

Abstract
Excitotoxicity resulting from excessive Ca2+ influx through glutamate receptors contributes to neuronal injury after stroke, trauma, and seizures. Increased cytosolic Ca2+ levels activate a family of calcium-dependent proteases with papain-like activity, the calpains. Here we investigated the role of calpain activation during NMDA-induced excitotoxic injury in embryonic (E16–E18) murine cortical neurons that (1) underwent excitotoxic necrosis, characterized by immediate deregulation of Ca2+ homeostasis, a persistent depolarization of mitochondrial membrane potential (Δψm), and insensitivity to bax-gene deletion, (2) underwent excitotoxic apoptosis, characterized by recovery of NMDA-induced cytosolic Ca2+ increases, sensitivity to bax gene deletion, and delayed Δψm depolarization and Ca2+ deregulation, or (3) that were tolerant to excitotoxic injury. Interestingly, treatment with the calpain inhibitor calpeptin, overexpression of the endogenous calpain inhibitor calpastatin, or gene silencing of calpain protected neurons against excitotoxic apoptosis but did not influence excitotoxic necrosis. Calpeptin failed to exert a protective effect in bax-deficient neurons but protected bid-deficient neurons similarly to wild-type cells. To identify when calpains became activated during excitotoxic apoptosis, we monitored calpain activation dynamics by time-lapse fluorescence microscopy using a calpain-sensitive Förster resonance energy transfer probe. We observed a delayed calpain activation that occurred downstream of mitochondrial engagement and directly preceded neuronal death. In contrast, we could not detect significant calpain activity during excitotoxic necrosis or in neurons that were tolerant to excitotoxic injury. Oxygen/glucose deprivation-induced injury in organotypic hippocampal slice cultures confirmed that calpains were specifically activated during bax-dependent apoptosis and in this setting function as downstream cell-death executioners.
Introduction
Overactivation of glutamate receptors (“excitotoxicity”) has been implicated in the pathophysiology of stroke (Zipfel et al., 1999) and several chronic neurodegenerative disorders (Rothstein et al., 1990; Lipton, 2007; Mattson, 2007). The excessive activation of NMDA and AMPA receptors significantly disrupt cellular ion homeostasis. As a result, a primarily Ca2+-dependent cell death occurs (Choi, 1987). After an intense and sustained period of glutamate receptor overactivation, the loss of ion homeostasis is frequently irreversible, ATP levels deplete, and excitotoxic necrosis is activated (Tymianski et al., 1993; Ankarcrona et al., 1995; Budd and Nicholls, 1996; White and Reynolds, 1996; Vergun et al., 1999; Ward et al., 2000). Conversely, transient ionic overloading often results in a less severe disturbance, allowing the cells to initially recover and restore their ionic gradients. Nevertheless, after the insult, neurons can either build on their initial recovery and survive or initiate a cell-death machinery, resulting in excitotoxic apoptosis (Ankarcrona et al., 1995; Lankiewicz et al., 2000; Luetjens et al., 2000). A number of studies addressed the differential activation of apoptosis and necrosis, in particular during ischemic brain injury (Manabat et al., 2003; Liu et al., 2004; Wei et al., 2004). Previous studies have shown excitotoxic apoptosis to induce the release of pro-apoptotic factors from mitochondria, including cytochrome c and apoptosis-inducing factor (AIF) (Budd et al., 2000; Lankiewicz et al., 2000; Luetjens et al., 2000; Cregan et al., 2002; Wang et al., 2004; Ward et al., 2006). The mitochondrial apoptosis pathway is controlled by pro- and anti-apoptotic Bcl-2 family proteins and either overexpression of anti-apoptotic bcl-2 or bcl-xL, or gene deficiency in the pro-apoptotic bax or bim genes to prevent excitotoxic apoptosis (Xiang et al., 1998; Wang et al., 2004; Dietz et al., 2007; Semenova et al., 2007; Concannon et al., 2010). In most cells, the release of cytochrome c activates a family of cysteine proteases, the caspases, by binding to apoptotic protease-activating factor-1 (APAF-1) (Liu et al., 1996; Zou et al., 1997). In neurons, however, caspase activation is frequently suboptimal despite the engagement of the mitochondrial apoptosis pathway (Budd et al., 2000; Lankiewicz et al., 2000). This phenomenon may be attributable to high expression levels of the caspase inhibitor XIAP (X-linked inhibitor of apoptosis protein) (Potts et al., 2003) or reduction in protein levels of APAF-1 during neuronal maturation (Wright et al., 2007).
Of note, the activation of the Ca2+-activated neutral cysteine protease calpain I has also been implicated in excitotoxic neuron death (Siman et al., 1989; Brorson et al., 1995; Lankiewicz et al., 2000). Similar to caspases, calpain I cleaves a variety of cytoskeletal proteins, enzymes, and transcription factors and may induce morphological alterations that mimic caspase-dependent apoptosis (Croall and Demartino, 1991; Wolf et al., 1999). Furthermore, calpain has been shown to cleave and inactivate pro-caspase 9, pro-caspase-3, and APAF-1 (Chua et al., 2000; Lankiewicz et al., 2000; Reimertz et al., 2001). Because calpains are activated by significant increases in cytosolic Ca2+, calpains may specifically play a role in cell demolition during excitotoxic necrosis, but this has not yet been experimentally tested. Using a combined biochemical, pharmacological, and single-cell imaging approach, we demonstrate here that calpains are required for the execution of bax-dependent excitotoxic apoptosis but surprisingly play no significant role during excitotoxic necrosis.
Materials and Methods
Materials.
Fetal calf serum, fetal bovine serum, horse serum, B27 supplement, minimal essential medium (MEM), Neurobasal medium, tetramethylrhodamine methyl ester (TMRM), Fluo-4 AM, and 1,2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM) were from Invitrogen (Bio Sciences). Calpeptin was purchased from ENZO Life Sciences and ionomycin from Merck Biosciences. All other chemicals, including NMDA and MK-801, came in analytical grade purity from Sigma-Aldrich.
Gene-targeted mice.
The generation and genotyping of bid−/− mice has been described previously (Kaufmann et al., 2007). Several pairs of heterozygous breeder pairs of bax-deficient mice were obtained from The Jackson Laboratory and maintained in house. The genotype of bax−/− mice was confirmed by PCR as described by The Jackson Laboratory (http://jaxmice.jax.org/protocolsdb/f?p=116:2:863695382966767::NO:2:P2_MASTER_PROTOCOL_ID,P2_JRS_CODE:250,002994). The bid−/− mice was generated on an inbred C57BL/6 background, using C57BL/6-derived ES cells. The bax−/− mice were originally generated on a mixed C57BL/6×129SV genetic background, using 129SV-derived ES cells but had been backcrossed for >12 generations onto the C57BL/6 background.
DNA extraction and genotyping.
DNA was extracted from tail snips using High Pure PCR Template Preparation Kit (Roche). Genotyping was performed using three specific primers as follows: 5′GGTCTGTGTGGAGAGCAAAC3′ (common), 5′TCAGGTGCCAGTGGAGATGAACTC3′ [wild-type (WT) allele-specific], and 5′GAGTCATACTTACTTCCTCCGAC3′ (mutant allele-specific) for bid; and 5′GTTGACCAGAGTGGCGTAGG3′ (common), 5′GAGCTGATCAGAACCATCATG3′ (WT allele-specific), and 5′CCGCTTCCATTGCTCAGCGG3′ (mutant allele-specific) for bax. The sizes of the expected PCR products are illustrated in Figure 4A.
Figure 4.
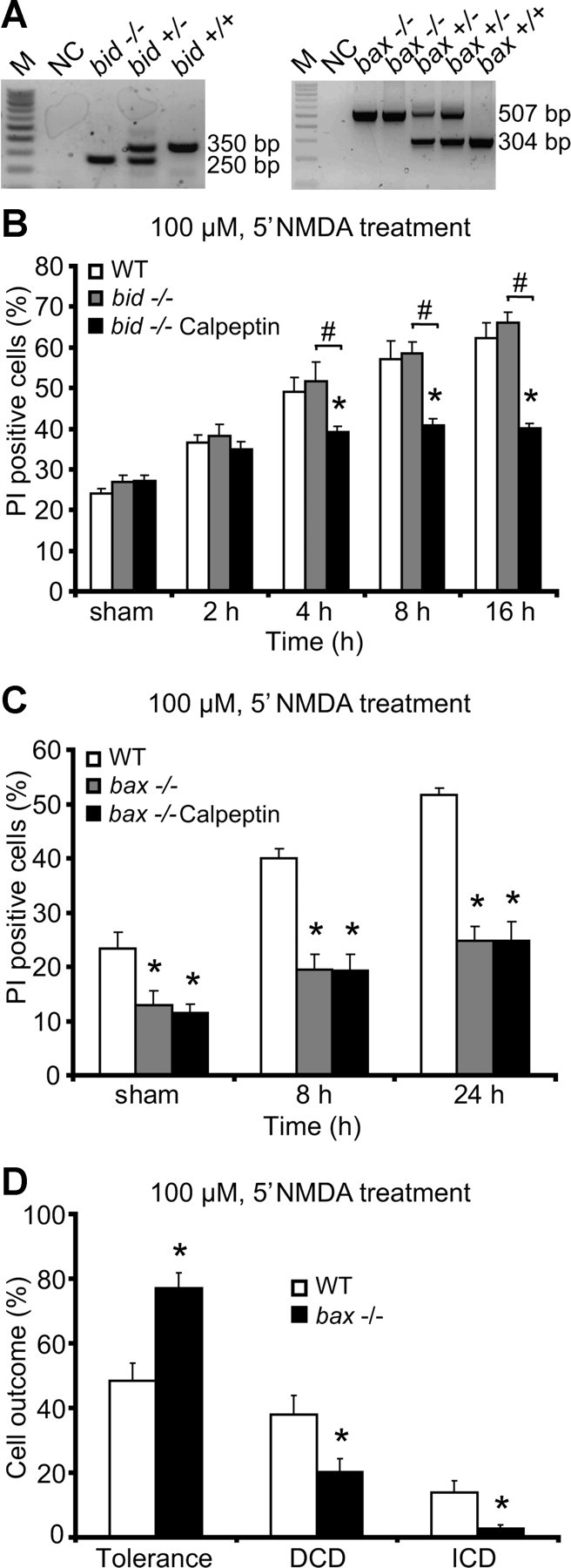
Protection by calpeptin depends on a functional mitochondrial apoptosis pathway. A, Representative standard PCR analysis of genomic DNA shows indicated strain and expected PCR bands sizes for bid and bax genotyping. M, 100 bp ladder; NC, negative control without the tail DNA template. B, C, Neocortical neurons from bid−/− mice, bax−/− mice, and WT controls were treated with 100 μm NMDA for 5 min over 24 h or sham conditions for 5 min in the presence or absence of calpeptin. Cell death was assessed by Hoechst and PI staining, and the results were quantified as a percentage of total neurons in the field. Each field contained ∼300–400 neurons, three fields were captured per well, and at least three wells were analyzed per time point. Means ± SEM are shown. *p ≤ 0.05 compared with NMDA-treated WT controls. #p ≤ 0.05 compared with NMDA-treated knock-out controls (ANOVA, post hoc Tukey's test). D, Quantification of cellular responses in neurons from bax−/− mice (n = 373 neurons) and WT controls (n = 275 neurons) treated with 100 μm NMDA for 5 min. Data were obtained from at least four independent experiments. Means ± SEM are shown. *p ≤ 0.05 compared with NMDA-treated WT controls (ANOVA, post hoc Tukey's test).
Preparation of mouse neocortical neurons.
Primary cultures of cortical neurons were prepared from E16 to E18 as described previously (Concannon et al., 2010). To isolate the cortical neurons, hysterectomies of the uterus of pregnant female mice were performed using an abdominal injection of 40 mg/kg pentobarbital (Dolethal) as lethal anesthesia. The cerebral cortices were pooled in a dissection medium on ice (PBS with 0.25% glucose and 0.3% bovine serum albumin). The tissue was incubated with 0.25% trypsin–EDTA at 37°C for 15 min. After the incubation, the trypsinization was stopped by the addition of medium containing sera. The neurons were then dissociated by gentle pipetting, and, after centrifugation (800 × g for 3 min), the medium containing trypsin was aspirated. Neocortical neurons were then resuspended in fresh plating medium (MEM containing 5% fetal calf serum, 5% horse serum, 100 U/ml penicillin/streptomycin, 0.5 mm l-glutamine, and 0.6% d-glucose). Cells were plated at 2 × 105 cells/cm2 on poly-lysine-coated plates and incubated at 37°C, 5% CO2. The plating medium was exchanged with 50% feeding medium (Neurobasal medium embryonic containing 100 U/ml Pen/Strep, 2% B27, and 0.5 mm l-glutamine) and 50% plating medium with additional cytosine arabinofuranoside (600 nm). Two days later, the medium was again exchanged for complete feeding medium. All experiments were performed on days in vitro (DIV) 8–11. All animal work was performed with ethics approval and under licenses granted by the Irish Department of Health and Children.
Cell lines.
Human SH-SY5Y neuroblastoma cells were grown in DMEM/Ham's F-12 (1:1 mixture) culture medium (Lonza) supplemented with 15% fetal calf serum and 100 U/ml penicillin/streptomycin. The motor neuron-like cell line NSC34 was maintained in DMEM (Lonza) supplemented with 10% fetal bovine serum, 2 mm l-glutamine, and 100 U/ml penicillin/streptomycin.
Preparation of organotypic hippocampal slice cultures.
Organotypic hippocampal slices cultures (OHSCs) were prepared and cultured according to the modified procedure (Kristensen et al., 2001). The brain from postnatal day 10 mouse pups was isolated and transferred to dissection medium containing HBSS (Invitrogen), 20 mm HEPES, 100 U/ml penicillin/streptomycin, and 0.65% glucose. Isolated hippocampi were placed on a McIlwain tissue chopper (Mickle Laboratory Engineering) and cut into 450-μm-thick sections. The slices were then transferred into fresh dissection medium and selected for clear hippocampal morphology (intact CA regions and dentate gyrus) and placed on porous (0.4 μm) membrane of Millicell inserts (Millipore). The inserts were placed in six-well tissue culture plates with 1 ml of culture medium consisting of MEM supplemented with 25% horse serum, 4 mm l-glutamine, 6 mg/ml d-glucose, 2% B27, 50 U/ml penicillin G, and 50 μg/ml streptomycin. The slices were maintained in a humidified incubator with 5% CO2 at 35°C with media changes every second day. All experiments were performed at DIV 10.
Determination of neuronal injury: Hoechst and propidium iodide staining of nuclear chromatin.
Neocortical neurons on 24-well plates were stained live with 1 μg/ml Hoechst 33258 (Sigma) and 5 μm propidium iodide (PI) (Sigma) dissolved in culture medium. Hoechst 33342 stains the condensed chromatin in apoptotic cells more brightly than the chromatin in healthy cells. The intact membrane of living cells excludes cationic dyes, such as PI. PI, a cell-impermeable red fluorescent dye, intercalates with nucleic acids in cells with membrane leakage. Nuclear morphology was assessed using an Eclipse TE 300 inverted microscope (Nikon) with 20×, 0.43 NA phase-contrast objective using the appropriate filter set for Hoechst, PI, and a charge-coupled device camera (SPOT RT SE 6; Diagnostic Instruments). All experiments were performed at least three times with independent cultures, and, for each time point, images of nuclei were captured in three subfields containing ∼300–400 neurons each and repeated in triplicate. The number of PI-positive cells was expressed as a percentage of total cells in the field. Resultant images were processed using NIH Image J (Wayne Rasband, National Institutes of Health, Bethesda, MD).
Oxygen–glucose deprivation in OHSCs.
Slices were tested for viability with PI (5 μm) before the oxygen–glucose deprivation (OGD) experiments. Healthy slices from WT and bax−/− mice were transferred to the hypoxic chamber (COY Lab Products). The hypoxic chamber had an atmosphere comprising 1.5% O2, 5% CO2, and 85% N2, and the temperature was maintained at 35°C. The slices were transferred to wells containing pre-equilibrated and deoxygenated OGD medium (bubbled with N2 for 1 h before use). The OGD medium consisted of the following (in mm): 2 CaCl2, 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 MgSO4, and 10 sucrose, pH 6.8. After 45 min of OGD, the slices were transferred to fresh oxygenated culture medium and placed in normoxic conditions (21% O2 and 5% CO2), and PI uptake was observed over a 24 h period. Control slices were free from OGD. Images were acquired with an Eclipse TE 300 inverted microscope (Nikon) and a 4× dry objective with Wasabi Software version 5.0 (Hamamatsu Photonics). The CA1 region was selected from WT and bax−/− hippocampal slices after OGD treatment, and the mean fluorescent intensity was determined. The regional gray-level intensity was measured using the Wasabi interactive tool, background gray-level was corrected, and the average intensity was plotted for nine slices per treatment. Neuronal injury was expressed as percentage of total injury (OGD performed for 4 h and recovery for 24 h).
After each time point, slices were placed into a six-well plate without media and 90 μl of lysis buffer [2% SDS (w/v), 67.5 mm Tris/Cl, pH 6.8, and 10% glycerol] was added to each insert containing three slices. Slices were removed by using a pipette and then transferred into an Eppendorf 1 ml tube. The protein was then stored at −80°C.
SDS-PAGE and Western blotting.
Preparation of cell lysates and Western blotting was performed as described previously (Reimertz et al., 2003). The resulting blots were probed with the following: a mouse monoclonal α-fodrin (αII-Spectrin) antibody (clone AA6; Millipore) diluted 1:1000; a rabbit polyclonal calpain large subunit antibody (Cell Signaling Technology) diluted 1:500; a rabbit polyclonal green fluorescent protein (GFP) antibody (peptide 26–39; Calbiochem) diluted 1:1000; and a mouse monoclonal β-actin antibody (clone DM 1A; Sigma) diluted 1:5000. Horseradish peroxidase-conjugated secondary antibodies diluted 1:10,000 (Pierce) were detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce) and imaged using a FujiFilm LAS-3000 imaging system (FujiFilm).
Plasmids, transfections, and shRNA.
Neocortical neurons (DIV 6) and NSC34 cells were transfected using Lipofectamine 2000 (Invitrogen), whereas SH-SY5Y cells were transfected with TurboFect In Vitro Transfection Reagent (Fermentas). The membrane-targeted calpain-sensitive Förster resonance energy transfer (FRET) probe pSCLPaX, containing the calpain-specific spectrin substrate linker, was prepared as follows. Complementary oligonucleotides 5′GCAG CAGGAGGTGTACGGAGGTAC3′ and 5′CTCCGTACACCTCCTGCTGCAGCT3′ were annealed and ligated into SacI and KpnI sites between enhanced cyan fluorescent protein (ECFP) and Venus in the FRET probe subcloning vector pTK-reverse SCAT (Hellwig et al., 2008). This generated a cassette consisting of a coding sequence for the calpain substrate sequence QQEVYG flanked at the N terminus by ECFP, a serine tripeptide, and an SacI site (encoding glutamate–leucine) and at the C terminus by a KpnI site (encoding glycine–threonine), a flexible serine–glycine–serine tripeptide, and Venus. The oligonucleotide sequences immediately adjacent to the overhangs were designed to destroy the SacI site during ligation with corresponding vector overhangs and generate a diagnostic PvuII site to facilitate exclusion of constructs with multiple concatenated oligonucleotides. The resulting pTK-reverse SCLPa vector was digested with BsrGI to generate a BsrGI site-flanked DNA fragment including calpain substrate motif and Venus coding sequence. This was inserted in-frame into the BsrGI site of the membrane-targeted ECFP expression vector pECFP–CAAX to generate the plasmid pSCLPaX. pECFP–CAAX was generated by replacing the coding sequence for EGFP in the plasmid pEGFP–CAAX (Hongisto et al., 2008) with the coding sequence for ECFP, using the enzymes AgeI and BsrGI. For overexpression of calpastatin, cells were transfected with a vector expressing GFP–calpastatin (MG209946; OriGene). For inhibition of calpain, cells were transfected with a vector expressing either a commercial shRNA targeting the large subunit of calpain I (SHCLNG-NM_007600, TRCN0000030664; Sigma) or the large subunit of calpain II (SHCLNG-NM_009794, TRCN0000030672; Sigma) or a scramble control vector (SHC001; Sigma). A plasmid with an enhanced GFP (pEGFP–N1; Clontech) was used to allow the identification of transfected neurons. Cells were used for experiments 36 h after transfection.
Real-time live-cell imaging.
Primary neocortical neurons on Willco dishes were coloaded with Fluo-4 AM (3 μm) and TMRM (20 nm) for 30 min at 37°C (in the dark) in experimental buffer containing the following (in mm): 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 HEPES, 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, and 15 glucose, pH 7.4. The cells were washed and bathed in 2 ml of experimental buffer containing TMRM, and a thin layer of mineral oil was added to prevent evaporation. Neurons were placed on the stage of an LSM 510 Meta confocal microscope equipped with a 63×, 1.3 NA oil-immersion objective and a thermostatically regulated chamber (Carl Zeiss). After 30 min equilibration time, neurons were exposed to NMDA-induced excitotoxic injury (30 μm NMDA for 5 min, 100 μm NMDA for 5 min, or 300 μm NMDA for 60 min), and glycine (10 μm) with MK-801 (5 μm) was added to block NMDA receptor activation as required. TMRM was excited at 543 nm, and the emission was collected with a 560 nm long-pass filter; Fluo-4 AM was excited at 488 nm, and the emission was collected through a 505–550 nm barrier filter. Images were captured every 30 s during NMDA excitation and every 5 min during the rest of the experiments. In separate experiments, neurons, transfected with the calpain-sensitive FRET probe and loaded with TMRM (20 nm) in experimental buffer, were placed on the stage of an LSM 5Live duoscan confocal microscope equipped with a 40×, 1.3 NA oil-immersion objective and a thermostatically regulated chamber set at 37°C (1080; Carl Zeiss). After a 30 min equilibration time, drug dissolved in experimental buffer was added to the medium. TMRM was excited at 561 nm, and the emission was collected by a 575 nm long-pass filter. CFP was excited at 405 nm, and emission was collected at 445–505 and 505–555 nm for FRET. Yellow fluorescent protein (YFP) was excited directly using the 489 nm laser diode and detected with the same bandpass filter used for FRET. Images were captured every 5 min throughout these experiments. All microscope settings, including laser intensity and scan time, were kept constant for the whole set of experiments. Control experiments under these conditions were also performed and showed that photo toxicity had a negligible impact. The cell-permeable Ca2+ chelator BAPTA-AM (500 nm) was used for TMRM/Fluo-4 AM and calpain-FRET probe experiments and was added to the medium on stage 4 h after the NMDA exposure. The donor unquenching control experiment was performed in neurons transfected with the FRET probe on the LSM 5live duoscan using the same mode of detection as stated above, except the missing delay between consecutive scans. Bleach scans were performed using the point scanner of the microscope to scan predefined regions only with the 489 nm diode-pumped solid-state laser acousto-optic tunable filter transmission set to 100% and using an 80/20 beam splitter to bleach Venus (see Fig. 6C). All images were processed and analyzed using MetaMorph Software version 7.5 (Universal Imaging), and the data were presented normalized to the baseline response.
Figure 6.
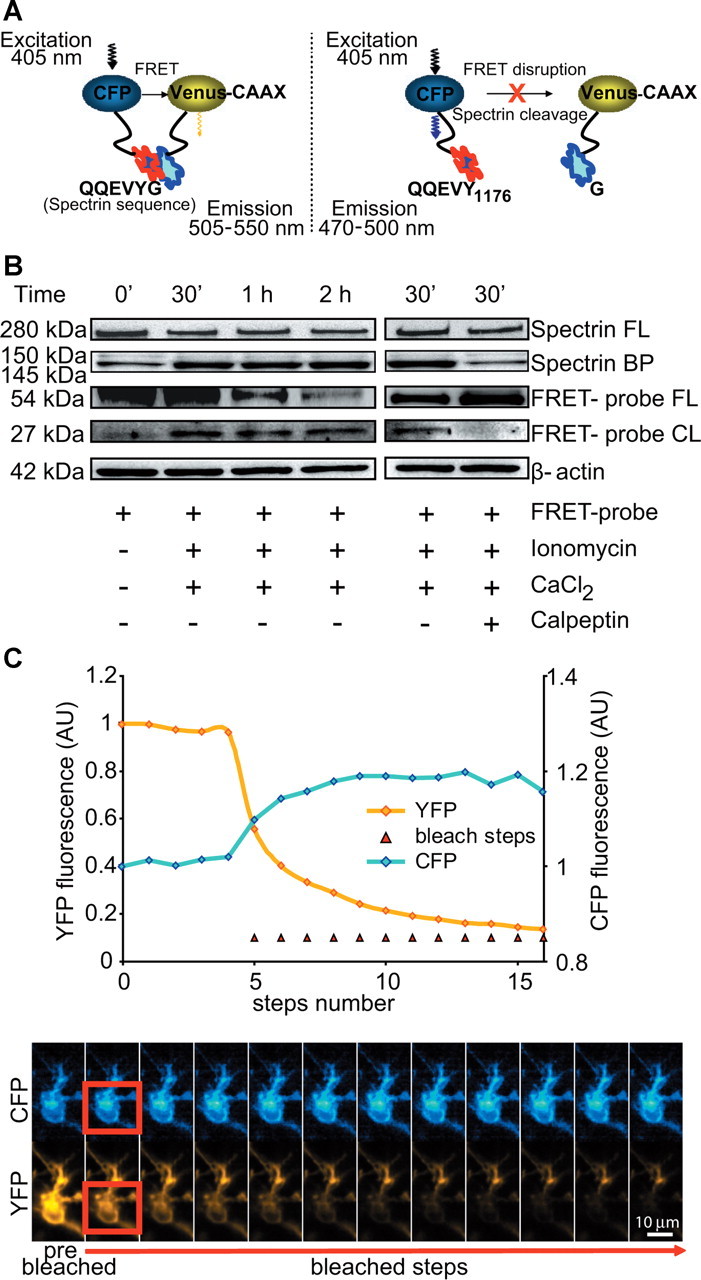
FRET-based single-cell analysis of calpain activity. A, The calpain-sensitive FRET probe consisted of CFP and Venus fluorophores linked together by a region containing the calpain-substrate spectrin sequence. The Venus fluorophore is fused to a CAAX motif. During cleavage of the spectrin linker, the FRET between CFP and Venus is disrupted. This results in decreased FRET fluorescent intensity and increased CFP fluorescent intensity. B, FRET probe expression and cleavage in vitro was validated by Western blotting in SH-SY5Y cells using an anti-GFP antibody. After FRET-probe transfection, the cells were treated with ionomycin (5 μm) and CaCl2 (10 mm) for the indicated times in the presence (+) or absence (−) of calpeptin. β-Actin was used as loading control. BP, Breakdown product; CL, cleavage; FL, full-length. C, The Venus bleaching and correlating CFP unquenching kinetics were performed on the LSM 5live duoscan in control neurons transfected with the FRET probe. From the fifth scan onward, each scan of a time series was followed by a bleach scan, as indicated by red triangles. On the bottom, corresponding time-frame images of the cell are illustrated. Bleached region, used to calculate the average intensity kinetics, is indicated in red and segmented for the cell area using the CFP channel. Scale bar, 10 μm.
Statistics.
Data are given as means ± SEM. For statistical comparison, one-way ANOVA followed by Tukey's post hoc test was used. p values <0.05 were considered to be statistically significant.
Results
Treatment with the calpain inhibitor calpeptin reduces delayed excitotoxic apoptosis in cortical neurons
To explore the role of calpains in excitotoxic neuronal injury, we used three models of NMDA-induced excitotoxicity in primary mouse cortical neurons. Cortical neurons were exposed to either NMDA (30 or 100 μm) for 5 min (Fig. 1A) or 300 μm NMDA for 60 min (Fig. 1B). Controls were sham exposed to experimental buffer. Confocal imaging of individual Ca2+ responses using Fluo-4 AM demonstrated that neurons (1) completely and persistently recovered their cytosolic Ca2+ to stable basal levels after termination of the NMDA exposure (Fig. 1C), (2) recovered their cytosolic Ca2+ levels after the NMDA exposure but showed a delayed Ca2+ deregulation (DCD) (Fig. 1D), or (3) failed to recover their NMDA-induced increase in intracellular Ca2+ and showed an immediate and sustained Ca2+ deregulation (ICD) (Fig. 1E). We classified neurons according to these Ca2+ responses into three groups designated “tolerant,” “apoptotic/DCD,” and “necrotic/ICD” (Ankarcrona et al., 1995; Ward et al., 2007). Parallel imaging of mitochondrial membrane potential (Δψm) changes demonstrated that DCD was associated with a late mitochondrial depolarization (Fig. 1D), whereas neurons undergoing ICD showed an early and persistent depolarization (Fig. 1E). In neurons exposed to 100 μm for 5 min, DCD occurred always subsequent to the delayed mitochondrial membrane potential depolarization within a period of 30 ± 2 min. Some neurons characterized by an ICD response still showed nuclear pyknosis (data not shown), suggesting that cells with an ICD response did not show a homogenously “necrotic” nuclear morphology. Neurons displaying an ICD response after a 60 min exposure to 300 μm NMDA were insensitive to bax-gene deletion (Fig. 1G). Neurons undergoing DCD showed chromatin condensation primarily in the absence of nuclear fragmentation (Fig. 2A) and may therefore be classified as “type II” apoptotic neurons (Leist and Jäättelä, 2001).
Figure 1.
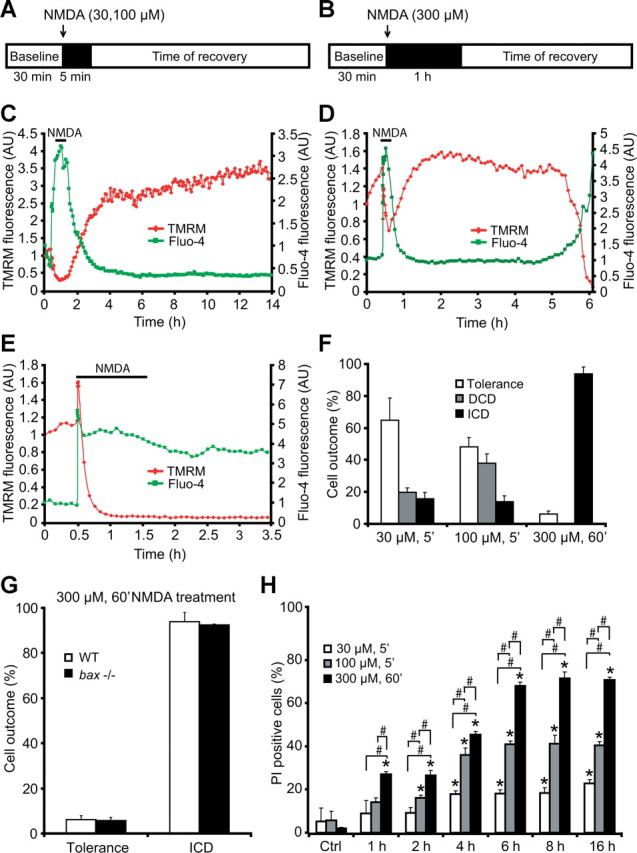
NMDA receptor overactivation produces distinct types of neuronal responses as evidenced by single-cell imaging: tolerance, apoptosis, and necrosis. A, B, Models of excitotoxic NMDA receptor overactivation. The neurons were exposed to 30 μm NMDA for 5 min, 100 μm NMDA for 5 min, or 300 μm NMDA for 60 min, after which alterations in Δψm and intracellular Ca2+ were monitored in single cells using time-lapse confocal fluorescence microscopy (LSM 510 Meta). C–E, Three neuronal responses were readily identifiable (Ward et al., 2007): C, DCD/tolerant neuron with recovery of neuronal Ca2+ overloading and extensive hyperpolarization of Δψm; D, excitotoxic apoptosis in which neuronal mitochondria transiently recovered their energetics and a late Δψm depolarization and DCD occurred hours after the initial excitation; E, ICD/necrosis with an acute and ICD associated with a rapid collapse of membrane potential. F, Quantification of cellular responses for the three injury models (n = 102, n = 109, n = 129 for 30 μm/5 min, 100 μm/5 min, and 300 μm/60 min treatments, respectively). Data were obtained from at least three independent experiments. G, Quantification of cellular responses in neurons from bax−/− mice (n = 61 neurons) and WT controls (n = 129 neurons) treated with 300 μm NMDA for 60 min. H, NMDA induces dose-dependent excitotoxicity over time. Cortical neurons were treated with 30 μm NMDA for 5 min, 100 μm NMDA for 5 min, 300 μm NMDA for 60 min, or sham conditions for 5 min and allowed to recover as indicated. Cell death was assessed by Hoechst and PI staining. Three subfields containing 300–400 neurons each were captured, and at least three wells were analyzed per time point. PI-positive nuclei were scored as dead neurons and expressed as a percentage of the total population. Data are means ± SEM from n = 3 separate cultures. Ctrl, Control. *p ≤ 0.05 compared with NMDA-treated control. #p ≤ 0.05 between NMDA treatments (ANOVA, post hoc Tukey's test).
Figure 2.

Calpain inhibitor calpeptin protects neurons against excitotoxic apoptosis. A, Representative images of phase-bright and the merged image of PI- and Hoechst-stained neurons exposed to sham conditions or 100 μm NMDA for 5 min in the presence and absence of the calpain inhibitor calpeptin. Images were taken at selected time points (8 and 24 h after treatment). Scale bar, 10 μm. B, Dose–response assay. Neurons were treated with 100 μm NMDA for 5 min in the presence (5 or 20 μm) or absence of calpeptin for the indicated times. The number of PI-positive cells was expressed as a percentage of total cells in the field. Data represent means ± SEM from n = 3 separate cultures. *p ≤ 0.05 compared with NMDA-treated controls. #p ≤ 0.05 between NMDA-treated neurons in the presence of calpeptin (5 or 20 μm) (ANOVA, post hoc Tukey's test). C, E, Cortical neurons were treated with 100 μm NMDA for 5 min over 24 h (C) or 300 μm NMDA for 60 min over 4 h (E) in the presence or absence of calpeptin, and the extent of injury was assessed with Hoechst and PI. Data represent means ± SEM from n = 3 separate cultures. *p ≤ 0.05 compared with NMDA-treated controls (ANOVA, post hoc Tukey's test). D, F, Western blot and densitometric analysis comparing the levels of spectrin cleavage in neurons after NMDA treatments: 100 μm NMDA for 5 min over 24 h (D) or 300 μm NMDA for 60 min over 4 h (F) in the presence or the absence of calpeptin. β-Actin was used as loading control. Experiments were repeated three times with different preparations with similar results. Densitometric data are expressed as a ratio of the 145 kDa spectrin breakdown product (BP) and the 280 kDa full-length (FL) protein normalized to β-actin. *p ≤ 0.05 compared with NMDA-treated controls. #p ≤ 0.05 between NMDA-treated neurons in the presence or absence of calpeptin (ANOVA, post hoc Tukey's test).
Quantification of the individual Ca2+ responses in relation to the different intensities of NMDA receptor overactivation demonstrated that the mild NMDA exposure of 30 μm for 5 min [peak Fluo-4 fluorescence, 1.94 ± 0.13 arbitrary units (AU)] primarily produced neuronal tolerance, whereas the 5 min exposure to 100 μm NMDA (peak Fluo-4 fluorescence, 2.47 ± 0.14 AU) produced a mix of DCD and ICD responses, with DCD responses being more prominent. In contrast, prolonged exposure of 300 μm NMDA for 60 min induced primarily ICD (Fig. 1F). Quantification of PI-positive cells, a marker of plasma membrane leakage occurring after primary or secondary necrosis, likewise indicated that prolonged exposure of 300 μm NMDA for 60 min caused significant amounts of immediate cell death within 1 h, whereas cell death induced by a 5 min exposure to 30 or 100 μm NMDA showed a more delayed profile with a maximal onset at 4 h (Fig. 1H).
Because the highest fraction of DCD and ICD were detected with 100 μm/5 min and 300 μm/60 min NMDA, respectively, additional experiments were conducted in these two injury models. To explore the role of calpains in mediating excitotoxic injury, neurons were pretreated with the selective calpain inhibitor calpeptin (5–20 μm) for 2 h and then exposed to NMDA. When neurons were exposed to conditions that favored DCD, calpeptin treatment exerted a robust dose-dependent and long-term neuroprotective effect as detected by phase-contrast microscopy, Hoechst 33258 staining, and quantification of PI uptake (Fig. 2A–C). Treatment with calpeptin also reduced the accumulation of the calpain-specific, 145 kDa spectrin breakdown product that accumulated in NMDA-exposed cultures as determined by quantitative Western blot analysis. Significant increases in spectrin cleavage were observed at 4–8 h compared with the sham-treated cultures and reduced in cultures pretreated with calpeptin (Fig. 2D). Surprisingly, however, pretreatment with calpeptin failed to show a significant protection under conditions that favor ICD, as induced by the exposure to 300 μm NMDA for 60 min (Fig. 2E). This was confirmed by the analysis of the accumulation of the 145 kDa, calpain-specific spectrin breakdown product that accumulated only marginally in the neuron cultures (Fig. 2F).
Gene silencing of calpain I and II or overexpression of calpastatin confirms the selective requirement for calpain activation in delayed excitotoxic apoptosis
Because pharmacological agents may have off-target effects, we next investigated whether the selective requirement of calpains in delayed excitotoxic apoptosis could be confirmed using genetic approaches. Gene silencing of the large subunit of calpain I and II using two distinct shRNA constructs was validated in NSC34 cells after 36 h of transfection when compared with cultures transfected with a scramble vector as determined by Western blot analysis (Fig. 3A). Of note, transfection with either shRNA targeting the large calpain subunit significantly reduced cell death in response to 100 μm/5 min NMDA compared with cells transfected with a scrambled shRNA sequence (Fig. 3B). In contrast, immediate cell death in response to 300 μm/60 min NMDA was not reduced by gene silencing of the large calpain subunit (Fig. 3C).
Figure 3.

Calpain inhibition using RNA interference technology and overexpression of the endogenous calpain inhibitor calpastatin attenuate NMDA-induced excitotoxic apoptosis. A, NSC34 cells were transfected with shRNA plasmids against the large subunit of calpain I (shRNA I), calpain II (shRNA II), or scramble shRNA. Reduced expression levels of calpain I and II after shRNA expression compared with scramble were assessed by Western blotting 36 h after transfection. Probing for β-actin served as a loading control. Experiments were repeated three times with similar results. B, C, Cell-death analysis after 100 μm NMDA treatment for 5 min assayed over 24 h (B) and 300 μm NMDA for 60 min treatment assayed over 4 h (C). Cortical neurons were transfected with shRNA plasmids as described above. A plasmid expressing GFP allowed for the identification of transfected neurons. The number of PI-positive cells in transfected neurons was quantified (n = 125–160 cells per time point quantified). Experiments were performed in triplicate in three separate cultures. *p ≤ 0.05 compared with NMDA-treated, scramble transfected controls (ANOVA, post hoc Tukey's test). D, E, Cortical neurons were transfected with a plasmid expressing the endogenous calpain inhibitor, calpastatin fused with GFP, or a plasmid expressing GFP alone as a control. Neurons were treated with 100 μm NMDA for 5 min and assayed over 24 h (D) and 300 μm NMDA for 60 min and assayed over 4 h (E). The number of PI-positive cells in transfected neurons was quantified (n = 142–173 cells per time point quantified). *p ≤ 0.05 compared with NMDA-treated controls (ANOVA, post hoc Tukey's). Experiments were repeated three times with independent culture preparations with similar results.
Inhibition of calpain activity can also be achieved by overexpression of the endogenous calpain inhibitor calpastatin. Neurons were transfected with a mammalian expression vector for calpastatin–GFP or with an EGFP control vector. Analysis of GFP-positive neurons indicated that transfection of calpastatin–GFP significantly reduced excitotoxic cell death in response to 100 μm/5 min NMDA (Fig. 3D). Again, immediate cell death in response to 300 μm/60 min NMDA was not reduced by calpastatin–GFP (Fig. 3E). These results confirmed that calpains played a predominant role in excitotoxic apoptosis.
The protective activity of calpain inhibition requires a functional mitochondrial apoptosis pathway
We next explored whether calpain acted on Bcl-2 family proteins to promote excitotoxic apoptosis. One possibility was that calpains activated pro-apoptotic Bcl-2 family proteins of the Bcl-2 homology 3 domain-only family. Bid represents a prominent candidate for such an activity. Bid is involved in the so-called extrinsic apoptosis pathway and is proteolytically activated by caspase-8 to generate a truncated form (tBid) (Li et al., 1998; Luo et al., 1998). It has also been suggested that calpain-mediated cleavage of Bid generates Bid fragments that, similar to tBid, may activate the mitochondrial apoptosis pathway (Chen et al., 2001; Polster et al., 2005). However, cortical neurons from bid-deficient mice were equally sensitive to the NMDA-induced delayed excitotoxic cell death as WT neurons (Fig. 4A,B). Furthermore, calpeptin also exerted significant neuroprotection in bid-deficient neurons, demonstrating that calpains were not acting on Bid to promote cell death (Fig. 4B).
In contrast, bax-deficient cortical neurons were highly resistant to NMDA-induced excitotoxic apoptosis (Fig. 4C). Moreover, calpeptin failed to provide a significant protection in bax-deficient mice, suggesting that deficiency in bax was dominant over the pro-apoptotic activity of calpain and that calpains required a functional mitochondrial apoptosis pathway to mediate excitotoxic apoptosis.
Quantification of the individual Ca2+ responses in response to a 100 μm/5 min NMDA exposure showed increased neuronal tolerance in the bax-deficient mice compared with the WT controls, as evidenced by decreased DCD responses (Fig. 4D). Of note, bax gene deletion also led to a significant reduction in ICD responses after the exposure to 100 μm/5 min NMDA, suggesting that ICD may accompany several cell-death modes or that bax gene deletion has an effect on neuronal Ca2+ handling.
Calpains are activated during bax-dependent neuronal injury induced by OGD
To validate our hypothesis that activation of calpains played a predominant role in bax-dependent cell death in a clinically more relevant setting, we performed experiments using OHSCs subjected to OGD. OGD is a widely used in vitro model of ischemic injury that also produces apoptotic (Kalda et al., 1998) and necrotic (Goldberg and Choi, 1993; Gwag et al., 1995) cell-death phenotypes. OHSCs derived from WT and bax-deficient mice were exposed to OGD for 45 min in the absence or presence of calpeptin (20 μm) and allowed to recover under normoxic conditions over 24 h. As shown in Figure 5B, a 45 min OGD treatment was sufficient to induce CA1 damage in WT OHSCs, whereas the neuronal injury in bax-deficient slice cultures was significantly attenuated. Calpeptin treatment exerted a significant neuroprotective effect in WT OGD-treated OHSCs but not in the bax-deficient slice cultures (Fig. 5A,B). Western blot analysis revealed an increased accumulation of the calpain-specific, 145 kDa spectrin breakdown product in the WT OHSCs but not in the bax-deficient slice cultures (Fig. 5C).
Figure 5.
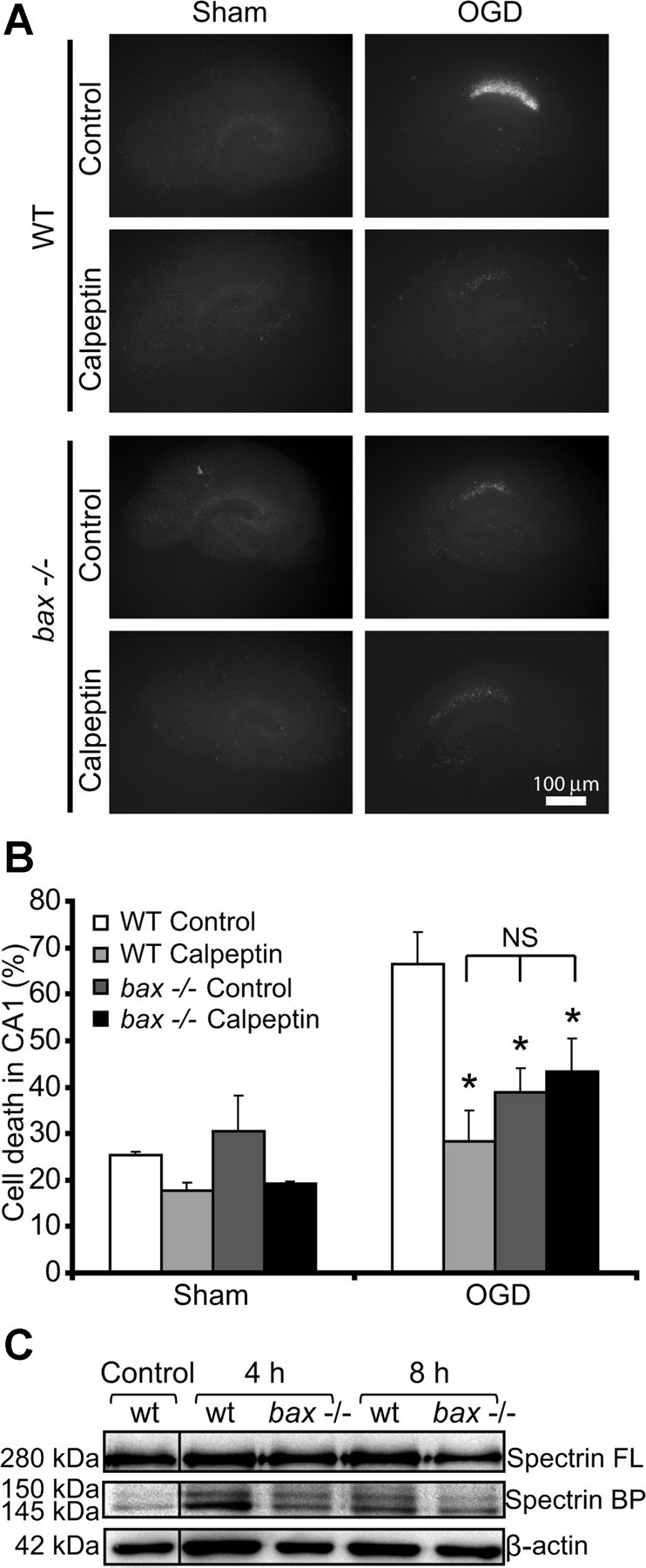
OHSCs isolated from bax-deficient mice show reduced calpain activation in response to OGD. A, Representative images of OHSCs derived from WT and bax−/− mice. The slices were sham exposed or subjected to OGD conditions for 45 min in the absence (control) or presence of calpeptin (20 μm) and allowed to recover for 24 h. Scale bar, 100 μm. B, Hippocampal slice cultures from WT and bax−/− mice were treated as described in A, and quantification of injury was assessed by PI staining 24 h after treatment (n = 5 slice cultures for each condition). Means ± SEM are shown. *p ≤ 0.05 compared with OGD-treated WT control (ANOVA, post hoc Tukey's test). C, Whole-cell extracts were prepared from WT and bax−/− hippocampal slice cultures treated to 45 min OGD and allowed to recover under normoxic conditions for the indicated time points. Spectrin cleavage was assessed by Western blotting. β-Actin served as loading control. BP, Breakdown product; FL, full-length.
Calpain activation occurs late during excitotoxic apoptosis
Our experiments demonstrated a functional role of calpains during delayed excitotoxic apoptosis but did not provide detailed information when during the cell-death signaling cascade calpains became activated. To provide spatiotemporal information on calpain activation and its relation to cell death during excitotoxic injury, we designed a calpain-sensitive FRET probe comprising CFP and Venus as FRET donor and acceptor, respectively, and an 18 aa linker that incorporated the QQEVYG calpain-cleavage site of spectrin. The intact FRET probe was anchored to the plasma membrane by a CAAX targeting sequence (for details, see Materials and Methods). During calpain activation, FRET probe cleavage of the linker peptide results in FRET disruption that can be detected by confocal time-lapse microscopy (Fig. 6A).
To confirm that the FRET probe was reporting calpain cleavage, we transfected human neuroblastoma SH-SY5Y cells with the FRET probe. After this, cells were exposed to ionomycin and CaCl2, a treatment that results in significant and rapid calpain activation in SH-SY5Y cells (Reimertz et al., 2001). Western blot analysis of cytosolic extracts demonstrated that endogenous full-length spectrin was cleaved with similar kinetics than the intact FRET probe (detected using a GFP antibody) during addition of ionomycin and CaCl2. Moreover, we detected a similar increase in the accumulation of the 145 kDa, calpain-spectrin cleavage product and the cleaved FRET probe (Fig. 6B, left). Cleavage of the FRET probe was also sensitive to a treatment with calpeptin (Fig. 6B, right).
To explore the calpain-sensitive FRET probe efficiency, we next performed acceptor photobleaching experiments in control neurons. After establishment of a baseline, repeated bleaching steps were recorded and allowed a clear detection of donor unquenching (using similar image acquisition settings as in the following time-lapse experiments). The unquenching was complete after five consecutive bleaching steps as indicated in Figure 6C.
Having established that the FRET probe was functional and reported calpain activity, we transfected the probe into cortical neurons and exposed them to 100 μm NMDA for 5 min (conditions that favor the induction of delayed excitotoxic apoptosis) or 300 μm NMDA for 60 min (conditions that favor ICD; see Fig. 1). Because ICD and DCD were associated with mitochondrial membrane potential depolarization (Fig. 1), we detected in parallel changes in mitochondrial membrane potential using the membrane-permeant cationic fluorescent probe TMRM. Interestingly, in neurons that showed a delayed excitotoxic apoptosis, we detected significant FRET probe cleavage. However, this occurred always subsequent to the delayed mitochondrial depolarization (Fig. 7A), within a period of 42 ± 8 min (Fig. 7C). This suggested that calpains were predominantly activated during the execution phase of delayed excitotoxic apoptosis. Calpain activity lasted on average 58 ± 13 min. Onset of cell death, detected by the sudden loss of cytosolic CFP fluorescence attributable to plasma membrane leakage, occurred always after onset of the FRET probe cleavage and with an average delay of 96 ± 8 min (Fig. 7C). In contrast, cells exposed to 300 μm NMDA for 60 min did not show significant FRET probe cleavage either before or during ICD (Fig. 7B). Analysis of all cells monitored and classified according to their type of response (tolerant, DCD, or ICD) revealed that calpain FRET probe cleavage was selectively detected in cells that showed DCD (Fig. 7D). In contrast, neither tolerant cells nor cells undergoing ICD showed a detectable FRET probe cleavage.
Figure 7.
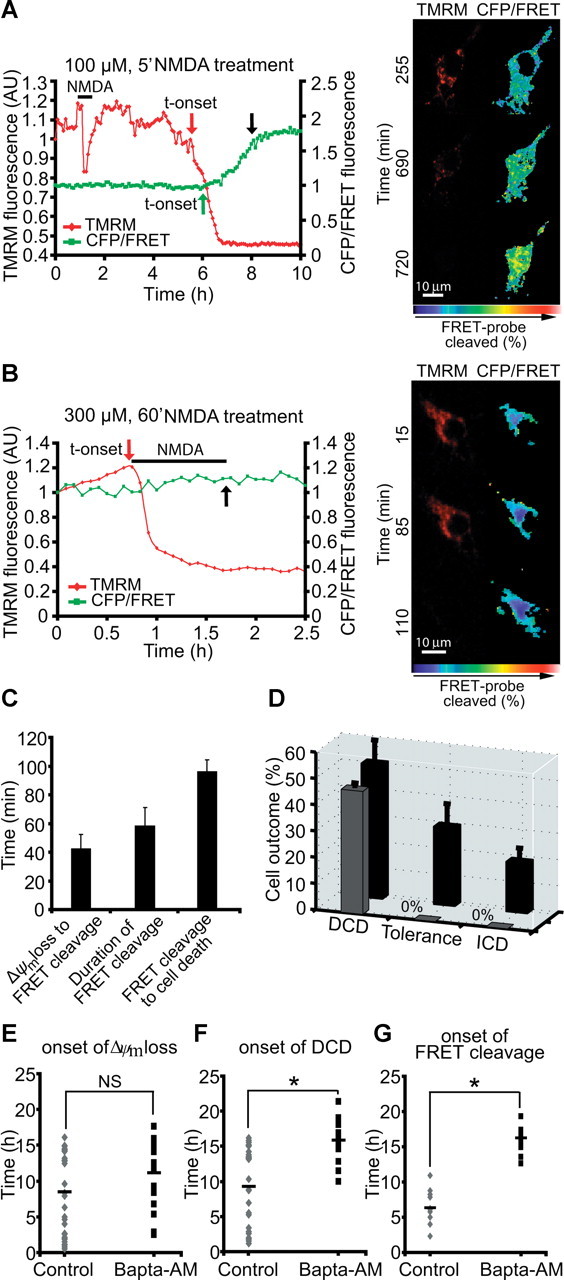
Calpain activation is selectively associated with excitotoxic apoptosis. A, B, Cortical neurons were transfected with the calpain-sensitive FRET probe and after 24 h mounted on the stage of a LSM 5Live duoscan, and neurons were treated with 100 μm NMDA for 5 min and assayed over 24 h (A) or 300 μm NMDA and assayed over 4 h (B). Fluorescent measurements were recorded for TMRM, FRET, CFP, and YFP by time-lapse confocal microscope. FRET probe imaging data are expressed as a ratio of CFP/FRET. TMRM was used as a Δψm indicator in the nonquenched mode. In neurons that showed a delayed excitotoxic apoptosis (A), we observed significant calpain activation (FRET probe cleavage) subsequent to delayed Δψm depolarization. In neurons that showed an immediate excitotoxic ICD/necrosis (B), no significant FRET cleavage was observed. The onset of these events (in red and green) and cell death (in black) is indicated by arrows (A, B), and representative time-frame images are illustrated (left, pseudocolored CFP/FRET emission ratio; red, TMRM images). Scale bar, 10 μm. C, Analysis of FRET probe cleavage and its relation to cellular outcome in cells treated with 100 μm NMDA for 5 min. Data shown represent the mean time taken between the onset of FRET probe disruption and onset of Δψm loss; the mean time taken for complete probe cleavage and the mean time taken between onset of FRET probe cleavage and onset of cellular collapse. Data were obtained from 18 separate experiments from 11 independent cultures (n = 22 neurons). D, Quantification of the number of cells in 100 μm NMDA/5 min treated neurons with FRET probe disruption (in black, total number of cells; in gray, cells with FRET probe disruption). Neurons were classified according to their type of cellular response (DCD, tolerance, ICD). Data were obtained from 18 separate experiments from 11 independent cultures (n = 47 neurons). E, F, Cortical neurons were exposed to 100 μm NMDA for 5 min and the Ca2+ chelator BAPTA-AM (500 nm) was added on stage 4 h after NMDA addition. Alterations in Δψm and intracellular Ca2+ were monitored in single cells using confocal fluorescence microscopy. Onset of Δψm loss (E) and onset of DCD (F) was analyzed in neurons in the absence (control, n = 29 neurons) or the presence of BAPTA-AM (n = 19 neurons). *p ≤ 0.05 compared with NMDA-treated control. G, Cortical neurons transfected with the calpain-sensitive FRET probe were treated as described in E and F and the onset of the FRET probe cleavage was analyzed in transfected neurons in the absence (control, n = 11 neurons) or presence of BAPTA-AM (n = 11 neurons). *p ≤ 0.05 compared with NMDA-treated control neurons.
To investigate whether the DCD was a trigger for calpain activation in this system, we treated murine cortical cultures on stage with the Ca2+ chelator BAPTA-AM (500 nm). BAPTA-AM is a membrane-permeable chelator that has been shown to be neuroprotective against excitotoxicity and is more selective and faster compared with other Ca2+ chelators, such as EGTA and EDTA (Tymianski et al., 1994). Excitotoxic injury was induced in cortical neurons by an exposure to 100 μm NMDA for 5 min, and BAPTA-AM (500 nm) was added 4 h after termination of the NMDA exposure. Analysis of the onset of Δψm depolarization and DCD using TMRM and Fluo-4-based confocal fluorescence microscopy showed that BAPTA-AM did not produce a considerable delay in the onset of the Δψm loss (Fig. 7E) but resulted in a significant delay in DCD (Fig. 7F). Furthermore, BAPTA-AM produced a robust delay in the onset of FRET probe cleavage compared with NMDA-exposed control neurons (Fig. 7G).
Discussion
In the present study, we have set out to characterize the role of calpains in response to transient and prolonged glutamate receptor overactivation in mouse neocortical neurons. Our data suggest that, surprisingly, calpains do not contribute to excitotoxic injury under conditions that are associated with ICD and excitotoxic necrosis. Using pharmacological, genetic, and single-cell imaging approaches, we demonstrate that calpains rather play a critical role in bax-dependent, excitotoxic apoptosis and are here activated as cell-death effectors during the execution stage.
Our study supports the concept that inhibition of calpain activity provides neuroprotection against excitotoxic neuronal injury. One of the limitations in our study may be that calpain inhibition slowed down cell death rather than arresting it. However, similar neuroprotective effects were observed in hippocampal and cortical neurons subjected to excitotoxic and OGD-induced neuronal injury in vitro, using the calpain inhibitors calpeptin, MDL-28170, and E-64 (Siman et al., 1989; Brorson et al., 1995; Rami et al., 1997; Lankiewicz et al., 2000). Similarly, neuroprotection was also observed using genetic approaches. Knockdown of μ-calpain (calpain I) in an in vitro model of OGD was able to prevent AIF nuclear translocation and increased cell survival (Cao et al., 2007). Moreover, calpain inhibition was neuroprotective in in vivo models of excitotoxic injury (Wu et al., 2004), ischemic stroke, and neurodegeneration (Lee et al., 1991; Rami and Krieglstein, 1993; Bartus et al., 1994; Hong et al., 1994). It has also been reported that knockdown of calpain II (m-calpain) increased survival of primary hippocampal neurons after NMDA excitotoxicity (Bevers et al., 2009). Importantly, previous studies did not fully address the question under which cell-death conditions calpain inhibition is neuroprotective and when in the cell death cascade calpains became activated. In this study, we demonstrate that calpains are specifically involved in bax-dependent excitotoxic apoptosis. Although treatment with calpeptin failed to show any significant protection in neurons undergoing ICD, it provided a long-term protection in neurons undergoing DCD/excitotoxic apoptosis. These findings were confirmed using genetic approaches, including gene silencing of calpain and overexpression of the endogenous inhibitor calpastatin (Fig. 3). Western blot data also suggested that, under conditions that favor ICD, calpain-specific spectrin proteolysis was not significantly elevated, whereas neurons exposed to conditions that favor DCD showed significant calpain activation (Fig. 2). These biochemical data were confirmed by single-cell imaging experiments, in which no significant cleavage of the calpain-sensitive FRET probe was detected in the soma of neurons that underwent ICD, whereas neurons undergoing DCD showed a full-blown calpain activity (Fig. 7).
These data bring up the question why ICD did not trigger a prominent calpain activation on neuronal somata despite the very significant increase in cytosolic Ca2+. A prolonged glutamate excitation is characterized by a rapid collapse of cellular bioenergetics and rapid ATP depletion (Tymianski et al., 1993; Ankarcrona et al., 1995; Budd and Nicholls, 1996; Vergun et al., 1999; Luetjens et al., 2000; Ward et al., 2007). It is possible that the activation of calpains requires an energy-dependent process and a substantial amount of ATP to be present. Alternatively, ICD-associated membrane rupture and cell death may occur too rapidly to allow for a controlled calpain activation. We cannot exclude that calpains were still activated locally and in defined structures, such as dendrites, or were activated at undetectable levels. However, our data demonstrate that any such activity did not contribute to cell death in neurons that underwent ICD.
Our experiments also demonstrate that delayed excitotoxic apoptosis required a functional, bax-dependent mitochondrial apoptosis pathway and that calpains work within this pathway as cell-death executioners. Our data did not show a bid requirement for excitotoxic apoptosis or for calpain-dependent cell death, because bid-deficient and WT mice were similarly sensitive to NMDA-induced apoptosis, and calpain inhibition was equally protective in bid-deficient and WT mice (Fig. 4). Polster et al. (2005) suggested in isolated mitochondria studies that calpain-dependent Bid cleavage triggers the release of pro-apoptotic factors from mitochondria. In intact neurons, such a mechanism may not operate in the setting of excitotoxic apoptosis.
Notably, our study demonstrates that calpain activation occurred downstream of mitochondrial engagement during excitotoxic apoptosis. These findings and the observation that excitotoxic apoptosis is bax-dependent elicit two related and important questions: (1) why does excitotoxic injury not trigger a prominent caspase activation, and (2) why do calpains represent the cell-death executioners during bax-dependent excitotoxic apoptosis whereas in other models of bax-dependent neuronal apoptosis, such as staurosporine exposure, caspases, but not calpains, function as executioners (Lankiewicz et al., 2000)? A key factor may be the extent of mitochondrial Ca2+ overloading in such paradigms. After the initial NMDA-induced cytosolic Ca2+ increase, neurons not only pump Ca2+ back into the extracellular compartment but also reestablish their cytosolic Ca2+ levels by mitochondrial Ca2+ uptake (Nicholls and Scott, 1980; Ward et al., 2005). A bax-dependent mitochondrial outer membrane permeabilization (MOMP) will, however, cause mitochondrial depolarization as a result of the loss of cytochrome c (Goldstein et al., 2000; Luetjens et al., 2000). This may trigger a Ca2+ release from the mitochondrial matrix, capable of activating calpains. Indeed, previous studies have shown that DCD is absent when MOMP is blocked (Concannon et al., 2010). Our calpain–FRET imaging data support such a hypothesis: activation of calpains occurred downstream of Δψm depolarization and with a delay similar to that of DCD (Figs. 1–7). Furthermore, delayed application of the Ca2+ chelator BAPTA-AM after NMDA exposure led to significant delay in DCD and calpain activation. However, this does not rule out a possible contribution of other factors, such as growth factor- and MAPK-dependent calpain activation (Zadran et al., 2010). Because other neuronal apoptosis paradigms do not induce a similar mitochondrial Ca2+ overloading, activation of calpains may be less pronounced or absent (Lankiewicz et al., 2000). Once activated, calpains inhibit the ability of cytochrome c to activate caspases by cleavage of procaspase-3 and procaspase-9, as well as APAF-1 (Chua et al., 2000; Lankiewicz et al., 2000; Reimertz et al., 2001). As a consequence, a caspase-independent excitotoxic apoptosis ensues that is mediated by calpains. Calpains cleave a variety of proteins that are required for neuronal function, including cytoskeletal proteins and transcription factors (Croall and Demartino, 1991). Calpains also process and trigger AIF release from mitochondria and may thereby induce cell death (Cregan et al., 2002; Wang et al., 2004; Cao et al., 2007). Calpains may also further increase neuronal Ca2+ overloading by cleavage and inactivation of the Na+/Ca2+ exchanger (Bano et al., 2005). Indeed, the overexpression of calpastatin or of a noncleavable Na+/Ca2+ exchanger has been shown to inhibit DCD during excitotoxic neuronal injury (Bano et al., 2005). Activation of calpains may therefore initiate a self-amplifying Ca2+-dependent cell death cascade, eventually resulting in the destruction of the neuron.
In conclusion, our study demonstrates that calpains play a critical role in bax-dependent excitotoxic apoptosis but contribute little to the destruction of neuron during excitotoxic necrosis. This mechanism may be of particular importance for the treatment of neuronal injury associated with submaximal glutamate receptor overactivation as evident in the ischemic penumbra or during chronic neurodegeneration.
Footnotes
This work was supported through the National Biophotonics and Imaging Platform, Ireland, funded by the Irish Government's Programme for Research in Third Level Institutions, Cycle 4 and Ireland's European Union Structural Funds Programmes 2007–2013, by Science Foundation Ireland Grant 08/INV1/1949, and by Academy of Finland Grant 127717. We thank Prof. Andreas Strasser (The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia) for the gift of the bid-deficient mice.
The authors declare no conflict of interest.
References
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Baker KL, Heiser AD, Sawyer SD, Dean RL, Elliott PJ, Straub JA. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:537–544. doi: 10.1038/jcbfm.1994.67. [DOI] [PubMed] [Google Scholar]
- Bevers MB, Lawrence E, Maronski M, Starr N, Amesquita M, Neumar RW. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J Neurochem. 2009;108:1237–1250. doi: 10.1111/j.1471-4159.2008.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brorson JR, Marcuccilli CJ, Miller RJ. Delayed antagonism of calpain reduces excitotoxicity in cultured neurons. Stroke. 1995;26:1259–1266. doi: 10.1161/01.str.26.7.1259. discussion 1267. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci U S A. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducin factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Tuffy LP, Weisov á P, Bonner HP, Dávila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase mediated activation of the BH3-only protein Bim couples energy depletion to stressinduced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croall DE, DeMartino GN. Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiol Rev. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- Dietz GP, Dietz B, Bähr M. Bcl-xL protects cerebellar granule neurons against the late phase, but not against the early phase of glutamate-induced cell death. Brain Res. 2007;1164:136–141. doi: 10.1016/j.brainres.2007.06.025. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calciumindependent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygenglucose deprivation in vitro. Neuroscience. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- Hellwig CT, Kohler BF, Lehtivarjo AK, Dussmann H, Courtney MJ, Prehn JH, Rehm M. Real-time analysis of TRAIL/CHX-induced caspase activities during apoptosis initiation. J Biol Chem. 2008;283:21676–21685. doi: 10.1074/jbc.M802889200. [DOI] [PubMed] [Google Scholar]
- Hong SC, Lanzino G, Goto Y, Kang SK, Schottler F, Kassell NF, Lee KS. Calcium-activated proteolysis in rat neocortex induced by transient focal ischemia. Brain Res. 1994;661:43–50. doi: 10.1016/0006-8993(94)91178-9. [DOI] [PubMed] [Google Scholar]
- Hongisto V, Vainio JC, Thompson R, Courtney MJ, Coffey ET. The Wnt pool of GSK-3β is critical for trophic deprivation induced neuronal death. Mol Cell Biol. 2008;28:1515–1527. doi: 10.1128/MCB.02227-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Eriste E, Vassiljev V, Zharkovsky A. Medium transitory oxygen-glucose deprivation induced both apoptosis and necrosis in cerebellar granule cells. Neurosci Lett. 1998;240:21–24. doi: 10.1016/s0304-3940(97)00914-2. [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, Johnstone RW, Dixit VM, Strasser A. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Kristensen BW, Noraberg J, Zimmer J. Comparison of excitotoxic profiles of ATPA, AMPA, KA and NMDA in organotypic hippocampal slice cultures. Brain Res. 2001;917:21–44. doi: 10.1016/s0006-8993(01)02900-6. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci U S A. 1991;88:7233–7237. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- Liu CL, Siesj ö BK, Hu BR. Pathogenesis of hippocampal neuronal death after hypoxia-ischemia changes during brain development. Neuroscience. 2004;127:113–123. doi: 10.1016/j.neuroscience.2004.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Luetjens CM, Bui NT, Sengpiel B, Münstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Scott ID. The role of mitochondria in the regulation of calcium ion transport in synaptosomes. Biochem Soc Trans. 1980;8:264–266. doi: 10.1042/bst0080264. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basañez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- Potts PR, Singh S, Knezek M, Thompson CB, Deshmukh M. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol. 2003;163:789–799. doi: 10.1083/jcb.200307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A, Krieglstein J. Protective effects of calpain inhibitors against neuronal damage caused by cytotoxic hypoxia in vitro and ischemia in vivo. Brain Res. 1993;609:67–70. doi: 10.1016/0006-8993(93)90856-i. [DOI] [PubMed] [Google Scholar]
- Rami A, Ferger D, Krieglstein J. Blockade of calpain proteolytic activity rescues neurons from glutamate excitotoxicity. Neurosci Res. 1997;27:93–97. doi: 10.1016/s0168-0102(96)01123-6. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Lankiewicz S, Poppe M, Prehn JH. Ca2+-induced inhibition of apoptosis in human SH-SY5Y neuroblastoma cells: degradation of apoptotic protease activating factor-1 (APAF-1) J Neurochem. 2001;78:1256–1266. doi: 10.1046/j.1471-4159.2001.00503.x. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Semenova MM, Mäki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9:1579–1590. doi: 10.1523/JNEUROSCI.09-05-01579.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. J Neurophysiol. 1994;72:1973–1992. doi: 10.1152/jn.1994.72.4.1973. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurons. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW, Kushnareva Y, Greenwood S, Connolly CN. Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J Neurochem. 2005;92:1081–1090. doi: 10.1111/j.1471-4159.2004.02928.x. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rehm M, Duessmann H, Kacmar S, Concannon CG, Prehn JH. Real time single cell analysis of Bid cleavage and Bid translocation during caspase-dependent and neuronal caspase-independent apoptosis. J Biol Chem. 2006;281:5837–5844. doi: 10.1074/jbc.M511562200. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisová P, Düssmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Ying DJ, Cui L, Langsdorf J, Yu SP. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004;1022:54–61. doi: 10.1016/j.brainres.2004.06.080. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf BB, Goldstein JC, Stennicke HR, Beere H, Amarante-Mendes GP, Salvesen GS, Green DR. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood. 1999;94:1683–1692. [PubMed] [Google Scholar]
- Wright KM, Smith MI, Farrag L, Deshmukh M. Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J Cell Biol. 2007;179:825–832. doi: 10.1083/jcb.200708086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci. 2010;30:1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel GJ, Lee JM, Choi DW. Reducing calcium overload in the ischemic brain. N Engl J Med. 1999;341:1543–1544. doi: 10.1056/NEJM199911113412011. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]