

Abstract
Alterations of learning and memory in mice with deregulated neuron-specific nuclear factor κB (NF-κB) activity support the idea that plastic changes of synaptic contacts may depend at least in part on IκB kinase (IKK)/NF-κB-related synapse-to-nucleus signaling. There is, however, little information on the molecular requirements and mechanisms regulating this IKK/NF-κB-dependent synapse development and remodeling. Here, we report that the NF-κB inducing IKK kinase complex is localized at the postsynaptic density (PSD) and activated under basal conditions in the adult mouse brain. Using different models of conditional genetic inactivation of IKK2 function in mouse principal neurons, we show that IKK/NF-κB signaling is critically involved in synapse formation and spine maturation in the adult brain. IKK/NF-κB blockade in the forebrain of mutant animals is associated with reduced levels of mature spines and postsynaptic proteins PSD95, SAP97, GluA1, AMPAR-mediated basal synaptic transmission and a spatial learning impairment. Synaptic deficits can be restored in adult animals within 1 week by IKK/NF-κB reactivation, indicating a highly dynamic IKK/NF-κB-dependent regulation process. We further identified the insulin-like growth factor 2 gene (Igf2) as a novel IKK/NF-κB target. Exogenous Igf2 was able to restore synapse density and promoted spine maturation in IKK/NF-κB signaling-deficient neurons within 24 h. This process depends on Igf2/Igf2R-mediated MEK/ERK activation. Our findings illustrate a fundamental role of IKK/NF-κB–Igf2–Igf2R signaling in synapse formation and maturation in adult mice, thus providing an intriguing link between the molecular actions of IKK/NF-κB in neurons and the memory enhancement factor Igf2.
Introduction
In the CNS, synapses are essential units for neuronal communication. Not only in the developing, but also in the adult brain, the regulation of synapse number, shape, and strength is mediated by the concerted action of multiple signaling cascades (Scheiffele, 2003; Waites et al., 2005; Williams et al., 2010). After initial formation, presynaptic and postsynaptic specializations undergo a maturation process as monitored by specific morphological and physiological changes of the newly established contacts.
The formation and maturation of the postsynaptic compartment of glutamatergic synapses is mainly organized by cell adhesion and scaffolding proteins such as the Neuroligins (Varoqueaux et al., 2006), the ProSAP/Shank family, Homer (Sala et al., 2001; Roussignol et al., 2005; Grabrucker et al., 2011), and membrane-associated guanylate kinases (MAGUKs) (El-Husseini et al., 2000). However, the signal transduction machinery promoting these maturational processes, and thus controlling the number of functional synapses, remains largely unknown. In addition, MAGUKs like PSD95, SAP97, and PSD93, tightly control the insertion of AMPA receptors (AMPARs) into the postsynaptic membrane, thereby modulating the strength of mature synapses in an activity-dependent fashion (Zamanillo et al., 1999; Mack et al., 2001; Elias et al., 2006; Howard et al., 2010). Several studies support a crucial role for the nuclear factor κB (NF-κB) family of transcription factors in the regulation of synaptic plasticity, learning, and memory (Fridmacher et al., 2003; Meffert et al., 2003; O'Mahony et al., 2006; O'Riordan et al., 2006; Ahn et al., 2008; Kaltschmidt and Kaltschmidt, 2009).
NF-κB transcription factors (p65/RelA, RelB, c-Rel, p100/p52, and p105/p50) form dimeric complexes that are retained in the cytoplasm by interaction with inhibitory IκB proteins. Upon activation, the IκB kinase (IKK) complex composed of IKK1 (IKKα), IKK2 (IKKβ), and NEMO (IKKγ), phosphorylates IκB proteins, thereby initiating their proteasomal degradation followed by the release and nuclear translocation of active NF-κB complexes (Ghosh and Karin, 2002; Scheidereit, 2006). The canonical NF-κB pathway depends on IKK2 and NEMO function, and dominant-negative versions of IKK2 have been shown to efficiently block this pathway in vivo (Herrmann et al., 2005; Baumann et al., 2007).
Recent studies provide evidence that IKK/NF-κB signaling is directly involved in spinogenesis and synapse formation (Russo et al., 2009; Boersma et al., 2011; Christoffel et al., 2011). To unravel underlying molecular mechanisms, we used mouse models with either a conditional expression of a dominant-negative acting IKK2 allele or a specific deletion of IKK2 in excitatory neurons. Thus, we could demonstrate that, in the adult mouse brain, synaptic IKK/NF-κB signaling is an essential prerequisite for the efficient formation and functional maturation of synaptic contacts. Strikingly, we found that insulin-like growth factor 2 (Igf2)—recently identified as memory enhancement factor in rats (D. Y. Chen et al., 2011)—is a novel neuronal NF-κB target gene. Moreover, our findings reveal a novel hitherto unknown molecular IKK/NF-κB–Igf2–Igf2R signaling module that essentially contributes to the highly dynamic establishment and maintenance of glutamatergic synapses.
Materials and Methods
Animals
Mice were kept in a specific pathogen-free animal facility at Ulm University. All mouse procedures were performed in compliance with the guidelines for the welfare of experimental animals issued by the Federal Government of Germany and approved by the District Government. Generation of luciferase-(Ptetbi)-VSV-IKK2-DN and luciferase-(Ptetbi)-ΔN-IκBα mice was described previously (Lavon et al., 2000; Herrmann et al., 2005). To achieve the conditional expression of the IKK2-DN and ΔNIκBα transgene in neurons, these lines were crossed to Tg(Camk2a-tTA)1Mmay animals (Mayford et al., 1996). To eventually generate IKK2nfl/fl mice, IKKβfl/fl mice (Park et al., 2002) were crossed to CaMKIIα iCre BAC mice (Casanova et al., 2001). Camk2a-tTA × IKK2-DN were bred in NMRI background, and Camk2a-tTA × ΔNIκBα and CaMKIIα iCre BAC × IKKβfl/fl animals in C57BL/6 background. Wild type, single-transgenic, and IKKβfl/fl were used as control groups. The day of vaginal plug detection was designated as E0.5, and the day of birth as P0. The IVIS200 in vivo imaging system (Caliper Life Sciences) was used for the detection of luciferase activity in the living animal. After intraperitoneal injection of d-luciferin (100 mg/kg body weight; PBS), mice were sedated by isoflurane inhalation and imaged with a collection time of 10 s. The resulting light emission was quantified using Living Image software (Caliper Life Sciences) and shown as an overlay picture or as photons/second/centimeter2/steradian. Mice were exposed to doxycycline as indicated. Doxycycline (1 mg/ml; Sigma-Aldrich) was supplied as drinking water containing 1% sucrose.
Antibodies
The following primary antibodies were used in this study: c-Rel, phospho-p42/44 MAPK (p-Erk1/2), phospho-IKK1/2 (all Cell Signaling), Luciferase (Novus Biologicals), Erk2, Igf2, IKK1/2, NEMO, RelA, RelB, SAP97, IGF-IIR (all Santa Cruz), MAP2, vesicular stomatitis virus (VSV) (both Sigma-Aldrich), Bassoon (Enzo Life Sciences), GluA1, GluN1 (NR1), PSD95, Synaptophysin (all Synaptic Systems), and ProSAP1/Shank2 (Grabrucker et al., 2011). Secondary antibodies were as follows: Alexa 488 and Alexa 568 (both Invitrogen).
Biochemical analysis
Protein extraction.
The homogenized tissue or cell pellets were resuspended in three packed volumes of buffer C (20 mm HEPES, 25% glycerol, 0.42 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 1 mm DTT, 1 mm PMSF, protease inhibitor mixture, Complete Mini; Roche) and subjected to three cycles of freezing in liquid nitrogen and subsequent thawing on ice. After centrifugation, the supernatant was used as total protein extract.
Western blot analysis.
Thirty to 50 μg of protein extract were separated by SDS-PAGE, transferred onto PVDF membranes, probed with specific antibodies, and developed by enhanced chemiluminescence. For semiquantitative analysis, the films were scanned and the gray value of each band was analyzed by NIH ImageJ (http://rsb.info.nih.gov/ij/) and normalized to Erk2.
Electrophoretic mobility shift assay.
The preparation of nuclear extracts was performed on freshly isolated hippocampi from control and IKK2nDN mice. Tissue cut in small pieces was washed in PBS and incubated for 15 min (4°C) in five packed tissue volumes of buffer A (10 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, 1 mm DTT, 1 mm PMSF, protease inhibitor mixture, Complete Mini; Roche). Tissue was then homogenized by douncing, and nuclei were pelleted and washed twice with buffer A. Nuclear proteins were extracted for 1 h in two packed cell volumes of buffer C. After centrifugation, the supernatant was used as nuclear extract for electrophoretic mobility shift assay (EMSA) experiments. Nuclear protein extracts (5 μg) were incubated for 20 min at room temperature with 3 μg of poly(dI/dC), 10 μg of BSA, in buffer containing 50 mm NaCl, 1 mm DTT, 10 mm Tris-HCl, 1 mm EDTA, 5% glycerol, and radiolabeled double-stranded oligonucleotides as previously described (Azoitei et al., 2005). The formed DNA–protein complexes were then separated from free oligonucleotides on a native 4% polyacrylamide gel.
ELISA.
The mIgf2 developer kit from R&D Systems was used according to the manufacturers' protocol.
Subfractionation protocol.
To obtain subcellular fractions from mouse brain including the PSD fraction, a subcellular fractionation procedure was performed as described previously (Schmeisser et al., 2009) with minor modifications. In brief, whole mouse forebrains at the age of 3–4 months were homogenized [homogenate (Ho)] in HEPES-buffered sucrose (320 mm sucrose, 5 mm HEPES, pH 7.4) containing protease inhibitor mixture (Roche) and centrifuged at 1000 × g for 10 min at 4°C to remove cell debris and nuclei. The supernatant was spun for 20 min at 12,000 × g to obtain the crude synaptosomal fraction (P2). After washing, P2 was further fractionated by sucrose density gradient centrifugation (0.8/1.0/1.2 sucrose) at 200,000 × g for 2 h at 4°C. Synaptosomes (Syn) were collected at the 1.0–1.2 m interface. To obtain the PSD fraction, the synaptosomes were diluted with 5 vol of 1 mm Tris, pH 8.1, and stirred on ice for 30 min. After centrifugation for 30 min at 33,000 × g, the pellet P3 was resuspended in 5 mm Tris, pH 8.1, and once again fractionated by centrifugation in a sucrose gradient for 2 h at 200,000 × g. The 1.0/1.2 m interphase (synaptic junctions) was suspended in 320 mm sucrose, 0.5% Triton X-100, 5 mm Tris, pH 8.1, stirred on ice for 15 min and centrifuged for 30 min at 33,000 × g, resulting in the one Triton-extracted PSD pellet (PSD).
Culture of primary neurons
Primary hippocampal neurons were prepared from E17.5 mouse embryos and cultured in B27-supplemented Neurobasal medium as described previously (Grabrucker et al., 2011).
Electron microscopy
Mice at 4 months of age were transcardially perfused with fixative (2% paraformaldehyde, 2.5% glutaraldehyde, 1% saccharose in 0.1 m cocadylate buffer, pH 7.3), and brains were dissected out and postfixed overnight (2% glutaraldehyde, 1% saccharose in 0.1 m cocadylate buffer). After dehydration and staining with 2% uranyl acetate, the material was embedded in epoxy resin. Ultrathin sections were cut using an ultramicrotome. After lead citrate staining, the sections (CA1 hippocampus) were examined using an EM 10 electron microscope.
Electrophysiology
Slice preparation.
Adult (>P60) control and IKK2nDN mice were killed with Suprane (Baxter), and the brains were removed. Transverse slices (400 μm) were cut from the middle and dorsal portion of each hippocampus with a vibroslicer in artificial CSF (ACSF) (4°C, bubbled with 95% O2–5% CO2, pH 7.4) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 1 CaCl2, 26 NaHCO3, and 12 glucose. Slices were placed in an interface chamber exposed to humidified gas at 28–32°C and perfused with ACSF containing 2 mm CaCl2. In some of the experiments, AP5 (dl-2-amino-5-phosphopentanoic acid) (50 μm; Sigma-Aldrich) was added to the ACSF to block NMDA receptor (NMDAR)-mediated synaptic plasticity.
Synaptic transmission and synaptic excitability.
Orthodromic synaptic stimuli (<300 μA, 0.1 Hz) were delivered through a tungsten electrode placed in stratum radiatum in the CA1 region. The presynaptic volley and the field EPSP (fEPSP) were recorded by a glass electrode (filled with ACSF) placed in the synaptic layer (stratum radiatum), while another electrode placed in the pyramidal cell body layer (stratum pyramidale) monitored the population spike. Following a period of at least 20 min with stable responses, we stimulated the afferent fibers with increasing strength (increasing the stimulus duration in steps of 10 μs from 0 to 90 μs, five consecutive stimulations at each step). Before the input/output (I/O), the strength was adjusted so that a population spike appeared in the soma layer recording electrode in response to 40, 50, 60, or 70 μs in each experiment to define the range. To assess synaptic transmission, we measured the amplitudes of the presynaptic volley and the fEPSP at the different stimulation strengths. During the analysis, care was taken to use extrapolated, raw measurements that were within the apparently linear part of the I/O curves. Values from individual experiments outside the linear part of the I/O curves (prevolley vs fEPSP) were omitted when pooling the data. The population spike amplitude was measured as distance between the maximal population spike peak and a line joining the maximum prespike and postspike fEPSP positivities. Data were pooled across mice of the same genotype.
LTP.
Orthodromic synaptic stimuli (50 μs, <300 μA) were delivered alternately through two tungsten electrodes, one situated in the stratum radiatum and another in the stratum oriens of the hippocampal CA1 region. Extracellular synaptic responses were monitored by two glass electrodes (filled with ACSF) placed in the corresponding synaptic layers. After obtaining stable synaptic responses in both pathways (0.1 Hz stimulation) for at least 10–15 min, one of the pathways was tetanized (a single 100 Hz tetanization for 1 s). As standardization, the stimulation strength used for tetanization was just above threshold for generation of a population spike in response to a single test shock.
Synaptic efficacy was assessed by measuring the slope of the fEPSP in the middle third of its rising phase. Six consecutive responses (1 min) were averaged and normalized to the mean value recorded 1–4 min before tetanization. Data were pooled across animals of the same genotype.
Golgi staining
Brains of adult IKK2nDN mice and control littermates as well as doxycycline-treated animals were immersed for 21 d in Golgi-Cox solution (1% potassium dichromate; 1% mercuric chloride; 0.8% potassium chromate in Milli-Q water). Subsequently, the brains were rinsed four times in Milli-Q water and dehydrated. Two hundred micrometer coronal sections were cut using a sledge microtome. The Golgi-Cox staining was developed by incubation in 16% ammonia for 30 min and then fixated in 1% sodium thiosulfate for 7 min. The sections were dehydrated and embedded in glycerin-gelatin. Z-staples images were taken using an upright Axio Scope microscope (100× oil objective) equipped with a Zeiss CCD camera (16 bits; 1280 × 1024 pixels per image) and analyzed using AxioVision software (Zeiss). Three to six animals were analyzed per condition. For quantification of spine density and analysis of spine morphology, CA1 hippocampal pyramidal neurons were analyzed per animal. Three secondary dendrites were randomly chosen from each neuron, their length was measured, and the spines were counted. Mushroom-like spines were defined by the observer when they resembled the typical mushroom shape. The total number of spines or the number of mushroom-like spines is depicted as quantity per 10 μm dendritic length.
Immunocytochemistry
When indicated, doxycycline (50 ng/ml; Sigma-Aldrich), mIgf1 (4 ng/ml; ED50 is 2 ng/ml in cell proliferation assay), mIgf2 (0.5, 2, 5, 10, 20 ng/ml; ED50 is 10 ng/ml in cell proliferation assay), Igf2 antibody (0.25, 1, 4 μg/ml), and IKK2 inhibitor BI 5700 (4, 16 μm) were added to the culture medium right after cell seeding or 24 h before fixation, respectively. The standard concentration of Igf2 (20 ng/ml) and anti-Igf2 (1 μg/ml) was used in the experiments, if not otherwise stated. When added for 21 d in vitro, the reagents were renewed every second day. Doxycycline, mIgf1, mIgf2, anti-Igf2, anti-Igf2R, and normal goat serum (gIgG) were all purchased from R&D Systems. Fluorescence images were taken using an upright Axio Scope microscope (40× objective) equipped with a Zeiss CCD camera (16 bits; 1280 × 1024 pixels per image), and analyzed using AxioVision (Zeiss) and Adobe Photoshop software (Adobe Systems). For the quantification of synapse density, cells were counterstained with Bassoon/ProSAP1 or Bassoon/MAP2, respectively. For each group, 5–20 neurons from three to five independent experiments were randomly chosen, and four dendrites were analyzed per neuron. The total number of synapses is depicted as quantity per 10 μm dendritic length as indicated in the descriptive statistical panels.
Kinase inhibitors
The inhibitors used in this study were taken from the Screen-Well Kinase Inhibitor Library (Enzo Life Sciences) that contains 80 known kinase inhibitors of well defined activity. Inhibitors are supplied dissolved in DMSO at 10 mm and used at 10 μm (final concentration): CaMKII, KN-62 (4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenyl isoquinolinesulfonic acid ester); tyrosine kinases, Tyrphostin AG 1288 [(3,4-dihydroxy-5-nitrobenzylidene)malononitrile]; p38 MAPK, SB 203580 (4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine); GSK-3β, Indirubin-3′-monoxime; MEK, PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one].
Microarray analysis
Total cellular RNA was isolated from control and IKK2nDN primary cultures at 21 d in vitro (DIV21). Preparation of in vitro transcription products was performed according to Affymetrix protocols, and probes were hybridized to GeneChip Mouse Genome 430 2.0 Arrays (Affymetrix) at Ulm University Chip Facility (Medical Faculty/Interdisciplinary Center for Clinical Research). Data were analyzed with the GeneSifter microarray data analysis system (Geospezia; http://genesifter.net/web/) using RMA normalization algorithm.
Morris water maze
The experimental subjects were 3-month-old male mice (Co group, n = 8; IKK2nDN, n = 8), which were housed individually in a 12 h light/dark schedule for 2 weeks before the Morris water maze (MWM) task. The experiment was performed as described by Vorhees and Williams (2006). Before the MWM task, a visible platform test was performed to exclude motor and visual acuity impairment. The platform was marked with a flag, and tracking length and latency to reach the platform were recorded. Position, tracking length, swim speed, and latency of finding the platform were recorded automatically by using the Viewer II software (Biobserve GmbH).
RNA isolation, reverse transcription, RT-PCR, and real-time RT-PCR
Total RNA from cultivated neurons and brain tissue (homogenized under liquid nitrogen using a pestle and mortar) was isolated using peqGOLD TriFast (peQlab). cDNA was synthesized from 2 μg of total RNA using Transcriptor High-Fidelity cDNA Synthesis Kit (Roche). PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) and a LightCycler 480 Instrument (Roche) using the Fast-Start Universal Probe Master and LNA probes from the Universal Probe Library (Roche) according to the manufacturers' instructions. Negative controls comprised replacement of either cDNA or reverse transcriptase with water. β-Actin, Rpl13a, PBGD, and HPRT1 were used as housekeeping genes. Primer sequences and probes used for real time and RT-PCR were as follows: β-actin (5′-GGA TGC AGA AGG AGA TTA CTG C-3′, 5′-CCA CCG ATC CAC ACA GAG TA-3′, probe no. 63), PBGD (5′-ACA AGG GTT TTC CCG TTT-3′, 5′-TCC CTG AAA GAT GTG CCTAC-3′, probe no. 79), Rpl13a (5′-CCT GCT GCT CTC AAG GTT GT-3′, 5′-GGT ACT TCC ACC CGA CCT C-3′, probe no. 41), HPRT1 (5′-GGA GCG GTA GCA CCT CCT-3′, 5′-CCT GGT TCA TCA TCA CTA ATC-3′, probe no. 69), and Igf2 (5′-CGC TTC AGT TTG TCT GTT CG-3′, 5′-GCA GCA CTC TTC CAC GAT G-3′, probe no. 40). For RT-PCR the following primers were used: Igf2 (5′-GAT CCC AGT GGG GAA GTC GAT GTT GG-3′, 5′-GAT GGT TGC TGG ACA TCT CCG AAG AGG-3′), IKK2-DN transgene (5′-CAG CCT GCA CCT GAG GAG TGA ATT C-3′, 5′-GGC GCT GGA ACT GTT GCC CAA G-3′), β-actin (5′-GGA TGC AGA AGG AGA TTA CTG C-3′, 5′-CCA CCG ATC CAC ACA GAG TA-3′).
Statistical analysis
Measured data were exported to Excel software (Microsoft). All data are shown as mean ± SEM. Data were tested for normality using SPSS software. Statistical significances were determined by using Student's t test or a linear mixed model (SAS9.2) (*p < 0.05; **p < 0.01; ***p < 0.001).
Transfections
Transient transfections of HeLa cells were performed as described previously (Wacker et al., 2011).
Results
The NF-κB-inducing IKK complex is enriched and activated at the PSD
Learning and memory deficits in mice with impaired neuronal NF-κB signaling indicated that plastic changes of synaptic contacts depend at least in part on synaptic NF-κB signaling (Meffert and Baltimore, 2005; Kaltschmidt and Kaltschmidt, 2009). To address the question whether NF-κB is directly activated at the synapse, an important prerequisite for synapse-to-nucleus signaling, we analyzed crude homogenate, synaptosomes, and the PSD fraction for the presence of IKK/NF-κB family members. Our analysis revealed that IKK1 and IKK2 are enriched in the PSD as well as phosphorylated IKK indicating active kinases (Fig. 1A). Interestingly, RelB was particularly enriched within the synaptosomal fraction, whereas c-Rel was almost exclusively found in the PSD (Fig. 1A). RelA was clearly detectable in the PSD but did not exhibit enriched levels. The quality of biochemical fractionation was confirmed by analyzing Synaptophysin (Syp) and ProSAP1/Shank2 expression (Fig. 1B).
Figure 1.
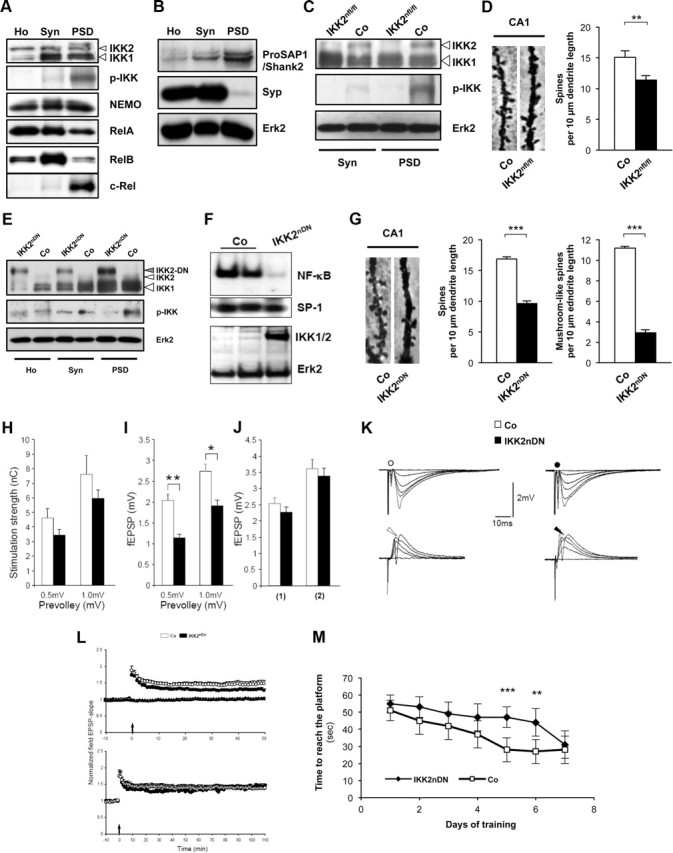
Inhibition of synaptic IKK/NF-κB signaling interferes with spine formation, synaptic transmission, and spatial learning. A, Representative immunoblot analysis of biochemical fractions from adult mouse forebrain probed with specific antibodies as indicated. B, Immunoblots of biochemical fractions from adult mouse forebrain. Purification of synaptic fractions was controlled by anti-ProSAP1/Shank2 (postsynaptic marker) and anti-Synaptophysin (Syp) (presynaptic marker) antibodies. Loading control, Erk2. C, Representative immunoblot analysis of biochemical fractions from control and transgenic IKK2nfl/fl mouse forebrains probed with specific antibodies as indicated. D, Representative images (10-μm-long segments) of secondary dendrites from CA1 pyramidal neurons of adult control and transgenic IKK2nfl/fl brains (Golgi-Cox, left panel). Quantification of spine density from CA1 pyramidal neurons of control and IKK2nfl/fl brains (right panel). n = 5–10 cells from three independent male littermate pairs. E, Representative immunoblot analysis of biochemical fractions from control and transgenic IKK2nDN mouse forebrains probed with specific antibodies as indicated. F, EMSA (top panel) and immunoblot (bottom panel) of hippocampal nuclear extracts from control (Co) and IKK2nDN mice. SP-1 served as a quality control in EMSA; Erk2 was used as loading control in immunoblot analysis. G, Representative images (10-μm-long segments) of secondary dendrites from CA1 pyramidal neurons of adult control and IKK2nDN brains (Golgi-Cox, left panel). Quantification of spine density from CA1 pyramidal neurons of adult control and IKK2nDN brains. n = 5–10 cells from six independent male littermate pairs (middle panel). Quantification of mushroom-like spines from CA1 pyramidal neurons of adult control and IKK2nDN brains. n = 5–10 cells from six independent male littermate pairs (right panel). H, Stimulation strengths necessary to elicit a prevolley of the indicated amplitudes in slices from control (open columns) and IKK2nDN (black columns) mice. Control (4.6 ± 0.6 nC, n = 40; 7.6 ± 1.3 nC, n = 32); IKK2nDN (3.4 ± 0.4 nC, n = 40; 5.9 ± 0.6 nC, n = 40); five animals per group. I, fEPSP amplitudes in the two genotypes as a function of the two prevolley amplitudes. Control (2.0 ± 0.1 mV, n = 40; 2.7 ± 0.2 mV, n = 32); IKK2nDN (1.1 ± 0.1 mV, n = 40; 1.9 ± 0.1 mV, n = 40); five animals per group. J, The fEPSP amplitudes in slices from the two genotypes necessary to elicit a just detectable population spike (1) and a population spike of 2 mV amplitude (2). (1) Control (2.5 ± 0.2 mV; n = 40); (1) IKK2nDN (2.3 ± 0.2 mV; n = 40); (2) Control (3.6 ± 0.3 mV; n = 32); (1) IKK2nDN (3.4 ± 0.2 mV; n = 24); five animals per group. K, Top row, Each trace is the mean of five consecutive synaptic responses in stratum radiatum elicited by different stimulation strengths in slices from control (left) and IKK2nDN (right) mice. The prevolleys preceding the fEPSPs are indicated by circles. Bottom row, Recordings from stratum pyramidale elicited by paired-pulse stimulation (50 ms interstimulus interval). The arrowheads indicate the population spike threshold. L, Top panel, Normalized and pooled fEPSP slopes evoked at CA3-to-CA1 synapses in slices from control and IKK2nDN mice. LTP was induced by a single tetanization. For the sake of clarity, only the nontetanized control pathway in IKK2nDN mice is shown. The arrow at the abscissa indicates the time of tetanic stimulation. Control (n = 28); IKK2nDN (n = 27); six animals per group. Bottom panel, As above, but fEPSPs were followed for 110 min after the tetanization. Control (n = 10); IKK2nDN (n = 9); five animals per group. M, Delayed performance of IKK2nDN mice during the 7 d testing period in the MWM as deduced from the latency time before reaching the hidden platform; eight animals per group. A–M, Co, Control; Ho, homogenate; Syn, synaptosomes; PSD, postsynaptic density fraction. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Recent studies implied that spinogenesis of medium spiny GABAergic neurons in the nucleus accumbens depends on IKK2 activity, while RelA function was shown to control synaptogenesis in the developing hippocampus (Russo et al., 2009; Boersma et al., 2011). Our biochemical data show that IKK activation takes place directly at the PSD under basal conditions in adult mice. To address its cellular consequences, we established IKK2 conditional knock-out mice (CaMKIIα iCre BAC × IKK2fl/fl) named IKK2nfl/fl, in which IKK2 is specifically deleted in excitatory neurons of the forebrain. These mice show decreased levels of phospho-IKK2 in synaptic fractions (Fig. 1C). In addition, IKK2nfl/fl animals exhibit a prominent decrease in spine density of CA1 neurons (Fig. 1D), indicating that the proper formation of hippocampal dendritic spines depends on synaptic IKK activity.
IKK inhibition decreases mature spine numbers and reduces synaptic transmission
It was unclear whether IKK-dependent regulation of spine density is restricted to a distinct developmental time window (Boersma et al., 2011) or whether it is extending into adulthood. We therefore used a different transgenic mouse model (Camk2a-tTA × IKK2-DN), named IKK2nDN, which allows the expression of a dominant-negative allele of the Ikbkb gene (IKK2-DN, N-terminally VSV-tagged) in forebrain neurons in a doxycycline (DOX)-dependent manner (Herrmann et al., 2005; Baumann et al., 2007). Biochemical fractionation shows an enrichment of IKK2-DN at synaptic sites causing a suppression of endogenous IKK2 activity, in particular at the PSD, as evidenced by attenuated p-IKK levels (Fig. 1E). On the functional level, expression of the IKK2-DN protein in the hippocampus induced an extinction of the basal level of NF-κB DNA-binding activity (Fig. 1F). The evaluation of dendritic spines in the hippocampal CA1 region of adult mice again showed a reduction of the total spine density and a decrease of mature mushroom-like spines in IKK2nDN mutants (Fig. 1G).
To assess changes in excitatory synaptic transmission and synaptic excitability, we recorded simultaneously in the apical dendritic and soma layers in the CA1 region of hippocampal slices and measured the fiber volley, the fEPSP, and the population spike as a function of different stimulation strengths (Fig. 1H–K). The stimulation strengths necessary to elicit fiber volleys of given amplitudes (0.5 and 1.0 mV) were similar (Fig. 1H) and did not suggest substantial changes in fiber density or number of afferent fibers in the two genotypes. In IKK2nDN mice, evoked fEPSPs for presynaptic fiber volleys of 0.5 and 1.0 mV, however, were severely reduced and reached only 55 and 70% of control values (Fig. 1I,K), a finding that is best explained by the reduced number of mature synapses. The fEPSP threshold for generating a population spike and the fEPSP magnitude necessary to elicit a population spike of 2 mV were similar in IKK2nDN and control mice, suggesting an unchanged postsynaptic excitability (Fig. 1J,K). We further examined LTP at hippocampal CA3-to-CA1 synapses in slices taken from adult mice. Although LTP in IKK2nDN mice was slightly smaller in magnitude, it was not statistically different compared with controls (Fig. 1L). Consistent with functional field long-term potentiation in the dorsal hippocampus the performance in the MWM task improved in both IKK2nDN and control mice over 7 d of testing. Although IKK2nDN mice significantly differed from control mice at days 5 and 6 of testing, they were finally able to find the hidden platform with a similar latency time as controls (Fig. 1M).
IKK/NF-κB signaling regulates spine formation and maturation in vivo
These findings prompted us to analyze the molecular composition of synaptosomes (Fig. 2A) and PSDs (Fig. 2B) from IKK2nDN and control mice. We found no significant differences in the levels of Synaptophysin or ProSAP1/Shank2 (Fig. 2A,B). However, a reduction in the level of the AMPAR subunit GluA1 was detected in synaptosomes from IKK2nDN mutants. This was associated with a moderate increase in GluN1, the obligatory subunit of the NMDAR (Fig. 2A). Interestingly, specific isoforms of the MAGUKs PSD95 (PSD95α) and SAP97 (SAP97β) were virtually absent in the PSD fraction from IKK2nDN mice (Fig. 2B). Remarkably, no significant changes in the overall expression of GluA1, GluN1, PSD95, and SAP97 isoforms were detected in total forebrain lysates from IKK2nDN and control animals (Fig. 2C). A very recent study demonstrated that knockdown of PSD95 results in a loss of entire patches of PSD material identified by electron microscopy analysis (X. Chen et al., 2011). Consistent with this finding, the synaptic lack of PSD95α in IKK2nDN mice might also lead to the patchy losses of PSD material seen in the CA1 hippocampus (Fig. 2D). The biochemical analyses indicate that the synaptopathic phenotype caused by IKK/NF-κB inhibition might be associated with a subcellular redistribution defect of PSD95α and SAP97β, proteins directly involved in synapse and spine maturation.
Figure 2.

Molecular and ultrastructural disruption of synapses in IKK2nDN brains. A, Representative immunoblot analysis of synaptosomal fractions from control and IKK2nDN mouse forebrain as indicated (left panel). Quantification of protein levels. n = 3 samples per group with each sample being a combined pool of 10 forebrains (right panel). B, Representative immunoblot analysis of PSD fractions from control and IKK2nDN mouse forebrain as indicated. The filled arrowheads mark α-isoforms, and the framed arrowheads mark β-isoforms of PSD95 and SAP97 (left panel). Quantification of protein levels. n = 3 samples per group with each sample being a combined pool of 10 forebrains (right panel). C, Representative immunoblot analysis of homogenates from control and IKK2nDN mouse forebrain (left panel). Quantification of protein levels is shown. n = 3 samples per group with each sample being a combined pool of 10 forebrains (right panel). D, Representative electron micrographs depicting CA1 synapses with presynaptic specializations (framed arrowheads) and postsynaptic spines (asterisks). The filled arrowhead marks the patchy loss within the PSD of IKK2nDN synapses (left panel). Shown is quantification of total CA1 synapses from control (n = 48 synapses from 3 mice) and IKK2nDN (n = 30 synapses from 3 mice) mice showing either segmented (patchy) or nonsegmented (nonpatchy) postsynaptic densities (right panel). A–D, Co, Control. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. *p < 0.05.
IKK/NF-κB signaling dynamically modulates synapse formation ex vivo
Consistent with our in vivo findings, we also detected all members of the IKK complex enriched and activated at postsynaptic sites of primary hippocampal neurons in vitro by immunofluorescence microscopy (Fig. 3A). To elucidate molecular mechanisms underlying the synaptic defects in IKK2nDN animals, we made use of dissociated hippocampal cultures. Initial characterization of primary hippocampal neurons from IKK2nDN mice revealed stable transgene expression over a culture time of DIV21 that is localized to dendrites as indicated by coimmunostaining of epitope (VSV)-tagged IKK2-DN (Fig. 3B,C) (Herrmann et al., 2005). When analyzing synapse density in mature IKK2nDN and control cultures (DIV17), we found a 30–40% reduction of synaptic contacts measured either by Bassoon/MAP2 (all synaptic contacts) or by Bassoon//ProSAP1/Shank2 (excitatory contacts) coimmunostainings in mutant neurons (Fig. 3D). A more detailed analysis of excitatory synapse development in vitro at DIV7, DIV10, DIV14, DIV17, and DIV21 (Grabrucker et al., 2009) showed a significant reduction of presynaptic and postsynaptic specializations in IKK2nDN cultures at all time points (Fig. 3D). Interestingly, the difference in synapse number between wild-type and IKK2nDN increased over time, supporting a maturational defect rather than the perturbation of early synaptogenesis (Fig. 3D, ratio Co/IKK2nDN). To confirm the IKK2-DN mediated reduction in synaptic contacts, synapse density in cultures expressing a degradation-resistant, transdominant IκBα protein (ΔNIκBα) that specifically interferes with NF-κB activation downstream of IKK action, was analyzed (Lavon et al., 2000). Mature primary hippocampal neurons from double-transgenic Camk2a-tTA × ΔNIκBα mice exhibited a similar reduction of synapse number as IKK2nDN cultures (Fig. 3E).
Figure 3.
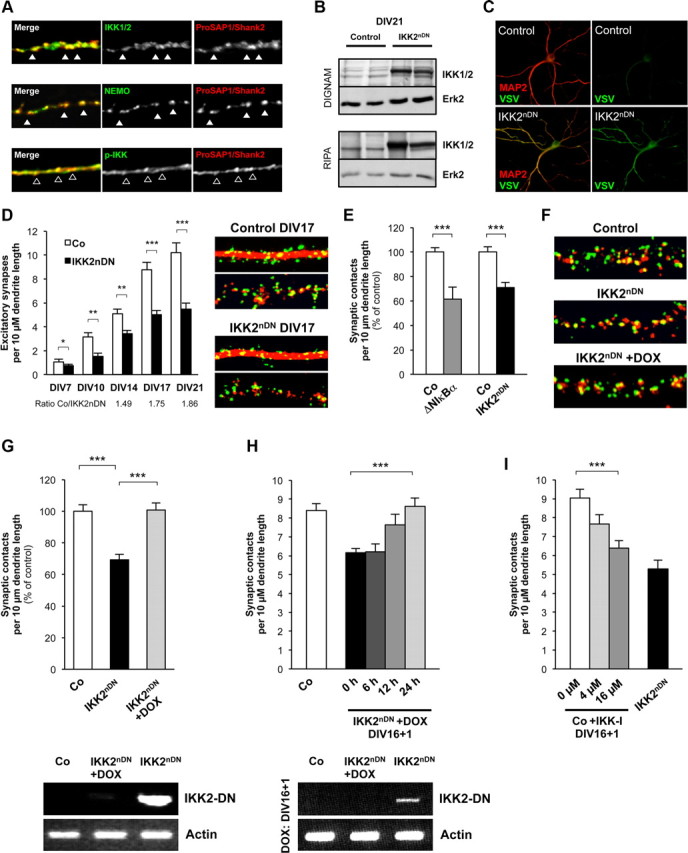
Synapse formation is dynamically regulated by IKK/NF-κB signaling ex vivo. A, Coimmunolabeling of DIV21 hippocampal cultures from wild-type mice with IKK1/2, NEMO, p-IKK (all green), and the postsynaptic marker ProSAP1/Shank2 (red). Colocalization with ProSAP1/Shank2 is marked by filled arrowheads in case of IKK1/2 and NEMO and by framed arrowheads in case of p-IKK. B, IKK2-DN transgene expression in cultured neurons (DIV21) from control and IKK2nDN animals. Immunoblot from two independent hippocampal cultures is shown. Top panel, DIGNAM extracts, Bottom panel, RIPA extracts. RIPA extract indicates IKK2-DN localization in membrane-associated fractions. Loading controls, Erk2. C, Coimmunolabeling of DIV21 hippocampal neurons from control (Co) and IKK2nDN mice for VSV (green) and MAP2 (red). D, Quantification of excitatory synapse numbers at the indicated time points of hippocampal in vitro synaptogenesis from control and IKK2nDN mice (left panel) and representative images of secondary dendrites (15-μm-long segments) from control and IKK2nDN DIV17 hippocampal neurons coimmunolabeled either for Bassoon (presynaptic specializations, green-colored dot-like structures) and MAP2 (dendrite, red-filled structure) or Bassoon (green) and ProSAP1/Shank2 (postsynaptic specializations, red-colored dot-like structures) (right panel). E, Direct comparison of reduced synapse numbers from hippocampal cultures expressing a transdominant IκBα (ΔNIκBα) or IKK2-DN (IKK2nDN) protein. F, Representative images of secondary dendrites (15-μm-long segments) from control, IKK2nDN and DOX-treated IKK2nDN hippocampal neurons coimmunolabeled for Bassoon (green) and ProSAP1/Shank2 (red) (left panel). G, Quantification of synapse numbers from the conditions depicted in F. H, Quantification of synapse numbers from DIV16 IKK2nDN hippocampal neurons treated with DOX for the indicated time periods. G, H, RT-PCR analysis of mRNA from control (Co), untreated, and DOX-treated IKK2nDN hippocampal neurons (bottom panels). Duration of treatment was as indicated. IKK2-DN indicates transgene expression. Loading controls, Actin. I, Quantification of synapse numbers from DIV16 control hippocampal neurons after 24 h treatment with IKK2 inhibitor (BI 5700) at the indicated concentrations. A–I, Co, Control. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Importantly, synapse numbers could be completely restored by blocking IKK2-DN expression with DOX in ex vivo cultures (Fig. 3F,G). Transgene expression was monitored by RT-PCR (Fig. 3G, bottom panel). This excludes that synaptic defects are already manifested during embryonic development. Our findings rather suggest that the lack of IKK/NF-κB signaling may hinder newly built synapses to reach and maintain a mature stage. To address this issue, we reactivated IKK/NF-κB at later stages of ex vivo differentiation. Intriguingly, at 12 h after inhibition of transgene expression, a partial rescue was already detectable and complete restoration was reached after 24 h when transgene expression was switched off (Fig. 3H). To confirm that synapse numbers can be rapidly regulated via active IKK, we inhibited IKK2 activity for 24 h using the IKK2-selective inhibitor BI 5700 (Huber et al., 2010). This treatment induced a dose-dependent reduction of synaptic contacts similar to that seen in IKK2nDN primary neurons (Fig. 3I). In summary, our ex vivo studies suggest that the IKK/NF-κB-dependent regulation of synapse formation/maturation is reversible and highly dynamic.
Igf2 is a novel NF-κB target gene
A critical question to solve was whether IKK/NF-κB affects synapse formation/maturation via an intrinsic or rather via an autocrine/paracrine mechanism. For this, we tested whether secreted factors might be involved and exchanged conditioned media from control and IKK2nDN neurons at DIV16 for 24 h. Synapse density in mutant cultures incubated with medium from control cells was completely reconstituted. In contrast, control cultures incubated with IKK2nDN medium exhibited a decrease in synapse number (Fig. 4A). This argues for an autocrine/paracrine regulatory mechanism and suggests that the lack of IKK/NF-κB-dependent target gene expression leads to impaired synapse formation/maturation in IKK2nDN neurons. To identify candidate molecules, we generated global gene expression profiles from control and IKK2nDN neurons at DIV21. Among the genes with the highest level of repression in IKK2nDN hippocampal neurons, we found Igf2 (Table 1). RT-PCR confirmed the downregulation of Igf2 mRNA in IKK2nDN cultures (Fig. 4B), and quantification by qRT-PCR revealed a reduction by 80–90% (Fig. 4B). Igf2 is expressed in neurons and virtually not in astrocytes (Fig. 4C). Notably, Igf2 is localized within the somatodendritic compartment, while expression was markedly reduced in IKK2nDN neurons (DIV21) (Fig. 4D). Decreased Igf2 levels were also found in the supernatant of mutant cultures as evaluated by ELISA (Fig. 4E).
Figure 4.
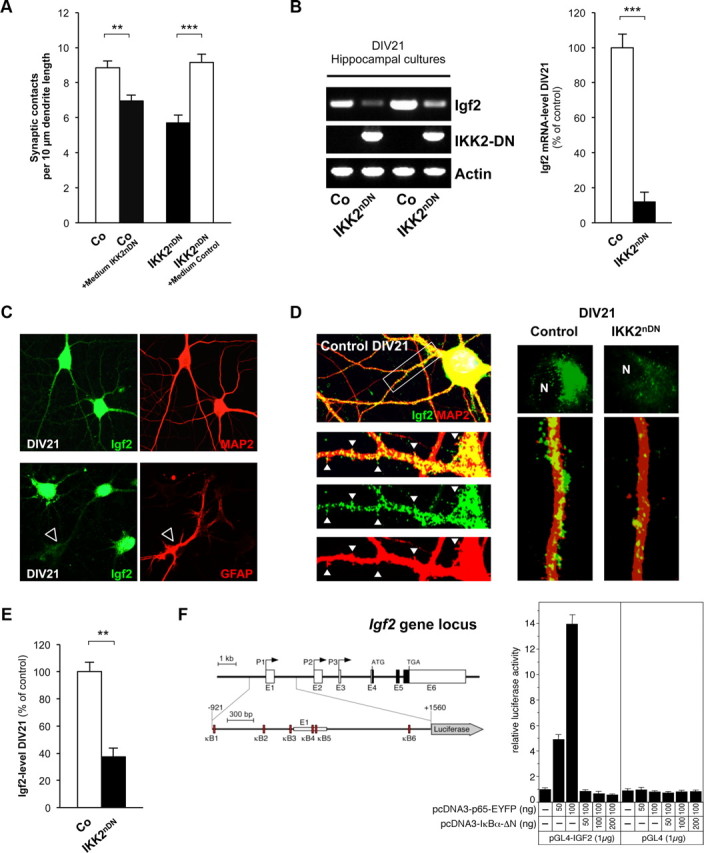
Igf2 is novel downstream effector of IKK/NF-κB signaling in neurons. A, Quantification of synapse numbers from DIV16 control hippocampal neurons treated with conditioned IKK2nDN medium and from DIV16 IKK2nDN hippocampal neurons treated with conditioned control medium for 24 h. B, RT-PCR analysis of Igf2 mRNA in hippocampal cultures from control and IKK2nDN animals at DIV21. IKK2-DN indicates transgene expression levels. Loading control, Actin (left panel). Quantitative RT-PCR analysis of Igf2 mRNA in control and IKK2nDN hippocampal cultures at DIV21 (right panel). C, Representative images of DIV21 wild-type hippocampal neurons colabeled either for Igf2 (green) and MAP2 (neurons, red) or GFAP (astrocyte, red, marked by arrowhead) as indicated. D, Representative images of a DIV21 wild-type hippocampal neuron coimmunolabeled for Igf2 (green) and MAP2 (dendrite, red). Insets, The white arrows mark sites of dendritic protrusions (left panel). Subcellular distribution of Igf2 (green) in control and IKK2nDN hippocampal neurons (green, Igf2; red, MAP2-stained dendrites). “N ” marks the nucleus. Note the reduced Igf2 intensity in IKK2nDN neurons (right panel). E, Quantification of Igf2 levels by ELISA in the supernatant of control and IKK2nDN hippocampal cultures at DIV21. F, Schematic presentation of the murine Igf2 gene locus composed of six exons, termed E1 to E6 (Gene ID 16002/ENSMUST00000105936). P1–P3 indicate alternative promoters driving transcription of three unique leader exons (E1–E3, untranslated exons, open boxes) that are spliced to the common protein coding exons E4–E6. Schematic presentation of the Igf2-specific reporter construct pGL4-IGF2, used in this study (base pairs −921 to +1560 relative to the transcription start at exon 1). The potential κB binding sites (κB1 to κB6) are shown as red boxes (left panel). Transient transfections of HeLa cells with a pGL4-IGF2 reporter construct together with a RelA expression vector (pcDNA3-p65-EYFP) either in the absence or presence of an expression vector for IκBα-ΔN (pcDNA3-IκBα-ΔN). Negative control, Empty pGL4 reporter gene construct (right panel). A–E, Co, Control. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. **p < 0.01; ***p < 0.001.
Table 1.
Differentially regulated genes in primary hippocampal neurons (IKK2nDN vs controls)
| Gene ID | Gene identifier | Fold change | p value |
|---|---|---|---|
| Rgs5 | BF585144 | 3.062 | 0.0299 |
| Col8a1 | NM_007739 | 3.062 | 0.0375 |
| Mfap4 | BC022666 | 2.837 | 0.0265 |
| — | NM_133736 | 2.823 | 0.0020 |
| Hmcn1 | BB024475 | 2.628 | 0.0162 |
| Rgs5 | BF585144 | 2.239 | 0.0462 |
| Ddit4l | AV251625 | 2.098 | 0.0189 |
| — | BB088582 | 1.964 | 0.0004 |
| Hist2h3c2 | BC015270 | 1.925 | 0.0454 |
| Arsk | AK013194 | 1.907 | 0.0397 |
| — | BG297038 | 1.866 | 0.0244 |
| Xist | L04961 | 1.838 | 0.0042 |
| Emid1 | NM_080595 | 1.827 | 0.0019 |
| Bmpr2 | NM_007561 | 1.804 | 0.0026 |
| — | BB185152 | 0.554 | 0.0217 |
| — | AV375653 | 0.553 | 0.0384 |
| Nfs1 | AI662441 | 0.533 | 0.0121 |
| Nupr1 | NM_019738 | 0.521 | 0.0037 |
| — | BB354985 | 0.519 | 0.0076 |
| Col3a1 | AW550625 | 0.517 | 0.0348 |
| Rasgrf1 | AI850720 | 0.514 | 0.0259 |
| Gjb6 | BC016507 | 0.507 | 0.0359 |
| — | AI506321 | 0.496 | 0.0318 |
| — | C76213 | 0.474 | 0.0115 |
| Ddx3y | AA210261 | 0.450 | 0.0031 |
| Kdm5d | AF127244 | 0.439 | 0.0000 |
| Rasgrf1 | NM_011245 | 0.433 | 0.0019 |
| Fam62b | BI102044 | 0.429 | 0.0105 |
| Eif2s3y | NM_012011 | 0.422 | 0.0271 |
| Ddx3y | AA210261 | 0.413 | 0.0186 |
| Ddx3y | AA210261 | 0.384 | 0.0035 |
| Uty | BB742957 | 0.373 | 0.0189 |
| — | BQ175646 | 0.367 | 0.0263 |
| Igf2 | NM_010514 | 0.341 | 0.0329 |
| — | BB181225 | 0.290 | 0.0038 |
| Tgfbi | NM_009369 | 0.287 | 0.0223 |
| — | AK018172 | 0.047 | 0.0135 |
The regulation of the Igf2 gene expression is highly complex. The murine Igf2 gene consists of six exons (Fig. 4F), and gene expression is regulated by three adjacent promoters, termed P1, P2, and P3 (Sasaki et al., 1992). Each transcript derived contains its own unique leader exon (E1, E2, and E3) connected with exons 4–6 encoding the Igf2 precursor protein. The gene is imprinted and only expressed from the paternal allele (DeChiara et al., 1991). We examined the regulatory regions of the Igf2 locus by Genomatix software and identified at least six potential NF-κB binding sites located in the P1 and P2 region (Fig. 4F, left panel). To test NF-κB responsiveness, we cloned an Igf2-luciferase reporter gene construct (pGL4-IGF2) containing these sites. Transient transfection of HeLa cells revealed a strong upregulation of the pGL4-IGF2 reporter gene upon RelA cotransfection. Cotransfection of RelA together with the inhibitor IκBα-ΔN blocked RelA-dependent transcription, implicating that Igf2 expression is directly regulated by IKK/NF-κB signaling (Fig. 4F, right panel).
Igf2 rapidly regulates synapse formation and spine maturation downstream of IKK/NF-κB signaling
To further investigate the role of Igf2 in synapse formation and spine maturation, we asked whether Igf2 is able to rescue the synaptic defects in IKK2nDN neurons. Recombinant Igf2 was able to restore synapse numbers in IKK2nDN neurons (Fig. 5A) and did so in a dose-dependent manner within 24 h (Fig. 5B). Furthermore, neutralizing Igf2 antibodies were able to phenocopy the IKK2-DN effect of Igf2 in control cultures also in a dose-dependent manner within the same time frame (Fig. 5C). Notably, the density (Fig. 5D) and maturation status of dendritic spines (Fig. 5E) in IKK2nDN cultures was completely normalized by Igf2, indicating a rapid and dynamic effect of Igf2 on the structural remodeling of dendritic spines and synaptic connections. Interestingly, Igf2 also enhanced the number of synapses and mature spines in control cultures (Fig. 5A,D,E). We finally determined whether the DOX-dependent rescue effect in IKK2nDN neurons was mediated by Igf2. Intriguingly, the synaptic defects were not reconstituted after coapplication of neutralizing anti-Igf2 antibodies and DOX for 24 h in IKK2nDN cultures, confirming the direct link between IKK/NF-κB and Igf2-mediated synapse formation/maturation (Fig. 5F).
Figure 5.
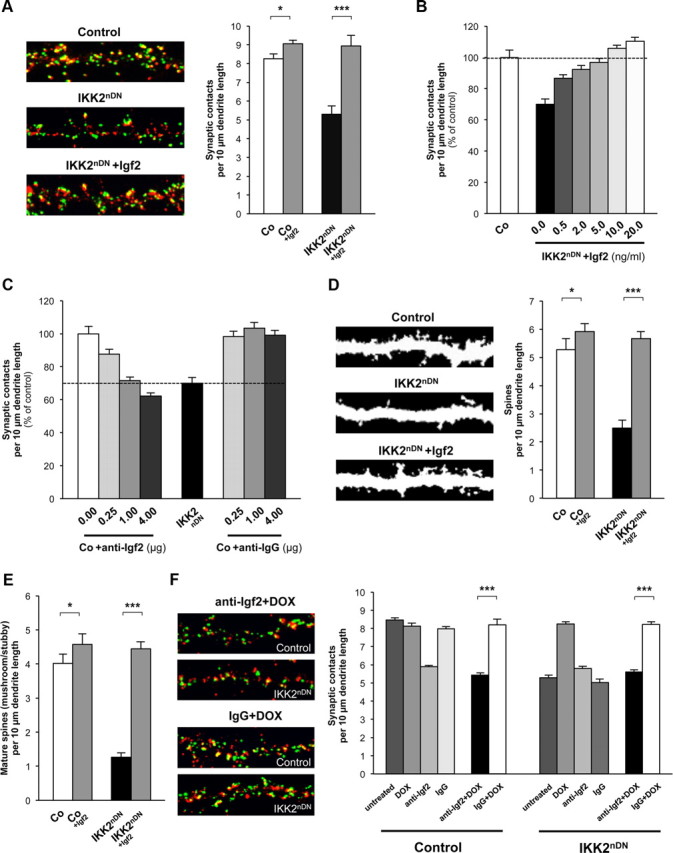
Igf2 rapidly regulates synapse density and spine maturation. A, Representative images of secondary dendrites (20-μm-long segments) from control, IKK2nDN and Igf2-treated (DIV16 + 1) IKK2nDN hippocampal neurons at DIV17. Coimmunolabeling for Bassoon (green) and ProSAP1/Shank2 (red; left panel). Quantification of synapse numbers from the indicated conditions (right panel). B, Quantification of synaptic contacts at DIV17 from control (Co) and IKK2nDN hippocampal neurons either left untreated or treated with increasing concentrations of Igf2 as indicated (DIV16 + 1). The dashed line marks the mean synapse density level of untreated control neurons. C, Quantification of synaptic contacts at DIV17 from control (Co) and IKK2nDN hippocampal neurons either left untreated or treated with increasing amounts of anti-Igf2 or anti-IgG antibodies as indicated (DIV16 + 1). The dashed line marks the mean synapse density level of untreated IKK2nDN neurons. D, Representative images of dendritic segments (length, 20 μm each) from control, IKK2nDN and Igf2-treated (DIV16 + 1) IKK2nDN hippocampal neurons at DIV17. Spines are labeled by MAP2 immunostaining (left panel). Quantification of total spine density (right panel) from the indicated conditions. E, Quantification of the density of mature (mushroom/stubby) spines from the indicated conditions. F, Representative images of secondary dendrites (20-μm-long segments) from control and IKK2nDN hippocampal neurons at DIV17 cotreated (DIV16 + 1) with anti-Igf2+DOX or IgG+DOX. Coimmunolabeling for Bassoon (green) and ProSAP1/Shank2 (red) (left panel). Shown is quantification of synaptic contacts at DIV17 from control and IKK2nDN hippocampal neurons treated (DIV16 + 1) as indicated (right panel). ***p < 0.001. A–F, Co, Control. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Igf2 action in primary neurons is mediated via Igf2R and MEK/ERK signaling
Igf2 is related to Igf1, which is more widely expressed in postnatal and adult life. Like Igf1, Igf2 can bind to and signal via the Igf1 receptor (Igf1R), which contains a tyrosine kinase domain (Chiu and Cline, 2010). We therefore initially asked whether Igf1 was able to mimic Igf2-mediated effects in IKK2nDN cultures. When Igf1 or Igf2 were added to mutant cultures, Igf1 exhibited only a moderate effect (Fig. 6A). Whereas Igf1 specifically signals via the Igf1R, Igf2 can bind and activate Igf1R, insulin receptors, and a specific Igf2 receptor (Igf2R) that we could detect within dendrites (MAP2 coimmunostaining) and at synaptic sites (Bassoon coimmunostaining; Fig. 6B). Consistent with this result, application of antagonistic Igf2R antibodies interfered with Igf2 action, abrogated the DOX-mediated rescue effect in IKK2nDN neurons and also decreased synapse numbers in control cultures (Fig. 6C). We therefore conclude that Igf2 action at the synapse is mediated predominantly via Igf2R signaling.
Figure 6.
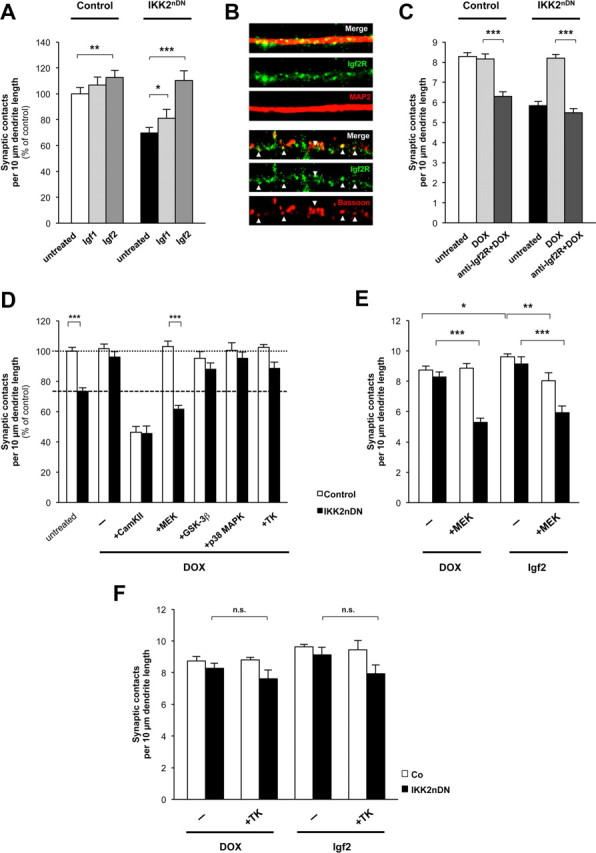
Igf2-induced synapse formation predominantly depends on Igf2R-mediated MEK/ERK signaling. A, Quantification of synaptic contacts from control and IKK2nDN hippocampal neurons treated as indicated. B, Representative images of secondary dendrites (15-μm-long segments) from wild-type hippocampal neurons at DIV17 coimmunolabeled either for Igf2R (green) and MAP2 (red, top panel) or Igf2R (green) and Bassoon (red, bottom panel). C, Quantification of synaptic contacts at DIV17 from control and IKK2nDN hippocampal neurons treated (DIV16 + 1) as indicated. D, Quantification of synaptic contacts at DIV17 from control and IKK2nDN hippocampal neurons either left untreated, treated with DOX (−), or cotreated with DOX and specific kinase inhibitors (10 μm f.c.) as indicated. Dashed line, Mean synapse density of IKK2nDN neurons; dotted line, mean synapse density of IKK2nDN+DOX (DIV16 + 1). E, Quantification of synaptic contacts at DIV17 from control and IKK2nDN hippocampal neurons treated with MEK inhibitor (PD98059, 10 μm f.c.) as indicated (DIV16 + 1). F, Quantification of synaptic contacts at DIV17 from control and IKK2nDN hippocampal neurons either treated with DOX or Igf2 in the absence or presence of Tyrphostin AG 1288 (tyrosine kinase inhibitor) as indicated (DIV16 + 1). A–F, Co, Control. All quantitative data are shown as mean ± SEM. All p values are derived from Student's unpaired, two-tailed t test. *p < 0.05; **p < 0.01; ***p < 0.001.
The signaling cascades downstream of Igf2R are not well characterized. Therefore, we reactivated the IKK/NF-κB/Igf2 signaling axis in IKK2nDN cultures by DOX application in combination with inhibitors of kinases known to be involved in synaptic differentiation (Fig. 6D). As a control, we repressed CaMKII activity as a known kinase critically involved in synaptic maturation and plasticity (Vaillant et al., 2002; Steiner et al., 2008). In addition, we blocked MEK activity as an integral component of the Ras–Raf–MEK–ERK pathway (Harvey et al., 2008), GSK3β activity, as this is a proposed kinase acting downstream of Igf2R (D. Y. Chen et al., 2011), and p38 activity, which was shown to preferentially act on presynaptic differentiation (Nakata et al., 2005). Furthermore, we used a pleiotropic tyrosine kinase inhibitor to address insulin receptor and Igf1 receptor signaling. The DOX-mediated rescue effect in mutant neurons was prevented in the presence of the CaMKII and MEK inhibitors, whereas inhibition of p38, GSK3β, and tyrosine kinase signaling did not interfere with the recovery of synaptic contacts (Fig. 6D). The application of the CaMKII inhibitor induced a massive decrease in synapse number, both in control and in IKK2nDN cultures (Fig. 6D), confirming that CaMKII is acting at multiple steps during synaptic differentiation. In contrast, the MEK inhibitor specifically prevented the DOX-dependent restoration of synapse numbers in IKK2nDN cultures, indicating an important function downstream of Igf2R. To validate this, we extended our analysis in the presence of exogenous Igf2 (Fig. 6E,F). The pleiotropic tyrosine kinase inhibitor Tyrphostin AG 1288 did not show any significant effect on synapse number in both mutant and control neurons, thus excluding classical receptor tyrosine kinase signaling (Fig. 6F). In contrast, inhibition of MEK–ERK signaling again blocked the reconstitution of synapse number in DOX-treated IKK2nDN cultures and also prevented the rescue effect exhibited by exogenous Igf2 (Fig. 6E). Furthermore, Igf2-mediated enhancement of synapse density in control neurons was also blocked by MEK inhibition (Fig. 6E). Since MEK inhibition did not affect synapse number in control neurons, but only interfered with the Igf2-mediated enhancement effect, argues against a pleiotropic inhibition of MEK/ERK-regulated cellular functions by the MEK inhibitor (Fig. 6D,E). Together, our findings indicate that Igf2 is able to regulate the number of synaptic contacts via Igf2R-mediated MEK–ERK activation within 24 h ex vivo.
The IKK/NF-κB/Igf2 axis dynamically regulates synapse formation in vivo
Finally, we analyzed whether our ex vivo findings from hippocampal cell culture can be translated to in vivo. Interestingly, Igf2R was enriched in the PSD from both control and IKK2nDN forebrains (Fig. 7A). In addition, we found reduced Erk activity in all IKK2nDN-derived fractions evident from the attenuated phospho-Erk1/2 levels (Fig. 7A). Furthermore, hippocampal Igf2 mRNA expression levels from IKK2nDN as well as IKK2nfl/fl animals were strongly repressed (Fig. 7B). Importantly, DOX treatment restored the reduced Igf2 levels in hippocampi from IKK2nDN animals (Fig. 7C). We therefore tested whether a short-term DOX application is able to reconstitute the synaptic deficits in IKK2nDN mice. For this purpose, IKK2nDN and control animals were administered DOX for 1 week while transgene expression was monitored by in vivo measurement of luciferase activity until day 4, when reporter gene activity already dropped by a factor of 100 (Fig. 7D). Three days later, we biochemically isolated synaptic fractions from forebrains and analyzed the levels of the transgene (Fig. 7D, inset) and defined synaptic proteins (Fig. 7E). Strikingly, we found a complete restoration of GluA1, GluN1, isoform-specific PSD95, SAP97, and p-Erk1/2 levels (Fig. 7E). Our findings on the molecular level were reflected by a complete restoration of CA1 hippocampal total and mushroom-like spine numbers in IKK2nDN animals from the same cohort (Fig. 7F).
Figure 7.

The IKK/NF-κB/Igf2 signaling system regulates synapse formation in vivo. A, Representative immunoblots of biochemical fractions from adult control and IKK2nDN mouse forebrain as indicated. Loading control, Erk2. B, Quantitative RT-PCR analysis of hippocampal Igf2 mRNA expression of control and IKK2nDN or control and IKK2nfl/fl animals. **p < 0.01; ***p < 0.001. C, Representative immunoblots of hippocampal Igf2 protein levels of control and IKK2nDN animals left untreated or treated with DOX. IKK2-DN indicates transgene expression, loading control: Erk2. D, Quantification of in vivo luciferase activity (in photons/second/centimeter2/steradian) in control and IKK2nDN brains before (0 h), 12, 36, and 96 h after DOX application. Inset, Immunoblots of synaptic fractions from control and IKK2nDN forebrains after 1 week of DOX application probed with anti-IKK1/2 antibodies. The arrowheads mark IKK2-DN transgene levels. E, Representative immunoblots of synaptosomal (left panel) and PSD fractions (right panel) from control and IKK2nDN forebrains 7 d after DOX application. Samples from 10 pooled forebrains are shown per lane. F, Quantification of spine density from secondary dendrites of CA1 hippocampal neurons from IKK2nDN and control mice. n = 5–10 cells from six independent male littermate pairs (left panel). Shown is quantification of mushroom-like spine density. n = 5–10 cells from six independent male littermate pairs (right panel). A–F, Co, Control; Ho, homogenate; Syn, synaptosomes; PSD, postsynaptic density fraction.
Discussion
IKK/NF-κB signaling has recently been shown to promote the development and remodeling of synaptic connections (Russo et al., 2009; Boersma et al., 2011; Christoffel et al., 2011). However, NF-κB-dependent mechanisms and target genes that mediate the structural changes finally resulting in formation, maturation, and stabilization of synapses, were hitherto poorly characterized.
We detected NF-κB signaling components at postsynaptic densities in vitro and in vivo. Inhibition of synaptic IKK activity resulted in a reduction of dendritic spines and reduced AMPA-mediated basal synaptic transmission in adult mice. This correlated with reduced levels of synaptic PSD95, SAP97, and GluA1. Strikingly, these changes could be reversed by reactivating IKK function. Similarly, synaptic defects recurred ex vivo in mature primary hippocampal cultures from IKK2nDN mutants and were reconstituted within 24 h. We found Igf2 directly targeted by IKK/NF-κB in vitro and in vivo and identified an IKK/NF-κB–Igf2–Igf2R signaling axis tightly controlling the number of synaptic connections in hippocampal cultures.
Our study has three major findings: First, we revealed a novel mechanism of highly dynamic IKK/NF-κB-dependent structural remodeling of synaptic connections within the adult brain. Second, we identified Igf2 as a novel IKK/NF-κB target gene proposing a synaptic Igf2–Igf2R module as a potent regulator of synapse formation/maturation. Third, we showed that the MEK/ERK signaling cascade is coupled to the downstream machinery of the Igf2 receptor in neurons.
Our results reveal that impaired IKK/NF-κB signaling is associated with low Igf2 levels and decreased amounts of the MAGUK SAP97β at the PSD. In contrast to SAP97α, SAP97β is involved in the trafficking of GluA1 to the postsynaptic membrane in response to neuronal activity (Schlüter et al., 2006). Therefore, loss of SAP97β may be one molecular reason for synaptic reduction of GluA1, thus contributing to reduced AMPAR-mediated basal synaptic transmission. Interestingly, a gain-of-function mouse model for NF-κB activation exhibited the reverse effect of increased neuronal excitatory network activity and synchrony (Shim et al., 2011). In both mouse models, hippocampal LTP remained intact, indicating a homosynaptic scaling of AMPAR responses mediated by IKK/NF-κB signaling.
PSD95 is another MAGUK regulating synaptic AMPAR accumulation at the PSD during synapse maturation. The reduction of PSD95α within the synaptic compartment seen in our model might therefore also account for the changes in GluA1-type AMPARs and the corresponding electrophysiological alterations. Recently, PSD95 expression was shown to be NF-κB dependent during critical periods of neuronal differentiation (DIV16), but not at later stages (DIV21) (Boersma et al., 2011). Our data from adult IKK2nDN forebrain fractionations support these observations, as we did not observe any changes of either PSD-95α or β in total homogenates of adult mice. However, we found that PSD95α is almost completely missing from the PSD fraction. N-terminal palmitoylation of PSD95α is an essential step during PSD-95-mediated AMPAR accumulation at the PSD and cannot be compensated by PSD95β (El-Husseini et al., 2002). As SAP97β is known to compensate for AMPAR deficits at synapses lacking other PSD-MAGUKs (Howard et al., 2010) and both PSD95α and SAP97β are lacking in the PSD of IKK2nDN mutants, we hypothesize that the IKK/NF-κB–Igf2–Igf2R signaling axis tightly regulates the presence of both molecules at the synapse, thereby contributing to the maturation and maintenance of synaptic connections.
Igf2 belongs to a family of secreted growth factors including insulin-like growth factor 1 and 2, both able to activate receptor tyrosine kinases that are expressed in the CNS (Hetts et al., 1997; Scolnick et al., 2008). In contrast to Igf1, which plays a role in axonal growth and pathfinding, neuronal progenitor proliferation, and synaptic homeostasis, little is known about the physiological function of Igf2 in the nervous system (Chiu et al., 2008; Chiu and Cline, 2010). Igf2 was proposed to regulate development and turnover of neuromuscular synapses (Ishii, 1989) and was recently identified as critical component in the extinction of fear memories via promoting survival and maturation of newborn hippocampal neurons in an Igf1R-dependent manner (Agis-Balboa et al., 2011). Our results introduce Igf2 as potent modulator of synapse density and spine morphology in mature hippocampal neurons. Interestingly, this function is independent of receptor tyrosine kinases such as the insulin and/or the Igf1R, but is transduced via the Igf2R/mannose-6-phosphate receptor. The latter is specifically activated by Igf2 and is thought to associate with Gαq and phospholipase-Cβ (PLCβ) (Maeng et al., 2009). In line with the proposed signaling cascade downstream of PLCβ leading to the activation of the Ras–MEK/ERK pathway, we detected an abrogation of the Igf2-induced effects in hippocampal cultures by MEK inhibition. As a consequence, less Erk1/2 activity was observed in synaptic fractions from IKK2nDN mice. It is well known that Ras–MEK/ERK signaling drives synaptic distribution of AMPARs during learning and memory processes (Zhu et al., 2002; Harvey et al., 2008; Patterson et al., 2010). We propose that this scenario is also initiated by the IKK/NF-κB–Igf2–Igf2R–MEK/ERK signaling axis during synapse formation/maturation/maintenance.
Recent data proposed Igf2 as novel target for cognitive enhancement therapies and suggested that increased Igf2 levels in the hippocampus account for an Igf2R-dependent insertion of GluA1 type AMPARs into the synaptic membrane, thereby augmenting synaptic plasticity (D. Y. Chen et al., 2011). Notably, we show that exogenous Igf2 application to wild-type neurons also increases the number of mature spines and synapses. Therefore, we extend the hypothesis of Igf2-mediated cognitive enhancement to the morphological correlate of enhanced synapse formation and spine maturation, which is inevitably coupled to enhanced GluA1-containing AMPAR accumulation at the PSD (Hall and Ghosh, 2008).
In addition to the GluA1-dependent changes in synaptic strength, we found that the GluA1-independent spatial reference learning (Reisel et al., 2002) is impaired in IKK2nDN mice during the acquisition phase of the Morris water maze task, thus giving further support for the role of NF-κB-regulated Igf2 signaling in learning. Other mouse models using very similar genetic tools (e.g., expression of the constitutive active form of CaMKIIα in excitatory forebrain neurons) exhibit a similar impairment during spatial reference memory acquisition (Mayford et al., 1996). Whether this impairment in spatial reference learning is mediated by hippocampal and/or cortical activation of NF-κB signaling in neurons, remains to be resolved and needs very brain region-specific inactivation of NF-κB signaling. The presence of the field LTP at the CA3-to-CA1 synapses in IKK2nDN mice cannot be used as indication for the function of the hippocampus in spatial reference memory since the direct correlation between the presence of CA3-to-CA1 LTP and spatial reference memory formation was disproven in several studies (Zamanillo et al., 1999; Neves et al., 2008). The IKK2nDN mice learned the position of the platform after 7 d of training providing evidence that spatial reference memory formation itself is NF-κB independent or can be compensated by the remaining NF-κB activity or other transcription factors. The delay during memory acquisition in the water maze task can be explained by memory interference induced by the pretraining in a hippocampus-independent task in the flagged pool and might not necessarily indicate a slower formation of a spatial map in the hippocampus as suggested in early experimental studies (O'Keefe and Nadel, 1978). Different mouse models with impaired NF-κB signaling in the CNS revealed more prominent deficits in spatial memory formation in the radial eight-armed maze (Meffert et al., 2003) and Morris water maze (Kaltschmidt et al., 2006) compared with IKK2nDN mice. However, these deficits were found again during the memory acquisition periods and, consistent with our results, could be overcome by prolonged training.
We detected Igf2 within mature hippocampal neurons and further located Igf2R within dendrites and at synaptic sites. These observations implicate an autocrine or local paracrine synaptotrophic mechanism regulating synapse formation/maturation via Igf2 secretion and Igf2R activation as it is known for the neurotrophin BDNF and TrkB receptors (Huang and Reichardt, 2003; Yoshii and Constantine-Paton, 2010). Remarkably, a study by Cao et al. (2011) identified activity-dependent, Synaptotagmin-10-controlled exocytosis of Igf1 secretion from somatodendritic vesicles in mitral neurons of the olfactory bulb. Whether a similar kind of mechanism underlies Igf2 secretion in hippocampal neurons and whether this process is directly influenced by IKK activity, still remain open.
The imprinted genes Igf2 and Igf2R were proposed to undergo epigenetic regulation during memory enhancement and consolidation (D. Y. Chen et al., 2011). Notably, a report by Lubin and Sweatt revealed that nuclear IKK1 activity is important for chromatin remodeling and accounts for memory reconsolidation in a fear conditioning paradigm (Lubin and Sweatt, 2007). As our results provide evidence that IKK1 is enriched at synaptic sites, this raises the question whether IKK1 might take part in activity-dependent, learning-related synapse-to-nucleus shuttling to achieve chromatin remodeling necessary for IKK/NF-κB-dependent Igf2 transcription.
Mental retardation disorders like Rett syndrome (Tropea et al., 2009; Colak et al., 2011), incontinentia pigmenti (Gautheron et al., 2010), and mutations in the TRAPPC9 gene (Philippe et al., 2009) are associated with an altered, but distinct IKK/NF-κB signaling signature. To understand how the newly identified IKK/NF-κB–Igf2–Igf2R axis is mechanistically involved in these diverse cases of mental retardation opens a promising future field of research especially as the need for cutting-edge therapeutic approaches will constantly rise within the field of neuropsychiatric diseases.
Footnotes
This study was supported by Deutsche Forschungsgemeinschaft Grants KFO167-P5 (B.B.), KFO142-P5 (T.W.), SFB497-B9 (F.O.), SFB497-B8 and BO 1718; 3-1 (T.M.B.), and The Letten Foundation (V.J., G.F.V., Ø.C.H.). M.J.S., A. Maqbool, and A. Magnutzki are members of the International Graduate School in Molecular Medicine at Ulm University. We thank A. Grabrucker, M. Schoen, and T. Wuttke for critical comments on this manuscript, and F. Greten and G. Schütz for providing IKKβfl/fl and CaMKIIα iCre BAC mice. We acknowledge the technical assistance of Ute Leschik, Rosi Rittelmann, and Renate Zienecker.
The authors declare no competing financial interests.
References
- Agis-Balboa RC, Arcos-Diaz D, Wittnam J, Govindarajan N, Blom K, Burkhardt S, Haladyniak U, Agbemenyah HY, Zovoilis A, Salinas-Riester G, Opitz L, Sananbenesi F, Fischer A. A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J. 2011;30:4071–4083. doi: 10.1038/emboj.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD. c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem. 2008;15:539–549. doi: 10.1101/lm.866408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoitei N, Wirth T, Baumann B. Activation of the IkappaB kinase complex is sufficient for neuronal differentiation of PC12 cells. J Neurochem. 2005;93:1487–1501. doi: 10.1111/j.1471-4159.2005.03148.x. [DOI] [PubMed] [Google Scholar]
- Baumann B, Wagner M, Aleksic T, von Wichert G, Weber CK, Adler G, Wirth T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117:1502–1513. doi: 10.1172/JCI30876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis. J Neurosci. 2011;31:5414–5425. doi: 10.1523/JNEUROSCI.2456-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca2+-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schütz G. A CaMKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis. 2001;31:37–42. doi: 10.1002/gene.1078. [DOI] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci. 2011;31:6329–6338. doi: 10.1523/JNEUROSCI.5968-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 2010;5:7. doi: 10.1186/1749-8104-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Chen CM, Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008;58:708–719. doi: 10.1016/j.neuron.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Al-Dhalaan H, Nester M, Albakheet A, Al-Younes B, Al-Hassnan Z, Al-Dosari M, Chedrawi A, Al-Owain M, Abudheim N, Al-Alwan L, Al-Odaib A, Ozand P, Inan MS, Kaya N. Genomic and transcriptomic analyses distinguish classic Rett and Rett-like syndrome and reveals shared altered pathways. Genomics. 2011;97:19–28. doi: 10.1016/j.ygeno.2010.09.004. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israël A, Mémet S. Forebrain-specific neuronal inhibition of nuclear factor-κB activity leads to loss of neuroprotection. J Neurosci. 2003;23:9403–9408. doi: 10.1523/JNEUROSCI.23-28-09403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J, Pescatore A, Fusco F, Esposito E, Yamaoka S, Agou F, Ursini MV, Courtois G. Identification of a new NEMO/TRAF6 interface affected in incontinentia pigmenti pathology. Hum Mol Genet. 2010;19:3138–3149. doi: 10.1093/hmg/ddq222. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Grabrucker A, Vaida B, Bockmann J, Boeckers TM. Synaptogenesis of hippocampal neurons in primary cell culture. Cell Tissue Res. 2009;338:333–341. doi: 10.1007/s00441-009-0881-z. [DOI] [PubMed] [Google Scholar]
- Grabrucker AM, Knight MJ, Proepper C, Bockmann J, Joubert M, Rowan M, Nienhaus GU, Garner CC, Bowie JU, Kreutz MR, Gundelfinger ED, Boeckers TM. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011;30:569–581. doi: 10.1038/emboj.2010.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BJ, Ghosh A. Regulation of AMPA receptor recruitment at developing synapses. Trends Neurosci. 2008;31:82–89. doi: 10.1016/j.tins.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Köhrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- Hetts SW, Rosen KM, Dikkes P, Villa-Komaroff L, Mozell RL. Expression and imprinting of the insulin-like growth factor II gene in neonatal mouse cerebellum. J Neurosci Res. 1997;50:958–966. doi: 10.1002/(SICI)1097-4547(19971215)50:6<958::AID-JNR6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Howard MA, Elias GM, Elias LA, Swat W, Nicoll RA. The role of SAP97 in synaptic glutamate receptor dynamics. Proc Natl Acad Sci U S A. 2010;107:3805–3810. doi: 10.1073/pnas.0914422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huber MA, Maier HJ, Alacakaptan M, Wiedemann E, Braunger J, Boehmelt G, Madwed JB, Young ER, Marshall DR, Pehamberger H, Wirth T, Kraut N, Beug H. BI 5700, a selective chemical inhibitor of IκB kinase 2, specifically suppresses epithelial-mesenchymal transition and metastasis in mouse models of tumor progression. Genes Cancer. 2010;1:101–114. doi: 10.1177/1947601910361749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii DN. Relationship of insulin-like growth factor II gene expression in muscle to synaptogenesis. Proc Natl Acad Sci U S A. 1989;86:2898–2902. doi: 10.1073/pnas.86.8.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol. 2009;1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S. NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol. 2006;26:2936–2946. doi: 10.1128/MCB.26.8.2936-2946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi BZ, Barshack I, Kopolovic J, Galun E, Bujard H, Ben-Neriah Y. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med. 2000;6:573–577. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack V, Burnashev N, Kaiser KM, Rozov A, Jensen V, Hvalby O, Seeburg PH, Sakmann B, Sprengel R. Conditional restoration of hippocampal synaptic potentiation in Glur-A-deficient mice. Science. 2001;292:2501–2504. doi: 10.1126/science.1059365. [DOI] [PubMed] [Google Scholar]
- Maeng YS, Choi HJ, Kwon JY, Park YW, Choi KS, Min JK, Kim YH, Suh PG, Kang KS, Won MH, Kim YM, Kwon YG. Endothelial progenitor cell homing: prominent role of the IGF2-IGF2R-PLCbeta2 axis. Blood. 2009;113:233–243. doi: 10.1182/blood-2008-06-162891. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappaB functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Nadel L. The hippocampus as a cognitive map. Oxford: Oxford UP; 1978. [Google Scholar]
- O'Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM, Kwon H, LeFevour A, Chakraborty-Sett S, Greene WC. NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol. 2006;26:7283–7298. doi: 10.1128/MCB.00510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, Desai P, Fitch O, Malone L, Ahn HJ, Liou HC, Sweatt JD, Levenson JM. Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci. 2006;26:4870–4879. doi: 10.1523/JNEUROSCI.4527-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- Patterson MA, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe O, Rio M, Carioux A, Plaza JM, Guigue P, Molinari F, Boddaert N, Bole-Feysot C, Nitschke P, Smahi A, Munnich A, Colleaux L. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am J Hum Genet. 2009;85:903–908. doi: 10.1016/j.ajhg.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci. 2005;25:3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, et al. Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29:3529–3537. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Jones PA, Chaillet JR, Ferguson-Smith AC, Barton SC, Reik W, Surani MA. Parental imprinting: potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (Igf2) gene. Genes Dev. 1992;6:1843–1856. doi: 10.1101/gad.6.10.1843. [DOI] [PubMed] [Google Scholar]
- Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Scheiffele P. Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci. 2003;26:485–508. doi: 10.1146/annurev.neuro.26.043002.094940. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ, Grabrucker AM, Bockmann J, Boeckers TM. Synaptic cross-talk between N-methyl-d-aspartate receptors and LAPSER1-beta-catenin at excitatory synapses. J Biol Chem. 2009;284:29146–29157. doi: 10.1074/jbc.M109.020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolnick JA, Cui K, Duggan CD, Xuan S, Yuan XB, Efstratiadis A, Ngai J. Role of IGF signaling in olfactory sensory map formation and axon guidance. Neuron. 2008;57:847–857. doi: 10.1016/j.neuron.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim DJ, Yang L, Reed JG, Noebels JL, Chiao PJ, Zheng H. Disruption of the NF-kappaB/IkappaBalpha autoinhibitory loop improves cognitive performance and promotes hyperexcitability of hippocampal neurons. Mol Neurodegener. 2011;6:42. doi: 10.1186/1750-1326-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker SA, Alvarado C, von Wichert G, Knippschild U, Wiedenmann J, Clauss K, Nienhaus GU, Hameister H, Baumann B, Borggrefe T, Knöchel W, Oswald F. RITA, a novel modulator of Notch signalling, acts via nuclear export of RBP-J. EMBO J. 2011;30:43–56. doi: 10.1038/emboj.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Williams ME, de Wit J, Ghosh A. Molecular mechanisms of synaptic specificity in developing neural circuits. Neuron. 2010;68:9–18. doi: 10.1016/j.neuron.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:304–322. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]