

Abstract
Mutations in the small heat shock protein HSPB1 (HSP27) are causative for Charcot-Marie-Tooth (CMT) neuropathy. We previously showed that a subset of these mutations displays higher chaperone activity and enhanced affinity to client proteins. We hypothesized that this excessive binding property might cause the HSPB1 mutant proteins to disturb the function of proteins essential for the maintenance or survival of peripheral neurons. In the present work, we explored this hypothesis further and compared the protein complexes formed by wild-type and mutant HSPB1. Tubulin came out as the most striking differential interacting protein, with hyperactive mutants binding more strongly to both tubulin and microtubules. This anomalous binding leads to a stabilization of the microtubule network in a microtubule-associated protein-like manner as reflected by resistance to cold depolymerization, faster network recovery after nocodazole treatment, and decreased rescue and catastrophe rates of individual microtubules. In a transgenic mouse model for mutant HSPB1 that recapitulates all features of CMT, we could confirm the enhanced interaction of mutant HSPB1 with tubulin. Increased stability of the microtubule network was also clear in neurons isolated from these mice. Since neuronal cells are particularly vulnerable to disturbances in microtubule dynamics, this mechanism might explain the neuron-specific CMT phenotype caused by HSPB1 mutations.
Introduction
Charcot-Marie-Tooth (CMT) disease represents a large group of genetically and clinically heterogeneous disorders affecting the peripheral nervous system. Clinically, CMT disease is characterized by a length-dependent degeneration of peripheral nerves resulting in weakness and wasting of distal muscles in lower limbs, feet and hands. So far, mutations in >40 genes have been linked to CMT neuropathy and related disorders (Barisic et al., 2008). In 2004, we found that dominant missense mutations in the small heat shock protein HSPB1 (also known as HSP27) were linked to Charcot-Marie-Tooth type 2F and distal hereditary motor neuropathy (HMN) (Evgrafov et al., 2004).
Small heat shock proteins (sHSPs) are ubiquitously expressed molecular chaperones characterized by the presence of a conserved α-crystallin domain. In contrast to heat shock proteins with an ATPase domain (e.g., HSP70), sHSPs do not have the intrinsic capacity to actively refold denatured proteins. However, they are capable of binding unfolded substrates and keeping them in a folding-competent state until these are cleared out by the ATP-dependent heat shock proteins (Haslbeck et al., 2005). HSPB1 is one of the most studied sHSPs due to its role in several human disorders including cancer, Alzheimer's disease, and heart disease (Sun and Macrae, 2005).
In a previous study, we addressed how mutations in HSPB1 might cause a peripheral neuropathy and characterized the biochemical consequences of CMT-causing mutations on the basic properties of this protein. In contrast to most other chaperonopathies, mutations in the α-crystallin domain localized close to the HSPB1 dimer interface (R127W, S135F, and R136W) lead to an increase, rather than a loss, of chaperone activity. This hyperactivation was corroborated by an increased affinity of the mutants for their client proteins. Two other mutations, one situated at the border of the α-crystallin domain (T151I) and one at the C terminus of the protein (P182L) did not show this hyperactivation (Almeida-Souza et al., 2010). We hypothesized that the excessive binding properties of the hyperactive mutants might disturb the function of HSPB1 client proteins that are essential for the survival of peripheral neurons.
To identify aberrant interactions caused by these hyperactive mutations, we compared the protein complexes formed by wild-type and mutant HSPB1 by tandem affinity purification. We found that hyperactive mutants exhibit enhanced interaction with tubulin and microtubules (MTs), which leads to a stabilization of the microtubule network. Importantly, the increased interaction of mutant HSPB1 with tubulin and the enhanced stability of the microtubule network were also obvious in neurons isolated from a mouse model that expresses mutant HSPB1 and recapitulates the typical CMT symptoms.
Materials and Methods
Stable cell line generation, constructs, and recombinant protein production.
Stable HEK293Flp-in cell lines were generated as described previously (Almeida-Souza et al., 2010) using constructs carrying HSPB1 C-terminally fused to a tandem affinity purification (TAP) tag composed of Flag, TEV, cleavage site, and Protein A (Tsai and Carstens, 2006). Stable HeLa cell lines were produced by lentiviral transduction according to the method described in Salmon and Trono (2006) and the constructs described previously (Almeida-Souza et al., 2010). TUBB3-GFP and EB1-GFP constructs were generated using Gateway recombination (Invitrogen). For recombinant protein production, the ORF from HSPB1 was amplified by PCR and cloned by topoisomerase cloning into the vector pCRT7-NT-TOPO (Invitrogen). Protein expression was induced in BL21 DE3 cells, and proteins were purified on a Ni-NTA column.
Dorsal root ganglion neuronal cultures.
Dorsal root ganglion (DRG) neurons, isolated from 3-month-old CO2-killed mice, were dissociated by incubation at 37°C with 0.5% collagenase for 45 min and 1.5% trypsin for 30 min. Cell suspensions were washed with DRGprep medium [DMEM medium supplemented with bovine serum (10%), nonessential amino acids (1%), l-glutamine (1%), and NaHCO3 (0.15%)], and centrifuged at 800 × g for 5 min. The pellet was resuspended in DRGprep and incubated for 50 min at 37°C and 7% CO2 on a bovine serum cushion. Next, cell suspension was centrifuged for 5 min at 800 × g, and the pellet was resuspended in DRG medium [DMEM:F12 medium supplemented with l-Glutamax (4 mm), bovine serum (10%), nonessential amino acids (1%), penicillin/streptomycin (50 U/ml), NGF (10 ng/ml; Millipore), and NaHCO3 (0.045%)]. DRG neurons were seeded at a density of 100 cells/well coated with poly-l-ornithine and laminin (Sigma), and incubated at 37°C and 7% CO2.
Tandem affinity purification and mass spectrometry.
TAPs for mass spectrometric protein identification were done by upscaling the protocol described before (Almeida-Souza et al., 2010). HEK293 stable cell lines were grown in 10× 150 cm2 dishes, harvested with a cell scraper, and washed two times with PBS. Cell pellets were lysed in 10 ml of TAP lysis buffer (50 mm Tris-HCl, pH 8.0, 10% glycerol, 1% NP40, 150 mm NaCl, 5 mm NaF, 5 μm ZnCl2, 1 mm Na3VO4, 10 mm EGTA, and Protease inhibitor cocktail (Roche). Equal amounts of cytosolic lysates from each stable cell line were incubated with 200 μl of IgG beads (GE Healthcare) for 4 h at 4°C with constant agitation. Beads were washed three times with washing buffer (20 mm Tris-HCl, pH 7.5, 5% glycerol, 0.1% NP40, and 150 mm NaCl) and three additional times with TEV protease cleavage buffer (10 mm Tris-HCl, pH 8, 150 mm NaCl, 0.1% NP40, and 0.5 mm EDTA). The washed IgG beads were then incubated overnight at 4°C and with constant agitation with 100 U of AcTEV protease (Invitrogen) in 500 μl of TEV protease cleavage buffer. After TEV cleavage, the supernatants containing the eluted complexes were incubated with 200 μl of anti-Flag beads (Sigma) for 4 h at 4°C with constant agitation. After incubation, beads were washed gently three times with washing buffer. Final elution was done by incubation of the Flag beads for 30 min at 4°C with 250 ng/μl of the 3xFlag peptide. Final eluates were run on NuPAGE gels (Invitrogen) and Coomassie stained. Bands of interest were excised and digested with sequencing grade-modified trypsin according to the manufacturer's instructions (Promega). The resulting protein digests were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Bruker HCT Esquire ion trap mass spectrometer, and protein identification was done using the Mascot algorithm (95% confidence level).
Immunoprecipitation from cells and mouse sciatic nerve.
Immunoprecipitations from HEK293 stable cell lines were performed using the same protocol as described for the TAP experiments until the first elution step after TEV protease cleavage and using only 5 mg of cell lysates. For the immunoprecipitations from mouse sciatic nerves, tissue homogenates were prepared using the extraction reagent from the ProFound Mammalian HA-tag IP/coIP kit (Thermo Fisher Scientific) with Lysing Matrix A (MP Biomedicals). Immunoprecipitation of the HA-tagged HSPB1 was performed according to manufacturer's instructions.
Ice-induced microtubule depolymerization assay.
HEK293Flp-in cells were seeded in 6-well plates in triplicate. Forty-eight hours after seeding, plates were placed on ice for the described time and subsequently lysed with MT stabilization buffer (20 mm Tris, pH 6.8, 2 mm EGTA, 1 mm MgCl2, 0,5% NP-40, 140 mm NaCl, and 4.7 μm taxol) (Bhattacharya and Cabral, 2004). After lysis, cell extracts were centrifuged at 20,000 × g for 10 min to separate the soluble from polymerized tubulin. Ratios between polymerized and unpolymerized tubulin were calculated by densitometric measurement of Western blot bands using ImageJ.
Microtubule network recovery and nocodazole washout experiments.
HeLa cells were seeded in 24-well plates containing coverslips and treated 48 h later with 10 μm nocodazole for 6 h. After treatment, media was changed and cells were allowed to recover for different periods. After recovery, soluble tubulin was washed away by detergent extraction (BRB80 containing 0.5% Triton X-100 for 30 s). Cells were then fixed with 3% paraformaldehyde (PFA) and stained for α-tubulin (ab7291, Abcam). Confocal image stacks from several cells were taken with a Zeiss LSM700 confocal microscope equipped with a 63× 1.4 NA objective with identical acquisition parameters. Total MT fluorescence intensity was determined by measuring average pixel intensities on maximum intensity projections for individual cells by use of ImageJ.
Neuronal microtubule depolymerization and repolymerization assay.
Assays were performed on DRG neurons at 2 d in vitro. At various time points during or after nocodazole (20 μm) treatment, neurons were fixed with 4% PFA after being subjected to detergent extraction and were subsequently stained for α-tubulin (ab7291, Abcam). Images were acquired on a Zeiss Imager M1 fluorescence microscope using a 20× 0.45 NA objective. MT network length was determined for the longest neurite per neuron using the Single Neurite Tracer Plugin for ImageJ. MT network length was normalized to the initial size before treatment.
HSPB1 and tubulin interaction measurements in cells.
Cos-1 cells were transiently transfected with HSPB1-V5 and TUBB3-GFP constructs, fixed with 3% PFA 48 h after transfection, and processed for immunofluorescence using a mouse monoclonal anti-HSPB1 antibody (SPA-803, Stressgen). Cos-1 cells were chosen for the in vivo colocalization experiments due to their low endogenous HSPB1 levels, high transfection efficiency, and large flat cytoplasmic extensions. Two-channel sequentially scanned images were acquired with a Zeiss LSM700 confocal microscope with 63× 1.4 NA objective. Pearson's correlation coefficient was calculated for 50 cytoplasmic regions (200 × 200 pixels) from 10 different cells using the JACoP ImageJ plugin (Bolte and Cordelières, 2006). Fluorescence intensity profile graphs were generated by using the plot profile feature from ImageJ from a randomly traced line in the cortical regions of cells. Fluorescence values were normalized by setting the highest value for each channel as 1.
Microtubule cosedimentation assay.
Cell extracts from HEK293Flp-in cells were diluted 1:1 in BRB80 (80 mm PIPES, 1 mm MgCl2, and 1 mm EDTA); supplemented with 1 mm GTP, 2 mm ATP, and 25 μm taxol; and incubated at 37°C for 30 min. After incubation, reactions were layered on a 40% glycerol cushion and centrifuged at 100,000 × g for 30 min at 37°C. Supernatant was separated into another tube, and the pellet was washed with BRB80 and resuspended in the same volume as the supernatant. The experiment was repeated three times, and the ratio between soluble and MT-bound HSPB1 was calculated by densitometric measurement of Western blot bands using ImageJ.
Microtubule dynamics measurements.
HeLa cells stably expressing different HSPB1 isoforms were cultivated in 35 mm glass-bottom dishes and transfected with the TUBB3-GFP or EB1-GFP constructs using lipofectamine LTX (Invitrogen). Cells were imaged 72 h after transfection for 2 min at 3 s/frame for TUBB3-GFP-transfected cells and at 2 s/frame for EB1-GFP-transfected cells on a Zeiss Axiovert 200 microscope with a microlens-enhanced dual spinning disc confocal system (UltraVIEW ERS; PerkinElmer), equipped with a 60× 1.4 NA objective. During imaging, cells were maintained at 37°C and 5% CO2. Individual TUBB3-GFP MTs were tracked manually using the Manual Tracking plugin of ImageJ, and dynamicity parameters were extracted from data by a custom-made Perl script. The MTs were considered static if they moved <0.3 μm/frame. The speed and distance traveled by EB1-GFP comets were calculated by automatic tracking using the Particle Tracker plugin of ImageJ.
Scratch-induced migration assay.
HEK293Flp-in cells were grown to confluence on 24-well plates and treated for 30 min with 5 μm cell tracker (Invitrogen) before being scratched with a pipette tip. Cells were imaged with a 10× objective by phase contrast and fluorescence video microscopy using a Zeiss Axiovert 200 microscope equipped with an incubation chamber at 37°C and 5% CO2. The migration history was plotted as the area covered by cells every 2 h, subtracted from the area occupied by the cells at the time 0 of the experiment.
In vitro microtubule polymerization assays.
Microtubule assembly was monitored by DAPI fluorescence according to the protocol described previously (Barron et al., 2003). Polymerization was performed using 10 μm microtubule-associated protein (MAP)-rich tubulin (Cytoskeleton Inc) in BRB80 buffer, 1 mm GTP, 10 μm DAPI, and recombinant HSPB1 at 5 μm when stated. The fluorescence of MT-bound DAPI was monitored with a BioTek plate reader every 30 s with the temperature set to 37°C and excitation/emission wavelengths set to 360/450 nm.
Binding assay by surface plasmon resonance.
Surface plasmon resonance (SPR) experiments were performed using a Biacore 3000 instrument. The surface of a CM5 sensor chip was activated with a 1:1 mixture of 0.1 m N-hydroxysuccinimide and 0.4 m 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. After activation of the surface, tubulin (50 μg/ml) in 10 mm sodium acetate, pH 4, was injected to flow cell 2 (FC2) to be immobilized on the sensor surface via primary amine groups. Residual active ester groups were blocked with 1 m ethanolamine-HCl, pH 8.5. As a control, FC1 was activated and deactivated as described for FC2. HSPB1WT and HSPB1S135F was flowed over the chip surface with concentrations ranging from 40 μm to 19.5 nm and 25.7 μm to 12.6 nm, respectively, in degassed HBS buffer (10 mm HEPES, 150 mm NaCl, 1 mm EDTA, 0.005% Tween20, pH 7.4) at a flow rate of 20 μl/min at 25°C during 2 min. Dissociation was done in HBS buffer over 30 min until the baseline level was reached. Data were evaluated using the software BIAeval (Biacore AB). The subtracted (FC2 − FC1) equilibrium signals were plotted versus HSPB1 concentrations, and a steady-state affinity model was fitted to the data, from which the dissociation constant KD was obtained.
Statistical analysis.
For all experiments, statistical significance for the different measurements was tested using a two-tailed t test, with the exception of the individual MT dynamics measurements, where statistical difference was evaluated by an ANOVA followed by a Bonferroni multiple test correction.
Results
CMT neuropathy-causing HSPB1 mutants show enhanced binding to tubulin and microtubules
To address how hyperactive HSPB1 mutations may cause CMT neuropathy, we compared the protein complexes formed by HSPB1WT and mutant isoforms. Parallel tandem affinity purifications from cells expressing equal levels of WT or mutant HSPB1 were performed, and interacting complexes were loaded on gel. A band running at ∼50 kDa was clearly more prominent in the lanes containing the α-crystallin HSPB1 mutants (Fig. 1A). Mass spectrometric identification revealed that this band corresponded to α- and β-tubulin. Immunoprecipitation with all the CMT disease-causing mutations previously identified in our group (Evgrafov et al., 2004) revealed that only the hyperactive HSPB1 mutations R127W, S135F, and R136W (Almeida-Souza et al., 2010) showed enhanced interaction to tubulin, while HSPB1T151I and HSPB1P182L showed a similar interaction compared with HSPB1WT (Fig. 1B).
Figure 1.

Hyperactive HSPB1 mutants bind to tubulin and microtubules. A, Coomassie staining of the final eluates from TAPs from HEK293Flp-in cells expressing TAP-tagged HSPB1-WT, R127W, and R136W. The prominent ∼50 kDa band was identified by mass spectrometry as α- and β-tubulin. B, Immunoprecipitations from HEK293Flp-in cells confirmed that the three hyperactive HSPB1 mutants (R127W, S135F, and R136W) displayed enhanced binding to tubulin. HSPB1 was detected using anti-flag antibody. Differences in HSPB1 size between input and the IP fractions are due to the removal of the Protein A tag during the elution step by TEV cleavage. C, Cosedimentation assay performed to evaluate HSPB1 binding to MTs. Cell extracts from HEK293Flp-in cells expressing different HSPB1 isoforms were supplemented or not with GTP and taxol, and incubated at 37°C to allow MT polymerization. MTs were then separated from tubulin by ultracentrifugation, and the amount of HSPB1 bound to MTs (visualized by anti-Flag Western blot) was determined by calculating the ratio between the amount of HSPB1 in the MT and the total amount of HSPB1 from the reactions supplemented with taxol (Graph, n = 3 independent experiments). Vertical thick black lines shown in Western blots correspond to divisions between gels. Data are presented as mean ± SEM. M, MT-containing fraction; S, soluble fraction. *p < 0.05; **p < 0.01.
Microtubules are cold-sensitive structures that rapidly depolymerize into tubulin monomers when placed on ice (Melki et al., 1989). Because the immunoprecipitations were performed on ice to prevent protein degradation, we decided to check whether the hyperactive HSPB1 mutants also bind to MTs. Tubulin from cellular extracts obtained from cells expressing different HSPB1 isoforms were polymerized and fractionated into soluble proteins and MTs by ultracentrifugation. Binding of HSPB1 to MTs was measured by calculating the ratio between the amount of HSPB1 protein in the MT-containing fraction and the total amount of HSPB1. As shown in Figure 1C, all HSPB1 isoforms bind to MTs with the hyperactive mutants displaying higher affinity to MTs compared to the HSPB1WT protein.
To visualize the binding of HSPB1 to MTs inside the cell, we transfected Cos-1 cells with constructs harboring V5-tagged HSPB1WT or HSPB1S135F together with a GFP-fused tubulin βIII construct (TUBB3-GFP). As shown in Figure 2A, HSPB1WT and HSPB1S135F showed a similar overall distribution with a merely cytoplasmic distribution. However, at higher magnification, it was clear that HSPB1S135F preferentially localized to MT-containing regions, while the HSPB1WT presented a more diffuse staining (Fig. 2B). Fluorescence intensity line plots and colocalization calculation with the Pearson's correlation coefficient confirmed these trends (Fig. 2C,D). Overall, the enhanced binding of hyperactive mutants to MTs did not cause any obvious change in the microtubule network (Fig. 2A).
Figure 2.
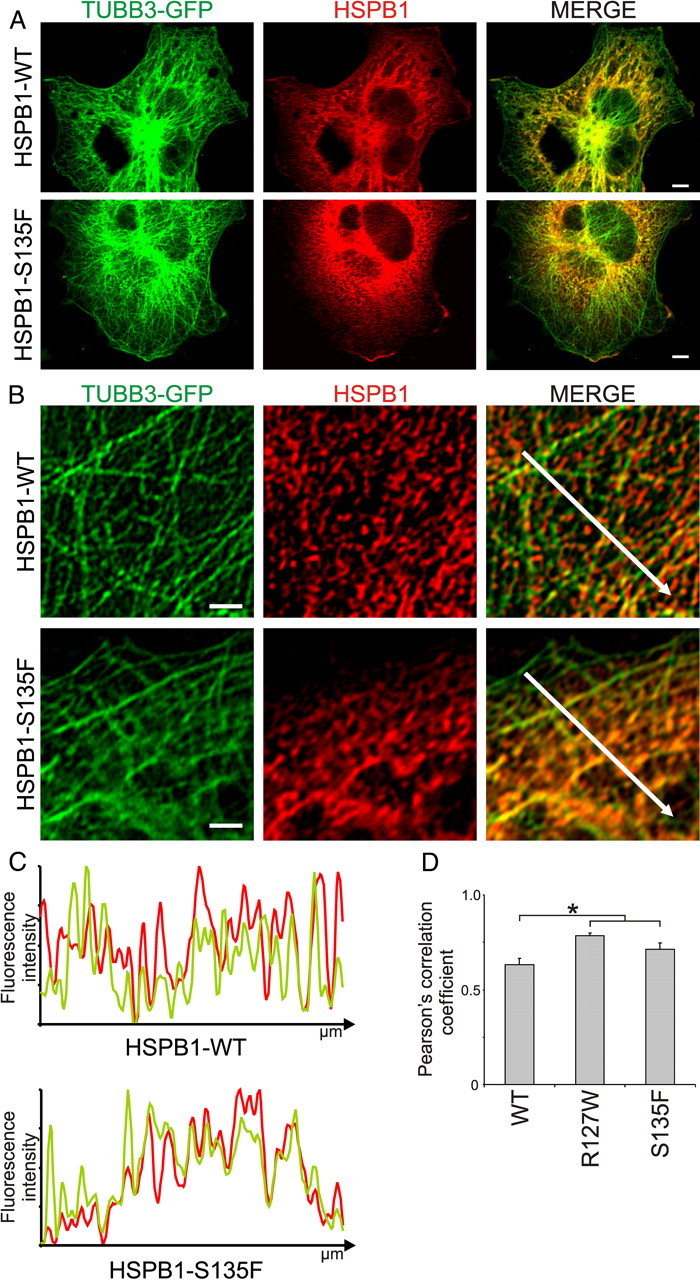
Colocalization of mutant HSPB1 and microtubules in cells. A, Overview image from Cos-1 cells transiently transfected with constructs harboring GFP-TUBB3 and V5-tagged HSPB1-WT or the HSPB1–S135F and stained for HSPB1. Scale bar, 5 μm. B, Representative pictures for the staining with anti-HSPB1 antibody showing preferential localization of the HSPB1–S135F mutant to MT regions (bottom) in contrast to the overall distribution of the HSPB1-WT (top). Scale bar, 2 μm. C, Normalized fluorescence intensity profiles for the white arrows traced in the merged pictures. The graphs for the HSPB1S135F mutant clearly show the correspondence between fluorescence peaks and valleys of HSPB1 and MT, while this correspondence is less clear for HSPB1WT. D, Pearson's correlation coefficient between HSPB1 staining and TUBB3-GFP was calculated for 50 independent areas from 10 different cells. Data are presented as mean ± SEM. *p < 0.05.
The microtubule cytoskeleton is a structure that is especially important for neurons, as evidenced by the fact that disturbances in MT dynamics are linked to several neurodegenerative diseases (Evans et al., 2005; Bunker et al., 2006; Gillardon, 2009; Tanabe and Takei, 2009; Cartelli et al., 2010; Tischfield et al., 2010). For this reason, we decided to explore the functional significance of this increased interaction further.
CMT neuropathy-causing HSPB1 mutants stabilize the microtubule network
Microtubules are dynamic structures that are regulated by a series of proteins (Baas et al., 2005). Among these regulators, MAPs from the MAP1 family stabilize MTs by binding along their length (Halpain and Dehmelt, 2006). To check whether the excessive binding properties of the hyperactive HSPB1 mutants might turn this protein into a MAP-like molecule and as such affect MT dynamics, we started by evaluating the effects of these mutations on depolymerization and repolymerization of the MT network. First, we performed a cold-induced MT depolymerization assay. Expression of hyperactive HSPB1 mutants rendered cells more resistant to cold-induced MT depolymerization, which was particularly evident at 12 min post-treatment (Fig. 3A,B). We next tested the repolymerization capacity of the MT network by treating cells with nocodazole and measuring the recovery of the MT network after nocodazole washout. In accordance with the previous result, expression of hyperactive HSPB1 mutants resulted in a faster recovery of the MT network when measured 30 min after nocodazole washout (Fig. 3C). In steady-state conditions, we never observed any obvious difference in fluorescence intensity between different genotypes (data not shown).
Figure 3.

Hyperactive HSPB1 mutants stabilize microtubules. A, B, Cold-induced MT depolymerization assay. HEK293Flp-in cells expressing different HSPB1 isoforms and EGFP as a negative control were placed on ice for different periods of time and separated into tubulin (T) and MT (M) fractions. The ratio of polymerized tubulin was calculated by dividing the tubulin signal intensity from M and T fractions. A, Graph showing the ratio of polymerized tubulin at different time points. B, Western blot showing tubulin and MT fractions at the zero and 12 min time points, and quantification of the ratio M/T (n = 3). C, MT network recovery experiment after nocodazole washout. HeLa cells stably expressing different HSPB1 isoforms were treated with 10 μm nocodazole for 6 h; allowed to recover for 0, 10, and 30 min; treated with detergent to remove soluble tubulin; and stained for tubulin. MT network recovery was measured as total fluorescence per cell (n = 58, 50, and 48 for WT; 53, 48, and 70 for R127W; and 53, 73, and 77 for S135F). Data points for HSPB1–R127W and HSPB1–S135F are statistically different from HSPB1-WT at 30 min (p < 0.01). D, Scratch-induced migration assay. HEK293Flp-in cells expressing different HSPB1 isoforms were grown until confluence and scratched with a pipette tip. Cells were imaged overnight, and migration was measured as the area migrated by cells every 2 h (n = 12, 14, 20, and 14 for WT, R127W, S135F, and T151I, respectively). Scale bar, 80 μm. Data points for HSPB1–R127W and HSPB1–S135F are statistically different from HSPB1-WT and HSPB1–T151I at 6, 8, 10, and 12 h (p < 0.01). Data are presented as mean ± SEM. E, Tubulin acetylation levels in HEK293Flp-in cells expressing different HSPB1 isoforms.
As an additional readout of MT network stability, we used a scratch-induced migration assay to assess the migration capacity of these cells. Cell migration is a process critically dependent on the reorganization of MTs and cell polarization (Vinogradova et al., 2009), and addition of MT stabilizing agents or knockdown of MT dynamics regulators has previously been shown to reduce cell migration (Grigoriev et al., 1999; Nakano et al., 2010). Confirming our previous results, a significant reduction in cell migration was detected only in cells expressing the hyperactive HSPB1 mutants (R127W and S135F), while cells expressing a CMT-causing HSPB1 mutation without enhanced tubulin binding (T151I) displayed a migration speed similar to cells expressing HSPB1WT (Fig. 3D).
Microtubule stabilization is commonly accompanied by the acetylation of a lysine residue (Lys40) in α-tubulin. Chemical inhibition of the tubulin deacetylase HDAC6 or treatment of cells with MT stabilizers was shown to increase tubulin acetylation (Matsuyama et al., 2002). Conversely, knockout of the tubulin acetyltransferase MEC-17 was shown to destabilize MT (Akella et al., 2010). Based on these data, we decided to check the tubulin acetylation levels of cells stably expressing different HSPB1 isoforms by Western blot. As shown in Figure 3E, the expression of the MT-stabilizing HSPB1 mutations did not change the acetylation pattern of tubulin. These results were confirmed in different cell types (data not shown). Together, our results show that hyperactive HSPB1 mutants are able to stabilize the MT network, however, without any increase in tubulin acetylation.
CMT neuropathy causing HSPB1 mutants disturb microtubule dynamics
To examine the effects of the hyperactive HSPB1 mutants on individual MTs, we used live cell imaging on HeLa cells stably expressing HSPB1 and transiently transfected with either a GFP-fused neuron-specific β-tubulin isoform (TUBB3-GFP) or GFP-fused end-binding protein 1 (EB1-GFP). The data for the MT dynamics extracted from the live cell movies are summarized in Table 1. In cells expressing the hyperactive HSPB1 mutants, MTs spent significantly less time in the shrinking or growth phase and more time in the stationary phase compared with the MTs from HSPB1WT-expressing cells. Accordingly, MTs from cells expressing hyperactive HSPB1 mutants also displayed lower catastrophe and rescue frequencies. No differences in growth speed of MTs were detected, which was confirmed by EB1 tracking (Table 2). These results are in accordance with what has been previously described for other MAPs (Moores et al., 2006; Faller and Brown, 2009) and suggest that HSPB1 mutants stabilize microtubules in a similar manner.
Table 1.
Hyperactive HSPB1 mutants disturb microtubule dynamics
| HSPB1WT (n = 86 MTs) | HSPB1R127W (n = 103 MTs) | HSPB1S135F (n = 104 MTs) | |
|---|---|---|---|
| Growth (%) | 0.165 ± 0.118 | 0.057 ± 0.067** | 0.067 ± 0.075** |
| Shrinkage (%) | 0.164 ± 0.143 | 0.059 ± 0.063)** | 0.068 ± 0.068** |
| Pause (%) | 0.671 ± 0.176 | 0.884 ± 0.097** | 0.865 ± 0.109** |
| Growth velocity (μm/s) | 0.257 ± 0.128 | 0.352 ± 0.210 ns | 0.377 ± 0.559 ns |
| Shrink velocity (μm/s) | 0.272 ± 0.120 | 0.349 ± 0.244** | 0.419 ± 0.326* |
| Catastrophe frequency (s−1) | 0.171 ± 0.104 | 0.091 ± 0.074** | 0.103 ± 0.106** |
| Rescue frequency (s−1) | 0.187 ± 0.098 | 0.087 ± 0.079** | 0.099 ± 0.089** |
TUBB3-GFP MTs were imaged at 3 s intervals in HeLa cells expressing HSPB1WT, HSPB1R127W, and HSPB1S135F. Individual TUBB3-GFP MTs were tracked manually from at least 10 different cells in seven independent movies. The MT life histories are represented as the percentage of time spent in growth, shrinkage, or pause. Catastrophe frequency is given as the frequency that MTs transitioned from growth (or pause) to shrinkage. Rescue frequency corresponds to the frequency at which MTs transitioned from shrinkage (or pause) to growth. Data are presented as average ± SD.
*p < 0.05;
**p < 0.01. ns, Nonsignificant.
Table 2.
Hyperactive HSPB1 mutants do not affect microtubule polymerization speed
| HSPB1WT (n = 21 cells) | HSPB1R127W (n = 23 cells) | HSPB1S135F (n = 33 cells) | |
|---|---|---|---|
| Comet velocity (μm/s) | 1.415 ± 0.145 | 1.424 ± 0.221 (ns) | 1.438 ± 0.214 (ns) |
| Distance traveled (μm) | 18.087 ± 2.154 | 17.061 ± 2.649 (ns) | 17.604 ± 2.629 (ns) |
The EB1-GFP comets were imaged at 2 s intervals from HeLa cells expressing HSPB1WT, HSPB1R127W, and HSPB1S135F. The EB1-GFP comets were tracked automatically with the ImageJ Particle Tracker plugin from at least 10 independent movies. Data is presented as average ± SD. ns, Nonsignificant.
Interestingly, MTs from cells expressing the hyperactive HSPB1 mutants showed faster depolymerization speeds compared with HSPB1WT-expressing cells. A closer analysis of the tracking data revealed that a significant fraction of the MTs from cells expressing the hyperactive HSPB1 mutants displayed very high depolymerization speeds when compared with cells expressing HSPB1WT (we defined “very high” as a depolymerization speed higher than the fastest depolymerization speed measured in the HSPB1WT-expressing cells (0.56 μm/s; HSPB1R127W = 12 of 82 MTs analyzed; HSPB1S135F = 21 of 84 MTs analyzed) (Fig. 4A,B). These observations suggest that the hyperactive HSPB1 mutants do not stabilize MTs permanently, but rather stabilize them in an aberrant and transient way by continuously counteracting depolymerization up to a certain threshold, at which point the depolymerizing forces are greater than the stabilization capacity of mutant HSPB1, causing the MTs to depolymerize very rapidly. We believe that this also explains why the increased stabilization is not reflected by an increased acetylation.
Figure 4.

Mutant HSPB1 stabilized microtubules depolymerize less often, but once they do, at higher speeds. A, Time-lapse microscopy from cells expressing HSPB1WT or HSPB1S135F and TUBB3-GFP. Red dots represent current position of MT tips, while yellow dots represent the position of the MT tips in the previous frame. MTs in cells expressing HSPB1S135F are more paused (shown here as the yellow and red dots overlapping on the same position), but once they depolymerize, they show a faster depolymerization speed than MT from cells expressing HSPB1WT. Scale bar, 2 μm. B, Graphical representation of life stories of MTs from cells expressing HSPB1WT and mutants. Note that, in general, MTs in mutant HSPB1-expressing cells move less (are more often on the horizontal axis representing the paused condition) and that, especially for the depolymerization phase, peaks—representing speed—are higher for mutants. Graph shows the concatenated life story of seven MTs for each genotype.
CMT causing HSPB1 mutants are able to stabilize microtubules in vitro by direct interaction
The data presented above show that increased binding of HSPB1 mutants stabilizes the MT network in cells. To test whether this effect is directly caused by HSPB1 binding or is mediated by other cell signals, we performed an in vitro MT polymerization assay using recombinant HSPB1WT or HSPB1S135F protein. In accordance with a stabilization effect by direct interaction, the presence of HSPB1S135F mutant in the polymerization reaction strongly facilitated tubulin polymerization when compared with the reactions containing tubulin alone or tubulin with HSPB1WT (Fig. 5A).
Figure 5.
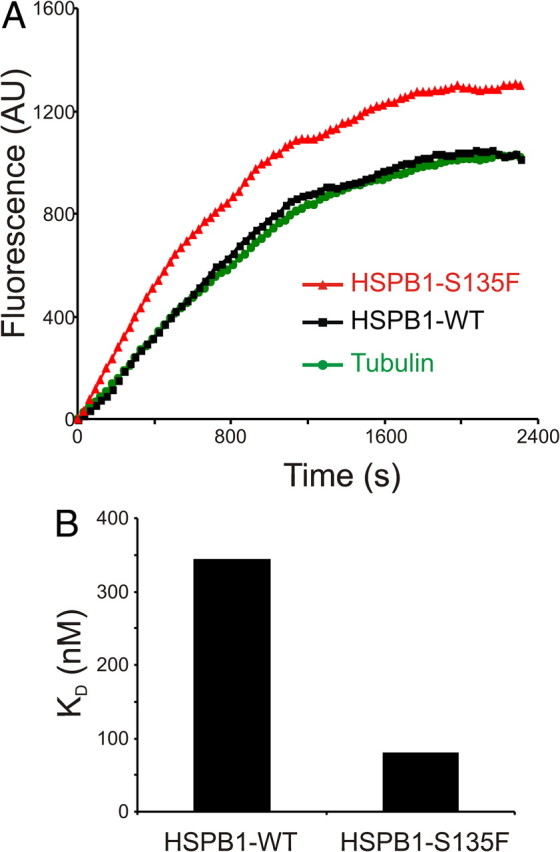
Hyperactive HSPB1 mutants stabilize microtubules in vitro. A, MT in vitro polymerization assay. Ten micromolar tubulin was polymerized alone or in the presence of 5 μm recombinant HSPB1WT or HSPB1S135F mutant. MT polymerization was measured as increasing DAPI fluorescence over time (see Experimental procedure section for details). B, Dissociation constants (KD) of HSPB1WT and HSPB1S135F to tubulin using surface plasmon resonance. Sensorgram data were fitted using a steady-state binding model, and saturation titration experiments were for the calculation of KD. AU, Arbitrary units.
To further investigate the differences in tubulin binding between HSPB1WT and HSPB1S135F, we measured the binding affinities of these proteins (KD) for tubulin by SPR. In line with our expectations, HSPB1S135F showed a threefold higher affinity for tubulin than HSPB1WT (KD: HSPB1S135F, 79 nm; HSPB1WT, 343 nm) (Fig. 5B). The stabilization effect by direct binding shown by the hyperactive HSPB1 mutants further strengthens the hypothesis that these mutants operate in a MAP-like manner.
Mutant HSPB1 stabilizes microtubules in peripheral neurons from HSPB1S135F transgenic mice
To check whether the MT-stabilizing effects seen on cells are relevant in vivo, we used a CMT2F mouse model that selectively expresses HSPB1S135F in postnatal neurons. These mice present a phenotype that recapitulates the symptoms seen in patients and have been described in detail previously (d'Ydewalle et al., 2011). We first investigated the differential interaction with tubulin by immunoprecipitating HSPB1 from sciatic nerves from 3-month-old (presymptomatic) mice expressing either HSPB1WT or HSPB1S135F isoforms. Corroborating our data using cell lines, HSPB1S135F was found to interact more strongly with tubulin than does HSPB1WT (Fig. 6A). Furthermore, the effect of this enhanced interaction on MT stability was confirmed in cultured primary DRG neurons by measuring the extent of the neuritic MT network during and after MT depolymerization treatment using nocodazole. In contrast to neurons isolated from HSPB1WT mice that suffered a drastic reduction in neuritic MT extent during the depolymerization treatment, neurons isolated from the HSPB1S135F mice displayed only a limited reduction in their neuritic MT extent (Fig. 6B,C). In the repolymerization phase, the MT network from HSPB1WT mice neurons showed a fast recovery to the initial length, while the MT network from HSPB1S135F mice neurons showed little recovery. These results confirm that also in peripheral neurons, HSPB1S135F leads to enhanced stability of the MT network, which might underlie disease formation in HSPB1S135F-expressing mice and possibly also in patients.
Figure 6.

The microtubule-stabilizing property of mutant HSPB1 is confirmed in HSPB1S135F transgenic mice. A, Immunoprecipitation of HSPB1 from sciatic nerves of mice expressing HA-tagged HSPB1WT, HSPB1S135F, and nontransgenic (Ntg) mice confirmed the differential interaction of HSPB1S135F to tubulin in vivo. B, MT stability measurement on DRG neurons from transgenic mice. MT stability was evaluated as the extent of the MT network within the DRG neurites from transgenic mice during (N) and after (R) nocodazole treatment (n = 40 neurites per time point). Data points for HSPB1S135F are statistically different from HSPB1WT at all time points, except for those at the start of the treatment (p < 0.01). Neuritic MT network extent was normalized to the initial size before treatment (N0). Data are presented as mean ± SEM. C, Representative pictures of DRG neurons from transgenic mice used at measurements shown in B. Neurons are stained for α-tubulin. Scale bar, 200 μm.
Discussion
The small heat shock protein HSPB1 is a multifunctional molecular chaperone that participates in a plethora of cellular processes (Huot et al., 1996; Charette et al., 2000; Cuesta et al., 2000; Paul et al., 2002). Despite its multifunctionality and ubiquitous expression, we found that mutations in HSPB1 affect specifically the peripheral nervous system by causing CMT2F and distal HMN (Evgrafov et al., 2004). In later work, we discovered that a subset of HSPB1 mutations are hyperactive and display increased affinity to their client proteins (Almeida-Souza et al., 2010). Here, we show that an enhanced binding of the CMT-causing HSPB1 mutants to tubulin and microtubules causes the molecule to function in a MAP-like manner, leading to a reduction in microtubule dynamics. This MT stabilization effect was shown in cells in vitro and, more importantly, in DRG neurons isolated from a CMT2F mouse model.
Disturbances in MT dynamics are a well established cause for neurodegeneration. Similar to our results, mutations in the GTPase dynamin 2 causing dominant intermediate CMT disease, were shown to decorate MTs and lead to MT stabilization (Tanabe and Takei, 2009). Recently, mutations in the neuron-specific tubulin βIII gene (TUBB3) were associated with type 3 congenital fibrosis of the extraocular muscles and led equally to MT stabilization. Interestingly, some of these patients presented with a peripheral neuropathy and were initially diagnosed as CMT patients (Tischfield et al., 2010). Mutations in the MT-severing protein spastin (SPG4) constitute the most common cause of hereditary spastic paraplegia (Depienne et al., 2007). Disease causing mutations in spastin were described to disturb its MT severing capacity by impairment of its ATPase activity (Evans et al., 2005). Microtubule dynamics defects were also linked to neurodegeneration in Alzheimer's disease (Bunker et al., 2006) and Parkinson's disease (Gillardon, 2009; Cartelli et al., 2010).
In addition to genetic mutations leading to disturbances in MT dynamics, acquired peripheral neuropathies also are known to be linked to a misregulation of the MT network. Chemotherapy-induced peripheral neuropathy is a very common and a major dose-limiting side effect of many chemotherapeutic drugs for cancer treatment (Lee and Swain, 2006; Wolf et al., 2008). Among the commonly used chemotherapeutic drugs, vinca alkaloids and taxol derivatives are, respectively, potent MT-depolymerizing and MT-stabilizing agents (Jordan et al., 1985; Yvon et al., 1999), and are believed to damage peripheral neurons by disturbing MT dynamics in axons (Meininger et al., 1990; Swain and Arezzo, 2008; Yang et al., 2009). This shows that a tight balance of MT dynamics seems especially important in the large axons of peripheral neurons.
In contrast to what is commonly described in the literature (Matsuyama et al., 2002; Tanabe and Takei, 2009), the MT stabilization caused by hyperactive HSPB1 mutants was shown to occur independently of any increase in MT acetylation. We believe that this difference can be explained by the following two facts: (1) the MT stabilization is not static, as shown by the occurrence of high-speed depolymerization events after long pause periods in mutant-expressing cells (Fig. 4); and (2) the MT stabilization effects caused by the mutants are not very pronounced (Fig. 3) and, as such, are in accordance with the mild, late-onset phenotype of this type of CMT disease.
Disturbances in MT dynamics are not by themselves the cause of neurodegeneration. Actually, these disturbances impair efficient transport of proteins, vesicles, and organelles along the cell. The highly polarized morphology of neurons makes them especially vulnerable to transport problems (Perlson et al., 2010). Looking from a broader perspective and taking axonal transport defects as a cause of length-dependent neurodegeneration, we can place MT dynamics-related disorders into a much larger group of diseases that encompass mutations in other players involved in intracellular transport, such as molecular motors (Puls et al., 2003) and vesicle recruitment factors (Verhoeven et al., 2003). The assignment of some HSPB1 mutations into the large group of neurodegenerative diseases caused by axonal transport defects represents an important step toward the development of therapies, as drugs designed to improve axonal transport may have beneficial effects for several diseases.
In a mouse model for CMT2F, we could confirm the enhanced interaction of mutant HSPB1 to tubulin and the concomitant increased stability of the microtubule network. This transgenic mouse line expresses HSPB1S135F postnatally in neurons and recapitulates all features of CMT2F (d'Ydewalle et al., 2011). Presymptomatically, at 3 months of age, the microtubule network in peripheral neurons isolated from these mice showed an enhanced stability, as reflected by an increased resistance to nocodazole-induced depolymerization. Interestingly, d'Ydewalle et al. (2011) observed a clear reduction in acetylation of microtubules, which is indicative of a lower stability of the microtubule network. In addition, they found that treatment with an HDAC6 inhibitor could rescue the disease phenotype. However, these experiments were performed after disease onset (8 months of age) when axonal loss became clearly apparent. The timing of these observations therefore suggests that the CMT2F symptoms might be triggered by the transition from an overstable MT network caused by the enhanced binding of mutant HSPB1 to tubulin to a destabilized MT network mediated by HDAC6 recruitment to microtubules.
At present, mutations in 10 different HSPB1 residues are known to cause CMT neuropathy (Houlden et al., 2008; Ikeda et al., 2009). We tested the five different mutations originally described by Evgrafov et al. (2004) and found that three of these mutations displayed an MT stabilization effect. Interestingly, two of the other mutated residues (R140 and K141), which we have not tested, are also present in the HSPB1 dimer interface and potentially have the same properties as our hyperactive mutants. On the other hand, the mutations (T151I and P182L) did not show any differential binding to tubulin or MTs. Therefore, a complete understanding of all cellular processes explaining the neuron-specific pathogenesis for all HSPB1 mutations remains elusive.
In conclusion, our work shows that hyperactive CMT causing HSPB1 mutations bind excessively to MTs, which leads to their stabilization. Based on our mouse data, we believe that this could be a potential pathomechanism for CMT2F, further underscoring the pivotal role of a fine-tuned regulation of the microtubule cytoskeleton in neuronal homeostasis.
Footnotes
This work was supported in part by the Methusalem program of the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), the Medical Foundation Queen Elisabeth, the “Association Belge contre les Maladies Neuromusculaires,” the American Muscular Dystrophy Association, the Hercules Foundation (providing funding for live cell imaging equipment), and the Interuniversity Attraction Poles P6/43 program of the Belgian Federal Science Policy Office. C.d'Y. and K.M. are supported by a PhD Fellowship of the Institute for Science and Technology and FWO-Flanders, respectively. S.J. and J.I. are postdoctoral researchers supported by the FWO-Flanders. H.R. is supported by a VIB PRJ Grant and the Odysseus program of the FWO-Flanders. We are grateful to A. Jacobs for technical assistance and E. Timmerman for the LC-MS/MS analyses. We also thank the VIB Protein Service Facility for recombinant protein production and the VIB Genetic Service Facility for sequencing support.
References
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an alpha-tubulin acetyltransferase. Nature. 2010;467:218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida-Souza L, Goethals S, de Winter V, Dierick I, Gallardo R, Van Durme J, Irobi J, Gettemans J, Rousseau F, Schymkowitz J, Timmerman V, Janssens S. Increased monomerization of mutant HSPB1 leads to protein hyperactivity in Charcot-Marie-Tooth neuropathy. J Biol Chem. 2010;285:12778–12786. doi: 10.1074/jbc.M109.082644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Karabay A, Qiang L. Microtubules cut and run. Trends Cell Biol. 2005;15:518–524. doi: 10.1016/j.tcb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Barisic N, Claeys KG, Sirotković-Skerlev M, Löfgren A, Nelis E, De Jonghe P, Timmerman V. Charcot-Marie-Tooth disease: a clinico-genetic confrontation. Ann Hum Genet. 2008;72:416–441. doi: 10.1111/j.1469-1809.2007.00412.x. [DOI] [PubMed] [Google Scholar]
- Barron DM, Chatterjee SK, Ravindra R, Roof R, Baloglu E, Kingston DG, Bane S. A fluorescence-based high-throughput assay for antimicrotubule drugs. Anal Biochem. 2003;315:49–56. doi: 10.1016/s0003-2697(02)00691-7. [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Cabral F. A ubiquitous beta-tubulin disrupts microtubule assembly and inhibits cell proliferation. Mol Biol Cell. 2004;15:3123–3131. doi: 10.1091/mbc.E04-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem. 2006;281:11856–11863. doi: 10.1074/jbc.M509420200. [DOI] [PubMed] [Google Scholar]
- Cartelli D, Ronchi C, Maggioni MG, Rodighiero S, Giavini E, Cappelletti G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+-induced neurodegeneration. J Neurochem. 2010;115:247–258. doi: 10.1111/j.1471-4159.2010.06924.x. [DOI] [PubMed] [Google Scholar]
- Charette SJ, Lavoie JN, Lambert H, Landry J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol. 2000;20:7602–7612. doi: 10.1128/mcb.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta R, Laroia G, Schneider RJ. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000;14:1460–1470. [PMC free article] [PubMed] [Google Scholar]
- Depienne C, Stevanin G, Brice A, Durr A. Hereditary spastic paraplegias: an update. Curr Opin Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17:968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, Dadali E, Auer-Grumbach M, Windpassinger C, Wagner K, Mitrovic Z, Hilton-Jones D, Talbot K, Martin JJ, Vasserman N, Tverskaya S, Polyakov A, Liem RK, Gettemans J, Robberecht W, De Jonghe P, Timmerman V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- Faller EM, Brown DL. Modulation of microtubule dynamics by the microtubule-associated protein 1a. J Neurosci Res. 2009;87:1080–1089. doi: 10.1002/jnr.21920. [DOI] [PubMed] [Google Scholar]
- Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—a point of convergence in parkinsonian neurodegeneration? J Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- Grigoriev IS, Chernobelskaya AA, Vorobjev IA. Nocodazole, vinblastine and taxol at low concentrations affect fibroblast locomotion and saltatory movements of organelles. Membr Cell Biol. 1999;13:23–48. [PubMed] [Google Scholar]
- Halpain S, Dehmelt L. The MAPI family of microtubule-associated proteins. Genome Biol. 2006;7:224. doi: 10.1186/gb-2006-7-6-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- Houlden H, Laura M, Wavrant-De Vrièze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56:273–279. [PubMed] [Google Scholar]
- Ikeda Y, Abe A, Ishida C, Takahashi K, Hayasaka K, Yamada M. A clinical phenotype of distal hereditary motor neuronopathy type II with a novel HSPB1 mutation. J Neurol Sci. 2009;277:9–12. doi: 10.1016/j.jns.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Himes RH, Wilson L. Comparison of the effects of vinblastine, vincristine, vindesine, and vinepidine on microtubule dynamics and cell proliferation in vitro. Cancer Res. 1985;45:2741–2747. [PubMed] [Google Scholar]
- Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol. 2006;24:1633–1642. doi: 10.1200/JCO.2005.04.0543. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meininger V, Binet S, Chaineau E, Fellous A. In situ response to vinka alkaloids by microtubules in cultured post-implanted mouse embryos. Biol Cell. 1990;68:21–29. doi: 10.1111/j.1768-322x.1990.tb00889.x. [DOI] [PubMed] [Google Scholar]
- Melki R, Carlier MF, Pantaloni D, Timasheff SN. Cold depolymerization of microtubules to double rings: geometric stabilization of assemblies. Biochemistry. 1989;28:9143–9152. doi: 10.1021/bi00449a028. [DOI] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Kappeler C, Kain S, Drummond D, Perkins SJ, Chelly J, Cross R, Houdusse A, Francis F. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006;25:4448–4457. doi: 10.1038/sj.emboj.7601335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y, Seguchi O, Higo S, Shintani Y, Asanuma H, Asakura M, Minamino T, Kaibuchi K, Mochizuki N, Kitakaze M, Takashima S. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol. 2010;12:583–590. doi: 10.1038/ncb2060. [DOI] [PubMed] [Google Scholar]
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol. 2002;22:816–834. doi: 10.1128/MCB.22.3.816-834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL. Retrograde axonal transport: pathways to cell death? Trends Neurosci. 2010;33:335–344. doi: 10.1016/j.tins.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Salmon P, Trono D. Production and titration of lentiviral vectors. Curr Protoc Neurosci Chapter. 2006;4 doi: 10.1002/0471142301.ns0421s37. Unit 4.21. [DOI] [PubMed] [Google Scholar]
- Sun Y, MacRae TH. The small heat shock proteins and their role in human disease. Febs J. 2005;272:2613–2627. doi: 10.1111/j.1742-4658.2005.04708.x. [DOI] [PubMed] [Google Scholar]
- Swain SM, Arezzo JC. Neuropathy associated with microtubule inhibitors: diagnosis, incidence, and management. Clin Adv Hematol Oncol. 2008;6:455–467. [PubMed] [Google Scholar]
- Tanabe K, Takei K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J Cell Biol. 2009;185:939–948. doi: 10.1083/jcb.200803153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A, Carstens RP. An optimized protocol for protein purification in cultured mammalian cells using a tandem affinity purification approach. Nat Protoc. 2006;1:2820–2827. doi: 10.1038/nprot.2006.371. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, Van Gerwen V, Wagner K, Hartung HP, Timmerman V. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova T, Miller PM, Kaverina I. Microtubule network asymmetry in motile cells role of Golgi-derived array. Cell Cycle. 2009;8:2168–2174. doi: 10.4161/cc.8.14.9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer. 2008;44:1507–1515. doi: 10.1016/j.ejca.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Yang IH, Siddique R, Hosmane S, Thakor N, Höke A. Compartmentalized microfluidic culture platform to study mechanism of paclitaxel-induced axonal degeneration. Exp Neurol. 2009;218:124–128. doi: 10.1016/j.expneurol.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]