

Abstract
Adult zebrafish show a remarkable capacity to regenerate their spinal column after injury, an ability that stands in stark contrast to the limited repair that occurs within the mammalian CNS post-injury. The reasons for this interspecies difference in regenerative capacity remain unclear. Here we demonstrate a novel role for Fgf signaling during glial cell morphogenesis in promoting axonal regeneration after spinal cord injury. Zebrafish glia are induced by Fgf signaling, to form an elongated bipolar morphology that forms a bridge between the two sides of the resected spinal cord, over which regenerating axons actively migrate. Loss of Fgf function inhibits formation of this “glial bridge” and prevents axon regeneration. Despite the poor potential for mammalian axonal regeneration, primate astrocytes activated by Fgf signaling adopt a similar morphology to that induced in zebrafish glia. This suggests that differential Fgf regulation, rather than intrinsic cell differences, underlie the distinct responses of mammalian and zebrafish glia to injury.
Introduction
Spinal cord injury (SCI) in mammals is characterized by a lack of spinal neuron regeneration through the lesion and subsequent functional impairment caudal to the affected region. A significant barrier to mammalian axonal regeneration during SCI is the aggressive astrocytic gliosis that is invariably initiated at the injury site and results in glial scar formation (Stichel and Müller, 1998; Silver and Miller, 2004; Hu et al., 2010). Reactive astrocytes seal the wound, repair the blood–brain barrier and consequently prevent an intense inflammatory response occurring at the injury (Faulkner et al., 2004). However, this process creates a physical barrier for axonal regeneration. Activated astrocytes that compose the glial scar also secrete cytokines and produce cell adhesion and extracellular matrix molecules, which create a tissue microenvironment that regenerating axons are unable to traverse (McKeon et al., 1991; Stichel et al., 1999; Asher et al., 2000). The overall complexity of the different inhibitory processes induced by injury of the mammalian spinal cord has made it difficult to identify specific proregenerative processes that could be targeted for therapeutic gain.
In contrast to the lack of spinal cord regeneration evident in mammals, some anamniote animals, such as fish and some amphibians, regenerate damaged nerves and recover locomotor ability in a remarkably short period following SCI (Simpson, 1968; Bernstein and Gelderd, 1970; Zottoli et al., 1994; Becker et al., 1997). This difference in regenerative capacity occurs even though similar cell types are present in the spinal cords of species both capable and incapable of regeneration (Naujoks-Manteuffel and Roth, 1989; Bastmeyer et al., 1993).
In adult zebrafish, glial scarring is not associated with brain injury models (Zupanc and Clint, 2001) and fish glia can actively support both fish and mammalian nerve regeneration (Bastmeyer et al., 1993; Bernhardt et al., 1996; Schweitzer et al., 2003; Zukor et al., 2011). Collectively, this raises the possibility that differential regulation of glia behavior contributes to the regenerative capacity of the teleost CNS, in addition to the intrinsic property of regenerating axons themselves.
This study examines the role of Fgf signaling in glia morphogenesis during spinal cord regeneration in zebrafish. We show that unlike mammalian models of SCI, zebrafish regeneration proceeds in the absence of a glial scar. In contrast to the dense network of glial processes generated from hypertrophic stellate glia in mammalian injury, zebrafish glia form an elongated morphology that joins the resected regions of the spinal cord. Furthermore, these elongated glia form a bridge, over which regenerating axons actively regrow to reinnervate regions caudal to the injury. We show that glial activation and the consequent regeneration permissive morphology that glia produce is controlled by Fgf signaling. Furthermore, mammalian astrocytes can be activated by Fgf to produce a similar morphology, suggesting that differential Fgf regulation could underlie the distinct responses of mammal and zebrafish to SCI.
Materials and Methods
Zebrafish strains and treatments.
We used animals of either sex from GFAP:GFPmi2001 (Bernardos and Raymond, 2006), nestin:GFPzf168 (Lam et al., 2009), Isl1:EGFPrw0 (Higashijima et al., 2000), mpeg1:GFP (Ellett et al., 2011) and mpx:GFP (Renshaw et al., 2006) transgenic lines to visualize distinct cell populations and hsp70l:dn-fgfr1-EGFPpd1 (Tsai et al., 2008) and spry4−/−fh117 (obtained from the Zebrafish International Resource Center) as models of Fgf inhibition or gain of function respectively. All experiments were conducted in accordance with Monash University guidelines. The dominant-negative form of Fgfr1 was induced by applying heat shock to hsp70l:dn-fgfr1-EGFP transgenic animals. As controls, wild-type animals were similarly heat shocked. Animals were exposed to an increased temperature from 26°C to 38°C (Lee et al., 2005) and remained at 38°C for 60 min, 4 h before SCI. Fish were exposed daily to this heat shock regime and spinal cords collected at indicated time points. For delayed heat shock, animals underwent the same procedure starting at 6 d post-SCI.
Spinal cord lesion.
Spinal cords of zebrafish were lesioned as described previously (Becker et al., 1997). Briefly, adult fish (3–6 months old, either sex) were anesthetized in 0.033% tricaine methanesulfonate (MS-222) in fish tank water, until respiratory movements of the opercula stopped (3–5 min). Halfway between the dorsal fin and the operculum, corresponding to the eighth vertebra (∼5 mm caudal to the operculum) of the spinal cord, a longitudinal incision was made through the muscle layer, and the vertebral column was exposed by holding the muscle tissue aside. Then the vertebral column was cut completely with micro-scissors. The wound was sealed with a drop of 3M Vetbond. The gills of the fish were flushed in a tank of fresh fish water by gently pulling the fish through the water. Fish resumed breathing within a few seconds.
Retrograde and anterograde axonal tracing.
A 1 μl drop of Fluoro-Ruby (tetra-methyl rhodamine dextran, molecular weight, 10,000 kDa) was air-dried onto pieces of Parafilm to obtain larger crystals (Becker et al., 1997). These crystals were inserted into the exposed spinal cord 3 mm downstream or upstream of the original lesion. Animals were killed and spinal cords processed 24 h after dye insertion.
Tissue preparation.
At different time points after SCI, fish were deeply anesthetized with an overdose of MS-222. The brain and spinal cord were exposed and fixed for 2 h in 4% PFA in PBS at room temperature. The brain and the spinal cord were subsequently dissected out and postfixed for 2–3 h at room temperature followed by immersion in 30% sucrose in PBS overnight at 4°C, before embedding in OCT. Spinal cords were cryostat sectioned at 20 μm thickness.
Immunohistochemistry.
Sections were labeled using standard immunohistochemical procedures to determine expression and localization of different proteins at the lesion site. Sections were postfixed for 10 min in 4% PFA, followed by incubation in blocking solution [PBS-Triton X-100 containing 5% normal goat serum; Invitrogen] for 1 h at room temperature. Primary antibodies were diluted in blocking solution and sections were incubated overnight at 4°C. After washing, sections were incubated for 2 h at room temperature with secondary antibodies diluted in blocking solution. Sections were mounted in Fluoromount (Dako). Primary antibodies used were: rabbit anti-FgfR2 (1:2000; Sigma), mouse anti-GFAP (1:1000; Invitrogen), rabbit anti-GFAP (1:1000; Dako), mouse anti-vimentin (1:500; Millipore), mouse anti-NeuN (1:1000; Millipore), rabbit anti-pMAPK (1:1000; cell signaling), rabbit anti-MAPK (1:1000; cell signaling), rabbit anti-GLAST (1:500; Abcam), mouse anti-bromodeoxyuridine (BrdU) (1:400, Roche), rabbit anti-GFP (1:500; Invitrogen), and rabbit anti-Ki67 (1:300, Thermo Scientific). Antigen retrieval was performed by incubating the sections for 15 min in 2 m HCl before blocking for BrdU immunohistochemistry. Secondary antibodies used were: goat anti-rabbit Alexa Fluor 488 (1:1000; Invitrogen), goat anti-rabbit Alexa Fluor 594 (1:1000; Invitrogen), goat anti-mouse Alexa Fluor 488 (1:1000; Invitrogen), and goat anti-mouse Alexa Fluor 594 (1:1000; Invitrogen).
SU5402 treatment.
A single injection of 1 μl of 5 μg/μl SU5402 (Calbiochem) in DMSO (Sigma) or DMSO only (controls) was injected to the lesion site straight after spinal cord transection.
BrdU application.
Intraperitoneal injections of 50 μl of BrdU (2.5 mg/ml; Sigma) were performed at 0, 2, or 4 d after lesion or in control nonlesioned fish. For quantitation of the post-injury proliferation time course, BrdU (2.5 mg/ml) was injected 24 h before fixation at each time point.
Fgf8 injections.
Recombinant human Fgf8 (0.14 mg/injection/fish; Yan et al., 2000) was injected intraperitoneally into Tg(GFAP:GFP) fish every second day starting immediately after SCI for 24 h or 5 d. Human Fgf8 shows 76% protein homology with zebrafish Fgf8.
Locomotor analysis.
A scale from 1 to 5 was designed to assess fish motor function improvement at different time points after spinal cord transaction: 1, body caudal to the lesion was paralyzed and the fish lay on the bottom of the tank with operative side upward; 2, fish were able to move forward only with body movement coordinated rostral to the lesion site while areas caudal to the lesion site were paralyzed; 3, turning was accomplished by a series of small jerky movements which brought the tail into line for the next series of forward movements; 4, fish were able to move musculature downstream of the lesion site and turns became more coordinated, including generating whole-body rotation to make turns; 5, swimming of the operated animals could not be distinguished from normal. When the movement of the swimming was coordinated correctly but the swimming was slower with the more notable reciprocal movement, the fish scored (4.5).
Probe generation and in situ hybridization.
Antisense mRNA templates were either amplified with Platinum Taq (Invitrogen) [Sprouty4 forward: CACGGTGCACTCCTTTGG, reverse: GACAGAAAGTGCAGCGAA; Fgf3 forward: CTGCTCTTGTTGTTACTGAGC, reverse: TGTCAGCCCTTCTGTTGTGG and cloned into pGEM-T Easy (Promega) or constructed from zebrafish EST clones (Pea3 IRBOp991G0430D Imagenes; Erm cb805 Imagenes; Fgf8 fb73a06 Addgene).
Plasmids were linearized, transcribed, and labeled, using T7 or SP6 polymerase (Roche) and a DIG RNA labeling mix (Roche). In situ hybridization was performed by standard procedures on 30 μm sections. Following staining, tissues were imaged using a Z1 AxioImager compound microscope. Before performing in situ hybridization, sections with cells expressing GFAP:GFP or with tetramethylrhodamine dextran (TMRD)-traced neurons were imaged allowing us to examine gene expression of the same glia or neuronal cells before and after in situ staining. In addition, colabeling with NeuN on fgf3 in situ were performed and imaged by confocal microscopy.
Microscopy.
Sections were examined by bright-field or fluorescence microscopy using a Z1 AxioImager (Zeiss) epifluorescence microscope. Photomicrographs (1300 × 1030 dpi) were obtained with various Plan-Neofluar objectives (Zeiss), and acquired as digital images using an AxioCam (Zeiss) digital camera with AxioVision software (v. 4.4; Zeiss). To confirm colocalization between different proteins, optical sections of the samples were acquired on a Z1 AxioImager (Zeiss) microscope equipped with the Apotome module and a 40× objective, using AxioVision software.
BrdU-positive cell counts.
The number of BrdU-labeled cells in the spinal cord sections were counted in a 200 μm2 grid located ∼500 μm proximal to the lesion site from both sides of the lesion in every second serial longitudinal 20 μm section. Results were expressed as the mean ± SEM (n = 4 per group).
p-MAPK intensity measurement in glia.
Average fluorescent intensity after immunostaining against p-MAPK was quantified using ImageJ. Glia with processes at the lesion site were outlined and fluorescent intensity was measured. Results are expressed as mean ± SEM.
Statistical analysis.
One-way ANOVA followed by multiple-comparisons using the Tukey's test was used to identify significant differences between groups in the experiments shown below in Figures 2b and 5f,h,q,t (*p < 0.05, **p < 0.001).
Figure 2.
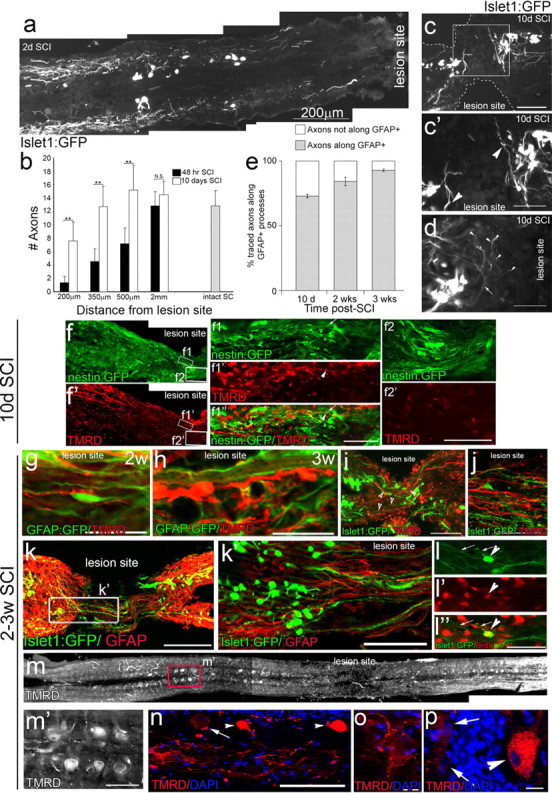
Axonal retraction and regeneration after SCI. a, After spinal cord lesion, injured axons initially retract as evident at 2 d post-SCI. b, Quantitation of the number of neuronal process at different distances from the lesion site reveals a significant increases in regeneration and sprouting toward the lesion site at 10 d (white bars) compared with 2 d (black bars) post-SCI at all distances analyzed (n = 7 per group, **p < 0.001, n.s., not significant). c, d, Examples of regenerating axons at 10 d post-SCI from neurons at the lesion edge. At this time, when glial bridges have not yet formed, new neurites present at the lesion are not yet aligned in the anterior–posterior axis, but instead growing perpendicular (arrowheads, c′, d). New small-bodied neurons near the lesion edge show extensive sprouting with multiple branches (arrowheads, d) arising from individual neurites (arrow). f, In nestin:GFP fish, low and high-power images of traced regenerated neurons, showing that regenerating axons at 10 d post-SCI have not yet entered the lesion site (f2). At the lesion, traced axons (arrowhead, f1) do occasionally align with glial processes (nestin positive—arrow, f1) (n = 5), whenever present. e, Quantitation shows that at all times post-SCI the majority of axons regenerate along glia processes. g–k, By 2 or 3 weeks post-SCI, the majority of TMRD-labeled neurites extend across the lesion site along GFAP-positive glial processes (2 weeks, n = 7; 3 weeks, n = 10) (g, h). New islet1:GFP-labeled neurons also regenerate along GFAP-positive glia processes (k, k′). Processes of islet1:GFP-labeled neurons, however, do not colabel with the tracer, suggesting that although they have grown across the lesion, they have not reached far enough downstream to contact the tracer injection site (n = 11) (i, j). l–l″, A proportion of new neurons with regenerating processes (arrows) at the lesion site incorporate BrdU (arrowhead). Retrograde label accumulates both in mature neuron cell bodies upstream of the lesion site (m, m′, n, p, arrowheads) and also in labeled processes terminating around unlabeled neuronal cell bodies (n, arrow; o). Scale bars: a, 200 μm; c, i, 100 μm; c′, d, f1–f1′, f2–f2′, i′, j–l′, m′, 50 μm; g, h, 25 μm; n–p, 10 μm.
Figure 5.
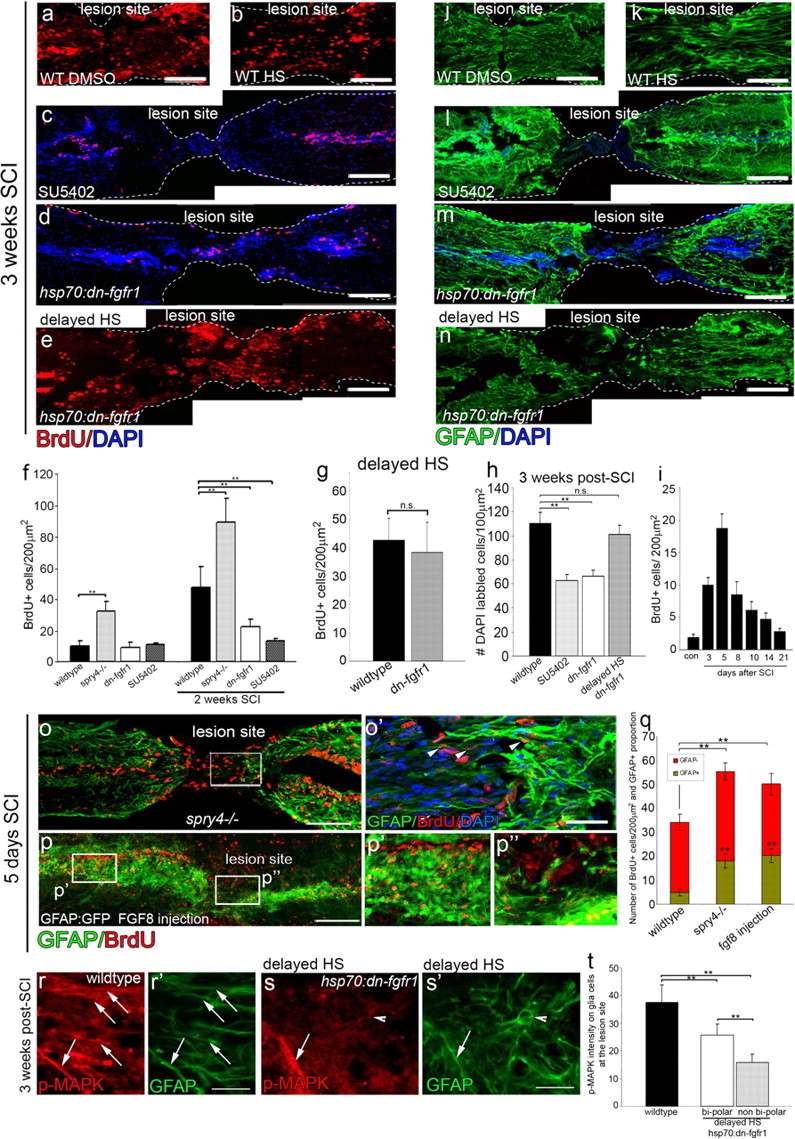
Fgf signaling regulates glia cell proliferation and migration. a–e, At 3 weeks post-SCI, Fgf inhibition by SU5402 injection (n = 6) (c) or heat shock of hsp70l:dn-fgfr1-EGFP (n = 7) (d) causes a significant reduction (f) in proliferation (assayed by BrdU incorporation) compared with WT DMSO (a) or wt heat shock (b) fish. In contrast, the delayed heat shock of hsp70l:dn-fgfr1-EGFP (initiated only at 6 d post-SCI) does not show a reduction in proliferation (e, g). h–n, Similarly, the resulting migration and accumulation of cells (DAPI labeled) at the lesion site is also reduced in SU5402-injected or heat-shocked hsp70l:dn-fgfr1-EGFP (h) fish compared with WT DMSO (n = 6) (j) or heat-shocked fish (n = 6) (k). Again delaying the heat shock by 5 d completely ameliorates the effect and there is no significant change in the number of cells that migrate and accumulate at the lesion in this treatment (n = 6) (h, n). i, Assessing the degree of proliferation across different time periods reveals that the bulk of the proliferative response occurs within the first 5 d post-SCI (n = 3 per time point). o–q, In contrast, Fgf signaling upregulation in spry4−/− mutants (o, n = 7) or Fgf8-injected fish (p, n = 6) results in a significant increase in proliferation already by 5 d post-SCI (q). Additionally, glia show an accelerated differentiation and earlier expression of GFAP:GFP in both of these increased Fgf signaling conditions (o–q). r–t, Differentiation of the bipolar glial morphology is correlated with MAPK signaling activity in wild-type and hsp70:dn-fgfr1 fish with delayed heat shock starting 6 d after SCI. Scale bars: a–e, j–n–p, 100 μm; o′, r, s′, 50 μm. Results are presented in f–i, q, and t as mean ± SEM, **p < 0.001, n.s., not significant.
For experiments with pairwise comparisons, the Student's t test was used (**p < 0.001). This includes experiments shown below in Figures 5g, 6f,j, and 7d,e.
Figure 6.
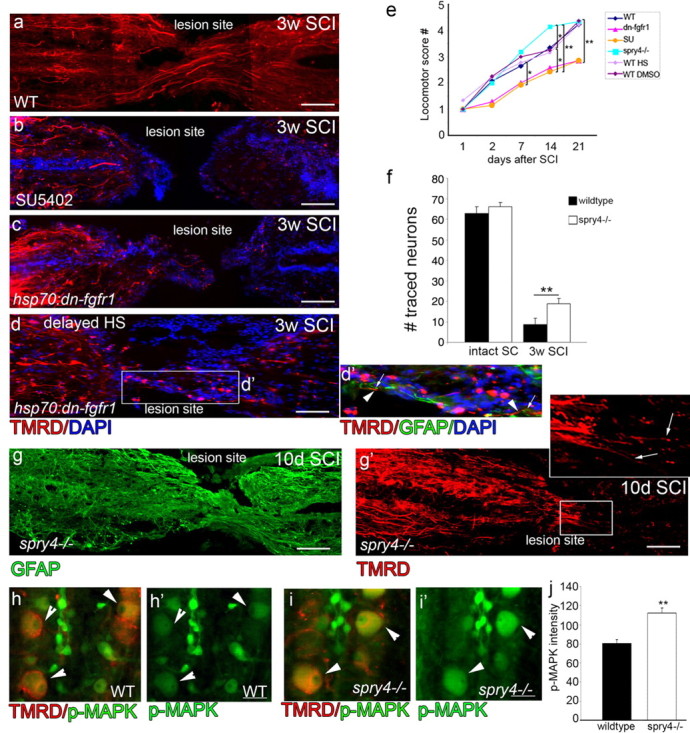
Axonal regeneration depends on Fgf-induced glial bridge formation. a–d, TMRD traced regenerated axons in different conditions. In WT, axons regenerate across the lesion site and take up the TMRD tracer (n > 15) (a). In contrast, in fish with Fgf signaling inhibition either by SU5402 injections (n = 6) or by heat shock of hsp70l:dn-fgfr1-EGFP fish (n = 10), no TMRD-labeled axons cross the lesion site. (d, d′) in delayed heat shock some TMRD-labeled axons (d′, arrowheads) are crossing along elongated glia (d′, arrows). e, Locomotor recovery assays (n > 10 animals from each genotype or treatment group; *p < 0.05, **p < 0.001) is significantly reduced in hsp70l:dn-fgfr1-EGFP and SU5402 compared with different wild-type controls and spry4−/−, which shows improved motor function 3 weeks after SCI. g, g′, At 10 d post-SCI, glia at the lesion express GFAP and exhibit bipolar morphology in spry4−/− fish(g).TMRD-labeled regenerating axons that are present at the lesion can enter the site aligned correctly in the anterior–posterior position (arrows in g′ inset) (n = 15). f, By 3 weeks post-SCI in spry4−/− fish there is a significant increase in axonogenesis with more neurons labeled by tracer injections (n = 8 from each genotype) (**p < 0.001). h–j, Compared with control (h), the increase in neural regeneration assessed by TMRD tracer injection in the spry4−/− fish (i) is matched by an increase in activated MAPK signaling in these traced neurons (h, i′, arrowheads) as quantified (j) (n = 4 from each genotype), **p < 0.001. Scale bars: a–e, g, g′, 100 μm; h, i′, 10 μm. Caudal side of the spinal cord is to the left in a–g′.
Figure 7.
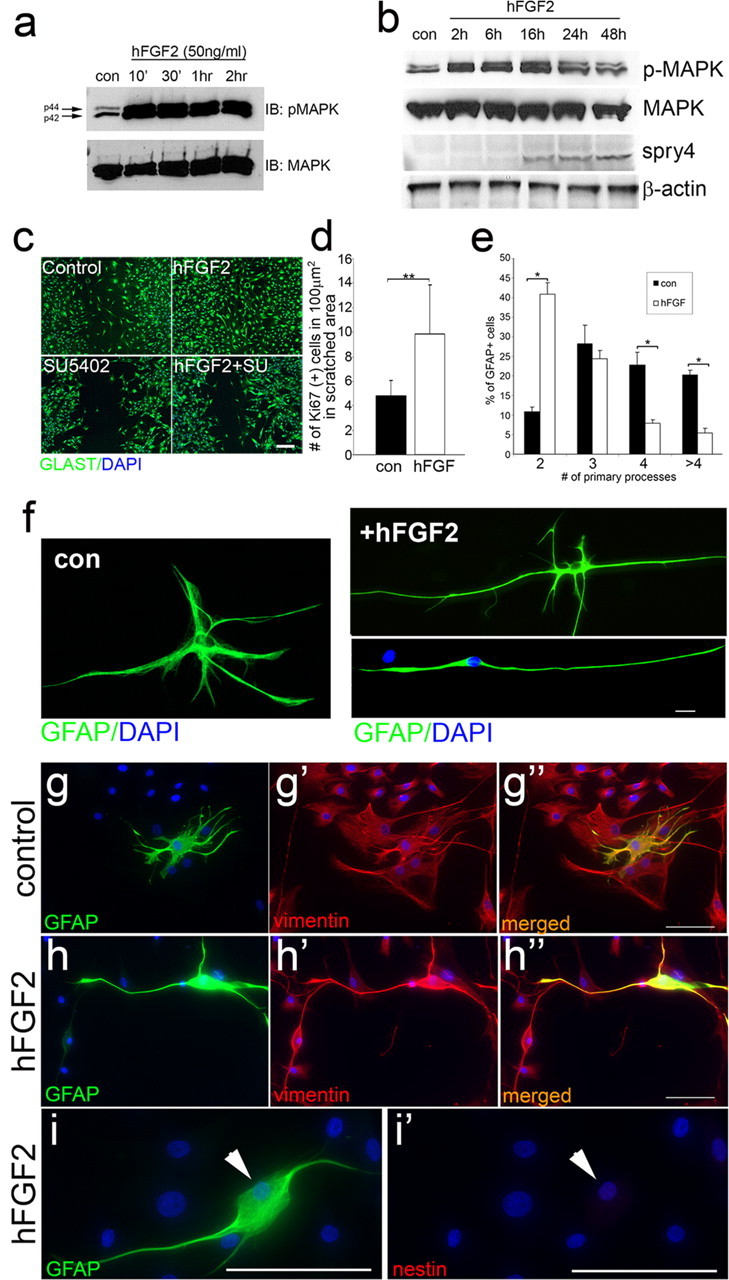
Fgf promotes radial glia morphology in primate astrocytes. a, b, Western blot analysis for p-MAPK activation during short term (a) or long-term (b) hFGF2 addition in primate astrocytes, shows increased levels of p-MAPK and Spry4 protein levels in these cells. β-Actin and MAPK are loading controls. c, Scratch wound assay on primate astrocytes visualized by GLAST labeling after 48 h. Addition of hFGF2 increases astrocyte migration into the wound area, whereas SU5402 and hFGF2+SU5402 treatment have little effect on astrocyte migration. d, Quantitation of the number of currently proliferating cells (mean ± SEM) shows that Fgf induces proliferation (n = 3 experiments, **p < 0.001). e–i, After 7 d in culture. e, Quantitation of the number (mean ± SEM) of primary processes extending from the cell body in control (con) and hFGF2-treated astrocytes (n = 3 experiments; *p < 0.001) shows an increased number of cells with bipolar morphology in hFGF2-treated cultures. f, Cellular morphology in control and hFGF2-treated astrocytes. High-power images of control astrocytes show normal multipolar stellate morphology compared with hFGF2-treated astrocytes, which have either two main long primary process with some short secondary extensions or just two long primary processes. g–i, All primate astrocytes in control and hFGF2 treatment express vimentin (g, h). Vimentin, but not nestin (i) is expressed in primate astrocytes after 7 d of hFGF2 treatment. Scale bars: c, g, h (low magnification), 50 μm; f, i (high magnification), 10 μm.
Behavioral analysis was performed blind and statistical analysis was performed using the nonparametric Mann–Whitney U test to identify significant differences between groups (*p < 0.05, **p < 0.001) in Figure 6e (see below).
Western blot analysis.
p-MAPK activation was analyzed via Western blotting in zebrafish 24 h post-SCI and in primate astrocytes with or without 50 ng/ml hFgf2 (R&D Systems) as described previously (Goldshmit et al., 2004; Goldshmit and Bourne, 2010). Analysis of p-MAPK activation by Western blotting was performed via dissection in ice-cold lysis buffer 1 mm on each side of the lesion. For MAPK activation in primate astrocyte cultures, cells were incubated in hFgf2 for the indicated time. Cells were prestarved of serum for 2 h and washed in cold PBS, and protein extracts were made.
Scratch wound and dedifferentiation assay.
Primary marmoset astrocyte cultures preparation and scratched wound assay was performed as previously described (Goldshmit and Bourne, 2010), with or without 50 ng/ml hFgf2 and/or SU5402 (25 μm; Calbiochem). Ki67-labeled cells were counted in a scratched area (100 mm2 frame) 48 h after plating.
For dedifferentiation assay cells were plated in low density (2 × 103 cells/well) as described in supplemental extended experimental procedures for 7 d. The number of cells with 2, 3, 4, or more than 4 primary processes extending from the cell body were counted from 3 different experiments, with two repetitions in each treatment per experiment. Results are expressed as the mean ± SEM. Significance of results was analyzed using the Student's t test.
Spinal cord transection by laser ablation.
Four dpf Tg(gfap:GFP) transgenic zebrafish were anesthetized in 0.033% tricaine and mounted in 1% low melting point agarose in E3 medium (Hall et al., 2007). A ridged silicone mounting plate was used to orient fish, such that the lateral surface was facing up [constructed using Sylgard 184 (Dow Corning) from a standard microinjection plate mold (TU-1; Adaptive Science Tools)]. Under a 10× dry objective (Zeiss Axioskop upright microscope), GFAP-GFP-positive spinal cords were transected using several rapid pulses of a 337 nm cartridge nitrogen laser (MicroPoint; Photonic Instruments Inc.), pumped through a 5 mm solution of coumarin 440 (Sigma-Aldrich) in methanol. The degree of ablation was controlled by monitoring the green channel. Following transection, animals were removed from the agarose, recovered from anesthesia and maintained in individual wells of a 24-well tissue culture plate.
Time-lapse photomicroscopy.
Following laser spinal cord transection, animals for time-lapse photomicroscopy were transferred to a 150 × 25 mm Petri dish containing 230 ml of E3 medium supplemented with 0.0013% tricaine. Individual fish were imaged on a Zeiss Z1 AxioImager upright microscope using a 20× immersion lens and Zeiss Axiovision software. At 10 min intervals, 16-plane Z-stack optical sections (64 μm range) were taken of the region of interest over a period of 24 h. The 16-bit images were exported as tif sequences and processed using Fiji (Schindelin et al., 2008) using the correct 3D drift plugin.
Reverse transcriptase-PCR of spinal cord tissue.
Approximately 0.5 g of brain, hindlimb muscle, liver and spinal cord tissues were dissected from C57BL/6 mice and RNA was extracted using TRI Reagent (Sigma). Total RNA was reverse transcribed using the Superscript III RT kit (Invitrogen) and the products were analyzed by reverse transcriptase (RT)-PCR. PCR products were amplified for 31 cycles and separated on a 2% agarose gel. GAPDH expression was evaluated as an internal control. The primer sequences used were Spry4 forward: CCTGTGAATCCCAGCTCAGT and reverse: GCAGGTAGCACAGCAGACAG; GAPDH forward: ACCACAGTCCATGCCATCAC and reverse: TCCACCACCCTGTTGCTGTA.
Spinal cord hemisections in mice.
As we described previously (Goldshmit et al., 2004), adult C57BL/6 mice (20–30 g) were anesthetized with a mixture of ketamine and xylazine (100 mg/kg and 16 mg/kg, respectively) in PBS, which was injected intraperitoneally. The spinal cord was exposed at the low thoracic to high lumbar area, at level T12. Fine forceps were used to remove the spinous process and lamina of the vertebrae and a left hemisection was made at T12. A fine scalpel was used to cut the spinal cord, which was cut a second time to ensure that the lesion was complete, on the left side of the spinal cord, and the overlying muscle and skin were then sutured. Sections were examined using standard immunohistochemical procedures. Primary antibodies used were rabbit anti-Spry4 (1:200; Santa Cruz Biotechnology); mouse anti-GFAP (1:1000; Invitrogen).
Results
Glial cells proliferate, migrate and differentiate to form a glial cell bridge after spinal cord injury in zebrafish
To examine the role of glial cells during spinal cord regeneration in zebrafish we established an adult spinal cord resection model. Glia at different stages of differentiation were distinguished using transgenic zebrafish expressing GFP under the control of either the GFAP promoter Tg(GFAP:GFP)mi2001 (Bernardos and Raymond, 2006) or nestin promoter Tg(nestin:GFP)zf168 (Lam et al., 2009). Mature, differentiated glia express high levels of GFAP and low levels of nestin. Upon stimulation, activated glia first lose their GFAP expression and upregulate nestin expression as they dedifferentiate to generate neurons or radial glia either directly or following proliferating (Reimer et al., 2008), after which they mature. Thus, these lines allow us to follow these distinct glia populations after spinal cord injury. In conjunction with BrdU labeling performed at a discrete early temporal window post-SCI we could identify four distinct phases of glial cell behavior post-SCI.
Phase 1: Proliferation and initial migration (3–5 d post-SCI)
This phase is characterized by an initial glial proliferative response. In control uninjured spinal cords and 24 h post-SCI, the vast majority (95.7%) of BrdU-positive proliferating cells are GFAP-positive radial glial cells located within the central canal (Fig. 1a–c). Three days post-SCI, proliferation immediately adjacent to the lesion site increases significantly occurring almost exclusively in GFAP-positive glial cells in and around the central canal (93% of BrdU-positive cells) of the spinal column (Fig. 1d), similar to the response evident in crush injury models in zebrafish (Hui et al., 2010). The few BrdU-positive cells found outside of the central canal are primarily macrophages (82.4%), and very rarely neutrophils, as quantified using transgenic lines marking macrophages (Tg(mpeg1:GFP); Ellett et al., 2011) or neutrophils (Tg(mpx:GFP); Renshaw et al., 2006). Five days post-SCI, newborn (BrdU positive) cells accumulating at the edge of the lesion express very low levels of GFAP (Fig. 1e). Some BrdU-negative cells also start accumulating at the lesion site. At this stage cells at the lesion express high levels of vimentin and are beginning to reduce their GFAP expression (arrowhead vs arrows, Fig. 1f). These cells also express high levels of nestin (Fig. 1g). This analysis reveals that both proliferative and nonproliferative cells at the lesion arise from radial glia and that they migrate out to the lesion site at this stage. Quantification at different time points post-SCI reveals that the vast majority of proliferating cells in the central canal are indeed GFAP-positive glia. By the time these newborn cells migrate to the lesion, they have decreased GFAP expression, but upregulate GFAP again, as they differentiate into mature glia at 3 weeks post-SCI, where the majority of newborn cells at the lesion once again express GFAP (Fig. 1h).
Figure 1.
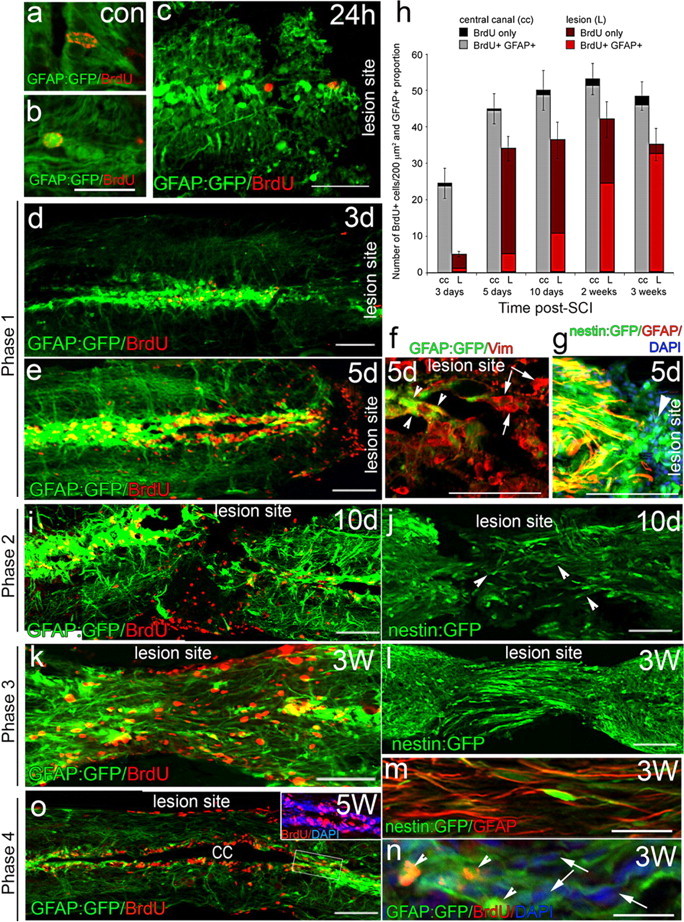
Radial glia cells along the central canal proliferate after injury and migrate to lesion site. a–e, BrdU is incorporated in a small number of radial glia (GFAP expressing) along the central canal in intact spinal cord (a, b) and 24 h (24 h) after SCI (c). Phase 1: Proliferation increases and BrdU-labeled cells migrate to the lesion site at 3 d (d) and 5 d (e) post-SCI. f, g, The cells that accumulate at the lesion site 5 d after SCI colabel with vimentin and nestin (g, arrowhead). Some of these cells still express GFAP (f, arrowheads), whereas others have lost GFAP expression (f, arrows). h, Quantitation of BrdU-expressing cells either along the central canal (cc) or at the lesion site (L) shows proliferation (increase in BrdU cells) primarily in the first 5 d post-SCI. The vast majority of proliferating cells in the central canal are glia (GFAP positive). Cells accumulating at the lesion site slowly reexpress GFAP as they differentiate into mature glia and by 3 weeks post-SCI the vast majority of cells at the lesion are GFAP positive again. Results are expressed as the mean ± SEM. for each group (at least n = 5). i, j, Phase 2: Glia at the lesion site that express nestin and start expressing GFAP at 10 d post-SCI have short bipolar processes aligned along the anterior–posterior axis (arrowheads). k–n, Phase 3: By 3 weeks post-SCI, the glia that fill the lesion site express GFAP (k, m, n) and still express nestin (l, m). Higher-power view shows that nestin and GFAP colocalize in the same cells (m) and that these cells now have a long bipolar morphology with their processes spanning the lesion site. n, Bipolar glia arise both from proliferative cells (BrdU labeled—arrowhead) and some from nonproliferative (BrdU negative—arrows) cells. o, Phase 4: By 5 weeks post-SCI, the lesion site is remodeled and the central canal reconnected. The lesion site is only distinguishable from neighboring regions by the BrdU-labeled cells. Caudal side of the spinal cord is to the left in all panels. At least n = 10 animals from each line for each time point. Scale bars: b, n, 25 μm; c, f, g, 50 μm; d, e, i, k, o, 100 μm.
Phase 2: Migration and differentiation (7–10 d post-SCI)
This phase is characterized by continued glial migration into the lesion site and initiation of glial bipolar morphology. Differentiating newborn glia (BrdU positive, GFAP negative) migrate to occupy the lesion site (Fig. 1i). These young glia express high levels of nestin and possess short processes with a bipolar morphology aligned in the anterior–posterior (AP) direction (Fig. 1j, arrowheads).
Phase 3: Glial bridge formation (2–3 weeks post-SCI)
The morphogenesis of this next phase departs dramatically from glial cell behavior in mammalian SCI models and is characterized by the formation of glial bridges across the lesion. Maturing glia (increasing GFAP and still express nestin) now exhibit long bipolar processes, which start to fully elongated along the AP axis to bridge across the two sides of the transected spinal cord (Fig. 1k–n). The elongated glia are generated from proliferative (BrdU positive) and also from nondividing glia within the central canal (Fig. 1n). The formation of this glial bridge defines phase 3 and constitutes the main difference between the zebrafish and mammalian response to SCI.
Phase 4: Remodeling (>4 weeks post-SCI)
The final phase of zebrafish spinal cord regeneration is characterized by considerable cellular remodeling at the lesion site resulting in reconnection of the central canal and axonogenesis across the lesion. Five weeks post-SCI the glial bridge is dismantled centrally and the spinal cord is fully reconstructed, including reconnection of the central canal. BrdU-positive glia remain evident within the central canal and outer surfaces of the spinal cord (Fig. 1o). The lesion site can now only be identified by the presence of BrdU-positive cells (Fig. 1o) Continuous time-lapse imaging of injured spinal cords of larval Tg(GFAP:GFP) zebrafish reveals that glial bridge formation during larval regeneration follows the same morphological process, albeit within a much accelerated time frame (Movie 1).
Movie 1.
Spinal cord lesions are bridged by glia within 24 h of injury in 4-d-old zebrafish. Time-lapse sequence illustrating GFAP-GFP-positive cell dynamics in a 4-d-old zesbrafish from time of ablation (0 h post-injury; hpi) to 24 h post-injury (lateral view, green channel). GFAP-GFP-expressing cells extend bipolar processes through the lesion site, thus forming a glial bridge between the two sides. Individual cells can be seen actively migrating into the zone of ablated tissue. During the course of the movie the distance between anterior and posterior sides of the spinal cord becomes progressively smaller, as the two sides converge.
Glial cell bridges provide a permissive environment for axonal regeneration
An analysis of axonal regeneration in response to spinal cord resection was performed in Tg(islet1:GFP)2rw012 transgenic zebrafish, in which all new and some mature motor and sensory neurons are labeled (Uemura et al., 2005, Reimer et al., 2008). Analysis of the initial response (2 d) post-SCI revealed that injured axons retract a considerable distance back from the lesion site—at least 500 μm (Fig. 2a). Axonogenesis then initiates, resulting in an increase of islet1:GFP-labeled neurites at various distances from the lesion (Fig. 2b). By 10 d post-SCI these regenerating axons can be detected at the edge of the resection zone (Fig. 2c,c′,d). Additionally to islet1:GFP-positive neurons from upstream (mature neurons), at 10 d post-SCI, we can also visualize new neurons (small islet1:GFP-positive somas at the lesion site), which contribute to axonogenesis (Fig. 2c,c′). Strikingly, the few axons that are present at the lesion site at this stage do not enter the lesion zone but are instead observed to grow perpendicular to the lesion edge, suggesting that at this phase of regeneration they are unable to enter and cross the lesion zone (Fig. 2c,d, arrowheads). Neurites from individual neurons can be seen branching and sprouting processes near the lesion (Fig. 2d; arrow shows primary neurite, arrowhead shows branches of the same neurite).
We next examined the relationship between axonal regeneration and glial bridge formation, by performing axonal tracer experiments in Tg(GFAP:GFP) and Tg(nestin:GFP) transgenic zebrafish. Axons were traced with TMRD, injected 3 mm rostral or caudal to the lesion site 10 d or 2/3 weeks post-SCI. Tracing at 10 d post-SCI, a phase post-injury when bipolar glial cells are present but have not yet bridged across the lesion site, reveals that descending and ascending axons that are still regenerating toward the lesion site as well as those that have already reached the lesion, grow along elongated nestin-positive glial processes, wherever these are present at the borders of the lesion zone (Fig. 2e,f,f1). Consistent with our observations in the Tg(islet1:GFP) zebrafish, regenerated axons present at the lesion fail to extend beyond glial processes into the lesion site (Fig. 2f2). By 2 weeks post-SCI, when bridging GFAP-positive glia are first detected at the lesion site, regenerating axons partially extend along bridging glia into the lesion (Fig. 2e,g). Furthermore, crossing axons were restricted exclusively to areas where bipolar glia bridges had already formed. Three weeks post-SCI, traced ascending and descending axons can be detected that traverse the injury site along glial bridges beyond the lesion site (Fig. 2e,h). Thus, regenerating axons pathfind directly through the lesion site in a manner that correlates both spatially with the presence of bipolar glial processes and also temporally, traversing into and through the lesion site only after glial bridges have been established.
Since we observe axonogenesis from both upstream mature neurons as well as new neurons at the lesion site (Fig. 2c,i), we next examined whether the initial wave of axonogenesis across newly formed glial bridges originated from new or preexisting neurons. At 3 weeks post-SCI, a significant level of neurogenesis (revealed by islet1 expression) occurs at the lesion site (Fig. 2i–l) consistent with observations from previous studies (Reimer et al., 2008, 2009). Axonogenesis from young neurons (islet 1 positive with small somas) and regenerating axons from mature neurons (anterogradely traced with TMRD) both extend across the lesion site along bipolar glial processes (Fig. 2i–l). Tracing by caudal TMRD injection reveals that regenerated axons that extend through and at least 2–3 mm further downstream of the lesion, belong exclusively to mature preexisting neurons with large somas, located distant from the lesion site (Fig. 2m,m′). These traced mature neurons do not colabel with islet:GFP, which is expressed primarily within small, newborn neurons (Fig. 2i,j). Thus, at 3 weeks post-SCI, the new axons from newborn neurons are present within the lesion, but have not extended enough to reach the site of the tracer injection, 2–3 mm downstream of the lesion. At this time, the new islet1:GFP-labeled neurites comprise ∼41 ± 7.1% SEM (n = 8) of processes present within the lesion site, while the remainder are TMRD labeled.
Tracer taken up caudal to the lesion reveals two distinct accumulation patterns. Labeling of regenerated descending axons accumulates in the neuronal cell bodies upstream of the lesion site (Fig. 2n,p, arrowheads), which are found among unlabeled neurons whose processes do not extend to the tracer injection site (Fig. 2n,o, arrows). Additionally, we observed tracer accumulating around neuronal cell bodies, in what appear to be axonal terminals (Fig. 2n,o), which we believe represent examples of ascending axons that have regenerated through the lesion site and terminated on target neurons.
Our data reveals that axonogenesis is initiated within the first 10 d from both new neurons near the lesion and mature neurons distant to the lesion site. The furthest extending regenerated axons at 3 weeks post-SCI primarily originate from mature neurons. However, regardless of the origins of regenerating axons, initial axonal regeneration of both ascending (caudal neurons) and descending (rostral neurons) track neurons occurs along glial bridges.
Fgf signaling is activated within glial cells at the lesion site
Our characterization of the unique set of morphogenetic processes that glia undergo after SCI in the zebrafish spinal column led us to examine the nature of the molecular signals triggered at the wound site that promote glial bridge formation. Previous studies have identified fgf3 as one of the genes induced at the site of SCI in zebrafish (Reimer et al., 2009). Fgf signaling presents as a good candidate pathway to modulate glial cell behavior, as Fgf2 has previously been implicated in locomotor recovery and the reduction of astrocytic gliosis after spinal cord contusion in rodents, although the mechanistic basis of this recovery has not been determined (Lee et al., 2008; Tsai et al., 2008; Kasai et al., 2010).
We examined the expression of known neural Fgf ligands, fgf8a and fgf3, and their downstream targets pea3, erm and spry4 after SCI. In uninjured spinal cords, fgf8a is expressed at a low level on GFAP-positive glial cells bordering the central canal (Fig. 3a–a⁗), whereas fgf3 and spry4 are expressed at a basal level within large neurons (large somas found more lateral to the central canal), but not glia (Fig. 3b,c). pea3 and erm are expressed at barely detectable levels within the intact spinal cord (Fig. 3d,e). Three days post-SCI, fgf8a, fgf3, spry4, pea3 and erm are strongly induced in GFAP-positive glial cells in the central canal adjacent to the lesion (Fig. 3f–j). Expression of fgf8a, pea and erm is maintained at 2 weeks post-SCI, mainly in glia positioned around the central canal within the regenerating lesion (Fig. 3k,k′). In contrast, at 2 weeks post-SCI, fgf3 and spry4 expression are decreased in glia bordering the central canal, while increased in neurons (NeuN positive, Fig. 3l,m). At 3 weeks, both fgf3 and spry4 are upregulated in regenerating neurons that are labeled by TMRD tracer injections (Fig. 3n,o).
Figure 3.
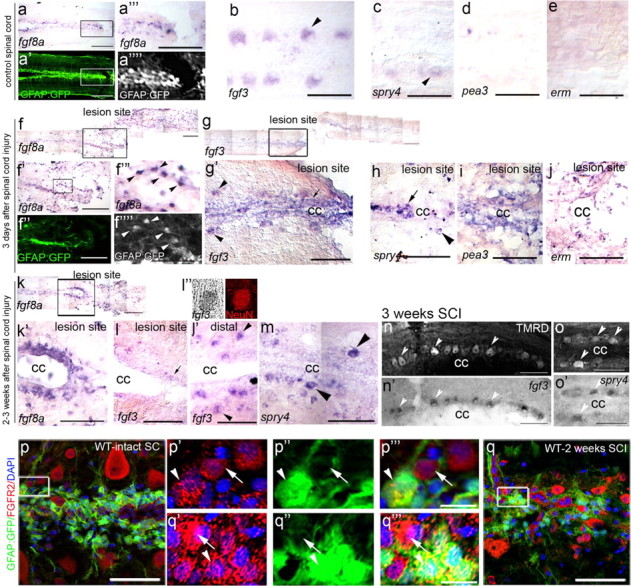
Fgf ligands and their downstream target genes are induced in glia and neurons after SCI. a–o, Sections from Tg(GFAP:GFP) transgenic fish documenting the location of GFAP-positive glia or traced neurons on each section before and after in situ hybridization. a–e, Intact control spinal cords. fgf8a is expressed at low levels on GFAP-positive glia cells in the ventricular zone (a‴ and a⁗ show boxed region in a, a′) and fgf3 is expressed at low levels on large neurons (b, arrowhead). spry4 is expressed at low levels in neurons (c, arrowhead), whereas pea3 (d) and erm (e) are almost undetectable in adjacent sections. f–j, Three days after SCI. fgf8a expression is significantly increased on GFAP-positive glia cells found at the lesion site (f′ is a higher magnification of boxed area in f and f‴ and f⁗ are higher magnifications of boxed region in f′). fgf3 expression increases in both neurons (g; g′, boxed area in g, arrowhead) and in glia (arrow) at the cc surrounding the lesion site. spry4 expression is high in neurons (h, arrowheads) and glia (arrows), while pea3 (i) and erm (j) expression is upregulated on glia cells. (k–o, 2–3 weeks after SCI. fgf8a is highly expressed (k; k′, boxed area in k) in glia cells at the lesion site and around the cc. fgf3 expression is decreased in glia cells at the lesion site (l, arrow), but increased in neurons upstream of the lesion (l′, arrowhead). (l″) fgf3-expressing cells are identified as neurons by colabeling with NeuN and by accumulation of TMRD axonal tracer labeling (n, arrowheads). spry4 expression is increased in neurons (m, arrowheads), which are also labeled by TMRD tracer (o, arrowheads). p, q, FgfR2 is upregulated on GFAP-positive glia cells (arrowheads) and neurons (arrows) 2 weeks post-SCI in injured (p; p′–p‴, boxed area in p) compared with intact (q; q′–q‴, boxed area in q) spinal cords. Caudal side of spinal cord to the left in all panels. cc, Central canal. Representative results from at least n = 4 fish in each condition. Scale bars: a, a′, f, f′, f″, g, k, 100 μm; in high-power magnification panels, including a‴, b–e, f‴, g′–j, k′–o′, p, q, 50 μm; p′–p‴, q′-q‴, 10 μm.
In line with these observations, the Fgf receptor2 (FgfR2) is expressed at a basal level on neurons and glia in the intact spinal cord (Fig. 3p, GFAP-positive glia—arrowhead, GFAP-negative neuron—arrow) and is upregulated in glia and neurons 2 weeks post-SCI (Fig. 3q, glia—arrowhead, neuron—arrow). Collectively, these results show an upregulation of fgf ligands, their associated receptors and their downstream target genes, post-SCI in glia and neurons specifically located adjacent to the lesion site.
We next examined the expression of p-MAPK at the lesion site, as it is the main downstream effector of Fgf signaling and a sensitive indicator as to which cells respond to Fgf signaling post-SCI. At 24 h post-SCI p-MAPK levels are dramatically increased specifically within GFAP-positive glia of the central canal, compared with intact spinal cord (Fig. 4a,b). At 3 d post-SCI, these cells upregulate nestin expression, in contrast to the low nestin expression evident in uninjured or 24 h post-SCI spinal cords (Fig. 4c–e). Upregulation of p-MAPK in these glia is maintained at least up to 3 weeks post-SCI, showing strong Fgf signaling within these cells across the phases of glial morphogenesis we describe here.
Figure 4.
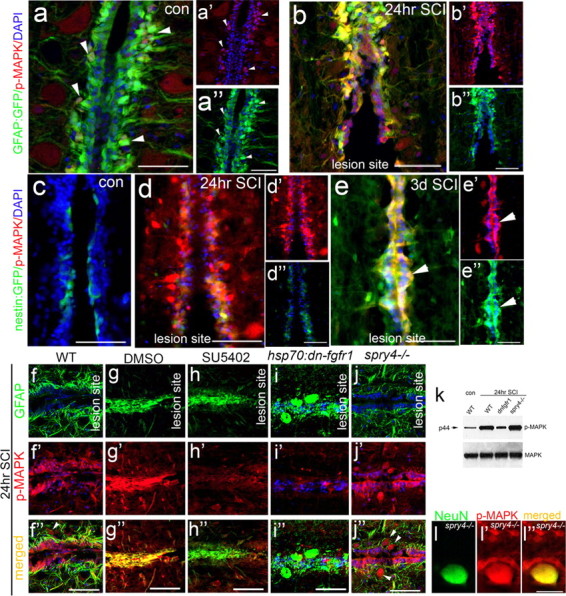
Fgf-dependent MAPK signaling is induced in glia and neurons after spinal cord injury. a, b, In intact control spinal cords p-MAPK levels are low in GFAP-expressing glia along the central canal (cc) (a–a″, arrowheads), but become strongly upregulated 24 h post-SCI (b) (n = 5). c–e, Nestin levels at the cc are also low (n = 4) in intact spinal cord, but both p-MAPK and nestin levels increase shortly after SCI (24 h, d; 3 d, e, arrowheads) in glia cells at the cc (n = 5). f–l, MAPK is activated by Fgf signaling after SCI as confirmed by Western blot analysis of protein derived from spinal cord lesion sites in wild-type, spry4−/− and dn-fgfr1 lines after SCI (k). In WT (f) and DMSO only control (g), p-MAPK levels increase 24 h post-SCI (f′, g′, k). When Fgf is inhibited by SU5402 injection (h) or heat shock of hsp70l:dn-fgfr1-EGFP fish (i), this increase in p-MAPK is abolished (h′, i′, k). Conversely, in spry4−/− fish p-MAPK activity is increased in GFAP-positive cells as in wild-type, and additionally in neurons (j″, arrowheads) that colabel with the neural NeuN marker (l–l″). Scale bars: a–j, 50 μm; l, 10 μm.
The Fgf pathway activates MAPK signaling during spinal cord regeneration
Given that Fgf signaling is specifically upregulated at the lesion site, we examined the role of Fgf signaling on different aspects of spinal cord regeneration using Fgf signaling inhibition and gain of function studies. For Fgf signaling inhibition two approaches were used. First, we used a transgenic line of zebrafish that expresses a dominant-negative form of the Fgf receptor from the heat shock promoter (hsp70l:dn-fgfr1-EGFP) (Lee et al., 2005). Daily heat shock treatments, applied for up to 3 weeks (vs heat shock on wild-type controls) allowed us to globally inhibit Fgf signaling at a specific a time period before and after SCI. Second, Fgf signaling was inhibited by a single injection of the small molecule inhibitor SU5402 (1 μl of 5 μg/μl DMSO vs DMSO-only controls) into the lesion site immediately following SCI to allow for spatial restriction of Fgf inhibition exclusively within this regenerating area.
Two independent approaches were also undertaken to look at the gain of Fgf function in the context of SCI. First we examined the response to SCI in homozygous spry4−/− mutants. Spry4 is a known cell autonomous inhibitor of Fgf signaling. Despite being homozygous for a truncating null mutation within the spry4 open reading frame, spry4−/− compared with the wild-type during the development of either glia or neurons (number of neurons at 2 weeks WT 42.3 ± 8.4 SEM; spry4−/− 43.1 ± 3.2 SEM per 200 μm2) or in adults (number of neurons WT 9.43 ± 2.45 SEM; spry4−/− 12.01 ± 3.2 SEM per 200 μm2). Furthermore, spry expression is low in the intact spinal cord (Fig. 3c) and the main effect of this mutant becomes relevant primarily after SCI, when spry is upregulated in WT spinal cord.
The second approach involved the injection of Fgf8 intraperitoneally into animals, which consequently undergo SCI. When Fgf signaling is inhibited using either heat shock induction of the dnFGFR or SU5402 injection, p-MAPK activation is dramatically decreased 24 h post-SCI compared with nontransgenic heat shocked and DMSO-only controls, which were equivalent to untreated controls (Fig. 4f–i,k). Conversely, in spry4−/− mutants where Fgf signaling is overactive, glia flanking the spinal canal exhibit higher levels of MAPK activation (Fig. 4f,j,k). MAPK is additionally activated in neurons, which are the large somas that also colabel with NeuN (Fig. 4j,l). In Fgf8 injections, p-MAPK is only increased on GFAP-expressing glia at the lesion site (data not shown). Thus, these studies reveal that Fgf signaling accounts for the majority of MAPK activation within glia post-SCI.
Fgf signaling regulates glial cell proliferation and migration.
We next examined the glial cell response to Fgf loss of function contexts focusing first on the initial proliferative and migratory phases (1 and 2).
By 3 weeks post-SCI in hsp70l:dn-fgfr1-EGFP heat shocked or SU5402-injected fish, glial cells within the central canal show a significantly reduced proliferative response (BrdU-positive cells, Fig. 5a–d,f) and less migration, resulting in a reduction of DAPI and GFAP-positive glia at the lesion compared with wild-type heat-shocked or DMSO only controls, which were equivalent to untreated controls (Fig. 5h,j–m). Because the peak of proliferation occurs in the first few days following SCI (Fig. 5i) as has also been demonstrated in a brain injury model of zebrafish (Kroehne et al., 2011), we additionally performed a delayed heat shock to assess the later effects of loosing Fgf function specifically on differentiation of glia. In the delayed heat shocked (initiated at 6 d post-SCI) of hsp70l:dn-fgfr1-EGFP fish, the initial proliferation is comparable to controls (Fig. 5e,g). Similarly, migration is normal, as the thickness of tissue at the lesion is comparable to control (Fig. 5n) and the number of cells at the lesion site is reduced only in SU5402-treated and early heat shocked hsp70l:dn-fgfr1-EGFP fish but not in the fish that had undergone delayed heat shock (Fig. 5h). Thus, Fgf signaling is necessary for proliferation and migration of glia post-SCI.
Next, we assessed the effect on proliferation in the context of Fgf gain of function. In spry4−/− fish, proliferation is significantly enhanced (Fig. 5o,q). In stark contrast to wild-type spinal cords, where glial cells accumulate at the lesion only by 10 d post-SCI, spry4−/− fish exhibit GFAP-positive glial cells prominent at the lesion site already by 5 d post-SCI (Fig. 5o,o′,q). This suggests that Fgf signaling is sufficient to accelerate both the proliferation, migration as well as the onset of differentiation (GFAP expression) of glial cells. In support of these observations glia cell differentiation was similarly accelerated within GFAP:GFP fish that had undergone intraperitoneal Fgf8 injections, and examined 5 d after SCI (Fig. 5p,p′,p″,q).
Fgf signaling regulates glial cell morphogenesis during glial bridge formation
Additionally we wanted to assess the role of Fgf signaling specifically on the later aspects of glia differentiation and morphogenesis post-SCI. In wild-type zebrafish, glial bridges span the lesion site by 2–3 weeks post-SCI (Fig. 1k–o). In heat-shocked hsp70l:dn-fgfr1-EGFP or SU5402-injected fish 3 weeks post-SCI, the majority of fish do not form glial bridges across the lesion (66% of hsp70l:dn-fgfr1-EGFP and 75% of SU5402-treated fish). The remaining fish possessed only thin connecting tissue between the two transected sides of the lesion (Fig. 5c,d,l,m). We again used the delayed heat shock strategy to conditionally inactivate Fgf function specifically during the late glia differentiation phase. Our results above have already demonstrated that in this experiment, the initial proliferation (which occurs primarily in the first 5 d post-SCI, Fig. 5i) and migration are comparable to WT under these conditions (Fig. 5e,g,h,n). Thus, any effects in this experiment are specifically due to perturbation of the late differentiation phase of the glial response. In this experiment, 3 weeks post-SCI, glial cells within heat-shocked hsp70l:dn-fgfr1-EGFP fish remained undifferentiated and expressed low levels of GFAP, despite migrating normally to the lesion zone. Furthermore, individual cells exhibited a multiprocess stellate morphology, and the vast majority of glia failed to produce the elongated morphology required to bridge the transected spinal column. The few elongated bipolar glia that did form, do not align in the anterior–posterior direction, and instead exhibit a random directional alignment (Fig. 5n,s′) in contrast to the well organized glial bridge evident in wild-type fish (Fig. 5k,r′). Moreover, p-MAPK signaling is significantly reduced specifically in those glia that do not exhibit a bipolar morphology (Fig. 5r–t). The disorganized morphology of activated glial cells in the delayed hsp70l:dn-fgfr1-EGFP is reminiscent of the glial scar formed in mammalian SCI, a feature which inhibits axonal regeneration through the lesion site. Thus, Fgf signaling is responsible for the morphological transformation of glia to form bipolar cells and glial bridges aligned along the AP axis that span the resection zone.
In contrast, glial bridge formation in either spry4−/− fish or after Fgf8 injections is dramatically accelerated. By 5 d post-SCI, glia at the lesion site of these fish already exhibit a bipolar morphology and express detectable levels of GFAP (Fig. 5o′,p″,q), in contrast to wild-type animals at this time point (Figs. 1e, 5q). These results suggest that Fgf signaling specifically drives the differentiation of glia into bipolar cells that form glial bridges that span the lesion site.
Glial bridge formation is required for axonal regeneration
As functional recovery ultimately depends on the regeneration of neurons across the lesion site, we examined the effect that Fgf signaling and interfering with glial differentiation might have on axonogenesis.
In the context of Fgf signaling inhibition immediately post-SCI (heat shock within hsp70l:dn-fgfr-EGFP fish or SU5402 injection) the lack of glial bridge formation correlated with a complete lack of axonal regeneration through the lesion site at 3 weeks post-SCI (Fig. 6a–c). Similarly, when heat shock in hsp70l:dn-fgfr-EGFP fish is delayed until 6 d post-SCI, which specifically prevents glial differentiation and bridge formation, the only axonal regeneration ever observed in these fish occurred when a rare bipolar glial cell did align correctly to span the lesion zone, allowing isolated axons to traverse along it (Fig. 6d,d′, axon—arrowheads, glia—arrows). Axonal regeneration does still occur when Fgf signaling is inhibited and upstream of the lesion (300 μm) a comparable number of neurites can be observed. Close to the lesion (100 μm) there is a small, significant reduction in regenerated neurites (14 ± 4.8 SEM hsp70l:dn-fgfr-EGFP vs 20.96 ± 5.4 SEM WT, p-value < 0.05), suggesting that there may be an additional effect on the rate of axonogenesis. However, for axons that have grown to the edge, the growth specifically through the lesion site depends not just on the presence of glial cells, but their differentiation to a specific bipolar morphology.
In contrast, at 10 d post-SCI, within spry4−/− fish, which possess a dramatically accelerated accumulation of glia and differentiation to form glial bridges across the lesion at 10 d post-injury (Fig. 6g), descending axons are already able to enter the lesion site (Fig. 6g′, arrows in inset) although overall axonal growth does not seem to be accelerated through the lesion site at this stage. However, by 3 weeks post-SCI significantly more axons are regenerating through the lesion site within spry4−/− mutant fish, as revealed by TMRD tracer injections, which label many more cell bodies upstream of the lesion site (lesion crossing axons), compared with those in wild-type fish (spry4−/− 28.4 ± 3.7% SEM; wild-type 13.9 ± 4.8% SEM; Fig. 6f). Neurons with traced TMRD-labeled processes also show a stronger activation of MAPK in spry4−/− fish (Fig. 6h–j, arrowheads). Thus, in both wild-type and in the Fgf enhanced context, glial bridges always form ahead of the onset of axonogenesis through the lesion, supporting the notion that glial bridges need to form before axons can reextend across the resection zone.
Locomotor function recovered at a rate consistent with the changes in glial morphogenesis and axonal regeneration we observed. The lack of glial bridge formation and axonogenesis resulting from Fgf signaling inhibition was accompanied by a failure to recover locomotor ability caudal to the lesion site (Fig. 6e). Collectively, these results suggest that Fgf signaling promotes gliogenesis and formation of a bipolar glial cell bridge, which facilitates axonal regrowth of resected axons.
Fgf signaling promotes primate astrocyte bipolarity
Given the critical role that Fgf signaling plays in glial cell morphogenesis during SC regeneration in zebrafish, we examined, whether Fgf could also influence the morphogenesis and differentiation of mammalian astrocytes. We therefore generated cultures of primate primary astrocytes, derived from 14-d-old marmoset cerebral cortex (Goldshmit and Bourne, 2010) and added human recombinant Fgf2 to these cells. Sustained MAPK phosphorylation was evident immediately after hFgf2 addition (Fig. 7a) and long-term p-MAPK activation (for up to 48 h), induced an increase of Spry4 protein levels, which leads to feedback inhibition of MAPK activation (Fig. 7b). These results suggest that primate astrocytes respond to Fgf2 addition by activation of Fgf signaling evident by the increase of downstream components. Furthermore, addition of hFgf2 in a scratch wound assay (Goldshmit and Bourne, 2010) results in the accelerated closure of the scratched area by 48 h, compared with that seen in an assay with control astrocytes which take 3–4 d (Fig. 7c). In contrast, addition of SU5402 completely inhibits the ability of astrocytes to close the scratch wound both in the presence and absence of Fgf addition (Fig. 7c). In this assay, Fgf stimulates proliferation and causes a significant increase in Ki67-positive proliferative cells (Fig. 7d) and migration (Fig. 7c) of primate astrocytes in a similar manner to that exhibited by zebrafish astrocytes.
To examine the effect of Fgf addition on the differentiation and morphology of astrocytes, hFgf2 was added to these cultures for 7 d. This results in GFAP-expressing astrocytes undergoing a dramatic morphological shift from their multipolar stellate shape to an elongated bipolar phenotype. Astrocytes showed a significant reduction in the number of processes with a >4-fold increase in the number of astrocytes with bipolar morphology (Fig. 7e,f). Furthermore, many of the Fgf-treated astrocytes with multiple processes also possess a highly elongated “bipolar” main process, from which smaller secondary processes emerge. Upon culturing for longer periods these resolved into true bipolar cells (Fig. 7f). Moreover, all astrocytes in culture showed vimentin expression, a marker of radial glia (Fig. 7g,h). However, Fgf treatment did not result in nestin (Fig. 7i) or pax6 (data not shown) expression in the cultured cells, which suggests the absence of any stem cells or multipotent progenitor cells in these cultures. Thus, Fgf signaling collectively regulates proliferation, migration and differentiation of primate astrocytes in vitro, in a manner similar to its role in driving migration and polarity of glial cells during zebrafish spinal cord regeneration in vivo.
Discussion
Fgf signaling is induced during zebrafish SCI and coordinates glial cell morphogenesis
Our results document a novel role for Fgf signaling in mediating glial cell morphogenesis and glial bridge formation, which is a critical step in facilitating axonal regeneration in the zebrafish SCI model (Fig. 8). Indeed, the process of glial bridge formation, which appears to be a common feature of fish and amphibian SCI models (Zukor et al., 2011), is in direct contrast to the response of glial cells to spinal cord injury in mammals. In mammals reactive astrocytes form a dense web of interdigitated processes that fill the space vacated by dead or dying cells and which prohibits axonal regeneration (McKeon et al., 1991; Stichel and Müller, 1998). Nevertheless, astrocytes are still one of the main available cellular pools capable of responding to different factors close to injured areas in the mammalian CNS and they can clearly undergo reversion to a proliferative, undifferentiated, stem cell like state (Duggal et al., 1997; Steindler and Laywell, 2003; Chen et al., 2005; Buffo et al., 2008; Yang et al., 2009; Barnabé-Heider et al., 2010). Our results show that like mammalian glial cells, fish glial cells are also the main proliferating cells after SCI that occupy the lesion site and seal the wound. However, in stark contrast to the mammalian cells, fish glial cells take up an alternate morphology (bipolar rather than stellate) that supports axonal regeneration across the lesion site rather than inhibiting this process by forming a dense glial scar. Hence, an ability to manipulate the differentiation potential of these cells at the site of injury to generate a more proregenerative phenotype, such as the glial cell bridge formation we describe here, has clear therapeutic implications (Davies et al., 2006; Fawcett, 2008).
Figure 8.
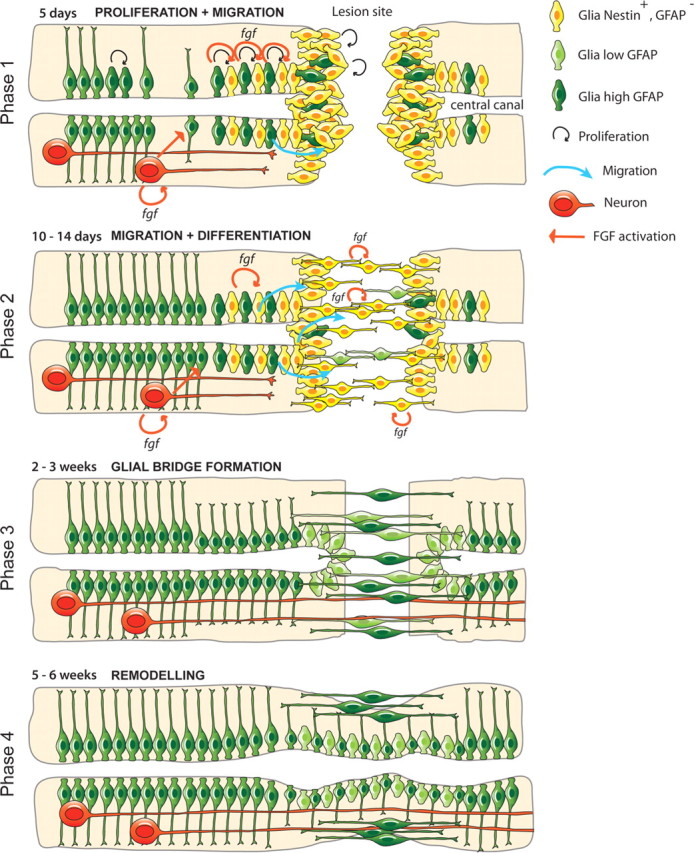
Model for zebrafish spinal cord regeneration. Phase 1: Proliferation and initial migration. At 5 d after SCI, initial transection of the spinal cord stimulates secretion of Fgf ligands in the central canal, which in turn induce glial cell dedifferentiation and proliferation to generate progenitor cells. These cells begin to migrate toward the lesion edge and cells at the lesion site possess low levels of GFAP expression and high levels of nestin expression at the lesion site. Fgf is also released by neuronal cells. Phase 2: Migration and Differentiation. By 10 d post-SCI, Nestin-positive cells increase GFAP levels and maintain Fgf expression. Migration initiates and glial cells begin to fill the lesion site and elongate into bipolar morphology, a process of differentiation that is also Fgf-dependent. Fgf expression is also upregulated in neuronal cells upstream of the lesion site. At this stage axonal regeneration toward, but not through the lesion occurs. Phase 3: Glial bridge formation. By 2–3 weeks after SCI, bipolar GFAP-expressing glia have completely bridged the gap between the two transected sides of the lesion, allowing axonal migration and regeneration. Phase 4: Remodeling. By 5 weeks, the central canal has been fully reconstructed and bipolar glia are only present at the surface. Axons now extend further across the lesion site.
Our studies also extend the results of previouly published work on the nature of neural regeneration in zebrafish that suggest that although both ascending and descending fibers are able to regenerate across the lesion site, ascending fibers are unable to reinnervate their target cells (Bunt and Fill-Moebs, 1984; Becker et al., 1997). In goldfish, successful regeneration of ascending axons has been reported (Hanna et al., 1998). Here, we directly demonstrate axonogenesis from new and mature neurons.
By 3 weeks post-SCI descending axons have already grown far enough downstream to be labeled by either rostral or caudal tracer injections. Caudal tracer application accumulates upstream of the lesion site in two distinct patterns. First, tracer accumulates in neuronal cell bodies through the labeling of regenerated descending axons. Second, tracer additionally accumulates in processes around neuronal cell bodies, which we believe are examples of regenerated ascending axons that have terminated around their target cells.
The inductive processes that guide astrocyte differentiation to become radial or progenitor glia in vivo and in vitro are surprisingly understudied. Interestingly, unipolar or bipolar GFAP-expressing cells (presumed radial glia) have been identified as the principal source of neurogenesis in the adult mouse (Garcia et al., 2004), revealing the proneurogenic effect of directing astrocyte differentiation into these radial glia. Experiments using astrocyte conditioned medium suggested that astrocytes secrete factors that may influence their ability to regain neuronal progenitor potential and dedifferentiate into radial glia (Yang et al., 2009) and TGF-α is one factor has been shown to drive astrocyte bipolar morphology in vitro (Zhou et al., 2001; White et al., 2011). EGF is an additional secreted factor that has been implicated in the dedifferentiation of rat astrocytes in culture (Yu et al., 2006). Another recent study showed that ErbB2 plays a role in the dedifferentiation of astrocyte into radial glia in vitro (Yang et al., 2011); however, until now, none of the factors that influence astrocyte differentiation in vivo have been determined. Our analysis implicates Fgf signaling in the control of this process in vivo and in vitro. Downstream effectors of Fgf signaling are induced within glial cells at the lesion site and loss or gain of Fgf signaling induces and prevents glial cell differentiation, respectively. Using a heat-inducible, dominant-negative form of the FgfR we can temporally distinguish these separate roles of Fgf in the initial proliferation of glial cell progenitors and their consequent differentiation to form bipolar cells that span the lesion site in the anterior–posterior direction in the form of glial bridges. Glial bridges form independently of regenerating axons and glial cells that occupy the lesion site form bipolar processes before axons reaching the lesion site.
These results may provide a molecular and cellular explanation for the enhanced regeneration seen in rodent SCI models administered with Fgf. Administration of Fgf to the lesion site following severe contusion in rats has been shown to significantly enhance functional recovery of hindlimb movements (Rabchevsky et al., 2000). Although this occurs in the absence of significant tissue sparing, this study did not examine the glial cell behavior at the site of injury. The neuronal regeneration marker, GAP43, is increased and GFAP levels were attenuated in animals treated with Fgf after spinal cord contusion (Tsai et al., 2008). A single Fgf2 injection into the lesion site after complete transection in rats induced the appearance of fibronectin-positive cells in cystic cavities, an observation suggestive of neurite growth (Kasai et al., 2010). However, the actual cellular basis for the functional recovery induced by Fgf treatment within rodent SCI models is yet to be defined.
Can the same process of regeneration be promoted within the mammalian spinal cord?
To examine the potential relevance to human brain and spinal cord repair, we studied the response of primate astrocytes to Fgf signaling in vitro. Our results demonstrate that Fgf signaling mediates both astrocyte proliferation and migration, and more importantly, the morphological changes that generate a bipolar cell morphology reminiscent of the shape of radial glia. Astrocytes from the adult rodent brain have been suggested to be able to generate progenitor cells in vitro and in vivo that possess the ability to reexpress nestin and produce neurospheres in culture (Duggal et al., 1997; Noctor et al., 2001; Steindler and Laywell, 2003; Chen et al., 2005; Mao et al., 2009; Yang et al., 2009). Intriguingly, injection of radial glia, derived from E13.5 rat neurospheres, into a rat spinal cord after contusion resulted in their adoption of a highly polarized morphology and formation of cellular bridges surrounding the lesion site. These transplanted cells also accumulate less chondroitin sulfate proteoglycans (CSPGs), which are glial scar components known to inhibit axonal regeneration (Hasegawa et al., 2005). These structures are highly reminiscent of the spinal cord regeneration process that we describe in zebrafish. Moreover, Hasegawa et al. (2005) showed that the injected cells initially expressed high levels of nestin and then increased GFAP levels, thereby mimicking the levels of expression of these proteins at the lesion site in the zebrafish spinal cord in vivo.
Fgf has been demonstrated to be secreted by astrocytes at the lesion site after SCI in rodents (Clarke et al., 2001) and amphibians (Fahmy and Moftah, 2010). Our preliminary examination of the spry4 transcript and protein expression in the spinal cord of the mouse shows that spry4 is expressed in intact spinal cord on astrocytes (Fig. 9a,b,b′). Four days post-SCI, Spry4 is upregulated on astrocytes at the lesion site (Fig. 9c,d′), suggesting that after SCI, Fgf signaling is activated in mouse astrocytes. In mouse, FgfR1 signaling, which is critical during brain development, is strongly downregulated after birth. This loss of Fgf signaling correlates with the reduced regenerative potential of adult compared with infant/juvenile brain (Vaccarino et al., 2007). These results suggest that it may be possible to manipulate both the levels and the temporal period of Fgf signaling post-SCI in mammals in a manner that can promote astrocyte and radial glial cell differentiation to a bipolar phenotype that may be proregenerative.
Figure 9.

Spry4 expression after spinal cord injury in mouse. a, RT-PCR of RNA extracted from the brain, liver, muscle and spinal cord of two uninjured mice shows spry4 expression primarily in the brain and spinal cord, with some expression detected in the liver. GAPDH was used as a PCR amplification control. b, b′, Spry4 is expressed in GFAP-expressing astrocytes in the gray matter of the intact spinal cord (arrowheads). c–c″, Four days post-SCI. Spry4 is upregulated at the lesion site on GFAP-positive reactive astrocytes. d, d′, High-power magnification of an astrocyte at the lesion site, which expresses high levels of GFAP and Spry4. Scale bars: b, d, 25 μm; c, 100 μm.
Collectively, these results show that increasing levels of Fgf signaling at the site of mammalian spinal cord injury may encourage radial glial cell differentiation in a manner that is proregenerative and that suppresses the formation of a glial scar. Success in directing proliferating astrocytes at the injury site to dedifferentiate to radial bipolar glia may encourage favorable conditions for axonal regeneration after spinal cord injury.
Footnotes
The Australian Regenerative Medicine Institute is supported by grants from the State Government of Victoria and the Australian Government. We thank Dr. Samuel McLenachan and Dr. Mirella Dottori for reading and commenting on the manuscript. We thank Dr. Felix Ellett for providing the Tg(mpeg1:GFP) and (Tg(mpx:GFP) transgenic lines. We thank Wouter Masselink for his assistance in statistics analysis.
The authors declare no competing financial interests.
References
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabé-Heider F, Göritz C, Sabelströom H, Takebayashi H, Pfrieger FW, Meletis K, Frisén J. Origin of new glial cells in intact and injured adult spinal cord. Cell Stem Cell. 2010;7:470–482. doi: 10.1016/j.stem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Bastmeyer M, Bähr M, Stuermer CA. Fish optic nerve oligodendrocytes support axonal regeneration of fish and mammalian retinal ganglion cells. Glia. 1993;8:1–11. doi: 10.1002/glia.440080102. [DOI] [PubMed] [Google Scholar]
- Becker T, Wullimann MF, Becker CG, Bernhardt RR, Schachner M. Axonal regrowth after spinal cord transection in adult zebrafish. J Comp Neurol. 1997;377:577–595. doi: 10.1002/(sici)1096-9861(19970127)377:4<577::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Raymond PA. GFAP transgenic zebrafish. Gene Expr Patterns. 2006;6:1007–1013. doi: 10.1016/j.modgep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Bernhardt RR, Tongiorgi E, Anzini P, Schachner M. Increased expression of specific recognition molecules by retinal ganglion cells and by optic pathway glia accompanies the successful regeneration of retinal axons in adult zebrafish. J Comp Neurol. 1996;376:253–264. doi: 10.1002/(SICI)1096-9861(19961209)376:2<253::AID-CNE7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Bernstein JJ, Gelderd JB. Regeneration of the long spinal tracts in the goldfish. Brain Res. 1970;20:33–38. doi: 10.1016/0006-8993(70)90151-4. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunt SM, Fill-Moebs P. Selection of pathways by regenerating spinal cord fiber tracts. Brain Res. 1984;318:307–311. doi: 10.1016/0165-3806(84)90035-x. [DOI] [PubMed] [Google Scholar]
- Chen J, Leong SY, Schachner M. Differential expression of cell fate determinants in neurons and glial cells of adult mouse spinal cord after compression injury. Eur J Neurosci. 2005;22:1895–1906. doi: 10.1111/j.1460-9568.2005.04348.x. [DOI] [PubMed] [Google Scholar]
- Clarke WE, Berry M, Smith C, Kent A, Logan A. Coordination of fibroblast growth factor receptor 1 (FGFR1) and fibroblast growth factor-2 (FGF-2) trafficking to nuclei of reactive astrocytes around cerebral lesions in adult rats. Mol Cell Neurosci. 2001;17:17–30. doi: 10.1006/mcne.2000.0920. [DOI] [PubMed] [Google Scholar]
- Davies JE, Huang C, Proschel C, Noble M, Mayer-Proschel M, Davies SJ. Astrocytes derived from glial-restricted precursors promote spinal cord repair. J Biol. 2006;5:7. doi: 10.1186/jbiol35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal N, Schmidt-Kastner R, Hakim AM. Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain Res. 1997;768:1–9. doi: 10.1016/s0006-8993(97)00588-x. [DOI] [PubMed] [Google Scholar]
- Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood. 2011;117:e49–e56. doi: 10.1182/blood-2010-10-314120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmy GH, Moftah MZ. Fgf-2 in astroglial cells during vertebrate spinal cord recovery. Front Cell Neurosci. 2010;4:129. doi: 10.3389/fncel.2010.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW. Bridging spinal cord injuries. J Biol. 2008;7:25. doi: 10.1186/jbiol89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Bourne J. Upregulation of EphA4 on astrocytes potentially mediates astrocytic gliosis after cortical lesion in the marmoset monkey. J Neurotrauma. 2010;27:1321–1332. doi: 10.1089/neu.2010.1294. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–10073. doi: 10.1523/JNEUROSCI.2981-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TE, Bryson-Richardson RJ, Berger S, Jacoby AS, Cole NJ, Hollway GE, Berger J, Currie PD. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc Natl Acad Sci U S A. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna GF, Nawar NN, Sharma SC. Regeneration of ascending spinal axons in goldfish. Brain Res. 1998;791:235–245. doi: 10.1016/s0006-8993(98)00105-x. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Chang YW, Li H, Berlin Y, Ikeda O, Kane-Goldsmith N, Grumet M. Embryonic radial glia bridge spinal cord lesions and promote functional recovery following spinal cord injury. Exp Neurol. 2005;193:394–410. doi: 10.1016/j.expneurol.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci. 2000;20:206–218. doi: 10.1523/JNEUROSCI.20-01-00206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Zhou J, Luo C, Lin J, Wang X, Li X, Bian X, Li Y, Wan Q, Yu Y, Feng H. Glial scar and neuroregeneration: histological, functional, and magnetic resonance imaging analysis in chronic spinal cord injury. J Neurosurg Spine. 2010;13:169–180. doi: 10.3171/2010.3.SPINE09190. [DOI] [PubMed] [Google Scholar]
- Hui SP, Dutta A, Ghosh S. Cellular response after crush injury in adult zebrafish spinal cord. Dev Dyn. 2010;239:2962–2979. doi: 10.1002/dvdy.22438. [DOI] [PubMed] [Google Scholar]
- Kasai M, Jikoh T, Fukumitsu H, Furukawa S. FGF-2-responsive and spinal cord-resident cells improve locomotor function after spinal cord injury. J Neurotrauma. 2010 doi: 10.1089/neu.2009.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroehne V, Freudenreich D, Hans S, Kaslin J, Brand M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development. 2011;138:4831–4841. doi: 10.1242/dev.072587. [DOI] [PubMed] [Google Scholar]
- Lam CS, März M, Strähle U. gfap and nestin reporter lines reveal characteristics of neural progenitors in the adult zebrafish brain. Dev Dyn. 2009;238:475–486. doi: 10.1002/dvdy.21853. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Chen CJ, Cheng CH, Huang WC, Kuo HS, Wu JC, Tsai MJ, Huang MC, Chang WC, Cheng H. Combined treatment using peripheral nerve graft and FGF-1: changes to the glial environment and differential macrophage reaction in a complete transected spinal cord. Neurosci Lett. 2008;433:163–169. doi: 10.1016/j.neulet.2007.11.067. [DOI] [PubMed] [Google Scholar]
- Lee Y, Grill S, Sanchez A, Murphy-Ryan M, Poss KD. Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development. 2005;132:5173–5183. doi: 10.1242/dev.02101. [DOI] [PubMed] [Google Scholar]
- Mao XG, Xue XY, Zhang X. The potential of the brain: plasticity implications for de-differentiation of mature astrocytes. Cell Mol Neurobiol. 2009;29:1105–1108. doi: 10.1007/s10571-009-9416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujoks-Manteuffel C, Roth G. Astroglial cells in a salamander brain (Salamandra salamandra) as compared to mammals: a glial fibrillary acidic protein immunohistochemistry study. Brain Res. 1989;487:397–401. doi: 10.1016/0006-8993(89)90849-4. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Turner AF, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances functional recovery following severe spinal cord injury to the rat. Exp Neurol. 2000;164:280–291. doi: 10.1006/exnr.2000.7399. [DOI] [PubMed] [Google Scholar]
- Reimer MM, Sörensen I, Kuscha V, Frank RE, Liu C, Becker CG, Becker T. Motor neuron regeneration in adult zebrafish. J Neurosci. 2008;28:8510–8516. doi: 10.1523/JNEUROSCI.1189-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer MM, Kuscha V, Wyatt C, Sörensen I, Frank RE, Knüwer M, Becker T, Becker CG. Sonic hedgehog is a polarized signal for motor neuron regeneration in adult zebrafish. J Neurosci. 2009;29:15073–15082. doi: 10.1523/JNEUROSCI.4748-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- Schindelin J. Fiji is just ImageJ (batteries included); ImageJ User and Developer Conference; Luxemburg. 2008. [Google Scholar]
- Schweitzer J, Becker T, Becker CG, Schachner M. Expression of protein zero is increased in lesioned axon pathways in the central nervous system of adult zebrafish. Glia. 2003;41:301–317. doi: 10.1002/glia.10192. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simpson SB., Jr Morphology of the regenerated spinal cord in the lizard, Anolis carolinensis. J Comp Neurol. 1968;134:193–210. doi: 10.1002/cne.901340207. [DOI] [PubMed] [Google Scholar]
- Steindler DA, Laywell ED. Astrocytes as stem cells: nomenclature, phenotype, and translation. Glia. 2003;43:62–69. doi: 10.1002/glia.10242. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Müller HW. The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res. 1998;294:1–9. doi: 10.1007/s004410051151. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Hermanns S, Luhmann HJ, Lausberg F, Niermann H, D'Urso D, Servos G, Hartwig HG, Müller HW. Inhibition of collagen IV deposition promotes regeneration of injured CNS axons. Eur J Neurosci. 1999;11:632–646. doi: 10.1046/j.1460-9568.1999.00466.x. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Shen LF, Kuo HS, Cheng H, Chak KF. Involvement of acidic fibroblast growth factor in spinal cord injury repair processes revealed by a proteomics approach. Mol Cell Proteomics. 2008;7:1668–1687. doi: 10.1074/mcp.M800076-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura O, Okada Y, Ando H, Guedj M, Higashijima S, Shimazaki T, Chino N, Okano H, Okamoto H. Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev Biol. 2005;278:587–606. doi: 10.1016/j.ydbio.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Fagel DM, Ganat Y, Maragnoli ME, Ment LR, Ohkubo Y, Schwartz ML, Silbereis J, Smith KM. Astroglial cells in development, regeneration, and repair. Neuroscientist. 2007;13:173–185. doi: 10.1177/1073858406298336. [DOI] [PubMed] [Google Scholar]
- White RE, Rao M, Gensel JC, McTigue DM, Kaspar BK, Jakeman LB. Transforming growth factor alpha transforms astrocytes to a growth-supportive phenotype after spinal cord injury. J Neurosci. 2011;31:15173–15187. doi: 10.1523/JNEUROSCI.3441-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HQ, Yu J, Kline AE, Letart P, Jenkins LW, Marion DW, Dixon CE. Evaluation of combined fibroblast growth factor-2 and moderate hypothermia therapy in traumatically brain injured rats. Brain Res. 2000;887:134–143. doi: 10.1016/s0006-8993(00)03002-x. [DOI] [PubMed] [Google Scholar]
- Yang H, Cheng XP, Li JW, Yao Q, Ju G. De-differentiation response of cultured astrocytes to injury induced by scratch or conditioned culture medium of scratch-insulted astrocytes. Cell Mol Neurobiol. 2009;29:455–473. doi: 10.1007/s10571-008-9337-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ling W, Vitale A, Olivera C, Min Y, You S. ErbB2 activation contributes to de-differentiation of astrocytes into radial glial cells following induction of scratch-insulted astrocyte conditioned medium. Neurochem Int. 2011;59:1010–1018. doi: 10.1016/j.neuint.2011.08.021. [DOI] [PubMed] [Google Scholar]
- Yu T, Cao G, Feng L. Low temperature induced de-differentiation of astrocytes. J Cell Biochem. 2006;99:1096–1107. doi: 10.1002/jcb.20979. [DOI] [PubMed] [Google Scholar]
- Zhou R, Wu X, Skalli O. TGF-alpha induces a stationary, radial-glia like phenotype in cultured astrocytes. Brain Res Bull. 2001;56:37–42. doi: 10.1016/s0361-9230(01)00591-3. [DOI] [PubMed] [Google Scholar]
- Zottoli SJ, Bentley AP, Feiner DG, Hering JR, Prendergast BJ, Rieff HI. Spinal cord regeneration in adult goldfish: implications for functional recovery in vertebrates. Prog Brain Res. 1994;103:219–228. doi: 10.1016/s0079-6123(08)61138-3. [DOI] [PubMed] [Google Scholar]
- Zukor KA, Kent DT, Odelberg SJ. Meningeal cells and glia establish a permissive environment for axon regeneration after spinal cord injury in newts. Neural Dev. 2011;6:1. doi: 10.1186/1749-8104-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupanc GK, Clint SC. Radial glia-mediated up-regulation of somatostatin in the regenerating adult fish brain. Neurosci Lett. 2001;309:149–152. doi: 10.1016/s0304-3940(01)02061-4. [DOI] [PubMed] [Google Scholar]