

Abstract
Small organophosphorus compounds stimulate Vγ9Vδ2 T cells if they serve as ligands of butyrophilin 3A1. Because the most potent natural ligand is (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP), which is the last intermediate in bacterial biosynthesis of isoprenoids that is not found in mammalian metabolism, activation of these T cells represents an important component of the immune response to bacterial infections. To identify butyrophilin ligands that may have greater plasma stability, and clinical potential, we have prepared a set of aryl phosphonamidate derivatives (9a-i) of the natural ligand. Testing of these new compounds in assays of T cell response has revealed that this strategy can provide compounds with high potency for expansion of Vγ9Vδ2 T cells (9f, EC50 = 340 pM) and interferon γ production in response to loaded K562 cells (9e, EC50 = 62 nM). Importantly, all compounds of this class display extended plasma stability (t1/2 > 24 h). These findings increase our understanding of metabolism of butyrophilin ligands and the structure-activity relationships of phosphonamidate prodrugs.
Keywords: phosphoantigen, phosphonamidate prodrug, butyrophilin, ligand, isoprenoid
Graphical Abstract
Introduction
After binding to the transmembrane protein butyrophilin 3A1 (BTN3A1), phosphoantigens such as the naturally occurring (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP, 1, Figure 1) are detected by Vγ9Vδ2 T cells through mechanisms that are not yet completely understood.1–6 From the standpoint of potential clinical application as anti-cancer or anti-infective agents, diphosphates such as HMBPP unfortunately suffer from less than desirable pharmacokinetic properties, which include susceptibility to phosphatase-mediated degradation that results in clearance from the plasma.7,8 Furthermore, the pKa values of HMBPP have been calculated to be approximately 1.8, 3.2, and 7.4, and so by virtue of the charged oxygen atoms at physiological pH it presents a high charge-to-mass ratio and low membrane permeability.9,10 To design an HMBPP analog with enhanced pharmacokinetic properties relative to the natural ligand 1, structural modifications which included the replacement of an O-P bond (a phosphate, i.e. HMBP, 1b) with a C-P bond (phosphonate, i.e. C-HMBP) intended to increase metabolic stability led to compound 2. Given that small highly-charged molecules generally have been associated with low membrane permeability,11 the most acidic positions of the free phosphonic acid form of compound 2, with calculated pKa values of 1.8 and 8.3,10 were masked with pivaloyloxymethyl (POM) protecting groups to increase the membrane permeability. Evaluation of the phosphoantigen prodrug 2 revealed stimulation of Vγ9Vδ2 T cell expansion with a half-maximal effective concentration (EC50) following 72 hours exposure of 5.4 nM, or within approximately 10–fold activity of HMBPP (EC50 = 0.51 nM under the same assay conditions). 9 Supporting our hypothesis, at shorter exposure times prodrug potency exceeds that of HMBPP, as following 2 hour exposure prodrug 2 stimulates T cell killing of cancer cells with an EC50 of 1.2 nM versus 19 nM for HMBPP.10,12 It is still unclear whether the phosphonate ligand undergoes phosphorylation within the cell to generate the active agent, but isothermal titration calorimetry (ITC) studies have shown that the salt of compound 2 does bind to the B30.2 domain of BTN3A1.9
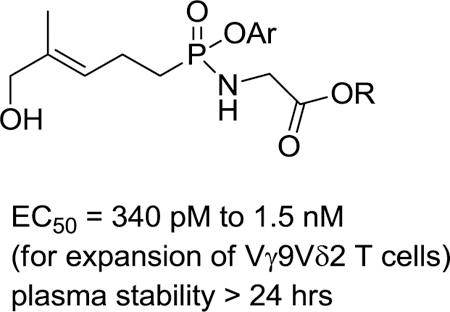
Figure 1.

A natural phosphoantigen (1) and some phosphonate prodrug analogs (2 and 3).
BTN3A1 is required for activation of Vγ9Vδ2 T cells by phosphoantigens,13,14 and several studies have shown that HMBPP and its biologically active analogs bind to the intracellular domain of BTN3A1.9,15,16 Relative to HMBPP, the structural modifications of phosphonate 2 provide a minimized charge-to-mass ratio that increases its effectiveness when extracellularly dosed, enabling the butyrophilin ligand to reach its intracellular target with exceptional potency in cell culture models. Use of the pivaloyloxymethyl (POM) ester increased the potency of this phosphonate ligand by 740-fold relative to the corresponding disodium salt, consistent with facile entry across the cell membrane and subsequent release of the parent ligand. In contrast, while the more metabolically stable dimethyl phosphonate ester presumably enters the cell just as readily, it is inactive at concentrations up to 10 μM, indicating that both crossing the cell membrane and hydrolysis to a charged species are necessary to stimulate proliferation. However, while POM2-C-HMBP does this, the plasma half-lives of bis-POM prodrugs are limited17 due to inherent susceptibility towards cleavage by non-specific esterases that are present in the blood.18 In addition, there has been concern about the impact of pivalic acid, a result of POM hydrolysis, on carnitine metabolism in mammals.18,19 These observations provided a basis for the rational exploration of alternative prodrug forms.
The pronucleotide approach pioneered by McGuigan and colleagues, initially was applied to nucleoside phosphates and then further developed for application to nucleoside phosphonates.20 When applied to phosphonates the key features of this strategy include a phosphonamide derived from a nitrogen-linked amino acid ester and a phosphonate ester derived from an aryl alcohol. The putative mechanism of biological deprotection of this general structure involves initial cleavage of the amino acid ester, catalyzed by carboxypeptidase Y (cathepsin A) or carboxylesterase I,21 to generate the corresponding carboxylate. Spontaneous cyclization then releases an equivalent of an aryl alcohol to afford the corresponding cyclic intermediate, which may be hydrolyzed subsequently to reveal a monoamidate intermediate.22 Cellular phosphoramidases such as Hint 123,24 may mediate the final hydrolysis25 to afford an equivalent of an amino acid along with the free drug. An advantage offered by the pronucleotide approach was demonstrated by Eisenberg et al. who masked the acyclic nucleoside phosphonate 9-(2-phosphonyl-methoxypropyl)adenine (PMPA), and determined that the monoalaninyl monophenoxy protected drug was stable in both whole blood and red blood cells.26 In vitro metabolic studies then demonstrated a greater half-life in human plasma (t1/2 = 90 min) than its commercially available counterpart tenofovir disoproxil fumarate.27 Assuming a comparable deprotection sequence for phosphoantigen phosphonamidates, we hypothesized that the preparation of compound 3, an aryloxy phosphonamidate of a BTN3A1 ligand, would function as a prodrug with increased plasma stability relative to its acyloxyalkyl ester counterpart.28
One potential drawback to any aryloxy phosphonamidate prodrug is that it will necessarily incorporate a phosphorus stereocenter. Even though each prodrug stereoisomer would release the same compound upon hydrolysis, the rate of release could differ as a function of stereochemistry at the phosphorus or at the α-carbon of the amino acid. While l-alanine is the amino acid most commonly employed in phosphonamidates, its use affords a mixture of diastereomers. Therefore our initial efforts were focused on glycine derivatives because this strategy would provide racemic mixtures rather than mixtures of diastereomers and minimize stereochemical complications. To evaluate the impact of the aryl moiety on our system, we chose to prepare the phenyl as well as both the 1– and 2–naphthyl derivatives. Here, we report the synthesis of a set of aryloxy phosphonamidates of a BTN3A1 ligand. Once these compounds were in hand, they allowed an experimental evaluation of the hypothesis that such compounds would function as prodrugs and demonstrate both increased cellular potency and enhanced plasma stability relative to bis(acyloxyalkyl) esters.
Results
Synthesis of phosphonamidate prodrugs of a butyrophilin ligand
Synthesis of the new phosphonamidates (Scheme 1) began with the known dimethyl ester 4.12 After treatment of compound 4 with oxalyl chloride to generate the intermediate phosphonic acid chloride 5, reaction with phenol, 1-naphthol, or 2-naphthol gave the corresponding mixed methyl aryl esters 6a,12 6b,12 and 6c, respectively. Upon exposure to NaI in acetonitrile, these mixed esters were cleaved selectively to afford the three sodium salts 7a–7c. Each of these three salts was then coupled with three glycine derivatives, the methyl, ethyl, and isopropyl glycine esters, under standard conditions to obtain the nine phosphonamidates 8a–8i. Finally, each of these phosphonamidates was treated with a catalytic amount of selenium dioxide and t-butyl hydroperoxide in pyridine/methanol to introduce the necessary E allylic hydroxyl group as a single olefin isomer.29 While this oxidation generally gave low yields, reserving it for the final step avoided the need for protection of the hydroxyl group during manipulations of the phosphorus substituents. Protection and deprotection of the allylic hydroxyl group has proven to be problematic in our earlier studies of bisacyloxy phosphonates9 as well as for others who have studied phosphoramidates of HMBPP.30 Furthermore, the selenium dioxide oxidation gives only the E-isomer, if the reaction is run to partial completion then separation of the product from unreacted starting material is straightforward, and by this strategy sufficient material was obtained in each case (9a–9i) to allow for the desired biological assays.
Scheme 1.

Synthesis of aryloxy phosphonamidate derivatives of a BTN3A1 ligand. Reagents and conditions: (a) (COCl)2, DMF (5 mol %), CH2Cl2, 0 °C to rt, overnight; (b) ArOH, Et3N, THF or toluene, 0 °C to rt; (c) NaI, H3CCN, reflux, overnight; (d) GlyOR·HCl, PPh3, 2,2’-dithiodipyridine, pyridine, 60 °C, overnight; (e) SeO2, 70% aqueous t-BuOOH, pyridine, methanol, 0 °C to rt, overnight.
Phosphonamidate prodrugs demonstrate enhanced plasma stability relative to acyloxyalkyl prodrugs
One critical limitation of the use of butyrophilin ligands for in vivo applications is the inherent instability of diphosphates in biological matrices,4 and the phosphoantigens HMBPP and BrHPP require continuous dosing or intramuscular injections to achieve biologically relevant concentrations in vivo.7,31 To assess stability of the prodrug forms, we employed an LC-MS based approach to examine the novel compounds 9a-i versus existing bis-acyloxyalkyl control compounds (e.g. 2). Test compounds were incubated in 50% pooled human plasma in PBS for various times, after which they were extracted into acetonitrile and the fraction of remaining compound was quantified by LC-MS peak integration following separation on a C18 column. We found that POM2-C-HMBP undergoes rapid plasma metabolism (Figure 2a), while the phosphonamidates were quite resistant to plasma metabolism (Figure 2b). The half-life of POM2-C-HMBP was calculated to be 8.4 minutes under these circumstances (Table 1). In stark contrast, excellent gains in plasma stability were observed in all of the tested phosphonamidates, which were so highly stable that it was not possible to estimate a half-life. All compounds had at least 97% remaining after 2 hours and in most cases over 90% remaining after 24 hours. In all three series, the methyl esters were the least stable. Taken together, the glycine phosphonamidate prodrugs are highly stable in human plasma and not susceptible to plasma esterase mediated deprotection.
Figure 2.

Plasma stability of phosphonamidate prodrugs. A) POM2-C-HMBP was exposed to 50% pooled human plasma in PBS for indicated time points. The graph indicates the mean fraction remaining and error bars represent standard deviations. Each data point was evaluated 2–4 independent times. B) The stability of the indicated phosphonamidates at 2 or 24 hour time points. Each data point was evaluated 2 independent times.
Table 1.
Stability of compounds in 50% pooled human plasma in PBS.
| Compound | 2 hour fraction remaining (SD) | 24 hour fraction remaining (SD) | t1/2 |
|---|---|---|---|
| POM2-C-HMBP (2) | 0.048 (0.034) | N.D. | 0.14 h |
| 9a | 0.97 (0.055) | 0.90 (0.11) | >24 h |
| 9b | 1.0 (0.042) | 0.96 (0.12) | >24 h |
| 9c | 0.99 (0.029) | 0.92 (0.050) | >24 h |
| 9d | 0.97 (0.035) | 0.76 (0.029) | >24 h |
| 9e | 1.0 (0.0071) | 0.89 (0.014) | >24 h |
| 9f | 1.0 (0.0071) | 0.95 (0.0071) | >24 h |
| 9g | 1.0 (0.0071) | 0.82 (0.0071) | >24 h |
| 9h | 1.0 (0.021) | 0.92 (0.035) | >24 h |
| 9i | 1.0 (0.085) | 0.95 (0.12) | >24 h |
Phosphonamidate prodrugs stimulate expansion of Vγ9Vδ2 T cells
In order to determine whether the increased stability of the compounds impacted their cellular activity, the novel compounds were evaluated for their ability to stimulate proliferation of human Vγ9Vδ2 T cells (Figure 3). Initial tests with the phenyl/GlyOiPr analog 9c were promising, and encouraged us to further characterize the activity of compounds 9a-i, which as a class demonstrated excellent cellular activity in initial screens in the proliferation assay (Figure 3a). Dose response experiments were then performed with each of the compounds (Figure 3b). The resulting data analysis including EC50 values is listed in Table 2. Of the nine compounds tested, all exhibited low nanomolar to mid picomolar potency for stimulating proliferation of the Vγ9Vδ2 T cells, ranging from an EC50 of 1.5 nM for the phenyl/GlyOMe analog 9a to an EC50 of 340 pM for the 1-naphthyl/GlyOiPr 9f. We had previously reported an EC50 of 510 pM for HMBPP in this assay. In the current study, four compounds (9b, 9d, 9e, and 9f) displayed greater efficacy, while the other compounds were active in a similar range. The test compounds generally showed maximal efficacy at concentrations of 100 nM. Some loss of efficacy at concentrations equal to or exceeding 10 µM was observed, which is typical of T cells exposed to high antigen concentrations and may result from an immune tolerance mechanism,32 although mild cytotoxicity of the test compound cannot be excluded in this case. When grouped together, the potency of the naphthyl compounds was greater than that of the phenyl compounds, with a slight potency preference to the 1-naphthyl over the 2-naphthyl substituent. Likewise, the compounds containing a glycine ethyl ester were more potent on average than the compounds containing either a methyl or isopropyl ester. Taken together, these phosphonamidate prodrugs potently stimulate proliferation of human Vγ9Vδ2 T cells with activity exceeding that of the bis-POM esters in all cases.
Figure 3.

Expansion of Vγ9Vδ2 T cells from PBMCs by phosphonamidate prodrugs. A) Following 3 days of compound exposure and 11 days of proliferation, the number of Vγ9Vδ2 T cells was assessed. Data are representative of 3 independent experiments using a concentration of 100 nM of each positive control and each test compound. B) Compounds were assessed for activity in dose response experiments, in comparison to non-stimulated cells (NS) and the positive controls HMBPP (HM) and POM2-C-HMBP (POM2) at 100 nM. Data shown is from three independent experiments each using a minimum of two different human donors.
Table 2.
Activity of test compounds for expansion of Vγ9Vδ2 T cells from peripheral blood mononuclear cells following 72 hour compound exposure.
| Compound | cLogPa | EC50 [µM] (95% CI) | Fold difference vs cmpd 10 | Fold difference vs cmpd 11 |
|---|---|---|---|---|
| 109 | -0.24 | 4.0 | NAa | NA |
| 2 (POM2-C-HMBP9 | 3.42 | 0.0054 | 740 | NA |
| 119 | 0.31 | >10 | NA | NA |
| 1212 | 3.56 | 0.014 | 290 | NDa |
| 1312 | 4.72 | 0.00079 | 5100 | 6.8 |
| 9a | 1.67 | 0.0015 (0.00038 to 0.0057) | 2700 | 3.6 |
| 9b | 2.05 | 0.00036 (0.00022 to 0.00059) | 11000 | 15 |
| 9c | 2.41 | 0.0011 (0.00066 to 0.0017) | 3600 | 4.9 |
| 9d | 2.83 | 0.00044 (0.00026 to 0.00076) | 9100 | 12 |
| 9e | 3.21 | 0.00036 (0.00024 to 0.00053) | 11000 | 15 |
| 9f | 3.57 | 0.00034 (0.000095 to 0.0012) | 12000 | 16 |
| 9g | 2.86 | 0.00058 (0.00029 to 0.0012) | 6900 | 9.3 |
| 9h | 3.23 | 0.00053 (0.000052 to 0.0054) | 7500 | 10 |
| 9i | 3.60 | 0.00082 (0.00048 to 0.0014) | 4900 | 6.6 |
| Mean Ar = phenyl |
NA | 0.00099 | NA | NA |
| Mean Ar = 1-naphthyl |
NA | 0.00038 | NA | NA |
| Mean Ar = 2-naphthyl |
NA | 0.00064 | NA | NA |
| Mean R = methyl |
NA | 0.00084 | NA | NA |
| Mean R = ethyl |
NA | 0.00042 | NA | NA |
| Mean R = isopropyl |
NA | 0.00075 | NA | NA |
cLogP values were determined from http://www.molinspiration.com/cgi-bin/properties, ND = not determined, NA = not applicable.
Select phosphonamidate prodrugs are mildly toxic to K562 leukemia cells
Novel butyrophilin ligands are of interest in part due to their potential to trigger an anti-cancer immune response driven by activation of Vγ9Vδ2 T cells. To model this response in vitro, we utilize co-cultures of the K562 acute myeloma leukemia cell line, which can be pre-loaded with test compounds, then mixed with primary purified Vγ9Vδ2 T cells. Prior to evaluating the mixed co-culture system, it was necessary to determine the potential cell toxicity of 9a-i to assess the possibility of direct anti-cancer activity. Thus K562 cells were treated with various doses of the test compounds and the viability of the cells was examined after 72 hours of treatment (Figure 4). As a class, only low levels of direct toxicity to K562 cells were observed, with all IC50 values above 23 µM (Table 3), and most above 100 µM. The IC50 values for phenol, 1-naphthol, and 2-naphthol were all above 100 µM. While no obvious patterns of activity were observed, it was interesting to note that the naphthyl/GlyOEt esters 9e and 9h displayed IC50 values of 24 and 28 µM, while the phenyl/GlyOEt 9b was relatively non-toxic (IC50 >100 µM). Similarly, the phenyl/GlyOiPr 9c had an IC50 of 23 µM, while both naphthyl/GlyOiPr forms 9f and 9i were relatively non-toxic (IC50 > 100 µM). Thus, with some mild exceptions, most of the nine tested compounds did not display strong cytotoxicity towards the K562 cells.
Figure 4.

K562 cell toxicity of phosphonamidate prodrugs. K562 cells were treated with test compounds for 72 hours and viability was assessed. Each compound was assessed in three independent experiments.
Table 3.
72 hour cytotoxicity of test compounds against K562 cells.
| Compound | IC50 [µM] |
|---|---|
| 9a | >100 |
| 9b | >100 |
| 9c | 23 |
| 9d | 40 |
| 9e | 24 |
| 9f | >100 |
| 9g | >100 |
| 9h | 28 |
| 9i | >100 |
| phenol | >100 |
| 1-naphthol | >100 |
| 2-naphthol | >100 |
Phosphonamidate prodrug loaded K562 cells trigger cytokine production by Vγ9Vδ2 T cells
Having established that direct cytotoxicity was low even during 72 hour exposure times, we next evaluated the compounds for their ability to stimulate Vγ9Vδ2 T cell cytokine production in response to K562 cells pre-loaded for only 4 hours with the test compounds (Figure 5). The compounds were active in this assay, with EC50 values ranging from 0.46 µM (compound 9a) to 0.062 µM (compound 9e) (Table 4), though the activity was reduced relative to the proliferation assays. Again, we observed that both naphthyl series were more potent than the phenyl series, in this case by 3–4 fold. Furthermore, the GlyOEt series was more potent than the GlyOMe and GlyOiPr series. Importantly, the compounds showed strong activity in this assay, even though they were only exposed to the K562 cells for 4 hours, and never directly exposed to the Vγ9Vδ2 T cells. The nanomolar activity of the compounds in this assay was also much lower than the mid micromolar direct toxicity observed in the 72 hour K562 viability assays. Therefore, the compounds trigger K562 cells to activate Vγ9Vδ2 T cell cytokine production without causing direct toxicity to the malignant cells.
Figure 5.

K562 cells loaded with phosphonamidate prodrugs stimulate production of interferon γ by Vγ9Vδ2 T cells. Each compound was assessed in three independent experiments using a minimum of two different human donors.
Table 4.
Interferon γ production by Vγ9Vδ2 T cells in response to K562 cells loaded for 4 hours with test compounds.
| Compound | EC50 [µM] (95% CI) |
|---|---|
| 9a | 0.46 (0.29 to 0.71) |
| 9b | 0.17 (0.070 to 0.42) |
| 9c | 0.74 (0.42 to 1.3) |
| 9d | 0.13(0.11 to 0.15) |
| 9e | 0.062(0.037 to 0.11) |
| 9f | 0.29 (0.14 to 0.59) |
| 9g | 0.12 (0.056 to 0.26) |
| 9h | 0.093 (0.069 to 0.13) |
| 9i | 0.21 (0.15 to 0.30) |
| Mean Ar = phenyl |
0.46 |
| Mean Ar = 1-naphthyl |
0.16 |
| Mean Ar = 2-naphthyl |
0.14 |
| Mean R = methyl |
0.24 |
| Mean R = ethyl |
0.11 |
| Mean R = isopropyl |
0.41 |
Discussion
Prior studies had suggested that acyloxyalkyl prodrugs are in general susceptible to biological deprotection by non-specific esterases found in human plasma.18,33,34 Our own investigations of phosphoantigen prodrugs had agreed with this conclusion, given that we had observed a fluorescent analog of compound 2 had a half-life of just 6–8 minutes in human plasma.35 Furthermore, as measured here, the bis-POM prodrug 2 has a half-life of ~8 minutes and has undergone more than 95% hydrolysis after just two hours. While such compounds might still have value for experiments conducted on cell systems, and as a tool to enhance oral absorption, their rapid plasma hydrolysis would limit their utility in any animal studies beyond that of oral uptake. In contrast to the limited plasma stability of the acyloxy protected prodrugs, all nine of the phosphonamidates reported here show substantially higher stability, with plasma half-lives greater than 24 hours in all nine cases (Table 1).
As shown in Table 2, when tested for stimulation of Vγ9Vδ2 T cell proliferation all nine of these new phosphonamidates displayed EC50 values of 1.5 nM or lower, By comparison, the corresponding dimethyl ester 11 which we reported earlier,9 was inactive at concentrations up to 10 μM. Because simple alkyl esters of phosphonates are generally believed to have high metabolic stability,18 and because compound 11 (Figure 6) does not stimulate proliferation of Vγ9Vδ2 T cells, it is reasonable to assume that the phosphonamidates undergo hydrolysis after they have entered the cell to release the active ligand. The mechanism of that hydrolysis has not been explored for these phosphonamidates, but for acyclic nucleoside phosphonates and phosphates Cathepsin A and possibly carboxylesterase 1 (CES1) are believed to be involved in initial hydrolysis of the amino acid ester.36
Figure 6.

A potent mixed aryl acyloxyalkyl butyrophilin ligand (13) and some control compounds.
Arguably the most prominent phosphoramidate is sofosbuvir, which has become a first line treatment for hepatitis C and has demonstrated a very high cure rate.37 That prodrug differs from those reported here in several respects, with perhaps the most obvious difference being the release of a phosphate metabolite from sofosbuvir rather than a phosphonate as described herein.24 During the SAR studies that led to the development of sofosbuvir it was reported that varying the alkyl ester of the amino acid side chain had an impact on the activity of the prodrug.38 Our own studies reported here also show a difference in activity between alkyl ester substituents of the glycine-based component, but the most potent compound (9f, EC50 = 340 pM) bears an isopropyl ester (Table 2). Furthermore, the mean activity of the three ethyl esters is approximately twice that of the three methyl esters or the three isopropyl esters.
Additional SAR studies leading to sofosbuvir revealed a substantial difference in activity between phenyl and 1-naphthyl phosphate esters with EC90 values of 0.91 μM and 0.09 μM, respectively.38 Our own studies also demonstrate a difference in activity between aryl phosphonate esters, with either the 1- or 2-naphthyl esters granting an increase in activity with respect to the corresponding phenyl ester. The mean EC50 value of the phenyl compounds is roughly three times higher than that of the 1-naphthyl compounds, with the 2-naphthyl compounds roughly midway between. Significantly, prior to these studies the most potent phosphoantigen prodrug known was the mixed 1-naphthyl/pivaloyloxymethyl ester 13 (Figure 6), which displayed an EC50 of 790 pM when tested for its ability to promote expansion of Vγ9 Vδ2 T cells from human peripheral blood mononuclear cells under the same conditions.39 Six of the nine phosphonamidates reported here displayed lower EC50 values, with the most potent compound (9f) showing an EC50 more than 2-fold lower (340 pM). All six of the more active compounds have lower cLogP values than compound 13, but a more definitive relationship between hydrophobicity and activity is not apparent.
All of these phosphonamidates showed high plasma stability and ultimately release the same active ligand, which suggests that any difference in potency traces back to other factors such as the ability to cross the cell membrane or undergo hydrolysis once inside the cell. Both the 1- and 2-naphthyl derivatives would be expected to be more hydrophobic than the phenyl derivatives, and this may be reflected in their consistently greater potency. However, both isomeric naphthyl derivatives should be very similar in terms of their hydrophobicity and cLogP values. There also is little difference in their acidity, although 1-naphthol has been reported to be slightly more acidic that 2-naphthol40 so the 1-naphthyl group might be a slightly better leaving group in terms of chemical reactivity. In terms of biochemical reactivity there could be more significant differences between these two isomers, but even though an understanding of their ability to undergo enzymatic hydrolysis might shed light on the basis for some differences in potency, those studies are beyond the scope of the present effort.
Finally, all nine new phosphonamidates were tested for their direct cytotoxicity to K562 cells, a human-derived myelogenous leukemia line, and little direct toxicity was observed (Table 3). Five of the nine compounds showed an IC50 value greater than 100 μM, the highest concentration tested, while the other four were in the 20–40 μM range. It is possible that the phenol or naphthol released upon prodrug hydrolysis contributes to the observed mild toxicity at high concentrations but it is unlikely a significant factor at concentrations near the EC50.
In an assay designed to measure interferon γ production by Vγ9Vδ2 T cells in response to exposure to K562 cells treated with the phosphonamidates, all nine phosphonamidates displayed EC50 values below 1 μM (Table 4). In this assay, the most potent compound was phosphonamidate 9e with an EC50 = 62 nM. As shown in Table 4, when viewed collectively both the 1- and 2-naphthyl compounds were more effective than the phenyl compounds, but there was little difference between compounds bearing either of the naphthyl isomers. In this assay, the glycine ethyl esters were twice as effective as the methyl esters, and four-fold more effective than the isopropyl esters. Because both the potency levels and the pattern of activity differed between the K562 cells and PBMC, the cell types may differ in their ability to metabolize compounds of this type. A potency difference of this magnitude was not previously observed in our prior bis-ester compounds such as compound 10. Therefore the phosphonamidates described here may represent a more selective way to activate Vγ9Vδ2 T cells.
Conclusions
These studies have established a synthetic route to phosphonamidate prodrug forms of an important butyrophilin ligand, and surveyed the impact of some variations in the amino acid ester and the phosphonate aryl ester. All compounds tested showed significantly enhanced plasma stability relative to the acyloxy esters previously reported, and four of the new compounds displayed greater potency than the best earlier prodrug form of this ligand. The combination of significant plasma stability and high potency suggests that these compounds would be appropriate for in vivo studies, although additional studies would be needed to establish the intracellular concentration of the parent ligand. The activity of these phosphoantigen phosphonamidates also provides a clear example that phosphonamidates can be applied beyond nucleosides or nucleoside analogues.
Experimental
Chemical Synthesis
General Experimental Procedures.
Acetonitrile was distilled from calcium hydride prior to use and dimethylformamide (DMF), pyridine, and triethylamine (Et3N) were dried over 4 Å molecular sieves (5% w/v). All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions in non–aqueous solvents were conducted in flame–dried glassware under a positive pressure of argon and with magnetic stirring. For TLC analyses, pre-coated silica polyester backed plates (200 μm thickness, UV254 indicator) were visualized under both short-wave ultraviolet light (254 nm) and by heating post exposure to p-anisaldehyde stain (93 parts 200 proof ethanol: 3.5 parts sulfuric acid: 1 part acetic acid: 2.5 parts p-anisaldehyde). Flash column chromatography was carried out using silica gel (60 Å, 40–63 μm). Glass columns were slurry-packed using the appropriate eluent with the sample either being loaded as a concentrated solution in the same eluent or pre-adsorbed onto silica gel. Fractions containing the product were identified by TLC, combined and the solvent was removed under reduced pressure. The purity of the final compounds was corroborated by HPLC analysis using an Agilent 1120 infinity LC solvent delivery system with a variable wavelength UV detector. Compounds to be used for bioassay were eluted from a C18 column (either 5 µm, 250 × 10 mm or 8 µm, 250 × 10.0 mm) as analytical columns at a flow rate of 2.0 mL/min using 100% HPLC grade methanol (isocratic, 12 minutes). Compounds for bioassay were >95% pure at 254 nm. All NMR spectra were obtained at either 400 or 500 MHz for 1H, 100 or 125 MHz for 13C, and 161 or 202 MHz for 31P with internal standards of (CH3)4Si (1H, 0.00 ppm) or CDCl3 (1H, 7.27; 13C, 77.2 ppm) or CD3OD (1H, 3.31; 13C, 49.0 ppm) or CD3C(O)CD3 (1H, 2.05; 13C, 206.3 ppm) or CD3CN (1H, 1.94; 13C, 118.3 ppm) for non–aqueous samples or D2O (1H, 4.80 ppm) for aqueous samples.41 The 31P chemical shifts are reported in ppm relative to 85% H3PO4 (external standard). High-resolution mass spectra were obtained by TOF MS ES+ at the University of Iowa Mass Spectrometry Facility.
Methyl naphthalene-2-yl (4-methylpent-3-en-1-yl)phosphonate (6c).
A solution of 2-naphthol (1.91 g, 13.3 mmol) and triethylamine (1.84 mL, 13.3 mmol) in toluene (10 mL) was added dropwise to a solution of the acid chloride 512 (5.3 mmol) in toluene (10 mL) and allowed to react for 15 hours. The reaction then was diluted with diethyl ether (30 mL) and quenched by addition of brine (5 mL). The organic portion was then washed four times with 1 M NaOH (5 mL), dried (MgSO4), filtered through celite, and concentrated in vacuo. The resulting reddish yellow oil was purified via chromatography (silica, 100% hexanes – 40% EtOAc in hexanes) and the product was concentrated to a yellow oil in 82% yield (1.32 g): 1H NMR (400 MHz, CDCl3) δ 7.79–7.78 (m, 3H), 7.68 (s, 1H), 7.46–7.41 (m, 2H), 7.35 (d, J = 8.8 Hz, 1H), 5.13 (t, J = 6.8 Hz, 1H), 3.81 (d, JPH = 11.2 Hz, 3H), 2.47–2.35 (m, 2H), 2.00–1.94 (m, 2H), 1.67 (s, 3H), 1.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 148.3 (d, JPC = 9.3 Hz), 134.0, 133.3, 130.9, 129.9, 127.7, 127.5, 126.7 125.4, 122.7 (d, JPC = 17.7 Hz), 120.5 (d, JPC = 4.4 Hz), 116.8 (d, JPC = 4.2 Hz), 52.8 (d, JPC = 6.1 Hz), 25.6, 25.6 (d, JPC = 136.9 Hz), 21.1 (d, JPC = 4.7 Hz), 17.7; 31P (161 MHz, CDCl3) δ +30.5; HRMS (ES+, m/z) calcd. for (M+H)+ C17H22O3P: 305.1307; found: 305.1304.
Synthesis of Ethyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-phenoxy-phosphoryl]amino]acetate (9b) from the mixed phosphonate ester 6a.12,42,43 General procedure for phosphonamidate preparation.
The mixed ester 6a12 (1.1 g, 4.3 mmol) was dissolved in freshly distilled acetonitrile (14 mL) and added as a solution to solid, flame-dried sodium iodide (645 mg, 4.3 mmol). The resultant solution was heated at reflux overnight, allowed to cool to room temperature, and then concentrated under reduced pressure to reveal a pale yellow to white solid (7a). Glycine ethyl ester HCl (1.1 g, 7.7 mmol) was added followed by anhydrous pyridine (21 mL) and then triethylamine (6.4 mL, 45.6 mmol) and the resulting solution was stirred. In a separate flask 2,2’-dithiodipyridine (6.9 g) and PPh3 (5.9 g) were dissolved in anhydrous pyridine (21 mL) and the resultant solution was stirred for 20 minutes. This solution was added to the solution of monosodium salt and the mixture was stirred overnight at 60 °C. The reaction mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc and filtered. The filtrate was concentrated under reduced pressure and the residue subjected to silica gel chromatography (0–10% EtOAc in Et2O) to provide the desired monoamidate 8b as a clear to pale yellow oil. In a separate flask, SeO2 (89 mg, 0.8 mmol) and pyridine (0.5 mL, 6.0 mmol) were dissolved in 70% aqueous tert-butyl hydroperoxide solution (0.9 mL), stirred for 30 minutes at room temperature and cooled to 0 °C.44 The aforementioned monoamidate oil was dissolved in MeOH (2.5 mL), added to the solution of oxidant and the reaction mixture was stirred for 18 hours. The solution was concentrated under reduced pressure and the residue was dissolved in EtOAc, washed with aqueous potassium carbonate (2x) and then brine, dried with MgSO4, and filtered. The filtrate was concentrated under reduced pressure and the residue subjected to silica gel chromatography (0–20% acetone in CH2Cl2) to provide compound 9b (30 mg, 8% over three steps) as a yellow oil: 1H NMR (500 MHz, CD3OD) δ 7.36–7.33 (m, 2H), 7.22–7.16 (m, 3H), 5.49 (t, J = 7.1 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.93 (s, 2H), 3.79–3.62 (m, 2H), 2.51–2.43 (m, 2H), 2.06–1.99 (m, 2H), 1.69 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 172.9, 151.9 (d, JCP = 9.4 Hz), 137.3, 130.7 (2C), 125.9, 125.1 (d, JCP = 17.6 Hz), 121.9 (d, JCP = 4.5 Hz, 2C), 68.6, 62.2, 43.2, 28.9 (d, JCP = 129.1 Hz), 21.6 (d, JCP = 4.0 Hz), 14.5, 13.7; 31P NMR (202 MHz, CD3OD) δ +35.0; HRMS (ES+, m/z) calcd. for (M+H)+ C16H25NO5P: 342.1470; found: 342.1462.
Methyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-phenoxy-phosphoryl]amino]acetate (9a).
The mixed ester 6a12 (1.1 g, 4.6 mmol) was treated according to the general procedure to afford the intermediates 7a and 8a, and then compound 9a (25 mg, 5% over three steps) as a yellow oil: 1H NMR (500 MHz, CD3OD) δ 7.37–7.33 (m, 2H), 7.22–7.15 (m, 3H), 5.49 (t, J = 7.1 Hz, 1H), 3.93 (s, 2H), 3.81–3.70 (m, 2H), 3.68 (s, 3H), 2.50–2.43 (m, 2H), 2.06–1.98 (m, 2H), 1.69 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 173.4 (d, JPC = 4.3 Hz), 151.9 (d, JPC = 9.6 Hz), 137.3, 130.7 (2C), 125.9, 125.1 (d, JPC = 17.1 Hz), 121.9 (d, JPC = 3.9 Hz, 2C), 68.6, 52.5, 43.0, 28.9 (d, JPC = 129.8 Hz), 21.6 (d, JPC = 4.2 Hz), 13.7; 31P NMR (161 MHz, CD3OD) δ +35.1; HRMS (ES+, m/z) calcd. for (M+H)+ C15H23NO5P: 328.1314; found:328.1322.
Isopropyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-phenoxy-phosphoryl]amino]acetate (9c).
The mixed ester 6a12 (384 mg, 1.5 mmol) was treated according to the general procedure for prodrug preparation to afford phosphonamidate 9c (99 mg, 19% over three steps) as a yellow oil, along with 88 mg of the corresponding aldehyde: 1H NMR (400 MHz, CD3C(O)CD3) δ 7.34 (t, J = 7.3 Hz, 2H), 7.26 (d, J = 7.7 Hz, 2H), 7.15 (t, J = 7.5 Hz, 1H), 5.49 (td, J = 7.2, 1.1 Hz, 1H), 4.97 (sept, J = 6.2 Hz, 1H), 4.57–4.51 (m, 1H), 3.92 (s, 2H), 3.83–3.62 (m, 2H), 2.49–2.39 (m, 2H), 2.01–1.93 (m, 2H), 1.65 (s, 3H), 1.20 (d, J = 6.2, Hz, 3H), 1.20 (d, J = 6.3, Hz, 3H); 13C NMR (125 MHz, CD3CN) δ 171.7 (d, JCP = 5.0 Hz), 151.8 (d, JCP = 8.9 Hz), 137.3 (d, JCP = 1.4 Hz), 130.6 (2C), 125.4, 124.2 (d, JCP = 16.1 Hz), 121.8 (d, JCP = 4.0 Hz, 2C), 69.5, 68.0, 43.4, 28.6 (d, JCP = 128.7 Hz), 22.0 (2C), 21.3 (d, JCP = 4.4 Hz), 13.8; 31P (202 MHz, CD3CN) δ + 32.9; HRMS (ES+, m/z) calcd. for (M+Na)+ C17H26NNaO5P: 378.1446; found: 378.1448.
Methyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(1-naphthyloxy)phosphoryl]amino]acetate (9d).
The mixed ester 6b12 (1.012 g, 3.3 mmol) was treated according to the general procedure to obtain phosphonate 9d (86 mg, 14% over three steps) as an amber oil: 1H NMR (400 MHz, CD3OD) δ 8.17–8.15 (m, 1H), 7.89–7.86 (m, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.57–7.51 (m, 2H), 7.50 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.9 Hz, 1H), 5.51 (t, J = 6.4 Hz, 1H), 3.92 (s, 2H), 3.80–3.66 (m, 2H), 3.61 (s, 3H), 2.56–2.50 (m, 2H), 2.20–2.12 (m, 2H), 1.65 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 173.3 (d, JCP = 4.0 Hz), 147.8 (d, JCP = 9.6 Hz), 137.4, 136.3, 128.9, 128.2, 127.7, 127.4, 126.6, 125.7, 125.0 (d, JCP = 17.6 Hz), 122.8, 116.7 (d, JCP = 3.8 Hz), 68.6, 52.5, 43.0, 28.9 (d, JCP = 131.3 Hz), 21.7 (d, JCP = 4.1 Hz), 13.6; 31P NMR (161 MHz, CD3OD) δ +35.6; HRMS (ES+, m/z) calcd. for (M+Na)+ C19H24NNaO5P: 400.1290; found: 400.1289.
Ethyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(1-naphthyloxy)phosphoryl]amino]acetate (9e).
The mixed ester 6b12 (932 mg, 3.1 mmol) was treated according to the general procedure to afford phosphonamidates 9e (40 mg, 9% over three steps) as an amber oil: 1H NMR (400 MHz, CD3OD) δ 8.17–8.15 (m, 1H), 7.89–7.87 (m, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.57–7.50 (m, 2H), 7.49 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 5.51 (td, J = 7.1, 1.1 Hz, 1H), 4.08 (q, J 7.1 Hz, 2H), 3.92 (s, 2H), 3.82–3.61 (m, 2H), 2.59–2.46 (m, 2H), 2.20–2.12 (m, 2H), 1.65 (s, 3H), 1.18 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 172.8 (d, JCP = 4.0 Hz), 147.8 (d, JCP = 9.5 Hz), 137.4, 136.3, 128.9, 128.2 (d, JCP = 4.8 Hz), 127.7, 127.4, 126.6, 125.7, 125.0 (d, JCP = 17.9 Hz), 122.8, 116.7 (d, JCP = 3.8 Hz), 68.6, 62.1, 43.2, 28.9 (d, JCP = 127.7 Hz), 21.7 (d, JCP = 4.4 Hz), 14.4, 13.6; 31P NMR (161 MHz, CD3OD) δ +35.6; HRMS (ES+, m/z) calcd. for (M+Na)+ C20H26NNaO5P: 414.1446; found: 414.1445.
Isopropyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(1-naphthyloxy)phosphoryl]amino]acetate (9f).
The mixed ester 6b12 (915 mg, 3.0 mmol) was treated according to the general procedure to give phosphonamidates 9f (103 mg, 18% over three steps) as an amber oil: 1H NMR (400 MHz, CD3OD) δ 8.17–8.15 (m, 1H), 7.88–7.86 (m, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.57–7.50 (m, 2H), 7.49 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 5.51 (td, J = 7.2, 1.2 Hz, 1H), 5.00–4.91 (sept, J = 6.2 Hz, 1H), 3.92 (s, 2H), 3.79–3.58 (m, 2H), 2.57–2.48 (m, 2H), 2.20–2.12 (m, 2H), 1.65 (s, 3H), 1.18 (d, J = 4.5 Hz, 3H), 1.17 (d, J = 4.5 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 172.3 (d, JCP = 4.3 Hz), 147.8 (d, JCP = 9.6 Hz), 137.4, 136.3, 128.9, 128.2 (d, JCP = 5.4 Hz), 127.7, 127.4, 126.6, 125.6, 125.0 (d, JCP = 17.4 Hz), 122.7, 116.6 (d, JCP = 3.8 Hz), 70.0, 68.5, 43.4, 28.9 (d, JCP = 128.4 Hz), 22.0, 21.9, 21.7 (d, JCP = 4.4 Hz), 13.7; 31P NMR (161 MHz, CD3OD) δ +35.6; HRMS (ES+, m/z) calcd. for (M+Na)+ C21H28NNaO5P: 428.1603; found: 428.1599.
Methyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(2-naphthyloxy)phosphoryl]amino]acetate (9g).
The mixed ester 6c (961 mg, 3.2 mmol) was treated according to the general procedure to afford the phosphonamidate 9g (59 mg, 11% over three steps) as an amber oil: 1H NMR (400 MHz, CD3OD) δ 7.86–7.83 (m, 2H), 7.80 (d, J = 8.1 Hz, 1H), 7.69 (s, 1H), 7.48 (td, J = 6.9, 1.2 Hz, 1H), 7.43 (td, J = 6.9, 1.1 Hz, 1H), 7.37–7.35 (m, 1H), 5.51 (td, J = 7.3, 1.0 Hz, 1H), 3.94 (s, 2H), 3.83–3.65 (m, 2H), 3.61 (s, 3H), 2.55–2.43 (m, 2H), 2.12–2.03 (m, 2H), 1.69 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 173.3 (d, JPC = 3.9 Hz), 149.5 (d, JPC = 9.6 Hz), 137.3, 135.4, 132.3, 130.8, 128.7, 128.4, 127.8, 126.5, 125.1 (d, JPC = 17.4 Hz), 122.0 (d, JPC = 4.4 Hz), 118.3 (d, JPC 4.9 Hz), 68.6, 52.5, 43.0, 28.9 (d, JPC = 129.7 Hz), 21.6 (d, JPC = 4.2 Hz), 13.7; 31P NMR (161 MHz, CD3OD) δ +35.4; HRMS (ES+, m/z) calcd. for (M+Na)+ C19H24NNaO5P: 400.1290; found: 400.1288.
Ethyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(2-naphthyloxy)phosphoryl]amino]acetate (9h).
The mixed ester 6c (990 mg, 3.3 mmol) was treated according to the general procedure to afford compound 9h (58 mg, 9% over three steps) as an amber oil: 1H NMR (500 MHz, CD3OD) 7.88–7.85 (m, 2H), 7.81 (d, J = 8.3 Hz, 1H), 7.69 (s, 1H), 7.51–7.48 (m, 1H), 7.46–7.43 (m, 1H), 7.37–7.35 (m, 1H), 5.51 (t, J = 6.6 Hz, 1H), 4.09 (q, J = 7.1 Hz, 2H), 3.94 (s, 2H), 3.81–3.63 (m, 2H), 2.55–2.44 (m, 2H), 2.12–2.05 (m, 2H), 1.70 (s, 3H), 1.18 (t, J = 8.0 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 172.8, 149.4 (d, JCP = 10.1 Hz), 137.3, 135.4, 132.3, 130.8, 128.7, 128.4, 127.8, 126.5, 125.0 (d, JCP = 17.2 Hz), 122.1 (d, JCP = 4.1 Hz), 118.2 (d, JCP = 4.3 Hz), 68.6, 62.1, 43.2, 28.9 (d, JCP = 128.6 Hz), 21.6 (d, JCP = 4.1 Hz), 14.4, 13.7; 31P NMR (202 MHz, CD3OD) δ +35.4; HRMS (ES+, m/z) calcd. for (M+Na)+ C20H26NNaO5P: 414.1446; found: 414.1447.
Isopropyl 2-[[[(E)-5-hydroxy-4-methyl-pent-3-enyl]-(2-naphthyloxy)phosphoryl]amino]acetate (9i).
The mixed ester 6c (999 mg, 3.3 mmol) was treated according to the general procedure to afford phosphonamidates 9i (137 mg, 10% over three steps) as an amber oil: 1H NMR (500 MHz, CD3OD) δ 7.85 (t, J = 8.4 Hz, 2H), 7.80 (d, J = 8.0 Hz, 1H), 7.69 (s, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.44 (t, J = 7.3 Hz, 1H), 7.36 (d, J = 8.7 Hz, 1H), 5.51 (t, J = 7.0 Hz, 1H), 4.95 (sept, J = 6.2 Hz, 1H), 3.94 (s, 2H), 3.77–3.62 (m, 2H), 2.53–2.47 (m, 2H), 2.11–2.05 (m, 2H), 1.69 (s, 3H), 1.18 (d, J = 7.0 Hz, 3H), 1.18 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 172.5 (d, JCP = 4.1 Hz), 149.6 (d, JCP = 10.1 Hz), 137.5, 135.5, 132.5, 130.9, 128.9, 128.6, 127.9, 126.6, 125.2 (d, JCP = 20.2 Hz), 122.1 (d, JCP = 4.1 Hz), 118.4 (d, JCP = 4.4 Hz), 70.2, 68.7, 43.6, 29.1 (d, JCP = 128.3 Hz), 22.1 (2C), 21.8 (d, JCP = 4.3 Hz), 13.9; 31P NMR (161 MHz, CD3OD) δ +35.4; HRMS (ES+, m/z) calcd. for (M+H)+ C21H29NO5P: 406.1783; found: 406.1782.
Biological assays.
Materials and supplies.
Human peripheral blood mononuclear cell (PBMCs) were isolated from buffy coat obtained from Research Blood Components (Boston, MA). K562 cells were from the American Type Culture Collection. The FITC-conjugated anti-γδ-TCR (5A6.E91) antibody and pooled human plasma was purchased from Fisher (Waltham, MA). The phycoerythrin conjugated anti-CD3 (UCHT1) antibody and interferon γ enzyme-linked immunosorbent assay kit were purchased from Biolegend (San Diego, CA). The CellQuanti-Blue Cell Viability Assay Kit was purchased from BioAssay Systems (Hayward, CA). HMBPP was purchased from Echelon (Salt Lake City, UT). The TCRγ/δ+ T Cell Isolation Kit was from Miltenyi (Bergisch Gladbach, Germany). POM2-C-HMBP was synthesized as described previously.9
Expansion of Vγ9Vδ2 T cells.
All compounds were evaluated for their ability to promote growth of human Vγ9Vδ2 T cells from peripheral blood as described previously.9,45 In each experiment, 100 nM of HMBPP and 100 nM of POM2-C-HMBP were used as positive controls. Negative controls contained cells with interleukin 2 in the absence of test compounds. EC50 values were determined as the concentration that induced 50% of the maximum proliferative effect. All experiments were performed at least three times using cells from at least two different donors.
ELISA for interferon γ.
Interferon γ was measured by ELISA as previously described and according to manufacturer’s directions.10,45 Briefly, K562 cells were treated with test compounds for 4 hours, washed, then mixed with Vγ9Vδ2 T cells that had been purified by negative selection. Each well contained a 3:1 effector: target ratio in 200 µL. After 20 hours, the concentration of interferon γ in the supernatant was determined.
Cell viability.
Viability assays were performed using K562 cells with various concentrations of test compounds. K562 cells (0.5 × 104 cells in 100 µL of RPMI media) were distributed into each well of a 96-well plate. Phosphonamidates were added for 72 hours, during the last 2 hours the cell-QB reagent was added, following which signals were quantified with a fluorescence plate reader. Viable cells were expressed as a percentage of untreated control cells after subtraction of a media-only blank.
Stability studies.
Pooled human plasma was diluted to 50% with phosphate buffered saline at pH 7.5. Test compounds were added at a final concentration of 100 µM in a volume of 100 µL. Compounds were incubated for various times as indicated in the text, then extracted with 300 µL of LCMS grade acetonitrile and vigorous mixing. Insoluble debris was pelleted by centrifugation at 10,000 rcf for 2 minutes. 10 µL of the extract was evaluated by LCMS with a Waters Synapt G2-Si Mass Spectrometer in positive mode using a gradient of water and acetonitrile and a C18 column. The gradient started at 25% acetonitrile then increased to 80% acetonitrile over 9 minutes and held there for 1 minute before re-equilibration. The retention times for all compounds were as follows (POM2-C-HMBP, tR = 5.43; 9a, tR = 3.33; 9b, tR = 3.72; 9c, tR = 4.11; 9d, tR = 4.16; 9e, tR = 4.51; 9f, tR = 4.87; 9g, tR = 4.21; 9h, tR = 4.55; 9i, tR 4.91 minutes). For all phosphonamidates tested, masses corresponding to the molecular ion [M+H]+, the sodium adduct [M + Na]+, and the dehydration product [M - OH]+ were observed at the reported retention time. The integrated values of these peaks were compared to those of t = 0 minutes in plasma for each test compound and expressed as a fraction of the initial compound that was remaining at a given time point.
Supplementary Material
Acknowledgments
We appreciate the assistance of Dr. Jeremy Balsbaugh and the University of Connecticut Proteomics & Metabolomics Facility with the LMCS analysis. We thank the Center for Biocatalysis and Bioprocessing for a fellowship (B.J.F.) through the Predoctoral Training Program in Biotechnology (T32 GM008365). Research reported in this publication was supported by the National Cancer Institute of the United States National Institutes of Health under Award Number R01CA186935 (A.J.W., P.I.), a grant from the Herman Frasch Foundation for Chemical Research (HF17, A.J.W., P.I.), and by a Research Program of Excellence Award from the Roy J. Carver Charitable Trust (D.F.W., P.I.).
Abbreviations Used
- BTN
butyrophilin
- HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl diphosphate
- BrHPP
4-bromo-3-hydroxy-3-methylbutyl diphosphate
- POM
pivaloyloxymethyl
- ITC
isothermal titration calorimetry
Footnotes
The other authors have no financial conflicts of interest.
Supporting Information
Molecular Formula Strings and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Rhodes DA; Reith W; Trowsdale J Regulation of Immunity by Butyrophilins [DOI] [PubMed]
- (2).Kabelitz D; Lettau M; Janssen O Immunosurveillance by Human Gammadelta T Lymphocytes: The Emerging Role of Butyrophilins F1000Research 2017, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gu S; Borowska M; Boughter CT; Adams EJ Butyrophilin3a Proteins and Vgamma9vdelta2 T Cell Activation Semin Cell Dev Biol [Online early access] DOI: 10.1016/j.semcdb.2018.02.007 . Published Online: March 9 2018 10.1016/j.semcdb.2018.02.007https://www.sciencedirect.com/science/article/pii/S1084952116304360?via%3Dihub. Published Online: March 9 2018 https://www.sciencedirect.com/science/article/pii/S1084952116304360?via%3Dihub (accessed July 26, 2018) [DOI] [PMC free article] [PubMed]
- (4).Wiemer DF; Wiemer AJ Opportunities and Challenges in Development of Phosphoantigens as Vgamma9vdelta2 T Cell Agonists Biochem. Pharmacol 2014, 89, 301–312. [DOI] [PubMed] [Google Scholar]
- (5).Boutin L; Scotet E Towards Deciphering the Hidden Mechanisms That Contribute to the Antigenic Activation Process of Human Vγ9vδ2 T Cells Frontiers in Immunology 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Morita CT; Jin C; Sarikonda G; Wang H Nonpeptide Antigens, Presentation Mechanisms, and Immunological Memory of Human Vgamma2vdelta2 T Cells: Discriminating Friend from Foe through the Recognition of Prenyl Pyrophosphate Antigens [DOI] [PubMed]
- (7).Sicard H; Ingoure S; Luciani B; Serraz C; Fournie JJ; Bonneville M; Tiollier J; Romagne F In Vivo Immunomanipulation of V Gamma 9v Delta 2 T Cells with a Synthetic Phosphoantigen in a Preclinical Nonhuman Primate Model J. Immunol 2005, 175, 5471–5480. [DOI] [PubMed] [Google Scholar]
- (8).Fournie JJ; Sicard H; Poupot M; Bezombes C; Blanc A; Romagne F; Ysebaert L; Laurent G What Lessons Can Be Learned from Gammadelta T Cell-Based Cancer Immunotherapy Trials? Cellular & molecular immunology 2013, 10, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hsiao CH; Lin X; Barney RJ; Shippy RR; Li J; Vinogradova O; Wiemer DF; Wiemer AJ Synthesis of a Phosphoantigen Prodrug That Potently Activates Vgamma9vdelta2 T-Lymphocytes Chem. Biol 2014, 21, 945–954. [DOI] [PubMed] [Google Scholar]
- (10).Kilcollins AM; Li J; Hsiao CH; Wiemer AJ Hmbpp Analog Prodrugs Bypass Energy-Dependent Uptake to Promote Efficient Btn3a1-Mediated Malignant Cell Lysis by Vgamma9vdelta2 T Lymphocyte Effectors J. Immunol 2016, 197, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kornberg RD; Mcconnel Hm; Mcnamee MG Measurement of Transmembrane Potentials in Phospholipid Vesicles Proc. Natl. Acad. Sci. U. S. A 1972, 69, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Foust BJ; Poe MM; Lentini N; Hsiao CHC; Wiemer AJ; Wiemer DF Mixed Aryl Phosphonate Prodrugs of a Butyrophilin Ligand ACS Med. Chem. Lett 2017, 8, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Riano F; Karunakaran MM; Starick L; Li J; Scholz CJ; Kunzmann V; Olive D; Amslinger S; Herrmann T Vgamma9vdelta2 Tcr-Activation by Phosphorylated Antigens Requires Butyrophilin 3 A1 (Btn3a1) and Additional Genes on Human Chromosome 6 Eur. J. Immunol 2014, 44, 2571–2576. [DOI] [PubMed] [Google Scholar]
- (14).Harly C; Guillaume Y; Nedellec S; Peigne CM; Monkkonen H; Monkkonen J; Li J; Kuball J; Adams EJ; Netzer S; Dechanet-Merville J; Leger A; Herrmann T; Breathnach R; Olive D; Bonneville M; Scotet E Key Implication of Cd277/Butyrophilin-3 (Btn3a) in Cellular Stress Sensing by a Major Human Gammadelta T-Cell Subset Blood 2012, 120, 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sandstrom A; Peigne CM; Leger A; Crooks JE; Konczak F; Gesnel MC; Breathnach R; Bonneville M; Scotet E; Adams EJ The Intracellular B30.2 Domain of Butyrophilin 3a1 Binds Phosphoantigens to Mediate Activation of Human V Gamma 9v Delta 2 T Cells Immunity 2014, 40, 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rhodes DA; Chen HC; Price AJ; Keeble AH; Davey MS; James LC; Eberl M; Trowsdale J Activation of Human Gammadelta T Cells by Cytosolic Interactions of Btn3a1 with Soluble Phosphoantigens and the Cytoskeletal Adaptor Periplakin J. Immunol 2015, 194, 2390–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Arimilli MN; Kim CU; Dougherty J; Mulato A; Oliyai R; Shaw JP; Cundy KC; Bischofberger N Synthesis, in Vitro Biological Evaluation and Oral Bioavailability of 9-[2-(Phosphonomethoxy)Propyl]Adenine (Pmpa) Prodrugs Antivir Chem Chemoth 1997, 8, 557–564. [Google Scholar]
- (18).Wiemer AJ; Wiemer DF Prodrugs of Phosphonates and Phosphates: Crossing the Membrane Barrier Top Curr Chem 2015, 360, 115–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Abrahamsson K; Holme E; Jodal U; Lindstedt S; Nordin I Effect of Short-Term Treatment with Pivalic Acid-Containing Antibiotics on Serum Carnitine Concentration - a Risk Irrespective of Age Biochem. Mol. Med 1995, 55, 77–79. [DOI] [PubMed] [Google Scholar]
- (20).Mehellou Y; Balzarini J; McGuigan C Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells ChemMedChem 2009, 4, 1779–1791. [DOI] [PubMed] [Google Scholar]
- (21).Birkus G; Kutty N; He GX; Mulato A; Lee W; McDermott M; Cihlar T Activation of 9- (R)-2- (S)- (S)-1-(Isopropoxycarbonyl)Ethyl Amino Phenoxyphosphinyl - Methoxy Propyl Adenine (Gs-7340) and Other Tenofovir Phosphonoamidate Prodrugs by Human Proteases Mol. Pharmacol 2008, 74, 92–100. [DOI] [PubMed] [Google Scholar]
- (22).Lonnberg T; Ora M; Lonnberg H Hydrolytic Reactions of Nucleoside Phosphoramidates: Kinetics and Mechanisms Mini-Rev Org Chem 2010, 7, 33–43. [Google Scholar]
- (23).Furman PA; Murakami E; Niu CR; Lam AM; Espiritu C; Bansal S; Bao HY; Tolstykh T; Steuer HM; Keilman M; Zennou V; Bourne N; Veselenak RL; Chang W; Ross BS; Du JF; Otto MJ; Sofia MJ Activity and the Metabolic Activation Pathway of the Potent and Selective Hepatitis C Virus Pronucleotide Inhibitor Psi-353661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Murakami E; Tolstykh T; Bao HY; Niu CR; Steuer HMM; Bao DH; Chang W; Espiritu C; Bansal S; Lam AM; Otto MJ; Sofia MJ; Furman PA Mechanism of Activation of Psi-7851 and Its Diastereoisomer Psi-7977 J. Biol. Chem 2010, 285, 34337–34347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Freel Meyers CL; Borch RF Activation Mechanisms of Nucleoside Phosphoramidate Prodrugs J. Med. Chem 2000, 43, 4319–4327. [DOI] [PubMed] [Google Scholar]
- (26).Eisenberg EJ; He GX; Lee WA Metabolism of Gs-7340, a Novel Phenyl Monophosphoramidate Intracellular Prodrug of Pmpa, in Blood Nucleosides Nucleotides & Nucleic Acids 2001, 20, 1091–1098. [DOI] [PubMed] [Google Scholar]
- (27).Lee WA; He GX; Eisenberg E; Cihlar T; Swaminathan S; Mulato A; Cundy KC Selective Intracellular Activation of a Novel Prodrug of the Human Immunodeficiency Virus Reverse Transcriptase Inhibitor Tenofovir Leads to Preferential Distribution and Accumulation in Lymphatic Tissue Antimicrob. Agents Chemother 2005, 49, 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Birkus G; Kutty N; He GX; Mulato A; Lee W; McDermott M; Cihlar T Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)Ethyl]Amino] Phenoxyphosphinyl]-Methoxy]Propyl]Adenine (Gs-7340) and Other Tenofovir Phosphonoamidate Prodrugs by Human Proteases Mol Pharmacol 2008, 74, 92–100. [DOI] [PubMed] [Google Scholar]
- (29).Bhalerao UT; Rapoport H Stereochemistry of Allylic Oxidation with Selenium Dioxide - Stereospecific Oxidation of Gem-Dimethyl Olefins J. Am. Chem. Soc 1971, 93, 4835–4840. [Google Scholar]
- (30).Davey MS; Malde R; Mykura RC; Baker AT; Taher TE; Le Duff CS; Willcox BE; Mehellou Y Synthesis and Biological Evaluation of (E)-4-Hydroxy-3-Methylbut-2-Enyl Phosphate (Hmbp) Aryloxy Triester Phosphoramidate Prodrugs as Activators of V Gamma 9/V Delta 2 T-Cell Immune Responses J. Med. Chem 2018, 61, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ali Z; Shao L; Halliday L; Reichenberg A; Hintz M; Jomaa H; Chen ZW Prolonged (E)-4-Hydroxy-3-Methyl-but-2-Enyl Pyrophosphate-Driven Antimicrobial and Cytotoxic Responses of Pulmonary and Systemic Vgamma2vdelta2 T Cells in Macaques J. Immunol 2007, 179, 8287–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schwartz RH T Cell Anergy Annu. Rev. Immunol 2003, 21, 305–334. [DOI] [PubMed] [Google Scholar]
- (33).Farquhar D; Khan S; Srivastva DN; Saunders PP Synthesis and Antitumor Evaluation of Bis[(Pivaloyloxy)Methyl] 2’-Deoxy-5-Fluorouridine 5’-Monophosphate (Fdump) - a Strategy to Introduce Nucleotides into Cells J. Med. Chem 1994, 37, 3902–3909. [DOI] [PubMed] [Google Scholar]
- (34).Dickson JK; Biller SA; Magnin DR; Petrillo EW; Hillyer JW; Hsieh DC; Lan SJ; Rinehart JK; Gregg RE; Harrity TW; Jolibois KG; Kalinowski SS; Kunselman LK; Mookhtiar KA; Ciosek CP Orally Active Squalene Synthase Inhibitors: Bis((Acyloxy)Alkyl) Prodrugs of the Alpha-Phosphonosulfonic Acid Moiety J. Med. Chem 1996, 39, 661–664. [DOI] [PubMed] [Google Scholar]
- (35).Wiemer AJ; Shippy RR; Kilcollins AM; Li J; Hsiao CHC; Barney RJ; Geng ML; Wiemer DF Evaluation of a 7-Methoxycoumarin-3-Carboxylic Acid Ester Derivative as a Fluorescent, Cell-Cleavable, Phosphonate Protecting Group Chembiochem 2016, 17, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mehellou Y; Rattan HS; Balzarini J The Protide Prodrug Technology: From the Concept to the Clinic J. Med. Chem 2018, 61, 2211–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hill A; Khoo S; Fortunak J; Simmons B; Ford N Minimum Costs for Producing Hepatitis C Direct-Acting Antivirals for Use in Large-Scale Treatment Access Programs in Developing Countries Clin. Infect. Dis 2014, 58, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sofia MJ; Bao D; Chang W; Du JF; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang PY; Zhang HR; Bansal S; Espiritu C; Keilman M; Lam AM; Steuer HMM; Niu CR; Otto MJ; Furman PA Discovery of a Beta-D-2 ‘-Deoxy-2 ‘-Alpha-Fluoro-2 ‘-Beta-C-Methyluridine Nucleotide Prodrug (Psi-7977) for the Treatment of Hepatitis C Virus J. Med. Chem 2010, 53, 7202–7218. [DOI] [PubMed] [Google Scholar]
- (39).Foust BJ; Allen C; Holstein SA; Wiemer DF A New Motif for Inhibitors of Geranylgeranyl Diphosphate Synthase Bioorg. Med. Chem 2016, 24, 3734–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bryson A; Matthews RW Effects of Substituents on Pka Values of Meta Substituted 1- and 2-Naphthols Aust. J. Chem 1963, 16, 401–410. [Google Scholar]
- (41).Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI Nmr Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist Organometallics 2010, 29, 2176–2179. [Google Scholar]
- (42).Mackman RL; Ray AS; Hui HC; Zhang LJ; Birkus G; Boojamra CG; Desai MC; Douglas JL; Gao Y; Grant D; Laflamme G; Lin KY; Markevitch DY; Mishra R; McDermott M; Pakdaman R; Petrakovsky OV; Vela JE; Cihlar T Discovery of Gs-9131: Design, Synthesis and Optimization of Amidate Prodrugs of the Novel Nucleoside Phosphonate Hiv Reverse Transcriptase (Rt) Inhibitor Gs-9148 Bioorg. Med. Chem 2010, 18, 3606–3617. [DOI] [PubMed] [Google Scholar]
- (43).Camps F; Coll J; Parente A Selenium Dioxide Oxidation of Substrates with Acid Labile Groups Synthesis-Stuttgart 1978, 215–216.
- (44).Tang X; Demiray M; Wirth T; Allemann RK Concise Synthesis of Artemisinin from a Farnesyl Diphosphate Analogue Bioorg. Med. Chem. Lett 2018, 26, 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shippy RR; Lin X; Agabiti SS; Li J; Zangari BM; Foust BJ; Poe MM; Hsiao CC; Vinogradova O; Wiemer DF; Wiemer AJ Phosphinophosphonates and Their Tris-Pivaloyloxymethyl Prodrugs Reveal a Negatively Cooperative Butyrophilin Activation Mechanism J. Med. Chem 2017, 60, 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.