

Abstract
Rapid eye movement (REM) sleep behavior disorder (RBD) is a neurological disease characterized by loss of normal REM motor inhibition and subsequent dream enactment. RBD is clinically relevant because it predicts neurodegenerative disease onset (e.g., Parkinson's disease) and is clinically problematic because it disrupts sleep and results in patient injuries and hospitalization. Even though the cause of RBD is unknown, multiple lines of evidence indicate that abnormal inhibitory transmission underlies the disorder. Here, we show that transgenic mice with deficient glycine and GABA transmission have a behavioral, motor, and sleep phenotype that recapitulates the cardinal features of RBD. Specifically, we show that mice with impaired glycine and GABAA receptor function exhibit REM motor behaviors, non-REM muscle twitches, sleep disruption, and EEG slowing—the defining disease features. Importantly, the RBD phenotype is rescued by drugs (e.g., clonazepam and melatonin) that are routinely used to treat human disease symptoms. Our findings are the first to identify a potential mechanism for RBD—we show that deficits in glycine- and GABAA-mediated inhibition trigger the full spectrum of RBD symptoms. We propose that these mice are a useful resource for investigating in vivo disease mechanisms and developing potential therapeutics for RBD.
Introduction
Rapid eye movement (REM) sleep is paradoxical because muscle tone and movement are largely absent, but overall brain activity is maximal (Jouvet, 1967). The disconnection between the REM-active brain and skeletal motor system may function to ensure motoric quiescence during periods of unconsciousness. The mechanisms that allow the REM-active brain to disconnect itself from the motor system are unknown. Determining how muscle tone is controlled during sleep is clinically relevant because abnormal REM motor control is the defining feature of human REM sleep behavior disorder (RBD).
Patients with RBD are neurologically and motorically normal during waking—it is only during sleep that their primary disease symptoms emerge (Mahowald and Schenck, 2005a). RBD is typified by REM motor activation and violent dream enactment; however, muscle twitches and limb jerks during non-REM (NREM) sleep, sleep disruption, and slowing of the EEG are also defining disease symptoms (Sforza et al., 1997; Olson et al., 2000; Schenck and Mahowald, 2002).
RBD is clinically important for two reasons. First, REM motor activation and repetitive NREM muscle jerks disrupt sleep continuity, and violent dream enactment often results in bodily injuries—lacerations, fractures, and hospital visits are common in RBD (Schenck et al., 1986). Second, a majority of RBD patients develop Parkinson's, multiple system atrophy, or dementia with Lewy bodies within 12 years of initial diagnosis (Schenck et al., 1996; Iranzo et al., 2006). RBD is therefore a harbinger of neurodegenerative disease, particularly those stemming from synucleinopathies.
Although the cause of RBD is unknown, abnormal glycine and GABA neurotransmission could be involved. First, glycine- and GABA-mediated inhibition of skeletal motoneurons is partly responsible for suppression of muscle activity during normal REM sleep (Morrison et al., 2003; Chase and Morales, 2005; Brooks and Peever, 2008b). Second, strokes and lesions that affect brainstem regions containing glycine and GABA neurons trigger motor activation during REM sleep (Schenkel and Siegel, 1989; Lu et al., 2006; Boeve et al., 2007). Third, patients with impaired glycine and GABA transmission often experience heightened motor activity during sleep (de Groen and Kamphuisen, 1978; Martinelli et al., 1996). Last, drugs that enhance GABAergic function (e.g., clonazepam and melatonin) are the most common and effective treatment for RBD motor symptoms (Olson et al., 2000; Schenck and Mahowald, 2002).
There is no genetic model of RBD, but such a resource would advance investigation of disease cause and progression. The current study was designed to determine whether genetic impairment of inhibitory neurotransmission could trigger RBD symptoms. To do this, we used infrared videography and electrophysiology to record sleep–wake behavior, motor activity, and muscle tone in transgenic mice with deficient glycine and GABAA receptor function. We show that these mice have a behavioral, motor, and sleep phenotype that mimics human RBD. We propose that abnormal glycine and GABA function could, at least in part, underlie human RBD symptoms. We assert that these transgenic mice could be a valuable resource for pinpointing RBD disease mechanisms.
Materials and Methods
Animals
Transgenic mice were previously generated for determining how reduced glycine and GABA inhibition affects the startle response (Becker et al., 2002). Transgenic mice were engineered to express a mutant glycine receptor α1 subunit 271Q controlled by the Thy-1 promoter. The mutation substitutes a glutamine for an arginine at position 271 in the extracellular domain of the glycine receptor α1 subunit. Mutant receptors are expressed throughout the transgenic CNS. In vitro cell recordings show that transgenic spinal neurons not only experience a 70% reduction in glycine receptor-mediated inhibition, but also exhibit a 91% reduction in GABAA receptor-mediated inhibition. The exact cause of reduced GABAA-mediated inhibition is unknown. Therefore, the transgenic mice used in this study experience a potent reduction in both glycine- and GABAA-mediated inhibition (Becker et al., 2002).
Current experiments used 23 male transgenic mice (23 ± 0.3 g) and 23 male wild-type littermates (29 ± 0.6 g). Mouse lines were bred and handled as previously described (Becker et al., 2002). Animals were both phenotyped and genotyped. Transgenics were visually phenotyped by holding them by the tail, which resulted in clenching of the hindfeet (Becker et al., 2002). In contrast, wild types spread their hindfeet when held by their tails. Transgenic (n = 8) and wild-type (n = 8) mice were also genotyped using PCR (35 cycles, 55°C annealing temperature) with glycine receptor α1-specific primers designed based on the endogenous murine receptor (5′-TGCAAAACCCACAAGAACAA-3′ and 5′-TGGCATTTGTAAGGGTGTGA-3′; common to both wild-type and transgenic animals) and mutant human receptor (5′-TATTCCCAGCCTGCTCATTG-3′; 5′-CGCCCTTGACTGAGATGCCA-3′; only present in transgenic mice). All procedures and experimental protocols were approved by the Animal Care Committee at the University of Toronto and were in accordance with the Canadian Council on Animal Care.
Surgical implantation of EEG and EMG electrodes
Mice were anesthetized with isoflurane (1–2%) and then implanted with EEG and EMG electrodes as described previously (Burgess et al., 2010). EEG recordings were obtained using four stainless steel microscrews (1 mm anterior ±1.5 mm lateral to bregma; 3 mm posterior ±1.5 mm lateral to bregma). EMG electrodes were made from multistranded stainless steel wires (AS131, Cooner Wire), which were sutured onto masseter, hindlimb, and neck muscles. All electrodes were attached to a microstrip connector (CLP-105-02-L-D, Electrosonic), which was affixed onto the animal's head with dental cement (Ketac-cem, 3M). After surgery, mice were given ketoprofen (3 mg/kg) and 5% dextrose in saline. Mice were individually housed in a sound-attenuated and ventilated chamber on a 12:12 light–dark cycle [110 lux; lights on 0700h (7:00 A.M.), lights off 1900h (7:00 P.M.)] for 10–15 d after surgery. Food and water were available ad libitum.
Data acquisition and experimental design
Electrophysiological recordings.
EEG and EMG activity were recorded by attaching a light-weight cable to the plug on the mouse's head, which was connected to a Super-Z head-stage amplifier and BMA-400 AC/DC Bioamplifier (CWE). The EEG was amplified 1000 times and bandpass filtered between 1 and 100 Hz. EMG signals were amplified 1000 times and bandpass filtered between 30 and 500 Hz. All electrophysiological signals were digitized at 1000 Hz (Spike 2 Software, 1401 Interface, CED) and monitored and stored on a computer. Infrared video recordings were captured (Sony DCR-HC28) and synchronized with the electrophysiological recordings using Spike 2 software.
Experimental protocols.
At the start of each experiment, animals were transferred to a round Plexiglas cage (diameter: 20 cm) inside a sound-attenuated, ventilated, and illuminated (110 lux) chamber. Each mouse was given 24 h to habituate to this environment. Animals were then tethered, using a lightweight tether attached to the Raturn system (BAS). Mice were given another 24 h to habituate to the recording tether. After habituation, we recorded 24 h of undisturbed video, EEG, and EMG activity.
Drug preparation and treatment
Clonazepam.
Clonazepam is a benzodiazepine that is used to treat human RBD. It rapidly alleviates RBD motor symptoms in 90% of patients (Lapierre and Montplaisir, 1992; Schenck et al., 1993; Olson et al., 2000). To investigate the effects of clonazepam on the motor phenotype of transgenic mice, a subset of wild-type and transgenic mice received intraperitoneal injections of 0.3 mg/kg clonazepam (Roche) dissolved in 0.9% saline. All injections were administered at 1:00 P.M. The volume of drug given to each animal was determined before each injection based on the animal's weight, and this volume was topped up with saline such that each animal received 0.3 mg/kg clonazepam in a 0.2 ml bolus. EEG and EMG activity were quantified for the 3 h following the injection. Effects of clonazepam on sleep and motor activity were compared to each animal's own pretreatment levels.
Melatonin.
Long-term melatonin treatment is used to alleviate RBD symptoms (Takeuchi et al., 2001; Boeve et al., 2003; Kunz and Mahlberg, 2010). To chronically treat wild-type and transgenic mice, a melatonin (Sigma-Aldrich) solution was prepared in ethanol and dissolved in the drinking water (12.5 μg/ml tap water, in 0.07% ethanol) for 2–4 weeks. The water bottle containing melatonin was protected from light throughout the experiment. A fresh melatonin solution was prepared twice per week. Both wild-type and transgenic mice drank an average of 4 ± 0.3 ml per 24 h; this volume is consistent with a previous report (Johnson et al., 2003). Although the volume of water intake varied among animals, melatonin was administered at ∼2 mg/kg/d. A group of wild-type and transgenic mice were treated with melatonin for 2–4 weeks, and then EEG and EMG activity was quantified over a 24 h period. This activity was compared to their untreated counterparts.
Data analysis
Behavioral state.
We classified four behavioral states. Active wake was characterized by high-frequency, low-voltage EEG signals coupled with high levels of EMG activity (i.e., chewing, grooming, drinking). Quiet wake was characterized by high-frequency, low-voltage EEG signals, but in the absence of overt motor activity. NREM sleep was characterized by high-amplitude, low-frequency EEG signals and minimal EMG activity. REM sleep was characterized by low-amplitude, high-frequency theta-like EEG activity and REM atonia interspersed by periodic muscle twitches. Sleep states were visually identified and analyzed in 5 s epochs using the Sleepscore version 1.01 script (CED).
EMG analysis.
Raw EMG signals were full-wave rectified, integrated, and quantified in arbitrary units. Average EMG activity for masseter, hindlimb, and neck muscle activity was averaged during 5 s epochs for each behavioral state. In REM sleep, EMG activity was further quantified by separating REM into its tonic (e.g., REM atonia) and phasic (e.g., muscle twitches) components (Brooks and Peever, 2008b; Burgess et al., 2008).
Sleep–wake architecture.
The proportion of time spent in each sleep–wake state was calculated across a 24 h period and compared between wild-type and transgenic mice. The number of state transitions (i.e., arousals from NREM and REM sleep, NREM to REM transitions) was also quantified.
EEG spectral analysis.
EEG spectral analysis was calculated in 1 Hz bins using fast Fourier transformation of each 5 s epoch, yielding a power spectra profile from 0 to 16 Hz. A mean EEG spectrum profile was obtained for each epoch and then, to minimize nonspecific differences in absolute power between individuals, EEG power in each frequency bin was expressed as a percentage of the total EEG power in the epoch. The spectral profiles of each behavioral state were then compared between wild-type and transgenic mice.
Statistical analyses
All statistical analyses used SigmaStat (SPSS) and applied a critical α value of 0.05. In all comparisons, the Kolmogorov–Smirnov test was used to test for normality. Differences in EMG activity between wild-type and transgenic mice were determined using t tests (or Mann–Whitney rank sum test for nonparametric data). The proportion of time spent in each sleep–wake state and total EEG power in each state were compared between wild-type and transgenic mice using ANOVA with repeated measures (RM ANOVA), and post hoc comparisons were performed using the Student–Newman–Keuls (SNK) test. All data are expressed as mean ± SEM.
Results
Transgenic mice demonstrate RBD behaviors
Normal REM sleep is marked by postural motor atonia that is punctuated by minor and infrequent muscle twitches (Brooks and Peever, 2008b; Burgess et al., 2008). RBD patients suffer a breakdown in this normal process and experience powerful muscle activation and hence motor behaviors during REM sleep (Schenck et al., 1986). Our first goal was to determine whether transgenic mice experience such behaviors.
We found that 100% of transgenic mice (n = 16) exhibited motor behaviors that mimic RBD symptoms. During REM sleep, mutants displayed gross body and limb movements—running, jerking, and chewing were common behaviors. Even though uncoordinated muscle twitches and jerks occurred, most motor events were characterized by coordinated limb and head movements. Such motor behaviors influenced sleep posture. Unlike wild-type mice, which slept in a typical curled position, transgenics slept on their side during REM sleep. Overt motor behaviors occurred throughout REM sleep episodes; however, there were also defined periods of motor quiescence. Motor behaviors and generalized body movements occurred during all REM episodes in all transgenic mice.
REM motor behaviors in transgenics were due to a robust increase in overall muscle activity (Fig. 1). EMG levels in the masseter, limb, and neck muscles were elevated by 107–119% during REM sleep (transgenic vs wild type; masseter: t test, t(28) = 4.58, p < 0.001; limb: t test, t(5) = 3.33, p = 0.021; neck: Mann–Whitney, U = 114.0, n1 = 8, n2 = 12, p = 0.023) (Fig. 1). Exaggerated muscle twitches that typify normal REM sleep were the primary trigger of heightened muscle activity and hence RBD behaviors. We found that the frequency, duration, and amplitude of muscle twitches during REM sleep episodes increased by 268%, 86%, and 66%, respectively (frequency: t test, t(28) = 7.99, p < 0.001; duration: t test, t(28) = 6.31, p < 0.001; amplitude: t test, t(28) = 3.87, p < 0.001) (Fig. 2A,B). Excessive muscle twitch activity during REM sleep is reminiscent of human RBD and therefore recapitulates the primary disease symptom.
Figure 1.

Transgenic mice exhibit an RBD phenotype. Typical raw EMG and EEG traces from a wild-type (top trace) and a transgenic (bottom trace) mouse during an episode of REM sleep. In contrast to wild-type mice, transgenics display elaborate motor behaviors that closely mimic RBD symptoms. While periods of motor quiescence still occur, transgenic mice exhibit overt motor behaviors throughout REM sleep. Group data for wild-type (Wt; white bars) and transgenic (Tg; black bars) mice demonstrating that transgenics have significantly higher muscle activity in the masseter (Wt: n = 16; Tg: n = 14), limb (Wt: n = 3; Tg: n = 4), and neck (Wt: n = 12; Tg: n = 8) during REM sleep. *p < 0.05; a.u., arbitrary units. All values are mean ± SEM.
Figure 2.

Exaggerated muscle twitches during REM sleep trigger RBD behaviors. A, EMG and EEG traces showing that phasic masseter muscle twitches are potently increased in transgenic versus wild-type mice. B, During REM sleep, transgenic mice (Tg; n = 16) have more frequent twitches (267% increase) that are longer-lasting (86% increase) and are of greater magnitude (65% increase) than wild types (Wt; n = 19). C, Transgenic and wild-type mice have similar levels of tonic muscle tone (i.e., atonia) during REM sleep. *p < 0.05; a.u., arbitrary units. All values are mean ± SEM.
In addition to elevated phasic motor activity, some RBD patients also experience REM sleep without atonia (Schenck and Mahowald, 2002). Even though we found that transgenic mice had increased phasic motor activity (Fig. 2B), they had normal levels of muscle tone, i.e., atonia, during REM sleep (transgenic vs wild type: Mann–Whitney, U = 246.0, n1 = 14, n2 = 16, p = 0.236) (Fig. 2C). Therefore, in transgenics REM sleep atonia remains intact but phasic motor activity is heightened. This phenotype is consistent with the defining disease feature—i.e., heightened muscle activity and motor behavior in REM sleep.
Because REM behaviors were wake-like in nature, we wanted to verify that such behaviors occurred during bona fide REM sleep episodes and not during waking. First, we showed that REM sleep is electrophysiologically different from wakefulness in transgenics. Quantitative spectral analysis demonstrates unequivocal differences in EEG frequencies between identified periods of wakefulness and REM sleep in transgenic mice (waking vs REM: RM ANOVA, F(1,14) = 14.71, p < 0.001) (Fig. 3A). We also found that REM sleep episodes were significantly shorter than periods of wakefulness (Mann–Whitney, U = 134.0, n1 = n2 = 16, p < 0.001) (Fig. 3B). Second, we showed that REM sleep characteristics (except motor activation) are similar between transgenic and wild-type mice. We found that transgenic and wild-type mice spend the same amount of time in REM sleep (SNK, q = 0.63, p = 0.658) (Fig. 3C), and we showed that REM sleep EEG frequencies are similar in the two types of mice (Fig. 3D).
Figure 3.
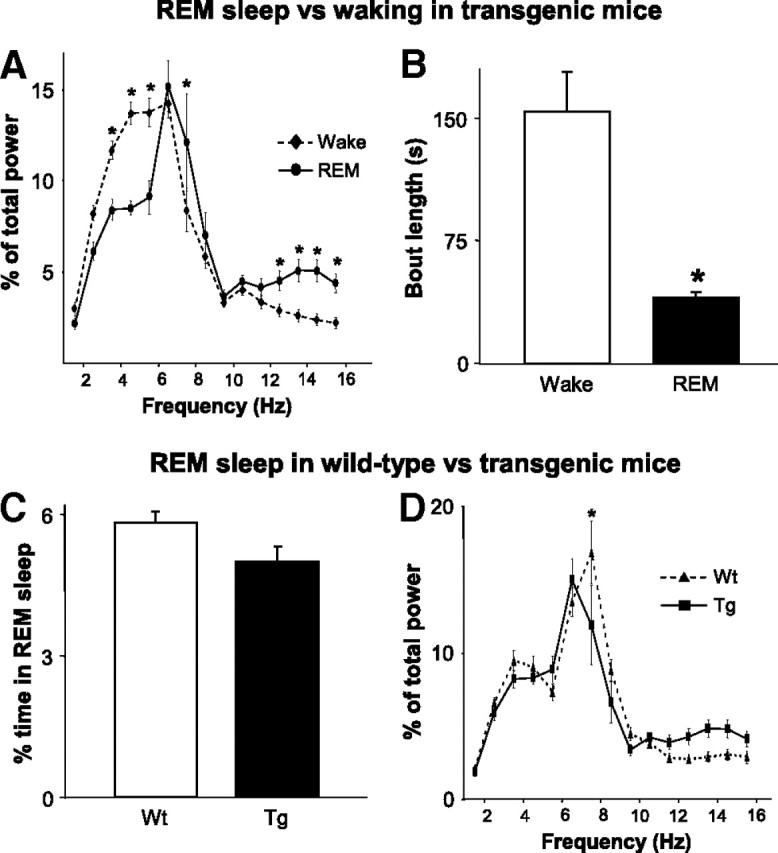
Verification that RBD behaviors occur during REM sleep. Because RBD behaviors are reminiscent of waking activity, we used post hoc analyses to confirm that such behaviors occur during REM sleep. A, In transgenic mice (Tg; n = 16), we show that there is a marked difference in the distribution of spectral power between periods of identified wakefulness and REM sleep. B, We also show that the length of identified REM sleep episodes is significantly shorter than periods of waking. C, Compared to wild-type mice (Wt; n = 19), transgenic mice spend the same amount of time in REM sleep. D, Also similar to wild-type mice, EEG spectral power is concentrated in the theta range during periods of identified REM sleep in transgenic mice. *p < 0.001. All values are mean ± SEM.
Although primary RBD symptoms appear during sleep (Schenck and Mahowald, 2002), subtle motor and gait slowing can also occur during waking (Postuma et al., 2006). Therefore, we wanted to characterize transgenic motor behavior during wakefulness. Transgenic mice were motorically normal during waking—they walked, ran, ate, drank, and groomed in a comparable fashion to their wild-type littermates. Even though transgenics engaged in normal waking behaviors, they had a somewhat stiffened gait when moving quickly. This motor phenotype was confirmed by analyzing overall levels of EMG tone during waking. We found that both masseter and hindlimb tone in transgenics were elevated, by 68% and 118%, respectively, above wild-type levels (masseter: t test, t(28) = 3.36, p = 0.002; hindlimb: t test, t(5) = 3.25, p = 0.023) (Fig. 4). Neck muscle tone during waking was similar between wild types and transgenics (Mann–Whitney, U = 88.0, n1 = 8, n2 = 12, p = 0.787) (Fig. 4).
Figure 4.

Muscle tone is increased during waking in transgenic mice. EMG and EEG traces from a wild-type (top left trace) and a transgenic (top right trace) mouse during waking. Group data for wild-type (Wt; white bars) and transgenic (Tg; black bars) mice demonstrating that transgenics have significantly higher muscle activity during waking in the masseter (Wt: n = 16; Tg: n = 14) and limb (Wt: n = 3; Tg: n = 4), but not the neck (Wt: n = 12; Tg: n = 8). *p < 0.05; a.u., arbitrary units. All values are mean ± SEM.
Transgenic mice exhibit RBD symptoms during NREM sleep
Repetitive muscle twitches/jerks during NREM sleep, especially in the limbs, are also a cardinal feature of RBD (Schenck and Mahowald, 2002). Therefore, we aimed to determine whether transgenic mice experience abnormal motor activity during NREM sleep. We found that 100% of transgenic mice (n = 16) experienced brief, repetitive EMG twitches and limb jerks during NREM sleep (Fig. 5). Twitches generally occurred simultaneously in each recorded muscles and were of sufficient magnitude to cause overt body movement and postural changes. This motor behavior is in sharp contrast to NREM activity in wild-type littermates, which is characterized by complete motor quiescence and inactivity.
Figure 5.

Transgenic mice exhibit RBD symptoms during NREM sleep. Typical raw EMG and EEG traces from a wild-type (top left trace) and a transgenic (top right trace) mouse during NREM sleep. Group data for wild-type (Wt; white bars) and transgenic (Tg; black bars) mice for the masseter (Wt: n = 16; Tg: n = 14), limb (Wt: n = 3; Tg: n = 4), and neck (Wt: n = 12; Tg: n = 8) during NREM sleep. While muscle tone is not elevated during this state, all transgenic mice experience brief, repetitive muscle twitches/jerks during NREM sleep (arrows). Such repetitive myoclonic twitching is not seen in wild-type mice (inset). a.u., Arbitrary units. All values are mean ± SEM.
We found that transgenic mice had an average of 2.5 ± 0.4 twitches/NREM episode (Fig. 5, inset). The jerks/twitches that dominate NREM sleep in transgenics were episodic in nature, with 70% occurring approximately every 20 s (Fig. 5). The episodic nature of NREM twitches is different from REM motor activity, which is characterized by clustered flurries of phasic muscle activity. Despite episodic muscle twitches, overall EMG tone was unaffected during NREM sleep in transgenic mice (transgenic vs wild type; masseter: t test, t(28) = 1.63, p = 0.115; limb: t test, t(5) = 2.16, p = 0.083; neck: t test, t(18) = 1.92, p = 0.071) (Fig. 5), which is consistent with reports from RBD patients (Olson et al., 2000; Schenck and Mahowald, 2002). Transgenic mice therefore exhibit a NREM motor phenotype that mimics human RBD.
Sleep is disrupted in transgenic mice
Sleep disruption is a hallmark of human RBD. Therefore, we wanted to determine whether reduced glycine and GABA receptor function affected sleep–wake behavior in transgenic mice. To do this, we quantified sleep–wake amounts and architecture across a 24 h period in both transgenic (n = 16) and wild-type (n = 19) mice. We found that unlike wild-type mice, transgenics suffer from pronounced sleep fragmentation (Fig. 6A). They awoke 135% and 34% more from NREM and REM sleep episodes than wild types did (wild type vs transgenic; NREM: Mann–Whitney, U = 407.5, n1 = 16, n2 = 19, p < 0.001; REM: t test, t(33) = 2.34, p = 0.025) (Fig. 6B).
Figure 6.

Sleep is disrupted in transgenic mice. A, Hypnograms showing that unlike wild-type mice, transgenics have markedly fragmented sleep. The white and black bars under each hypnogram indicate the light and dark periods, respectively. A zoomed-in half-hour portion of the hypnogram (shaded, outlined region) is shown for each mouse depicting the increased number of state transitions occurring in transgenic versus wild-type mice. B, Compared to wild-type mice (Wt; white bars, n = 19), transgenics (Tg; black bars, n = 16) have more arousals from NREM and REM sleep. W, Wake; N, NREM; R, REM. *p < 0.01. All values are mean ± SEM.
Increased arousals had an impact on sleep–wake amounts (RM ANOVA, F(1,2) = 10.16, p < 0.001) and sleep architecture in transgenic mice. Compared to wild types, mutants spent more time awake and less time in NREM sleep (transgenic vs wild type: wake: 64 ± 1% vs 57 ± 2%; SNK, q = 5.22, p < 0.001; NREM: 28 ± 1% vs 36 ± 2%; SNK, q = 5.80, p < 0.001) (Fig. 7A). Reduced NREM sleep amounts were caused by sleep fragmentation. Although the number of NREM episodes increased by 89% (transgenic vs wild type: t test, t(33) = 6.54, p < 0.001) (Fig. 7B), overall NREM amounts decreased as indicated by a decrease in the length of each NREM episode (45% below wild types; t test, t(33) = 7.83, p < 0.001) (Fig. 7B). Fragmentation of NREM sleep was not caused by NREM motor activation because only 10% of arousals were preceded (within 5 s) by muscle twitches, i.e., 90% arousals were triggered by nonmotor events. REM sleep amounts were unchanged in transgenics (transgenic vs wild type: 5 ± 0.3% vs 6 ± 0.2%; SNK, q = 0.63, p = 0.658) (Fig. 7A).
Figure 7.

Sleep is disrupted in transgenic mice. A, Sleep disruptions experienced by transgenic mice (Tg; black bars, n = 16) increase the amount of wakefulness and decrease NREM sleep in these mice compared to their wild-type littermates (Wt; white bars, n = 19). REM sleep amounts are unchanged. The increase in wakefulness is due to an increase in the amount of time spent in quiet wake (inset). B, The decrease in NREM sleep is due to a decrease in the length, not number, of NREM episodes. *p < 0.01. All values are mean ± SEM.
The increase in waking amounts was caused by transgenics spending significantly more time in quiet waking (transgenic vs wild type: 22 ± 2% vs 10 ± 0.6%; SNK, q = 8.85, p < 0.001), but less time in active waking (transgenic vs wild type: 42 ± 2% vs 47 ± 2%; SNK, q = 3.83, p = 0.008) (Fig. 7A, inset). Because quiet waking often includes transitions into NREM sleep and because transgenics have fragmented NREM sleep, we assert that increased quiet waking is further demonstration that transgenics suffer from sleep disruption.
Cortical activation is impaired in transgenic mice
A neurological marker of RBD is impaired cortical activation. Analysis of EEG activity shows that relative to healthy controls, RBD patients have slower cortical EEG frequencies, which is an index of impaired cortical activation (Gagnon et al., 2006b). EEG slowing also occurs in Parkinson's disease and dementia (Soikkeli et al., 1991; Briel et al., 1999). Because transgenic mice have both sleep and motor deficits that mimic human RBD, we wanted to determine whether they also have altered EEG activity.
Gross EEG examination revealed no difference between wild-type (n = 15) and transgenic (n = 11) mice in any state. However, spectral analysis demonstrates a shift in EEG power to lower frequencies (i.e., 5–7 Hz) in transgenic mice during waking and NREM sleep—an indication of impaired cortical activation (wake: RM ANOVA, F(1,14) = 4.66, p < 0.001; NREM: RM ANOVA, F(1,14) = 3.83, p < 0.001) (Fig. 8). Conversely, REM sleep EEG was largely unaffected in transgenic mice, except for a slight reduction in power in the lower alpha range (i.e., 8–10 Hz) (Fig. 3D).
Figure 8.
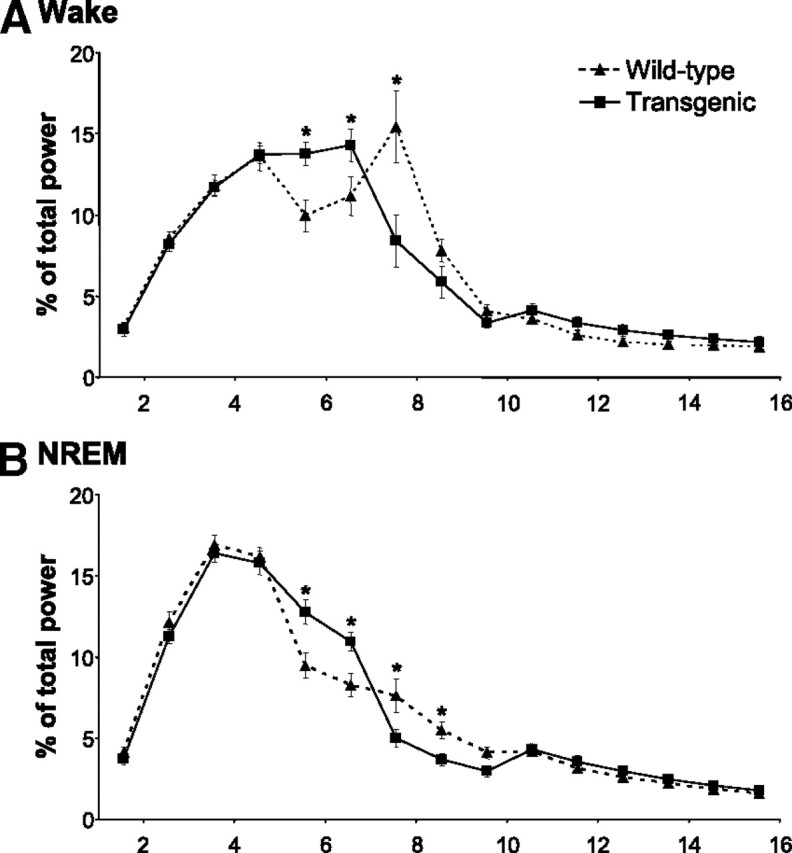
EEG slowing in transgenic mice. EEG spectral profiles for wild-type (n = 15; dotted line) and transgenic (n = 11; solid line) mice for wake (A) and NREM (B) sleep. Transgenics have more power in the lower frequency ranges and less power in the higher frequency ranges, resulting in an overall EEG slowing in these states. *p < 0.01. All values are mean ± SEM.
Clonazepam and melatonin rescue the RBD phenotype
Clonazepam is the most common and effective treatment for RBD (Schenck and Mahowald, 2002). Clonazepam functions to strengthen GABAergic inhibition by acting on benzodiazepine receptors. Even though GABAA-mediated inhibition is reduced in transgenics, they nonetheless have functional GABAA receptors (Becker et al., 2002). Therefore, we wanted to determine whether clonazepam could strengthen inhibition and thereby rescue the RBD phenotype in mutant mice.
We found that a single dose of clonazepam reduced overall EMG masseter tone by 26 ± 4% during REM sleep in transgenic mice (n = 7; control vs clonazepam: paired t test, t(6) = 5.17, p = 0.002) (Fig. 9). While clonazepam did not reduce EMG tone to wild-type levels (Mann–Whitney, U = 125.0, n1 = 7, n2 = 16, p = 0.007), the effects on muscle activity are in line with the reduction in phasic EMG activity that has been reported in clonazepam-treated RBD patients (Lapierre and Montplaisir, 1992). Clonazepam also reduced the muscle twitches occurring in NREM sleep by 51 ± 15% (control vs clonazepam: paired t test, t(5) = 3.03, p = 0.029) (Fig. 9A, inset); however, it had only minor effects on waking levels of muscle tone (paired t test, t(6) = 2.48, p = 0.048; data not shown). Unlike clonazepam treatment in transgenics, this intervention had no effect on motor activity in wild-type mice (n = 10; control vs clonazepam: wake: paired t test, t(8) = 2.05, p = 0.075; NREM: paired t test, t(8) = 0.66, p = 0.526; REM: paired t test, t(8) = 0.17, p = 0.871; data not shown).
Figure 9.

Clonazepam and melatonin rescue the RBD phenotype. A, In transgenic mice, a single dosage of clonazepam (3 mg/kg, i.p.) reduced masseter muscle tone by 26% in REM sleep. B, Long-term oral melatonin treatment (2 mg/kg/d for 2–4 weeks) also suppressed muscle tone during REM sleep, reducing it by 43% in transgenics. Clonazepam, but not melatonin, reduced NREM muscle twitches in transgenics (insets). *p < 0.05; a.u., arbitrary units. All values are mean ± SEM.
Long-term melatonin treatment is also used to suppress RBD symptoms (Kunz and Mahlberg, 2010). Like clonazepam, melatonin potentiates GABAergic function (Coloma and Niles, 1988; Rosenstein et al., 1989; Wu et al., 1999). Here, we aimed to determine whether melatonin could reduce RBD symptoms. We found that a 2–4 week melatonin treatment reduced masseter muscle tone in REM sleep by 43% in transgenic mice [control (n = 14) vs melatonin treatment (n = 7): t test, t(19) = 2.71, p = 0.014] (Fig. 9B). This reduction in muscle tone restored EMG activity in transgenics to untreated wild-type levels (Mann–Whitney, U = 104.0, n1 = 7, n2 = 16, p = 0.193). Despite the powerful effects on REM sleep EMG levels, melatonin had no effect on the frequency of muscle twitches in NREM sleep (t test, t(11) = 1.13, p = 0.284) (Fig. 9B, inset). Even though melatonin treatment reduced waking levels of muscle tone by 32%, this was not statistically significant (t test, t(19) = 1.97, p = 0.063; data not shown). Finally, melatonin treatment had no effect on muscle tone during either waking or sleep in wild-type mice [control (n = 16) vs melatonin treatment (n = 3): wake: t test, t(17) = 1.82, p = 0.087; NREM: t test, t(17) = 1.36, p = 0.192; REM: t test, t(17) = 1.04, p = 0.315; data not shown].
Although clonazepam reduced RBD motor symptoms in transgenic mice, it did not improve sleep amounts or alleviate sleep fragmentation. Compared to controls, we found that clonazepam dosing had no effect on amounts of waking or NREM or REM sleep (RM ANOVA, F(1,2) = 0.56, p = 0.582) (Fig. 10A). We also found that clonazepam treatment did not improve sleep fragmentation. Transgenic mice awoke from both NREM (paired t test, t(7) = 1.68; p = 0.137) and REM (paired t test, t(7) = 0.383, p = 0.713) sleep as frequently during clonazepam dosing as they did under control conditions (Fig. 10A, insets).
Figure 10.
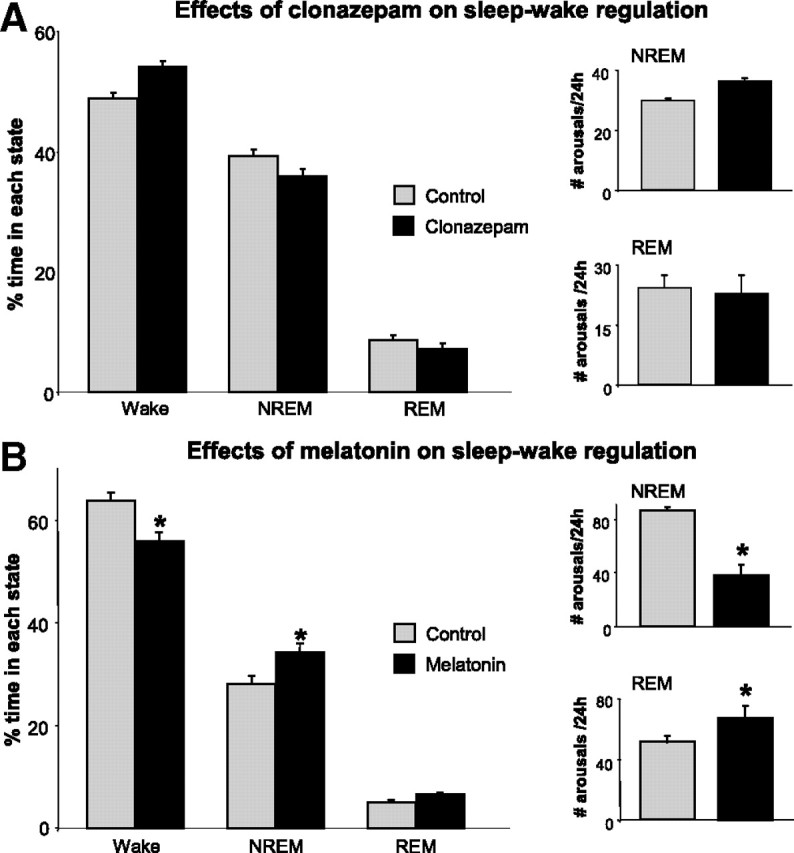
Melatonin, but not clonazepam, improves sleep and alleviates sleep fragmentation in transgenic mice. A, Clonazepam treatment (3 mg/kg, i.p.) does not affect amounts of wake, NREM, or REM sleep in treated versus untreated (control) transgenic mice (n = 7). Clonazepam treatment does not reduce the number of arousals out of NREM or REM sleep, i.e., sleep fragmentation (insets). B, Melatonin treatment (2 mg/kg/d for 2–4 weeks) reduced wakefulness and increased NREM sleep in treated (n = 7) versus untreated (control; n = 14) transgenic mice. Treatment had no effect on REM sleep amounts. Melatonin treatment relieved the sleep fragmentation associated with NREM sleep, but it did not reduce REM sleep disruption (insets). *p < 0.05. All values are mean ± SEM.
Unlike clonazepam, melatonin treatment improved sleep (RM ANOVA, F(1,2) = 12.78, p < 0.001) and reduced sleep fragmentation (Fig. 10B). Compared to untreated transgenics, we found that melatonin reduced waking time (SNK, q = 5.55, p < 0.001) (Fig. 10B) while significantly increasing the amount of time spent in NREM sleep (SNK, q = 4.37, p = 0.003) (Fig. 10B). In fact, melatonin treatment restored sleep–wake amounts to untreated wild-type levels (ANOVA, F(1,2) = 0.29, p = 0.753). REM sleep amounts were unaffected by melatonin treatment (SNK, q = 1.29, p = 0.428) (Fig. 10B). Importantly, we found that melatonin treatment significantly reduced sleep fragmentation by decreasing the number of arousals from NREM sleep by 55% (t test, t(21) = 2.50, p = 0.021) (Fig. 10B, inset); however, it did not reduce REM sleep disruption (t test, t(21) = 2.14, p = 0.043) (Fig. 10B, inset).
Discussion
RBD is a public health concern because it forecasts neurodegenerative disease, disrupts sleep, and results in patient injuries and hospitalization (Mahowald and Schenck, 2005a). Transgenic mice with deficient glycine and GABAA receptor function are the first model of RBD that successfully recapitulates the behavioral, motoric, and sleep features that define the disorder—i.e., REM behaviors, NREM myoclonic jerks, sleep fragmentation, and EEG slowing (Table 1). Importantly, the RBD phenotype in transgenic mice can be rescued by drugs that treat human disease symptoms. Our findings are the first to indicate that deficits in glycine- and GABAA-mediated inhibition trigger the full spectrum of RBD symptoms. We propose that these mice are a powerful resource for investigating in vivo disease mechanisms and developing potential therapeutics for RBD.
Table 1.
Impaired glycine and GABA neurotransmission in transgenic mice triggers the defining features of human RBD
| REM sleep behavior disorder | Transgenic mice |
|---|---|
| Abnormal REM sleep behaviors (e.g., vigorous/violent movements, limb or body jerking, complex motor activity) | Gross body and limb movements during REM sleep. Running, jerking and chewing are common motor activities in REM sleep. |
| Elevated phasic EMG tone in REM sleep | Excessive phasic twitches in masseter, neck, and hindlimb muscles during REM sleep |
| Repetitive limb movements during NREM sleep | Periodic myoclonic twitches during NREM sleep |
| Sleep fragmentation | Increased arousals from NREM and REM sleep |
| Reduced cortical activation, e.g., EEG slowing | Increased EEG power in the lower frequency ranges |
| Clonazepam and melatonin relieve motor symptoms | Both clonazepam and melatonin alleviate REM sleep motor symptoms |
Impaired inhibitory transmission triggers RBD motor symptoms
Human RBD can be triggered by different factors. Idiopathic RBD is the most common and clinically concerning form because the majority of cases lead to neurodegenerative disorders (e.g., Parkinson's) (Schenck and Mahowald, 2002). RBD can be pharmacologically induced; e.g., antidepressants that affect CNS noradrenergic/serotonergic tone can cause reversible RBD behaviors (Winkelman and James, 2004; Gagnon et al., 2006a). Last, RBD onset has been associated with stroke or trauma-induced brainstem lesions (Kimura et al., 2000; Tippmann-Peikert et al., 2006).
Even though RBD has multiple triggers, a common mechanism could nonetheless underlie the disorder. Our current results show that reduced glycine- and GABAA-mediated inhibition induces RBD motor symptoms in transgenic mice. Accordingly, we assert that impaired inhibitory neurotransmission plays a central role in triggering RBD motor behaviors during sleep. Multiple lines of clinical and experimental evidence support this assertion.
Motor atonia and intermittent muscle twitches are hallmarks of REM sleep (Brooks and Peever, 2008a). Glycinergic and GABAergic inhibition act to suppress motoneuron function, and therefore muscle activity, during normal REM sleep. Blockade of glycine and GABAA receptors on motoneurons markedly increases muscle twitches during REM sleep in rats (Brooks and Peever, 2008b). This motor phenotype is reminiscent of REM motor activity in both RBD patients and transgenic mice. Lesions in glycinergic/GABAergic brainstem regions elicit RBD-like motor behaviors in cats and rats (Schenkel and Siegel, 1989; Holmes and Jones, 1994; Lu et al., 2006). Further, microstructural changes in inhibitory brainstem circuitry are associated with human RBD (Kimura et al., 2000; Tippmann-Peikert et al., 2006; Unger et al., 2010). We assert that reduced CNS inhibition could therefore underlie REM motor behaviors in RBD.
Muscle tone is minimal during normal NREM sleep, but in RBD it is punctuated by repeated myoclonic twitches. Impaired inhibition in transgenic mice yields a motor phenotype similar to the NREM muscle jerks/twitches in human RBD. Studies show that glycine and GABA normally inhibit motoneurons and muscle tone during NREM sleep in rats (Morrison et al., 2003; Brooks and Peever, 2008b). Genetic disorders that affect glycine or GABA transmission also cause abnormal motor activity in NREM sleep (Solimena et al., 1990; Shiang et al., 1993; Dinkel et al., 1998). For example, patients with hyperekplexia—a disorder linked to glycine receptor mutations—have NREM myoclonic twitches and REM sleep motor behaviors (de Groen and Kamphuisen, 1978; Martinelli et al., 1996). Together these observations suggest that impaired inhibitory function triggers NREM and REM motor behaviors in RBD.
Abnormal inhibition underlies sleep disruption in RBD
GABA and glycine neurons form part of the neurocircuitry that generates and regulates sleep (Sherin et al., 1996, 1998; Szymusiak et al., 1998). Lesion-induced loss of brainstem and hypothalamic GABA neurons destabilizes sleep and disrupts normal NREM–REM architecture (Lu et al., 2000; Schmidt et al., 2000). Our current findings indicate that glycine and GABA transmission are also important for normal sleep regulation. Specifically, we show that genetic impairment of glycine and GABAA receptor function causes sleep disruption by fragmenting NREM and REM sleep.
Sleep is disrupted in human RBD. Patients not only arouse more from sleep, they can also experience more stage 1 NREM sleep (Schenck et al., 1987; Sforza et al., 1988). REM motor behaviors and NREM limb jerks may elicit sleep disruption; however, most arousals from sleep are not associated with preceding motor activity (Schenck et al., 1993). Current results show that most arousals from sleep (i.e., 90%) are not caused by NREM motor activity in transgenic mice, suggesting that other factors elicit sleep disruption in RBD. Because glycine and GABA regulate normal sleep and muscle tone, we assert that sleep disruption in RBD is caused not only by motor events, but also by abnormal inhibitory transmission. This hypothesis is supported by the fact that drugs that enhance inhibition also improve sleep in both transgenic mice and RBD patients (Kunz and Bes, 1999).
Inhibition and EEG slowing in RBD
RBD patients experience reduced cortical activation, i.e., EEG slowing during waking and NREM sleep (Fantini et al., 2003; Massicotte-Marquez et al., 2008). EEG slowing is clinically concerning because it also occurs in Parkinson's and dementia with Lewy bodies (Soikkeli et al., 1991; Briel et al., 1999). Reduced EEG activity in RBD is characterized by increased theta and delta power in frontal, temporal, and occipital regions. We show that transgenic mice also exhibit reduced cortical activation, which is manifested by increased EEG power in the theta range (i.e., 5–7 Hz) during waking and NREM sleep.
The cause of EEG slowing in RBD is unknown. However, imaging studies show reduced blood flow and glucose metabolism in frontal, parietal, and temporal regions in RBD (Shirakawa et al., 2002; Caselli et al., 2006; Mazza et al., 2006). These areas also have reduced EEG activity during waking and NREM in RBD patients. Subcortical regions that contain GABA neurons are affected in RBD (Gagnon et al., 2006b; Boeve et al., 2007). These same GABA-rich regions modulate cortical activation and project to the cortical regions affected by EEG slowing (Jones, 2005). We suggest that changes in inhibitory subcortical–cortical circuits could underlie EEG slowing in both human and murine RBD.
Increasing inhibitory tone improves RBD symptoms
Drugs that strengthen inhibitory activity effectively alleviate RBD symptoms. Clonazepam and melatonin are the most common treatments for RBD (Schenck and Mahowald, 2002)—both function to enhance GABAergic transmission (Skerritt and Johnston, 1983; Coloma and Niles, 1988; Rosenstein et al., 1989; Wu et al., 1999). We found that clonazepam and melatonin also alleviated RBD symptoms in transgenic mice. Both drugs reduced motor activation and behavior during REM sleep, but only clonazepam suppressed NREM muscle twitches/jerks. We also showed that melatonin, but not clonazepam, effectively improved sleep in mutant mice. These drugs may improve motor function and sleep by restoring GABAergic tone to sleep and motor circuits that are deficient in transgenic mice. Because melatonin and clonazepam have differential effects on sleep and motor function, each may act at unique sites within the brainstem circuitry.
RBD motor symptoms can be pharmacologically induced—e.g., by serotonin reuptake inhibitors and monoamine oxidase inhibitors (Mahowald and Schenck, 2005b; Gagnon et al., 2006a). Motor symptoms subside with the cessation of drug treatment, suggesting that such compounds affect the inhibitory brainstem circuits that normally suppress REM motor activity. For example, fluoxetine, a serotonergic antidepressant, can cause drug-induced RBD (Winkelman and James, 2004). Fluoxetine also suppresses glycine receptor efficacy (Ye et al., 2008), which functions to inhibit REM motor activity. We assert that iatrogenic RBD, like idiopathic RBD, may result from disturbances in normal inhibitory tone and neurocircuitry.
Inhibitory link between RBD and Parkinson's disease
RBD is a harbinger of neurodegenerative disorders. For example, 40–65% of patients develop Parkinson's disease within 12 years of the initial RBD diagnosis (Schenck et al., 2003; Iranzo et al., 2006; Postuma et al., 2009). Although Parkinson's is typically considered a dopamine system disease, it also involves degeneration of GABA and glycine function (Lloyd et al., 1977; Rinne et al., 1978; Nishino et al., 1988). In fact, loss of nondopaminergic cells precedes dopamine cell loss in disease progression (Braak et al., 2003). We therefore suggest that early inhibitory dysfunction may explain why RBD develops before Parkinson's disease onset.
RBD motor symptoms typically worsen with time (Iranzo et al., 2009b), suggesting that progressive neuronal degeneration contributes to RBD symptoms. While there is no clear evidence that dopaminergics influence RBD symptoms (Iranzo et al., 2009a), imaging studies nonetheless indicate that dysfunction of the nigrostriatal dopamine system is associated with RBD (Albin et al., 2000; Eisensehr et al., 2000). We suggest that initial RBD onset is triggered by loss of GABAergic/glycinergic function and that progressive dopamine cell degeneration not only causes Parkinson's symptoms but also worsens RBD.
A mouse model of RBD
The underlying cause of RBD is unknown. Current results show that impaired inhibitory transmission triggers the hallmark features of RBD in transgenic mice. Although our results show that abnormal glycine/GABAA receptor activity elicits an RBD phenotype, other proteins mediating inhibition (e.g., potassium chloride transporter 2, KCC2) could also be involved in triggering RBD symptoms. Determining the specific inhibitory mechanism(s) underlying RBD is therefore of immediate clinical importance.
Our findings not only identify a potential mechanism for RBD, they also indicate that transgenic mice could serve as a resource for determining RBD pathogenesis. Although previous animal studies identify brainstem circuits underlying abnormal REM motor control (Jouvet and Delorme, 1965; Hendricks et al., 1982; Friedman and Jones, 1984; Schenkel and Siegel, 1989; Shouse and Siegel, 1992; Holmes and Jones, 1994; Sanford et al., 2001; Lu et al., 2006; Vetrivelan et al., 2009), this new model represents an important advance because it recapitulates all primary RBD features stemming from a defined genetic mutation. We propose that these transgenic mice are a useful model for determining how glycine and GABA contribute to RBD symptoms. Current results also emphasize the need to determine whether impaired inhibitory transmission contributes to human RBD.
Footnotes
This study was funded by the Canadian Institutes of Health Research, the National Science and Engineering Research Council of Canada, the Canadian Foundation for Innovation, and the Parker B. Francis Foundation. We thank Dr. Hans Weiher for donating the founder line of transgenic mice. We also thank University of Toronto support staff for providing animal care.
References
- Albin RL, Koeppe RA, Chervin RD, Consens FB, Wernette K, Frey KA, Aldrich MS. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410–1412. doi: 10.1212/wnl.55.9.1410. [DOI] [PubMed] [Google Scholar]
- Becker L, von Wegerer J, Schenkel J, Zeilhofer HU, Swandulla D, Weiher H. Disease-specific human glycine receptor α1 subunit causes hyperekplexia phenotype and impaired glycine- and GABAA-receptor transmission in transgenic mice. J Neurosci. 2002;22:2505–2512. doi: 10.1523/JNEUROSCI.22-07-02505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Ferman TJ. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med. 2003;4:281–284. doi: 10.1016/s1389-9457(03)00072-8. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–2788. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Briel RC, McKeith IG, Barker WA, Hewitt Y, Perry RH, Ince PG, Fairbairn AF. EEG findings in dementia with Lewy bodies and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1999;66:401–403. doi: 10.1136/jnnp.66.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PL, Peever JH. Unraveling the mechanisms of REM sleep atonia. Sleep. 2008a;31:1492–1497. doi: 10.1093/sleep/31.11.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PL, Peever JH. Glycinergic and GABAA-mediated inhibition of somatic motoneurons does not mediate rapid eye movement sleep motor atonia. J Neurosci. 2008b;28:3535–3545. doi: 10.1523/JNEUROSCI.5023-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Tse G, Gillis L, Peever JH. Dopaminergic regulation of sleep and cataplexy in a murine model of narcolepsy. Sleep. 2010;33:1295–1304. doi: 10.1093/sleep/33.10.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess C, Lai D, Siegel J, Peever J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep–wake cycle. J Neurosci. 2008;28:4649–4660. doi: 10.1523/JNEUROSCI.0334-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli RJ, Chen K, Bandy D, Smilovici O, Boeve BF, Osborne D, Alexander GE, Parish JM, Krahn LE, Reiman EM. A preliminary fluorodeoxyglucose positron emission tomography study in healthy adults reporting dream-enactment behavior. Sleep. 2006;29:927–933. doi: 10.1093/sleep/29.7.927. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. Control of motoneurons during sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Ed 3. Philadelphia: Saunders; 2005. pp. 154–168. [Google Scholar]
- Coloma FM, Niles LP. Melatonin enhancement of [3H]-γ-aminobutyric acid and [3H]muscimol binding in rat brain. Biochem Pharmacol. 1988;37:1271–1274. doi: 10.1016/0006-2952(88)90781-2. [DOI] [PubMed] [Google Scholar]
- de Groen JH, Kamphuisen HA. Periodic nocturnal myoclonus in a patient with hyperexplexia (startle disease) J Neurol Sci. 1978;38:207–213. doi: 10.1016/0022-510x(78)90067-9. [DOI] [PubMed] [Google Scholar]
- Dinkel K, Meinck HM, Jury KM, Karges W, Richter W. Inhibition of gamma-aminobutyric acid synthesis by glutamic acid decarboxylase autoantibodies in stiff-man syndrome. Ann Neurol. 1998;44:194–201. doi: 10.1002/ana.410440209. [DOI] [PubMed] [Google Scholar]
- Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus FJ, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder—comparison with Parkinson's disease and controls. Brain. 2000;123:1155–1160. doi: 10.1093/brain/123.6.1155. [DOI] [PubMed] [Google Scholar]
- Fantini ML, Gagnon JF, Petit D, Rompré S, Décary A, Carrier J, Montplaisir J. Slowing of electroencephalogram in rapid eye movement sleep behavior disorder. Ann Neurol. 2003;53:774–780. doi: 10.1002/ana.10547. [DOI] [PubMed] [Google Scholar]
- Friedman L, Jones BE. Study of sleep-wakefulness states by computer-graphics and cluster-analysis before and after lesions of the pontine tegmentum in the cat. Electroencephalogr Clin Neurophysiol. 1984;57:43–56. doi: 10.1016/0013-4694(84)90007-5. [DOI] [PubMed] [Google Scholar]
- Gagnon JF, Postuma RB, Montplaisir J. Update on the pharmacology of REM sleep behavior disorder. Neurology. 2006a;67:742–747. doi: 10.1212/01.wnl.0000233926.47469.73. [DOI] [PubMed] [Google Scholar]
- Gagnon JF, Postuma RB, Mazza S, Doyon J, Montplaisir J. Rapid-eye-movement sleep behaviour disorder and neurodegenerative diseases. Lancet Neurol. 2006b;5:424–432. doi: 10.1016/S1474-4422(06)70441-0. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Morrison AR, Mann GL. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res. 1982;239:81–105. doi: 10.1016/0006-8993(82)90835-6. [DOI] [PubMed] [Google Scholar]
- Holmes CJ, Jones BE. Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular-formation for sleep-wake states studied by cytotoxic lesions in the cat. Neuroscience. 1994;62:1179–1200. doi: 10.1016/0306-4522(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Molinuevo JL, Santamaría J, Serradell M, Martí MJ, Valldeoriola F, Tolosa E. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. 2006;5:572–577. doi: 10.1016/S1474-4422(06)70476-8. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Santamaria J, Tolosa E. The clinical and pathophysiological relevance of REM sleep behavior disorder in neurodegenerative diseases. Sleep Med Rev. 2009a;13:385–401. doi: 10.1016/j.smrv.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Ratti PL, Casanova-Molla J, Serradell M, Vilaseca I, Santamaria J. Excessive muscle activity increases over time in idiopathic REM sleep behavior disorder. Sleep. 2009b;32:1149–1153. doi: 10.1093/sleep/32.9.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RF, Beltz TG, Thunhorst RL, Johnson AK. Investigations on the physiological controls of water and saline intake in C57BL/6 mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R394–R403. doi: 10.1152/ajpregu.00130.2003. [DOI] [PubMed] [Google Scholar]
- Jones BE. Basic mechanisms of sleep-wake states. In: Kryger MH, Roth T, Dement WC, editors. Principles and practices of sleep medicine. Philadelphia: Saunders; 2005. pp. 136–153. [Google Scholar]
- Jouvet M. Neurophysiology of the states of sleep. Physiol Rev. 1967;47:117–177. doi: 10.1152/physrev.1967.47.2.117. [DOI] [PubMed] [Google Scholar]
- Jouvet M, Delorme F. Locus coeruleus et sommeil paradoxal. C R Soc Biol. 1965;159:895–899. [Google Scholar]
- Kimura K, Tachibana N, Kohyama J, Otsuka Y, Fukazawa S, Waki R. A discrete pontine ischemic lesion could cause REM sleep behavior disorder. Neurology. 2000;55:894–895. doi: 10.1212/wnl.55.6.894. [DOI] [PubMed] [Google Scholar]
- Kunz D, Bes F. Melatonin as a therapy in REM sleep behavior disorder patients: an open-labeled pilot study on the possible influence of melatonin on REM-sleep regulation. Mov Disord. 1999;14:507–511. doi: 10.1002/1531-8257(199905)14:3<507::aid-mds1021>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res. 2010;19:591–596. doi: 10.1111/j.1365-2869.2010.00848.x. [DOI] [PubMed] [Google Scholar]
- Lapierre O, Montplaisir J. Polysomnographic features of REM sleep behavior disorder: development of a scoring method. Neurology. 1992;42:1371–1374. doi: 10.1212/wnl.42.7.1371. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, Shemen L, Hornykiewicz O. Distribution of high affinity sodium-independent [3H]gamma-aminobutyric acid ([3H]GABA) binding in human-brain—alterations in Parkinson's disease. Brain Res. 1977;127:269–278. doi: 10.1016/0006-8993(77)90540-6. [DOI] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Mahowald MW, Schenck CH. Insights from studying human sleep disorders. Nature. 2005a;437:1279–1285. doi: 10.1038/nature04287. [DOI] [PubMed] [Google Scholar]
- Mahowald MW, Schenck CH. REM sleep parasomnias. In: Kryger MH, Roth T, Dement WC, editors. Principles and practices of sleep medicine. Philadelphia: Saunders; 2005b. pp. 897–916. [Google Scholar]
- Martinelli P, Nassetti S, Minardi C, Macrì S, Ippoliti M. Electrophysiological evaluation of the stiff-man syndrome: further data. J Neurol. 1996;243:551–553. doi: 10.1007/BF00886879. [DOI] [PubMed] [Google Scholar]
- Massicotte-Marquez J, Décary A, Gagnon JF, Vendette M, Mathieu A, Postuma RB, Carrier J, Montplaisir J. Executive dysfunction and memory impairment in idiopathic REM sleep behavior disorder. Neurology. 2008;70:1250–1257. doi: 10.1212/01.wnl.0000286943.79593.a6. [DOI] [PubMed] [Google Scholar]
- Mazza S, Soucy JP, Gravel P, Michaud M, Postuma R, Massicotte-Marquez J, Decary A, Montplaisir J. Assessing whole brain perfusion changes in patients with REM sleep behavior disorder. Neurology. 2006;67:1618–1622. doi: 10.1212/01.wnl.0000242879.39415.49. [DOI] [PubMed] [Google Scholar]
- Morrison JL, Sood S, Liu H, Park E, Liu X, Nolan P, Horner RL. Role of inhibitory amino acids in control of hypoglossal motor outflow to genioglossus muscle in naturally sleeping rats. J Physiol. 2003;552:975–991. doi: 10.1113/jphysiol.2003.052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino N, Fujiwara H, Noguchi-Kuno SA, Tanaka C. GABAa receptor but not muscarinic receptor density was decreased in the brain of patients with Parkinson's disease. Jpn J Pharmacol. 1988;48:331–339. doi: 10.1254/jjp.48.331. [DOI] [PubMed] [Google Scholar]
- Olson EJ, Boeve BF, Silber MH. Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 2000;123:331–339. doi: 10.1093/brain/123.2.331. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Lang AE, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology. 2006;66:845–851. doi: 10.1212/01.wnl.0000203648.80727.5b. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72:1296–1300. doi: 10.1212/01.wnl.0000340980.19702.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne UK, Koskinen V, Laaksonen H, Lönnberg P, Sonninen V. GABA receptor binding in the parkinsonian brain. Life Sci. 1978;22:2225–2228. doi: 10.1016/0024-3205(78)90575-1. [DOI] [PubMed] [Google Scholar]
- Rosenstein RE, Estévez AG, Cardinali DP. Time-dependent effect of melatonin on glutamic acid decarboxylase activity and 36Cl− influx in rat hypothalamus. J Neuroendocrinol. 1989;1:443–447. doi: 10.1111/j.1365-2826.1989.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Sanford LD, Cheng CS, Silvestri AJ, Tang X, Mann GL, Ross RJ, Morrison AR. Sleep and behavior in rats with pontine lesions producing REM without atonia. Sleep Res Online. 2001;4:1–5. [Google Scholar]
- Schenck CH, Mahowald MW. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120–138. doi: 10.1093/sleep/25.2.120. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Ettinger MG, Mahowald MW. Chronic behavioral disorders of human REM sleep—a new category of parasomnia. Sleep. 1986;9:293–308. doi: 10.1093/sleep/9.2.293. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Patterson AL, Mahowald MW. Rapid eye movement sleep behavior disorder. A treatable parasomnia affecting older adults. JAMA. 1987;257:1786–1789. [PubMed] [Google Scholar]
- Schenck CH, Hurwitz TD, Mahowald MW. Symposium: normal and abnormal REM sleep regulation: REM sleep behaviour disorder: an update on a series of 96 patients and a review of the world literature. J Sleep Res. 1993;2:224–231. doi: 10.1111/j.1365-2869.1993.tb00093.x. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388–393. doi: 10.1212/wnl.46.2.388. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Mahowald MW. REM behavior disorder (RBD): delayed emergence of parkinsonism and/or dementia in 65% of older men initially diagnosed with idiopathic RBD, and an analysis of the minimum and maximum tonic and/or phasic electromyographic abnormalities found during REM sleep. Sleep. 2003;26:A316–A316. [Google Scholar]
- Schenkel E, Siegel JM. REM-sleep without atonia after lesions of the medial medulla. Neurosci Lett. 1989;98:159–165. doi: 10.1016/0304-3940(89)90503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MH, Valatx JL, Sakai K, Fort P, Jouvet M. Role of the lateral preoptic area in sleep-related erectile mechanisms and sleep generation in the rat. J Neurosci. 2000;20:6640–6647. doi: 10.1523/JNEUROSCI.20-17-06640.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sforza E, Zucconi M, Petronelli R, Lugaresi E, Cirignotta F. REM sleep behavioral disorders. Eur Neurol. 1988;28:295–300. doi: 10.1159/000116288. [DOI] [PubMed] [Google Scholar]
- Sforza E, Krieger J, Petiau C. REM sleep behavior disorder: clinical and physiopathological findings. Sleep Med Rev. 1997;1:57–69. doi: 10.1016/s1087-0792(97)90006-x. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Shirakawa S, Takeuchi N, Uchimura N, Ohyama T, Maeda H, Abe T, Ishibashi M, Ohshima Y, Ohshima H. Study of image findings in rapid eye movement sleep behavioural disorder. Psychiatry Clin Neurosci. 2002;56:291–292. doi: 10.1046/j.1440-1819.2002.00961.x. [DOI] [PubMed] [Google Scholar]
- Shouse MN, Siegel JM. Pontine regulation of REM-sleep components in cats: integrity of the pedunculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM-sleep. Brain Res. 1992;571:50–63. doi: 10.1016/0006-8993(92)90508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerritt JH, Johnston GAR. Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur J Pharmacol. 1983;89:193–198. doi: 10.1016/0014-2999(83)90494-6. [DOI] [PubMed] [Google Scholar]
- Soikkeli R, Partanen J, Soininen H, Pääkkönen A, Riekkinen P., Sr Slowing of EEG in Parkinson's disease. Electroencephalogr Clin Neurophysiol. 1991;79:159–165. doi: 10.1016/0013-4694(91)90134-p. [DOI] [PubMed] [Google Scholar]
- Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABAergic neurons and pancreatic beta-cells in stiff-man syndrome. N Engl J Med. 1990;322:1555–1560. doi: 10.1056/NEJM199005313222202. [DOI] [PubMed] [Google Scholar]
- Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998;803:178–188. doi: 10.1016/s0006-8993(98)00631-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi N, Uchimura N, Hashizume Y, Mukai M, Etoh Y, Yamamoto K, Kotorii T, Ohshima H, Ohshima M, Maeda H. Melatonin therapy for REM sleep behavior disorder. Psychiatry Clin Neurosci. 2001;55:267–269. doi: 10.1046/j.1440-1819.2001.00854.x. [DOI] [PubMed] [Google Scholar]
- Tippmann-Peikert M, Boeve BF, Keegan BM. REM sleep behavior disorder initiated by acute brainstem multiple sclerosis. Neurology. 2006;66:1277–1279. doi: 10.1212/01.wnl.0000208518.72660.ff. [DOI] [PubMed] [Google Scholar]
- Unger MM, Belke M, Menzler K, Heverhagen JT, Keil B, Stiasny-Kolster K, Rosenow F, Diederich NJ, Mayer G, Möller JC, Oertel WH, Knake S. Diffusion tensor imaging in idiopathic REM sleep behavior disorder reveals microstructural changes in the brainstem, substantia nigra, olfactory region, and other brain regions. Sleep. 2010;33:767–773. doi: 10.1093/sleep/33.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q, Lu J. Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci. 2009;29:9361–9369. doi: 10.1523/JNEUROSCI.0737-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JW, James L. Serotonergic antidepressants are associated with REM sleep without atonia. Sleep. 2004;27:317–321. doi: 10.1093/sleep/27.2.317. [DOI] [PubMed] [Google Scholar]
- Wu FS, Yang YC, Tsai JJ. Melatonin potentiates the GABA(A) receptor-mediated current in cultured chick spinal cord neurons. Neurosci Lett. 1999;260:177–180. doi: 10.1016/s0304-3940(98)00983-5. [DOI] [PubMed] [Google Scholar]
- Ye ZY, Lu YG, Sun H, Cheng XP, Xu TL, Zhou JN. Fluoxetine inhibition of glycine receptor activity in rat hippocampal neurons. Brain Res. 2008;1239:77–84. doi: 10.1016/j.brainres.2008.08.055. [DOI] [PubMed] [Google Scholar]