

Abstract
Regulation of NMDA receptor trafficking is crucial to modulate neuronal communication. Ca2+/calmodulin-dependent protein kinase phosphorylates the tail domain of KIF17, a member of the kinesin superfamily, to control NMDA receptor subunit 2B (GluN2B) transport by changing the KIF17–cargo interaction in vitro. However, the mechanisms of regulation of GluN2B transport in vivo and its physiological significance are unknown. We generated transgenic mice carrying wild-type KIF17 (TgS), or KIF17 with S1029A (TgA) or S1029D (TgD) phosphomimic mutations in kif17−/− background. TgA/kif17−/− and TgD/kif17−/− mice exhibited reductions in synaptic NMDA receptors because of their inability to load/unload GluN2B onto/from KIF17, leading to impaired neuronal plasticity, CREB activation, and spatial memory. Expression of GFP-KIF17 in TgS/kif17−/− mouse neurons rescued the synaptic and behavioral defects of kif17−/− mice. These results suggest that phosphorylation-based regulation of NMDA receptor transport is critical for learning and memory in vivo.
Introduction
Trafficking of cargo to synapses is essential for establishment and maintenance of synaptic specialization (Bredt and Nicoll, 2003; Wenthold et al., 2003; Hirokawa and Takemura, 2005; Hirokawa et al., 2010). In neurons, molecular motor proteins, such as myosin, dynein and the kinesin superfamily proteins (KIFs), are required to transport synaptic cargos to their site of action (Hirokawa, 1998; Vale, 2003). Trafficking of proteins is regulated at multiple stages of transport; motor–cargo binding and motor activation can be regulated during the early stage of transport, microtubule-based translocation during the middle stage, and cargo release and retention at the final destination during the late stage. The molecular processes involved in this regulation are only just beginning to be understood (Arimura and Kaibuchi, 2007; Guillaud et al., 2008; Wang et al., 2008; Arimura et al., 2009; Wang and Schwarz, 2009).
We previously showed that KIF17 transports NMDA receptor subunit 2B (GluN2B)-containing vesicles in dendrites (Setou et al., 2000; Guillaud et al., 2003), and KIF17-mediated GluN2B transport has been implicated in learning and memory (Wong et al., 2002; Yin et al., 2011). KIF17 interacts with the PDZ domain of Mint1 through its tail domain, and this complex in turn binds the NMDA receptor GluN2B subunit through the intermediate adaptor proteins CASK and Velis (Setou et al., 2000). We have also demonstrated that CaMKII phosphorylates Ser1029 of the C-terminal tail domain (CTD) of KIF17, and that this phosphorylation attenuates the interaction between Mint1 and KIF17, possibly as a result of the release of GluN2B-containing vesicles from the molecular motor (Guillaud et al., 2008). Recent findings demonstrate that the number and subunit composition of synaptic NMDA receptors changes dynamically in a cell-specific and synapse-specific manner (Roche et al., 2001; Tovar and Westbrook, 2002; Washbourne et al., 2002; Kennedy and Ehlers, 2006; Lau and Zukin, 2007). The trafficking of NMDA receptor subunits directly affects their recruitment to, and retention within, synaptic compartments and, thus, the magnitude of the synaptic potential.
Many aspects of the regulation of NMDA receptor transport in neurons remain to be elucidated, including the functional implications of such regulation on synaptic plasticity, as well as any implications the dysregulation of NMDA receptor transport may have in mammalian brain disease. Here, we focused on the significance of the KIF17–cargo interaction in vivo by establishing and analyzing various transgenic mouse lines. We demonstrate that alteration of the KIF17–cargo interaction results in changes in neuronal plasticity and spatial memory formation.
Materials and Methods
Site-directed mutagenesis.
S1029A and S1029D point mutations were introduced into the KIF17 CTD using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). PCR was performed under the following conditions: denaturation at 95°C for 1 min, followed by 16 cycles at 95°C for 30 s, 58°C for 1 min and 68°C for 12 min, and then a final extension step at 68°C for 7 min. The primers used for mutagenesis were as follows: for the S1029A mutation, (forward) 5′-GCC AAG CGT AAA AAA gcc AAA AAC AGC TTC GG-3′, (reverse) 5′-CGG TTC GCA TTT TTT cgg TTT TTG TCG AAG CC-3′; for the S1029D mutation, (forward) 5′-GCC AAG CGT AAA AAA gac AAA AAC AGC TTC GG-3′, (reverse) 5′-CGG TTC GCA TTT TTT ctg TTT TTG TCG AAG CC-3′. Reaction mixtures were cooled on ice and digested further with 1 μl of DpnI (10 U/μl) for 1 h at 37°C to cleave parental DNA. The PCR product was then precipitated with ethanol, and the entire pellet was resuspended and transformed into 100 μl of competent Escherichia coli cells. Plasmids were isolated using a mini-prep, and positive mutants were selected by sequencing.
Generation of transgenic mice.
Genes encoding GFP and KIF17 with or without mutations (GFP-KIF17, GFP-KIF17 S1029A, GFP-KIF17 S1029D, 3.9 kb) were fused and subcloned into the NotI site of the pNN279 vector, downstream of the CaMKIIα promoter (8.5 kb) (Mayford et al., 1996; Wong et al., 2002). For microinjection, the vector was linearized by digestion with SalI and purified using a QIAEX gel extraction kit (Qiagen).
Transgenic founder mice were produced by microinjection of the linearized DNA fragment into the pronuclei of BDF1-derived zygotes using standard techniques (Krestel et al., 2001). The founder mice were backcrossed to wild-type C57B/6J mice to generate the transgenic mouse lines. Backcrossing was repeated >10 times to generate an almost pure C57B/6J background.
To generate Tg+/kif17−/− mice, mice expressing GFP-KIF17 (with or without mutation) fusion proteins were bred with kif17−/− mice (Yin et al., 2011) to generate mice carrying the gfp-kif17 gene on a kif17−/− background. The number of founders we made was 5, 6, and 6 for TgS, TgA, and TgD transgenic mice. Then, three lines for each genotype were selected to intercross with kif17−/− mice to generate TgS/kif17−/−, TgA/kif17−/−, and TgD/kif17−/− mice, respectively. The genotypes of these mice were determined by PCR using the following primers: for the gfp transgene, 5′-GCG GAG AGG GTG AAG GTG ATG C-3′ and 5′-CAG GGC CGT CGC CGA TTG G-3′; for the endogenous kif17 gene, 5′-CGG GAC AAA GGA GGC ACA TTC-3′ and 5′-TGG GAT TAT CAA AGG CAG GAC C-3′; for the neo transgene, 5′-TGG GCA CAA CAG ACA ATC GG-3′ and 5′-ACT TCG CCC AAT AGC AGC CAG-3′.
Antibodies.
KIF17 polyclonal antibody (pAb) (1:300, Yin et al., 2011), KIF5B pAb (1:500, Kanai et al., 2000), GluN2A and GluN2B pAbs (1:200, Invitrogen), GluR1 pAb (1:300, Millipore), Mint1 monoclonal antibody (mAb) (1:100, Transduction Laboratories), PSD-95 mAb (1:500, BD Transduction Laboratories), tubulin DM1A mAb (1:1000, Sigma), synaptophysin mAb (1:300, Sigma), GFP pAb (1:200, Invitrogen), MAP2 mAb (1:200, Sigma), cAMP-response element-binding protein (CREB) pAb (1:500, New England Biolabs), and phospho-CREB (pCREB) pAb (1:1000, New England Biolabs). The Alexa Fluor 488 and Alexa Fluor 568 fluorescent secondary antibodies (1:300) were obtained from Invitrogen, and horseradish peroxidase-conjugated secondary antibodies (1:10,000) were obtained from GE Healthcare .
Immunoprecipitation and immunoblotting.
Mouse hippocampi were dissected on ice and placed in ice-cold RIPA buffer (50 mm Tris, pH 8.0, 1% Triton X-100, 0.1% SDS, 150 mm NaCl) containing Protease Inhibitor Cocktail tablets (Roche Applied Science). The tissue was homogenized and centrifuged at 13,000 × g for 20 min at 4°C, and protein concentration was determined by BCA protein assay (Pierce). Hippocampal homogenate (500 μg) was incubated with 1 μg of anti-KIF17/anti-GFP antibodies at 4°C for 2 h with agitation. Protein-G-coupled Sepharose beads (Pharmacia) were added and the mixture was incubated at 4°C for a further 2 h. The beads were spun down at 13,000 × g for 20 s, washed three times in RIPA buffer, resuspended in 2× sample loading buffer, and boiled for 5 min. Proteins were separated by SDS-PAGE, and probed with specific primary antibodies and corresponding horseradish peroxidase-conjugated secondary antibodies. Western blot signals were detected using an enhanced chemiluminescence procedure (GE Healthcare). Densitometry analysis was performed using ImageJ software (NIH).
Morris water maze task.
Male mice (10–12 weeks old, n = 8 for each genotype) were trained in a 120 cm diameter open field water maze containing opaque water at 25 ± 1°C. The escape platform (10 cm in diameter) was placed 1 cm under the surface of the water. An overhead camera- and computer-assisted tracking system recorded the navigation of mice in the maze. After 4 d of non-spatial pretraining, animals were subjected to 6 consecutive days of spatial training, with four trials per day, followed by a probe trial 24 h later (Bannerman et al., 1995; Sakimura et al., 1995; Yin et al., 2011). During the non-spatial pretraining task, the pool was surrounded with white curtains to deprive mice of visual cues beyond the maze. A fixed platform was hidden in a different location during each trial and mice were trained to find the platform using distal cues.
Electrophysiology.
Transverse slices (400 μm) of acutely dissected hippocampi were prepared from mice of postnatal day 25–35. Slices were incubated in an interface chamber at 34°C, superfused with oxygenated artificial CSF (ACSF, containing, in mm: 119 NaCl, 2.6 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, and 11 d-glucose), and allowed to equilibrate for at least 60 min.
To record field EPSPs (fEPSPs), both the stimulating and recording electrodes were placed in the stratum radiatum of the CA1 area. Stimulus intensity (test pulse duration of 0.1 ms) was set to evoke 35–40% maximal fEPSP at a frequency of 0.1 Hz. After recording a stable 20 min baseline of fEPSPs for 20 min, long-term potentiation (LTP) was induced by tetanic stimulation (100 Hz, 1 s).
EPSCs in CA1 pyramidal neurons were recorded using the whole-cell patch-clamp technique. Recording electrodes (3–5 MΩ) were filled with intracellular solution containing (in mm): 122.5 Cs gluconate, 17.5 CsCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 Mg-ATP, 0.3 Na3-GTP, pH 7.2 (290–300 mOsm). Stimulus intensity was set to evoke half-maximal AMPA responses (at −90 mV). To isolate NMDA-mediated EPSCs, neurons were voltage-clamped at +40 mV in the presence of 10 μm NBQX (Tocris Bioscience), an AMPA receptor antagonist. Both NBQX and d-2-amino-5-phosphonovaleric acid (d-APV, Tocris Bioscience) were diluted in ACSF and applied to the bath. All membrane potential values were corrected for liquid junction potentials of −11 mV. Access resistance was monitored throughout experiments, and ranged from 10 to 20 MΩ. Unstable recordings (access resistance changed by >10%) during experiments were excluded from further analysis. Records were filtered at 2 kHz, digitized at 10 kHz, and analyzed using Clampfit 9.2 software (Molecular Devices).
Neuronal cultures, transfection, and live imaging.
Primary cultures of hippocampal neurons were prepared as described previously (Goslin et al., 1991). Briefly, hippocampi were isolated from embryonic day 16 mice and dissociated with 0.25% trypsin at 37°C for 15 min. Hippocampal neurons were then plated on glass coverslips or plastic Petri dishes coated with polyethyleneimine (Sigma) and poly-l-lysine (Sigma).
For live-cell imaging, hippocampal neurons were used at 7–10 d in vitro (DIV). GluN1–1a (untagged)/GluN2B-RFP or GluN1–1a (untagged)/GluN2A-RFP vectors were introduced into cultured neurons using a modified Ca2+-phosphate transfection method (Jiang and Chen, 2006). Forty-eight to 72 h after transfection, living neurons were observed under an LSM 5 Duo confocal laser-scanning microscope (Zeiss). Cells were kept in minimal essential medium (Invitrogen) warmed to 37°C in the presence of 1 mm kynurenate, 1 μm strychnine, and 1 μm tetrodotoxin. Movement of GluN2B and GluN2A clusters along dendrites was monitored over time, and images were acquired every 5 s. Images were processed using Photoshop 7.0 (Adobe) and further edited as a video file using After Effects (Adobe). The analysis and graphical representation were performed using ImageJ and GraphPad Prism.
To visualize the dynamic post-Golgi release of GluN2B or GluN2A vesicles, GluN1–1a (untagged)/GluN2B-RFP or GluN1–1a (untagged)/GluN2A-RFP were initially expressed in hippocampal cells in the presence of brefeldin A (BFA; 10 μg/ml, Wako Pure Chemical Industries Ltd.) and were observed using an LSM 5 Duo confocal laser-scanning microscope (Zeiss) after extensive washing to remove BFA. Quantification was performed at 1 h after BFA washout. Three regions of interest were drawn at peripheral locations, and the corresponding mean fluorescence values were averaged. This value represents an estimation of GluN2B/2A-RFP at non-Golgi locations. Golgi-associated fluorescence was determined from perinuclear regions. To ascertain the percentage of post-Golgi release of GluN2B/2A-RFP, we divided fluorescence intensity outside the Golgi area by that inside the total cell area. Background was subtracted using the fluorescence intensity outside the Golgi area.
Immunocytochemistry.
After 16–21 DIV, cultured hippocampal cells were fixed in methanol for 10 min at −20°C and incubated with primary antibodies in 3% bovine serum albumin overnight at 4°C, followed by incubation with Alexa Fluor 568 and Alexa Fluor 647 secondary antibodies for 1 h at room temperature. Images were acquired using an LSM510 confocal laser-scanning microscope (Zeiss) and were then analyzed using Adobe Photoshop and ImageJ software.
Surface biotinylation assay.
Biotinylation assay was performed using a Pierce cell surface protein isolation kit (Pierce Biotechnology), as described in the manufacturer's protocol. Briefly, at 16–21 DIV, hippocampal neuron cultures were washed twice with ice-cold PBS and labeled with 0.25 mg/ml sulfo-NHS-SS-biotin in PBS for 30 min at 4°C with gentle agitation. Cells were subsequently lysed with lysis buffer and labeled proteins were isolated with NeutrAvidin Agarose. Bound proteins were released in SDS-PAGE sample buffer containing 50 mm DTT and immunoblotted with antibodies against GluN2B, GluN2A, and GluR1.
Semiquantitative reverse transcriptase PCR.
Total RNA was extracted from hippocampi of four mice of each genotype using an Agilent Total RNA Isolation Mini Kit (Agilent Technologies), and semiquantitative reverse transcriptase PCR (RT-PCR) was performed as described previously (Yin et al., 2011). Briefly, mRNA (1 μg) was used to synthesize first-strand complementary DNA with ReverTra Ace reverse transcriptase (Toyobo). Diluted cDNA series were subjected to amplification with AmpliTaq DNA Polymerase (Applied Biosystems) in a programmable thermal cycler with denaturation performed at 94°C for 30 s, annealing at 60–65°C for 30 s, and extension at 72°C for 30 s for 30 cycles. The primers were as follows: for kif17, 5′-GTT CTT AGT GCG GGC TTC CTA TC-3′ and 5′-GGA GTC CTT ATT CAT CAG GGT GTA GC-3′; for GRIN2B, 5′-GCG ATT TGG TTA CTC TGG GGT C-3′ and 5′-GTC TCT GGA ACT TCT TGT CAC TCA GG-3′; for GRIN2A, 5′-CGG GTC TCA TTT CAG TCT CTT ACG-3′ and 5′-GGT TGT CAT CTG GCT CAC AGT CAG-3′; for GRIN1, 5′-GGA TAC CAG ATG TCC ACC AGA CTA AAG-3′ and 5′-AAC GCA GAA GCC ATA ACA GCA C-3′; and for tubulin, 5′-GGC CAG ATG CCA AGT-3′ and 5′-AGC TGT GGA AAA CCA AGA-3′.
Turnover assay.
Cultured hippocampal neurons (7–10 DIV) were treated with 20 μg/ml cycloheximide (CHX; Sigma) for 0, 10, 20, and 30 h. Cells were harvested and probed with anti-GluN2A, -GluN2B, and -tubulin antibodies. Values are given as average signal intensities (mean ± SEM) for GluN2A or GluN2B compared with that for tubulin, and normalized to 100% at time 0.
Degradation analysis.
Cultured hippocampal neurons (7–10 DIV) were incubated with CHX (20 μg/ml) for 24 h, in the presence or absence of lysosomal inhibitor (100 μg/ml leupeptin, 200 μm chloroquine, Sigma) or proteasomal inhibitor (10 μm lactacystin, 10 μm MG132, Calbiochem). After treatment, cells were lysed and subjected to Western blot analysis.
CREB activation.
Adult mice (10–12 weeks old, n = 4 mice/genotype) were trained in the water maze task with four trials/d for 10 d, including 4 d of non-spatial pretraining and 6 d of hidden platform training. After a probe trial on day 11, the mice were killed immediately. Both hippocampi were dissected and placed in ice-cold RIPA buffer containing phosphatase and protease inhibitors (50 mm Tris, pH 7.5, 1% NP-40, 0.5% SDS, 150 mm NaCl, 1 mm DTT, 1 mm Na3VO4, 1 mm NaF, and Complete protease inhibitor). The extracts were separated by SDS-PAGE and probed with anti-pCREB, anti-CREB, anti-GluN2A, anti-GluN2B, anti-KIF17 and anti-KIF5B antibodies.
Statistical analysis.
Data were analyzed as indicated, and the statistical significance was assessed using one-way ANOVA, two-tailed unpaired Student's t test, or the χ2 test.
Results
In vivo association of KIF17 with Mint1 and GluN2B
In vitro, KIF17 transports GluN2B-containing vesicles by binding to Mint1, and GluN2B release is triggered by CaMKII-dependent phosphorylation of the Ser1029 residue of KIF17 (Setou et al., 2000; Guillaud et al., 2008). To study the role of KIF17 phosphorylation in vivo, two mutations (S1029A and S1029D) were introduced at Ser1029 of the KIF17 CTD by PCR mutagenesis, to mimic various phosphorylation states (Fig. 1A; Guillaud et al., 2008). S1029A mimics an unphosphorylated form of KIF17, which is able to bind to GluN2B-containing vesicles but prevents GluN2B release. S1029D mimics a permanently phosphorylated form of KIF17, which prevents KIF17 from binding and transporting GluN2B-containing vesicles. Using these mutant and wild-type KIF17 cDNAs, we generated three transgenic mouse lines: TgS (carrying normal KIF17 cDNA), TgA (carrying KIF17 with the S1029A mutation) and TgD (carrying KIF17 with the S1029D mutation). All mouse lines expressed GFP-fused KIF17 proteins in the postnatal forebrain under the control of the CaMKIIα promoter (Fig. 1B–D). Transgenic mice of each genotype were then intercrossed with kif17−/− mice to generate three new transgenic mouse lines with a disrupted endogenous kif17 gene: TgS/kif17−/−, TgA/kif17−/− and TgD/kif17−/− (Fig. 1E). PCR was used to detect gfp and endogenous kif17 genes in genomic DNA extracted from mouse tails (Fig. 1F).
Figure 1.
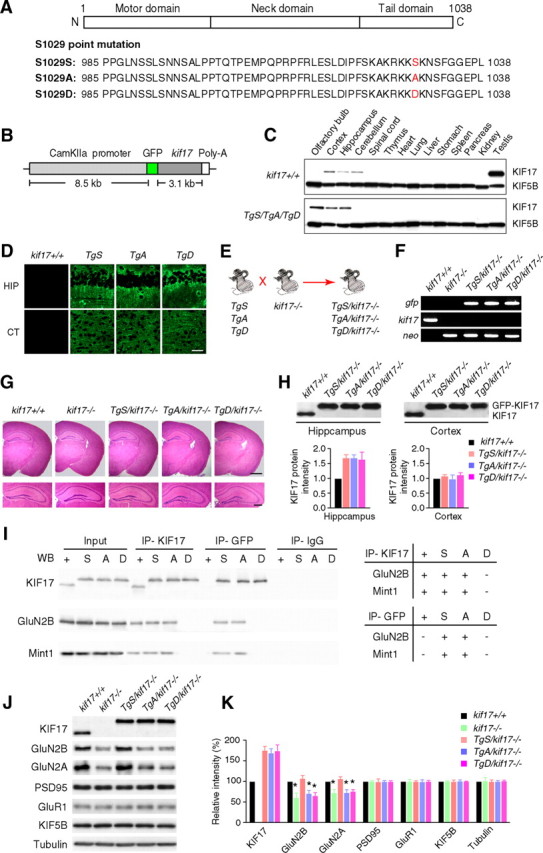
Generation and biochemical characterization of KIF17 transgenic mouse lines. A, Point mutation of the C-terminal domain KIF17 at Ser1029. The C-terminal domain of KIF17 is critical for cargo binding. Ser1029 is phosphorylated by CaMKII to regulate the KIF17–Mint1 interaction. S1029S: wild-type KIF17. S1029A: substitution of Ser1029 by Ala, which mimics the unphosphorylated state. S1029D: substitution of Ser1029 by Asp, which mimics the phosphorylated state. B, The construct for generation of KIF17 transgenic mice. KIF17 expression was restricted to the postnatal forebrain because of the use of CaMKIIα promoter. C, Expression of KIF17 in various tissues. KIF5B was used as a control. In transgenic mice, GFP-KIF17 was restricted to the postnatal forebrain (olfactory bulb, hippocampus, and cortex) by using the CaMKIIα promoter. D, Immunohistochemistry to examine KIF17 localization in forebrain regions: hippocampal CA1 (HIP) and cortex (CT) subregions. Scale bar, 100 μm. E, Schematic illustration of the procedure used to generate Tg+/kif17−/− mice. KIF17 transgenic mice (TgS, TgA, and TgD) were bred with kif17−/− mice to generate three new transgenic mouse lines with disrupted endogenous kif17 gene (TgS/kif17−/−, TgA/kif17−/−, and TgD/kif17−/−). F, PCR screening using primers for gfp, neo, and endogenous kif17. G, Paraffin sections of the mouse brain stained with hematoxylin and eosin. Scale bars, 500 μm (top right) and 200 μm (bottom right). H, Western blot analysis and quantification of GFP-KIF17 in the hippocampi and cortices of Tg+/kif17−/− mice. The lower bands indicate endogenous KIF17, and the upper bands indicate GFP-KIF17 fusion proteins. I, Immunoprecipitation of mouse hippocampal extracts using anti-KIF17 and anti-GFP antibodies; anti-IgG antibody was used as a negative control. J, K, The levels of KIF17, GluN2B and other associated proteins in the hippocampus of transgenic mice. Data from three independent experiments are expressed as mean ± SEM (*p < 0.01; one-way ANOVA and post hoc comparison test).
All Tg+/kif17−/− transgenic mice showed apparently normal growth. Histological examinations of their brains revealed no obvious morphological abnormalities (Fig. 1G). In the hippocampi of Tg+/kif17−/− mice, levels of GFP-KIF17 fusion proteins were ∼1.5- to 2-fold greater than levels of endogenous KIF17 in wild-type hippocampus (Fig. 1H). Immunoprecipitation studies showed that both GluN2B and Mint1 associated with GFP-KIF17 and GFP-KIF17 S1029A, but not with GFP-KIF17 S1029D, suggesting that Ser1029 is the critical site mediating the KIF17–Mint1/GluN2B association in vivo (Fig. 1I). Western blot analysis of hippocampal lysates from each mouse line revealed a reduction in the level of GluN2B and GluN2A in kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice, compared with wild-type controls (Fig. 1J,K). No significant difference was observed in the hippocampal levels of GluN2B and GluN2A in kif17+/+ and TgS/kif17−/− mice (Fig. 1J,K).
Spatial memory in Tg+/kif17−/− mice
We assessed the spatial learning abilities of transgenic, kif17−/− and kif17+/+ mice using the Morris water maze task (Bannerman et al., 1995; Sakimura et al., 1995; Yin et al., 2011). All groups of mice displayed similar escape latencies during non-spatial pretraining (Fig. 2A), and learned to navigate to the platform over the course of 6 spatial training days, as indicated by the progressive decrease in escape latencies (Fig. 2B). However, consistent with our previous report (Yin et al., 2011), kif17−/− mice were deficient in learning this task compared with control mice. They consistently exhibited longer mean daily escape latencies than kif17+/+ mice (Fig. 2B). In the probe trial, wild-type mice exhibited a clear spatial preference for the target quadrant in which the platform was previously located, whereas kif17−/− mice did not display a preference for a particular quadrant, as measured by either target quadrant searching time or platform crossings (Fig. 2C–E). In contrast to kif17−/− mice, TgS/kif17−/− mice showed no obvious defects during spatial training or probe testing (Fig. 2B–E), suggesting that expression of GFP-KIF17 rescued the defects in spatial memory observed in kif17−/− mice. TgA/kif17−/− and TgD/kif17−/− mice showed similar defects in spatial memory as observed in kif17−/− mice. They exhibited longer escape latencies than kif17+/+ mice and loss of spatial preference for the target quadrant (Fig. 2B–E). These results indicate that the KIF17–Mint1/GluN2B interaction is required for spatial memory formation.
Figure 2.

Performance in the Morris water maze task. A, Escape latencies during non-spatial pretraining (four trials per day). B, Escape latencies during spatial training. C, D, Probe test performed 24 h after the last training sessions. The escape platform was removed, and mice were allowed to swim for 60 s. Target quadrant searching time (C) and platform crossings (D, the number of times the mice cross the exact location of the platform) were calculated to evaluate the spatial learning ability of mice. TQ, Target quadrant; AL, adjacent left; OP, opposite; AR, adjacent right. The quadrant time (percentage) in TQ was as follows: kif17+/+, 44.8 ± 7.4%; kif17−/−, 25.4 ± 3.5%; TgS/kif17−/−, 40.6 ± 3.8%; TgA/kif17−/−, 30.4 ± 3.1%; TgD/kif17−/−, 27.9 ± 4.4%. The platform crossings were as follows: kif17+/+, 3.9 ± 0.3%; kif17−/−, 2.1 ± 0.3%; TgS/kif17−/−, 3.6 ± 0.6%; TgA/kif17−/−, 2.3 ± 0.4%; TgD/kif17−/−, 2.1 ± 0.3%. Values represent mean ± SEM (n = 8 mice/genotype, *p < 0.05; one-way ANOVA and post hoc test). E, Representative searching strategy during the probe test. Kif17+/+ and TgS/kif17−/− mice focused their search in the target quadrant, while kif17−/−, TgA/kif17−/−, and TgD/kif17−/− mice navigated over the entire area of the pool.
NMDA-mediated synaptic activity and plasticity in Tg+/kif17−/− mice
To gain insight into the function of NMDA receptors in Tg+/kif17−/− mice, we studied LTP evoked at CA3-to-CA1 synapses in acute hippocampal slices (Bliss and Collingridge, 1993; Shors and Matzel, 1997). Tetanic stimulation (100 Hz, 1 s) of Schaffer collateral/commissural fibers in stratum radiatum elicited almost identical, robust, and lasting potentiation in slices prepared from kif17+/+ and TgS/kif17−/− mice (LTP magnitude at 60 min, kif17+/+: 154.7 ± 11.4% versus TgS/kif17−/−: 159.4 ± 13.0%, n = 10 slices/10 mice/group) (Fig. 3A). In contrast, in slices from kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice, field EPSP (fEPSP) slopes revealed a continuous decline throughout the duration of recording, compared with slices from wild-type mice (LTP magnitude at 60 min, kif17+/+: 154.7 ± 11.4%, kif17−/−: 111.2 ± 14.0%, TgA/kif17−/−: 119.5 ± 9.0%, TgD/kif17−/−: 119.0 ± 10.0%, n = 10 slices/10 mice/group) (Fig. 3A,B).
Figure 3.

Synaptic NMDA receptor-mediated LTP and EPSCs. A, B, LTP induced by a single train of tetanus (100 Hz for 1 s, arrow) in hippocampal CA1 neurons of kif17+/+, kif17−/−, and Tg+/kif17−/− mice. Sample traces show typical fEPSPs recorded 5 min before and 60 min after LTP induction. Expression of LTP was attenuated in kif17−/−, TgA/kif17−/− and TgD/kif17−/− slices. C, D, AMPA- and NMDA-mediated EPSCs recorded in CA1 pyramidal neurons of kif17+/+, kif17−/−, and Tg+/kif17−/− mice. NMDA/AMPA ratios in neurons from kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice were decreased, consistent with a decrease in NMDA-mediated currents (mean ± SEM, *p < 0.01; one-way ANOVA and post hoc test). NMDA/AMPA ratios in neurons of kif17+/+ and TgS/kif17−/− mice were not different. E, Input–output curves plotting the fEPSP slopes against their corresponding presynaptic fiber volley amplitudes. Each symbol represents a set of experiments from a single slice. F, Paired-pulse facilitation of fEPSPs was measured using pairs of presynaptic fiber stimulation pulses separated by 20, 50, 100, and 200 ms. For each group, the mean ± SEM is indicated. G, Current–voltage relationship of NMDA receptor channel currents recorded in hippocampal slices. Current amplitudes were normalized to the values at +40 mV EPSC. Values are mean ± SEM.
NMDA-mediated EPSCs recorded in CA1 pyramidal cells showed decreased NMDA/AMPA ratios in neurons of kif17−/−, TgA/kif17−/− and TgD/kif17−/− compared with kif17+/+ mice (Fig. 3C,D). The NMDA receptor-mediated currents of TgS/kif17−/− mouse neurons were comparable to those in neurons of kif17+/+ mice (percentage current in kif17+/+: 71.5 ± 4.8%, kif17−/−: 40.1 ± 2.3%, TgS/kif17−/−: 69.3 ± 4.6%, TgA/kif17−/−: 42.3 ± 4.5%, TgD/kif17−/−: 41.7 ± 3.5%, n = 7 slices/5 mice/group) (Fig. 3C,D). The NMDA-mediated EPSCs were eliminated in slices after application of d-APV (50 μm), an antagonist for all subtypes of NMDA receptors.
We also examined basal synaptic function in the hippocampal CA1 region. The input–output curves and paired-pulse facilitation (Fig. 3E,F) were virtually identical in slices from any of the five genotypes, indicative of intact release probability and presynaptic function. Moreover, the current–voltage relationship for NMDA components was unaltered by the disruption of KIF17–cargo interaction (Fig. 3G).
Together, these results suggest that mutation of the KIF17 CTD leads to defective NMDA receptor-dependent synaptic activity and plasticity.
Dynamics and cellular localization of GluN2 subunits in Tg+/kif17−/− mouse neurons
To assess how KIF17 phosphorylation might mediate the effects at the molecular and cellular level shown above, we first characterized the dynamics of NMDA receptor trafficking in live hippocampal neurons. Cultured hippocampal cells expressing GluN2B-RFP or GluN2A-RFP with untagged GluN1 (splice variant GluN1–1a) were imaged using confocal laser scanning microscopy (Yin et al., 2011). GluN2B receptor clusters fell into four categories based on their motility: anterograde, retrograde, vibrating, and stationary. In neurons from kif17+/+, TgS/kif17−/− and TgA/kif17−/− mice, GluN2B clusters were highly dynamic, and almost one third of the GluN2B clusters moved in an anterograde direction at a comparable velocity to KIF17 (Guillaud et al., 2003; Yin et al., 2011) (percentage of anterograde movement, kif17+/+: 39 ± 8%, TgS/kif17−/−: 36 ± 6%, TgA/kif17−/−: 32 ± 5%; anterograde transport velocity, kif17+/+: 0.69 ± 0.18 μm/s, TgS/kif17−/−: 0.67 ± 0.17 μm/s, TgA/kif17−/−: 0.62 ± 0.21 μm/s, n = 48 neurons/three mice/genotype) (Fig. 4A–D; Movie 1). In contrast, in neurons from kif17−/− and TgD/kif17−/− mice, GluN2B clusters were much less mobile, traveling anterogradely at a much slower velocity compared with those in neurons from wild-type mice (percentage of anterograde movement, kif17−/−: 12 ± 5%, TgD/kif17−/−: 14 ± 4%; anterograde transport velocity, kif17−/−: 0.24 ± 0.1 μm/s, TgD/kif17−/−: 0.25 ± 0.12 μm/s, n = 48 neurons from three mice for each genotype) (Fig. 4A–D; Movie 1). However, time-lapse recordings showed similar dynamic properties for GluN2A clusters in neurons from the mice of all five genotypes (Fig. 4E–H; Movie 2).
Figure 4.
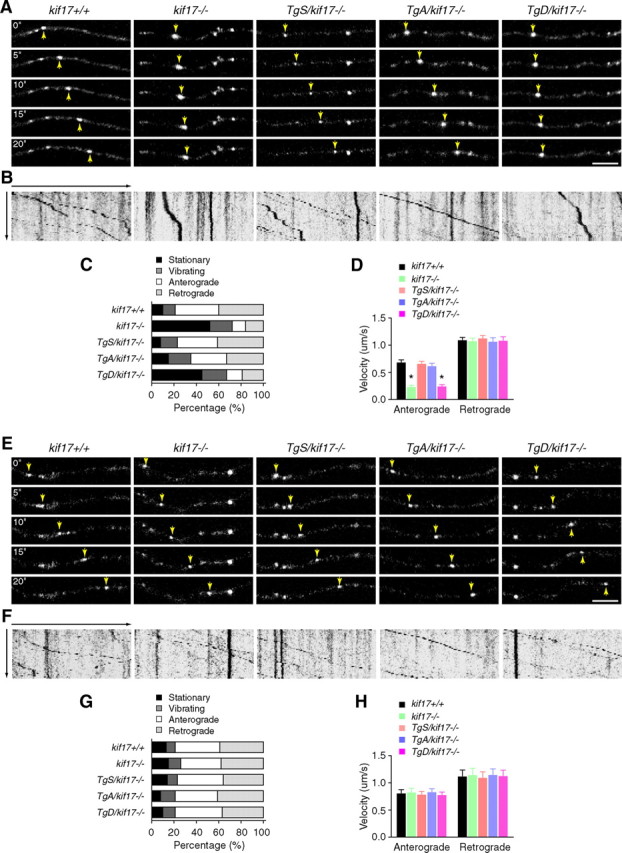
Dynamics of GluN2 subunits in live hippocampal neurons. A–D, Analysis of GluN2B motility. A, Time-lapse images of movement of GluN2B-RFP clusters in dendrites. Arrows point to anterogradely moving clusters. Scale bar, 5 μm. B, Kymograph showing the motility of GluN2B clusters. C, Classification of motility (p < 0.0001; χ2 test). D, Average velocities for anterograde and retrograde movement of GluN2B clusters. E–H, Analysis of GluN2A motility. E, Time-lapse images of movement of GluN2A-RFP clusters in dendrites. Arrows point to anterogradely moving clusters. Scale bar, 5 μm. F, Kymograph showing the motility of GluN2A clusters. G, Classification of motility (p = 0.90, χ2 test). H, Average velocities for anterograde and retrograde movement of GluN2A clusters. Data are expressed as mean ± SEM (n = 48 neurons from three animals/genotype, *p < 0.05; one-way ANOVA and post hoc test).
Movie 1.
Movement of GluN2B clusters in hippocampal neurons. GluN1–1a (untagged) and GluN2B-RFP were coexpressed in hippocampal neurons and observed with an LSM-510 confocal system. A 20 μm area on the dendrite was monitored every 5 s for 5 min. Capture rates were 5 frames per second.
Movie 2.
Movement of GluN2A clusters in hippocampal neurons. GluN1–1a (untagged) and GluN2A-RFP were coexpressed in hippocampal neurons and observed with an LSM-510 confocal microscope. A 20 μm area on the dendrite was monitored every 5 s for 5 min. Capture rates were 5 frames per second.
To further investigate the dynamic process of NMDA receptor transport, we observed the ER-to-Golgi and post-Golgi dynamics of NMDA receptors in neurons expressing GluN1/GluN2B-RFP or GluN1/GluN2A-RFP. GluN2B-RFP was diffusely distributed in the somatodendritic area when expressed in the presence of BFA (Fig. 5A). At 1 h after BFA washout, GluN2B-RFP accumulated in the Golgi apparatus in neurons from mice of each genotype (Fig. 5A,B), suggesting that ER-to-Golgi transport of GluN2B was not affected and that KIF17-mediated GluN2B trafficking is a post-Golgi event (Wenthold et al., 2003; Lau and Zukin, 2007). Consistently and importantly, the release of GluN2B-RFP from the Golgi apparatus was significantly impaired, and most GluN2B signals tended to be retained in the Golgi region in neurons from kif17−/− and TgD/kif17−/− mice (Fig. 5A,C). In contrast, our findings revealed no difference in the post-Golgi release of GluN2A-RFP in neurons from the mice of all five genotypes (Fig. 5D–F).
Figure 5.

ER-to-Golgi and post-Golgi transport of GluN2 subunits in neurons. A–C, Dynamics of GluN2B-RFP after BFA washout. A, Hippocampal cultures were cotransfected with untagged GluN1–1a together with GluN2B-RFP vectors. After BFA washout, images were acquired at indicated time points. Scale bar, 20 μm. B, Fluorescence intensity of the Golgi and non-Golgi were expressed as ratios at 1 h after BFA washout (mean ± SEM, p > 0.05; one-way ANOVA and post hoc test). C, Percentage of released GluN2B-RFP signals from the Golgi region (mean ± SEM, *p < 0.05; one-way ANOVA and post hoc test). For quantification, 15 neurons from three mice of each genotype were examined. D–F, Dynamics of GluN2A-RFP after BFA washout. D, Hippocampal cultures were cotransfected with untagged GluN1–1a together with GluN2A-RFP vectors. After BFA washout, images were acquired at indicated time points. Scale bar, 20 μm. E, Fluorescence intensity of Golgi and non-Golgi were expressed as ratios at 1 h after BFA washout (mean ± SEM, p > 0.05; one-way ANOVA and post hoc test). F, Percentage of released GluN2A-RFP signals from the Golgi region (mean ± SEM, p > 0.05; one-way ANOVA and post hoc test). For quantification, 15 neurons from three mice of each genotype were examined.
Next we focused on a possible change in the synaptic localization of GluN2 subunits. Primary cultured hippocampal neurons were double-stained with antibodies against GluN2B/synaptophysin or GluN2B/microtubule-associated protein 2 (MAP2). Signals for GFP-KIF17 were acquired concomitantly. In neurons from kif17+/+mice, GluN2B clusters were mostly colocalized with presynaptic terminals labeled by the anti-synaptophysin antibody. The density of both synaptic GluN2B (which overlapped with synaptophysin signal) and dendritic GluN2B (which overlapped with MAP2 signal) was significantly reduced in neurons from kif17−/− and TgD/kif17−/− compared with wild-type mice (Fig. 6A–E). Interestingly, in neurons from TgA/kif17−/− mice, the density of synaptic GluN2B clusters was selectively decreased, whereas the density of dendritic GluN2B clusters was unchanged (Fig. 6A–E). Notably, comparison of neurons from TgA/kif17−/− and TgD/kif17−/− mice revealed a marked difference in the pattern of GluN2B expression. In the dendritic shafts of TgA/kif17−/− neurons, GluN2B colocalized with GFP-KIF17 S1029A. However, in TgD/kif17−/− neurons, it did not colocalize with GFP-KIF17 S1029D (Fig. 6A). These data suggest that GluN2B-containing vesicles are delivered into dendrites, and associate with GFP-KIF17 S1029A in the dendritic shafts of neurons from TgA/kif17−/− mice. By contrast, in neurons from TgD/kif17−/− mice, most GluN2B-containing vesicles remained in the Golgi apparatus because of the defective post-Golgi transport of GluN2B (Fig. 5). Immunocytochemical analysis showed that the density of GluN2A-positive clusters was decreased both in synaptic regions and in dendritic shafts in neurons from kif17−/−, TgA/kif17−/− and TgD/kif17−/− compared with kif17+/+ mice (Fig. 7A–E). Neurons from TgS/kif17−/− mice showed normal levels of GluN2B- and GluN2A-positive cluster densities (Figs. 6, 7). Consistently, levels of surface-expressed endogenous GluN2B and GluN2A were reduced in neurons from kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice, but not in those from TgS/kif17−/− mice, compared with the levels in neurons from wild-type mice (Fig. 8A,B).
Figure 6.

Intracellular localization of GluN2B. A, B, Primary cultures of hippocampal neurons were double-stained with anti-GluN2B/synaptophysin (A) or anti-GluN2B/MAP2 (B) antibodies. Transgenically expressed KIF17 (green) was present in TgS/kif17−/−, TgA/kif17−/− and TgD/kif17−/− neurons. Arrows indicate colocalization of GFP-KIF17 and GluN2B in dendritic shafts. Scale bar, 10 μm. C, Quantification of the synaptic density of GluN2B-positive clusters (colabeled with anti-synaptophysin) in dendrites. D, Quantification of the density of GluN2B clusters in dendritic shafts (colabeled with anti-MAP2). E, Comparison of the distribution of GluN2B clusters in dendritic shafts and synapses. Twenty neurons from three animals were examined for each genotype. Data are expressed as mean ± SEM (*p < 0.01; one-way ANOVA and post hoc test).
Figure 7.

Intracellular localization of GluN2A. A, B, Primary cultures of hippocampal neurons were double-stained with anti-GluN2A/synaptophysin (A) or anti-GluN2A/MAP2 (B) antibodies. GFP-fused KIF17 (green) was present in TgS/kif17−/−, TgA/kif17−/− and TgD/kif17−/− neurons. Scale bar, 10 μm. C, Quantification of the synaptic density of GluN2A-positive clusters (colabeled with anti-synaptophysin) in dendrites. D, Quantification of the density of GluN2A clusters in dendritic shafts (colabeled with anti-MAP2). E, Comparison of the distribution of GluN2A clusters in dendritic shafts and synapses. Twenty neurons from three animals were examined for each genotype. Data are expressed as mean ± SEM (*p < 0.01; one-way ANOVA and post hoc test).
Figure 8.

Surface expression of endogenous GluN2B and GluN2A in hippocampal neurons. Surface-expressed proteins in cultured neurons were labeled and isolated, and then resolved by SDS-PAGE. A, Immunoblots of levels of GluN2B, GluN2A, and GluR1 in total extracts and surface components. B, Quantification of surface expression experiments. The intensities of GluN2B and GluN2A (total and surface) were normalized to GluR1. Data are expressed as mean ± SEM from three independent experiments. *p < 0.05; one-way ANOVA and post hoc test.
Together, these experiments show that disruption of the KIF17–Mint1 interaction accounts for reduced levels of functional GluN2B and GluN2A in neurons from kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice.
NMDA receptor protein degradation in Tg+/kif17−/− mice
Changes in NMDA receptor levels could arise because of modulation of protein synthesis, trafficking, or degradation, and/or by regulated removal/loss of synaptic proteins (Yashiro and Philpot, 2008). Previous studies have revealed that changes in the intracellular dynamics of NMDA receptors affect the turnover of NMDA receptors themselves (Monyer et al., 1994; Rezvani et al., 2007; Yi and Ehlers, 2007; Tai and Schuman, 2008; Yin et al., 2011). To address the possibility of an alteration in the turnover of NMDA receptors in Tg+/kif17−/− mouse neurons, we first measured the mRNA levels of NMDA receptors in the hippocampus. The level of GRIN2B mRNA was decreased in the hippocampi of kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice, compared with that of control animals, but no difference was observed in the level of GRIN2A mRNA in the hippocampi of these mice (Fig. 9A,B). We subsequently compared the half-life of GluN2 subunits in primary hippocampal cultures treated with CHX, a translational inhibitor (Hatanaka et al., 2006). This turnover assay revealed a robust, accelerated turnover of GluN2A protein in neurons from kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice, compared with that in neurons from wild-type mice (Fig. 9C,D). Similar levels of turnover of GluN2A were observed in neurons from kif17+/+ and TgS/kif17−/− mice (Fig. 9C,D). The rate of loss of GluN2B protein was similar in neurons from all mouse lines (Fig. 9C,E).
Figure 9.
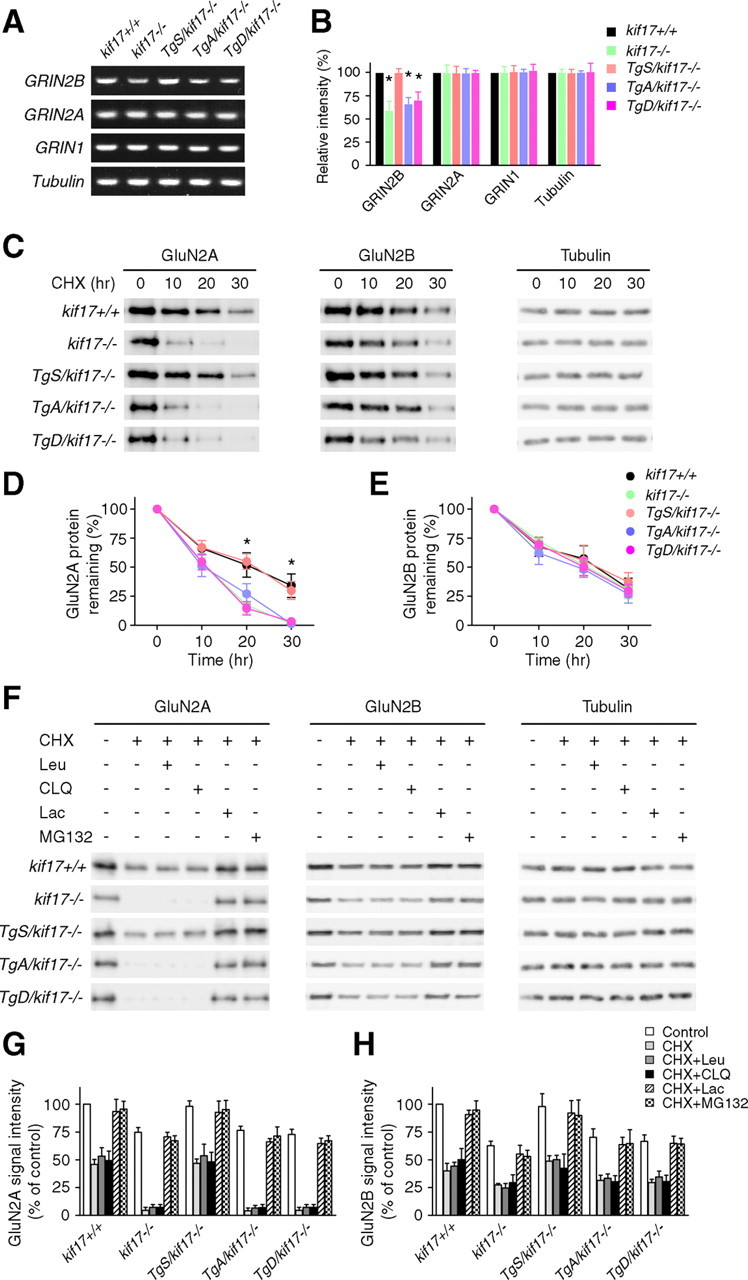
Receptor degradation analysis in hippocampal neurons. A, B, RT-PCR analysis of NMDA receptors in hippocampal extracts. Data are representative of three separate experiments. Values are expressed as mean ± SEM (*p < 0.05; one-way ANOVA and post hoc test). C, Representative immunoblots of levels of GluN2A and GluN2B in neurons treated with CHX (20 μg/ml) for 0, 10, 20, and 30 h, respectively. D, E, Quantification of GluN2A/2B degradation. Data were obtained from three independent experiments. Values are the average signal intensities (mean ± SEM) for GluN2A and GluN2B standardized to the signal intensity of tubulin and normalized to 100% at time 0 (*p < 0.05; one-way ANOVA and post hoc test). F, Representative immunoblots of levels of GluN2 subunits in neurons incubated with CHX for 20 h, in the presence or absence of lysosomal inhibitors (Leu, leupeptin, 100 μg/ml; CLQ, chloroquine, 200 μm), or proteasomal inhibitors (Lac, lactacystin, 10 μm; MG132, 10 μm). G, H, Quantification of changes in GluN2 subunits as shown in F, based on three independent experiments. Data are expressed as mean ± SEM.
We next sought to determine changes in GluN2A degradation rates in the presence of proteasome inhibitors (lactacystin or MG132) or lysosome inhibitors (leupeptin or chloroquine). GluN2A degradation was efficiently blocked in kif17−/−, TgA/kif17−/− and TgD/kif17−/− neurons treated with proteasome inhibitors, but not lysosome inhibitors (Fig. 9F–H), thereby suggesting that like in kif17−/− mice, loss of GluN2A occurs through the degradation dependent on ubiquitin-proteasome system in TgA/kif17−/− and TgD/kif17−/− mice.
CREB phosphorylation
CREB phosphorylation is considered a molecular marker of memory processing for spatial learning in the hippocampus (Mizuno et al., 2002; Colombo et al., 2003; Porte et al., 2008). CREB-dependent upregulation of GRIN2B and kif17 gene transcription has also been reported (Wong et al., 2002; Yin et al., 2011). Thus, we wanted to investigate whether CREB activation is altered in Tg+/kif17−/− mice. After training in the water maze, the expression levels of pCREB, CREB, GluN2A, GluN2B, KIF17, and KIF5B in the hippocampi of all five genotypes of mice were determined (Fig. 10). Significantly, water maze training induced an increase in the levels of pCREB, GluN2A, and GluN2B in kif17+/+ and TgS/kif17−/− mice, whereas no increase was detected in kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice (Fig. 10A,B). Levels of total CREB and KIF5B were not affected by the water maze training in mice of all genotypes (Fig. 10A,B). Interestingly, the expression level of KIF17 was increased only in kif17+/+ mice, reflecting that only endogenous kif17 had a CREB binding site in its promoter region (Fig. 10A,B).
Figure 10.
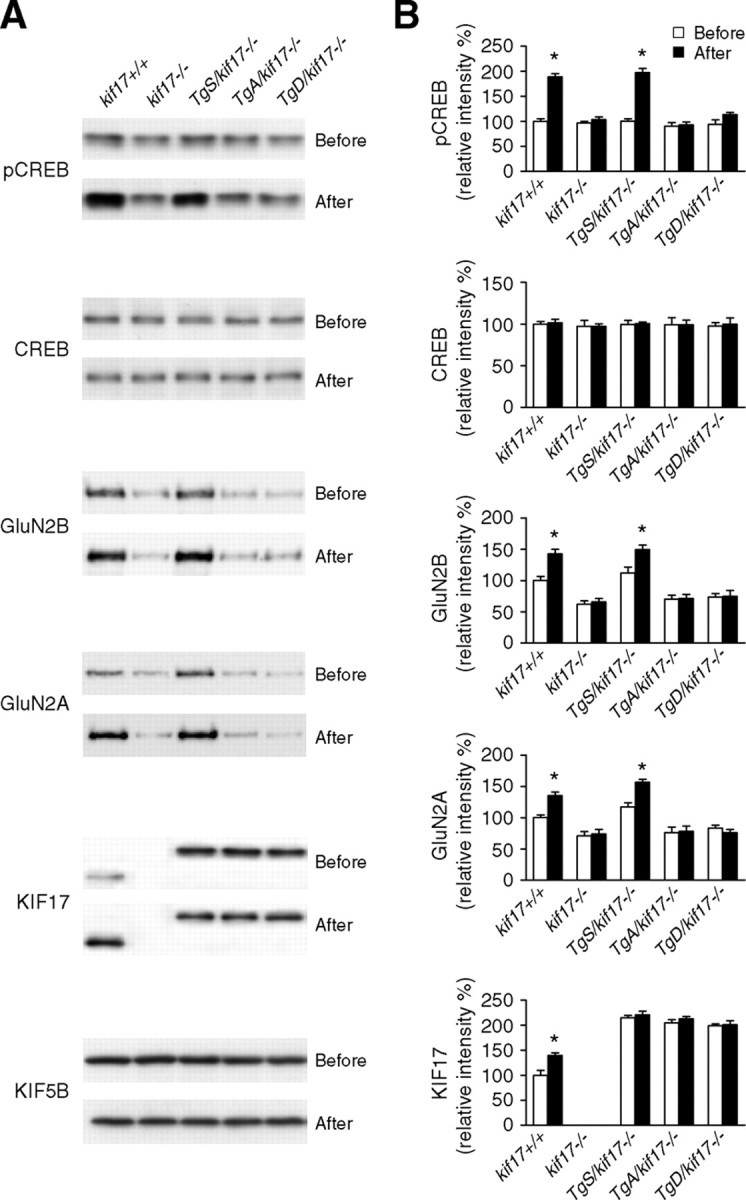
CREB activation. A, Immunoblots of p-CREB, CREB, GluN2B, GluN2A, KIF17 and KIF5B in hippocampal homogenates from kif17+/+, kif17−/−, TgS/kif17−/−, TgA/kif17−/− and TgD/kif17−/− mice before and after water maze training. B, Quantification of immunoblots and mean values normalized to control (KIF5B). Data (mean ± SEM) were obtained from four independent experiments (*p < 0.05; one-way ANOVA and post hoc test).
Together, these findings reveal that phosphorylation of CREB and CREB-dependent gene activation (GRIN2B and kif17) are attenuated in kif17−/−, TgA/kif17−/−, and TgD/kif17−/− mice.
Discussion
In the present study, we generated genetically modified mice in which the KIF17–Mint1 interaction was disrupted in postnatal forebrain. In TgA/kif17−/− mice, KIF17 was expressed with a S1029A mutation, which mimics KIF17 in an unphosphorylated state at Ser1029. In TgD/kif17−/− mice, KIF17 was expressed with a S1029D mutation, which mimics KIF17 in a phosphorylated state at Ser1029. Immunoprecipitation experiments showed that the KIF17 S1029A mutant bound to the Mint1/GluN2B complex, but the KIF17 S1029D mutant did not (Fig. 1I), suggesting that Ser1029 is a pivotal site of regulation for the association of KIF17 and Mint1/GluN2B in vivo.
We previously suggested that the memory disturbance of the kif17−/− mice might reflect changes in NMDA receptor channel activity, synaptic plasticity and/or levels of synaptic NMDA receptors (Yin et al., 2011). The present study revealed impairments in incrementally acquired spatial memory, but not in performing nonspatial learning tasks, in both TgA/kif17−/− and TgD/kif17−/− mice (Fig. 2). Kif17−/− mice bear similar phenotypic profiles to TgA/kif17−/− and TgD/kif17−/− mice. The reduction of NMDA receptor-mediated currents, as well as the impaired LTP magnitude observed in TgA/kif17−/−, TgD/kif17−/− and kif17−/− mice (Fig. 3) could potentially underlie their behavioral phenotype. Notably, the expression of GFP-KIF17 in TgS/kif17−/− mouse brain rescued the deficits in spatial performance and neuronal plasticity in kif17−/− mice (Figs. 2, 3).
The mechanism by which KIF17 regulates NMDA receptor transport through Mint1 remains to be established. Neurons from TgA/kif17−/− and TgD/kif17−/− mice exhibited reduced synaptic accumulation of GluN2B (Fig. 6) because of defective post-Golgi transport (Figs. 4A–D, 5A–C; Movie 1). Importantly, neurons from TgA/kif17−/− mice showed a selective decrease of GluN2B clusters in synapses, but the density of GluN2B clusters in dendritic shafts remained unchanged. In contrast, most GluN2B clusters tended to aggregate in the Golgi apparatus in neurons from TgD/kif17−/− mice instead of being transported into dendritic shafts (Fig. 6). This distinct pattern of mislocalization of GluN2B is considered to result from the different properties of GFP-KIF17 S1029Aand GFP-KIF17 S1029D: the former retains the ability to transport GluN2B vesicles within dendritic shafts, but the Ser to Ala substitution at residue 1029 inhibits the release of GluN2B, and the latter is not capable of interacting with Mint1 to transport GluN2B vesicles in the post-Golgi pathway. Together, we conclude that both binding of cargo to a molecular motor protein and cargo release from this complex are essential processes for GluN2B recruitment to the synaptic destination.
Our further analysis revealed that transcriptional regulation by the KIF17–GluN2–CREB loop could contribute to the synaptic loss of GluN2B in neurons of TgA/kif17−/− and TgD/kif17−/− mice (Figs. 9A,B, 10A,B) (Wong et al., 2002; Dhar and Wong-Riley, 2011; Yin et al., 2011). Recent studies have begun to identify the specific roles of some KIF members in cell signaling, which is independent of the ATPase activity required for conventional motor function. For instance, KIF4 controls the survival of postmitotic neurons by regulating PARP-1 activity (Midorikawa et al., 2006), and KIF26A represses GDNF-Ret signaling during enteric nervous system development (Zhou et al., 2009). Together with previous reports (Wong et al., 2002; Yin et al., 2011), the findings described herein suggest that KIF17 regulates NMDA-related memory processing through CREB activity.
Our findings also revealed that KIF17–Mint1/GluN2B regulates the steady-state level of GluN2A at synapses. We showed a decrease in the level of GluN2A in neurons from TgA/kif17−/− and TgD/kif17−/− mice (Figs. 7, 8), which was primarily due to proteasomal-controlled degradation (Fig. 9C–H). Based on the data presented herein, together with our findings in kif17-null mice (Yin et al., 2011), this effect could result from the loss of GluN2B function and altered neuronal activity (Barria and Malinow, 2002; Ehlers, 2003; Lavezzari et al., 2004; Scott et al., 2004; Kim et al., 2005; Yashiro and Philpot, 2008).
Based on our findings, we propose the following model for the KIF17-mediated regulation of NMDA receptor trafficking (Fig. 11). In the case of TgS/kif17−/− mice, GFP-KIF17 could transport GluN2B within dendrites, and release the cargo in the vicinity of synapses after CaMKII-dependent phosphorylation, maintaining normal learning-related synaptic plasticity. However, in TgA/kif17−/− and TgD/kif17−/− mice, reduced synaptic receptor abundance caused by impairments in binding and release of GluN2B to/from KIF17, and the degradation of GluN2A by proteasomal activity, would ultimately lead to impaired performance in spatial information processing.
Figure 11.

Model for the regulation of KIF17-cargo interaction. A, In wild-type neurons, KIF17 binds to the scaffolding Mint1 complex to transport GluN2B-containing vesicles from the cell body to synaptic terminals in dendrites. CaMKII-dependent phosphorylation of KIF17 releases the NMDA receptor, facilitating its insertion at synapses. Synaptic NMDA receptor-mediated Ca2+ influx triggers LTP, activates signaling pathways and enhances CREB-dependent gene transcription. B, In kif17−/− neurons, lack of KIF17-mediated GluN2B transport leads to downregulation of GluN2B and GluN2A via different pathways, contributing to defects in synaptic plasticity and memory in kif17−/− mice. C, In TgS/kif17−/− neurons, GFP-KIF17 protein efficiently transports GluN2B-containing vesicles along dendrites; thus, the deficiencies in kif17−/− mice are rescued by the expression of GFP-KIF17 protein. D, In TgA/kif17−/− neurons, GFP-KIF17 S1029A binds to Mint1 and enables the transport of GluN2B vesicles from cell body to dendrites, but inability of CaMKII to phosphorylate GFP-KIF17 S1029A prevents the unloading and further synaptic recruitment of GluN2B vesicles. E, In TgD/kif17−/− neurons, GFP-KIF17 S1029D does not transport GluN2B vesicles, resulting in the accumulation of GluN2B subunits in the Golgi apparatus. Collectively, the defective transport of GluN2B, and the attenuated CREB responses result in a reduction in the level of synaptic GluN2B in both TgA/kif17−/− and TgD/kif17−/− neurons. The loss of GluN2B function appears to be a triggering factor for the proteasomal control of GluN2A degradation. In conclusion, TgA/kif17−/− and TgD/kif17−/− mice show impairments in synaptic plasticity and spatial memory because of insufficient levels of synaptic NMDA receptors.
Recently, several in vitro studies have suggested that the modulation of synaptic cargo trafficking at the level of motor–cargo binding can occur through changes in local Ca2+ levels (Chang et al., 2006; Wang et al., 2008; Wang and Schwarz, 2009) or Rab GTPase activity (Pfeffer and Aivazian, 2004; Niwa et al., 2008), or alternatively, through CaMKII-dependent signaling (Hoogenraad et al., 2007; Guillaud et al., 2008). Here, for the first time, our study demonstrates in vivo regulation of protein transport to synapses through modulation of the loading/unloading of cargo to/from a molecular motor using multidisciplinary systems analysis of genetically engineered mice.
Footnotes
This work was supported by a Grant-in-Aid for specially promoted research to N.H. from the Ministry of Education, Culture, Science, Sports and Technology of Japan. We thank M. Hollman (Ruhr University) and S. Okabe (The University of Tokyo) for providing the GluN1 cDNA, and T. Hensch (RIKEN) and H. Katagiri (RIKEN) for their help and suggestions regarding electrophysiology. We also thank all other members of the Hirokawa Laboratory for their help and discussion.
The authors declare no competing financial interests.
References
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kimura T, Nakamuta S, Taya S, Funahashi Y, Hattori A, Shimada A, Ménager C, Kawabata S, Fujii K, Iwamatsu A, Segal RA, Fukuda M, Kaibuchi K. Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell. 2009;16:675–686. doi: 10.1016/j.devcel.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RGM. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature. 1995;378:182–186. doi: 10.1038/378182a0. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo PJ, Brightwell JJ, Countryman RA. Cognitive strategy-specific increases in phosphorylated cAMP response element-binding protein and c-Fos in the hippocampus and dorsal striatum. J Neurosci. 2003;23:3547–3554. doi: 10.1523/JNEUROSCI.23-08-03547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Wong-Riley MT. The kinesin superfamily protein KIF17 is regulated by the same transcription factor (NRF-1) as its cargo NR2B in neurons. Biochim Biophys Acta. 2011;1813:403–411. doi: 10.1016/j.bbamcr.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Cambridge, MA: MIT; 1991. pp. 251–281. [Google Scholar]
- Guillaud L, Setou M, Hirokawa N. KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J Neurosci. 2003;23:131–140. doi: 10.1523/JNEUROSCI.23-01-00131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- Hatanaka K, Ikegami K, Takagi H, Setou M. Hypo-osmotic shock induces nuclear export and proteasome-dependent decrease of UBL5. Biochem Biophys Res Commun. 2006;350:610–615. doi: 10.1016/j.bbrc.2006.09.093. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Feliu-Mojer MI, Spangler SA, Milstein AD, Dunah AW, Hung AY, Sheng M. Liprinalpha1 degradation by calcium/calmodulin-dependent protein kinase II regulates LAR receptor tyrosine phosphatase distribution and dendrite development. Dev Cell. 2007;12:587–602. doi: 10.1016/j.devcel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Jiang M, Chen G. High Ca2+-phosphorate transfection efficiency in low-density neuronal cultures. Nat Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel RA. GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 2001;29:E39. doi: 10.1093/nar/29.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Baranes D, Podsypanina K, Kandel ER. The 3′-untranslated region of CaMKIIα is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci U S A. 1996;93:13250–13255. doi: 10.1073/pnas.93.23.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa R, Takei Y, Hirokawa N. KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell. 2006;125:371–383. doi: 10.1016/j.cell.2006.02.039. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Maekawa N, Saito K, Seishima M, Nabeshima T. CREB phosphorylation as a molecular marker of memory processing in the hippocampus for spatial learning. Behav Brain Res. 2002;133:135–141. doi: 10.1016/s0166-4328(01)00470-3. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Niwa S, Tanaka Y, Hirokawa N. KIF1Bβ- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat Cell Biol. 2008;10:1269–1279. doi: 10.1038/ncb1785. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- Porte Y, Buhot MC, Mons N. Alteration of CREB phosphorylation and spatial memory deficits in aged 129T2/Sv mice. Neurobiol Aging. 2008;29:1533–1546. doi: 10.1016/j.neurobiolaging.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Shim D, De Biasi M. Nicotine regulate multiple synaptic proteins by inhibiting proteasomal activity. J Neurosci. 2007;27:10508–10519. doi: 10.1523/JNEUROSCI.3353-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor ε1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Matzel LD. Long-term potentiation: what's learning got to do with it? Behav Brain Sci. 1997;20:597–614. doi: 10.1017/s0140525x97001593. [DOI] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wong RW, Setou M, Teng J, Takei Y, Hirokawa N. Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:14500–14505. doi: 10.1073/pnas.222371099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Yin X, Takei Y, Kido MA, Hirokawa N. Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron. 2011;70:310–325. doi: 10.1016/j.neuron.2011.02.049. [DOI] [PubMed] [Google Scholar]
- Zhou R, Niwa S, Homma N, Takei Y, Hirokawa N. KIF26A is an unconventional kinesin and regulates GDNF-Ret signaling in enteric neuronal development. Cell. 2009;139:802–813. doi: 10.1016/j.cell.2009.10.023. [DOI] [PubMed] [Google Scholar]