

Abstract
Injured neurons become dependent on trophic factors for survival. However, application of trophic factors to the site of injury is technically extremely challenging. Novel approaches are needed to circumvent this problem. Here, we unravel the mechanism of the emergence of dependency of injured neurons on brain-derived neurotrophic factor (BDNF) for survival. Based on this mechanism, we propose the use of the diuretic bumetanide to prevent the requirement for BDNF and consequent neuronal death in the injured areas. Responses to the neurotransmitter GABA change from hyperpolarizing in intact neurons to depolarizing in injured neurons. We show in vivo in rats and ex vivo in mouse organotypic slice cultures that posttraumatic GABAA-mediated depolarization is a cause for the well known phenomenon of pathological upregulation of pan-neurotrophin receptor p75NTR. The increase in intracellular Ca2+ triggered by GABA-mediated depolarization activates ROCK (Rho kinase), which in turn leads to the upregulation of p75NTR. We further show that high levels of p75NTR and its interaction with sortilin and proNGF set the dependency on BDNF for survival. Thus, application of bumetanide prevents p75NTR upregulation and neuronal death in the injured areas with reduced levels of endogenous BDNF.
Introduction
While intact mature central neurons do not require brain-derived neurotrophic factor (BDNF) for survival (Giehl et al., 2001; Li et al., 2008; Shulga et al., 2008; Rauskolb et al., 2010), under pathophysiological conditions neuronal populations become dependent on the trophic support provided by BDNF (Klöcker et al., 2000; Schäbitz et al., 2000; Giehl et al., 2001; Shulga et al., 2008, 2009). However, supplying the affected neurons with this trophic factor in vivo has proven to be technically extremely challenging (Thoenen and Sendtner, 2002), and novel strategies are needed to circumvent this problem.
The mechanism of posttraumatic dependency on BDNF is not clear. BDNF signaling is mediated through two receptors, TrkB and neurotrophin receptor p75 (p75NTR). The signaling of the latter has been frequently associated with cell death (Huang and Reichardt, 2001). Trauma is well known to increase the level of p75NTR expression in neurons and glia (Chao, 2003). Apart from promoting cell death, high p75NTR levels increase the affinity and specificity of Trk receptors to their cognate ligands (Huang and Reichardt, 2001; Chao, 2003); this could enhance the trophic efficiency of available BDNF. Thus, we hypothesized that, after trauma, the fate of the cell could depend on the balance between BDNF survival-promoting signaling through TrkB and the opposing signaling of p75NTR. High levels of p75NTR expression in axotomized neurons could set the requirement for BDNF trophic support, leaving the survival outcome at the mercy of the accessibility of endogenous BDNF. If this is the case, discovering the way to prevent pathological p75NTR upregulation would provide the possibility for the development of novel therapeutic strategies.
Although downstream signaling, processing, and function of p75NTR is a subject of intense investigation (Underwood and Coulson, 2008), much less is known about the regulation of p75NTR expression in nontraumatic (Gao et al., 2007) and traumatic (Dubreuil et al., 2003; Peterson and Bogenmann, 2003; Ramos et al., 2007) conditions. p75NTR is widely expressed in the developing CNS, downregulated in the adult brain, and reexpressed after injury (Underwood and Coulson, 2008). Interestingly, the changes in the expression level of p75NTR are paralleled by functional changes in the GABAergic inhibitory neurotransmission: the developmental and posttraumatic shifts in GABAA receptor-mediated responses (Blaesse et al., 2009). GABA-induced responses are depolarizing and able to induce intracellular Ca2+ ([Ca2+]i) increase during development, but become hyperpolarizing in mature neurons due to the upregulation of K+–Cl− cotransporter KCC2 (Blaesse et al., 2009). After trauma, GABAA-mediated responses become again depolarizing due to functional downregulation of KCC2 and accumulation of intracellular Cl− through the activity of Na+–K+–2Cl− cotransporter NKCC1 (Nabekura et al., 2002; Blaesse et al., 2009). In this context, an interesting question arises: Are parallel changes in p75NTR expression and the shift in polarity of GABAA-mediated responses interrelated?
Using in vivo and ex vivo models, we show that GABAA-mediated [Ca2+]i increase is essential for the posttraumatic upregulation of p75NTR. We also show that the induction of p75NTR expression renders injured neurons dependent on the trophic support by BDNF. Diuretic drug bumetanide is a specific inhibitor of NKCC1 (Blaesse et al., 2009), and thus it is able to prevent the posttraumatic shift in the polarity of GABA-evoked responses. We show that, through this action, bumetanide prevents the upregulation of p75NTR and consequent neuronal death, and suggest bumetanide as a novel therapeutic strategy for the injured neurons.
Materials and Methods
Organotypic slice cultures
Hippocampal slices from 8- to 9-d-old male and female NMRI mice were prepared according to the method by Stoppini et al. (1991). Transverse 350-μm-thick slices were cut from the hippocampi with a McIlwain tissue chopper. They were immediately placed on sterile Millicell-CM membranes (Millipore) in six-well culture trays with 1 ml of plating medium (Neurobasal, with B27 supplement; Invitrogen) containing antibiotic (Primocin; InvivoGen). Slices were kept at 37°C under 5% CO2 in air. One day after plating, the medium was changed for antibiotic-free medium and renewed again 4 d after plating.
Ex vivo axotomy model
All experiments were performed 4 d after plating. A lesion between CA3 and CA1 regions was made using a piece of a razor blade (cutting edge, 4 mm) under light stereomicroscope. The lesion traverses the whole slice thickness in the vertical direction, completely separating CA3 and CA1; the dentate gyrus region is not affected, and the slice is not cut into two pieces (see Fig. 1a). We have previously reported that unlesioned slices of this age possess GABA- and BDNF-evoked responses characteristic for the intact mature nervous tissue, and that this ex vivo axotomy model gives similar results as observed in the in vivo axotomy model (Shulga et al., 2008). TrkB-Fc (recombinant human TrkB-Fc chimera; 200 ng/ml; R&D Systems), Fc (human IgG Fc fragment; 230 ng/ml; Millipore), bumetanide (10 μm; Tocris), Rex antibody (1:250; a kind gift from Dr. Louis Reichardt, University of California, San Francisco, San Francisco, CA), antibody to p75 ECD (1:500 for function blocking; AB1554; Millipore), plasmin (45 nm; American Diagnostica), antibodies to proNGF and proBDNF (1:100; Alomone Labs), BAPTA-AM (100 μm; Invitrogen; was added to the medium and loaded three times on top of the slices every 10 min; thereafter, slices were transferred to the medium without BAPTA-AM), TTX (2 μm; Tocris), KCl (10 mm), Ni2+/Cd2+ (100 μm), muscimol (50/100 μm; Tocris), bicuculline (20 μm; Sigma-Aldrich), 4-aminopyridine (4-AP) (100 μm; Tocris), nimodipine (20 μm; Tocris), and (1R,4r)-4-((R)-1-aminoethyl)-N-(pyridin-4-yl)cyclohexanecarboxamide (Y-27632) (100 μm; Sigma-Aldrich) were added to slices immediately after the lesion procedure; unlesioned slices were treated at the same time. All slices were collected for analyses 1 d after the experiment, unless stated otherwise in the text.
Figure 1.

Axotomy induces the trophic requirement for endogenous BDNF. a, Ex vivo axotomy model. Left, Representative fluorescent picture of a lesioned slice stained immunohistochemically for NeuN. Schematic drawing: CA3 neurons (yellow) are axotomized and CA1 neurons (blue) are denervated. Scale bar, 100 μm. Right, A representative confocal image taken from the middle of CA3 region for the stereological counting of neurons. Scale bar, 20 μm. b, Zoomed parts of confocal images from the CA3 region of the lesioned slices with and without TrkB-Fc (the scavenger of the endogenous BDNF) stained immunohistochemically for NeuN (green) and cleaved caspase-3 (red). White arrows, Cleaved caspase-3-positive neurons. Scale bar, 10 μm. c, Ratio of cleaved caspase-3-positive (=apoptotic) neuronal number to total neuronal number obtained by blind stereological counting, percentage of unlesioned control ratio value. Deprivation of endogenous BDNF with TrkB-Fc causes significant increase in apoptotic neuronal number in the lesioned, but not unlesioned, slices at 1 d after the injury. Control human Fc fragment had no effect. n = 50 slices/4880 neurons; ***p < 0.001, one-way ANOVA and Bonferroni's post hoc analyses. Error bars indicate SEM.
In vivo axotomy
All animal experiments were approved by the Local Ethics Committee for Animal Research at the University of Helsinki. Adult male Wistar rats (240–330 g) were used. One hour before the surgery, rats received an intraperitoneal injection of bumetanide (30 mg/kg; Tocris; diluted in saline containing 0.25% NaOH). Control animals received an intraperitoneal injection of saline at the same time. Animals were anesthetized with isoflurane. A hemilaminectomy was performed at the lumbar L4–L6 region of the spinal cord. The dura was opened, and the corresponding L4–L6 ventral roots were identified under microscopic control, isolated, and rhizotomized with fine microscissors. The divided roots were then left in the spinal canal. The wounds were closed with sutures. After surgery, the animals were allowed to survive for 3 d and then perfused with 4% paraformaldehyde. The final identification of rhizotomized spinal cord segments was performed in each animal after fixation. Spinal cords were postfixed for 24 h and submerged in 30% sucrose in PBS; thereafter, 14 μm cryosections were prepared and stained immunohistochemically.
Knock-out animals
Pups were used from a mouse line with disruption in the Slc12a2 gene encoding the Na+-K+-2Cl− cotransporter isoform 1 (NKCC1) (Pace et al., 2000). Genotyping was performed on tail biopsy DNA by PCR using standard protocols (Pfeffer et al., 2009). Sortilin-deficient and wild-type parental mice were genotyped as described previously (Jansen et al., 2007). p75+/+ and p75−/− mice (Lee et al., 1992) were perfusion-fixed with 4% paraformaldehyde, postfixed for 24 h, and submerged in 30% sucrose in PBS; thereafter, 50 μm cryosections were prepared and stained immunohistochemically according to the same protocol as organotypic hippocampal slices (see below).
Basal forebrain primary cultures
Rat embryonic septa were dissected on embryonic day 17. The meninges were carefully removed, and the tissue was incubated in papain solution for 10 min before trituration by pipetting. The cells were plated at the density of 200,000 cells/well on poly-l-lysine-coated coverslips in 24-well culture plates. The neurons were cultured in Neurobasal medium (Invitrogen) containing 1% penicillin–streptomycin, 1% l-glutamine, and 2% B27 supplement (Invitrogen). One-third of the medium was changed on day 3 after plating. On day 7 in vitro, the cells were treated overnight with proNGF (1 ng/ml) or proBDNF (5 ng/ml) in the absence or presence of antibodies against proNGF or proBDNF (1:100). The following day, the neurons were rinsed with PBS and fixed 15 min with 4% paraformaldehyde (PFA) solution.
Immunohistochemistry
Organotypic slice cultures.
Slices remained on the Millicell-CM membranes during the whole staining procedure. They were fixed in 4% PFA overnight at 4°C. Thereafter, the slices were washed in PBS and dehydrated through a graded series of methanol in PBS (30, 50, 80%). Next, the slices were incubated in Dent's fixative (80% methanol, 20% DMSO; Sigma-Aldrich), washed in TBSTD (TBS, 0.1% Tween, 5% DMSO), and placed in 5% bovine serum albumin (BSA) (Sigma-Aldrich) plus 0.4% sheep serum (Sigma-Aldrich) in TBSTD overnight at 4°C. Thereafter, slices were incubated with primary antibodies diluted in the blocking solution at 4°C for 48 h. Rabbit anti-p75NT ECD Ab (1:500; AB1554; Millipore), mouse anti-NeuN Ab (1:400; Millipore), rabbit anti-cleaved caspase-3 Ab (1:200; Cell Signaling Technology), rabbit anti-p-cofilin (1:100; Santa Cruz Biotechnology), and goat anti-cleaved caspase-6 (1:100; Santa Cruz Biotechnology) were used. After washing in TBSTD, slices were incubated overnight with Alexa Fluor 488-conjugated donkey anti-mouse or rabbit IgG, Alexa Fluor 568-conjugated goat anti-rabbit or goat IgG (1:400; Invitrogen), and Cy5-conjugated donkey anti-mouse IgG (1:400; Jackson ImmunoResearch Laboratories). Slices were then washed in TBSTD, incubated in Hoechst (1:1000; Invitrogen), rinsed with TBSTD, and mounted with membranes on Superfrost Plus slides in ProLong Gold mounting medium (Invitrogen).
Basal forebrain primary cultures.
After the fixation, coverslips were washed with PBS, treated with 0.2% Triton X-100 in PBS (TXP), and incubated in 10% sheep serum in TXP. Neurons were left in chicken anti-ChAT antibody (1:200; Abcam) in 10% sheep serum in TXP overnight at +4°C, washed with TXP, and incubated with Alexa Fluor 568-conjugated goat anti-chicken IgG (1:400; Invitrogen) in TXP at room temperature. Coverslips were then washed in TXP containing Hoechst (1:1000; Invitrogen), rinsed in MilliQ water, and mounted with ProLong Gold antifade reagent (Invitrogen).
Cryosections from ventral root axotomized rats.
Sections were allowed to melt in the room temperature, washed in PBS, and incubated with 5% BSA. Thereafter, sections were incubated overnight with primary antibodies [p75NTR: 1:500; Millipore; MAB365; NeuN: 1:400; Millipore; KCC2 (Ludwig et al., 2003): 1:500; cleaved caspase-3: 1:600; Cell Signaling Technology] in 5% goat serum, 5% BSA, and 0.3% Triton X-100 in PBS. The next day, sections were washed in PBS and incubated with secondary antibodies (Alexa Fluor 568-conjugated goat anti-rabbit or anti-mouse and Alexa Fluor 488-conjugated donkey anti-mouse or anti-rabbit; 1:400; Invitrogen) diluted in 0.3% Triton X-100 in PBS. Thereafter, sections were washed in PBS and mounted with ProLong Gold antifade reagent (Invitrogen).
Image acquisition and analyses
Image stacks consisting of 11 images from 10 and 20 μm below the organotypic slice surface were taken with a Leica TCS SP5 confocal microscope (Leica Microsystems) with an HC PL APO 20×, 0.7 NA objective using glycerol immersion, with zoom factor 4.5–5. Two image stacks were taken from each slice, one from the middle of the CA3 and the other from the middle of the CA1 region (see Fig. 1a, left). Three-dimensional reconstruction images from the stacks were analyzed in Bitplane Imaris software. Image stacks had a volume of 155 × 155 × 10 μm, the majority of the 3D images containing 70–160 neurons (see Fig. 1a, right). Background extraction was performed blindly and in a similar way for every 3D image, using the image histogram. For the analysis of cleaved caspase-3 or -6-positive neurons, isosurface tool in Imaris software was used to mask the caspase-3 or -6 channel with NeuN channel to visualize only the neuronal cells. Neurons that had cleaved caspase-3 or -6 staining colocalizing with the NeuN-defined nucleus were considered as positive. For the analysis of p75NTR-positive neurons, InMotion tool and a high-magnification factor in Imaris software were used for enhanced detail visibility. Neurons that had p75NTR staining around a NeuN-defined nucleus were considered as positive. The amount of p75NTR-positive or cleaved caspase-3-positive neurons and the total number of neurons were analyzed by blind manual stereological counting, and the ratio was calculated.
For the analyses of p-cofilin immunostaining of organotypic slices and ChAT immunostaining of basal forebrain primary cultures, fluorescence images were acquired with Olympus BX61 microscope using 4, 10, or 20× objectives. For the analysis of immunostaining of cryosections from the ventral root axotomized rats, fluorescence images were acquired with Olympus BX61 microscope using 10 or 20× objectives. For the analysis of p75NTR, total amount of p75NTR-positive neurons for each animal from four sections per animal was counted by blind stereological counting. Total amount of neurons (identified by NeuN) was counted from each animal from four sections consecutive to each p75NTR-stained section, and total p75NTR-positive neuronal number was divided by total NeuN-positive neuronal number for each animal. Criteria for analysis were area of min 3900 arbitrary units (for the images taken with 10× objective) in ImagePro software (corresponding to ca 600 μm2) to identify the motoneurons, and the density of p75NTR or cleaved caspase-3 staining of min 13 arbitrary units. For the analysis of KCC2, KCC2 immunostaining intensity was measured from each neuron in ImagePro software (selection criteria as above, identified by double staining with NeuN) and the average for each section was quantified.
Western blotting
Slices were homogenized in homogenization buffer (0.3 m sucrose, 10 mm HEPES, 1 mm EDTA, pH 7.2, containing protease inhibitors; Complete Mini; Roche) and incubated with Laemmli loading buffer for 45 min at 45°C. The solution was separated in 8–15% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Blots were probed with antibody 9651 to p75NTR (Huber and Chao, 1995) (1:2000), proNGF (1:500; Alomone Labs), phospho-Akt Ser73 (1:2000; Cell Signaling Technology), pan-Akt (1:1000; Cell Signaling Technology), KCC2 (Ludwig et al., 2003) (1:2000), phosphotyrosine (clone 4G10; 1:1000; Millipore), or NKCC1 (T4; Developmental Studies Hybridoma Bank; 1:500), and developed using ECL-plus kit (GE Healthcare). After that, membranes were stripped using DTT/SDS in Tris-HCl, pH 7.0, and probed with mouse anti-mNGF (Millipore; 1:200), mouse anti-α-tubulin Ab (Sigma-Aldrich; 1:2000), or rabbit anti-β-tubulin (Covance; 1:3000). Optical densities of the bands were analyzed with the AIDA imaging software.
Immunoprecipitation
GammaBind G-Sepharose (GE Healthcare) was blocked with 5% milk, 1% BSA in PBS overnight and incubated with KCC2 antibody (Ludwig et al., 2003) (or rabbit serum for negative control) overnight at +4°C. Slices were homogenized in RIPA buffer (150 mm NaCl, 1% Triton-X, 0.5% DOC, 0.1% SDS, 50 mm Tris, pH 8) containing protease and phosphatase inhibitors (Complete Mini and PhosSTOP; Roche). One milligram of protein in 1 ml of RIPA was preclarified by G-Sepharose and then incubated with KCC2 antibody (Ludwig et al., 2003) attached to G-Sepharose overnight at +4°C. Sepharose beads were washed with RIPA buffer and eluted in 30 μl of Laemmli buffer, heated 5 min at +95°C. The supernatants were loaded to 8% SDS-PAGE gel and separated by standard Western blot protocol described above.
Real-time PCR
Total RNA was isolated with RNeasy kit (QIAGEN) and reverse transcribed with SuperScript III RNase H-Reverse Transcriptase (Invitrogen). cDNA samples were amplified using iQ SYBR Green Supermix (Bio-Rad) and detected with the Bio-Rad CFX96 real-time PCR detection system. Primers were designed on PrimerQuest software and tested with OligoAnalyzer software (eu.idtdna.com) and contained an intronic sequence in between. Sequences were as follows: AGGGCACATACTCAGATGAAGCCA (p75NTR FW), GGGCGTAGACCTTGTGATCCAT (p75NTR RW), CTCCGCTTTCATGTAGAGGAAG (Pgk1 FW), and GACATCTCCTAGTTTGGACAGTG (Pgk1 RW) (Willems et al., 2006). Results were analyzed on Bio-Rad CFX Manager software and normalized to Pgk1.
Fluo-4[Ca2+]i imaging
Neurons were loaded with Fluo-4 acetoxyl methyl (10 μm; Invitrogen) in extracellular standard solution containing the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 15 d-glucose, 2 CaCl2; 5% CO2/95% O2. Slices were then laid on the glass bottom of a submerged-type chamber, and this was placed on a microscope stage and continuously perfused with standard solution gassed with 95% O2/5% CO2 at a rate of 2–3 ml/min. Fluo-4 fluorescence was visualized using a Leica TCS SP5 confocal system with a 20× objective lens (HCX APO L 20×/0.90 W; Leica) mounted on a DM5000 microscope (Leica). For excitation, a blue (OPSL 488 nm/270 mW) laser was used through a DD 488/561 beam splitter. Data were stored for off-line analysis by means of image-processing software LAS AF (Leica). Variations in [Ca2+]i were observed as changes in intensities at 488 nm. Changes were monitored in the CA3 pyramidal neurons.
Statistical analysis
Statistical analysis was performed on PASW Statistic 18 software (SPSS). The normality of distribution was tested by Kolmogorov–Smirnov and Shapiro–Wilk tests (α level, 0.05). Comparison of mean values was performed by t test or one-way ANOVA followed by Bonferroni's post hoc analyses (α level, 0.05). Data are presented as mean ± SEM. Data are normalized to the values of unlesioned control slices, unless otherwise specified in the text. The average of raw values (percentage of cleaved caspase-3 or p75NTR-positive neurons of total neuronal number) for the unlesioned control slices from all the experiments conducted during this current study were as follows: cleaved caspase-3 positive, 18 ± 2% (CA3)/21 ± 2% (CA1); p75NTR-positive, 20 ± 1.5% (CA3)/20 ± 2% (CA1).
Results
Axotomized central neurons become dependent on endogenous BDNF
Survival of axotomized mature neurons is reduced upon withdrawal of endogenous BDNF, while intact mature neurons do not require BDNF for survival (Shulga et al., 2008). We used an ex vivo axotomy model in which an organotypic hippocampal slice is lesioned between CA3 and CA1 regions (Fig. 1a). We applied TrkB-Fc to withdraw endogenous BDNF. To assess the level of neuronal cell death, we performed double immunostaining for cleaved caspase-3 [a marker for cells committed to die through apoptosis (Logue and Martin, 2008)] and NeuN (a pan-neuronal marker) 24 h after the axotomy (Fig. 1b), and quantified the ratio of cleaved caspase-3-positive neurons to total NeuN-positive neuronal number. The lesion itself did not increase the number of apoptotic neurons (Fig. 1c). TrkB-Fc had no effect on the unlesioned slices; however, in the lesioned slices, it significantly increased the number of apoptotic neurons (Fig. 1c). Thus, decrease of endogenous BDNF after axotomy triggers the activation of apoptotic caspase-3. This indicates that hippocampal neurons become dependent on BDNF for survival upon trauma.
Emergence of posttraumatic BDNF requirement depends on p75NTR function
We next asked what mediates the posttraumatic dependency on BDNF. p75NTR is known to be upregulated after various kinds of injury (Chao, 2003), compete with Trk receptor signaling, and enhance the affinity and specificity of Trk receptors to their cognate ligands (Huang and Reichardt, 2001; Chao, 2003). To test whether p75NTR is involved in the generation of dependency on BDNF after axotomy, we blocked p75NTR with the function-blocking antibody recognizing the extracellular portion of p75NTR (AB1554). This antibody completely prevented the significant increase in cleaved caspase-3-positive neuronal number upon TrkB-Fc application to the lesioned slices (Fig. 2a,b). The effect was corroborated by the previously characterized Rex antibody known to inhibit p75NTR function (Weskamp and Reichardt, 1991) (Fig. 2b). Thus, when p75NTR was blocked, BDNF deprivation did not cause death of the axotomized neurons.
Figure 2.
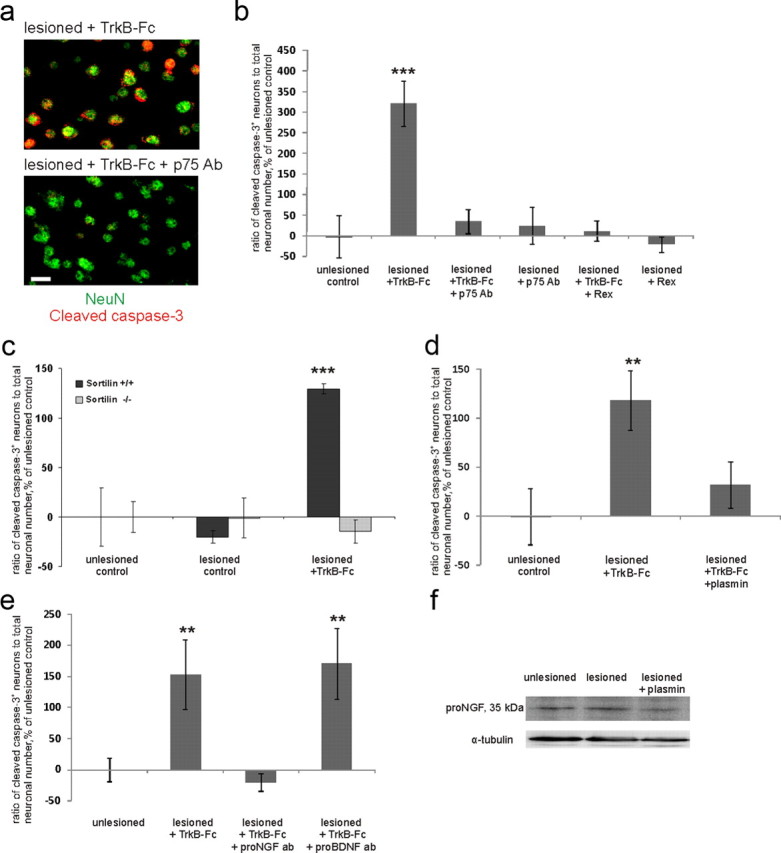
Posttraumatic emergence of the requirement for BDNF is caused by p75NTR. a, Images of axotomized TrkB-Fc-treated neurons with and without p75NTR-blocking antibodies obtained similarly to those in Figure 1b. Scale bar, 10 μm. b, Ratio of cleaved caspase-3-positive (=apoptotic) neuronal number to total neuronal number obtained by blind stereological counting, percentage of unlesioned control ratio value. Treatment with two different function-blocking antibodies directed against the extracellular domain of p75NTR [p75 Ab; AB1554; Millipore; and Rex (Weskamp and Reichardt, 1991)] abolished the increase in the number of apoptotic neurons promoted by TrkB-Fc. n = 46 slices/4750neurons; ***p < 0.001, one-way ANOVA and Bonferroni's post hoc analyses. c, TrkB-Fc treatment did not induce caspase activation in sortilin knock-out mice. Quantification was performed as in b; n = 48 slices/4600 neurons; ***p < 0.001, one-way ANOVA and Bonferroni's post hoc analyses. d, Treatment with plasmin abolished the TrkB-Fc-induced increase in the number of apoptotic neurons. Quantification was performed as in b; n = 33 slices/3350 neurons; **p = 0.017, one-way ANOVA and Bonferroni's post hoc analyses. e, Treatment with antibodies toward proNGF completely prevented TrkB-Fc-induced death induction, whereas antibodies toward proBDNF had no effect. n = 41 slices/3420 neurons; **p = 0.05, one-way ANOVA and Bonferroni's post hoc analyses. f, Western blot analyses with the antibody to proNGF. proNGF expression was upregulated by lesion and diminished by plasmin. n = 3 experiments/36 slices; **p = 0.041, one-way ANOVA and Bonferroni's post hoc analyses. For full image of the representative blot, see Figure 3a. Error bars indicate SEM.
To further confirm the role of p75NTR in the apoptotic death of the neurons in our model, we turned to sortilin-deficient mice, as proneurotrophins elicit cell death through p75NTR only upon its association with sortilin coreceptor (Willnow et al., 2008). We applied TrkB-Fc to the lesioned organotypic slices prepared from sortilin−/− and wild-type mice of the same strain. In the wild-type slices, TrkB-Fc application again robustly increased the number of cleaved caspase-3-positive neurons, whereas in the sortilin−/− slices there was no effect of TrkB-Fc (Fig. 2c). In conclusion, the presence of functionally active p75NTR and sortilin is needed for the posttraumatic dependency of the neurons on BDNF.
It is known that p75NTR can promote apoptosis upon proneurotrophin binding (Chao, 2003); however, it is not clear to which extent neurotrophins are released in pro- form under physiological or pathological conditions (Matsumoto et al., 2008; Yang et al., 2009). To test whether endogenous proneurotrophins might activate signaling through p75NTR, we aimed to decrease their concentration by application of plasmin, which cleaves extracellular proBDNF and proNGF to their mature forms (Lee et al., 2001). Plasmin significantly diminished the number of apoptotic neurons in TrkB-Fc-treated slices (Fig. 2d). Although plasmin has several targets, one possibility is that enhanced conversion of proneurotrophins to mature neurotrophins blocks the activation of the apoptotic cascade produced by the scavenging of endogenous BDNF. To test this idea, we applied blocking antibodies toward proNGF and proBDNF to lesioned slices treated with TrkB-Fc. Verification of the functional blocking effect of these antibodies was performed on basal forebrain primary cultures (Fig. 3). Here, we obtained similar results as previously shown by Volosin et al. (2006). Strikingly, the use of proNGF antibodies on hippocampal organotypic cultures completely abolished TrkB-Fc-induced death, while proBDNF antibodies had no effect (Fig. 2e). This result is in line with the previous data on the role of proNGF as an activating ligand for p75NTR in the in vivo axotomy model (Harrington et al., 2004). To verify that proNGF is present in the slices, we performed Western blot analyses with the same antibodies to proNGF that were used for functional blockage (Figs. 2f, 3). ProNGF expression was increased in the lesioned slices (119 ± 5% of unlesioned control). As expected, proNGF content was decreased by plasmin (Lee et al., 2001) (90 ± 5% of unlesioned control; Fig. 2f), which is in line with the antiapoptotic effect of this agent (Fig. 2d).
Figure 3.
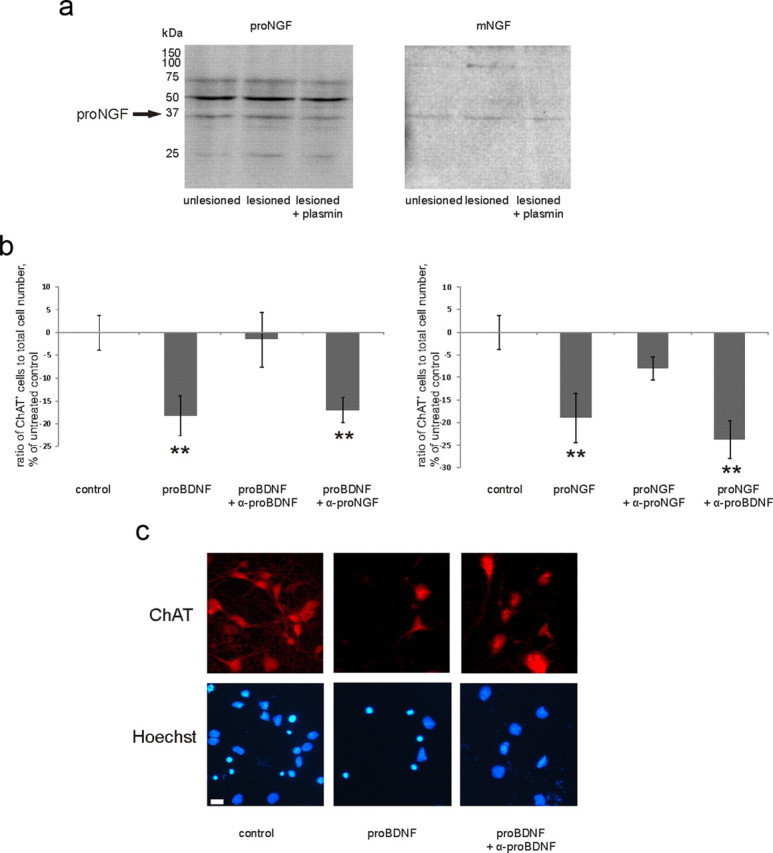
a, Representative Western blot with antibody to proNGF (left), reblotted with the antibody to mature NGF (right). Band row at 35 kDa is recognized by both antibodies, which verifies it as proNGF. For quantification, see Figure 2f. b, To verify the function-blocking specificity of antibodies to proNGF and proBDNF, we applied proNGF and proBDNF in the absence or presence of antibodies to basal forebrain primary cultures (for details, see Materials and Methods). Each antibody specifically diminished the reduction in the number of ChAT-positive neurons induced by its cognate proneurotrophin, not having an effect on the response to another proneurotrophin. **p = from left to right: 0.027, 0.046, 0.015, 0.001, one-way ANOVA and Bonferroni's post hoc analyses; n = 5670 cells. Scale bar, 10 μm. c, Representative pictures from the experiment described in b. Error bars indicate SEM.
In conclusion, proNGF as well as p75NTR and sortilin are essential for the induction of posttraumatic dependency on BDNF trophic support, since elimination of any of these factors renders injured neurons nonresponsive to endogenous BDNF withdrawal.
Specific inhibition of NKCC1 with bumetanide prevents BDNF withdrawal-induced death and posttraumatic upregulation of p75NTR
Postaxotomy induction of dependency on BDNF can be prevented by the NKCC1-specific (Blaesse et al., 2009) inhibitor bumetanide, which prevents posttraumatic GABA-induced intracellular Ca2+ increase (Shulga et al., 2008). The mechanism behind nonresponsiveness to BDNF withdrawal in bumetanide-treated neurons is an open question. Here, we tested whether neuronal death is prevented by bumetanide in BDNF-deprived injured neurons. As expected, bumetanide application prevented the increase in the number of apoptotic neurons in TrkB-Fc-treated lesioned slices (Fig. 4a).
Figure 4.

The NKCC1-blocking drug bumetanide prevents the posttraumatic emergence of requirement for BDNF and blocks the postaxotomy upregulation of p75NTR. a, Bumetanide abolishes the posttraumatic cell death induction in neurons deprived of BDNF by TrkB-Fc application. n = 33 slices/3350 neurons; **p = 0.021, one-way ANOVA and Bonferroni's post hoc analyses. b, Quantification of real-time PCR analyses for p75NTR mRNA, percentage of unlesioned control, at 1, 2, and 6 h after axotomy. At 6 h after axotomy, p75NTR mRNA was upregulated, but not in the presence of bumetanide. n = 72 slices; ***p < 0.001, one-way ANOVA and Bonferroni's post hoc analyses. c, Fragments of representative confocal images from the CA3 region of unlesioned, lesioned, and lesioned bumetanide-treated slices stained immunohistochemically for NeuN (blue) and p75NTR (red). Scale bar, 10 μm. d, Ratio of p75NTR-positive neuronal number to total neuronal number, obtained by blind stereological counting in the CA3 region, percentage of unlesioned control ratio value. The amount of p75NTR-positive neurons was increased after axotomy with and without application of TrkB-Fc; the increase is fully prevented by bumetanide. n = 53 slices/5800 neurons. **p = 0.05 (lesioned control)/0.028 (lesioned plus TrkB-Fc), one-way ANOVA and Bonferroni's post hoc analyses. e, p75NTR is not upregulated in the lesioned slices from NKCC1 knock-out mice. Data were obtained as in d; n = 37 slices/3540 neurons. **p = 0.02, one-way ANOVA and Bonferroni's post hoc analyses. Error bars indicate SEM.
Since interfering with p75NTR function and applying bumetanide had the same effect, we next asked whether bumetanide affects p75NTR expression. We measured p75NTR mRNA expression from the whole slice. There was no significant difference in p75NTR mRNA levels at 1–2 h (Fig. 4b), but 6 h after axotomy p75NTR mRNA levels were significantly increased (Fig. 4b; 136 ± 1%). Strikingly, this increase was not present in the bumetanide-treated slices (Fig. 4b; 85 ± 1%).
To monitor the neuronal, region-specific expression of p75NTR, we performed immunohistochemical stainings with an antibody targeted against the extracellular portion of p75NTR, which inhibits p75NTR function (AB1554; see above). The specificity of this antibody has been previously verified by the lack of immunostaining in basal forebrain neurons of p75−/− animals (Gehler et al., 2004; Dhanoa et al., 2006) (Fig. 5a). We made high-magnification confocal image stacks and counted the ratio of p75NTR-positive neurons to total neuronal number from 3D reconstruction images (Fig. 4c) (see Materials and Methods). There was a significant increase in the number of p75NTR-positive neurons after the lesion both in the absence and in the presence of TrkB-Fc (Fig. 4d). However, there was a significant increase in apoptotic neuronal number [as indicated by cleaved caspase-6 (Wang et al., 2001; Troy et al., 2002) colocalization with neuronal nuclei] in the pool of p75NTR-positive neurons only in the presence of TrkB-Fc (Fig. 5b,c).
Figure 5.
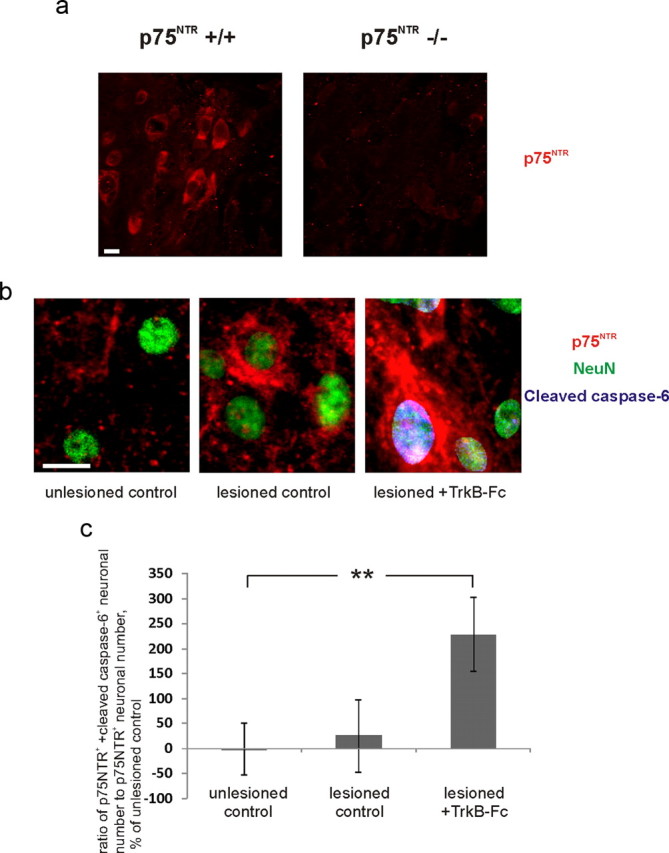
Increase in caspase-6/p75NTR-positive neurons in the presence of Trkb-Fc after axotomy. a, Immunostainings of the basal forebrain showing the specificity of immunohistochemical staining with the antibody targeted against the extracellular portion of p75NTR AB1554 in wild type but not in p75NTR−/− animals. Scale bar, 10 μm. b, Fragments of representative confocal images from unlesioned, lesioned, and lesioned TrkB-Fc-treated hippocampal organotypic slices stained immunohistochemically for NeuN (green), p75NTR (red), and cleaved caspase-6 (blue). Scale bar, 10 μm. c, Ratio of p75NTR plus cleaved caspase-6-positive neuronal number to p75NTR-positive neuronal number, obtained by stereological counting in the CA3 region, percentage of unlesioned control ratio value. Cleaved caspase-6 channel was masked with NeuN channel (see Materials and Methods). n = 12 slices/87 neurons; **p = 0.05, two-tailed t test versus unlesioned control. Error bars indicate SEM.
Strikingly, bumetanide treatment completely abolished the increase in the number of p75NTR-positive neurons (Fig. 4d). Also, genetic deletion of NKCC1 fully prevented posttraumatic upregulation of p75NTR (Fig. 4e). Bumetanide did not have a significant effect on p75NTR expression in the unlesioned slices (Fig. 4d).
We also measured p75NTR expression by Western blot analyses from whole slice homogenates with an antibody to the extracellular portion of p75NTR (Huber and Chao, 1995) (9651) and observed postaxotomy upregulation of p75NTR (136 ± 2% of unlesioned control), which was prevented by bumetanide (lesioned plus bumetanide: 102 ± 11% of unlesioned control; Fig. 6a). Thus, inhibition of NKCC1-dependent chloride accumulation blocked the lesion-induced increase in p75NTR expression and death produced by the reduction of endogenous BDNF.
Figure 6.

a, Western blot analyses with antibody 9651 (Huber and Chao, 1995) from whole-slice homogenates show postaxotomy upregulation of p75NTR, which is prevented by bumetanide. p75NTR is normalized to α-tubulin. n = 3 experiments/36 slices; **p = 0.028, one-way ANOVA and Bonferroni's post hoc analyses. b, pAkt/Akt ratio is decreased in lesioned TrkB-Fc-treated slices, whereas treatment with antibody to p75NTR or bumetanide returns it to the level of the unlesioned control. Phospho-Akt is normalized to pan-Akt; n = 3 experiments/60 slices; **p = 0.007 by two-tailed t test versus unlesioned control. Additional bands in lesioned plus p75Ab-treated sample result from the staining of rabbit-anti p75Ab used for function blocking with secondary anti-rabbit antibody. Error bars indicate SEM.
Increased cell death is reflected in the decrease of survival signaling. p75NTR signaling is known to interfere with PI-3 kinase survival pathway downstream Trk receptors in cell lines (Huang and Reichardt, 2001). To investigate whether interfering with p75NTR function or applying bumetanide enhances survival signaling in neurons, we measured phosphorylation of Akt (also known as PKB), a major survival-promoting kinase in the neurons (Huang and Reichardt, 2001), from the whole-slice homogenates. Akt phosphorylation was significantly reduced in TrkB-Fc-treated lesioned slices; treatment with either blocking antibody to p75NTR or bumetanide prevented this decrease (Fig. 6b). There was no significant change in Akt expression (lesioned control: 91 ± 4%; lesioned plus TrkB-Fc: 106 ± 12%; lesioned plus TrkB-Fc plus bumetanide: 113 ± 26%; lesioned plus TrkB-Fc plus p75NTR Ab: 135 ± 27%; n = 60 slices; pan-Akt normalized to α-tubulin; here and elsewhere in the text, the data are presented as percentage of unlesioned control slices ratio value, unless otherwise specified). ProNGF has been shown to prevent the ability of BDNF to induce phosphorylation of Akt (Volosin et al., 2006) through phosphatase PTEN (phosphatase and tensin homolog deleted on chromosome 10) (Song et al., 2010). Thus, it is possible that, due to the low concentration of TrkB-Fc used in these experiments, the level of endogenous BDNF, which is lowered dramatically by TrkB-Fc, becomes nonetheless sufficient to maintain the phosphorylation of Akt in the absence of the opposing signaling of proNGF through p75NTR.
Posttraumatic depolarizing shift in GABAA-mediated responses occurs shortly after injury
To determine whether a change in GABAA-mediated responses is established at an early stage after injury, we monitored [Ca2+]i evoked by the GABAA-specific agonist muscimol in the CA3 region of organotypic cultures 3–4 h after injury (Fig. 7). A larger proportion of monitored neurons responded with [Ca2+]i increase to muscimol in injured slices than in uninjured controls (100 μm; unlesioned control: 3 ± 1% of total recorded neurons; n = 172 pyramidal cells; lesioned: 61 ± 9% of total recorded neurons; n = 269 pyramidal cells). This effect was completely inhibited by blocking intracellular chloride accumulation with the NKCC1 inhibitor bumetanide (10 μm; lesioned: 1 ± 0.3% of total recorded neurons; n = 244 pyramidal cells). All monitored cells responded to glutamate (100 μm). These results are consistent with our previous finding (Shulga et al., 2008). The presence of depolarizing GABAA-mediated responses as soon as 3 h after axotomy reinforces the causal relationship between this event and the upregulation of p75NTR.
Figure 7.

GABAA-mediated responses become depolarizing shortly after axotomy. [Ca2+]i was monitored with Fluo-4 in hippocampal organotypic cultures. a, Loading of Fluo-4 in the CA3 of an axotomized organotypic culture. Scale bar, 250 μm. b–d, Representative recordings from unlesioned and lesioned slices. Muscimol could evoke [Ca2+]i increase in lesioned (c) but not in unlesioned (b) slices already 3 h after axotomy. Application of NKCC1 inhibitor bumetanide to the perfusion was able to block this effect (d). Glutamate application was used to ensure the viability of the slice (b–d). n = 3–4 slices per experimental group.
We found no changes in NKCC1 (111 ± 24% of unlesioned control; n = 3 experiments/24 slices; p > 0.1 by two-tailed t test) and in KCC2 protein expression (126 ± 17% of unlesioned control; n = 3 experiments/24 slices; p > 0.1 by two-tailed t test) 4 h after axotomy by Western blot analyses (Fig. 8a,b). KCC2 and NKCC1 are regulated also at the functional level (e.g., the proportion of membrane-bound transporters and/or the change in the intrinsic rate of ion transport can be affected). Recent evidence shows that phosphorylation at tyrosine residues of the KCC2 protein decreases membrane stability, which results in reduced chloride extrusion efficiency (Lee et al., 2010). Intriguingly, we observed a significant increase in KCC2 phosphorylation 4 h after axotomy (Fig. 8c,d). Thus, the present data indicate that an early posttraumatic reduction in functional KCC2 expression could be crucially involved in the changes in GABAA-mediated responses.
Figure 8.

Western blot analyses from the whole slices 4 h after axotomy. No change in protein expression of KCC2 (a) or NKCC1 (b) was observed. c, Representative Western blot from lysates immunoprecipitated with anti-KCC2 antibody, stained for phosphotyrosine and KCC2. d, Ratio of tyrosine-phosphorylated KCC2 to total KCC2, percentage of unlesioned control. n = 3 experiments/96 slices; **p = 0.05, two-tailed t test. Error bars indicate SEM.
Posttraumatic upregulation of p75NTR depends on membrane depolarization and activation of voltage-gated Ca2+ channels
The results above show that, after trauma, bumetanide can block GABAA agonist-induced increase in [Ca2+]i (Shulga et al., 2008), which is mediated by GABA-induced depolarization and consequent opening of voltage-gated Ca2+ channels (Blaesse et al., 2009). We tested whether intracellular Ca2+ and activation of voltage-gated Ca2+ channels are important for the posttraumatic p75NTR upregulation. We chelated intracellular Ca2+ by application of BAPTA-AM (100 μm). This treatment completely abolished the posttraumatic increase in the number of p75NTR-positive neurons (Fig. 9a). We next blocked voltage-gated Ca2+ channels with Ni2+/Cd2+ (100 μm), which also completely abolished posttraumatic increase in p75NTR (Fig. 9a). Inhibition of L-type voltage-gated Ca2+ channels with nimodipine (20 μm) gave a similar outcome (lesioned: 356 ± 102%; lesioned plus nimodipine: 94 ± 34% of unlesioned control; n = 21 slices; p = 0.05 by one-way ANOVA and Bonferroni's post hoc analyses). These results pinpoint L-type Ca2+ channel activation as an important step in the mechanism of trauma-induced p75NTR upregulation.
Figure 9.
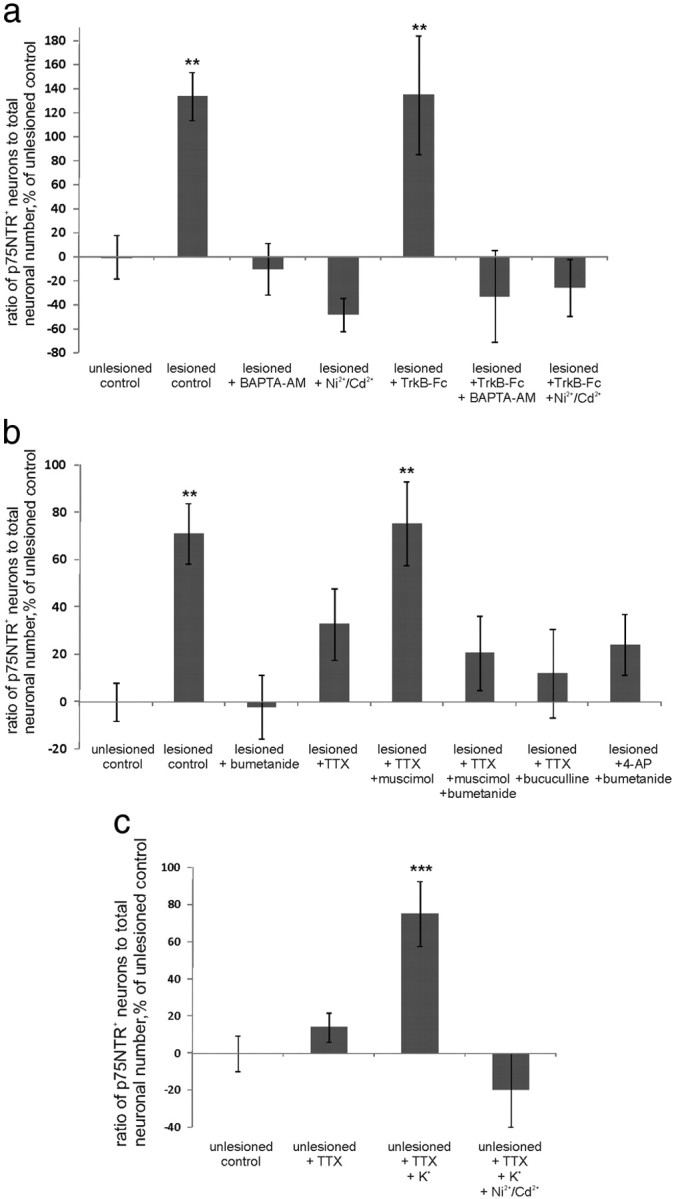
p75NTR is upregulated by a mechanism requiring membrane depolarization and opening of voltage-gated Ca2+-channels. a–c, Data obtained as in Figure 4d. a, Application of BAPTA-AM (intracellular calcium chelator) as well as of Ni2+/Cd2+ (blocker of voltage-gated calcium channels) prevented posttraumatic upregulation of p75NTR. n = 29 slices/3290 neurons; **p = 0.041 (lesioned control)/0.039 (lesioned plus TrkB-Fc), one-way ANOVA and Bonferroni's post hoc analyses. b, Application of voltage-gated Na+ channel blocker TTX partly reduced the upregulation of p75NTR. Adding GABAA agonist muscimol in the presence of TTX restored the level of p75NTR to the level of lesioned control; the effect of muscimol could be abolished by bumetanide. Adding GABAA antagonist bicuculline in the presence of TTX completely abolished the increase in p75NTR. Applying K+ channel blocker 4-AP in the presence of bumetanide did not result in the significant increase in the number of p75NTR-positive neurons; n = 128 slices/13,600 neurons; **p = 0.037 (lesioned control)/0.019 (lesioned plus TTX plus muscimol), one-way ANOVA and Bonferroni's post hoc analyses. c, Application of KCl and TTX to induce membrane depolarization in the absence of action potential firing upregulated p75NTR in nonaxotomized slices; the effect could be blocked by Ni2+/Cd2+; n = 48 slices/5490 neurons; ***p = 0.001, one-way ANOVA and Bonferroni's post hoc analyses. Error bars indicate SEM.
GABA-mediated depolarization is able to upregulate p75NTR in the absence of action potential firing
GABAA-mediated depolarization in axotomized neurons could increase the probability of action potential firing. This in turn could increase Ca2+-regulated vesicular release of GABA (Farrant and Kaila, 2007). Consequently, the effect of bumetanide on p75NTR could be mediated not only through direct prevention of depolarization-induced activation of voltage-gated Ca2+ channels but also through a reduction in excitability, neuronal activity, and GABA release. To discriminate between these possibilities, we looked at p75NTR expression after application of voltage-gated Na+ channel blocker TTX (2 μm), GABAA agonist muscimol (50 μm), and voltage-gated K+ channel blocker 4-AP (100 μm). To test whether GABAA-mediated depolarization can upregulate p75NTR in the absence of action potential firing, we applied TTX to the lesioned slices. Although TTX reduced the amount of p75NTR-positive neurons to a level that did not differ significantly from the unlesioned control values, it did not, unlike bumetanide, fully block the posttraumatic p75NTR upregulation (Fig. 9b). Increasing the concentration of TTX from 2 to 10 μm did not result in the increase of TTX efficiency (lesioned control, 216 ± 8%; lesioned plus 10 μm TTX, 157 ± 50%; n = 12 slices/1490 neurons). Thus, reduction in activity does not fully account for the effect of bumetanide. TTX could reduce Ca2+-regulated release of GABA (Farrant and Kaila, 2007); to address the question whether this could explain the TTX-induced reduction in the amount of p75NTR-positive neurons, we added muscimol to compensate for this effect. In the lesioned TTX-treated slices, muscimol increased the number of p75NTR-positive neurons to the level of the lesioned control values, and the effect of muscimol could be blocked with bumetanide. On the contrary, addition of the GABAA antagonist bicuculline (20 μm) in the presence of TTX completely abolished lesioned-induced increase in p75NTR expression, further verifying the idea that GABAA receptors are crucial for posttraumatic p75NTR upregulation, and that incomplete blockade of this upregulation by TTX results from the incomplete blockade of GABA release (TTX does not block the spontaneous release of GABA) (Fig. 9b). Thus, GABAA-mediated depolarization upregulates p75NTR in the absence of neuronal activity. To enhance neuronal activity in the absence of GABA-mediated depolarization, we applied 4-AP in the presence of bumetanide. This did not result in a significant increase in the amount of p75NTR-positive neurons (Fig. 9b).
We also tested whether p75NTR upregulation can be induced in nonaxotomized slices by conditions mimicking posttraumatic GABA-mediated depolarization in the absence of enhanced activity. Slices were subjected to prolonged exposure (24 h) to high potassium (10 mm) in the presence of TTX. A modest increase in extracellular K+ reverses KCC2-mediated K+–Cl+ transport leading to accumulation of intracellular chloride (Farrant and Kaila, 2007). TTX was added 30 min and 15 min before potassium chloride (KCl) on top of the slice and after that added to the medium together with KCl. The treatment resulted in a significant increase in the number of p75NTR-positive neurons, the magnitude of the effect being comparable with the effect of the lesion. The effect of KCl could be blocked by Ni2+/Cd2+, suggesting that membrane depolarization upregulates p75NTR through opening of voltage-gated Ca2+ channels (Fig. 9c).
Posttraumatic [Ca2+]i increase activates Rho kinase to upregulate p75NTR
After spinal cord injury, there is an increase in the activation of the small GTPase Rho. Interestingly, Rho antagonist halts the posttraumatic increase in p75NTR (Dubreuil et al., 2003). In non-neuronal cells, the activation of Rho and its downstream effector Rho kinase (ROCK) is induced by depolarization-induced activation of voltage-gated Ca2+ channels (Sakurada et al., 2003; Liu et al., 2005); RhoA is also activated downstream the complex of p75NTR with NgR and LINGO1 receptors in neurons (McKerracher and Higuchi, 2006). To test whether ROCK has a role in the posttraumatic p75NTR upregulation in the present injury model, we applied the ROCK inhibitor Y-27632. Strikingly, this treatment completely abolished the rise in the amount of p75NTR-positive neurons after axotomy both in the presence and in the absence of TrkB-Fc (Fig. 10a). ROCK inhibitor also prevented the KCl-induced upregulation of p75NTR (mediated by opening of voltage-gated Ca2+ channels; see above) in intact CA3 neurons (Fig. 10a).
Figure 10.
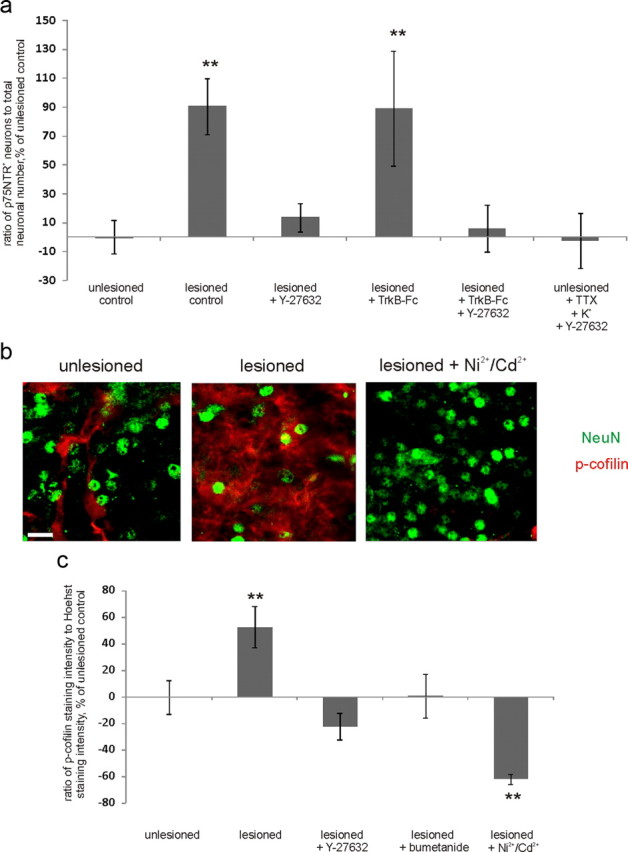
Posttraumatic [Ca2+]i increase induces ROCK activation to increase p75NTR expression. a, ROCK inhibitor Y-27632 completely abolished posttraumatic p75NTR upregulation in the presence and in the absence of TrkB-Fc, as well as KCl-induced upregulation of p75NTR; n = 58 slices/5750 neurons; **p = 0.006 (lesioned control) and 0.024 (lesioned plus TrkB-Fc), one-way ANOVA and Bonferroni's post hoc analyses. b, Fragments of representative confocal images from the CA3 region of unlesioned, lesioned, and lesioned Ni2+/Cd2+-treated slices stained immunohistochemically for NeuN (green) and p-cofilin (red). Scale bar, 10 μm. c, Quantification of p-cofilin staining intensity measured from the middle of the CA3 region, normalized to Hoechst staining intensity. P-cofilin staining intensity is induced by lesion, but not in the presence of Y-27632, bumetanide, or Ni2+/Cd2+; n = 46 slices; **p = 0.024 (lesioned control)/0.003 (lesioned plus Ni2+/Cd2+), one-way ANOVA and Bonferroni's post hoc analyses. Error bars indicate SEM.
To elucidate whether ROCK is acting downstream or upstream of the posttraumatic [Ca2+]i increase, we monitored [Ca2+]i at 3–4 h after axotomy (when GABAA agonist already is able to induce [Ca2+]i increase; see above), in the presence of Y-27632, which was applied at the same time with the lesion. We observed no reduction in the ability of muscimol to induce [Ca2+]i increase after axotomy by Y-27632 (65.3 ± 7.2% of total recorded neurons; n = 256 pyramidal cells) compared with lesioned control slices (see above). Thus, we concluded that ROCK is acting downstream of the [Ca2+]i increase.
To further verify this idea, as well as to test whether ROCK can be activated by Ca2+ not only in non-neuronal cells (Sakurada et al., 2003; Liu et al., 2005) but also in neural tissue, we used phospho-cofilin as a marker of ROCK activation (Maekawa et al., 1999). We normalized total p-cofilin immunostaining intensity measured from the middle of the CA3 region to total number of cells in the same region (Fig. 10b,c). We observed a significant increase in total p-cofilin staining intensity after axotomy. This increase was not present upon application of Y-27632, which verifies that axotomy induces an increase in ROCK activation. Notably, posttraumatic increase in cofilin phosphorylation was also prevented by bumetanide and Ni2+/Cd2+, indicating the crucial role of posttraumatic GABA-mediated depolarization and [Ca2+]i increase through voltage-gated Ca2+ channels in the activation of ROCK. Thus, GABAA-induced [Ca2+]i increase leads to upregulation of p75NTR through the activation of Rho kinase.
Denervation and axotomy-induced cell death share similar mechanisms
While in the ex vivo lesion model the CA3 region is axotomized, the CA1 region is subjected to denervation. To investigate the cell death mechanism in the denervated neurons, we quantified the CA1 region of slices from the key experiments described above. The data are summarized in Figure 11. Interestingly, in the CA1 region there was a significant increase in the number of p75NTR-positive neurons after axotomy only in the presence of TrkB-Fc (Fig. 11e); there was also no activation of ROCK in the CA1 region of the lesioned slices in the absence of TrkB-Fc (as indicated by p-cofilin staining intensity normalized to Hoechst staining intensity; 89 ± 23% of unlesioned control; n = 16 slices).
Figure 11.

Cell death in denervated neurons in the CA1 region is induced by similar mechanism as in the axotomized neurons. a–d, Ratio of cleaved caspase-3-positive neuronal number to total neuronal number obtained by blind stereological counting in the CA1 region, percentage of unlesioned control ratio value. a, TrkB-Fc induces a significant increase in apoptotic neuronal number in the lesioned, but not unlesioned, slices at 1 d after the injury; n = 43 slices/4800 neurons; **p = 0.013, one-way ANOVA and Bonferroni's post hoc analyses. b, Treatment with function-blocking antibodies to p75NTR (Fig. 2b) abolished the increase in the number of apoptotic neurons promoted by TrkB-Fc; n = 46 slices/4890 neurons; **p = 0.002, one-way ANOVA and Bonferroni's post hoc analyses. c, Treatment with plasmin abolished the TrkB-Fc-induced increase in the number of apoptotic neurons; n = 33 slices/3990 neurons; **p = 0.004, one-way ANOVA and Bonferroni's post hoc analyses. d, Bumetanide abolishes the death induction in neurons deprived of BDNF; n = 33 slices/3750 neurons; **p = 0.007, one-way ANOVA and Bonferroni's post hoc analyses. e, f, Ratio of p75NTR-positive neuronal number to total neuronal number, obtained by blind stereological counting in the CA1 region, percentage of unlesioned control ratio value. e, The amount of p75NTR-positive neurons was increased after axotomy only in the presence of TrkB-Fc; the increase was fully blocked by bumetanide; n = 56 slices/7800 neurons; **p = 0.032, one-way ANOVA and Bonferroni's post hoc analyses. f, ROCK inhibitor Y-27632 completely abolished posttraumatic p75NTR upregulation in the presence of TrkB-Fc; n = 58 slices/5840 neurons; **p = 0.002 (lesioned plus TrkB-Fc), one-way ANOVA and Bonferroni's post hoc analyses. Error bars indicate SEM.
Posttraumatic upregulation of p75NTR and cell death induction is blocked by bumetanide in vivo
Finally, we tested whether the proposed mechanism of p75NTR upregulation is relevant in vivo. We used a model in which spinal motoneurons are axotomized at the level of the ventral root (Risling et al., 1992) (Fig. 12a) (see also Materials and Methods). Strikingly, this treatment resulted in the profound upregulation of p75NTR in injured motoneurons, which was blocked by intraperitoneal administration of bumetanide (30 mg/kg). Results of the quantification of p75NTR-positive neurons normalized to total neuronal number are presented in Figure 12, b and c. Results without normalization are as follows: lesioned control, 928 ± 141%; lesioned plus bumetanide, 222 ± 41% of unlesioned control (quantified as in Fig. 12c; p = 0.002 by ANOVA and Bonferroni's post hoc analysis). We also observed a significant increase in the amount of cleaved caspase-3-positive neurons in the spinal cords of lesioned control rats, which was blocked by bumetanide administration (Fig. 12e,f). We have also quantified the number of NeuN-positive motoneurons as follows (see Materials and Methods): unlesioned, 108 ± 6 neurons; lesioned, 28 ± 7 neurons; lesioned plus bumetanide, 67 ± 11 neurons in 4 sections (p = 0.005 by one-way ANOVA). KCC2 immunostaining intensity was significantly reduced in injured neurons (Nabekura et al., 2002) (80 ± 3% of unlesioned control; Fig. 12d; identified by the same criteria as p75NTR-positive neurons) (see Materials and Methods). Thus, blockade of NKCC1 prevents posttraumatic upregulation of p75NTR and cell death in vivo.
Figure 12.
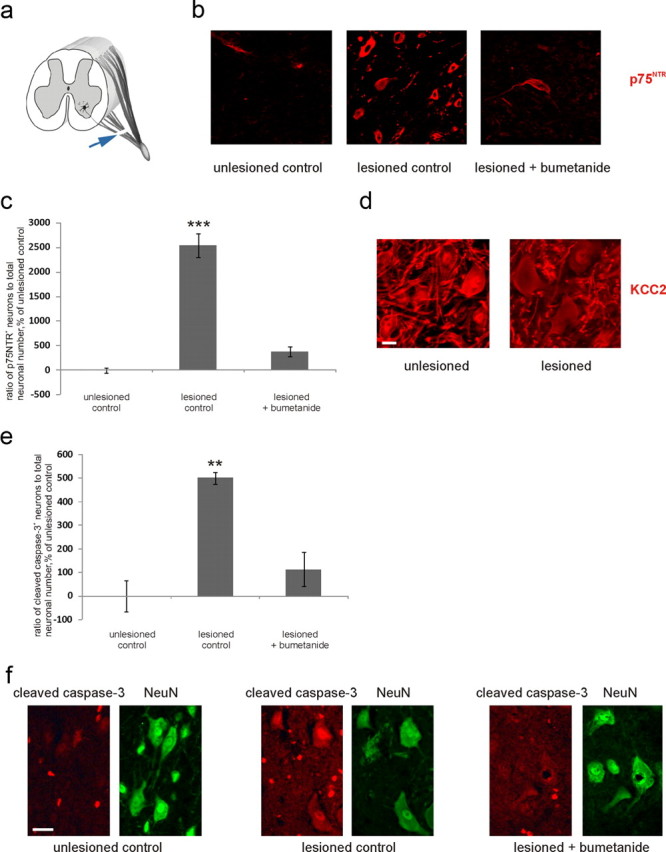
Bumetanide prevents upregulation of p75NTR and cell death induction after in vivo ventral root axotomy. a, Schematic representation of the injury. Axotomy of spinal motoneurons was performed at the ventral root between the spinal cord and the junction with the dorsal root (for details, see Materials and Methods). Arrow, Lesion site. b, Representative images from the sections of the spinal cords of unlesioned, lesioned (by ventral root axotomy), and lesioned bumetanide-treated rats at the level of the injury stained immunohistochemically for p75NTR. Scale bar, 30 μm. c, Quantification of p75NTR-positive neurons from the similar sections as in b. Total amount of p75NTR-positive neurons from four sections of the spinal cord and total amount of neurons (NeuN-defined) from four sections consecutive to each p75NTR-stained section was counted by blind stereological counting from ventral horns (for selection criteria, see Materials and Methods) and the p75NTR/NeuN ratio for each animal quantified. Data are presented as percentage of unlesioned control ratio. n = 3 unlesioned, 2 lesioned, 3 lesioned bumetanide-treated rats. ***p < 0.001, one-way ANOVA and Bonferroni's post hoc analyses. d, Representative images of sections as in b stained immunohistochemically for KCC2. Scale bar, 15 μm. KCC2 immunostaining intensity, measured from images similar as in d, is reduced in neurons in the ventral horn of the lesioned animals. n = 18 sections/5 animals; **p = 0.004, one-way ANOVA and Bonferroni's post hoc analyses. e, Quantification of cleaved caspase-3-positive neurons performed as in c from similar sections as in f. NeuN was detected from the same sections as cleaved caspase-3; n = 3 unlesioned, 2 lesioned, 3 lesioned bumetanide-treated rats; **p = 0.012, one-way ANOVA and Bonferroni's post hoc analyses. f, Representative images from the sections of the spinal cords of unlesioned, lesioned, and lesioned bumetanide-treated rats at the level of the injury stained immunohistochemically for cleaved caspase-3 and NeuN. Scale bar, 30 μm. Error bars indicate SEM.
Discussion
p75NTR is upregulated under various pathological conditions, including seizures, ischemia, neurodegeneration, osmotic swelling, and mechanical damage, with levels of reexpression being comparable with levels seen during early development (Dechant and Barde, 2002; Chao, 2003; Peterson and Bogenmann, 2003). These insults are also followed by functional downregulation of KCC2 and the consequent accumulation of intracellular chloride through NKCC1 and depolarizing responses to GABA (Blaesse et al., 2009). We show here ex vivo and in a clinically relevant in vivo model (Risling et al., 1992; Carlstedt, 2008) that the axotomy-induced upregulation of p75NTR can be prevented by blocking GABAA-mediated depolarization or subsequent activation of voltage-gated Ca2+ channels. Therefore, our results strongly indicate that postaxotomy reexpression of p75NTR is induced by the shift in the polarity of GABA-mediated responses. Our data open the theoretical possibility that similar mechanism regulates p75NTR also in other pathological conditions such as seizures and neurodegeneration.
After trauma, GABAA-mediated [Ca2+]i increase induces the dependency on BDNF trophic support (Shulga et al., 2008). Endogenous BDNF is not required for the survival of intact adult neurons (Giehl et al., 2001; Li et al., 2008; Shulga et al., 2008; Rauskolb et al., 2010). However, there is a positive therapeutic effect of BDNF in injured adult neurons (Klöcker et al., 2000; Schäbitz et al., 2000; Shulga et al., 2008, 2009). The exact reason for this shift is not known. The results in this study indicate that upregulation of p75NTR is responsible for setting the dependency on BDNF for survival.
In the in vivo axotomy model in which corticospinal neurons are lesioned, neuronal death occurs without BDNF scavenger (Giehl et al., 2001; Harrington et al., 2004; Shulga et al., 2008). However, scavenging endogenous BDNF in these neurons results in further neuronal loss (Shulga et al., 2008), whereas application of exogenous BDNF completely prevents axotomy-induced neuronal loss and atrophy (Giehl and Tetzlaff, 1996). Thus, appearance of postaxotomy dependency on BDNF trophic support for survival, as well as shortage of endogenous BDNF and the resultant neuronal death, are phenomena that occur in vivo. Accordingly, in the corticospinal neuron lesion model (Giehl et al., 2001; Harrington et al., 2004; Shulga et al., 2008) and in ventral root axotomy model used in this work, neuronal death occurs most probably due to lack of endogenous BDNF in vivo. Blocking p75NTR with Rex antibody (Giehl et al., 2001) or using p75−/− (Harrington et al., 2004) or sortilin−/− animals (Jansen et al., 2007) prevents axotomy-induced neuronal death in corticospinal neurons. In the organotypic culture axotomy model, death of axotomized CA3 region neurons occurs only in the presence of TrkB-Fc (Shulga et al., 2008), most probably because endogenous BDNF that is highly expressed in the DG and CA3 regions (Conner et al., 1997) triggers survival signaling that overwhelms the death signaling from p75NTR. Thus, the ex vivo model used in this work allows monitoring the requirement for namely endogenous BDNF.
To elucidate the mechanism regulating p75NTR levels in more detail, we applied the voltage-gated Na+ channel blocker TTX and the voltage-gated K+ channel blocker 4-AP and observed that GABAA-mediated depolarization was able to increase the p75NTR expression in the absence of action potential firing, whereas enhancement of activity in the absence of GABAA-mediated depolarization did not have an effect on p75NTR levels. Interestingly, the inhibitor of ROCK, the downstream effector of small GTPase Rho, was as effective as bumetanide in preventing the posttraumatic upregulation of p75NTR. It has been shown that inhibiting Rho prevents upregulation of p75NTR after spinal cord injury (Dubreuil et al., 2003). We now show that the activation of one of its effectors ROCK is the mechanism coupling GABAA-mediated [Ca2+]i increase and the posttraumatic p75NTR upregulation. In non-neuronal cells, Rho–ROCK pathway has been shown to be activated through membrane depolarization-induced opening of voltage-gated Ca2+ channels (Sakurada et al., 2003; Liu et al., 2005), and here we show that ROCK can be activated by the same mechanism also in neural tissue.
The proposed mechanism of the emergence of neuronal dependency for BDNF trophic support after axotomy is summarized in Figure 13. The responses to axonal injury in the present ex vivo model strongly correlate to responses in several in vivo models studied in this and other works. Thus, this study justifies further preclinical and clinical studies on the therapeutic potential of bumetanide for injured neurons. According to our results, neuronal, neurotrophin-dependent survival after trauma can be profoundly affected by pharmacological agents acting on the GABAergic system and not on the neurotrophin system itself. This opens the possibility of developing more novel and well tolerated treatments than local supply of neurotrophic factors. On the basis of our results, bumetanide can be proposed as a potential treatment for the adult injured CNS. Notably, administration of GABAA agonists ethanol, benzodiazepines, and barbiturates to neonatal rats during the period of developmental synaptogenesis triggers widespread neuronal apoptosis (Ikonomidou et al., 2000): thus, in the future, understanding not only posttraumatic but also developmental coupling of GABAA-mediated depolarization and p75NTR expression could be of clinical significance.
Figure 13.

Proposed mechanism of the emergence of trauma-induced dependency on BDNF.
Footnotes
This work was supported by the Academy of Finland, Sigrid Juselius Foundation (E.C., U.A., C.R.), Helsinki Biomedical Graduate School and the Finnish Brain Foundation, (A.S.) and Biocentrum Helsinki (C.R.). We thank M. Palviainen and O. Nikkilä for excellent technical assistance, P. Uvarov for the help with primer design, S. Soni for language editing, and M. Kjølby for perfusing p75−/− and wt animals. We thank the Division of Pharmacology and Toxicology for providing the facilities for in vivo experiments.
References
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Carlstedt T. Root repair review: basic science background and clinical outcome. Restor Neurol Neurosci. 2008;26:225–241. [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75NTR: novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Dhanoa NK, Krol KM, Jahed A, Crutcher KA, Kawaja MD. Null mutations for exon III and exon IV of the p75 neurotrophin receptor gene enhance sympathetic sprouting in response to elevated levels of nerve growth factor in transgenic mice. Exp Neurol. 2006;198:416–426. doi: 10.1016/j.expneurol.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Gao X, Daugherty RL, Tourtellotte WG. Regulation of low affinity neurotrophin receptor (p75NTR) by early growth response (egr) transcriptional regulators. Mol Cell Neurosci. 2007;36:501–514. doi: 10.1016/j.mcn.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler S, Gallo G, Veien E, Letourneau PC. p75 neurotrophin receptor signaling regulates growth cone filopodial dynamics through modulating RhoA activity. J Neurosci. 2004;24:4363–4372. doi: 10.1523/JNEUROSCI.0404-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Giehl KM, Röhrig S, Bonatz H, Gutjahr M, Leiner B, Bartke I, Yan Q, Reichardt LF, Backus C, Welcher AA, Dethleffsen K, Mestres P, Meyer M. Endogenous brain-derived neurotrophic factor and neurotrophin-3 antagonistically regulate survival of axotomized corticospinal neurons in vivo. J Neurosci. 2001;21:3492–3502. doi: 10.1523/JNEUROSCI.21-10-03492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Mörl K, Meyer M, Hempstead BL, Yoon SO, Giehl KM. Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc Natl Acad Sci U S A. 2004;101:6226–6230. doi: 10.1073/pnas.0305755101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LJ, Chao MV. Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor gene occur by different mechanisms. Dev Biol. 1995;167:227–238. doi: 10.1006/dbio.1995.1019. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O, Sjoegaard SS, Breiderhoff T, Gotthardt M, Lin F, Eilers A, Petersen CM, Lewin GR, Hempstead BL, Willnow TE, Nykjaer A. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat Neurosci. 2007;10:1449–1457. doi: 10.1038/nn2000. [DOI] [PubMed] [Google Scholar]
- Klöcker N, Kermer P, Weishaupt JH, Labes M, Ankerhold R, Bähr M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B signaling. J Neurosci. 2000;20:6962–6967. doi: 10.1523/JNEUROSCI.20-18-06962.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zuo J, Pertens E, Helli PB, Janssen LJ. Regulation of Rho/ROCK signaling in airway smooth muscle by membrane potential and [Ca2+]i. Am J Physiol Lung Cell Mol Physiol. 2005;289:L574–L582. doi: 10.1152/ajplung.00134.2005. [DOI] [PubMed] [Google Scholar]
- Logue SE, Martin SJ. Caspase activation cascades in apoptosis. Biochem Soc Trans. 2008;36:1–9. doi: 10.1042/BST0360001. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- McKerracher L, Higuchi H. Targeting rho to stimulate repair after spinal cord injury. J Neurotrauma. 2006;23:309–317. doi: 10.1089/neu.2006.23.309. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace AJ, Lee E, Athirakul K, Coffman TM, O'Brien DA, Koller BH. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl− cotransporter. J Clin Invest. 2000;105:441–450. doi: 10.1172/JCI8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S, Bogenmann E. Osmotic swelling induces p75 neurotrophin receptor (p75NTR) expression via nitric oxide. J Biol Chem. 2003;278:33943–33950. doi: 10.1074/jbc.M302376200. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Stein V, Keating DJ, Maier H, Rinke I, Rudhard Y, Hentschke M, Rune GM, Jentsch TJ, Hübner CA. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A, Ho WC, Forte S, Dickson K, Boutilier J, Favell K, Barker PA. Hypo-osmolar stress induces p75NTR expression by activating Sp1-dependent transcription. J Neurosci. 2007;27:1498–1506. doi: 10.1523/JNEUROSCI.4806-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risling M, Fried K, Lindå H, Cullheim S, Meier M. Changes in nerve growth factor receptor-like immunoreactivity in the spinal cord after ventral funiculus lesion in adult cats. J Neurocytol. 1992;21:79–93. doi: 10.1007/BF01189007. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of rho and rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates bax and bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–2217. doi: 10.1161/01.str.31.9.2212. [DOI] [PubMed] [Google Scholar]
- Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, Yan Q, Kaila K, Saarma M, Rivera C, Giehl KM. Posttraumatic GABAA-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J Neurosci. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga A, Blaesse A, Kysenius K, Huttunen HJ, Tanhuanpää K, Saarma M, Rivera C. Thyroxin regulates BDNF expression to promote survival of injured neurons. Mol Cell Neurosci. 2009;42:408–418. doi: 10.1016/j.mcn.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ. ProNGF induces PTEN via p75NTR to suppress trk-mediated survival signaling in brain neurons. J Neurosci. 2010;30:15608–15615. doi: 10.1523/JNEUROSCI.2581-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci. 2002;5(Suppl):1046–1050. doi: 10.1038/nn938. [DOI] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem. 2002;277:34295–34302. doi: 10.1074/jbc.M205167200. [DOI] [PubMed] [Google Scholar]
- Underwood CK, Coulson EJ. The p75 neurotrophin receptor. Int J Biochem Cell Biol. 2008;40:1664–1668. doi: 10.1016/j.biocel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci. 2006;26:7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C. Characterization of a p75NTR apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–33820. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- Willems E, Mateizel I, Kemp C, Cauffman G, Sermon K, Leyns L. Selection of reference genes in mouse embryos and in differentiating human and mouse ES cells. Int J Dev Biol. 2006;50:627–635. doi: 10.1387/ijdb.052130ew. [DOI] [PubMed] [Google Scholar]
- Willnow TE, Petersen CM, Nykjaer A. VPS10P-domain receptors—regulators of neuronal viability and function. Nat Rev Neurosci. 2008;9:899–909. doi: 10.1038/nrn2516. [DOI] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]