

Abstract
Presynaptic inhibition via G-protein-coupled receptors (GPCRs) and voltage-gated Ca2+ channels constitutes a widespread regulatory mechanism of synaptic strength. Yet, the mechanism of intermolecular coupling underlying GPCR-mediated signaling at central synapses remains unresolved. Using FRET spectroscopy, we provide evidence for formation of spatially restricted (<100 Å) complexes between GABAB receptors composed of GB1a/GB2 subunits, Gαoβ1γ2 G-protein heterotrimer, and CaV2.2 channels in hippocampal boutons. GABA release was not required for the assembly but for structural reorganization of the precoupled complex. Unexpectedly, GB1a deletion disrupted intermolecular associations within the complex. The GB1a proximal C-terminal domain was essential for association of the receptor, CaV2.2 and Gβγ, but was dispensable for agonist-induced receptor activation and cAMP inhibition. Functionally, boutons lacking this complex-formation domain displayed impaired presynaptic inhibition of Ca2+ transients and synaptic vesicle release. Thus, compartmentalization of the GABAB1a receptor, Gβγ, and CaV2.2 channel in a signaling complex is required for presynaptic inhibition at hippocampal synapses.
Introduction
G-protein-coupled receptors (GPCRs), G-proteins, and N- and P/Q-type high-voltage-gated Ca2+ (CaV) channels (Nowycky et al., 1985) represent key signaling elements controlling Ca2+ flux at nerve terminals (Dunlap et al., 1995). Since the first work of Dunlap and Fischbach (1978), numerous studies suggest that GPCR activation induces inhibition of CaV channels via direct interaction with Gβγ subunits of G-proteins (for review, see Hille, 1994; Zamponi and Snutch, 1998; Catterall, 2000; Dolphin, 2003; De Waard et al., 2005). This membrane-delimited mechanism of CaV channel inhibition has been proposed to enable precise inhibition of neurotransmitter release in space and time.
Despite several decades of intensive research, the intermolecular coupling mechanism underlying GPCR-mediated presynaptic inhibition at central synapses remains unresolved. According to collision coupling theory assuming free lateral diffusion of receptors, G-proteins, and effectors within the cell membrane, only receptors activated by agonist are capable of interacting with G-proteins (Orly and Schramm, 1976; Tolkovsky and Levitzki, 1978). As this model encounters difficulty explaining signaling specificity at the single-cell level, alternative models were proposed that assume coupling between the signaling units without agonist (precoupling) and formation of predetermined signaling microdomains (Neubig et al., 1988; Neubig, 1994). However, currently there is no experimental evidence supporting any of these models in central synapses.
Recent advances in molecular biology and optical imaging enabled real-time monitoring of intermolecular dynamics in living cells by resonance energy transfer technology (Lohse et al., 2008). These studies in heterologous expression systems, although boosting our understanding of receptor/G-protein/effector coupling mechanisms, yielded conflicting results. Some studies have suggested existence of precoupled complexes between GPCRs, G-proteins, and effectors such as G-protein-activated inwardly rectifying K+ (GIRK) channels (Nobles et al., 2005; Galés et al., 2006; Riven et al., 2006; Fowler et al., 2007), and diffusion-determined collision coupling has been proposed by others (Hein et al., 2005). The disparity between these results emphasizes the need to study intermolecular dynamics in native environments and at specialized subcellular compartments.
Among various presynaptic GPCRs, GABAB receptors are widely expressed in the brain as autoreceptors and heteroreceptors (Wu and Saggau, 1997; Bettler et al., 2004). GABAB receptors are obligatory heterodimers, requiring two homologous subunits, GB1 and GB2, to function (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Kuner et al., 1999). Molecular diversity of GABABRs arises from two pharmacologically indistinguishable GB1 isoforms, 1a and 1b (Bettler et al., 2004), and from auxiliary KCTD subunits (Schwenk et al., 2010). Transgenic mice with targeted isoform-specific genetic deletions suggest that GB1a-containing receptors (GB1aRs) are predominantly localized at glutamatergic boutons, mediating presynaptic inhibition of glutamate release (Vigot et al., 2006; Guetg et al., 2009). Activation of presynaptic GABAB receptors induces inhibition of synaptic vesicle release through suppression of Ca2+ flux (Wu and Saggau, 1995; Takahashi et al., 1998; Laviv et al., 2010), although several Ca2+-independent mechanisms have been proposed as well (Scanziani et al., 1992; Parnas et al., 2000; Sakaba and Neher, 2003). Recent quantitative proteomic study on molecular composition of CaV2 channel nano-environment suggests that GABAB receptors strongly interact with CaV2.2 channels in the brain (Müller et al., 2010).
Although much information has been gained on the structure and function of GABAB receptors, several key questions remain unresolved. First, does assembly of the presynaptic GB1aR–Gβγ–CaV2.2 complex require synaptic activity or, alternatively, are the signaling units precoupled in a macromolecular complex regardless of GABA-induced activation? Second, how does GABA affect the microarchitecture of signaling complexes at individual synaptic boutons? Third, are GB1aRs required for the complex formation? And finally, if GB1aRs are indeed compartmentalized with Gβγ subunits and CaV2.2 channels, how does it impact the function of central synapses? To address these questions, we integrated fluorescence resonance energy transfer (FRET) spectroscopy, optical imaging of vesicle exocytosis, and presynaptic calcium transients at individual presynaptic boutons in cultured hippocampal neurons (Laviv et al., 2010). Our findings suggest that GB1aRs, G-protein heterotrimers and CaV2.2 channels are precoupled in presynaptic hippocampal boutons. We identified the proximal C-terminal domain of the GB1a protein as an essential molecular domain mediating the GB1aR–Gβγ–CaV2.2 signaling complex assembly but as dispensable for the receptor and Gαi/o activation. The structural segregation of ligand-binding and complex-formation domains allowed us to isolate the impact of signaling compartmentalization on presynaptic function. Our findings suggest that ligand-induced receptor activation is necessary but insufficient for presynaptic inhibition and propose that precoupling of GB1aRs, Gβγ subunits, and CaV2.2 channels in a signaling nano-domain is required for a proper negative regulation of basal synaptic vesicle release at hippocampal synapses.
Materials and Methods
Hippocampal cell culture.
Primary cultures of CA3-CA1 hippocampal neurons were prepared from newborn Wistar rats and 1a−/−, 1b−/−, WT (BALB/c background) mice on postnatal days 0–2, as described previously (Slutsky et al., 2004). The generation of the 1a−/− and 1b−/− mice has been described previously (Vigot et al., 2006). All animal experiments were approved by the Tel Aviv University Committee on Animal Care.
Molecular biology.
Construction of fusion proteins has been described previously: GB1aCFP, GB1aYFP, and GαoCFP (Fowler et al., 2007); pHluorin-GB1a (Guetg et al., 2009); Gβ1YFP, Gγ2CFP (Riven et al., 2006); CFP-Epak-YFP (van der Krogt et al., 2008); and CaV2.2CFP (Altier et al., 2006). Wild type and mutants of GB1a proteins used throughout the study were constructed and expressed in peYFP-N1 under control of CMV promoter. Nontagged GB1a-WT protein was engineered by digestion of peYFP-N1-GB1a-WT with AgeI/NotI, and then blunting and religation. GB1a-Δ21-YFP was created by overlap-extension PCR and subcloning into peYFP-N1-GB1a-WT using BamHI/AgeI. Nontagged GB1a-Δ21 was created by digestion peYFP-N1-GB1a-Δ21 with AgeI/NotI, and then blunting and religation. Nontagged GB1a-ΔSD receptor was engineered by overlap-extension PCR and subcloning into EcoRI/BamHI sites of peYFP-N1 plasmid bearing the nontagged GB1a-WT receptor gene. GB1a-S269A-YFP was created by overlap-extension PCR and subcloning into ApaI/AgeI sites of peYFP-N1-GB1a-WT. Nontagged GB1a-S269A was constructed by digestion of peYFP-N1-GB1a-S269A with AgeI/NotI, and then blunting and religation. Nontagged GB1a-Δ103 was created by PCR and then subcloned into BamHI/NotI sites of peYFP-N1-GB1a-WT. Nontagged GB1a-Δ74 and GB1a-Δ39 were described previously (Boyer et al., 2009).
Transient cDNA transfections have been performed using Lipofectamine-2000 reagents and neurons were typically imaged 18–24 h after transfection.
Confocal imaging.
Hippocampal neurons were imaged using a Zeiss LSM510 META confocal microscope (Carl Zeiss) using a 40 × 1.2 NA water-immersion objective and FV1000 spectral Olympus confocal microscope using a 60 × 1.2 NA water-immersion objective. The experiments were conducted at room temperature in extracellular Tyrode solution containing the following (in mm): NaCl, 145; KCl, 3; glucose, 15; HEPES, 10; MgCl2, 1.2; CaCl2, 1.2; pH adjusted to 7.4 with NaOH. To isolate miniature synaptic activity, TTX (1 μm) was added to extracellular solution.
FRET imaging.
FRET imaging was carried as described previously (Laviv et al., 2010). For spectral analysis, CFP was excited at 405 nm (Zeiss) or at 442 nm (Olympus) and fluorescence emission was measured between 400 and 700 nm, with a 10 nm λ step size. To reduce phototoxicity and photobleaching, most of the FRET experiments were performed using a narrowed emission spectrum (460–560 nm) composed of CFP peak (486 ± 10 nm) and a YFP peak (534 ± 10 nm) containing YFP emission due to FRET, direct YFP excitation at 405 nm, and CFP emission tail. YFP was imaged at 514 nm (excitation) and 525–560 nm (emission). Photobleaching of YFP was performed with 514 nm laser line, at 2.3 mW of laser output. We used a single point activation module for rapid and efficient multiregion bleaching. We typically photobleached two to six boutons per imaged axon: bleach duration was 35 ms per bouton with a 5 ms interval between boutons.
Image acquisition parameters were optimized for maximal signal-to-noise ratio and minimal phototoxicity: 700 V photomultiplier voltage; 4 μs/pixel scan speed, 0.05–0.18 mW (514 nm) or 0.15–0.2 (440 nm) laser output; 90–130 μm pinhole; 512 × 512 pixels image size. Z-stacks were collected from 3–4 μm optical slice at 0.6–0.8 μm steps; images were then stacked using maximal intensity projection per pixel algorithm and converted to a single 2D image for analysis. Images were acquired without averaging.
Calculation of FRET efficiency.
Donor dequenching due to the desensitized acceptor was measured from CFP emission (460–500 nm) before and after the acceptor photobleaching. FRET efficiency, E, was then calculated using the equation E = 1 − IDA/ID, where IDA is the peak of donor emission in the presence of the acceptor and ID is the peak after acceptor photobleaching. To exclude potential contribution of donor/acceptor ratio to FRET efficiency measurements, all FRET experiments were performed under saturation conditions of acceptor over donor. Detection of CFP/YFP signals was done using custom-written scripts in MATLAB (MathWorks) as described previously (Laviv et al., 2010). Briefly, regions of interest (ROIs) were marked at boutons that underwent YFP photobleaching. Average intensity of ROIs was subtracted from background ROI intensity in close proximity to the bouton. All the boutons that exhibited YFP photobleaching by >90% of initial fluorescence intensity were included in the analysis. Nonbleached boutons at the same image area were analyzed to ensure lack of nonspecific photobleaching due to image acquisition.
Detecting presynaptic calcium transients.
Fluorescent calcium indicator Calcium Green 488 BAPTA-1 AM was dissolved in DMSO to yield a concentration of 1 mm. For cell loading, cultures were incubated at 37°C for 30 min with 3 μm of this solution diluted in standard extracellular solution. Extracellular solution contained 20 μm DNQX to block recurrent activity and 50 μm APV to block calcium flux through NMDA receptors. Imaging was performed using FV1000 Olympus confocal microscope, under 488 nm (excitation) and 510–570 nm (emission), using 500 Hz line scanning. Ca2+ transients were quantified following averaging of 10 traces. Integral was calculated for ΔF/F per bouton before and after baclofen application. Integration time window was 300 ms, starting from the end of the stimulus.
FM-based imaging and analysis.
Activity-dependent FM1-43 and FM4-64 styryl dyes have been used to estimate basal synaptic vesicle exocytosis. Action potentials have been elicited by passing 50 mA constant current for 1 ms (∼50% above the threshold for eliciting action potential) through two platinum wires, separated by ∼7 mm and close to the surface of the coverslip. The extracellular medium contained nonselective antagonist of ionotropic glutamate receptors (kynurenic acid, 0.5 mm) to block recurrent neuronal activity. Synaptic vesicles were loaded with 15 μm FM4-64 in all the experiments with GFP/CFP/YFP transfection, and 10 μm FM1-43 was used in all the nontransfected neurons. FM was loaded by bathing the cultures in a medium containing dye. FM was present 5 s before and 30 s after the electrical stimulation (600 stimuli at 20 Hz). After dye loading, external dye was washed away in Ca2+-free solution containing ADVASEP-7 (0.1 mm) to scavenge membrane-bound FM. The fluorescence of individual synapses was determined from the difference between images obtained after staining and after destining (ΔF). For detection of FM+ puncta, ΔF images have been analyzed (only the puncta exhibiting ≥90% destaining were subjected to analysis). Detection of signals has been done using custom-written scripts in MATLAB as described previously (Abramov et al., 2009). Briefly, the following criteria were used for signal detection: the fluorescence intensity was 2 SDs above the mean background and the area of puncta was between 0.1 and 2 μm2.
Chemical reagents.
FM4-64 (SynaptoRed C2), FM1-43 (SynaptoGreen C4), and Advasep-7 were purchased from Biotium; baclofen, DNQX, CGP35348, and CGP54626 from Tocris Bioscience; TTX from Alomon Labs; TeTx, APV, and kynurenic acid from Sigma-Aldrich; and PTX from Calbiochem.
Statistical analysis.
Error bars shown in the figures represent SEM. The number of boutons is defined by n and the number of experiments (cultures) by N. All the experiments were repeated at least in three different batches of cultures. One-way ANOVA with post hoc Dunnett's or Bonferroni's tests was used to compare several conditions. Student's unpaired t tests were used in the experiments where two populations of synapses were compared. Student's paired t tests were used when the same population of synapses was tested before and after treatment. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., nonsignificant.
Results
GABABRs, G-proteins, and CaV2.2 channels are precoupled at hippocampal boutons
In presynaptic terminals, the GABABR is a heterodimer of GB1a/GB2 subunits that mediates GABA-dependent inhibition of voltage-gated N-type Ca channels (Wu and Saggau, 1995; Dittman and Regehr, 1996). Inhibition of CaV2.2 channels occurs through a G-protein-dependent mechanism (Takahashi et al., 1998). Using FRET to detect intermolecular associations between CFP/YFP-tagged proteins, we examined whether presynaptic GABAB receptors interact with CaV2.2 channels and G-proteins at individual hippocampal boutons. We investigated possible associations between GB1aRs, Gαoβ1γ2 G-protein subunits, and CaV2.2 channels at boutons of pyramidal hippocampal neurons using the following tagged proteins (Fig. 1A): (1) YFP-tagged GB1a receptor subunit (GB1aYFP) (Fowler et al., 2007); (2) CFP-tagged α1 subunit of CaV2.2 channel (CaV2.2CFP) (Altier et al., 2006); (3) CFP-tagged Gαo subunit, where CFP is internally inserted after E94 (GαoCFP) (Fowler et al., 2007); (4) Gβ1 G-protein subunit N-terminally tagged to YFP (Gβ1YFP) (Riven et al., 2006); and (5) Gγ2 G-protein subunit N-terminally tagged to YFP (Gγ2YFP) (Riven et al., 2006). All of the tagged proteins were functionally characterized previously (Altier et al., 2006; Riven et al., 2006; Fowler et al., 2007; Laviv et al., 2010). Presynaptic localization of tagged GABAB receptors in boutons was confirmed previously by colocalization of GB1aYFP with CFP-tagged synapsin Ia protein and with FM4-64 dye (Laviv et al., 2010). We first examined the basal FRET efficiency (E), determined by donor (CFP) dequenching following acceptor (YFP) photobleaching (Laviv et al., 2010), produced by spontaneous miniature synaptic activity in the presence of tetrodotoxin (+TTX). We detected significant FRET efficiencies between GB1aYFP/CaV2.2CFP (0.11 ± 0.006, n = 33), GB1a/Gαo94CFP (0.14 ± 0.007, n = 53), GB1aCFP/Gβ1YFP (0.22 ± 0.005, n = 45), GB1aCFP/Gγ2YFP (0.22 ± 0.007, n = 21), and CaV2.2CFP/Gβ1YFP (0.22 ± 0.006, n = 31). On average, FRET efficiencies for all the tested protein pairs were significantly higher (p < 0.0001, mean E varied from 0.12 to 0.23 between the pairs) than nonspecific FRET between CFP-tagged proteins of interest and a nonrelated tumor necrosis factor receptor 2, C-terminally tagged with YFP (TNFR2YFP) (<2%) (Fig. 1B). Furthermore, background enhancement of CFP emission, assessed by photobleaching at 514 nm in neurons expressing only GB1aCFP, was negligible (0.016 ± 0.002, n = 26) (Fig. 1B). FRET efficiency did not depend on the donor-to-acceptor ratio (Fig. 1C). Therefore, FRET measurements suggest close association of the receptor and G-proteins (GB1a/Gαo94CFP; GB1aCFP/Gβ1YFP; GB1aCFP/Gγ2YFP), the channel and G-proteins (CaV2.2CFP/Gβ1YFP) and, interestingly, between the receptor and channel (GB1aYFP/CaV2.2CFP), in hippocampal boutons under miniature synaptic activity.
Figure 1.

GB1aRs, Gαoβ1γ2 G-protein subunits, and CaV2.2 channels are precoupled at single hippocampal boutons. A, Representative confocal images of pyramidal neuron axons in hippocampal cultures that were cotransfected with GB1aYFP/CaV2.2CFP, GB1aYFP/GαoCFP, GB1aCFP/Gβ1YFP and CaV2.2CFP/Gβ1YFP. Scale bars, 2 μm. B, FRET was detected between GB1aYFP/CaV2.2CFP (n = 33, N = 7), GB1aYFP/Gαo*94CFP (n = 53), GB1aCFP/Gβ1YFP (n = 45, N = 10), GB1aCFP/Gγ2YFP (n = 21, N = 4), CaV2.2CFP/Gβ1YFP (n = 31, N = 6) proteins under miniature synaptic activity at single hippocampal boutons. To verify FRET specificity, E was measured between the CFP-tagged proteins of interest and nonrelated TNFR2YFP. Error bars indicate SEM. ***p < 0.001. C, FRET efficiency is plotted for individual presynaptic boutons as function of CFP/YFP intensity ratio (FCFP/FYFP). No correlation was found: Spearman r is 0.14, 0.11, −0.08, and 0.05 for GB1aYFP/CaV2.2CFP, GB1aYFP/Gαo*94CFP, GB1aCFP/Gβ1YFP, and CaV2.2CFP/Gβ1YFP, respectively (p > 0.5). D, FRET was detected between GB1aYFP/CaV2.2CFP (n = 8, N = 3), GB1aYFP/GαoCFP (n = 9, N = 3), GB1aCFP/Gβ1YFP (n = 8, N = 3), and CaV2.2CFP/Gβ1YFP (n = 12, N = 4) proteins in nonreleasing TeTx-pretreated hippocampal boutons. E, FRET was detected between GB1aYFP/CaV2.2CFP (n = 9, N = 3), GB1aYFP/GαoCFP (n = 9, N = 3), GB1aCFP/Gβ1YFP (n = 10, N = 3), and CaV2.2CFP/Gβ1YFP (n = 9, N = 3) proteins in nonreleasing immature (4–5 DIV) hippocampal neurons.
To assess whether miniature synaptic activity is required for induction of GB1aR–G-protein–CaV2.2 channel associations, we examined FRET efficiencies within the tagged proteins of interest under resting conditions in boutons that are incapable of vesicle recycling and, therefore, lack vesicular GABA release. To accomplish this, we measured FRET in tetanus toxin (TeTx) treated neurons, in which SNARE-mediated vesicle release is inhibited (Fig. 1D) and in immature boutons of young (4–5 DIV) neurons (Fig. 1E). Notably, basal FRET increased by ∼50% between GB1aYFP/GαoCFP (p < 0.01) and GB1aYFP/CaV2.2CFP (p < 0.05), and it decreased by ∼45% between GB1aCFP/Gβ1YFP (p < 0.0001) and by ∼35% between CaV2.2CFP/Gβ1YFP (p < 0.001) proteins in both young and TeTx-treated neurons. Together, these data suggest that (1) GB1a, Gαoβ1g2 G-protein, and CaV2.2 channel are preassembled in the absence of GABA release; and (2) miniature GABA release triggers rearrangement in the signaling complex, promoting FRET between precoupled GB1a/Gβ1g2 and CaV2.2/Gβ1 proteins, while reducing FRET between the GB1a/CaV2.2 channel and GB1a/Gαo proteins.
Agonist-induced rearrangements within the GB1aR signaling complex
To further assess activity-dependent intermolecular conformational changes within the GB1aR signaling complex, we explored effects of GABABR agonist and antagonist on FRET between the tested protein pairs. For the receptor and channel, FRET between GB1aYFP and CaV2.2CFP proteins decreased by ∼60% following application of 10 μm baclofen (n = 13–20, p < 0.0001) (Fig. 2A, left). Conversely, the GABAB receptor antagonist CGP54626 (1 μm) triggered a ∼40% increase in GB1aYFP/CaV2.2CFP FRET (n = 14, p < 0.01) (Fig. 2A, right). CGP54626 did not affect E in the absence of neurotransmitter release (n = 8, p > 0.6). We next examined the effect of pertussis toxin (PTX), which uncouples the Gαi/o subunit from the receptor. Baclofen did not alter FRET between GB1aYFP/CaV2.2CFP proteins in PTX-treated neurons (0.12 ± 0.012 vs 0.115 ± 0.011 for control and PTX-treated neurons, respectively, n = 7–8, p > 0.8), indicating functional coupling with the receptor was required for baclofen effect. For the receptor and G-protein, baclofen significantly decreased GB1aYFP/GαoCFP FRET (0.05 ± 0.002, n = 12, p < 0.0001) (Fig. 2B, left), similar to a previous study (Frank et al., 2005). Pretreatment with PTX abolished baclofen-induced FRET changes (0.13 ± 0.013 vs 0.14 ± 0.02 for control and PTX-treated neurons, respectively, n = 13, p > 0.2). CGP35348 (1 μm) induced a ∼70% increase in FRET (0.23 ± 0.009, n = 13, p < 0.0001) (Fig. 2B, right) but had no effect in the absence of neurotransmitter release (n = 8–16, p > 0.4). Together, these data suggest that quantal GABA release weakens association between precoupled CaV2.2 channel/GB1a receptor subunit and GB1a/Gαo proteins.
Figure 2.

Agonist-induced structural rearrangements in the GB1aR/G-protein/CaV2.2 channel complex. A, E between GB1aYFP/CaV2.2CFP was reduced by baclofen (10 μm, left, n = 13–29, N = 4–6, ***p < 0.0001), but was increased by CGP54626 (1 μm, right, n = 14, N = 4, **p < 0.01). B, E between GB1aYFP/GαoCFP was reduced by baclofen (10 μm, left, n = 12–17, N = 4, ***p < 0.0001), but was increased by GABABR antagonist CGP35348 (1 μm, right, n = 13–18, N = 4, ***p < 0.0001). C, E between GB1aCFP/Gβ1YFP was increased by baclofen (10 μm, left, n = 12–15, N = 4, ***p < 0.0001), but was decreased by GABABR antagonist CGP54626 (1 μm, right, n = 15, N = 4, ***p < 0.0001). D, E between Gβ1YFP/CaV2.2CFP was increased by baclofen (10 μm, left, n = 14–27, N = 4–6, ***p < 0.0001), but was decreased by CGP54626 (1 μm, right, n = 14–17, N = 4–5, ***p < 0.0001). Error bars indicate SEM. E, Fluorescence intensity of pHluorin tagged to GB1a does not change under miniature activity, by application of 10 μm baclofen and as function of stimulation frequency (10 and 100 Hz). Slope of linear fit is 1.04, 0.98, 1.01, and 0.99 for miniature activity, baclofen application, 10 and 100 Hz, respectively.
In contrast to GB1a/CaV2.2 and GB1a/Gαo interactions, agonist promoted FRET between GB1a/Gβ1 and CaV2.2/Gβ1 proteins. Baclofen produced a ∼30% increase in GB1aCFP/Gβ1YFP FRET (n = 14–27, p < 0.0001) (Fig. 2C, left), and CGP54626 reduced FRET by ∼48% (n = 14–17, p < 0.0001) (Fig. 2C, right). FRET efficiency between CaV2.2CFP and Gβ1YFP proteins was increased by baclofen by ∼32% (n = 14–15, p < 0.0001) (Fig. 2D, left), whereas it was decreased by CGP54626 antagonist by ∼47% (n = 16–22, p < 0.0001) (Fig. 2D, right). These data indicate that basal GABA promotes GB1a/Gβ1 and CaV2.2/Gβ1 associations in hippocampal boutons.
To test whether a reduction in FRET between GB1a/CaV2.2 or between GB1a/Gαo by GABA might be explained by receptor internalization, we used pHluorin, a pH-sensitive GFP, tagged to the N terminus of GB1a protein to monitor surface expression of the GB1a under physiological conditions. Fluorescence of pHluorin-GB1a was unchanged by stimulation frequencies of 10 Hz or even 100 Hz spikes, or by maximal receptor activation (10 μm baclofen; Laviv et al., 2010) (Fig. 2E). These results suggest that reduction in the number of membrane receptors is unlikely to occur under our experimental conditions.
GB1a is required for Gβγ/CaV2.2 association
Having observed that binding of GABA to the GB1a subunit is not essential for the precoupling of the GB1aR signaling complex, suggesting the formation of a receptor–G-protein–channel complex, we investigated whether the GB1a protein is essential for Gβγ/CaV2.2 interactions. Therefore, we measured possible FRET between Gβ1YFP and CaV2.2CFP in boutons of hippocampal neurons prepared from GB1a knock-out (Vigot et al., 2006) (1a−/−) mice (Fig. 3A). Surprisingly, boutons lacking GB1a protein revealed no specific FRET between CaV2.2CFP and Gβ1YFP proteins under miniature synaptic activity (0.03 ± 0.004, n = 31) (Fig. 3B). Furthermore, neither block of synaptic activity nor agonist application induced specific FRET signals in 1a−/− boutons (n = 10–18) (Fig. 3B). We also observed no specific FRET between CaV2.2CFP and either N-terminally tagged Gγ2N′-YFP (0.02 ± 0.01, n = 10) or C-terminally tagged Gγ2C′-YFP (0.03 ± 0.006, n = 9) G-proteins (Fig. 3C). By contrast, significant FRET (p < 0.0001) was observed between Gγ2C′-YFP/CaV2.2CFP (0.12 ± 0.006, n = 10) and between Gγ2N′-YFP/CaV2.2CFP (0.23 ± 0.005, n = 15) proteins in boutons of WT neurons, suggesting that there were no constraints on fluorophore mobility. Notably, CaV2.2CFP/Gβ1N′-YFP FRET was not altered in GB1b knock-out (1b−/−) boutons (0.18 ± 0.01, n = 7), consistent with the idea that GB1a and not GB1b is targeted to excitatory presynaptic boutons (Vigot et al., 2006). To confirm that FRET disruption was specific to the GB1a deletion, we examined CaV2.2CFP/Gβ1YFP interactions following ectopic expression of GB1a protein in 1a−/− neurons. Indeed, expression of GB1a protein rescued the CaV2.2CFP/Gβ1YFP FRET in 1a−/− boutons (0.15 ± 0.01, n = 17) (Fig. 3B). Moreover, FRET between GB2YFP receptor subunit and CaV2.2CFP channel was abolished in 1a−/− compared with WT neurons (n = 8, p < 0.0001) (Fig. 3D). Thus, deletion of the GB1a protein disrupts association of key signaling molecules, CaV2.2 channel and Gβ1γ2 (Fig. 3E).
Figure 3.

Disruption of FRET between the receptor, CaV2.2 channel, and Gβγ at hippocampal boutons of 1a−/− neurons. A, Confocal images of axonal part of 1a−/− hippocampal pyramidal neuron that was cotransfected with CaV2.2CFP and Gβ1YFP. Scale bar, 2 μm. B, Lack of specific FRET between CaV2.2CFP and Gβ1N′-YFP in 1a−/− boutons: under miniature synaptic activity (Cnt, n = 31, N = 6), in the presence of 10 μm baclofen (n = 16, N = 4), and in TeTx-treated (n = 14, N = 3) and young (n = 14, N = 4) neurons. Transfection of 1a−/− neurons with GB1a resulted in rescue of CaV2.2CFP/Gβ1N′-YFP FRET (n = 17, N = 4, ***p < 0.0001). C, Lack of specific FRET between CaV2.2CFP and either Gγ2N′-YFP (n = 10, N = 3) or Gγ2C′-YFP (n = 9, N = 3). D, Disruption of specific FRET between CaV2.2CFP and GB2YFP protein in 1a−/− neurons (n = 8, N = 3, ***p < 0.0001). E, Diagram illustrating disruption of CaV2.2CFP/Gβ1YFP FRET in boutons of 1a−/− neurons. One-way ANOVA analysis with post hoc Bonferroni's multiple comparison tests (B) and paired t test (C, D) indicated significance. Error bars indicate SEM.
Proximal C-terminal GB1a domain controls Gβγ–CaV2.2 channel interaction
Next, we searched for the molecular domain in the GB1a protein that mediates assembly of the GB1aR–Gβγ–CaV2.2 channel signaling complex. We created a series of GB1a deletions/truncations and tested whether expression of genetically modified GB1a versus wild-type GB1a (GB1a-WT) proteins in 1a−/− neurons disrupts CaV2.2CFP/Gβ1YFP FRET. To rule out a possible role for the endogenous GB1a subunit, we examined all of the truncations in the 1a−/− cultures. First, we examined whether the GB1a N-terminal sushi domains, functioning as axonal targeting signals (Biermann et al., 2010), mediate the Gβγ/CaV2.2 interaction. Deletion of two sushi domains in the GB1a (G28-Q157, GB1a-ΔSD) (Fig. 4A) did not abolish Gβ1YFP/CaV2.2CFP FRET (0.13 ± 0.009, n = 15) (Fig. 4B). In addition, S269A mutation in the GB1a protein (GB1a-S269A), which was shown to decrease by >10-fold the affinity toward GABA (Galvez et al., 2000), resulted in CaV2.2CFP/Gβ1YFP FRET as well (0.13 ± 0.01, n = 14) (Fig. 4B). These results confirm our previous data showing significant FRET between CaV2.2 and Gβγ in the absence of GABA binding (Fig. 1D,E) and suggest that agonist-induced activation of the receptor is not essential for the CaV2.2/Gβγ association.
Figure 4.

Proximal C-terminal domain of the GB1a protein is essential for Gβγ/CaV2.2 channel association. A, Schematics show GB1a constructs used to examine the domain responsible for Gβγ/CaV2.2 association. SD, Two sushi domains; LBD, ligand-binding domain; 7TM, seven-transmembrane domain; PCT, proximal C-terminal domain; CC, coiled–coiled domain; DCT, distal C-terminal domain. B, Mean FRET for the indicated transfection conditions in 1a−/− neurons: GB1a-WT (n = 66, N = 11), GB1a-Δ103 (n = 44, N = 8, **p < 0.01), GB1a-Δ21 (n = 24, N = 6, **p < 0.0001), GB1a-Δ74 (n = 14, N = 4, p > 0.05), GB1a-Δ39 (n = 17, N = 4, p > 0.05), GB1a-ΔSD (n = 15, N = 4, p > 0.05), and GB1a-S269A (n = 14, N = 4, p > 0.05). One-way ANOVA analysis with post hoc Dunnett's multiple-comparison tests relative to 1a−/− boutons transfected with GB1a-WT indicated significance. C, Effect of 10 μm baclofen on the GB1aYFP/CaV2.2CFP FRET in 1a−/− boutons transfected with GB1a-WT (n = 7–20, N = 3–5, **p < 0.01) or GB1a-Δ21 (n = 23–24, N = 5–6, p > 0.05). D, Effect of 10 μm baclofen on the GB1aYFP/GαoCFP FRET in 1a−/− boutons transfected with GB1a-WT (n = 12–15, N = 3–4, *p < 0.05) or GB1a-Δ21 (n = 14–16, N = 3–5, *p < 0.05, paired t test). Error bars indicate SEM.
Next, we tested whether the C terminus of GB1a is responsible for the complex assembly. Truncation of the entire C terminus (at R857; GB1a-Δ103) abolished CaV2.2CFP/Gβ1YFP FRET (0.024 ± 0.002, n = 44) (Fig. 4B). To precisely identify the site in GB1a C terminus that mediates CaV2.2/Gβγ association, we created a series of C-terminal deletions/truncations: deletion of the proximal domain of C terminus (ΔR857-S877; GB1a-Δ21), truncation at the coiled–coiled domain (K886; GB1a-Δ74), and truncation of the distal part of C terminus (L921; GB1a-Δ39). Notably, GB1a-Δ21 abolished CaV2.2CFP/Gβ1YFP FRET (0.036 ± 0.007, n = 24) (Fig. 4B). In contrast, neither GB1a-Δ74 nor GB1a-Δ39 deletions prevented FRET between Gβ1YFP and CaV2.2CFP (0.12 ± 0.02, n = 14; and 0.10 ± 0.01, n = 17, respectively). Together, these results suggest that the proximal C-terminal GB1a domain is required for Gβγ–CaV2.2 channel association at presynaptic boutons.
To assess whether the proximal C-terminal domain of GB1a is essential for intermolecular associations between the receptor and CaV2.2 channel, we measured FRET between YFP-tagged GB1a-WT or GB1a-Δ21 with CaV2.2CFP. The GB1a-Δ21 deletion resulted in ∼54% reduction of basal FRET (from 0.13 ± 0.02, n = 20, in GB1a-WT to 0.06 ± 0.008, n = 24, p < 0.001) (Fig. 4C) and abolished baclofen-induced decrease of FRET, which is typically observed in GB1a-WT (Fig. 4C). In contrast, deletion of the proximal C-terminal GB1a domain did not affect either basal FRET between GB1a-Δ21YFP and GαoCFP or baclofen-induced FRET reduction (n = 12–16, p < 0.05) (Fig. 4D). These results suggest that the proximal C-terminal GB1a domain mediates GB1aR–CaV2.2 channel interaction, whereas it is dispensable for association of the GB1a with Gαo G-protein subunit.
Proximal C-terminal GB1a domain does not affect agonist-induced GB1aR activation and cAMP inhibition
Next, we explored whether proximal C-terminal GB1a domain affects agonist-induced activation of the GB1aR. Our previous work suggests that agonist-induced increase in FRET between the C-terminally tagged GB1a/GB2 receptor subunits reflects receptor activation (Laviv et al., 2010). Therefore, we measured the potency of baclofen to induce conformational changes in the GB1aYFP/GB2CFP receptor expressed in presynaptic boutons, comparing changes in receptors containing YFP-tagged GB1a-WT versus GB1a-Δ21 proteins. Baclofen dose–response curve for FRET efficiency between GB1aCFP and GB2YFP revealed no significant difference in ED50 between GB1a-WT and GB1a-Δ21 proteins (GB1a-WT: 0.82 ± 0.003 μm, n = 10–21; GB1a-Δ21: 0.58 ± 0.05 μm, n = 11–21) (Fig. 5A).
Figure 5.
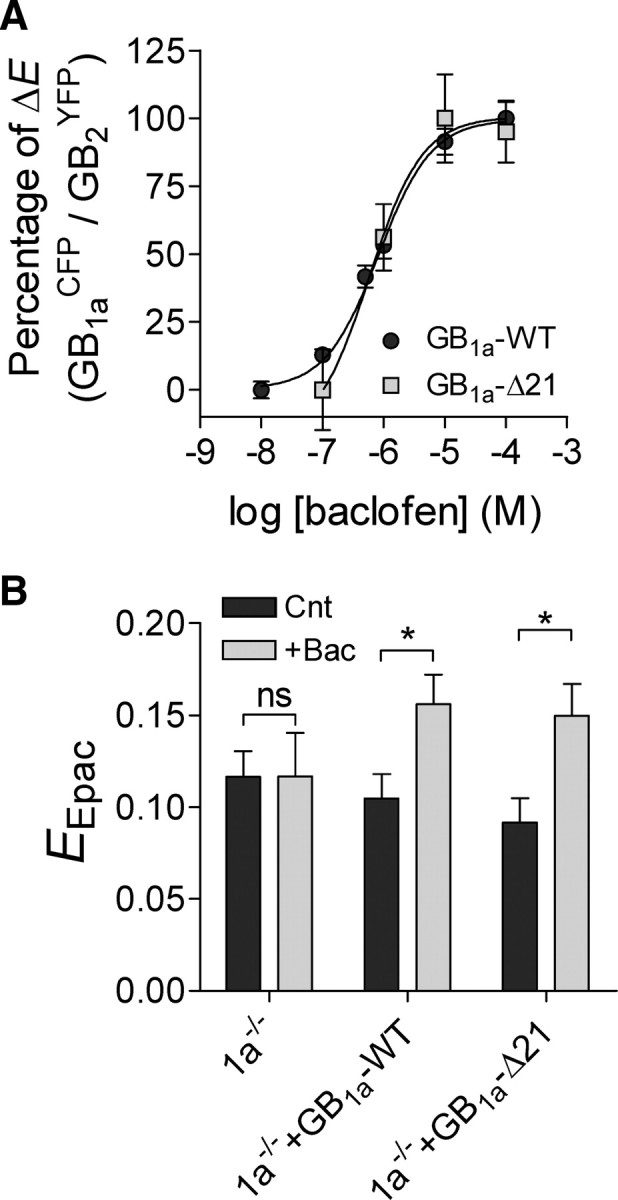
The GB1a proximal C-terminal domain does not affect baclofen-induced GB1a/GB2 receptor activation and cAMP inhibition. A, Dose–response curves of baclofen on GB1aCFP/GB2YFP FRET efficiency for GB1a-WT (n = 10–21, N = 5, ED50 = 0.82 ± 0.003 μm) and GB1a-Δ21 (n = 11–21, N = 3–5, ED50 = 0.58 ± 0.013 μm) proteins. E at 100 μm baclofen was set as 100%. B, Effect of 10 μm baclofen on CFP–Epac–YFP FRET efficiency (EEpac), reporting cAMP level, in 1a−/− boutons and in 1a−/− boutons expressing GB1a-WT or GB1a-Δ21 proteins. Baclofen increased EEpac in GB1a-WT-expressing (n = 30–32, N = 6, *p < 0.05) and GB1a-Δ21-expressing (n = 27–35, N = 5–6, *p < 0.05) boutons, but did not affect EEpac in 1a−/− boutons (n = 25–26, N = 5, p > 0.05) One-way ANOVA with post hoc Bonferroni's multiple-comparison tests indicated significance. Error bars indicate SEM.
As deletion of the proximal C-terminal domain did not affect GB1aYFP/GαoCFP FRET, we assessed the ability of the GB1a-Δ21 mutant to activate Gαi/o protein by measuring effect of baclofen on cAMP levels at individual synapses using CFP-Epac-YFP FRET reporter (van der Krogt et al., 2008). Baclofen induced ∼50% increase in CFP-Epac-YFP FRET, indicating inhibition of cAMP level in GB1a-WT-expressing boutons (n = 30–32, p < 0.05) (Fig. 5B). In contrast, baclofen did not affect CFP-Epac-YFP FRET in 1a−/− boutons (n = 25–26, p > 0.05) (Fig. 5B). In GB1a-Δ21-expressing boutons, baclofen induced a ∼60% increase in FRET (n = 27–35, p < 0.05) (Fig. 5B). Together, these results suggest that the proximal C-terminal GB1a domain does not affect agonist-induced GB1aR activation and Gαi/o-dependent signaling.
Proximal C-terminal GB1a domain is essential for agonist-induced presynaptic inhibition
Having established the necessity of GB1a protein for CaV2.2–Gβγ association, we examined the functional role of the GB1a protein and its proximal C-terminal domain in presynaptic inhibition of Ca2+ flux and synaptic vesicle release. First, we compared inhibitory effect of baclofen on presynaptic Ca2+ transients in functional boutons of 1a−/− neurons versus boutons expressing GB1a-WT or GB1a-Δ21 proteins. Presynaptic Ca2+ transients evoked by low-frequency stimulation were measured by high-affinity fluorescent calcium indicator Oregon Green 488 BAPTA-1 AM (Laviv et al., 2010). Baclofen affected the size of action-potential-dependent fluorescence transients (ΔF/F) in 1a−/− boutons by 10 ± 1.7% (n = 31), and it induced significantly higher reduction of calcium transients in GB1a-WT-expressing boutons (26.2 ± 1.7% inhibition, n = 17, p < 0.001) (Fig. 6A,B). Notably, GB1a-Δ21-expressing boutons displayed reduced sensitivity of Ca2+ transients to baclofen compared with GB1a-WT-expressing boutons (14.8 ± 3% inhibition, n = 19, p < 0.01) (Fig. 6A,B), suggesting that baclofen-induced inhibition of N-type calcium channels depends on the proximal C-terminal domain of the GB1a receptor subunit.
Figure 6.

The role of the GB1a proximal C-terminal domain on baclofen-induced inhibition of Ca2+ transients. A, Baclofen did not affect spike-dependent presynaptic Ca2+ transients (ΔF/F) evoked by 0.2 Hz stimulation in 1a−/− boutons and in GB1a-Δ21-expressing boutons, but reduced it in GB1a-WT-expressing boutons. Ca2+ transients were quantified as before (black) and after (gray) baclofen application (average of 10 traces). B, Average data on baclofen-induced modification in Ca2+ transients in 1a−/− (n = 31, N = 6, p > 0.05), GB1a-WT-expressing (n = 17, N = 4, ***p < 0.001, compared with 1a−/−), and GB1a-Δ21-expressing (n = 19, N = 5, p > 0.05, compared with 1a−/−) boutons. One-way ANOVA with post hoc Bonferroni's multiple-comparison tests indicated significance. Error bars indicate SEM.
Next, we assessed the role of the proximal C-terminal GB1a domain on baclofen-induced inhibition of synaptic vesicle exocytosis using FM4-64 dye (Laviv et al., 2010). The total pool of recycling vesicles was stained by maximal stimulation (600 APs at 10 Hz) and subsequently destained by 1 Hz stimulation. Baclofen (10 μm) profoundly decreased the destaining rate constant (measured as 1/τdecay, whereas τdecay is an exponential time course) in WT (N = 12, p < 0.001) (Fig. 7A,F), but it affected the destaining rate by a much lesser extent in WT neurons expressing membrane-targeted Gβγ scavenger, N-myristoylated phosducin (Rishal et al., 2005) (MyrPhd, N = 4, p < 0.001) (Fig. 7B,F). Furthermore, baclofen did not affect the destaining rate in 1a−/− (N = 9, p > 0.3) (Fig. 7C,F) boutons, complementing electrophysiological data on the lack of baclofen effect on basal synaptic transmission in 1a−/− CA3-CA1 synapses (Vigot et al., 2006). Transient expression of GB1a-WT protein together with GFP in 1a−/− neurons resulted in recovery of the inhibitory effect of baclofen (N = 6, p < 0.001, compared with GFP-expressing neurons) (Fig. 7D,F). Moreover, transient expression of the GB1a-Δ21 together with GFP in 1a−/− neurons significantly reduced baclofen effect on the destaining rate constant (N = 6, p < 0.0001, compared with expression of GB1a–WT + GFP-in 1a−/− neurons) (Fig. 7E,F). These results strongly suggest that (1) the GB1a protein is essential for baclofen-induced inhibition of synaptic vesicle release, (2) Gβγ is a mediator of baclofen-induced presynaptic inhibition, and (3) the proximal C-terminal GB1a domain is essential for baclofen-induced inhibition of synaptic vesicle release in hippocampal boutons.
Figure 7.

The GB1a proximal C-terminal domain is required for baclofen-induced inhibition of synaptic vesicle release. A–E, Representative FM destaining curves before and after application of 10 μm baclofen in WT cultures (n = 84, A), WT cultures transfected with MyrPhd and GFP (n = 112, B), 1a−/− cultures (n = 71, C), 1a−/− cultures transfected with GB1a-WT and GFP (n = 69, D), and 1a−/− cultures transfected with GB1a-Δ21 and GFP (n = 94, E). F, Summary of baclofen effect on FM destaining rate in WT (N = 12), GFP-expressing boutons (N = 3), WT cultures transfected with MyrPhd and GFP (N = 4), 1a−/− (N = 9), 1a−/− transfected with GB1a-WT and GFP (N = 6), and 1a−/− transfected with GB1a-Δ21 and GFP (N = 6) cultures. Error bars indicate SEM. (***p < 0.001, n.s. for p > 0.05, one-way ANOVA analysis with post hoc Bonferroni's multiple-comparison tests).
Discussion
In the current study, we discovered that the GABAB1a subunit plays a crucial role in creating a functional receptor–G-protein–channel complex in presynaptic boutons of hippocampal neurons. First, we found that the presynaptic GB1aR, heterotrimeric Go-protein and CaV2.2 channel are precoupled, forming macromolecular complex regardless of synaptic activity and agonist stimulation at individual hippocampal boutons. Second, basal GABA levels are sufficient to induce rearrangements within the complex. Third, intermolecular associations within the receptor, Gβγ, and CaV2.2 channel complex require expression of the GB1a protein. Fourth, we identified the proximal C-terminal domain in the GB1a protein as essential motif for the complex formation. This domain is required specifically for baclofen-induced presynaptic inhibition of Ca2+ transients and vesicle release, but not for the receptor activation and cAMP inhibition. Together, these findings suggest that compartmentalization of the presynaptic signaling complex, in addition to agonist-induced receptor activation, critically controls the GB1aR-mediated presynaptic inhibition at hippocampal boutons.
Presynaptic GABABR–G-protein–CaV2.2 channel signaling complex
Intermolecular interactions between receptors, G-proteins, and high-voltage-gated Ca2+ channels represent key events in inhibition of neurotransmitter release. Accumulating biochemical and electrophysiological evidence suggests that direct membrane-delimited interaction of Ca2+ channel α1 subunit with Gβγ mediates inhibition of presynaptic calcium currents and of synaptic vesicle exocytosis by GPCRs (Dascal, 2001; Dolphin, 2003; Tedford and Zamponi, 2006; Catterall and Few, 2008). Yet, the mechanisms of intermolecular coupling remain controversial. Although “physical scaffolding” of signaling components within the same G-protein-coupled signaling unit has been proposed to enhance signaling specificity (Neubig, 1994; Tsunoda et al., 1997), direct evidence for compartmentalization of GPCR signaling at central synapses is still missing.
To explore the microarchitecture and dynamics of proteins in specialized presynaptic compartments, FRET spectroscopy has been used. Our data provide direct evidence for the close (<100 Å) proximity between the tagged GB1aR, Gαoβ1γ2 G-protein heterotrimer, and CaV2.2 channel (Fig. 1). Specific FRET signals were detected in boutons lacking SNARE-mediated exocytosis, under block of receptor activation by antagonist, and in boutons expressing mutated GB1a-S269A protein with reduced affinity for GABA. These FRET-based data resonate with the recent proteomic study on molecular nano-environment of the CaV2 channels in the rodent brain (Müller et al., 2010). The authors demonstrated direct interaction between the GABAB receptor subunits and CaV2.2 channel under high stringency conditions. However, lack of interaction between the GABAB receptor and CaV2.2 channel was observed under resting conditions by coimmunoprecipitation in sensory dorsal root ganglion neurons (Puckerin et al., 2006), suggesting possible differences in signaling complex organization at central versus peripheral synapses. Furthermore, a direct interaction between CaV2.2 and Gβγ G-protein subunits in the CNS has been also demonstrated by proteomic studies (Khanna et al., 2007; Müller et al., 2010). Together, FRET-based and proteomic analyses strongly suggest assembly of the GABAB receptor presynaptic signaling complexes under resting conditions in central synapses.
What is the molecular mechanism underling the GB1aR-mediated presynaptic inhibition at hippocampal synapses? One possibility is that presynaptic inhibition arises from reduction in the number of available signaling molecules at the plasma membrane of boutons. For example, agonist-induced cointernalization of CaV2.2 channel with GPCRs such as opioid-like-receptor ORL1 and dopamine D1 receptor have been observed in earlier studies (Altier et al., 2006; Altier and Zamponi, 2008; Kisilevsky et al., 2008). To test whether the membrane fraction of GB1aRs depends on activity-dependent concentration of GABA in the vicinity of boutons, we measured fluorescence of pHluorin, N-terminally tagged to the GB1a protein as function of neuronal activity or agonist stimulation. We did not observe any change in the pHluorin fluorescence as function of stimulation frequency or agonist concentration (Fig. 2E), suggesting that under physiological conditions, reduction in the number of GB1aRs at the presynaptic membrane does not contribute to presynaptic inhibition at hippocampal boutons. In addition, agonist-induced, receptor-independent internalization of CaV2.2 channel might mediate presynaptic inhibition as has been suggested in dorsal root ganglion neurons (Tombler et al., 2006). However, this mechanism seems to be Gβγ-independent, and our results suggest that the GB1aR-mediated presynaptic inhibition in hippocampal synapses is mediated by Gβγ (Fig. 7F).
Our data favor the model based on agonist-induced structural rearrangements within the GB1aR–Gβγ–CaV2.2 channel complex as the mechanism mediating inhibition of basal synaptic vesicle exocytosis. Although synaptic activity was not essential for the complex formation, quantal synaptic transmission, an elementary unit of synaptic communication, induced structural rearrangements between the presynaptic signaling units (Fig. 2). Tonic activation of the GB1aR complex under miniature synaptic activity (in the presence of TTX) was relieved following application of the receptor antagonist. Thus, basal levels of GABA in the synaptic cleft are sufficient to activate GB1aRs through induction of conformation changes in the GB1a/GB2 heterodimer (Laviv et al., 2010), leading to activation of G-protein and closer association of precoupled Gβγ subunits and CaV2.2 channels (Fig. 2G). As a result of these conformational rearrangements, basal GABA induces reduction of presynaptic calcium flux (Fig. 6) and consequent inhibition of synaptic vesicle release in hippocampal boutons (Fig. 7).
Proximal GB1a C-terminal domain is required for the GB1aR complex formation
It first came as a surprise that FRET between CaV2.2/Gβγ was reduced to background level in 1a−/− boutons lacking GB1a protein (Fig. 3). Presynaptic expression of the tagged CaV2.2 channels and Gβγ subunits was not altered in 1a−/− neurons, suggesting a proper trafficking to the 1a−/− boutons. The lack of specific FRET was observed with both Gβ1 and Gγ2 proteins and did not depend on the position of fluorophore tagging, suggesting that there were no constraints on fluorophore mobility that could convert changes in orientation into substantial changes in FRET. Therefore, these results suggest that reduction in donor–acceptor distance, rather than dipole–dipole orientation of the donor and acceptor fluorophores, underlies FRET disruption. Neither alterations in synaptic activity nor agonist-induced stimulation triggered the complex formation. This association was recovered following transient expression of the GB1a-WT protein in 1a−/− neurons. Notably, partial rescue of CaV2.2/Gβγ association was observed following expression of the GB1a-S269A protein with >10-fold reduced affinity to GABA (Galvez et al., 2000). These results led to the conclusion that the GB1a protein is essential for precoupling of the receptor, Gβγ, and CaV2.2 channel.
What is the GB1a molecular domain mediating formation of the GB1aR signaling complex at hippocampal boutons? Previous work suggested that the C terminus of the GB1 receptor subunit is not essential for the GABAB receptor activation detected through GIRK channel activity (Margeta-Mitrovic et al., 2001). Based on these results, the authors concluded that the GB1 protein is not required for specific coupling to G-protein and its activation. Our findings confirm that C terminus of the GB1a protein is not required for activation of the receptor, receptor/Gαi/o interaction, and cAMP inhibition. Conversely, our data strongly suggest that the GB1a proximal C-terminal domain (R857-S877) is required for a tight association between the receptor, Gβγ, and CaV2.2 channel. Functionally, deletion of this GB1a domain impaired presynaptic inhibition of Ca2+ flux and of synaptic vesicle exocytosis. These results imply differential regulation of GIRK and CaV2.2 channels by the GB1a C terminus. Thus, in addition to agonist-induced receptor activation, preassembly of the GB1aR–Gβγ–CaV2.2 channel signaling complex is essential for agonist-induced presynaptic inhibition at hippocampal boutons. It will be interesting to explore functional significance of GB1 proximal C-terminal domain on regulation of voltage-gated Ca2+ channels and NMDA receptors in spines and dendrites (Chalifoux and Carter, 2010, 2011).
Does association of Gβγ and CaV2.2 channel result from constitutive activity of GB1aRs expressed at hippocampal boutons? Unfortunately, no information is available on constitutive activity of presynaptic GB1aRs in central synapses. Our data provide no evidence for constitutive GB1aR activity because CGP54626 antagonist, which works as a partial agonist at constitutively active GABABRs (Mukherjee et al., 2006), did not affect any tested association between the GB1aR, Gβγ subunits, and CaV2.2 channel in the absence of GABA (data not shown). Therefore, our results imply that GB1a plays an essential role in coordinating and integrating the complex assembly regardless of agonist-induced receptor activation.
Individual hippocampal boutons may express diverse GPCRs, in addition to GB1a/GB2 receptors, raising the question of the mechanisms of GPCR-mediated signaling in 1a−/− boutons lacking functional Gβ1γ2/CaV2.2 signaling complexes. Presynaptic inhibition mediated through adenosine (Vigot et al., 2006) and muscarinic (Vertkin and Slutsky, unpublished data) receptors remains functional in 1a−/− hippocampal synapses. These data imply that adenosine and muscarinic receptors are capable of presynaptic inhibition through distinct Ca2+-dependent and Ca2+-independent mechanisms. For example, adenosine-induced presynaptic inhibition is not limited to the N-type calcium channel (Wu and Saggau, 1994; Dittman and Regehr, 1996). Moreover, M2 muscarinic receptors can inhibit neurotransmitter release through direct block of release machinery (Parnas and Parnas, 2007). Further work is needed to understand the precise mechanisms underlying signaling specificity at synapses expressing multiple GPCRs.
Together, our data support the “physical scaffolding” model of GPCR, G-protein, and effector suggesting a precoupling of presynaptic G-protein signaling complexes in central synapses. Our study revealed the proximal C-terminal part of GB1a receptor subunit as essential domain for a tight association of the receptor, Gβγ subunits, and CaV2.2 channel. As the GB1a proximal C-terminal domain does not affect receptor activation, identification of this motif allowed us, for the first time, to isolate functional significance underlying precoupling of signaling units in a macromolecular complex. It remains to be seen whether compartmentalization of GPCRs, Gβγ subunits, and CaV2.2/1 channels constitutes a general mechanism underlying presynaptic inhibition at central synapses.
Footnotes
This work was supported by the Israel Science Foundation (I.S., Grants 993/08, 1925/08 and 170/08), the Binational Science Foundation (I.S. and P.A.S., Grant 2007199), and the Swiss Science Foundation (3100A0–117816). We thank Dr. N. Dascal and Dr. E. Reuveny for helpful discussions; Dr. E. Reuveny for providing TNFR2YFP, Gβ1YFP, and Gγ2YFP cDNAs; and Dr. K. Jalink for providing CFP-Epak-YFP cDNA.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-[beta] as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Altier C, Zamponi GW. Signaling complexes of voltage-gated calcium channels and G-protein-coupled receptors. J Recept Signal Transduct Res. 2008;28:71–81. doi: 10.1080/10799890801941947. [DOI] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Biermann B, Ivankova-Susankova K, Bradaia A, Abdel Aziz S, Besseyrias V, Kapfhammer JP, Missler M, Gassmann M, Bettler B. The sushi domains of GABAB receptors function as axonal targeting signals. J Neurosci. 2010;30:1385–1394. doi: 10.1523/JNEUROSCI.3172-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SB, Clancy SM, Terunuma M, Revilla-Sanchez R, Thomas SM, Moss SJ, Slesinger PA. Direct interaction of GABAB receptors with M2 muscarinic receptors enhances muscarinic signaling. J Neurosci. 2009;29:15796–15809. doi: 10.1523/JNEUROSCI.4103-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N. Ion-channel regulation by G proteins. Trends Endocrinol Metab. 2001;12:391–398. doi: 10.1016/s1043-2760(01)00475-1. [DOI] [PubMed] [Google Scholar]
- De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium ocmponent of sensory neurone action potentials. Nature. 1978;276:837–839. doi: 10.1038/276837a0. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- Fowler CE, Aryal P, Suen KF, Slesinger PA. Evidence for association of GABA(B) receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J Physiol. 2007;580:51–65. doi: 10.1113/jphysiol.2006.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Thümer L, Lohse MJ, Bünemann M. G protein activation without subunit dissociation depends on a G{alpha}(i)-specific region. J Biol Chem. 2005;280:24584–24590. doi: 10.1074/jbc.M414630200. [DOI] [PubMed] [Google Scholar]
- Galés C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor–G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Galvez T, Urwyler S, Prézeau L, Mosbacher J, Joly C, Malitschek B, Heid J, Brabet I, Froestl W, Bettler B, Kaupmann K, Pin JP. Ca2+ requirement for high-affinity gamma-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol. 2000;57:419–426. doi: 10.1124/mol.57.3.419. [DOI] [PubMed] [Google Scholar]
- Guetg N, Seddik R, Vigot R, Turecek R, Gassmann M, Vogt KE, Bräuner-Osborne H, Shigemoto R, Kretz O, Frotscher M, Kulik A, Bettler B. The GABAB1a isoform mediates heterosynaptic depression at hippocampal mossy fiber synapses. J Neurosci. 2009;29:1414–1423. doi: 10.1523/JNEUROSCI.3697-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P, Frank M, Hoffmann C, Lohse MJ, Bünemann M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005;24:4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Khanna R, Zougman A, Stanley EF. A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J Biochem Mol Biol. 2007;40:302–314. doi: 10.5483/bmbrep.2007.40.3.302. [DOI] [PubMed] [Google Scholar]
- Kisilevsky AE, Mulligan SJ, Altier C, Iftinca MC, Varela D, Tai C, Chen L, Hameed S, Hamid J, Macvicar BA, Zamponi GW. D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron. 2008;58:557–570. doi: 10.1016/j.neuron.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kuner R, Köhr G, Grünewald S, Eisenhardt G, Bach A, Kornau HC. Role of heteromer formation in GABAB receptor function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- Laviv T, Riven I, Dolev I, Vertkin I, Balana B, Slesinger PA, Slutsky I. Basal GABA regulates GABA(B)R conformation and release probability at single hippocampal synapses. Neuron. 2010;67:253–267. doi: 10.1016/j.neuron.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Nikolaev VO, Hein P, Hoffmann C, Vilardaga JP, Bünemann M. Optical techniques to analyze real-time activation and signaling of G-protein-coupled receptors. Trends Pharmacol Sci. 2008;29:159–165. doi: 10.1016/j.tips.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. Function of GB1 and GB2 subunits in G protein coupling of GABA(B) receptors. Proc Natl Acad Sci U S A. 2001;98:14649–14654. doi: 10.1073/pnas.251554498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee RS, McBride EW, Beinborn M, Dunlap K, Kopin AS. Point mutations in either subunit of the GABAB receptor confer constitutive activity to the heterodimer. Mol Pharmacol. 2006;70:1406–1413. doi: 10.1124/mol.106.024463. [DOI] [PubMed] [Google Scholar]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, Schulte U. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A. 2010;107:14950–14957. doi: 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- Neubig RR, Gantzos RD, Thomsen WJ. Mechanism of agonist and antagonist binding to alpha 2 adrenergic receptors: evidence for a precoupled receptor–guanine nucleotide protein complex. Biochemistry. 1988;27:2374–2384. doi: 10.1021/bi00407a019. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- Orly J, Schramm M. Coupling of catecholamine receptor from one cell with adenylate cyclase from another cell by cell fusion. Proc Natl Acad Sci U S A. 1976;73:4410–4414. doi: 10.1073/pnas.73.12.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas H, Parnas I. The chemical synapse goes electric: Ca2+- and voltage-sensitive GPCRs control neurotransmitter release. Trends Neurosci. 2007;30:54–61. doi: 10.1016/j.tins.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Parnas H, Segel L, Dudel J, Parnas I. Autoreceptors, membrane potential and the regulation of transmitter release. Trends Neurosci. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- Puckerin A, Liu L, Permaul N, Carman P, Lee J, Diversé-Pierluissi MA. Arrestin is required for agonist-induced trafficking of voltage-dependent calcium channels. J Biol Chem. 2006;281:31131–31141. doi: 10.1074/jbc.M605000200. [DOI] [PubMed] [Google Scholar]
- Rishal I, Porozov Y, Yakubovich D, Varon D, Dascal N. G-2-3-dependent and G-2-3-independent basal activity of G protein-activated K+ channels. J Biol Chem. 2005;280:16685–16694. doi: 10.1074/jbc.M412196200. [DOI] [PubMed] [Google Scholar]
- Riven I, Iwanir S, Reuveny E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron. 2006;51:561–573. doi: 10.1016/j.neuron.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, Tarusawa E, Kulik A, Unger A, Ivankova K, Seddik R, Tiao JY, Rajalu M, Trojanova J, Rohde V, Gassmann M, Schulte U, Fakler B, Bettler B. Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010;465:231–235. doi: 10.1038/nature08964. [DOI] [PubMed] [Google Scholar]
- Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44:835–849. doi: 10.1016/j.neuron.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J Neurosci. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM, Levitzki A. Mode of coupling between the beta-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry. 1978;17:3795. doi: 10.1021/bi00611a020. [DOI] [PubMed] [Google Scholar]
- Tombler E, Cabanilla NJ, Carman P, Permaul N, Hall JJ, Richman RW, Lee J, Rodriguez J, Felsenfeld DP, Hennigan RF, Diversé-Pierluissi MA. G protein-induced trafficking of voltage-dependent calcium channels. J Biol Chem. 2006;281:1827–1839. doi: 10.1074/jbc.M508829200. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker CS. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- van der Krogt GN, Ogink J, Ponsioen B, Jalink K. A comparison of donor–acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS One. 2008;3:e1916. doi: 10.1371/journal.pone.0001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigot R, Barbieri S, Bräuner-Osborne H, Turecek R, Shigemoto R, Zhang YP, Luján R, Jacobson LH, Biermann B, Fritschy JM, Vacher CM, Müller M, Sansig G, Guetg N, Cryan JF, Kaupmann K, Gassmann M, Oertner TG, Bettler B. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron. 2006;50:589–601. doi: 10.1016/j.neuron.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol. 1995;485(Pt 3):649–657. doi: 10.1113/jphysiol.1995.sp020759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]