolaryngol 2008;72(1):121–126. [DOI] [PubMed] [Google Scholar]
- 54. Cif L, Gonzalez V, Garcia‐Ptacek S, et al. Progressive dystonia in Mohr‐Tranebjaerg syndrome with cochlear implant and deep brain stimulation. Mov Disord 2013;28(6):737–738. [DOI] [PubMed] [Google Scholar]
- 55. Yasunaga S, Grati M, Cohen‐Salmon M, et al. A mutation in OTOF, encoding otoferlin, a FER‐1‐like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 1999;21(4):363–369. [DOI] [PubMed] [Google Scholar]
- 56. Marlin S, Feldmann D, Nguyen Y, et al. Temperature‐sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem Biophys Res Commun 2010;394(3):737–742. [DOI] [PubMed] [Google Scholar]
- 57. Runge CL, Erbe CB, McNally MT, et al. A novel otoferlin splice‐site mutation in siblings with auditory neuropathy spectrum disorder. Audiol Neurootol 2013;18(6):374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roux I, Safieddine S, Nouvian R, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 2006;127(2):277–289. [DOI] [PubMed] [Google Scholar]
- 59. Chen K, Liu M, Wu X, Zong L, Jiang H. Targeted next generation sequencing reveals OTOF mutations in auditory neuropathy spectrum disorder. Int J Pediatr Otorhinolaryngol 2018;115:19–23. [DOI] [PubMed] [Google Scholar]
- 60. Baig SM, Koschak A, Lieb A, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci 2011;14(1):77–84. [DOI] [PubMed] [Google Scholar]
- 61. Platzer J, Engel J, Schrott‐Fischer A, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell 2000;102(1):89–97. [DOI] [PubMed] [Google Scholar]
- 62. Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci 2003;23(34):10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schrauwen I, Helfmann S, Inagaki A, et al. A mutation in CABP2, expressed in cochlear hair cells, causes autosomal‐recessive hearing impairment. Am J Hum Genet 2012;91(4):636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruel J, Emery S, Nouvian R, et al. Impairment of SLC17A8 encoding vesicular glutamate transporter‐3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 2008;83(2):278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ryu N, Lee S, Park H‐J, et al. Identification of a novel splicing mutation within SLC17A8 in a Korean family with hearing loss by whole‐exome sequencing. Gene 2017;627:233–238. [DOI] [PubMed] [Google Scholar]
- 66. Ryu N, Sagong B, Park H‐J, et al. Screening of the SLC17A8 gene as a causative factor for autosomal dominant non‐syndromic hearing loss in Koreans. BMC Med Genet 2016;17(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Seal RP, Edwards RH. The diverse roles of vesicular glutamate transporter 3. Handb Exp Pharmacol 2006;175:137–150. [DOI] [PubMed] [Google Scholar]
- 68. Akil O, Seal RP, Burke K, et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 2012;75(2):283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schoen CJ, Emery SB, Thorne MC, et al. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila . Proc Natl Acad Sci U S A 2010;107(30):13396–13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Santarelli R, Rossi R, Scimemi P, et al. OPA1‐related auditory neuropathy: site of lesion and outcome of cochlear implantation. Brain 2015;138(3):awu378–awu576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McLean WJ, Smith KA, Glowatzki E, Pyott SJ. Distribution of the Na,K‐ATPase alpha subunit in the rat spiral ganglion and organ of corti. J Assoc Res Otolaryngol 2009;10(1):37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nayagam BA, Muniak MA, Ryugo DK. The spiral ganglion: connecting the peripheral and central auditory systems. Hear Res 2011;278(1–2):2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jeon E‐J, Xu N, Xu L, Hansen MR. Influence of central glia on spiral ganglion neuron neurite growth. Neuroscience 2011;177:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Adam MP, Ardinger HH, Pagon RA, et al. Deafness‐dystonia‐optic neuronopathy syndrome. 1993.
- 75. Bahmad F, Merchant SN, Nadol JB, Tranebjaerg L. Otopathology in Mohr‐Tranebjaerg syndrome. Laryngoscope 2007;117(7):1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zong L, Guan J, Ealy M, et al. Mutations in apoptosis‐inducing factor cause X‐linked recessive auditory neuropathy spectrum disorder. J Med Genet 2015;52(8):523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Simon M, Richard EM, Wang X, et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl‐tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet 2015;11(3):e1005097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bird TD. Charcot‐Marie‐Tooth (CMT) Hereditary Neuropathy Overview. 1998 Sep 28 [Updated 2019 Jan 24]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993‐2019. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK1358/
- 79. Starr A, Michalewski HJ, Zeng F‐G, et al. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145‐>Ser). Brain 2003;126(pt 7):1604–1619. [DOI] [PubMed] [Google Scholar]
- 80. Goswamy J, Bruce IA, Green KMJ, O'Driscoll MP. Cochlear implantation in a patient with sensori‐neural deafness secondary to Charcot‐Marie‐Tooth disease. Cochlear Implants Int 2012;13(3):184–187. [DOI] [PubMed] [Google Scholar]
- 81. Rance G, Fava R, Baldock H, et al. Speech perception ability in individuals with Friedreich ataxia. Brain 2008;131(pt 8):2002–2012. [DOI] [PubMed] [Google Scholar]
- 82. Mowry SE, King S. Cochlear implantation in chronic demyelinating inflammatory polyneuropathy. Cochlear Implants Int 2017;18(2):116–120. [DOI] [PubMed] [Google Scholar]
- 83. Veske A, Oehlmann R, Younus F. Autosomal recessive non‐syndromic deafness locus (DFNB8) maps on chromosome 21q22 in a large consanguineous kindred from Pakistan. Hum Mol Genet 1996;5(1):165–168. [DOI] [PubMed] [Google Scholar]
- 84. Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A. Insertion of β‐satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nature 2001;27(1):59–63. [DOI] [PubMed] [Google Scholar]
- 85. Bonne‐Tamir B, DeStefano AL, Briggs CE, et al. Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am J Hum Genet 1996;58(6):1254–1259. [PMC free article] [PubMed] [Google Scholar]
- 86. Weegerink NJD, Schraders M, Oostrik J, et al. Genotype–phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 2011;12(6):753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gao X, Yuan Y‐Y, Wang G‐J, et al. Novel mutations and mutation combinations of TMPRSS3 cause various phenotypes in one chinese family with autosomal recessive hearing impairment. Biomed Res Int 2017;2017(2):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guipponi M, Vuagniaux G, Wattenhofer M. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 2002;11(23):2829–2836. [DOI] [PubMed] [Google Scholar]
- 89. Guipponi M, Toh M‐Y, Tan J, et al. An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum Mutat 2007;29(1):130–141. [DOI] [PubMed] [Google Scholar]
- 90. Fasquelle L, Scott HS, Lenoir M, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem 2011;286(19):17383–17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu W, Löwenheim H, Santi PA, Glueckert R, Schrott‐Fischer A, Rask‐Andersen H. Expression of trans‐membrane serine protease 3 (TMPRSS3) in the human organ of Corti. Cell Tissue Res 2018;372(3):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV. Sensory neuron diversity in the inner ear is shaped by activity. Cell 2018;174(5):1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li Y, Peng A, Ge S, Wang Q, Liu J. miR‐204 suppresses cochlear spiral ganglion neuron survival in vitro by targeting TMPRSS3. Hear Res 2014;314(pt C):60–64. [DOI] [PubMed] [Google Scholar]
- 94. Miyagawa M, Nishio S, Sakurai Y. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation. Ann Otol Rhinol Laryngol 2015;124(1 suppl):193S–204S. [DOI] [PubMed] [Google Scholar]
- 95. Eppsteiner RW, Shearer AE, Hildebrand MS, et al. Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis. Hear Res 2012;292(1–2):51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Georg Thieme Verlag KG, Tropitzsch A, Knoblich N, et al. Cochlear implant performance in patients with TMPRSS3 mutations. Laryngo‐Rhino‐Otol 2018;97(S 02):10738. [Google Scholar]
- 97. Rehman AU, Santos‐Cortez RLP, Morell RJ, et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet 2014;94(1):144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Azaiez H, Booth KT, Bu F, et al. TBC1D24 mutation causes autosomal‐dominant nonsyndromic hearing loss. Hum Mutat 2014;35(7):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Falace A, Buhler E, Fadda M, et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6‐dependent pathway. Proc Natl Acad Sci U S A 2014;111(6):2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Delmaghani S, del Castillo FJ, Michel V, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet 2006;38(7):770–778. [DOI] [PubMed] [Google Scholar]
- 101. Ebermann I, Walger M, Scholl HPN, et al. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum Mutat 2007;28(6):571–577. [DOI] [PubMed] [Google Scholar]
- 102. Schwander M, Sczaniecka A, Grillet N, et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci 2007;27(9):2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Delmaghani S, Defourny J, Aghaie A, et al. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell 2015;163(4):894–906. [DOI] [PubMed] [Google Scholar]
- 104. Wu C‐C, Lin Y‐H, Liu T‐C, et al. Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine 2015;94(27):e1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Miyamoto RT, Kirk KI, Renshaw J, Hussain D. Cochlear implantation in auditory neuropathy. Laryngoscope 1999;109(2 pt 1):181–185. [DOI] [PubMed] [Google Scholar]
- 106. Trautwein PG, Sininger YS, Nelson R. Cochlear implantation of auditory neuropathy. J Am Acad Audiol 2000;11(6):309–315. [PubMed] [Google Scholar]
- 107. Leigh J, Rance G, Dettman S, Dowell R. Cochlear implant outcomes for children with auditory neuropathy spectrum disorder. Perspect Hear Hear Disord Child 2009;19(2):75. [Google Scholar]
- 108. Teagle HFB, Roush PA, Woodard JS, et al. Cochlear implantation in children with auditory neuropathy spectrum disorder. Ear Hear 2010;31(3):325–335. [DOI] [PubMed] [Google Scholar]
- 109. Roush P, Frymark T, Venediktov R, Wang B. Audiologic management of auditory neuropathy spectrum disorder in children: a systematic review of the literature. Am J Audiol 2011;20(2):159–170. [DOI] [PubMed] [Google Scholar]
- 110. Willaredt MA, Ebbers L, Nothwang HG. Central auditory function of deafness genes. Hear Res 2014;312:9–20. [DOI] [PubMed] [Google Scholar]
- 111. Libé‐Philippot B, Michel V, Boutet de Monvel J, et al. Auditory cortex interneuron development requires cadherins operating hair‐cell mechanoelectrical transduction. Proc Natl Acad Sci U S A 2017;121:201703408–201703410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Liming BJ, Carter J, Cheng A, et al. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: hearing loss in the pediatric patient. Int J Pediatr Otorhinolaryngol 2016;90:251–258. [DOI] [PubMed] [Google Scholar]
- 113. Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg 2015;153(2):175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]