

Abstract
We have identified the adenine nucleotide translocator (ANT) isoforms ANT1 and ANT2 that are present in the plasma membrane of mouse cerebellar neurons as novel binding partners of the cell adhesion molecule L1. The direct interaction between ANT and L1 is mediated by sites within the fibronectin type III domains of L1 and the first and third extracellular loops of the ANT proteins. We also show that L1 interacts with the ANT binding partner matrix metalloprotease 14 (MMP14) and that the ANT proteins bind directly to the L1 interaction partner glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Moreover, we provide evidence that the functional interplay between L1, ANT proteins, MMP14, and GAPDH at the plasma membrane mediates L1-induced neurite outgrowth of cerebellar neurons. Disruption of this interplay by ANT inhibitors, ANT-derived synthetic peptides, and/or function-blocking MMP14 and ANT antibodies leads to alterations in L1-dependent neurite outgrowth. Stimulation of L1-mediated signaling in cerebellar neurons triggers transient ATP secretion via ANT proteins and leads to transient src family-dependent tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14. Thus, our results indicate that plasma membrane-localized ANT1 and ANT2 regulate L1-mediated neurite outgrowth in conjunction with MMP14.
Introduction
A functional nervous system depends on a number of highly organized processes, such as cell proliferation, migration, differentiation, adhesion, and survival. Many of these processes are regulated by cell adhesion molecules, such as L1. L1 mediates or modulates many of these important events by homophilic or heterophilic interactions at the cell surface of one cell or of neighboring cells or between cells and the extracellular matrix (for review, see Maness and Schachner, 2007). Heterophilic interactions between L1 and other molecules modulate signal transduction pathways and cellular responses triggered by homophilic L1 interactions and play important roles in L1-dependent neuronal cell migration and differentiation, axon growth, guidance, and fasciculation, as well as myelination, synaptic plasticity, learning, and memory (Brümmendorf and Rathjen, 1995; Schachner, 1997; Haspel and Grumet, 2003; Loers and Schachner, 2007; Schmid and Maness, 2008; Zhang et al., 2008). However, for many of these functions it is not known which interacting partners are involved, and it is thus important to identify binding partners of L1. We recently identified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an extracellular binding partner of L1 and showed that GAPDH phosphorylates L1 and regulates L1-induced neurite outgrowth (Makhina et al., 2009). In the present study, we identified the adenine nucleotide translocator (ANT) isoforms ANT1 and ANT2 as binding partners of L1. ANT is also known as ADP/ATP carrier, translocase, or transporter, and four ANT isoforms are known: ANT1, which is highly expressed in differentiated tissues, such as skeletal muscle, heart, and brain; ANT2, which is predominantly expressed in proliferating, regenerating, or undifferentiated cells, such as lymphocytes or cancer cells; ANT3, which is ubiquitously expressed; and ANT4, which is mainly expressed in testis and germ cells (for review, see Dahout-Gonzalez et al., 2006; Klingenberg, 2008; Chevrollier et al., 2011). So far, all ANT isoforms have been considered to be mitochondrial carrier proteins that exchange ATP and ADP between the cytoplasm and the mitochondrial matrix. However, more recent studies indicate that ANT is also present in the plasma membrane of Leishmania (Detke and Elsabrouty, 2008) and cancer cells (Radichev et al., 2009), confirming a previous study on fibroblasts that suggests a plasma membrane localization of ANT (Sigal and Resh, 1993). In Leishmania, plasma membrane ANT has been proposed to function as an ATP sensor and shown to play a crucial role in chemotaxis (Detke and Elsabrouty, 2008). In cancer cells, plasma membrane ANT has been proposed to be involved in energy metabolism and chemotaxis (Radichev et al., 2009).
In our study, we demonstrated that ANT1 and ANT2 interact with L1 and GAPDH at the plasma membrane of cerebellar neurons and that they modulate L1-induced neurite outgrowth. In addition, we showed that L1 interacts with the ANT binding partner matrix metalloprotease 14 (MMP14) and that L1 stimulation of cerebellar neurons triggers ATP release and src-dependent tyrosine phosphorylation of L1, ANT, and MMP14. These results indicate a complex functional interplay between L1, ANT, GAPDH, and MMP14 at the neuronal cell surface.
Materials and Methods
Animals.
C57BL/6J mice of either sex were used as wild-type mice. L1-deficient mice (Rolf et al., 2001) of either sex were kept as heterozygous breeding pairs and were maintained on a mixed genetic background (129SVJ × C57BL/6 × Black Swiss). Mice were kept at standard laboratory conditions with food and water supply ad libitum and with an artificial 12 h light/dark cycle. All experiments were conducted in accordance with the German and European Community laws on protection of experimental animals, and all procedures used were approved by the responsible committee of the State of Hamburg.
Antibodies and reagents.
L1 antibodies and recombinant Fc fusion proteins (Makhina et al., 2009) as well as rabbit pan-ANT and ANT1- and ANT2-specific antibodies (Vyssokikh et al., 2001) have been described previously. Monoclonal antibody against GAPDH was from Millipore Bioscience Research Reagents, monoclonal MMP14 antibody LEM-2/63.1 was from Abcam, monoclonal L1 antibody 172-R and rabbit βIII-tubulin antibody were from Covance, mouse cofilin antibody was from BD Biosciences, mouse pan-ANT antibody SLC25A4 was from Abnova, and goat pan-ANT and rabbit Bcl-2 antibodies were from Santa Cruz Biotechnology. Streptavidin conjugated with horse radish peroxidase (HRP) and recombinant catalytically active MMP14 and MMP14 comprising the prodomain, the catalytic domain, and the hemopexin domain were from ThermoScientific. Human Fc fragment, nonimmune IgG from goat, rat, mouse, or rabbit, and all secondary antibodies were obtained from Dianova. GAPDH, atractyloside, and carboxyatractyloside were purchased from Sigma-Aldrich. The following synthetic peptides were from Schafer-N: peptide comprising amino acids 40–70, 144–180, 242–272, or 235–253 of mouse ANT2 and containing sequences of the first (HASKQITADKQYKGIIDCVVRIPKEQGVLSF), second (DVGKAGAEREFKGLGDCLVKIYKSDGIKGLYQGFNVS), or third (SGRKGTDIMYTGTLDCWRKIARDEGSKAFFK) extracellular loop; peptides containing the signature of mitochondrial carriers, RRRMMM, and 13 flanking amino acids (RRRMMMQSGRKGTDIMYTG), the N-terminal amino acids 1–28 of ANT1 (MGDQALSFLKDFLAGGIAAAVSKTAVAPIERVKLLLQV), or the N-terminal amino acids 2–13 of ANT1 (GDQALSFLKDF) or ANT2 (TDAAVSFAKDF) used for the generation of the ANT1- and ANT2-specific antibodies.
Preparation of brain homogenate and subfractions.
The preparation of brain homogenate and brain subfraction was described previously in detail (Makhina et al., 2009). Briefly, brains from 2- to 3-month-old C57BL/6J mice were homogenized in homogenization buffer (50 mm Tris/HCl, pH 7.4, 0.32 m sucrose, 1 mm CaCl2, 1 mm MgCl2). The homogenate was centrifuged at 17,000 × g, and the resulting pellet was subjected to centrifugation on a step gradient of 1.2, 1.0, and 0.8 m sucrose. The 1.0/1.2 m sucrose interface, which was enriched in synaptosomal membranes, was isolated and resuspended in PBS, pH 7.3, containing 1% Triton X-100 and taken as a detergent-solubilized synaptosomal membrane subfraction.
Coimmunoprecipitation and pull-down experiments.
For immunoprecipitation, primary antibodies and a detergent-solubilized synaptosomal membrane subfraction or brain homogenate solubilized in RIPA buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1% NP-40, protease inhibitor cocktail; Roche) were incubated with Protein A/G beads (ThermoScientific) overnight at 4°C. In pull-down experiments, L1-Fc and NCAM-Fc were incubated with brain homogenate and Protein A/G beads overnight at 4°C. The precipitates were washed three times with RIPA buffer and subjected to Western blot analysis.
Mass spectrometry and post-source decay analysis.
Coimmunoprecipitation using monoclonal L1 antibody, matrix-assisted laser desorption/ionization (MALDI) mass spectrometry, post-source decay analysis, and data analysis was performed as described in detail previously (Makhina et al., 2009). Briefly, L1 antibody 555 immobilized on Epoxy-270 magnetic beads (Invitrogen) and detergent-solubilized synaptosomal membrane subfraction were incubated. Then, bound proteins were eluted and subjected to SDS-PAGE followed by silver staining. Protein bands were excised from the gel, treated with DTT, alkylated with iodoacetamide, and washed with deionized water, 50% acetonitrile/water and acetonitrile. After in-gel digestion with trypsin (Promega), the samples were desalted and applied to C18 ZipTip pipette tips (Millipore) prewashed with 50% acetonitrile/water and equilibrated with 0.1% trifluoroacetic acid/water. After several washes of the ZipTip, tryptic peptides were eluted, MALDI mass spectra were recorded, and post-source decay analysis was performed.
Western blot analysis and chemical cross-linking.
Western blot analysis and cross-linking was performed as described previously (Makhina et al., 2009). Briefly, samples were separated by SDS-PAGE followed by transfer to nitrocellulose membrane (Protran; Schleicher and Schuell). Membranes were blocked with 5% nonfat dry milk powder in PBS and incubated with primary antibody, washed in PBS with 0.05% Tween 20 (PBS-T), and probed with HRP-conjugated secondary antibody. For the detection of biotinylated proteins, membranes were incubated with HRP-conjugated streptavidin. Detection was performed using chemiluminescence (Pierce).
For cross-linking, sulfo-SBED reagent (Perbio) dissolved in DMSO was incubated with L1-Fc and NCAM-Fc in PBS. After dialysis, Protein A magnetic beads (Invitrogen) were incubated with the dialyzed samples, washed in PBS, and incubated with BS-3 cross-linker (Pierce). After washing with Tris-buffered saline, pH 7.3, the beads were incubated with detergent-solubilized synaptosomal membrane subfraction and the samples were then treated with UV light (365 nm) using an ultraviolet cross-linker (GE Healthcare). After successive washing steps with PBS, PBS with 1% Triton X-100, 0.1% SDS, 0.5% SDS, or 1% SDS, proteins were eluted from the magnetic beads by boiling in SDS sample buffer (60 mm Tris/HCl, pH 6.8, 2% SDS, 1% β-mercaptoethanol, 10% glycerol, 0.02% bromophenol blue).
ELISA and label-free binding assay using photonic crystal optical biosensors.
ELISA was performed as described by Makhina et al. (2009). Briefly, GAPDH or ANT at concentrations of 5 μg/ml in PBS or synthetic peptides at a concentration of 50 μg/ml in PBS were coated on Maxisorb plastic modules (Nunc). After washing with PBS-T, the wells were incubated with 2% BSA in PBS, washed with PBS-T, and incubated with different concentrations of the solute protein in PBS containing 1% BSA, 1 mm CaCl2, 1 mm MgCl2, and 1 mm MnCl2. Wells were washed with PBS-T and incubated with primary antibodies, washed with PBS-T, and incubated with secondary antibodies coupled to HRP. Bound protein was detected by using ABTS reagent (Sigma) for color development and measuring absorption at 405 nm using an ELISA reader.
As an alternative to ELISA, photonic crystal optical biosensors were used to investigate direct protein–protein interactions. Optical biosensors based on photonic crystals measure the kinetic adsorption of biomolecular materials through their greater dielectric permissivity at optical wavelengths compared to water (Cooper, 2002). The biosensor used (SRU Biosystems) incorporates a narrowband guided-mode resonance reflectance filter, in which the reflected color is modulated by the attachment/detachment of biochemical material to the surface (Cunningham et al., 2004). This allows detection of protein–protein, e.g., antigen–antibody, interactions without labeling of proteins, thereby minimizing side effects und unspecific binding. Plates (384 well) with TiO surface (SRU Biosystems) were washed three times with Dulbecco's PBS containing Ca2+ and Mg2+ (PAA Laboratories) (PBS+) and coated overnight at 4°C with 125 ng of protein or peptide per well. Wells were then washed three times with PBS+ and the peak wavelength shift was measured (BIND PROFILER; SRU Biosystems) to control for sufficient coating of proteins. Wells were then blocked with 2% BSA in PBS+ for 3 h at room temperature. After three washes with PBS+, peak wavelength shift was determined, and different amounts of ligands in PBS+ were added to the wells. Binding of ligands to substrate-coat protein or peptide was measured every 30 s for 60 min. Values obtained from wells without ligands were set as background and subtracted from values obtained with ligand solution. All interactions were performed in triplicate.
Cell culture and cell surface biotinylation.
Primary cultures of cerebellar granule cells were prepared from 6- to 7-day-old C57BL/6J mice and cultured as described previously (Makhina et al., 2009). For cell surface biotinylation, cerebellar neurons were seeded at a density of 2 × 106 cells per well in 6-well plates (Corning) and maintained for 24 h. The cells were washed twice with ice-cold PBS-2+ (PBS, 0.5 mm CaCl2, 2 mm MgCl2) and incubated for 10 min on ice with 0.5 mg of sulfo-NHS-LS-biotin (Pierce) freshly dissolved in PBS-2+. After treating the cells with 20 mm glycine in PBS-2+ for 5 min on ice, cells were washed twice with ice-cooled PBS-2+ and lysed with RIPA buffer for 30 min at 4°C with mild shaking. Lysed cells were collected using a rubber scraper, centrifuged for 10 min at 1,000 × g and 4°C. Streptavidin-coupled magnetic beads (Invitrogen) were added to the lysate and incubated overnight at 4°C. After washing of magnetic beads, proteins were eluted by boiling in SDS sample buffer and subjected to Western blot analysis. Estimation of the amount of extracellular GAPDH relative to the total cellular GAPDH was done by analysis of the band intensity using the TINA software program (TINA version 2.10; Raytest).
Immunocytochemistry and confocal laser scanning microscopy.
Freshly dissociated cerebellar granule cells were seeded on coverslips coated with poly-l-lysine (PLL) (Sigma-Aldrich) at a density 1–2.5 × 105 cells per well in 24-well plates and maintained for 24 h in serum-free medium (Makhina et al., 2009). One hour before the experiment, the medium was exchanged, and cultures were placed on ice and incubated with primary antibodies for 15 min, washed carefully three times with fresh serum-free medium, fixed using 4% paraformaldehyde in PBS, blocked using 0.5% BSA in PBS, and incubated with the corresponding secondary antibodies coupled to the fluorescent dyes Cy2, Cy3, or Cy5. ANT-specific antibodies (3 μl) were preincubated with 10 μl of a 1 mg/ml peptide solution. For permeabilization, cells were treated with 0.3% Triton X-100 for 30 min on ice after fixation. Coverslips were embedded in Aqua PolyMount medium (Polysciences) and analyzed with a Zeiss LSM510 argon-krypton confocal laser scanning microscope.
Neuritogenesis of cerebellar neurons and ATP determination.
Primary cerebellar granule cells were seeded at a density of 2.5–5 × 104 cells/well in 48-well plates (Corning) coated with 10 μg/ml PLL or with 10 μg/ml PLL and 20 μg/ml L1-Fc, and cultured in serum-free medium. ANT inhibitors (5 μm), antibodies (10 μg IgG/ml; 10 μl antiserum/ml), or peptides (100 μg/ml) were added 1 h after the cell seeding. Cells were maintained for 24 h, washed carefully with prewarmed medium, fixed in 2.5% glutaraldehyde, and stained using 1% (w/v) toluidine blue and 1% methylene blue in 1% sodium tetraborate for 1 h at room temperature. Neurite outgrowth was analyzed by measuring the total length of neurites in the Axiovert microscope with AxioVision 4.6 imaging system (Zeiss).
For ATP determination, cell culture medium was removed upon treatment of cerebellar neurons and 100 μl per 48 wells of hot (95°C) 10 mm Tris, pH 7.4, and 0.1 mm EDTA was immediately added to the cells. After resuspension of the cells and transfer to a tube, cell lysates were boiled for 2 min and centrifuged at 14,000 × g for 2 min. An ATP determination kit (Perbio) and a luminometer (Victor3; PerkinElmer) were used to determine the ATP level by luminescence after addition of 100 μl ATP determination reagent to 10 μl of the sample.
Results
ANT binds to the extracellular domain of L1
Recently, we have identified GAPDH as a binding partner of L1 by mass spectrometric analysis after immunoprecipitation from mouse brain using the monoclonal L1 antibody 555, which recognizes an epitope in the third fibronectin type III domain (Makhina et al., 2009). In this immunoprecipitates, we also observed an ∼32 kDa protein band, which was not detectable in the immunoprecipitate obtained with a nonimmune control antibody (Fig. 1A). Mass spectrometry of peptides obtained after tryptic digestion of this protein revealed that four of the detected peptides could be assigned to the mouse ANT isoforms ANT1 and ANT2, while three peptides specifically matched to ANT1 and one peptide to ANT2 (Table 1). To verify that ANT1 and/or ANT2 interact with L1 in mouse brain, immunoprecipitations with monoclonal L1 antibody 172-R against an epitope in the intracellular domain and with L1 antibody 555 or polyclonal L1 antibody against extracellular domains were performed, and ANT antibodies were used for Western blot analysis of immunoprecipitates. A rabbit pan-ANT antibody detected ANT as a 32 kDa protein in all three L1 immunoprecipitates, but not in the immunoprecipitates obtained by the corresponding control antibodies (Fig. 1B). Conversely, ANT antibodies directed to N-terminal ANT1- or ANT2-specific sequences were used for immunoprecipitation, and the immunoprecipitates were probed by Western blot analysis with L1 antibodies. The monoclonal L1 antibody 555 and the polyclonal L1 antibody against extracellular epitopes detected an L1 fragment of ∼90 kDa in the immunoprecipitates obtained from a detergent-solubilized brain homogenate and synaptosomal membrane subfraction using the ANT1- and ANT2-specific antibody (Fig. 1C). No band was detectable when a nonimmune control antibody was used for immunoprecipitation (Fig. 1C). The 90 kDa L1 fragment was also immunoprecipitated in minor amounts by the monoclonal L1 antibody together with a 140 kDa L1 fragment and the 200 kDa full-length L1, while the polyclonal L1 antibody predominantly immunoprecipitated this fragment and minor amounts of a 140 kDa L1 fragment (Fig. 1C). In brain homogenate of wild-type mice, both L1 antibodies recognized mainly the 200 and 140 kDa L1 forms, while the 90 kDa fragment was detected only in small amounts (Fig. 1C). No L1 positive bands were detectable when brain homogenate of L1-deficient mice was used for immunoprecipitation (data not shown), proving that the 90 kDa band coimmunoprecipitated with ANT1 and ANT2 represents an L1-specific band. Western blot analysis with the L1 antibody 172-R against the intracellular domain revealed that a 90 kDa L1 fragment and small amounts of a 140 kDa fragment were detected in immunoprecipitates obtained with the rabbit pan-ANT as well as with the ANT1- and ANT2-specific antibodies, but not in the immunoprecipitates of the control antibody (Fig. 1D). In brain homogenate, this L1 antibody recognized the full-length 200 kDa L1, a 140 kDa L1 form and a 90 kDa L1 fragment (Fig. 1D). Since the 90 kDa L1 fragment coimmunoprecipitated by ANT1- and ANT2-specific antibodies was detected by L1 antibodies directed either to the extracellular or to the intracellular part of L1, one has to conclude that the 90 kDa fragment consists of part of the extracellular domain as well as of the transmembrane and intracellular domains. The combined results of the immunoprecipitation experiments indicate that the 32 kDa band that coimmunoprecipitated with L1 corresponds to ANT1 and ANT2, and that a 90 kDa L1 fragment, but not the full-length L1, interacts with ANT1 and ANT2.
Figure 1.
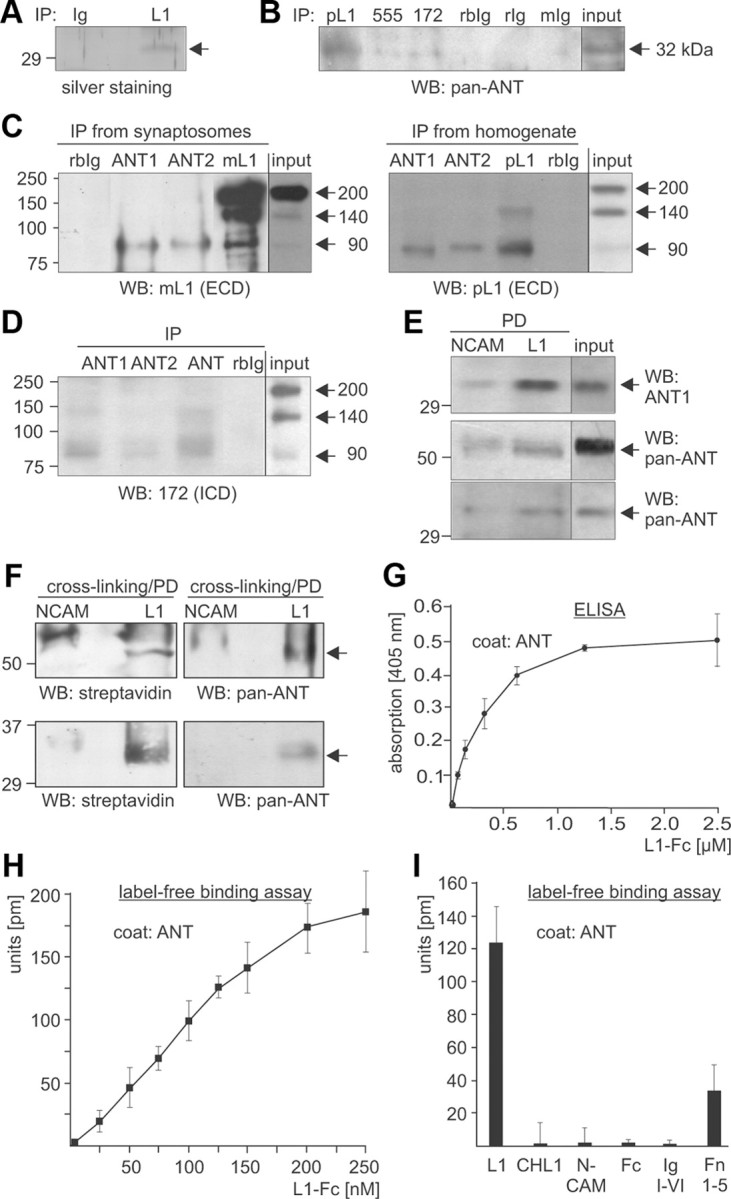
ANT is coimmunoprecipitated with L1 and binds to the extracellular domain of L1. A, Immunoprecipitation from a detergent-solubilized membrane subfraction using monoclonal L1 antibody 555 (L1) or nonimmune rat antibody (IgG) and subsequent SDS-PAGE and silver staining allowed the identification of ANT1 and ANT2 (arrow) as potential L1 interaction partners. B, A detergent-solubilized brain homogenate was subjected to IP using polyclonal rabbit L1 antibody (pL1), monoclonal rat L1 antibody 555 (555), and monoclonal mouse L1 antibody 172-R (172), or the corresponding nonimmune rabbit (rbIg), rat (rIg), and mouse (mIg) antibodies. Brain homogenate (input) and the immunoprecipitates (IP) were probed by Western blot (WB) analysis with rabbit pan-ANT antibody. C, A detergent-solubilized synaptosomal membrane subfraction (IPs from synaptosomes; left) or brain homogenate (IPs from homogenate; right panel) was subjected to IP using rabbit ANT1- and ANT2-specific antibodies, nonimmune rabbit (rbIg) antibody, and monoclonal L1 antibody 555 (mL1) or polyclonal L1 antibody (pL1). D, A detergent-solubilized brain homogenate was subjected to immunoprecipitation using ANT1- and ANT2-specific antibodies, rabbit pan-ANT antibody (ANT), or nonimmune rabbit antibody (rbIg). C, D, Brain homogenate or the synaptosomal membrane subfraction (input) and immunoprecipitates were subjected to WB analysis with L1 antibody 555 (mL1) or polyclonal L1 antibody (pL1) against epitopes in the extracellular domains (ECD) (C) or L1 antibody 172-R (172) against the intracellular L1 domain (ICD) (D). E, L1-Fc and NCAM-Fc were incubated with detergent-solubilized brain homogenate, and proteins bound to L1-Fc and NCAM-Fc were isolated by pull-down (PD) using Protein A beads. Brain homogenate (input) and pulled down proteins bound to L1-Fc (L1) or NCAM-Fc (NCAM) were probed by Western blot analysis with rabbit ANT1 antibody (top) or mouse pan-ANT antibody (middle, short exposure time; bottom, long exposure time). F, L1-Fc and NCAM-Fc were conjugated to the trifunctional cross-linker sulfo-SBED, immobilized to Protein A-coupled magnetic beads, and incubated with a detergent-solubilized synaptosomal membrane subfraction. Upon chemical cross-linking, proteins bound to L1-Fc (L1) or NCAM-Fc (NCAM) were isolated by PD and subjected to WB analysis using HRP-coupled streptavidin or rabbit pan-ANT antibody (top, short exposure time; bottom, long exposure time). G, Substrate-coated ANT purified from bovine heart was incubated with different concentrations of soluble L1-Fc, and binding of L1 to ANT was determined by ELISA using L1 antibody 555. H, Binding of L1 to ANT is shown by label-free binding assay using substrate-coated purified ANT and different concentrations of soluble L1-Fc. I, Label-free binding assay using substrate-coated purified ANT and equimolar amounts of soluble L1-Fc, CHL1-Fc, NCAM-Fc, Fc, or L1 fragments comprising the immunoglobulin-like domains I-VI (L1/Ig I-VI) or the fibronectin type III domains 1–5 (L1/Fn 1–5) fused to Fc. Mean values ± SEM (n = 3) are shown (G–I).
Table 1.
Mass spectrometry of the 32 kDa protein band isolated by immunoprecipitation with immobilized L1 antibody revealed seven and five masses that match with mouse ANT1 and ANT2, respectively
| m/z submitted | MH+ matched | ΔDa | Start | End | Missed cleavages | Peptide sequence | |
|---|---|---|---|---|---|---|---|
| ANT1 | 902.480 | 902.484 | −0.004 | 273 | 280 | 0 | GAWSNVLR |
| 1004.580 | 1004.552 | 0.028 | 97 | 105 | 0 | QIFLGGVDR | |
| 1169.580 | 1169.574 | 0.006 | 64 | 72 | 0 | EQGFLSFWRa | |
| 1205.600 | 1205.584 | 0.016 | 189 | 199 | 0 | AAYFGVYDTAK | |
| 1219.640 | 1219.668 | −0.028 | 11 | 23 | 0 | DFLAGGIAAAVSK | |
| 1446.640 | 1446.742 | −0.102 | 81 | 92 | 0 | YFPTQALNFAFK | |
| 1755.820 | 1755.935 | −0.115 | 281 | 296 | 1 | GMGGAFVLVLYDEIKK | |
| ANT2 | 902.480 | 902.484 | −0.004 | 273 | 280 | 0 | GAWSNVLR |
| 1121.580 | 1121.574 | 0.006 | 64 | 72 | 0 | EQGVLSFWRa | |
| 1219.640 | 1219.668 | −0.028 | 11 | 23 | 0 | DFLAGGIAAAVSK | |
| 1446.640 | 1446.742 | −0.102 | 81 | 92 | 0 | YFPTQALNFAFK | |
| 1755.820 | 1755.935 | −0.115 | 281 | 296 | 1 | GMGGAFVLVLYDEIKK |
aSequence was confirmed by post-source decay fragment ion mass analysis.
To investigate whether the interaction of L1 with ANT1 and/or ANT2 was mediated via its extracellular or intracellular domain, we performed pull-down experiments using brain homogenates and recombinant proteins comprising the intracellular domains of L1 and NCAM fused to a His tag or the extracellular domains of L1 and NCAM fused to human Fc (L1-Fc, NCAM-Fc). Western blot analysis with ANT antibodies showed that none of the intracellular domains pulled down ANT (data not shown). In contrast, L1-Fc precipitated significant amounts of a 32 kDa ANT protein, while NCAM-Fc precipitated only very small amounts of ANT (Fig. 1E), suggesting that ANT interacts specifically with the extracellular domain of L1. Interestingly, using a mouse pan-ANT antibody, we detected ANT predominantly as an ∼60 kDa protein in the L1-Fc precipitate, while the 32 kDa band was detectable by this antibody only after longer exposure times (Fig. 1E).
To further substantiate that the extracellular domain of L1 mediates the interaction with ANT, chemical cross-linking was performed. Recombinant L1-Fc and NCAM-Fc were coupled to the biotin-carrying cross-linker sulfo-SBED and to Protein A beads. These beads were incubated with a detergent-solubilized synaptosomal membrane subfraction. After exposure to UV light, which led to covalent attachment of proteins bound to L1-Fc and NCAM-Fc, bound proteins were eluted from the beads in the presence of reducing agents. Under these conditions, the disulfide bridge in the cross-linker was cleaved, resulting in detachment of bound proteins from NCAM-Fc and L1-Fc and a concomitant transfer of the biotin label to the bound proteins. Biotin-labeled L1-Fc or NCAM-Fc binding proteins were detectable by Western blot analysis using streptavidin-HRP. Biotin-labeled bands of 60 and 32 kDa were seen when L1-Fc was used for cross-linking, but not when NCAM-Fc was used (Fig. 1F). A 60 kDa band and a 32 kDa band were also detected by Western blot analysis using the mouse pan-ANT antibody (Fig. 1F). This result suggests that the biotin-labeled 32 and 60 kDa bands represent different forms of ANT and that the extracellular domains of L1 bind to ANT.
To test whether L1 interacts directly with ANT via its extracellular domain, an ELISA-based binding study with substrate-coated ANT purified from heart and soluble L1-Fc was performed. L1-Fc showed a concentration-dependent binding to substrate-coated ANT (Fig. 1G), while Fc alone did not bind to substrate-coated ANT (data not shown). A concentration-dependent binding of L1-Fc to substrate-coated ANT was also observed in a label-free binding assay using an optical biosensor system (Fig. 1H), while no binding of CHL1-Fc, NCAM-Fc, or Fc used as a control was detectable (Fig. 1I). To narrow down the binding site(s) for ANT on the extracellular domain of L1, recombinant L1 fragments comprising the immunoglobulin-like domains I–VI and the fibronectin type III domains 1–5 fused to Fc were used in a label-free binding assay with substrate-coated purified ANT. The L1 fusion protein comprising the immunoglobulin-like domains I-VI did not bind to ANT, whereas that comprising fibronectin type III domains 1–5 showed binding to ANT (Fig. 1I). In summary, the results show that L1 interacts with ANT via its extracellular fibronectin type III domains.
ANT is present in plasma membranes of cerebellar neurons
Since it has been reported that ANT is located at the plasma membrane of Leishmania, fibroblasts, and cancer cells (Sigal and Resh, 1993; Detke and Elsabrouty, 2008; Radichev et al., 2009), and since we demonstrated that ANT interacts with the extracellular domain of L1, we tested whether ANT1 and/or ANT2 are present at the cell surface of cultured cerebellar neurons. Therefore, live primary cultures of cerebellar neurons were subjected to cell surface biotinylation, and biotinylated proteins were isolated using streptavidin-conjugated beads. Western blot analysis with ANT1- and ANT2-specific antibodies showed that ANT1 and ANT2 were isolated from cerebellar neurons as biotinylated protein migrating with an apparent molecular weight of ∼60 kDa (Fig. 2A,B). In the input control, ANT1 and ANT2 were also seen as 60 kDa band (Fig. 2A). After long exposure times, very small signal intensities were obtained for 32 kDa ANT1 and ANT2 bands (Fig. 2A,B). Densitometric analysis of the signal intensities revealed that 0.6 and 2.5% of total ANT1 and ANT2, respectively, were biotinylated and thus, were located at the cell surface. Western blot analysis using an antibody against the cytoplasmic protein tubulin revealed that tubulin was found in large amounts in the cell lysate, but no biotinylated tubulin was detectable (Fig. 2C), showing the intactness of the neurons during cell surface biotinylation. The biotinylated 32 and 60 kDa protein bands were also detectable by streptavidin-HRP in immunoprecipitates obtained after surface biotinylation using the ANT1- and ANT2-specific antibodies (Fig. 2D). The results clearly demonstrate that ANT1 and ANT2 are present at the cell surface of cerebellar neurons and thus are located in the plasma membrane of these neurons.
Figure 2.
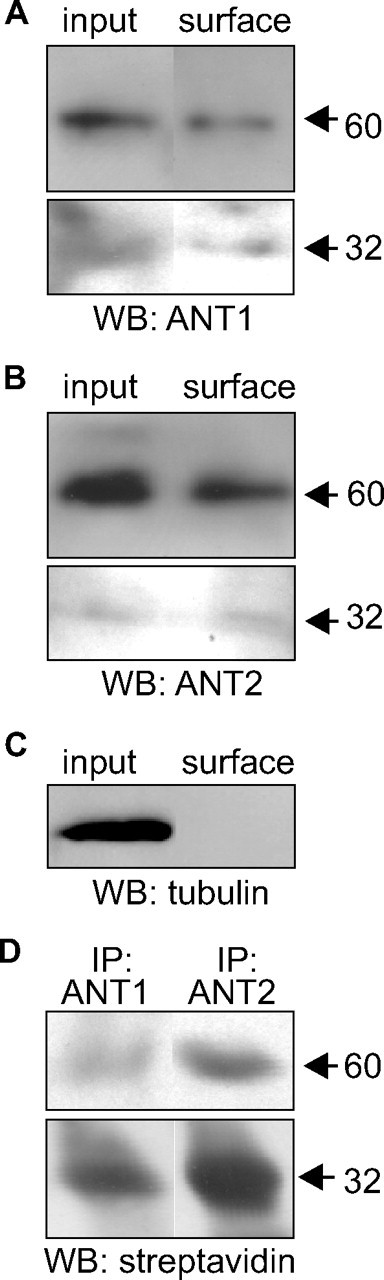
Cell surface localization of ANT1 and ANT2. A–D, After cell surface biotinylation of live cerebellar neurons, biotinylated cell surface proteins were isolated using streptavidin-coupled beads (A–C), and ANT1 and ANT2 were immunoprecipitated using ANT1- and ANT2-specific antibodies (D). Cell lysates (input), isolated biotinylated proteins (surface) and the ANT1 and ANT2 immunoprecipitates (IP) were subjected to Western blot (WB) analysis using either ANT1-specific (ANT1; A), ANT2-specific (ANT2; B), or tubulin (C) antibodies or using HRP-conjugated streptavidin (D). The 60 kDa ANT protein seen after short time exposure (top) and the 32 kDa ANT protein seen after long time exposure (bottom) are indicated.
To confirm the plasma membrane localization of ANT, ANT antibodies were used for immunocytochemical analysis of live cerebellar neurons. Confocal microscopic analysis using goat pan-ANT and L1 555 antibodies showed strong ANT and L1 immunoreactivity at the surface of neuronal cell bodies and superimposition of the stainings indicated a pronounced colocalization of ANT with L1 at the surface of neurons (Fig. 3A). A similar remarkable coimmunostaining of ANT and L1 at the cell surface of live cerebellar neurons was obtained when polyclonal L1 and rabbit pan-ANT antibodies were used and when mouse pan-ANT antibody was used in combination either with monoclonal L1 antibody 555 or rabbit polyclonal L1 antibody (data not shown).
Figure 3.
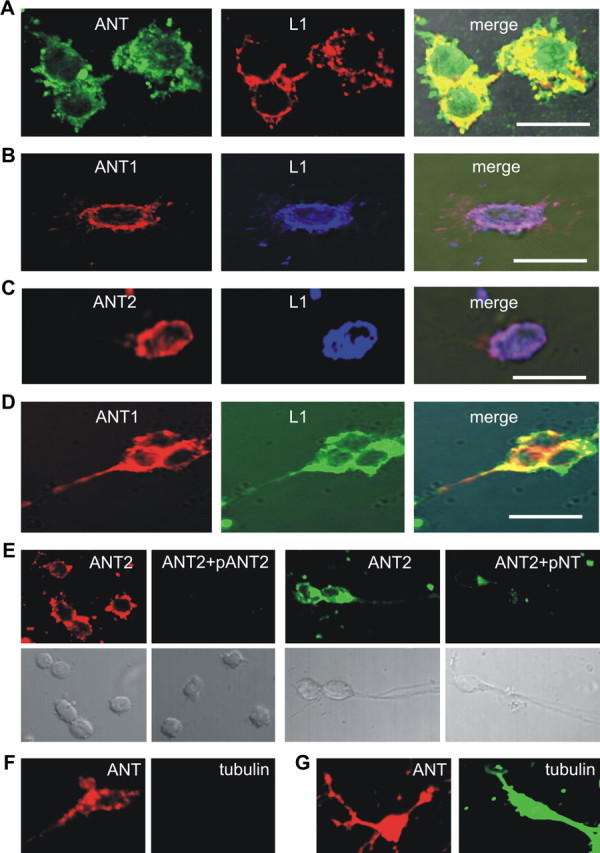
ANT1 and ANT2 colocalize with L1 at the cell surface of live cerebellar neurons. A–E, Live cerebellar neurons were stained with rat monoclonal L1 antibody 555 (A–D) and goat pan-ANT antibody (A), ANT1-specific (B, D) and ANT2-specific (C) antibodies, and ANT2-specific antibody that had been preincubated with peptides containing the N-terminal amino acids 2–13 of ANT2 (pANT2) or 1–28 of ANT1 (pNT) (E). F, G, Live neurons were stained with goat pan-ANT and tubulin antibodies (F), and fixed and permeabilized neurons were stained either with goat pan-ANT or tubulin antibodies (G). Confocal microscopy shows colocalization of ANT with L1 in the merged images (A–D). Scale bars: 10 μm.
Surprisingly, the rabbit ANT1- and ANT2-specific antibodies, which are directed against sequences at the extreme N termini that are proposed to be exposed to the cytoplasmic side of the plasma membrane (see Fig. 6A) also showed cell surface immunostaining on live neurons and strong coimmunostaining with the L1 antibody 555 on cell bodies and along neurites (Fig. 3B–D). To verify that the antibodies to the N termini of ANT1 and ANT2 react with their epitopes at the cell surface, we used ANT isoform-specific synthetic peptides to inhibit the binding of these ANT antibodies. ANT2 immunostaining of live cerebellar neurons was abolished after preincubation of the ANT2-specific antibody with the peptide to which the ANT2-specific antibody was directed and was reduced after incubation with the peptide comprising the N-terminal 28 amino acids of ANT1 (Fig. 3E). ANT1 immunostaining was also reduced in the presence of the peptide comprising the N-terminal 28 amino acids of ANT1 (data not shown). Using ANT antibodies and antibodies against the cytoplasmic proteins tubulin or cofilin or against the mitochondrial protein Bcl-2 for coimmunostaining of live cerebellar neurons, we observed ANT-positive immunostaining at the surface of live neurons, but no staining of tubulin (Fig. 3F), cofilin, or Bcl-2 (data not shown). When the antibodies were used for immunostaining of fixed and permeabilized neurons, an intense intracellular staining of ANT and tubulin (Fig. 3G) as well as of cofilin or Bcl-2 (data not shown) was found. These results demonstrate that ANT1 and ANT2 are present at the cell surface of live neurons in colocalization with L1 on cell bodies and along neurites. In addition, these results show that the extreme N termini of ANT1 and ANT-2 are located extracellularly.
Figure 6.
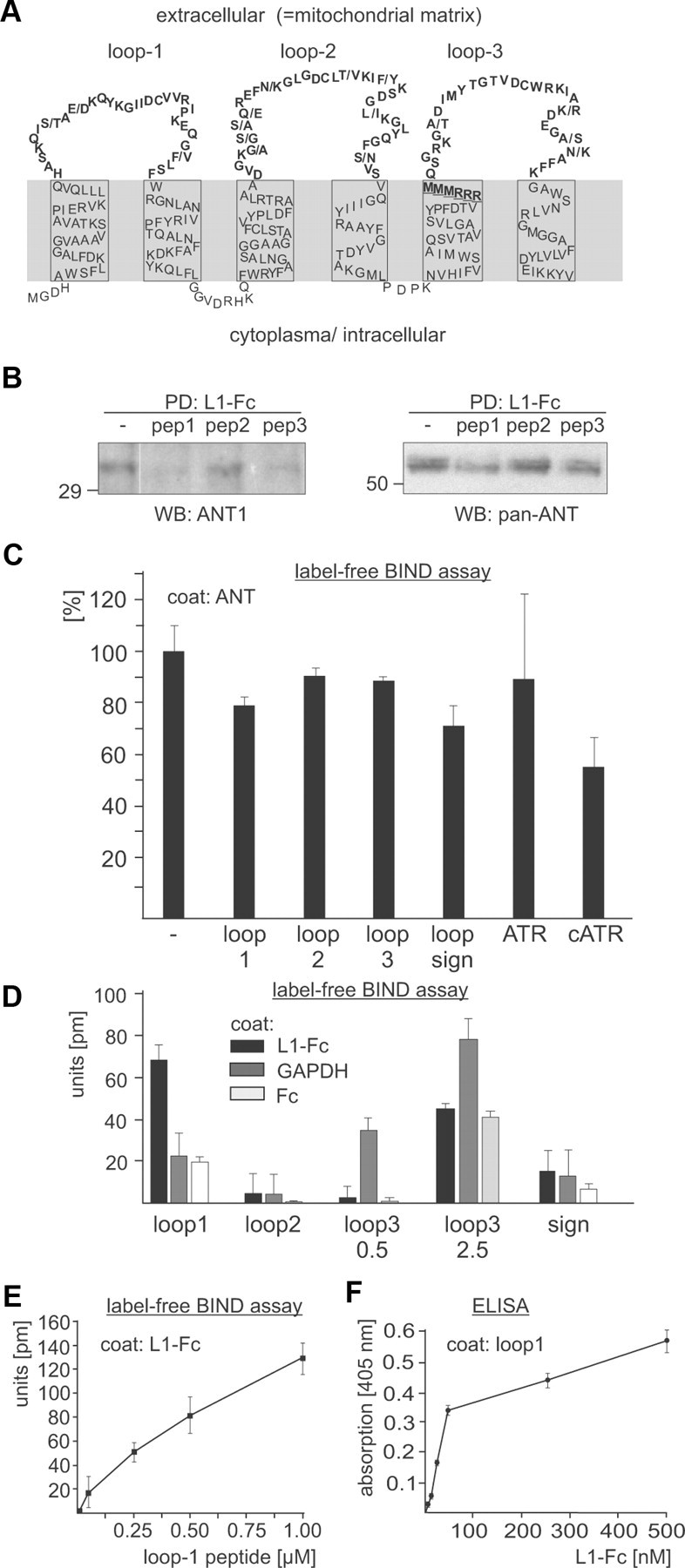
L1 binds directly to the first N-terminal loop of ANT. A, Schematic presentation of the ANT structure and topology. The murine ANT2 sequence is depicted and the differences between ANT1 and ANT2 sequences are indicated by dashes. The loops which are exposed to the mitochondrial matrix or the extracellular space are shown in bold. The RRRMMM motif is underlined. B, Synthetic peptides comprising loop 1, loop 2, and loop 3 of ANT were applied as competitors to a pull-down (PD) assay using L1-Fc and a detergent-solubilized brain homogenate. Protein precipitates were subjected to Western blot (WB) analysis with ANT1-specific or pan-ANT antibodies. C, L1-Fc was preincubated with synthetic peptides or the ANT inhibitors atractyloside (ATR) and carboxyatractyloside (cATR) and subjected to a label-free binding assay using substrate-coated ANT. Binding of L1 to ANT in the presence of inhibitors and peptides relative to binding in their absence, which was set to 100%, is shown. D, Label-free binding assay is shown using L1-Fc, Fc, and GAPDH as substrate coats and 0.5 or 2.5 μm peptide containing loop 1, loop 2, loop 3, or the signature motif (sign) of ANT. E, Binding of peptide comprising loop 1 to substrate-coated L1-Fc is shown by label-free binding assay. F, Binding of L1-Fc to substrate-coated peptide comprising loop 1 is shown by ELISA. Mean values ± SD (n = 3) are shown (C–F).
ANT binds to the L1 binding partner GAPDH, and L1 binds to the ANT binding partner MMP14
We showed previously that extracellular GAPDH binds to the extracellular domain of L1 (Makhina et al., 2009), and it has been reported that ANT2 binds to the matrix metalloprotease MMP14 (Radichev et al., 2009). Since we observed here that L1 interacts with ANT1 and ANT2, we examined whether the L1 binding partners ANT and GAPDH as well as the ANT binding partners L1 and MMP14 interact with each other. To address this question, we first performed immunoprecipitation experiments and found by Western blot analysis with a monoclonal GAPDH antibody that ANT1 and ANT2 antibodies coimmunoprecipitated GAPDH from a detergent-solubilized synaptosomal membrane subfraction, while a nonimmune control antibody did not immunoprecipitated GAPDH (Fig. 4A). Moreover, the polyclonal L1 antibody, but not a nonimmune control antibody, coimmunoprecipitated MMP14 from a detergent extract of mouse brain as seen in Western blot analysis with a monoclonal MMP14 antibody (Fig. 4B). By ELISA, we then analyzed whether ANT and GAPDH interact directly and observed a concentration-dependent and saturable binding of purified ANT to substrate-coated GAPDH (Fig. 4C), verifying a direct interaction between GAPDH and ANT. This direct interaction was confirmed by a label-free binding assay: a concentration-dependent binding of GAPDH to substrate-coated purified ANT was observed (Fig. 4D). In addition, a concentration-dependent binding of L1-Fc, but not Fc, to recombinant MMP14 was observed in a label-free binding assay (Fig. 4E). The direct interaction of MMP14 and L1 suggests that MMP14 could proteolytically cleave L1 and generate the 90 kDa fragment that interacts with ANT1 and ANT2. To test this idea, we incubated L1-Fc or L1 immunopurified from mouse brain without or with recombinant catalytically active MMP14. No significant cleavage of L1 by MMP14 was observed (data not shown), excluding that MMP14 cleaves L1.
Figure 4.

L1 interacts with MMP14 and GAPDH interacts with ANT. A, B, A detergent-solubilized brain homogenate was subjected to immunoprecipitation using ANT1- or ANT2-specific antibodies (A), polyclonal L1 (B), and nonimmune rabbit (rbIg) antibodies (A, B). Brain homogenate (input) and immunoprecipitates (IP) were subjected to Western blot (WB) analysis using monoclonal GAPDH (A) or MMP14 antibodies (B). C, D, Binding of GAPDH to ANT was determined by ELISA with substrate-coated GAPDH and different concentrations of soluble ANT (C) or by label-free binding assay using substrate-coated ANT and different concentrations of soluble L1-Fc (D). E, Binding of L1 to MMP14 was determined by label-free binding assay using substrate-coated MMP14 and different concentrations of soluble L1-Fc and of Fc control. Mean values ± SD from triplicates of a representative experiment are shown (C–E). The experiments were repeated twice with identical results.
To test the view that GAPDH and the ANT molecules are colocalized at the cell surface of neurons, and to confirm the colocalization of MMP14 and L1 at the neuronal cell surface, these molecules were analyzed by coimmunostainings of live cerebellar neurons. Using the MMP14 antibody and the L1 antibody 555, pronounced coimmunostaining of L1 and MMP14 was observed (Fig. 5A). Immunostaining with the GAPDH antibody and the ANT1- or ANT2-specific antibodies showed a partial colocalization of ANT1 and ANT2 with GAPDH at the cell surface of live neurons (Fig. 5B). These results show that L1 and MMP14 as well as ANT and GAPDH colocalize at the neuronal cell surface and support the view that they interact at the cell surface.
Figure 5.

ANT1 and ANT2 colocalize with GADPH and L1 colocalizes with MMP14 at the cell surface of live cerebellar neurons. A, B, Live cerebellar neurons were coimmunostained with L1 antibody 555 and MMP14 antibody (A) or with ANT1- or ANT2-specific antibodies and GAPDH (B) antibody. Confocal microscopy shows colocalization of L1 with MMP14 and of ANT with GAPDH in merged images. Scale bars: 10 μm.
Sequences comprising the first extracellular ANT loop or containing the signature motif RRRMMM mediate the interaction between L1 and ANT
To investigate whether the three loops of the ANT structure supposed to be extracellularly exposed (Detke and Elsabrouty, 2008) (Fig. 6A) mediate the interaction with L1, three loop-specific synthetic peptides were used as competitors in pull-down assays with L1-Fc. Western blot analysis of the precipitates with ANT antibodies revealed that the amount of the 32 and 60 kDa ANT proteins was reduced in the presence of ANT peptides comprising the first and third loop, while the amount of both proteins was not altered in the presence of the peptide comprising the second loop (Fig. 6B). These results suggest that sequences present in the first and/or third putative extracellular loops of ANT1 and ANT2 are involved in mediating the interaction between L1 and ANT1 or ANT2.
To examine whether the loops mediate the direct binding of L1 to ANT, the synthetic peptides comprising the extracellular ANT loops were used as competitors in a label-free binding assay using purified ANT as substrate-coat and L1-Fc as soluble binding partner. The presence of equimolar concentrations of peptide comprising the first loop reduced the binding of L1-Fc to ANT by ∼20%, while the peptide with the second and third loop did not significantly reduce the binding (Fig. 6C). Interestingly, carboxyatractyloside, but not atractyloside, also reduced the binding of L1-Fc to ANT by ∼45% when present in a 20-fold molar excess (Fig. 6C), indicating that carboxyatractyloside interferes with the binding of L1 to ANT. Since the signature of nucleotide carriers, the RRRMMM sequence, has not only been implicated in nucleotide transport but also in binding of carboxyatractyloside and possibly ADP (Pebay-Peyroula et al., 2003), we tested the possibility that L1 binds to this signature motif by using a synthetic peptide comprising this signature and 13 flanking amino acids of the third loop as competitor in the label-free binding assay. In parallel, we tested a synthetic peptide comprising the N-terminal 28 amino acids of ANT1 as competitor, since immunostaining suggested that the N-termini of ANT1 and ANT2 are oriented toward the extracellular space. In the presence of the peptide with the RRRMMM signature, the binding of L1-Fc to substrate-coated purified ANT was reduced by ∼30% (Fig. 6C), indicating that L1 binds to sequences containing the motif RRRMMM. In contrast, the peptide comprising the N-terminal sequence did not affect the binding of L1-Fc to ANT (data not shown). When the peptide with the signature was used as competitor in a pull-down assay, a similar reduction in the amount of ANT precipitated by L1-Fc was seen as before with the peptide comprising the first and third loop (data not shown). Using the synthetic peptides as direct binding partner of L1-Fc in the label-free binding assay, only the peptide comprising the sequences of the first loop showed a specific binding to substrate-coated L1-Fc when compared to unspecific binding to substrate-coated Fc (Fig. 6D). This peptide showed a concentration-dependent binding to coated L1-Fc, but not to Fc (Fig. 6E). Furthermore, when the peptide with the first loop was used as substrate coat in ELISA experiments, a concentration-dependent binding of L1-Fc was observed (Fig. 6F), although L1-Fc did not bind to the other ANT-derived peptides (data not shown). Interestingly, compared to its binding to substrate-coated Fc, only the peptide with the third loop showed a concentration-dependent binding to substrate-coated GAPDH (Fig. 6D). The combined results of the binding assays suggest that the direct interaction between L1 and ANT is mediated by sequences of the first extracellular loop of ANT1 and ANT2, and probably by the signature of nucleotide carriers, RRRMMM, while the third extracellular loop of ANT1 and ANT2 appears to mediate the interaction between ANT and GAPDH.
L1 stimulates src-dependent tyrosine phosphorylation of ANT and MMP14 and triggers ATP release via ANT
Since L1 stimulation activates nonreceptor tyrosine kinase src (Ignelzi et al., 1994; Schmid et al., 2000; Loers et al., 2005; Yeaney et al., 2009), and since L1 and its binding partners ANT and MMP14 are tyrosine phosphorylated by src kinases (Schaefer et al., 2002; Nyalendo et al., 2007; Feng et al., 2010), we investigated whether L1 stimulation of cerebellar neurons would lead to tyrosine phosphorylation of ANT and MMP14. After incubating cerebellar neurons without or with L1-Fc for different time periods, cells were lysed, and the tyrosine-phosphorylated proteins were immunoprecipitated and subjected to Western blot analysis using L1, ANT, and MMP14 antibodies. Tyrosine phosphorylation of full-length L1, ANT1, ANT2, and MMP14 was observed after 5 min, and this enhancement of tyrosine phosphorylation was still seen after 10 min (Fig. 7A). After 20, 30, and 60 min, a successive decline of the amount of tyrosine-phosphorylated L1, ANT1, and ANT2 to background values was observed. In case of MMP14, a continual increase of tyrosine-phosphorylated MMP14 was observed after 20 and 30 min (Fig. 7A), while no tyrosine-phosphorylated MMP14 was detectable after 60 min (Fig. 7A). The time-dependent increase of tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14 was reduced in the presence of the src kinase inhibitor PP2 (Fig. 7B). In this context, it is noteworthy to mention that the increase of tyrosine phosphorylation of MMP14 was not associated with an increase of the amounts of the 90 kDa L1 fragment or a decrease of the amounts of full-length L1 (data not shown), indicating that even tyrosine phosphorylation of MMP14 triggered by L1 stimulation does not result in an MMP14-mediated L1 cleavage. Treatment with Fc alone or treatment of L1-deficient cerebellar neurons with L1-Fc did not lead to tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14 (data not shown), indicating that L1 at the surface of neurons is required to mediate the L1-specific triggering of signal transduction leading to tyrosine phosphorylation of L1, ANT, and MMP14.
Figure 7.

Stimulation of L1 functions at the cell surface results in scr-dependent tyrosine phosphorylation of L1, ANT, and MMP14. A–C, Live cerebellar neurons were incubated without (−) or with L1-Fc for different time periods (A, B) in the absence (A) or presence (B) of the src kinase inhibitor PP2, or with L1-Fc for 15 min in the absence (−) or presence of the ANT-derived peptides comprising the first loop (+loop1) or containing the signature motif RRRMMM (+sign) (C). Cell lysates were prepared and subjected to immunoprecipitation using beads conjugated with antibodies directed against phosphotyrosine. The immunoprecipitates were subjected to Western blot analysis (WB) using polyclonal L1 antibody, ANT1- or ANT2-specific antibody, or MMP14 antibody.
Since the ANT-derived peptides comprising the first loop or containing the signature motif interfere with the interaction between L1 and ANT, we tested whether these peptides affect the tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14. In the presence of the peptide comprising the first loop, a pronounced inhibition of L1-induced tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14 was observed, while in the presence of the peptide containing the signature motif, only a slight or no inhibition was observed (Fig. 7C).
The observations that the ANT molecules are localized at the plasma membrane and that the extracellular domains of L1 and the ANT molecules interact raised the questions whether the ANT molecules transport ATP and ADP across the plasma membrane and whether L1 affects the transport activity of the ANT molecules at the cell surface. To address this question, cerebellar neurons were treated without or with L1-Fc for different times in the absence and presence of the ANT inhibitors atractyloside and carboxyatractyloside followed by determination of the intracellular ATP levels. Compared to the ATP level of neurons without treatment, a reduction by ∼30% was observed after incubation with L1-Fc for 2 and 5 min (Fig. 8A). After 10 min incubation with L1-Fc, the level was still reduced by ∼10%, while the level reached control level after incubation for 20 min (Fig. 8A). The L1-induced transient reduction of intracellular ATP levels was blocked in the presence of atractyloside (data not shown) and carboxyatractyloside (Fig. 8A), and was not observed when L1-deficient cerebellar neurons were used for stimulation with L1-Fc (data not shown). These results indicate that the reduction of the intracellular ATP level is due to an L1-triggered and L1-dependent ANT-mediated release of ATP from the cell. However, we could not detect a transient increase of the ATP levels in the cell culture supernatants upon stimulation with L1-Fc (data not shown), suggesting that the ATP is immediately degraded or used up upon release.
Figure 8.

Stimulation of L1 functions at the cell surface leads to a transient reduction of intracellular ATP levels. A, B, Live cerebellar neurons were incubated without or with L1-Fc for different time periods in the absence (−) or presence (+) of carboxyatractyloside (cATR) (A), or were incubated for 3 min without (−) or with L1-Fc (L1) in the presence of PP2 or ANT-derived peptides containing loop 1, loop 2, loop 3, the signature motif (sign), or N-terminal amino acids 1–28 of ANT1, ANT1- or ANT2-specific antibodies (anti-ANT1; anti-ANT2), or antibodies against MMP14 (anti-MMP) or GAPDH (anti-GAPDH) (B). ATP levels in cell lysates are shown relative to the level obtained in the absence of L1-Fc, which was set to 100%. Mean values ± SD from triplets of three independent experiments are shown, and values that are significantly different from the values obtained upon treatment of neurons with L1-Fc without additives are indicated by asterisks: *p < 0.001; **p < 0.0001; ***p < 0.00001 (Student's t test).
To test whether the L1-stimulated reduction of intracellular ATP levels depends on tyrosine phosphorylation, cerebellar neurons were pretreated with the src kinase inhibitor PP2 and then incubated for 3 min with L1-Fc in the presence of PP2. The L1-induced reduction of the ATP levels observed in the absence of PP2 was completely abolished by PP2 (Fig. 8B), indicating that the ANT-mediated reduction of the ATP levels depends on tyrosine phosphorylation.
The intracellular ATP levels were not reduced in the presence of the ANT-derived peptides comprising the first loop or containing the RRRMMM motif, while an L1-induced reduction of intracellular ATP levels was observed in the presence of the ANT-derived peptides comprising the second and third loops or the N-terminal 28 amino acids of ANT1 (Fig. 8B). Furthermore, the L1-induced reduction of intracellular ATP levels was inhibited in the presence of the MMP14 antibody, the GAPDH antibody, and the ANT2-specific antibody, while an L1-triggered reduction of the intracellular ATP level was observed in the presence of the ANT1-specific antibody (Fig. 8B). In summary, the results indicate that L1 stimulation results in transient tyrosine phosphorylation of L1, ANT1, ANT2, and MMP14 and in a subsequent or concomitant reduction of the intracellular ATP, which appears to be due to an ATP release via ANT. The ANT2-mediated ATP release appears to depend on the interaction between ANT2 with L1, ANT1, GAPDH, and/or MMP14.
ANT and MMP14 are involved in regulation of L1-induced neurite outgrowth of cerebellar neurons
We finally investigated whether plasma membrane ANT and MMP14 also affect the biological functions of L1 and analyzed the effects of ANT inhibitors, ANT antibodies, ANT-derived peptides, and MMP14 antibodies on L1-mediated neuroprotection (Chen et al., 1999; Loers et al., 2005) and L1-induced neurite outgrowth of cerebellar granule cells. None of the tested agents affected survival of cerebellar neurons upon hydrogen peroxide treatment (data not shown), implying that ANT and MMP14 do not play a role in L1-mediated neuroprotection. Analysis of neurite outgrowth showed that total lengths of neurites on L1-Fc relative to the lengths observed on PLL were enhanced and that this elevated neurite outgrowth on L1 was further increased in the presence of 5 μm atractyloside or carboxyatractyloside (Fig. 9A). The ANT inhibitors did not alter neurite outgrowth on PLL or Fc compared to that observed in the absence of inhibitors (Fig. 9A). ANT1- and ANT2-specific antibodies and a function-blocking MMP14 antibody, but not a control antibody (data not shown) or the mouse and rabbit pan-ANT antibodies (Fig. 9A), inhibited neurite outgrowth of cerebellar neurons maintained on L1-Fc substrate. The ANT antibodies and the MMP antibody showed only a slight or no inhibition on PLL substrate, respectively (Fig. 9A). In the presence of the ANT-derived peptide comprising the third extracellular loop, L1-promoted neurite outgrowth was also further enhanced, while it was reduced when the peptides containing the N-terminal amino acids 2–13 of ANT1 or ANT2 sequences, the N-terminal 28 amino acids of ANT1, the first extracellular loop of ANT1, or the signature motif of mitochondrial carriers were added (Fig. 9B). The peptide with the second extracellular loop did not alter L1-promoted neurite outgrowth, and none of the peptides affect neurite outgrowth on PLL substrate (Fig. 9B). Interestingly, neither the inhibitors nor the antibodies and peptides showed any effects on neurite outgrowth of L1-deficient cerebellar neurons maintained on L1-Fc or PLL substrates (data not shown), indicating that ANT-mediated effects on L1-induced neurite outgrowth depend on the interaction between L1 and ANT at the neuronal cell surface. Disturbance of this ANT/L1 interaction leads to dysregulation of L1-induced neurite outgrowth. Moreover, disturbance of the interaction of MMP14 with L1 and ANT also affects L1-dependent neurite outgrowth.
Figure 9.
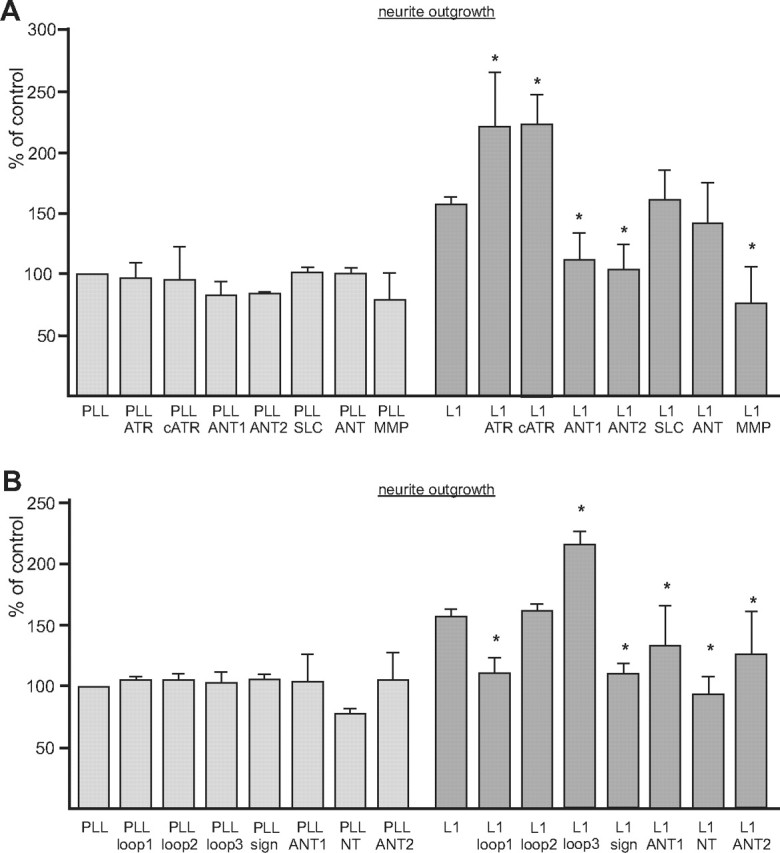
ANT modulates L1-induced neurite outgrowth of cerebellar granule cells. A, Live cerebellar neurons were maintained on substrate-coated PLL or PLL plus L1-Fc (L1) in the presence of atractyloside (ATR), carboxyatractyloside (cATR), ANT1-specific (ANT1), ANT2-specific (ANT2), mouse pan-ANT (SLC), rabbit pan-ANT (ANT), or MMP14 (MMP) antibodies. B, Neurons were maintained on PLL or L1-Fc (L1) in the presence of 100 μg/ml ANT peptides containing loop 1, loop 2, loop 3, the signature motif (sign), the N-terminal amino acids 2–13 of ANT1 and ANT2 or the N-terminal 28 amino acids of ANT1 (NT). Total lengths of neurites were measured, and values obtained for neurons on substrate-coated PLL without additives were set to 100%. Mean values ± SEM from three independent experiments are shown, and values that are significantly different from the value obtained for neurite length on substrate-coated L1 without any additives are indicated: *p < 0.05 (one-way ANOVA with Turkey's multiple comparison test).
Discussion
We have identified the ATP/ADP exchanging carrier proteins ANT1 and ANT2 as direct binding partners of the cell adhesion molecule L1. At the surface of neurons, ANT1 and ANT2 interact via their extracellular domains with the transmembrane cell surface adhesion molecule L1, with the transmembrane protease MMP14 and with extracellularly localized GAPDH. The interactions of these proteins at the cell surface evoke signal transduction pathways downstream of L1, lead to ATP secretion via ANT1 or ANT 2, and enhance neurite outgrowth.
Cell surface topology of ANT
So far, only three studies on Leishmania, murine fibroblasts, and breast carcinoma and fibrosarcoma cells provide solid evidence that ANT is not only localized in mitochondria, but also at the cell surface (Sigal and Resh, 1993; Detke and Elsabrouty, 2008; Radichev et al., 2009). Our study confirms these findings and shows that the three ANT loops that project into the lumen of mitochondria are exposed at the neuronal cell surface (Fig. 10A). Since antibodies against N-terminal ANT1 and ANT2 sequences showed a pronounced ANT1 and ANT2 surface staining of live cerebellar neurons, it is likely that the N-termini of ANT1 and ANT2, which are proposed to be exposed to the cytoplasm, are actually oriented toward the extracellular space (Fig. 10A). This orientation would request that the first transmembrane helix is not inserted into the membrane and instead is present in the extracellular space, implying that the topology of the plasma membrane ANT is different from that of mitochondrial ANT. A switch of the sorting mode from posttranslational mitochondrial import to cotranslational endoplasmic reticulum targeting and plasma membrane translocation is possibly responsible for the different localization and topology of ANT in distinct cellular subcompartments. We detected two molecular types of the ANT isoforms as 32 and 60 kDa proteins at the plasma membrane of neurons, depending on the antibody used for Western blot analysis or the conditions under which ANT was isolated. We propose that the 60 kDa form represents a stable homodimer or heterodimer consisting of ANT1 and/or ANT2, and that the 32 kDa form is the monomer of either ANT1 of ANT2. The mechanisms by which the ANT isoforms and their molecular forms reach the plasma membrane are yet not known.
Figure 10.

Schematic working model of the functional interplay between L1, ANT, GAPDH, and MMP14 at the plasma membrane. A, In a previous study, we showed that GAPDH binds to sites within the fourth and fifth fibronectin type III domain of full-length L1 (L1–200). In the present study, we show that (1) MMP14 directly interacts with unknown binding sites within the extracellular domain of L1, (2) a 90 kDa L1 fragment (L1–90) comprising at least the third, fourth, and fifth fibronectin type III domain interacts with the first N-terminal extracellular ANT loop, and (3) GAPDH binds to sites within the third extracellular loop of ANT. In addition, it has been reported that MMP14 interacts with ANT. L1–90, MMP14, GAPDH, and ANT may not only form heteromeric binary, but also ternary complexes, or a quaternary complex containing L1, MMP14, GAPDH, and ANT. B, Stimulation of L1-dependent signal transduction pathways by L1-Fc leads to transient src-dependent tyrosine phosphorylation of L1, MMP14, and ANT. Tyrosine phosphorylation, in particular of ANT, triggers transient ANT-mediated transport of ATP out of the cells into the extracellular space. Extracellular ATP may be used by extracellular kinases to phosphorylate proteins exposed or present extracellularly, such as GAPDH or L1. Extracellularly phosphorylated proteins may then play a crucial role in promoting neurite outgrowth.
Interactions between ANT, L1, MMP14, and GAPDH
Mass spectrometric analyses suggested that both ANT1 and ANT2 interact with L1. This assumption is supported by the finding that both isoforms coimmunoprecipitated with L1 and that they colocalized with L1 at the cell surface of live neurons. In binding assays, we observed direct binding of the first extracellular ANT loop to L1, indicating that sequences of this loop mediate the interaction between L1 and ANT (Fig. 10A). In addition, the signature of nucleotide carriers, the RRRMMM sequence in the ANT molecules, appears to be involved in the L1/ANT interaction, since the peptide containing this motif reduced the binding of ANT to L1 in label-free binding and pull-down assays. Since the peptide comprising the third ANT loop inhibited the interaction of L1-Fc with ANT in pull-down assays but did not show direct binding to L1-Fc, it is conceivable that the third ANT loop containing peptide interferes with the binding of other ANT binding partner(s), which in turn bind to L1, leading to a reduced precipitation of ANT by L1-Fc in the pull-down assay. Such a binding partner could be GAPDH to which the peptide within the third loop of ANT binds (Fig. 10A). GAPDH interacts with L1 (Makhina et al., 2009) and ANT, and thus it may be one of the L1 binding partners that modulate the interaction between L1 and ANT. Another L1 binding partner that could modulate the L1/ANT interaction is MMP14, which also binds to ANT (Radichev et al., 2009) (Fig. 10A).
Involvement of ANT in L1-mediated neuritogenesis and signaling
The finding that ANT antibodies, inhibitors, and peptides were active on wild-type neurons but not on L1-deficient neurons shows that the L1/ANT interaction is required for L1-triggered neurite outgrowth. The remarkable effects of the antibodies directed to the N-terminal sequences of ANT1 or ANT2 and of peptides comprising N-terminal ANT sequences on neurite outgrowth provide evidence that the N termini of ANT1 and ANT2 are oriented toward the extracellular space and are involved in neurite outgrowth (Fig. 10A). The functional contribution of the first extracellular ANT loop and the signature motif RRRMMM in L1-mediated neuritogenesis is documented by the findings that peptides comprising the first loop or containing this signature motif reduce binding of L1 to ANT and diminish L1-induced neurite outgrowth. The enhancement of L1-induced neurite outgrowth by the peptide comprising the third ANT loop could be due to disturbance of the interaction between ANT and its other binding partner(s), such as GADPH. Moreover, the fact that ANT inhibitors affect L1-induced neurite outgrowth further supports the notion that ANT modulates L1-mediated neurite outgrowth.
L1-mediated signaling events underlying neuritogenesis involve L1 binding partners and ATP
Stimulation of L1 signaling by L1-Fc leads to src-dependent transient tyrosine phosphorylation of L1 (for a review, see Maness and Schachner, 2007) (Fig. 10B). Here, we showed that stimulation of L1 signaling also triggers tyrosine phosphorylation of ANT1, ANT2, and MMP14 and reduces intracellular ATP levels, being associated with transient ANT-dependent release of ATP (Fig. 10B), which could act as a neurotransmitter or extracellularly phosphorylate neuritogenic molecules (Fields, 2011; Lalo et al., 2011) (Fig. 10B). Of note, L1 possesses kinase activity, and L1-induced neurite outgrowth is modulated by ATP and phosphorylation of L1 and/or GAPDH (Makhina et al., 2009). ANT2 is able to transport ATP into and ADP out of mitochondria (for references, see Chevrollier et al., 2011), while ANT1 transports ATP out of the mitochondria and ADP into the mitochondria. It is conceivable that plasma membrane ANT2 transports ATP out of the cell and that ANT1 transports ATP into the cell, leading to imbalances of ATP and ADP levels inside and outside of cells. This view is supported by our observation that reduction of intracellular ATP levels was blocked by an ANT2-specific antibody and ANT inhibitors. L1-stimulated reduction of intracellular ATP levels could be due to an ANT2-mediated transport of ATP to the extracellular space, which is also regulated by GAPDH, because treatment with GAPDH antibody blocks the L1-induced reduction of intracellular ATP levels. Regulation of the equilibrium between ADP and ATP will impact both intracellular and extracellular phosphorylation events. Stimulation of the neuritogenic functions of L1 not only leads to src-dependent phosphorylation of ANT and to ATP release via ANT, but also to transient tyrosine phosphorylation of MMP14, which is also involved in neuritogenesis (Zhang et al., 2007). It is noteworthy in this context that MMP14 antibody treatment inhibited L1-induced ATP release and neurite outgrowth. However, MMP14 does not modulate L1-triggered functions by proteolytic L1 processing, because L1 was not cleaved by MMP, and increase of tyrosine phosphorylation-mediated activation of MMP14 was not accompanied by changes in L1 proteolysis. Thus, direct binding of MMP14 to ANT and/or L1 may influence neurite outgrowth. Interestingly, ANT2 alters tumor cell migration and invasion by interacting with MMP14, which can thus affect cell functions via proteolytic and nonproteolytic mechanisms (Radichev et al., 2009). Since MMP14 is active as extracellular matrix-degrading enzyme and is released from cancer cells via exosomes, thus being delivered to the extracellular matrix (Hakulinen et al., 2008), we expect that L1 stimulation may lead to activation of MMP14 and proteolytic processing of extracellular matrix proteins to allow neurite outgrowth and migration of neurons.
In conclusion, our study shows that the interplay between the cell adhesion molecule L1, plasma membrane-bound ANT1 and ANT2, extracellular GAPDH, and metalloprotease MMP14 at the surface of neurons modulates L1-mediated functions, such as neurite outgrowth. It is expected that the functions of these interacting partners will not be limited to neuritogenesis in vitro, but will extend their importance also to mechanisms underlying synaptic functions and regeneration after injury in vivo, to which L1 has been shown to contribute.
Footnotes
This work was supported by the New Jersey Commission for Spinal Cord Research and the Li Kashing Foundation (M.S.), and by the Deutsche Forschungsgemeinschaft (SFB Transregio 19, C7). We are grateful to Barbara Ueberle (Deutsches Krebsforschungszentrum, Heidelberg, Germany) for mass spectrometry, Achim Dahlmann for genotyping, Eva Kronberg for excellent animal care, and Claus Schafer-Nielsen (Copenhagen, Denmark) for synthetic peptides.
References
- Brümmendorf and Rathjen, 1995.Brümmendorf T, Rathjen FG. Cell adhesion molecules 1: immunoglobulin superfamily. Protein Profile. 1995;2:963–1108. [PubMed] [Google Scholar]
- Chen et al., 1999.Chen S, Mantei N, Dong L, Schachner M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J Neurobiol. 1999;38:428–439. doi: 10.1002/(sici)1097-4695(19990215)38:3<428::aid-neu10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Chevrollier et al., 2011.Chevrollier A, Loiseau D, Reynier P, Stepien G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim Biophys Acta. 2011;1807:562–567. doi: 10.1016/j.bbabio.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Cooper, 2002.Cooper MA. Optical biosensors in drug discovery. Nat Rev Drug Discovery. 2002;1:515–528. doi: 10.1038/nrd838. [DOI] [PubMed] [Google Scholar]
- Cunningham et al., 2004.Cunningham BT, Li P, Schulz S, Lin B, Baird C, Gerstenmaier J, Genick C, Wang F, Fine E, Laing L. Label-free assays on the BIND system. J Biomol Screen. 2004;9:481–490. doi: 10.1177/1087057104267604. [DOI] [PubMed] [Google Scholar]
- Dahout-Gonzalez et al., 2006.Dahout-Gonzalez C, Nury H, Trézéguet V, Lauquin GJ, Pebay-Peyroula E, Brandolin G. Molecular, functional, and pathological aspects of the mitochondrial ADP/ATP carrier. Physiology (Bethesda) 2006;21:242–249. doi: 10.1152/physiol.00005.2006. [DOI] [PubMed] [Google Scholar]
- Detke and Elsabrouty, 2008.Detke S, Elsabrouty R. Leishmania mexicana amazonensis: Plasma membrane adenine nucleotide translocator and chemotaxis. Exp Parasitol. 2008;118:408–419. doi: 10.1016/j.exppara.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng et al., 2010.Feng J, Lucchinetti E, Enkavi G, Wang Y, Gehrig P, Roschitzki B, Schaub MC, Tajkhorshid E, Zaugg K, Zaugg M. Tyrosine phosphorylation by Src within the cavity of the adenine nucleotide translocase 1 regulates ADP/ATP exchange in mitochondria. Am J Physiol Cell Physiol. 2010;298:C740–C748. doi: 10.1152/ajpcell.00310.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, 2011.Fields RD. Nonsynaptic and nonvesicular ATP release from neurons and relevance to neuron-glia signaling. Semin Cell Dev Biol. 2011;22:214–219. doi: 10.1016/j.semcdb.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakulinen et al., 2008.Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem. 2008;105:1211–1218. doi: 10.1002/jcb.21923. [DOI] [PubMed] [Google Scholar]
- Haspel and Grumet, 2003.Haspel J, Grumet M. The L1CAM extracellular region: a multi-domain protein with modular and cooperative binding modes. Front Biosci. 2003;8:S1210–S1225. doi: 10.2741/1108. [DOI] [PubMed] [Google Scholar]
- Ignelzi et al., 1994.Ignelzi MA, Jr, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- Klingenberg, 2008.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Lalo et al., 2011.Lalo U, Verkhratsky A, Pankratov Y. Ionotropic ATC receptors in neuronal-glial communication. Semin Cell Dev Biol. 2011;22:220–228. doi: 10.1016/j.semcdb.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Loers and Schachner, 2007.Loers G, Schachner M. Recognition molecules and neural repair. J Neurochem. 2007;101:865–882. doi: 10.1111/j.1471-4159.2006.04409.x. [DOI] [PubMed] [Google Scholar]
- Loers et al., 2005.Loers G, Chen S, Grumet M, Schachner M. Signal transduction pathways implicated in neural recognition molecule L1 triggered neuroprotection and neuritogenesis. J Neurochem. 2005;92:1463–1476. doi: 10.1111/j.1471-4159.2004.02983.x. [DOI] [PubMed] [Google Scholar]
- Makhina et al., 2009.Makhina T, Loers G, Schulze C, Ueberle B, Schachner M, Kleene R. Extracellular GAPDH binds to L1 and enhances neurite outgrowth. Mol Cell Neurosci. 2009;41:206–218. doi: 10.1016/j.mcn.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Maness and Schachner, 2007.Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Nyalendo et al., 2007.Nyalendo C, Michaud M, Beaulieu E, Roghi C, Murphy G, Gingras D, Béliveau R. Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J Biol Chem. 2007;282:15690–15699. doi: 10.1074/jbc.M608045200. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula et al., 2003.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Radichev et al., 2009.Radichev IA, Remacle AG, Sounni NE, Shiryaev SA, Rozanov DV, Zhu W, Golubkova NV, Postnova TI, Golubkov VS, Strongin AY. Biochemical evidence of the interactions of membrane type-1 matrix metalloproteinase (MT1-MMP) with adenine nucleotide translocator (ANT): potential implications linking proteolysis with energy metabolism in cancer cells. Biochem J. 2009;420:37–47. doi: 10.1042/BJ20090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolf et al., 2001.Rolf B, Kutsche M, Bartsch U. Severe hydrocephalus in L1-deficient mice. Brain Res. 2001;891:247–252. doi: 10.1016/s0006-8993(00)03219-4. [DOI] [PubMed] [Google Scholar]
- Schachner, 1997.Schachner M. Neural recognition molecules and synaptic plasticity. Curr Opin Cell Biol. 1997;9:627–634. doi: 10.1016/s0955-0674(97)80115-9. [DOI] [PubMed] [Google Scholar]
- Schaefer et al., 2002.Schaefer AW, Kamei Y, Kamiguchi H, Wong EV, Rapoport I, Kirchhausen T, Beach CM, Landreth G, Lemmon SK, Lemmon V. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J Cell Biol. 2002;157:1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid and Maness, 2008.Schmid RS, Maness PF. L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr Opin Neurobiol. 2008;18:245–250. doi: 10.1016/j.conb.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid et al., 2000.Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal and Resh, 1993.Sigal CT, Resh MD. The ADP/ATP carrier is the 32-kilodalton receptor for an NH2-terminally myristylated src peptide but not for pp60src polypeptide. Mol Cell Biol. 1993;13:3084–3092. doi: 10.1128/mcb.13.5.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyssokikh et al., 2001.Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dörner A, Zorov DB, Brdiczka D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J. 2001;358:349–358. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaney et al., 2009.Yeaney NK, He M, Tang N, Malouf AT, O'Riordan MA, Lemmon V, Bearer CF. Ethanol inhibits L1 cell adhesion molecule tyrosine phosphorylation and dephosphorylation and activation of pp60(src) J Neurochem. 2009;110:779–790. doi: 10.1111/j.1471-4159.2009.06143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2007.Zhang Y, Klassen HJ, Tucker BA, Perez MT, Young MJ. CNS progenitor cells promote a permissive environment for neurite outgrowth via a matrix metalloproteinase-2-dependent mechanism. J Neurosci. 2007;27:4499–4506. doi: 10.1523/JNEUROSCI.0200-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2008.Zhang Y, Yeh J, Richardson PM, Bo X. Cell adhesion molecules of the immunoglobulin superfamily in axonal regeneration and neural repair. Restor Neurol Neurosci. 2008;26:81–96. [PubMed] [Google Scholar]