

Abstract
Liver X receptors (LXRs) regulate immune cell function and cholesterol metabolism, both factors that are critically involved in Alzheimer's disease (AD). To investigate the therapeutic potential of long-term LXR activation in amyloid-β (Aβ) peptide deposition in an AD model, 13-month-old, amyloid plaque-bearing APP23 mice were treated with the LXR agonist TO901317. Postmortem analysis demonstrated that TO901317 efficiently crossed the blood–brain barrier. Insoluble and soluble Aβ levels in the treated APP23 mice were reduced by 80% and 40%, respectively, compared with untreated animals. Amyloid precursor protein (APP) processing, however, was hardly changed by the compound, suggesting that the observed effects were instead mediated by Aβ disposal. Despite the profound effect on Aβ levels, spatial learning in the Morris water maze was only slightly improved by the treatment. ABCA1 (ATP-binding cassette transporter 1) and apolipoprotein E (ApoE) protein levels were increased and found to be primarily localized in astrocytes. Experiments using primary microglia demonstrated that medium derived from primary astrocytes exposed to TO901317 stimulated phagocytosis of fibrillar Aβ. Conditioned medium from TO901317-treated ApoE−/− or LXRα−/− astrocytes did not increase phagocytosis of Aβ. In APP23 mice, long-term treatment with TO901317 strongly increased the association of microglia and Aβ plaques. Short-term treatment of APP/PS1 mice with TO901317 also increased this association, which was dependent on the presence of LXRα and was accompanied by increased ApoE lipidation. Together, these data suggest that astrocytic LXRα activation and subsequent release of ApoE by astrocytes is critical for the ability of microglia to remove fibrillar Aβ in response to treatment with TO901317.
Introduction
Ample evidence suggests a role for innate immune function and brain cholesterol metabolism in Alzheimer's disease (AD) (Mulder and Terwel, 1998; Heneka and O'Banion, 2007). Both processes happen to be under the control of liver X receptors (LXRs) (Whitney et al., 2002; Zelcer and Tontonoz, 2006). Two types of LXRs exist, LXRα and LXRβ. LXRα is found at high levels in cholesterol-metabolizing tissues and its expression is acutely regulated (Whitney et al., 2001), while LXRβ is ubiquitously expressed (Baranowski, 2008). Both LXRs act as cholesterol sensors and are activated by hydroxylated forms of cholesterol, leading to induction of genes that encode for proteins directly responsible for cholesterol export from the cell, such as apolipoprotein E (ApoE), ATP-binding cassette transporters (ABCA1, ABCG1), and sterol regulatory element-binding protein 1c (SREBP1c) (Zelcer and Tontonoz, 2006). In humans ApoE is present as three different isoforms, ApoE2, -3, and -4. The fact that ApoE4 is the best described risk factor for sporadic AD (Roses, 2006) hints to a link between this disease and cholesterol metabolism. The control of innate immune cell function by LXRs is not directly related to cholesterol metabolism and involves interference of nuclear factor κB signaling (Zelcer and Tontonoz, 2006).
Because of the central role played by lipid metabolism in the development of atherosclerosis (Beaven and Tontonoz, 2006), agonists for LXRs are under development for therapy in this condition and are currently being evaluated in respective clinical trials. This encourages research into the application of LXR agonists for other diseases in which lipid metabolism is implicated, including AD. The fact that LXRs affect dual processes implicated in AD adds to the attraction to test these compounds in AD.
Several studies have addressed the effect of LXR stimulation in cellular or rodent models of AD. It has been shown that the LXR agonists TO901317 and GW3965 alter amyloid-β (Aβ) production in neuronal and non-neuronal cell cultures overexpressing amyloid precursor protein (APP) (Koldamova et al., 2003, 2005b; Kim et al., 2007).
Short-term administration of LXR agonists TO901317 and GW3965 to 5-month-old preplaque Tg2576 mice improved fear conditioning (Riddell et al., 2007; Jiang et al., 2008) and long-term treatment with TO901317 restored object recognition in plaque-bearing APPSLxPS1mut mice (Vanmierlo et al., 2009). Long-term treatment with GW3965 has been shown to reduce plaque formation by 50% in aged Tg2576 mice (Jiang et al., 2008). Short-term treatments with TO901317 have shown small reductions of soluble Aβ40 and -42 (Koldamova et al., 2005b), or Aβ42 only, in hippocampus of Tg2576 mice (Riddell et al., 2007).
While these studies elucidate the dependence of LXR agonist-mediated effects on different treatment protocols, ages, and disease stages, the underlying mechanism of LXR agonist protection in aged APP transgenic mice and, in particular, an effect on the removal of fibrillar Aβ remain unclear. In the present study we investigated the role of LXRs in microglial removal of fibrillar Aβ in vitro and after chronic stimulation of LXRs by TO901317 in aged APP23 mice for 7 weeks from 13 months of age onwards.
Materials and Methods
Animal experiments.
In the present study APP23 mice and littermate wild-type mice were used. In the APP23 mouse line, human APP751 carrying the Swedish double mutation (K670M/N671L) is overexpressed sevenfold over the endogenous mouse APP. The mice were generated on a C57BL/6 × DBA/2 background and were back-crossed into C57BL/6. APP23 mice were 13 months old at the beginning of the study. Deposition of Aβ in APP23 mice starts at 6 months of age and continuously increases (Sturchler-Pierrat et al., 1997), and at 12 months substantial plaque deposition has been demonstrated (Kuo et al., 2001). Of relevance to our present work, Sturchler-Pierrat et al. (1997) have described Aβ plaque-associated microglia already present at 12 months. Thus, at 13 months, when we initiated treatment with the LXR agonist, there was a substantial Aβ plaque deposition, and microglia associated with it, present in APP23 mice. Mice were housed separately in individually ventilated cages 3 weeks in advance of the experiment, under standard conditions (at 22°C, humidity 18%) with free access to food and water on a reverse dark/light cycle of 12 h light and 12 h darkness. APP23 and control mice received 50 mg/kg TO901317 or vehicle daily by oral gavage for 7 weeks. The vehicle was a 0.5% methylcellulose solution and the compound was suspended by sonication. In week 5 of treatment, behavioral habituation of the mice was assessed in the open field. In weeks 6 and 7, animals underwent spatial memory assessment using the Morris water maze. For blood and tissue collection the animals were shortly anesthetized by inhalation of isoflurane and received an injection of 50 μl of 0.5 m EDTA through the left heart chamber. Blood was drawn from the left heart chamber and stored at −80°C until compound determination. Subsequently, mice were transcardially perfused with ice-cold PBS and brains were removed. One hemisphere of the brain was fixed in ice-cold 4% paraformaldehyde, PBS overnight, and used for immunohistochemistry. The other hemisphere was separated into a hindbrain and a forebrain part. Brain parts were snap-frozen in liquid nitrogen and subsequently stored at −80°C until further determination of insoluble and soluble Aβ species, other APP processing products, and compound determination.
Additionally, 13-month-old APP/PS1/LXR−/− mice and APP/PS1/LXR+/− littermates on a C57BL/6 background were used. Parental strains of these mice are described by Jankowsky et al. (2004) and Alberti et al. (2000). The APP/PS1/LXR−/− and APP/PS1/LXR+/− mice were treated with TO901317 as described above, but only for 6 d. CSF was collected from the cisterna magna of these mice as previously described (DeMattos et al., 2002). Brain hemispheres were obtained as described for the APP23 mice and used for the immunohistochemical determination of the association of microglia with Aβ.
Animal care and handling was performed according to the declaration of Helsinki and approved by the local ethical committees.
Determination of TO901317 levels in blood and brain tissue.
The concentrations of TO901317 were determined in EDTA-treated blood and brain tissue (after homogenization in phosphate buffer, pH 7.2) by liquid chromatography/tandem mass spectrometry (LC/MS/MS). These analyses were performed on a TSQ mass spectrometer (ThermoFisher Scientific). The instrument was operated in the atmospheric pressure and negative electrospray ionization mode with selected reaction monitoring. Liquid chromatography was performed on a MaxRP Synergi column (50 × 2 mm, 2.5 μm pore size; Phenomenex), with a gradient elution (varying the percentages of water, methanol, and acetonitrile, and with 1% formic acid). Samples were prepared using protein precipitation with acetonitrile. The limit of quantitation was 8 pmol/ml in blood and 24 pmol/g tissue in brain.
Open field behavior.
The open field consisted of a 61 × 61 × 61 cm Perspex box with dark walls and a white floor and was dimly illuminated. The open field was virtually divided into corridors along the walls (10 cm wide; corners, 10 × 10 cm; and a center, 40 × 40 cm). Mice were put in the middle of the open field and tracked by a camera connected to a computer running on dedicated software (Ethovision, Noldus) for 10 min on 3 consecutive days. Vertical movements were registered, and distance, corridor, corner, and center times were determined.
Morris water maze.
The Morris water maze used consisted of a 61-cm-high plastic circular basin (diameter 1 m) approximately half filled with water, with an invisible platform of 15 × 15 cm just under water in the middle of one of four equal virtual segments (quadrants). A white curtain surrounded the basin. Three asymmetrically applied intra-maze cues were presented to the animals. The water was made turbid with white dispersion paint. The mice were subjected to 1 training session per day for 8 consecutive days. One training session consisted of 4 trials of 40 s each. Time between trials was 10 s. The starting position in each trial was quasi-random. If a mouse did not succeed in finding the platform, it would be put onto it. The mice were tracked by a computer operating on dedicated software (Ethovision; Noldus), which registered vertical movements and calculated distance laid back, latency to the platform, time in quadrant, and quadrant entries.
Gel electrophoresis and Western blotting.
Forebrains were weighed and homogenized by sonication in 10 volumes of Tris-buffered saline (TBS) with Complete Inhibitor Cocktail (Roche Diagnostics). Homogenates were run on 4–20% SDS-PAGE. C-terminal fragments were detected with rabbit antiserum APP-C8 raised against the C-terminal amino acids of APP (Schrader-Fischer and Paganetti, 1996). ABCA1 was detected with anti-ABCA1 antibody (1:200, Novus Biologicals, 100-1663), ApoE with anti-ApoE (1:1000, Novus Biologicals, 100-2040), and β-actin with anti-β-actin (1:5000, Sigma, A2228).
Nondenaturing gradient gel electrophoresis.
Samples of CSF were mixed 1:1 with native sample buffer and electrophoresed on 4–20% Tris-Glycine gels (Invitrogen). Proteins with known hydrated diameters were used as size standards (GE Healthcare). Proteins were transferred to a nitrocellulose membrane and probed with anti-goat ApoE antibody (Calbiochem, catalog #178479, 1:200) and secondary horse anti-goat IgG linked HRP (Santa Cruz Biotechnology, Sc 2020, 1:2500), followed by horse anti-goat IgG linked to HRP (Vector Laboratories, 1:1000). Bands were visualized with enhanced chemiluminescence (Pierce) and imaged with Syngene G:Box.
Aβ determination by sandwich immunoassay on MSD Multi-Array plate.
Brain regions were weighed and homogenized by sonication in 10 volumes of TBS (20 mm Tris-HCl, pH 7.6, 137 mm sodium chloride, and Complete Protease Inhibitor Cocktail; Roche Diagnostics). For determination of total Aβ40 and Aβ42, forebrain homogenates were extracted for 15 min at 4°C with 70% formic acid. The extracts were neutralized by addition of 19 volumes of 1 m Tris-base and centrifuged for 15 min at 20,000 × g. For determination of TBS-soluble Aβ, forebrain homogenates were centrifuged at 100,000 × g for 15 min. For determination of Triton X-100-soluble Aβ, Triton X-100 was added to the forebrain homogenate (final concentration 1%) and samples were extracted on ice for 15 min before centrifugation at 100,000 × g. Samples were mixed every 5 min. Human (6E10) Aβ40 or Aβ42 ultra-sensitive kits were used (Meso Scale Discovery). Triton X-100-soluble Aβ was diluted to a final concentration of 1:100 with 3% Blocker A. Triton X-100 extracts were diluted 1:100. Signals were measured on a SECTOR Imager 6000 reader (Meso Scale Discovery).
Determination of soluble APPα and soluble APPβ by sandwich immunoassay on MSD multiplex plate.
Supernatants were diluted with 1% Triton X-100, TBS (final dilution 1:2000), loaded on an MS6000 soluble APPα (sAPPα)/sAPPβ multiplex plate (Meso Scale Discovery), and further processed as described for Aβ.
Detection of Aβ aggregation.
FAM-Aβ1-42 (Peptide Specialty Laboratories) was solubilized as previously described (Teplow, 2006). In brief, FAM-Aβ1-42 was solubilized in 1,1,1–3,3,3-hexafluoroisopropanol, incubated for 10 min, and evaporated using a SpeedVac. FAM-Aβ1-42 was resolubilized to 221 μm in 10 mm NaOH, and aggregation was started by dilution to 25 μm using 50 mm Tris-HCl, pH 7. Samples were incubated for 18 h at 37°C and flash-frozen in liquid nitrogen. Samples were stored at −80°C afterward. Aggregation state was determined by sample separation on 4–12% NuPAGE gel and Western blot using antibody 6E10 (Covance), followed by enhanced chemiluminescence detection (Millipore) using HRP-conjugates (Jackson ImmunoResearch Laboratories). Signals were analyzed using the Chemidoc XRS documentation system (Bio-Rad).
Immunohistochemistry.
Free-floating 40-μm-thick serial sections were cut on a vibratome (Leica). Sections obtained were stored in 0.1% NaN3, PBS in a cold room. For immunohistochemistry, sections were treated with 50% methanol for 15 min. Then, sections were washed three times for 5 min in PBS and blocked in 3% BSA, 0.1% Triton X-100, PBS (blocking buffer) for 30 min, followed by overnight incubation with the primary antibody in blocking buffer. Next, sections were washed three times in 0.1% Triton X-100, PBS, incubated with Alexa 488- or Alexa 594-conjugated secondary antibodies (1:500, Invitrogen) for 90 min, and washed three times with 0.1% Triton X-100, PBS for 5 min. Finally, the sections were mounted on glasses in tap water and embedded in Mowiol solution with 0.1% 1,4-diazobicyclo[2.2.2]octan. The following primary antibodies were used with respective concentrations: rabbit polyclonal 2964 against Aβ (gift from Dr. Jochen Walter, University of Bonn, Bonn, Germany), rabbit polyclonal anti-ABCA1 (1:200, Novus Biologicals), goat polyclonal anti-ApoE (1:200, M-20, sc-6384, Santa Cruz Biotechnology), rabbit polyclonal anti-ApoE (1:200, ab20874, Abcam), rabbit polyclonal anti-GFAP (1:1000, Dako), rat monoclonal anti-mouse CD11b (1:200, Serotec), and rabbit polyclonal anti-Iba1 (1:500, Wako, Osaka, Japan). Fluorescence microscopy was performed using an Olympus BX61 microscope, and images were processed in Cell-P (Olympus).
Primary microglial cell culture.
Primary microglial cell cultures were prepared as previously described in detail (Giulian and Baker, 1986). Briefly, mixed glial cultures were prepared from newborn mice and cultured in DMEM supplemented with 10% FCS and 100 U · ml−1 penicillin/streptomycin. Microglial cells were used after 10–14 d of primary cultivation. They were harvested by shake-off, and replated and allowed to attach to the substrate for 30 min. Attached cells were extensively washed with culture medium and kept in culture for 1–2 d before being used. For the experiments, wild-type, ApoE−/− (Plump et al., 1992), LXRα−/−, LXRβ−/−, and LXRα−/−/β−/− (Alberti et al., 2000) on a C57BL/6 background were used.
Phagocytosis of FAM-labeled Aβ1-42 by microglial cells.
Microglial phagocytosis of FAM-labeled Aβ1-42 (FAM-Aβ) (AnaSpec) was measured by plate-based assay as previously described (Floden and Combs, 2006). In brief, cells were plated at a density of 50,000 per well. Cells were incubated with 500 nm Aβ for up to 4 h, starting 1 h after plating. Finally, the Aβ-containing medium was removed and extracellular Aβ was quenched with 100 μl of 0.2% trypan blue in PBS, pH 4.4, for 1 min. After aspiration, fluorescence was measured at 485 nm excitation/ 535 nm emission using a SpectrafluorPlus reader (Tecan). To be able to normalize for cell numbers, cells were incubated with 100 μl of 50 μg/ml Hoechst 33342 in PBS for 30 min and fluorescence was measured at 360 nm excitation/465 nm emission. Additionally, the occurrence of microglial Aβ phagocytosis was verified by confocal laser scanning microscopy. Cells were fixed with 4% formaldehyde for 10 min and immunostained using rat anti-mouse monoclonal antibody MCA711 for CD11b (Serotec), and lysosomes were stained using LysoTracker Red (Invitrogen). Confocal laser scanning microscopy was performed using an LSM 510 (Carl Zeiss). To validate the presence of aggregated Aβ within microglia, cells were incubated with aggregated Aβ1-42 overnight and stained with Thioflavin S. Fluorescence microscopy was performed using an Olympus BX61.
Determination of plaque-associated microglia.
Double-immunofluorescent staining for CD11b and Aβ was performed using the above-described antibodies. Fields of plaques were randomly selected in the cortex. Images were made in Cell-P with automatic illumination. The area of CD11b overlaying plaques was determined with the Colocalization Finder plugin in NIH ImageJ 1.44 and corrected for total plaque area determined with the subroutine Particle Analysis after background subtraction equal for all images and binarization. Only plaques with a diameter smaller than 30 μm were included in the analysis. Per-animal coverage of 50–200 plaques by microglia was determined. Animal number per group was 7.
Statistical analysis.
For statistical analysis, GraphPad Prism or SPSS was used. ELISA, Western blot data, or image analysis data were analyzed where appropriate by Student's t test or two-way ANOVA followed by Student's t tests if indicated. Behavioral data on learning in the Morris water maze were analyzed by three-way ANOVA, with transgene and treatment as the independent between-subject variables and day as within-subject variable, and distance or latency as the dependent variables. Integrated responses (area under the curve) were also analyzed for the same between-subject and dependent variables. Probe trial data were analyzed by three-way ANOVA with place (quadrant 1–4) as within-subject variable, transgene and treatment as between-subject variables, and time as dependent variable. Probe trial data were further analyzed with repeated-measures ANOVA and Tukey's post hoc test. Data on open field behavior were analyzed by three-way ANOVA with transgene and treatment as between-subject dependent variables, day as within-subject dependent variable, and time in corridor or center and distance laid back as dependent variables. Fluorescence measurements of uptake of labeled Aβ were analyzed by one-way ANOVA followed by Tukey's post hoc tests.
Results
Pharmacological treatment of APP23 mice with the LXR agonist TO901317
To test whether LXR activation by the pharmacological agonist TO901317 affects the deposition of Aβ, 13-month-old APP23 mice were treated for 7 weeks with TO901317 (50 mg/kg) by daily oral gavage. To determine the penetration of TO901317 into the brain, the compound concentrations in brain tissue and blood were assessed at 4 h after the last dosing by LC/MS/MS. Treatment with TO901317 for 7 weeks resulted in hind- and forebrain concentrations that were substantially higher (approximately threefold) than those found in the peripheral blood in both APP23 mice and wild-type controls (Fig. 1A). Concentrations of the compound in the brain reached values of ∼5 nmol/g fresh weight, while those in the blood were ∼2 nmol/ml. Assessment of total body weight over the entire observation period did not reveal any differences in response to drug treatment (Fig. 1B), which indicates that the drug was well tolerated. While serial inspection of livers of TO901317-treated and untreated mice revealed a tendency toward liver enlargement in response to treatment, it seems important to note that this phenomenon did not affect specific measures of animal well-being including ambulation, grooming, skin condition, open field behavior, or weight (see below). Notably, APP23 mice had a somewhat lower body weight compared with controls (two-way ANOVA, p < 0.001), in accordance with their reported hypermetabolic condition (Vloeberghs et al., 2008). The factor treatment did not affect open field behavior (three-way ANOVAs, p values > 0.05). Open field behavior, assessed in the fifth week of treatment, was not different between APP23 and wild-type mice, except for a slightly higher activity of the transgenic mice on the first day in the open field (interaction day × transgene in the three-way ANOVA and one-way ANOVA for the factor transgene on day 1, p values <0.05) (Fig. 1C). The effect of day was highly significant for all measures (three-way ANOVA, p values < 0.001), which means that the animals habituated to the open field.
Figure 1.
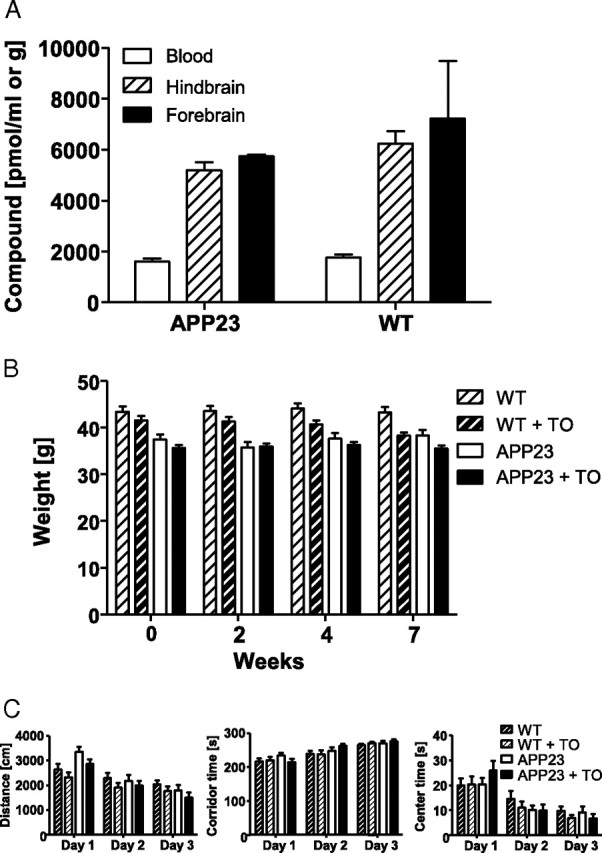
TO901317 accumulates in the brain and does not affect body weight or basal behavior of mice in the open field upon long-term treatment. A, Higher concentrations of TO901317 as determined with liquid chromatography tandem mass spectroscopy were found in the hind- and forebrain compared with the peripheral blood 4 h after the last administration of TO9 01317 (50 mg/kg per day for 7 weeks). Number of hindbrains per group was 19–22, number of forebrains per group was 3. B, Body weight of wild-type (WT) and APP23 mice treated with either vehicle or TO901317 (TO) was monitored at the indicated time points during the experimental period. Number of animals per group was 19–22. C, Distance laid back, corridor time, and center time of indicated groups of mice in the open field were not affected by treatment with TO901317. Number of animals per group was 12. Data represent means ± SEM.
Inverse effects of TO901317 on Aβ, ApoE, and ABCA1
Determination of forebrain Aβ levels revealed a decrease by 40% and 80% of both soluble and insoluble Aβ1-40 and Aβ1-42 levels, respectively, in response to TO901317 treatment (Fig. 2A,B). Analysis of APP processing in TO901317- and vehicle-treated mice revealed a slight but significant reduction of APP and soluble APPα. Soluble APPβ, C99, and C83 were not statistically different between groups (Fig. 2C). Additionally, TO901317 treatment did not alter the concentration of murine Aβ in wild-type mice (Fig. 2D). Immunohistochemical analysis with an antibody directed against fibrillar Aβ confirmed the Aβ-lowering effects of oral TO901317 treatment in aged APP23 mice (Fig. 2E). Quantification of Thioflavin S-stained Aβ in brain sections of TO901317-treated APP23 mice revealed a strong reduction compared with nontreated APP23 mice (Fig. 2F). Collectively, these data suggest that the reduction of Aβ by TO901317 was not mediated by modulation of the APP processing pathway.
Figure 2.
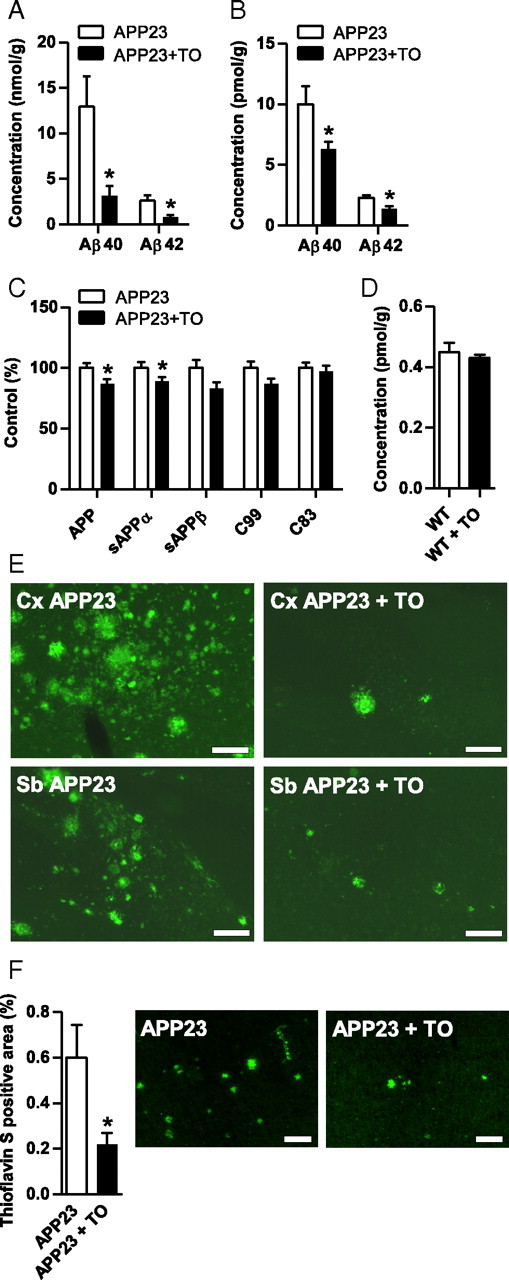
TO901317 reduces Aβ burden in the APP23 transgenic mouse model of AD. A, B, Concentrations of human Aβ40 and Aβ42 in formic acid extracts (A) or soluble fraction (B) in APP23 mice, as determined by sandwich ELISA, were strongly reduced in response to TO901317 (TO; 50 mg/kg per day for 7 weeks). 6E10 was used as a capture antibody and anti-Aβ40 as detection antibody. C, Sandwich immunoassay on Triton X-100 extracts or Western blot determination on brain homogenates showed a minor but significant reduction of APP and sAPPα upon TO901317 treatment. Capture antibodies in the sandwich immunoassay were anti-sAPPα and anti-sAPPβ; reporter was anti-APP. For Western blot, detection of APP and the C-terminal fragments 6E10 was used. D, Endogenous Aβ levels remained unaffected by drug treatment as demonstrated by ECL immunoassay. E, Representative micrographs of Aβ immunohistochemistry with antibody 2964, showing the frontal cortex (Cx) or subiculum (Sb) of TO901317- and vehicle-treated APP23 mice. Asterisks indicate significant differences (unpaired Student's t test, *p < 0.05, n = 12). Data represent means ± SEM. F, Thioflavin S-positive area is reduced in brain sections of APP23 mice treated with TO901317. Scale bars, 200 μm. WT, Wild type.
Treatment with TO901317 increased concentrations of ABCA1 and ApoE in APP23 and control mice (Fig. 3A–C). To investigate which brain cells are most likely to respond to TO901317, the distribution of two proteins of which the genes are targets for LXR, notably ABCA1 and ApoE, was investigated by immunohistochemistry. ABCA1 was distributed throughout the brain and could be shown to be present in astrocytes (Fig. 3D). Its presence in astrocytes could still be observed in the very fine ramifications where GFAP staining is very faint or absent (data not shown). ApoE was found in astrocytes as well (Fig. 3E), as observed by others (Fujita et al., 1999). ABCA1 and ApoE could not be found to colocalize with CD11b-positive microglia, regardless of their morphological phenotype, which here at the plaque site may reflect their response to Aβ (Fig. 3D,E), indicating very low levels or even the absence of these proteins in microglial cells in vivo. Immunofluorescence for ABCA1 and ApoE in astrocytes was not notably stronger in the vicinity of plaques, demonstrating that expression of these proteins is not a marker for activated astroglia. ABCA1 was present in astrocytes to the same extent in different brain areas, whereas this distribution differs greatly for GFAP. Treatment with TO901317 did not alter the cellular localization of ABCA1 and ApoE (data not shown). Immunostaining using an anti-ABCA1 antibody was absent in brain sections of ABCA1−/− mice (Fig. 3D).
Figure 3.
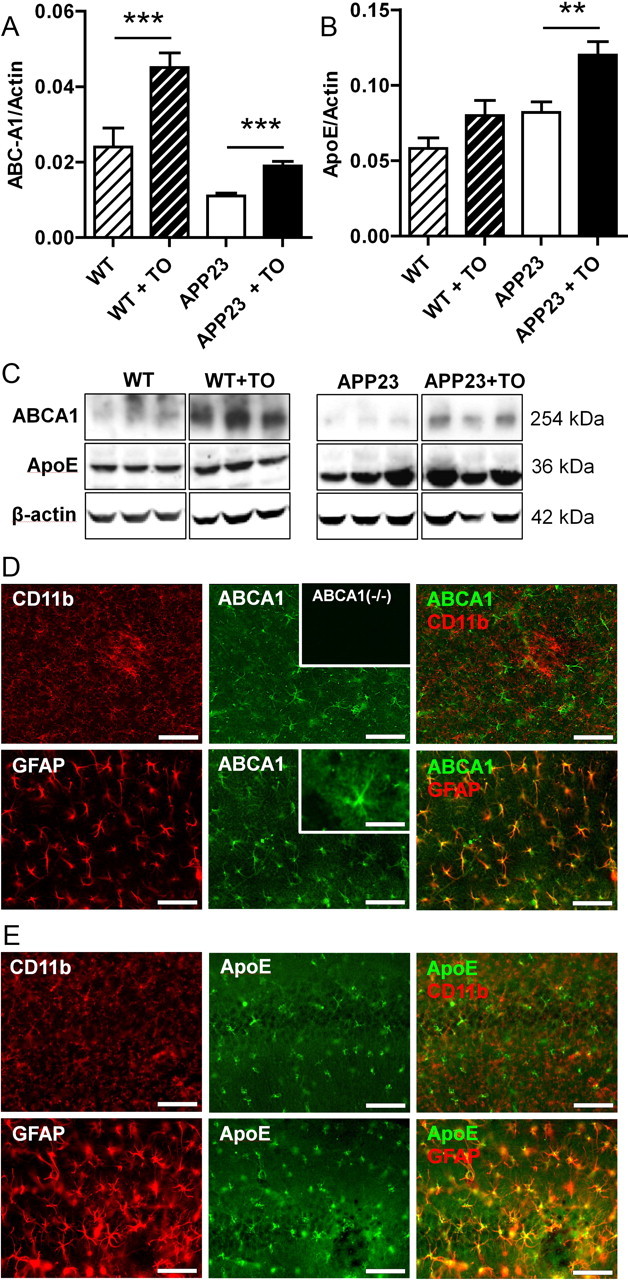
LXR activation positively regulates ApoE and ABCA1 expression in astrocytes. A, Densitometric quantification of Western blot detection of ApoE in brain lysates from APP23 and wild-type (WT) mice treated with either vehicle or TO901317 (TO). ApoE level was corrected for the level of the housekeeping protein actin. B, Densitometric analysis of Western blot detection of ABCA1 in brain lysates from APP23 and wild-type mice treated with either vehicle or TO901317. ABCA1 level was corrected for actin level. C, Representative images of ABCA1, ApoE, and β-actin Western blot detection in wild-type or APP23 mice treated with either vehicle or TO901317. D, E, Brain sections of vehicle-treated mice showing neocortex (D) or hippocampus (E) were immunostained for CD11b (microglial marker), GFAP (marker for astrocytic activation), and ApoE (D) or ABCA1 (E). The inset at the top of D shows absence of immunostaining in ABCA1 knock-out mice, while the inset at the bottom displays the ABCA1 immunostaining at higher magnification in the astrocytic arbors. Together, these immunostainings show that ApoE and ABCA1 expression is predominantly located in astrocytes but not in microglia in the murine brain. Asterisks indicate significant differences (ANOVA, Student's t test, n = 12, **p < 0.01, ***p < 0.001). Data represent means ± SEM. Scale bars, 100 μm, except inset, 25 μm.
Spatial learning of TO901317-treated APP23 transgenics
Spatial learning was assessed in the Morris water maze in the sixth to seventh week of treatment. A highly significant between-subject effect for the factor strain was observed on the dependent variables distance and latency (p < 0.001), indicating a spatial memory deficit in the APP23 mice compared with controls (Fig. 4A). In addition, the effect of the within-subject factor day was highly significant (p values < 0.01), indicating a learning effect. An effect for the factor treatment was not observed on these variables. In conclusion, APP23 mice exhibited a clear spatial memory deficit in the Morris water maze test compared with wild-type littermates.
Figure 4.
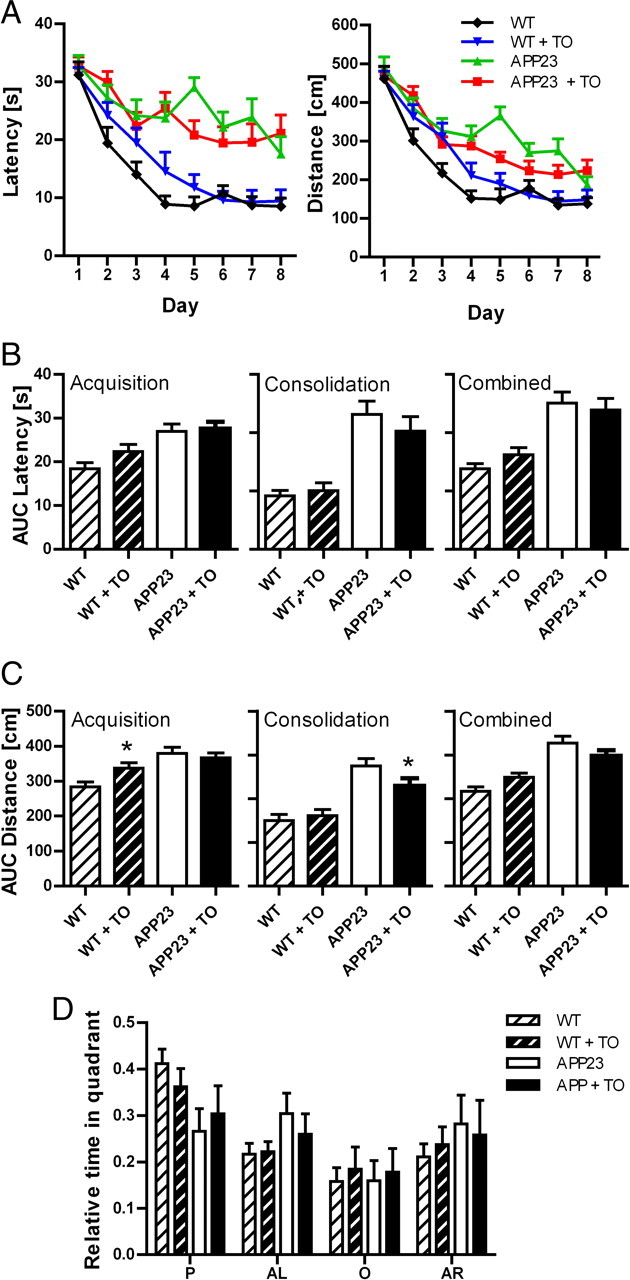
Spatial memory learning of APP23 mice treated with TO901317. A, Time needed to reach the hidden platform (latency in seconds) and distance traveled (in centimeters) in the Morris maze by vehicle or TO901317 (TO)-treated APP23 (n = 16) and wild-type (WT) mice (n = 16). B, C, Integrated distance traveled (area under the curve) (B) and time (C) were determined for the whole observation period or divided in an acquisition (day 1–4) and a consolidation (day 5–8) phase. Asterisks indicate significant differences (Student's t test, *p < 0.05). D, TO901317 treatment does not reinstate place preference in the Morris water maze in APP23 mice. P, AL, O, and AR represent platform quadrant, and quadrant to the left, the right, and opposite of P, respectively. Data represent means ± SEM. Groups consisted of 16 animals. For detailed description of the statistics, see Results.
Data were further analyzed with respect to overall performance (average distance and latency for days 1–8), acquisition (averages for day 1–4 of training), and consolidation (averages for day 5–8 of training). The effect of the transgene was again highly significant for performance overall, during acquisition and consolidation (Fig. 4B,C). Post hoc analysis revealed a minor worsening effect of the treatment during acquisition in the wild-type animals and a minor improvement during consolidation in the APP23 mice (Fig. 4B). Together, a beneficial effect was only detectable during the consolidation but not during the acquisition phase. Overall, the magnitude of improvement was small.
Three-way ANOVA of probe trial data revealed a significant effect for the factor place (quadrant) (p < 0.01) and an interaction between the factors place and transgene (p < 0.05) (Fig. 4D). Further statistical analysis revealed that these effects were accounted for by a place preference of the wild-type mice (repeated measures one-way ANOVA and Tukey's post hoc test, p < 0.001) and the absence thereof in the APP23 mice (one-way ANOVA, Tukey's post hoc test, p > 0.05). The probe trial revealed that the wild-type mice used a spatial strategy, while the APP23 mice failed to do so. No treatment effects were observed on probe trial performance.
Modulation of microglial Aβ1-42 phagocytosis by the LXR agonist TO901317
Phagocytosis of aged FAM-labeled Aβ1-42 by microglia in vitro was assayed as previously described by Floden and Combs (2006), and controlled by laser confocal microscopy and immunoprecipitation of Aβ in lysates of microglial cells. Aggregation of Aβ upon aging was verified by Western blot analysis (Fig. 5A) and detection of Thioflavin S-positive Aβ in exposed microglia (Fig. 5A). Direct treatment of microglial cells for 2 h with the LXR agonist TO901317 did not enhance Aβ phagocytosis (Fig. 5A). However, when Aβ was presented in medium derived from astrocytes that were preexposed to increasing concentrations of the LXR agonist (TO901317, 10 nm to 10 μm) for 24 h, uptake by microglia increased significantly in a dose-dependent manner, suggesting that LXR agonist stimulated astrocytes to secrete a product that positively regulates phagocytosis of Aβ by microglia (Fig. 5A). Importantly, generation of microglia conditioned media by preexposing microglial cells for up to 24 h to TO901317 did not enhance Aβ phagocytosis (data not shown).
Figure 5.
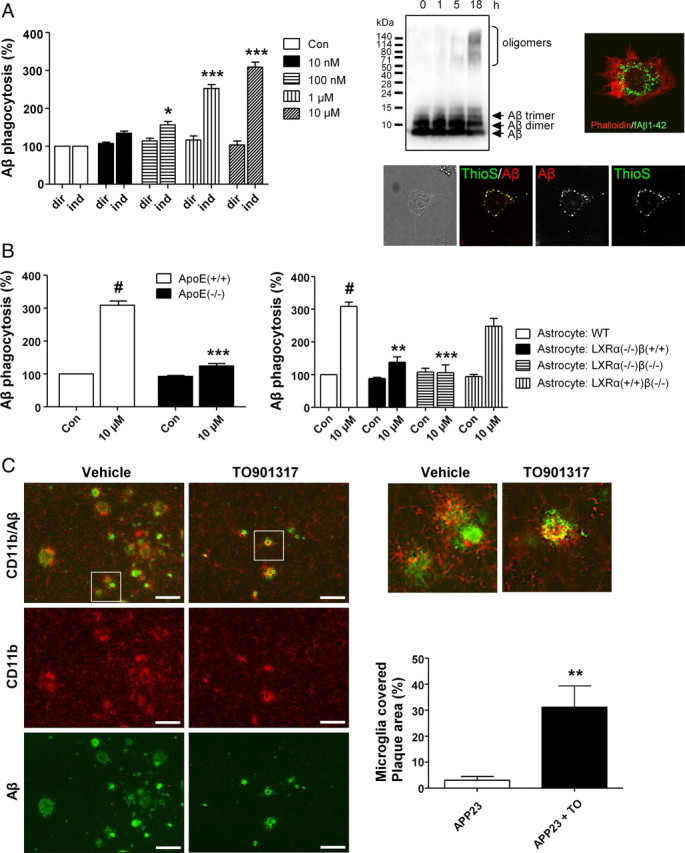
LXR agonist modulation of microglial Aβ1-42 phagocytosis requires the presence of LXRα and ApoE in astrocytes. A, Uptake of fibrillar Aβ by microglial cells exposed to medium derived from either microglia (dir) or astrocytes (ind) treated with increasing concentrations of the LXR agonist TO901317. Western blot analysis demonstrated the time-dependent (from 0 to 18 h) aggregation of Aβ used for the described experiments. Immunocytochemical experiments, using fluorescence detection of an anti-phalloidin antibody to visualize cell boundaries (red), together with FAM-Aβ, were used to verify the intracellular localization of Aβ within microglial cells. Thioflavin S staining detecting aggregated Aβ within microglia was performed. Data were analyzed by ANOVA followed by Tukey's post hoc test. Asterisks indicate significant differences from dir (n = 5, *p < 0.05, ***p < 0.001). B, Analysis of microglial phagocytosis after coincubation with media derived from either ApoE+/+ or ApoE−/− astrocytes exposed to TO901317 (10 μm) or vehicle (left), and analysis of microglial phagocytosis after coincubation with media derived from either wild-type, LXRα−/−/β+/+, or LXRα−/−/β−/− astrocytes exposed to TO901317 (10 μm) or vehicle (right). Data were analyzed by ANOVA followed by Tukey's post hoc test. Number sign (#) indicates difference from control (p < 0.001). Asterisks indicate differences from stimulated wild-type (**p < 0.01, ***p < 0.001). C, Microglia are more associated with plaques in APP23 mice after treatment with TO901317 (TO). Amyloid plaques were stained with rabbit polyclonal 2964 and microglia with monoclonal rat anti-CD11b. Data were analyzed by unpaired Student's t test (n = 7, **p < 0.01). Data represent means ± SEM. Scale bars, 100 μm.
Generation of primary astrocytic cultures from ApoE knock-out mice was performed to test whether ApoE is critical for the TO901317-mediated increase in phagocytosis of Aβ by microglia. Microglial cells that were exposed to conditioned media from TO901317-treated wild-type astrocytes showed normal Aβ phagocytosis. In strong contrast, Aβ phagocytosis was nearly absent when microglial cells were exposed to conditioned media from TO901317-treated ApoE knock-out astrocytes, suggesting that astrocyte-derived ApoE is indeed a critical factor (Fig. 5B). Control experiments exposing ApoE intact or knock-out microglia to TO901317 for an identical time period (24 h) showed that the presence or absence of ApoE in microglia did not exert any regulatory influence on subsequent Aβ phagocytosis (data not shown). A further set of experiments, using astrocyte cultures generated from LXRα knock-out, LXRβ knock-out, and LXRα/β knock-out mice, was performed to test whether the action of TO901317 depends on the presence of LXR. Aβ phagocytosis was analyzed after microglial cells were exposed to conditioned media obtained from TO901317-treated wild-type, LXRα knock-out, LXRβ knock-out, or LXRα/β knock-out astrocytes. Interestingly, Aβ phagocytosis was almost reduced to control levels when microglial cells were exposed to media from both LXRα knock-out and LXRα/β knock-out astrocytes, while media taken from LXRβ knock-out astrocytes did not affect the phagocytosis of Aβ, collectively suggesting that the presence of LXRα is a key requirement for astrocyte-mediated TO901317 action on microglial phagocytosis (Fig. 5B). Together, these data suggest that the stimulation of microglial Aβ phagocytosis by the LXR agonist TO901317 greatly depended on both astrocytic ApoE and LXRα.
To validate these in vitro findings, immunohistochemical costaining for microglia and Aβ was performed in sections from treated and untreated APP23 mice. Because of the reduced number of plaques, less activated microglia were found in TO901317-treated APP23 mice generally. However, remaining small plaques in TO901317-treated APP23 mice demonstrated a closer association with activated microglia compared with those in controls (Fig. 5C). The extent to which microglia were in close association with small plaques, as defined by yellow costaining of CD11b and Aβ, was quantified and turned out to be increased by tenfold (Fig. 5C). In addition we studied the effects of short-term treatment with TO901317 in 13-month-old APP/PS1/LXRα+/− and APP/PS1/LXRα−/− mice on association of microglia with Aβ in brain and lipidation of ApoE in CSF. Aβ was more covered by microglia in the presence of LXRα and TO901317, but this was not observed in the absence of LXRα (Fig. 6). Lipidation of ApoE was investigated using native gel electrophoresis. Migratory velocity of ApoE in the gel depends on lipidation status. ApoE from CSF of APP/PS1/LXRα−/− mice treated with TO901317 showed less lipidation than that of APP/PS1/LXRα+/− mice treated with TO901317. Note that the effect of TO901317 on ApoE concentration did not depend on LXRα.
Figure 6.
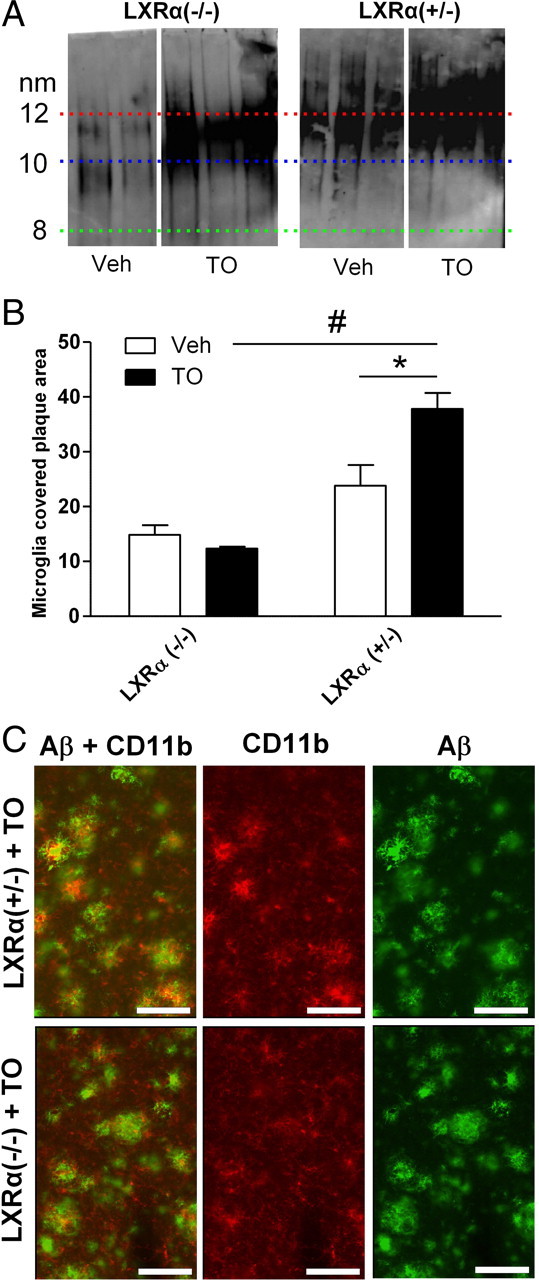
LXRα mediates ApoE lipidation and increased microglial Aβ plaque association. A, Native Western blot detection of ApoE in CSF derived from TO901317 (TO)- and vehicle (Veh)-treated APP/PS1/LXRα+/− and APP/PS1/LXRα−/− mice. B, Quantitative analysis of microglial plaque association from TO901317 and vehicle-treated APP/PS1/LXRα+/− and APP/PS1/LXRα−/− mice. C, Representative images showing CD11b-positive microglia associated with Aβ-positive plaques in neocortex of TO901317-treated APP/PS1/LXRα+/− and APP/PS1/LXRα−/− mice. Data represent means ± SEM. Data were analyzed by two-way ANOVA, followed by Student's t test (n = 4, *p < 0.05, #p < 0.01). Scale bars, 100 μm.
Discussion
In human AD brain as well as in APP transgenic mouse models, microglia respond to aggregated and misfolded Aβ most visible in the vicinity of Aβ plaques. Experimental evidence, however, found that they are inefficient in removing fibrillar Aβ (Bolmont et al., 2008), suggesting that either the deposition of Aβ itself or secondary metabolic or inflammatory events lead to a progressive impairment of microglial clearance function. Strategies to overcome this microglial paralysis are under intense investigation and include vaccination against full-length or truncated Aβ peptides or stimulation of Toll-like receptor signaling (Okura et al., 2008; Scholtzova et al., 2009). Here it is demonstrated that LXR activation is able to reset microglial function, in particular, the capacity for Aβ phagocytosis.
ApoE-dependent microglial phagocytosis of fibrillar Aβ contributes to LXR agonist-stimulated reduction in amyloid pathology
The central finding of the present study is the ApoE-dependent stimulation of microglial phagocytosis of fibrillar Aβ by the LXR agonist TO901317 in cell culture. Either the association of fibrillar Aβ with ApoE or an effect of ApoE on microglia may be responsible for the increased phagocytosis. Previously, it has been observed that the LXR agonist GW3965 stimulates degradation of soluble Aβ in astroglial and microglial cultures dependent on ApoE and its lipidation status (Jiang et al., 2008). These observations were proposed to explain the effect of long-term administration of GW3965 on plaque-load in Tg2576 mice. However, long-term treatment with TO901317 in 6-month-old APP23 mice did not have an effect on soluble Aβ (Lefterov et al., 2007), nor did it affect endogenous soluble Aβ in wild-type mice (this study). Moreover, the reduction of insoluble Aβ by 80% found in the present study seems to be too extensive to be caused by prevention of Aβ deposition alone, based on previously reported age-dependent changes of insoluble Aβ levels and disease progression in APP23 mice (Kuo et al., 2001). The hypothesis that ApoE positively regulates the phagocytosis of aggregated Aβ is further supported by findings from Giunta et al. (2008), who provided evidence that aged Aβ phagocytosis is increased by recombinant ApoE3. More generally, but in line with our hypothesis, Grainger et al. (2004) have shown that phagocytosis of apoptotic thymocytes was reduced in ApoE-deficient macrophages, and this effect was corrected by addition of ApoE. Together, these findings lend credit to a contributory role of microglial phagocytosis of fibrillar Aβ in vivo following LXR activation. This hypothesis is further strengthened by the observation that activated microglial cells were closely associated with remaining plaques in the TO901317-treated APP23 mice. Next to phagocytosis, however, other mechanisms, including release of degrading proteases and peripheral clearance, may also be involved in the reduction of Aβ (Jiang et al., 2008).
In addition to increased phagocytosis, an altered interaction between ApoE and Aβ, influenced by the ApoE lipidation status, might be partially responsible for the reduction of insoluble Aβ in TO901317-treated mice, similar to the proposed mechanism of plaque reduction in PDAPP mice overexpressing ABCA1 (Wahrle et al., 2008). In keeping with this theory, PDAPP mice deficient for ABCA1 reveal a plaque increase at advanced age (Koldamova et al., 2005a; Wahrle et al., 2005). Long-term treatment with TO901317 resulted in increased production of ApoE and ABCA1 in aged, plaque-bearing APP23 mice, and was likely to have led to increased amounts of lipidated ApoE. It is therefore tempting to hypothesize that increased association of lipidated ApoE with Aβ may have slowed down Aβ aggregation and contributed to the overall Aβ reduction. However, a paradoxical increase in soluble Aβ reported for young ABCA1-overexpressing PDAPP mice (Koldamova et al., 2005a; Wahrle et al., 2008) and indicative of an altered interaction between soluble ApoE and Aβ, was not observed either in young APP23 mice (Lefterov et al., 2007) or in control or aged APP23 mice (this study) after long-term TO901317 treatment, together making the alternative mechanism rather unlikely. However, a role for toxic non-lipidated or poorly lipidated ApoE as a catalyst of Aβ aggregation cannot be excluded entirely on the basis of the reported findings (Wahrle et al., 2005, 2008).
TO901317 resets microglia for phagocytosis of Aβ in an astrocyte- and LXRα-dependent fashion
Interestingly, microglial Aβ phagocytosis appeared to depend on LXRα and not LXRβ, since additional knock-out of LXRβ did not reduce the effect of TO901317 any further than LXRα knock-out only. This is in accordance with abstract data from Riddell et al. (2009) that show that only loss of LXRα blocked the ability of TO901317 to increase ApoE and reduce Aβ levels. Other examples can be found in the literature that LXRα and LXRβ are nonredundant and can mediate differential effects when both are present and stimulated by LXR agonists. For instance, it has been shown that the ability of TO901317 to induce expression of ABCA1 and ABCG1 is selectively impaired in LXRα−/− macrophages (Bischoff et al., 2010). Nonredundant roles of LXRα and LXRβ are also in line with different studies on LXRα and LXRβ knock-out mice that have revealed that LXRα has a more important role in cholesterol homeostasis, whereas LXRβ has functions in the immune system and neurons (Alberti et al., 2000, 2001; Wang et al., 2002; Steffensen et al., 2004; Andersson et al., 2005; Kim et al., 2008). Moreover, even though the affinity of TO901317 for LXRα and LXRβ may not be different, the biological effects of TO901317 often indicate a greater action through LXRα (Fiévet and Staels, 2009), whereas, at least in hepatocytes and astrocytes, the abundance of LXRβ is even greater than that of LXRα (Tobin et al., 2000; Abildayeva et al., 2006).
Colocalization of ApoE and ABCA1 supports central role of astrocytes in cholesterol metabolism and ApoE lipidation in the brain
ApoE is the primary cholesterol transporter in the brain and cholesterol is loaded onto ApoE by ABCA1. However, the reported distributions of ApoE and ABCA1 in the brain appear to mismatch. ApoE has been found in astrocytes, whereas ABCA1 has mainly been found in neurons in vivo (Koldamova et al., 2003). In the present study, it was observed that both ApoE and ABCA1 colocalized in astrocytes, matching their site of synthesis and suggesting that the levels of the respective proteins may be considerably higher in astrocytes compared with neurons and microglia. Therefore, the latter may contain some ABCA1 and ApoE, but are unlikely to provide the bulk of lipidated ApoE. Microglial ApoE synthesis has been observed in vivo under certain conditions, such as kainic acid treatment (Xu et al., 2006), but neither the presence of Aβ nor TO901317 treatment appears to stimulate ApoE production by microglia in vivo (this study).
Behavioral effects of TO901317 in AD models
The present study confirmed the spatial learning impairment generally observed in APP23 mice (Van Dam et al., 2003, 2008; Buxbaum et al., 2008). Although a major effect of the compound on plaque pathology was observed, the effect on behavior was minor. The treatment was started at a stage during which APP23 mice are known to already have a considerable plaque-load and show Aβ plaque-associated microglia. Possibly, the impairment in neuronal function caused by Aβ cannot be fully reversed at this stage. Aβ is toxic to synapses (Koffie et al., 2009), and synaptotoxicity may have caused synaptic dysfunction or even loss in aged APP23 mice that cannot be easily restored. Future therapeutic approaches should aim, therefore, at prevention or combine reduction of pathology with restoration of neuronal circuitry.
Different studies have been performed on the effect of long-term treatment with LXR agonists on amyloid-β and/or behavior in mice expressing mutant APP or APP and PS1. Jiang et al. (2008) found a reduction in plaque-load in 16-month-old Tg2576 mice, but tested behavior in 5-month-old Tg2576 mice. In contrast, Vanmierlo et al. (2009) did not find a reduction in plaque-load, but found a behavioral improvement in 23-month-old APPSLxPS1mut mice. In the present study, a reduction in plaque-load was observed in 13-month-old APP23 mice, but only a minor behavioral improvement. The differences between these studies are likely to be found in differences in animal models, ages, dosage of compound, type of compound, and routes and duration of administration used. For example, in the study by Lefterov et al. (2007) and in the present study, oral gavage of 50 mg/kg TO901317 per day was used, whereas Vanmierlo et al. (2009) administered the compound in the food at a lower dose (30 mg/kg per day). Vanmierlo et al. (2009) determined the optimal dose by short-term treatment regimens. However, Lefterov et al. (2007) observed that drug effects wear off with time of treatment. Positive effects were observed with relatively simple cognitive tests, such as fear conditioning in preplaque Tg2576 mice and object recognition in plaque-bearing APPSLxPS1mut mice (Riddell et al., 2007; Jiang et al., 2008; Vanmierlo et al., 2009). In the present study, a minor drug effect was observed in the Morris water maze task, probably because this task requires more complex cognitive modalities and therefore may be more demanding.
Conclusion
In conclusion, long-term TO901317 treatment in plaque-bearing APP23 mice is able to ameliorate amyloid pathology, but only mildly improves spatial memory performance. TO901317 may affect the clearance of insoluble Aβ, best explained by a stimulation of microglial phagocytosis of fibrillar Aβ. This effect appears to be selectively mediated through LXRα. To fully exploit the potential of LXR agonists in AD models, treatment probably should be initiated at the time Aβ starts to deposit and be sustained into old age.
Footnotes
The work was supported by grants from Deutsche Forschungsgemeinschaft and the Federal Ministry of Education and Research (BMBF) to M.T.H. (KFO 177, TP4), by National Institutes of Health Grant AG13956 (to D.M.H.), and by American Health Assistance Foundation Grant 2010613 (to P.B.V.).
References
- Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AH, Ramaekers FC, de Vente J, Groen AK, Wellington CL, Kuipers F, Mulder M. 24(S)-Hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem. 2006;281:12799–12808. doi: 10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- Alberti S, Steffensen KR, Gustafsson JA. Structural characterisation of the mouse nuclear oxysterol receptor genes LXRalpha and LXRbeta. Gene. 2000;243:93–103. doi: 10.1016/s0378-1119(99)00555-7. [DOI] [PubMed] [Google Scholar]
- Alberti S, Schuster G, Parini P, Feltkamp D, Diczfalusy U, Rudling M, Angelin B, Björkhem I, Pettersson S, Gustafsson JA. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J Clin Invest. 2001;107:565–573. doi: 10.1172/JCI9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson S, Gustafsson N, Warner M, Gustafsson JA. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc Natl Acad Sci U S A. 2005;102:3857–3862. doi: 10.1073/pnas.0500634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranowski M. Biological role of liver X receptors. J Physiol Pharmacol. 2008;59:31–55. [PubMed] [Google Scholar]
- Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu Rev Med. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, Li AC, Schulman IG. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res. 2010;51:900–906. doi: 10.1194/jlr.M900096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of A beta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (A beta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229–236. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- Fiévet C, Staels B. Liver X receptor modulators: effects on lipid metabolism and potential use in the treatment of atherosclerosis. Biochem Pharmacol. 2009;77:1316–1327. doi: 10.1016/j.bcp.2008.11.026. [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK. beta-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci. 2006;26:4644–4648. doi: 10.1523/JNEUROSCI.4822-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita SC, Sakura K, Tsuchiya R, Hamanaka H. Apolipoprotein E is found in astrocytes but not in microglia in the normal mouse brain. Neurosci Res. 1999;35:123–133. doi: 10.1016/s0168-0102(99)00075-9. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Zhou Y, Hou H, Rrapo E, Fernandez F, Tan J. HIV-1 TAT inhibits microglial phagocytosis of Abeta peptide. Int J Clin Exp Pathol. 2008;1:260–275. [PMC free article] [PubMed] [Google Scholar]
- Grainger DJ, Reckless J, McKilligin E. Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J Immunol. 2004;173:6366–6375. doi: 10.4049/jimmunol.173.10.6366. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Jenkins NA, Copeland NG, Borchelt DR. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol Aging. 2004;25:885–892. doi: 10.1016/j.neurobiolaging.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of A beta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Fan X, Gabbi C, Yakimchuk K, Parini P, Warner M, Gustafsson JA. Liver X receptor beta (LXRbeta): a link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson's dementia. Proc Natl Acad Sci U S A. 2008;105:2094–2099. doi: 10.1073/pnas.0711599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, Rahmanto AS, Kamili A, Rye KA, Guillemin GJ, Gelissen IC, Jessup W, Hill AF, Garner B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–2861. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, Isanski BA, DeKosky ST, Lazo JS. 22R-Hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem. 2003;278:13244–13256. doi: 10.1074/jbc.M300044200. [DOI] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005a;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005b;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, Kalback WM, Luehrs DC, Vishnivetskaya TA, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Weller RO, Roher AE. The evolution of A beta peptide burden in the APP23 transgenic mice: implications for A beta deposition in Alzheimer disease. Mol Med. 2001;7:609–618. [PMC free article] [PubMed] [Google Scholar]
- Lefterov I, Bookout A, Wang Z, Staufenbiel M, Mangelsdorf D, Koldamova R. Expression profiling in APP23 mouse brain: inhibition of A beta amyloidosis and inflammation in response to LXR agonist treatment. Mol Neurodegener. 2007;2:20. doi: 10.1186/1750-1326-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder M, Terwel D. Possible link between lipid metabolism and cerebral amyloid angiopathy in Alzheimer's disease: a role for high-density lipoproteins? Haemostasis. 1998;28:174–194. doi: 10.1159/000022429. [DOI] [PubMed] [Google Scholar]
- Okura Y, Kohyama K, Park IK, Matsumoto Y. Nonviral DNA vaccination augments microglial phagocytosis of beta-amyloid deposits as a major clearance pathway in an Alzheimer disease mouse model. J Neuropathol Exp Neurol. 2008;67:1063–1071. doi: 10.1097/NEN.0b013e31818b48db. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Roses AD. On the discovery of the genetic association of apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–366. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T. Induction of Toll-like receptor 9 signaling as a method for ameliorating Alzheimer's disease-related pathology. J Neurosci. 2009;29:1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader-Fischer G, Paganetti PA. Effect of alkalizing agents on the processing of the beta-amyloid precursor protein. Brain Res. 1996;716:91–100. doi: 10.1016/0006-8993(96)00002-9. [DOI] [PubMed] [Google Scholar]
- Steffensen KR, Neo SY, Stulnig TM, Vega VB, Rahman SS, Schuster GU, Gustafsson JA, Liu ET. Genome-wide expression profiling; a panel of mouse tissues discloses novel biological functions of liver X receptors in adrenals. J Mol Endocrinol. 2004;33:609–622. doi: 10.1677/jme.1.01508. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow DB. Preparation of amyloid beta-protein for structural and functional studies. Methods Enzymol. 2006;413:20–33. doi: 10.1016/S0076-6879(06)13002-5. [DOI] [PubMed] [Google Scholar]
- Tobin KA, Steineger HH, Alberti S, Spydevold O, Auwerx J, Gustafsson JA, Nebb HI. Cross-talk between fatty acid and cholesterol metabolism mediated by liver X receptor-alpha. Mol Endocrinol. 2000;14:741–752. doi: 10.1210/mend.14.5.0459. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- Van Dam D, Coen K, De Deyn PP. Cognitive evaluation of disease-modifying efficacy of donepezil in the APP23 mouse model for Alzheimer's disease. Psychopharmacology. 2008;197:37–43. doi: 10.1007/s00213-007-1010-x. [DOI] [PubMed] [Google Scholar]
- Vanmierlo T, Rutten K, Dederen J, Bloks VW, van Vark-van der Zee LC, Kuipers F, Kiliaan A, Blokland A, Sijbrands EJ, Steinbusch H, Prickaerts J, Lutjohann D, Mulder M. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Vloeberghs E, Van Dam D, Franck F, Serroyen J, Geert M, Staufenbiel M, De Deyn PP. Altered ingestive behavior, weight changes, and intact olfactory sense in an APP overexpression model. Behav Neurosci. 2008;122:491–497. doi: 10.1037/0735-7044.122.3.491. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci U S A. 2002;99:13878–13883. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Goodwin B, Galardi CM, Maglich JM, Wilson JG, Willson TM, Collins JL, Kliewer SA. Liver X receptor (LXR) regulation of the LMR alpha gene in human macrophages. J Biol Chem. 2001;276:43509–43515. doi: 10.1074/jbc.M106155200. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK, Wilson JG, Winegar DA, Kliewer SA. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol Endocrinol. 2002;16:1378–1385. doi: 10.1210/mend.16.6.0835. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]