

Abstract
Cholesterol is an essential membrane component enriched in plasma membranes, growth cones, and synapses. The brain normally synthesizes all cholesterol locally, but the contribution of individual cell types to brain cholesterol metabolism is unknown. To investigate whether cortical projection neurons in vivo essentially require cholesterol biosynthesis and which cell types support neurons, we have conditionally ablated the cholesterol biosynthesis in these neurons in mice either embryonically or postnatally. We found that cortical projection neurons synthesize cholesterol during their entire lifetime. At all stages, they can also benefit from glial support. Adult neurons that lack cholesterol biosynthesis are mainly supported by astrocytes such that their functional integrity is preserved. In contrast, microglial cells support young neurons. However, compensatory efforts of microglia are only transient leading to layer-specific neuronal death and the reduction of cortical projections. Hence, during the phase of maximal membrane growth and maximal cholesterol demand, neuronal cholesterol biosynthesis is indispensable. Analysis of primary neurons revealed that neurons tolerate only slight alteration in the cholesterol content and plasma membrane tension. This quality control allows neurons to differentiate normally and adjusts the extent of neurite outgrowth, the number of functional growth cones and synapses to the available cholesterol. This study highlights both the flexibility and the limits of horizontal cholesterol transfer in vivo and may have implications for the understanding of neurodegenerative diseases.
Introduction
Defects in cholesterol synthesis can have devastating effects on the developing nervous system, including corpus callosum agenesis (Waterham, 2002). Even during development, when the blood–brain barrier has not fully formed yet, the brain normally synthesizes the majority of its cholesterol locally (Tint et al., 2006). Cholesterol from the circulation can only be used by neuronal progenitors, when cholesterol biosynthesis has been inactivated in the ventricular zone (Saito et al., 2009). However, massive cell death in the cortical plate precludes further analysis of neuronal differentiation. It is possible that neocortical and hippocampal neurons synthesize cholesterol in the adult brain, as they express the required genes (Lein et al., 2007). In addition, neurons could also benefit from glial cholesterol that is transported within the brain by lipoprotein particles (Pfrieger, 2003; Hayashi et al., 2004). Mature cerebellar granule cells are even independent of cell-autonomous cholesterol synthesis (Fünfschilling et al., 2007). Also in vitro, the cholesterol requirements depend on the neuronal subtype (Ko et al., 2005; Steinmetz et al., 2006). It is unknown to date which cell types account for the cholesterol synthesis in the developing brain and to which degree cholesterol flux between cells can rescue cholesterol deficits.
To study brain cholesterol metabolism, we first have ablated cholesterol synthesis in postnatal forebrain projection neurons. The absence of defects confirmed that adult neurons can fully rely on exogenous cholesterol supply for function. Astrocytes were identified as the likely source of cholesterol. In a second approach, we ablated cholesterol biosynthesis in the same type of neurons (forebrain projection neurons) at the embryonic stage just after neuronal commitment starting at embryonic day 11.5 (E11.5). Despite apparent support from microglia, cortical projections were strongly affected. We conclude that, although glial cholesterol can support neurons at any stage of development, defective neuronal cholesterol biosynthesis can only be fully rescued in the adult brain.
Materials and Methods
Animal strains.
Experiments were in compliance with the animal policies of the Max Planck Institute of Experimental Medicine, approved by the German Federal State of Niedersachsen. Mouse strains were squalene synthase (SQS)-floxed mice (Saher et al., 2005), Nex-cre mice (Goebbels et al., 2006), and calcium/calmodulin-dependent protein kinase II (CaMKII)-cre transgenic mice (Minichiello et al., 1999). SQS/Nex-cre mutants with the genotype Fdft1flox/flox * Nex+/cre were controlled by littermates with the genotype Fdft1+/flox * Nex+/cre. In some experiments, mice additionally contained one allele yellow fluorescent protein Cre reporter Rosa26+/yfp (Srinivas et al., 2001). In the case of SQS/CaMKII-cre, mice were Fdft1flox/flox without (control) or littermates with one CaMKII-cre transgenic allele (mutant). Analyses were done using mice of either sex. Quantitative real-time PCR analysis on genomic DNA was done as described previously (Fünfschilling et al., 2007).
Cell culture.
Microisland cultures of hippocampal neurons were prepared as described previously (Jockusch et al., 2007). For continental hippocampal cultures, neurons derived from E17.5 mice were prepared and cultured according to Banker and colleagues (Dotti et al., 1988) in the absence or presence of 10 μg/ml (1 μg/ml in case of microisland cultures to ensure survival of astrocyte islands) cholesterol (Sigma-Aldrich) supplementation. Cholesterol was added from an ethanolic stock solution, and control cultures received equal amount of ethanol. Continental cultures had a purity of >98% based on immunolabeling for astrocytes and microglia. To ensure survival, cultures grown for 14 d in vitro (14 DIV) that were used to assess the ultrastructure of synapses were flipped onto a lawn of astrocytes (Dotti et al., 1988).
Growth cone collapse assays were done as described previously (Knöll et al., 2006). Briefly, after 3 DIV, hippocampal neurons were incubated for 30 min with 1 μg/ml EphrinA5-Fc chimeric protein or with human Fc (R&D Systems), both as complex with 10 μg/ml anti human Fc antibody (Sigma-Aldrich). In the case of cholesterol depletion assays, assay media also contained 0.5 mm methyl-β-cyclodextrin (CD) and/or 10 μm zaragozic acid A (ZA) (Sigma-Aldrich). Coverslips were then fixed and processed for immunolabeling. Data are expressed as percentage of collapsed neurites per cell in response to EphrinA5-Fc normalized to Fc.
Immunocytochemistry was performed using standard procedures. Briefly, fixed cells [10 min, 4% paraformaldehyde (PFA) in PBS] were permeabilized with 0.1% Triton X-100 for 5 min and incubated for 30 min in blocking solution (2% bovine serum albumin in PBS). Primary antibodies were diluted in blocking solution and applied overnight at 4°C. After washing with PBS, cells were incubated with secondary antibodies in blocking solution containing 0.2 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) or 50 μg/ml filipin for 1 h at room temperature. Quantification of filipin-derived fluorescence was done as described previously (Qin et al., 2006): after determining the area of the cell using the YFP fluorescence, the mean intensity of cellular filipin-derived fluorescence was measured.
For quantification of neurite lengths or of synaptic puncta, the NIH ImageJ software was used. Neurites were traced using images with βIII-Tubulin immunolabeling. Synaptic puncta were counted using binarized images of synaptophysin immunolabeling. Only those synaptophysin-positive puncta that were apposed to a MAP2-positive dendrite were included into the quantification. Data obtained from cell culture experiments were positively tested for normal distribution by the Kolmogorov–Smirnov test. Significance was then evaluated by one-way ANOVA or Student's t test (two-tailed uncoupled samples).
Biochemistry and biophysics.
SDS-PAGE and immunoblotting were performed according to standard methods (Saher et al., 2005). Cholesterol measurements were done as described previously (Saher et al., 2009). Mechanical properties of plasma membranes were measured by tether extraction using atomic force microscopy (AFM) (Sun et al., 2005). Using the AFM tip, a tether, a nanotube formed from membrane, was extracted from the plasma membrane. The force required to pull this tether from the cell (tether force) is a measure for plasma membrane bending rigidity and interaction of the membrane with the underlying cytoskeleton (Sheetz and Dai, 1996). An Asylum MFP3D AFM was placed onto a bright-field illuminated inverted optical microscope equipped with a 60×, 1.49 NA objective. First, a cell was selected with the optical microscope, and the AFM tip was positioned above the cell body. Then the tip was brought down to touch the cell at a force of ∼0.25 nN and pulled away for a distance of 5 μm. In ∼20% of all curves, a tether was extracted by the AFM tip, which was visible as constant force plateau when the tip was retracted. Since the tether force was measured at a relatively low pulling speed of 2 μm/s, the presented tether forces are 10–20% higher than the force F0 that would be required to hold the tether at a constant length (Sun et al., 2007). For the experiments, we used Olympus biolevers with a calibrated spring constant of ∼0.006 or ∼0.03 N/m. The experiments were performed at 24°C.
Histological and morphological analyses.
For histological analyses, mice were fixed in 4% PFA by perfusion (neonatal to adult) or immersion (embryonic tissue). Depending on the antibody, tissue sections were cut using a microtome, a cryostat, or a vibratome. Immunolabeling and histochemical stainings such as hematoxylin and eosin (H&E) and Gallyas silver impregnation were done as described previously (Saher et al., 2005). For quantification of Cre-positive cells, paraffin sections were immunolabeled for Cre. All nuclei touching a radial line through the cortical plate were counted with three lines per cross-section and n = 3 animals per genotype and age. Golgi silver impregnation (FD NeuroTechnologies) and Fluoro-Jade C staining (Millipore Bioscience Research Reagents) were done according to the manufacturers' instructions. In situ hybridization was performed as described previously (Saher et al., 2005). As hybridization probe, Apoe cDNA comprising exons 2–4 was used, transcribed in antisense and sense (control) direction. For DiI tracing, a small DiI crystal was placed in the motor cortex of newborn, PFA-perfused mice. Brains were stored at 37°C for 4 weeks, embedded, and cut on a vibratome. Specimens were analyzed by light microscopy (Zeiss Axiophot or using a confocal laser-scanning microscope, Leica DM RXA). Images were processed by using NIH ImageJ software.
Ultrastructural analysis of primary hippocampal neurons was done after 14 DIV as described previously (Connert et al., 2006). Briefly, cells were fixed overnight at 4°C in cacodylate buffer with 2.5% glutaraldehyde, 1% PFA, and 1% tannic acid. After postfixation with 1% OsO4 for 1 h, cells were stained with 1.5% uranyl acetate and 1.5% tunstphosphoric acid in 70% ethanol and embedded in Epon 812. Ultrathin sections of ∼60 nm were poststained using uranyl acetate and lead citrate and viewed with a Zeiss EM912 microscope.
Antibodies.
The following antibodies were used in this study: β-Actin (Sigma-Aldrich), Cntn2 (Developmental Studies Hybridoma Bank), Cre (Covance), Ctip2, Foxp2, Lmo4, Tbr1 (Abcam), GAPDH (BIOMOL), GFAP (Dako), YFP (Rockland), Iba1 (Wako), Mac3, SQS (BD Biosciences), Apolipoprotein E (ApoE), Map2, NeuN, Tau, βIII-Tubulin (Millipore Bioscience Research Reagents), Phalloidin–Rhodamine (Invitrogen), Smi31 (Sternberger Monoclonals), Synaptophysin (Synaptic Systems), Oct6/SCIP/Pou3f1 (generous gift from Dies Meijer, Amsterdam, The Netherlands), and Satb2 (generous gift from Victor Tarabykin, Göttingen, Germany).
Electrophysiology.
Hippocampal neurons of microisland cultures grown for 8–11 DIV were whole-cell voltage clamped at −70 mV with an Axoclamp amplifier under the control of the Clampex program 10.1. All analyses were performed using Axograph X. EPSCs were evoked by depolarizing cells from −70 to 0 mV. The readily releasable pool (RRP) size was measured after application of hypertonic sucrose solution (Jockusch et al., 2007). Miniature EPSCs (mEPSCs) were recorded in the presence of 300 nm TTX. The extracellular solution contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 CaCl2, and 4 MgCl2; 320 mOsmol/L, pH 7.3. The patch pipette solution for autaptic recordings contained the following (in mm): 136 KCl, 17.8 HEPES, 1 EGTA, 4.6 MgCl2, 4 NaATP, 0.3 Na2GTP, 15 creatine phosphate, and 5 U/ml phosphocreatine kinase; 315–320 mOsmol/L, pH 7.4. The patch pipette solution for paired recording of the postsynaptic cell contained the following (in mm): 5 QX-314, 10 HEPES, 0.25 EGTA, 1 MgCl2, 4 MgATP, 0.3 Na2GTP, 8 NaCl, 10 TEA-Cl, 15.5 CsCl, 117.5 cesium methane sulfonate; 300 mOsmol/L, pH 7.4. All extracellular solutions were applied with a custom-built fast flow system consisting of an array of flow pipes controlled by a stepper motor that allows complete and rapid solution exchange with time constants of ∼30 ms. All chemicals, except for TTX (Tocris Bioscience) and calcimycin (Calbiochem), were purchased from Sigma-Aldrich.
Data analysis.
All numerical values are shown as the mean ± SEM; n = 3–6, unless specified. Statistical significance was determined by Student's t test.
Results
Compensation of defective cholesterol biosynthesis in mature neurons
The enzyme SQS (Fdft1 gene) catalyzes the first step in the cholesterol biosynthetic pathway that is dedicated to the formation of sterols, and inactivation of Fdft1 abolishes cholesterol biosynthesis (Tozawa et al., 1999; Saher et al., 2005). To investigate whether cell-autonomous cholesterol biosynthesis is required to maintain the integrity of mature neurons, we genetically inactivated SQS in postnatal forebrain projection neurons (referred to as SQS/CaMKII-cre mice). Recombination was mediated by the CaMKII-cre transgene (Minichiello et al., 1999) that starts expressing around postnatal day 5 (P5) (Brinkmann et al., 2008) and shows robust recombination from P15 onward (Minichiello et al., 1999).
Conditional SQS/CaMKII-cre mutants were born at the expected mendelian ratio, were viable and fertile, and had a normal life span. Mutants could not be distinguished from littermate controls by physical examination and lacked neurological defects such as clasping, tremor, or convulsions. Nevertheless, by quantitative PCR analysis on genomic DNA of 1-year-old SQS/CaMKII-cre mutants, 39 ± 3% of cortical cells were recombined. This value indicates that the vast majority of projection neurons was targeted (Ren et al., 1992). Neuronal survival, shape, projections, maturation, or the maintenance of neuronal identity was normal as assessed by histological stainings (Gallyas for myelinated fiber tracts; Golgi for random labeling of individual neurons) and marker expression (NeuN for differentiated neurons; Ctip2 for layer 5/6 projection neurons) (Fig. 1a–d). Neither microglia activation nor astrogliosis was observed (Fig. 1e,f). When visualizing SQS on brain sections, we found moderate SQS expression in many cortical neurons in control animals (Fig. 1g), implying ongoing cholesterol biosynthesis. In contrast, mutant neurons completely lacked SQS staining. Astrocytes (identified based on their characteristic morphology), however, showed an enhanced SQS signal, despite the absence of astrogliosis. This redistribution of SQS expression in the cortex resulted in a comparable SQS content in conditional mutants and controls (Fig. 1h). It has previously been shown that ApoE, the major apolipoprotein of the CNS involved in lipid/cholesterol transport, can be upregulated to compensate for cholesterol synthesis deficits and CNS injury (Skene and Shooter, 1983; Saher et al., 2005). Indeed, conditional mutants showed robust increase in ApoE content, implying active cholesterol transport (Fig. 1h). Hence, defective cholesterol biosynthesis in mature cortical neurons can be compensated completely, likely by cholesterol transfer from astroglial cells.
Figure 1.
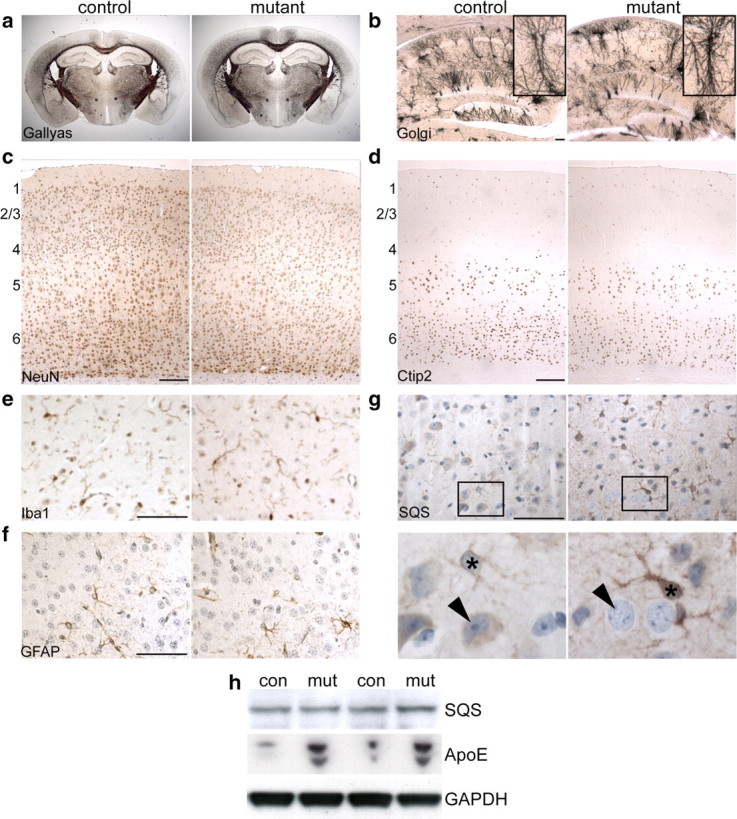
Rescue of conditional inactivation of SQS driven by CaMKII-cre. a, Gallyas silver impregnation shows normal myelinated fiber tracts in 16-month-old SQS/CaMKII-cre mutant animals. b, Morphology of hippocampal neurons as visualized by Golgi silver impregnation is unchanged. The insets show representative CA1 neurons. c–f, Immunohistochemical stainings on coronal brain sections detecting NeuN (c), Ctip2 (d), Iba1 (e), and GFAP (f) appear comparable in controls and mutants (6 months). The numbers indicate approximate position of neocortical layers. g, SQS immunolabeling in the cortex. While in control animals both neurons (arrowhead) and astrocytes (asterisk) are positive for SQS, neurons in conditional mutants fail to express SQS (arrowhead) and astrocytes show enhanced staining for SQS (asterisk). h, Western blot of cortical lysates of 2-month-old animals detecting SQS and ApoE. Comparable staining of GAPDH confirmed equal protein loading (4 representative animals shown). Scale bars: b–d, 100 μm; e–g, 50 μm.
Neonatal lethality after inactivation of SQS in embryonic neurons
Are embryonic neurons equally resistant to loss of cholesterol biosynthesis in vivo? These neurons could be more sensitive to cholesterol deprivation because neuronal membrane expansion is high in the embryonic brain. In addition, potential support from astrocytes is limited as most astrocytes are born postnatally (Vallejo, 2009). We analyzed SQS/Nex-cre mice in which Cre under the control of the Nex (also referred to as Neurod6) promoter mediates the inactivation of SQS in virtually all newly committed, postmitotic cortical projection neurons as they are born between E11.5 and E16.5 (Goebbels et al., 2006; Molyneaux et al., 2007). Where appropriate, mice also contained one allele Cre reporter Rosa26+/yfp (Srinivas et al., 2001), which allows identification of Nex-cre-expressing cells both in controls (Fdft1+/flox * Nex+/cre) and mutants (Fdft1flox/flox * Nex+/cre). In the cortex, the following structures are not targeted by Nex-cre: thalamic afferents, GABAergic neurons, Cajal–Retzius cells of the marginal zone, and neuronal progenitors of the ventricular zone.
Mutant mice were born in mendelian ratio but died neonatally probably due to respiratory distress and failure to feed (Fig. 2a). The body weight of mutant pups was slightly reduced (82 ± 6%). Surprisingly, sagittal brain sections of neonatal mice visualizing the Cre reporter YFP revealed that the gross brain morphology was preserved (Fig. 2b), showing only some disorganization. Brain sections from E16.5 mice immunolabeled for Cre showed similar localization of targeted cells in mutants and controls with only slight alterations, such as few ectopic Cre-positive cells in the marginal zone of mutants (Fig. 2c). In both mutants and controls, practically all cells of the cortical plate were positive for Cre (see below). When detecting the ER-resident SQS protein on consecutive sections, the cortical plate appeared heavily positive in controls but was virtually devoid of SQS in mutants (Fig. 2d). Compatible with the expression pattern of Nex-cre, the subventricular zone of mutants also showed some SQS reduction. In contrast, SQS staining was normal in cells lacking Nex expression, such as the Cajal–Retzius cells of the marginal zone and neuronal progenitors of the ventricular zone. The intermediate zone was also heavily positive for SQS protein, possibly because of SQS located in the axoplasmic reticulum of thalamic neurons. As assessed by Western blotting of cortical lysates, SQS protein was diminished by approximately one-third in mutants compared with control littermates (Fig. 2e). This finding matched the reduction of cholesterol in forebrain lysates from newborn mutants as measured by HPLC [4.07 ± 0.21 mg of cholesterol per gram wet weight in mutants compared with 6.01 ± 0.23 mg in controls; p < 0.001 (n = 4–6 animals)]. We conclude that SQS/Nex-cre mutant neurons form a remarkably normal cortex despite efficient ablation of neuronal SQS expression.
Figure 2.

Conditional inactivation of cholesterol biosynthesis. a, Newborn conditional mutants lack milk in their stomach (arrows) and die perinatally. b, Immunolabeling of the Cre reporter YFP on sagittal brain sections of control and mutant mice at P0 visualize targeted neurons in the cortex (Ctx), hippocampus (Hip), and ventral part of the accessory olfactory nucleus (AOV). Fiber tracts are also YFP positive, such as anterior commissure (ac) and the pyramidal tract (py). The neurons of the thalamus (Th) were not targeted. c, Immunohistochemistry for Cre visualizing targeted neurons in the cortex at E16. d, Immunohistochemistry detecting SQS at E16 demonstrates the loss of SQS in the cortical plate (CP) and a reduction in the subventricular zone (SVZ) of mutant animals. In both controls and mutants, SQS is present in the marginal zone (MZ), intermediate zone (IZ), and ventricular zone (VZ). Scale bars, 100 μm. e, The level of SQS protein (48 kDa) is reduced by 38 ± 3% in mutant cortical lysates shown by Western blots from E18.5 control and mutant animals (4 representative animals shown). Comparable staining of β-actin demonstrates equal loading of protein.
Microgliosis and premature neuronal death in vivo
Are glial cells also at this young age involved in preserving neuronal integrity of conditional mutants? Brain sections from E16.5 to P0 mice were immunostained for MAC-3, a marker for activated microglia, or Iba1, which detects all microglia (data not shown). In contrast to controls, cortices of mutant animals at P0 contained many activated microglia (Fig. 3a). Some microglia were recruited already at E16.5, and numbers of activated microglia increased dramatically from E17.5 to P0 with a preferential location in lower cortical layers, the subventricular zone, and the designated subcortical white matter (Fig. 3a,e). In contrast, astrogliosis was limited. While embryonic mutants completely lacked enhanced presence of GFAP-positive cells (data not shown), the few isolated astrocytes in neonatal mice were restricted to areas of massive microgliosis (Fig. 3b).
Figure 3.
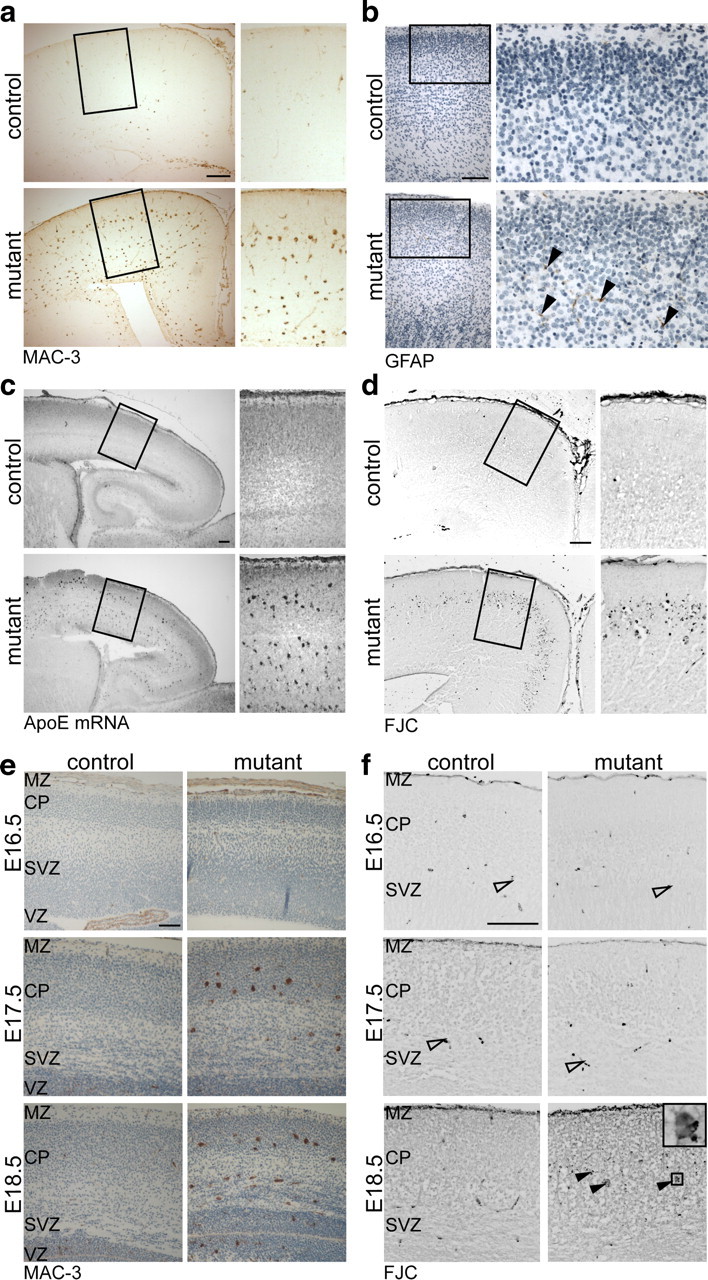
Impaired survival of a subset of mutant neurons and microgliosis. a, Immunohistochemistry detecting activated microglia (MAC-3) on coronal brain sections reveals massive microgliosis in neonatal mutant mice. b, Immunostaining for GFAP (positive cells marked by arrowheads) shows few astrocytes in mutant mice at P0. c, In situ hybridization detecting Apoe mRNA in neonatal mice shows substantial increase of Apoe-expressing cells specifically in the neocortex of mutants. d, Degenerating neurons were visualized by FJC (shown as inverted image). While on brain sections of control mice hardly any neurons are fluorescent, a band of degenerating neurons is present in the neocortex of mutant mice. e, Microglia activation visualized by MAC-3 begins around E16.5 in the neocortex of conditional mutants and increases with development. f, Enhanced neuronal degeneration in conditional mutants visualized by FJC (shown as inverted images) is not seen earlier than E18.5 (filled arrowheads; detail of boxed area on the right). The open arrowheads mark blood cells that are also stained by FJC. Scale bar, 100 μm.
Visualizing Apoe mRNA by in situ hybridization revealed a dramatic increase of ApoE-expressing cells in mutants (Fig. 3c). Judging by their morphology and location, these cells clearly resembled the Mac3-positive microglia. In contrast, untargeted brain regions including the caudate–putamen showed comparable Apoe expression, implying that the induction of Apoe was locally controlled (data not shown).
Neuronal degeneration was also evaluated from E16.5 to P0 by assessing condensed nuclei (data not shown) and by staining with Fluoro-Jade C (FJC), a fluorescent compound that accumulates in degenerating neurons. The cortex of control animals showed only negligible FJC-derived fluorescence at any age tested. In contrast, a few cortical neurons of mutant animals were degenerating starting in areas of microgliosis at E18.5 (Fig. 3f) with a massive increase at P0 (Fig. 3d). However, the vast majority of mutant neurons were not lost (yet), as the number of Cre-positive cells in the cortical plate was unchanged (number of Cre-positive nuclei in mutants compared with controls: 104 ± 6% at E16.5 and 97 ± 6% at P0; n = 3 animals). We cannot exclude the possibility that microglia-mediated clearance was too fast to detect neuronal degeneration before E18.5. However, these findings support the hypothesis that, in addition to their response to neurotoxic insults, microglia may actively support neighboring neurons: microglia are recruited at least 2 d before the onset of neuronal death (Fig. 3, compare e, f) and they are in the position to synthesize cholesterol and provide it in the form of ApoE-containing lipoprotein particles.
Cholesterol-dependent subcerebral projection neurons
What is the identity of the neurons that degenerate perinatally? Several neuronal transcriptional regulators were visualized on brain sections showing neocortical layer enriched expression patterns (Molyneaux et al., 2007), such as Satb2, Ctip2, Tbr1, NeuN and Oct6, FoxP2, Lmo4 (data not shown). In the sensorimotor area of the neocortex of newborn mutants and controls, we found staining of these markers in equivalent cortical areas (Fig. 4), confirming the preservation of cortical anatomy. Abnormalities in migration are obviously not a feature of SQS/Nex-cre mutants. Only rarely did we observe condensed nuclei positive for any of the tested markers, probably because of the fast degradation of these transcription factors in dying neurons. Subplate neurons and layer 6 neurons were affected only to a minor extent (Fig. 4d; data not shown). Surprisingly, only one subset of neurons, the presumptive subcerebral projection neurons located in layer 5 with large nuclei and strong expression of Ctip2 and Oct6 (Arlotta et al., 2005; Molyneaux et al., 2007), were practically absent in mutants (Fig. 4c, indicated with brackets).
Figure 4.
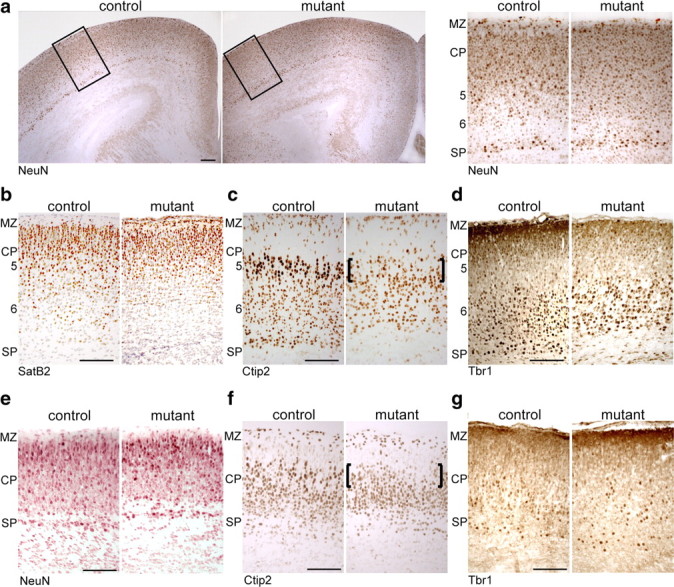
Reduced number of layer 5 neocortical neurons. Immunohistochemistry on coronal brain sections at P0 detecting NeuN (a), SatB2 (b), Ctip2 (c), Tbr1 (d), and at E16.5 detecting NeuN (e), Ctip2 (f), and Tbr1 (g). The brackets mark the absence of subcerebral projection neurons. The numbers indicate approximate position of cortical layers. MZ, Marginal zone; CP, cortical plate; SP, subplate. Scale bar, 100 μm.
The dramatic reduction of specifically subcerebral projection neurons could be explained by premature neuronal death and/or by impaired differentiation. To assess potential abnormalities in neuronal differentiation, we visualized the above-mentioned neuronal markers on brain sections of E16.5 mice, before the onset of neuronal degeneration and microglia activation (Fig. 4e–g). Already at E16.5, only the presumptive subcerebral projection neurons of layer 5 were clearly reduced in number (Fig. 4f). This demonstrates that, in addition to cell survival, cholesterol depletion affects the final specification of subcerebral projection neurons.
Shorter projections in vivo
As axons and dendrites harbor membrane domains with particular cholesterol demands (Tashiro et al., 2004), we examined the integrity of neurites in newborn SQS/Nex-cre mice (also containing the YFP Cre reporter). The thickness of the subplate was reduced in conditional mutants, but the formation of apical dendrites in the neocortex as assessed by MAP2 staining appeared quite normal (Fig. 5a).
Figure 5.
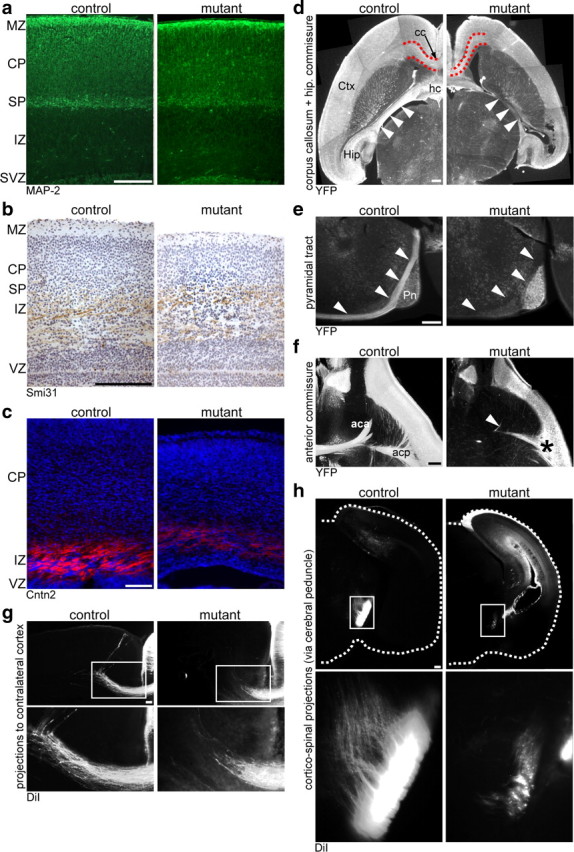
Projections in vivo. a–c, Coronal brain sections were immunolabeled detecting MAP2 (a), phosphorylated neurofilaments by the Smi31 antibody (b), and Contactin2 (Cntn2) (c). d–f, Immunohistochemistry detecting the Cre reporter YFP on horizontal (d, f) or sagittal (e) vibratome sections. d, The corpus callosum (cc), subcortical white matter (outlined in red), hippocampal commissure (hc), and fornix (arrowheads) contain less projections from targeted neurons in mutants compared with controls. e, Whereas the pyramidal tract (arrowheads) is immunostained in controls, equivalent projections in mutant mice are absent. f, While the anterior commissure with its anterior part (aca) and posterior part (acp) is clearly detectable in controls, the aca is completely absent in mutants (arrowhead) and the acp is diminished in size (asterisk). g, h, Coronal vibratome sections detecting DiI-labeled projections. In mutant animals, both callosal fibers (g) and corticospinal projections (h) are reduced in number and lengths. CP, Cortical plate; Ctx, cortex; Hip, hippocampus; IZ, intermediate zone; MZ, marginal zone; Pn, pons; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone. Scale bar, 100 μm.
In contrast, when cortical axons in the intermediate zone were stained by Contactin2, a marker of cortical efferents (Fukuda et al., 1997), staining intensity was severely reduced in mutants (Fig. 5c). The abundance of axons in the intermediate zone appeared unchanged when detecting the panaxonal Smi31 antigen staining targeted cortical efferents and nontargeted thalamic afferents (Fig. 5b). This highlights the specificity of defects for targeted neurons. Indeed, fiber tracts originating exclusively from targeted neurons were greatly reduced in size (corpus callosum, hippocampal commissure) or even absent (anterior part of the anterior commissure, pyramidal tract at the pons). This was evident when comparing H&E-stained sections (data not shown), visualizing the Cre reporter YFP (Fig. 5d–f) and DiI fiber tracing of crystals placed in the motor cortex (Fig. 5g,h). Axonal pathfinding was not affected, because misrouting of fiber tracts was never observed. We conclude that mutant neurons extend axons in vivo that often fail to reach their target in time. Impaired differentiation and degeneration of subcerebral projection neurons likely contribute to the lack of the pyramidal tract.
Rescue of mutant neurons by supplemented cholesterol in vitro
Why are embryonic neurons not rescued by glial support in vivo? Could the cholesterol supply by embryonic glial cells in vivo not suffice to meet the neuronal demand? To investigate whether external cholesterol can completely rescue embryonic neurons, we analyzed primary hippocampal neurons from E17 SQS/Nex-cre mice in vitro in the absence or presence of cholesterol supplementation. Targeted mutant and control neurons were identified by staining for the YFP Cre reporter.
First, we determined cholesterol dependency of neuronal survival (Fig. 6a). After 1 DIV, ∼45% of all DAPI-positive cells were YFP-positive neurons under all tested conditions, indicating that freshly isolated mutant neurons were viable. By 4 DIV, this percentage had dropped to ∼25% for control neurons regardless of cholesterol supplementation. In contrast, in cultures derived from mutants, only 5 ± 2% of cells were YFP-positive mutant neurons surviving in the absence of cholesterol. This massive loss of mutant neurons was completely rescued in the presence of exogenous cholesterol (23 ± 2%). We conclude that neurons require cholesterol for survival and mutant neurons can use exogenous cholesterol.
Figure 6.
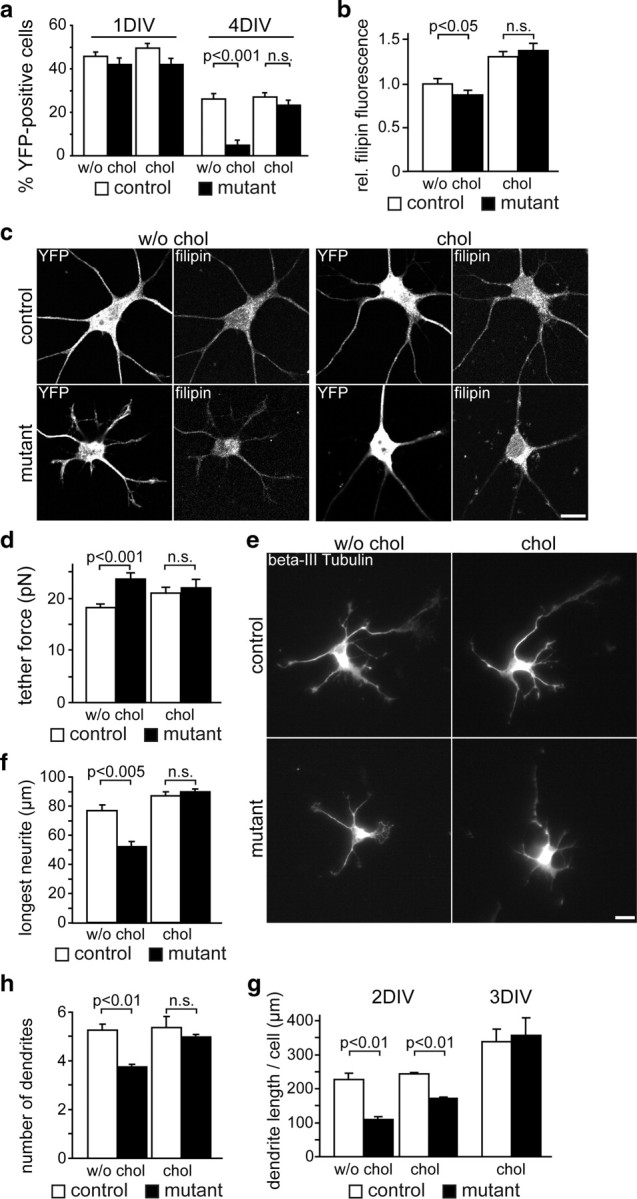
Cholesterol dependency of neurite outgrowth. a, Survival of hippocampal neurons derived from SQS/Nex-cre conditional mutants (Fdft1flox/flox * Nex+/cre * Rosa26+/yfp) and controls (Fdft1+/flox * Nex+/cre * Rosa26+/yfp) was assessed after 1 d (1 DIV) and 4 d (4 DIV) in culture supplemented with or without cholesterol by immunolabeling for YFP (n = 5 animals per genotype). b, Quantitative evaluation of filipin-derived fluorescence intensity that correlates with cellular free cholesterol content of neurons at 3 DIV (n = 21–24 cells per condition of two to three different embryos) from images as in c. c, Hippocampal neurons were cultured for 3 d with (chol) or without (w/o chol) cholesterol, and immunolabeled for YFP and filipin. Scale bar, 10 μm. d, AFM tether extraction measurements reveal that the mean tether force of mutant neurons is higher than that of the controls (mutants, 23.6 ± 1.2 pN; controls, 18.2 ± 0.5 pN) but normalizes after the addition of cholesterol (mutants, 21.9 ± 1.7 pN; controls, 20.9 ± 1.3 pN; n = 15–30 cells of 4 animals per genotype). e, Immunofluorescence of hippocampal neurons after 2 d detecting βIII-Tubulin. Tracings of neurites were taken from 80–91 cells per condition (n = 4–5 animals per genotype). Scale bar, 10 μm. f, The length of the longest neurite (axon) after 2 DIV without cholesterol supplementation was reduced to 68 ± 5% in mutants, which was rescued by cholesterol (103 ± 2%). g, The number of dendrites per cell was reduced to 71 ± 2% in mutants compared with controls after 2 DIV without cholesterol supplementation. This effect was rescued by the addition of cholesterol (93 ± 2%). h, After 2 DIV, the total dendrite length per cell was reduced to 48 ± 4% in mutants compared with controls in the absence of cholesterol supplementation. The addition of cholesterol increased the total dendrite length to 71 ± 2% after 2 d, and rescued it after 3 DIV (105 ± 15%). Error bars indicate SEM.
Second, cellular (free nonesterified) cholesterol content was determined by staining for cholesterol (filipin) and YFP. The mean filipin-derived fluorescence intensity (that correlates with the cholesterol content) (Qin et al., 2006) was quantified in the YFP-positive cellular area. After 3 DIV, mutant neurons showed only a slight decrease by 12 ± 3% compared with control values (Fig. 6b,c). This is surprising as cultured neurons treated with cholesterol biosynthesis inhibitors for 1–2 d show a drop in cellular cholesterol of at least 50% (Fan et al., 2002; Sierra et al., 2011). When grown in cholesterol-supplemented media, fluorescence intensity increased in mutant and control neurons reaching ∼130% of control values. Thus, cultured cells take up large amounts of cholesterol from the medium (when supplied in the nonphysiological form of an ethanolic solution). Plasma membrane cholesterol, which is crucial for the functionality of axons and dendrites, cannot be calculated from filipin-stained images as filipin also stains intracellular cholesterol. For that reason, we turned to an indirect but more precise experiment analyzing mechanical properties of plasma membranes by AFM. Plasma membrane tension was measured via the force that is required to pull a nanotube/tether from the plasma membrane. This plasma membrane tether force is mainly determined by the interaction of the membrane with the underlying cytoskeleton, and the membrane bending rigidity (Sheetz and Dai, 1996), both influenced by membrane cholesterol (Sun et al., 2007). Interestingly, the tether force of mutant neurons was slightly but significantly higher, and this difference was leveled out by the addition of cholesterol (Fig. 6d). Similar changes, however more dramatic, were measured in bovine aortic endothelial cells upon cyclodextrin treatment (Sun et al., 2007). Despite the robust increase of total cholesterol in case of cholesterol supplementation (increased filipin fluorescence) (see above), plasma membrane cholesterol remained normal (tether force unchanged in controls) or returned to normal (tether force reached control levels in mutants). Surplus cholesterol was thus located in intracellular stores. These findings indicate neurons tolerate only minimal changes of the plasma membrane cholesterol content.
Next, we assessed the ability of mutant neurons to form and extend processes in vitro (Fig. 6e–g). Neurite lengths were measured after 2 DIV, when death of mutant neurons was not prominent (80–91 cells from four to five animals per condition). In the absence of supplemented cholesterol, mutant neurons formed ∼30% shorter neurites (axon length, 68 ± 5%; mean dendrite length, 69 ± 6% of control). Moreover, the number of dendrites was reduced to 71 ± 2%, resulting in reducing the total dendritic length per neuron to 48 ± 4%. Polarization of neurons was unchanged based on normal distribution of respective marker proteins for axons (Tau) and dendrites (MAP2) (data not shown). While cholesterol supplementation had only minor impact on control neurons, it fully rescued the axon length and number of dendrites in mutants after 2 DIV. The length of dendrites also increased but reached control levels only after 3 DIV (Fig. 6g), reflecting the normal order of differentiation of cultured neurons (Dotti et al., 1988). Together, control neurons do not depend on external cholesterol for the formation of neurites, and endogenous cholesterol is not rate limiting. In mutant neurons, the cholesterol content of the plasma membrane is maintained in a physiological range at the expense of cellular growth. We hypothesize that plasma membrane quality control system permits membrane growth only when cholesterol content is above threshold (see also below) as it has been proposed for oligodendrocytes (Saher et al., 2005).
Cholesterol-dependent growth cone response
Cholesterol is a critical component of membrane lipid rafts (Lingwood and Simons, 2010). Hence, cholesterol could influence neurite extension by facilitating signaling events of receptors whose activity depends on membrane lipid rafts (Guirland et al., 2004; Pereira and Chao, 2007; Petrie et al., 2009; Qin et al., 2010). Since Eph receptors have a putative raft localization (Liu et al., 2008), we tested whether EphrinA5-induced Eph receptor signaling could be used as a model for cholesterol-dependent signaling events. To this end, we examined the responsiveness of neuronal growth cones of wild-type hippocampal neurons to the repulsive axon guidance molecule EphrinA5 under cholesterol deprivation. Induction of growth cone collapse by EphrinA5-Fc chimeric protein was controlled by cultures that received Fc without EphrinA5. In addition, cells received the following treatments: cholesterol was extracted using 0.5 mm CD, and cholesterol biosynthesis was abolished by the SQS inhibitor ZA. EphrinA5-Fc collapsed the same ratio of growth cones of untreated cells and of cells treated with ZA or CD alone (Fig. 7a). Only when cholesterol was extracted and its replenishment was inhibited at the same time, EphrinA5-induced signaling was abolished, collapsing merely 10 ± 22% of levels found for untreated cells (Fig. 7a). Importantly, the formation of growth cones in experiments without EphrinA5 was independent of the treatment (untreated, 79 ± 5%; CD, 64 ± 8%; ZA, 80 ± 6%; CD plus ZA, 68 ± 7%). Higher doses of CD disintegrated cells and caused growth cone collapse such that EphrinA5-mediated collapse could not be evaluated. Thus, EphrinA5-induced growth cone collapse is indeed cholesterol dependent, although only a profound depletion of the cholesterol content abolishes Eph receptor signaling.
Figure 7.
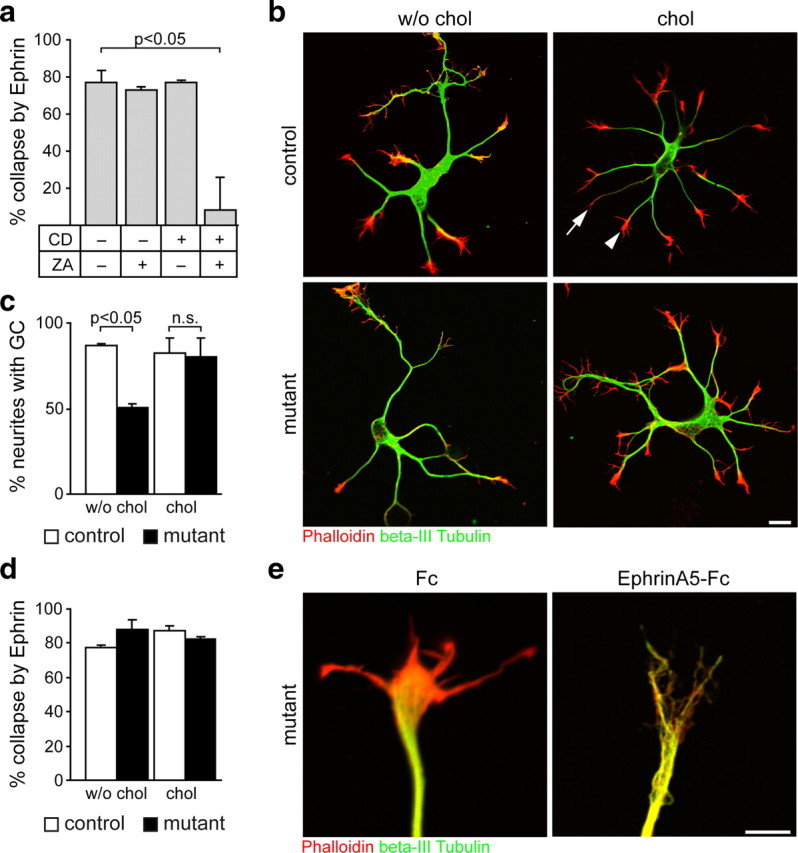
Formation of growth cones and EphrinA5-induced signaling. a, Wild-type hippocampal neurons after 3 DIV were either treated with 0.5 mm CD and/or 10 μm ZA or left untreated. Growth cone collapse was induced by addition of EphrinA5-Fc, while treatment with Fc alone served as control. Cells were stained detecting f-Actin (Phalloidin) and βIII-Tubulin and analyzed using 20 cells per condition (n = 4 animals). b, Hippocampal neurons derived from mutant and control animals after 3 DIV in the presence or absence of cholesterol were stained for f-Actin (Phalloidin), βIII-Tubulin, and YFP (data not shown). A growth cone was considered collapsed or underdeveloped when the f-Actin signal did not increase the neurite width visualized by βIII-Tubulin (arrow) compared with a normal growth cone (arrowhead). Scale bar, 10 μm. c, Percentage of neurites forming a growth cone (GC) in experiments as in b. Growth cones were analyzed using 24 cells per condition (n = 2 animals per genotype). d, Percentage of growth cone collapse was assessed in experiments as in e. Growth cones were analyzed using 24 cells per condition (n = 2 animals per genotype). e, Cultures as described in b were treated with EphrinA5-Fc or Fc. In the presence of Fc, mutant neurons formed a normal-appearing growth cone that could be collapsed by EphrinA5-Fc. Scale bar, 5 μm. Error bars indicate SEM.
Do growth cones of SQS/Nex-cre mutant neurons contain sufficient cholesterol to support Eph receptor signaling? Induction of growth cone collapse was observed in control neurons in the absence (77 ± 1%) or presence of cholesterol (87 ± 5%) (Fig. 7b–e). Surprisingly, the same efficiency of collapse was observed in mutant neurons (88 ± 3% without cholesterol; 82 ± 1% with cholesterol). Unlike cholesterol-deprived wild-type neurons (see above), the percentage of neurites with growth cones in mutants was reduced a priori (with Fc): 87 ± 1% neurites with a growth cone in control cells compared with 51 ± 2% in mutants (Fig. 7c). This reduction was leveled out completely by the addition of cholesterol. Together, in contrast to acutely deprived wild-type neurons, neurons with genetically ablated cholesterol biosynthesis produce fewer growth cones, but these contain sufficient cholesterol to be fully responsive to EphrinA5.
Normal synaptic transmission but fewer synapses
Acute cholesterol depletion by cyclodextrin treatment results in altered membrane properties that also affect synaptic transmission in Purkinje cells and wild-type hippocampal neurons (Wasser et al., 2007; Smith et al., 2010). Although spontaneous vesicle release is normal in time course and amplitude, the frequency of their release is increased. Therefore, we investigated the potential of mutant neurons to form functional synapses. As the mutants die perinatally, we examined spontaneous single synaptic fusion events, mEPSCs with microisland cultures of hippocampal neurons after 11–15 DIV. The mean amplitude and time course of mEPSCs were unchanged (Fig. 8a), demonstrating that quantal sizes were unaffected, and postsynaptic defects are unlikely in mutant neurons. However, the frequency of mEPSCs in mutant neurons was reduced to approximately one-third (Fig. 8a). When action potential-evoked EPSCs were recorded, the amplitudes were reduced to 41 ± 6% of control values (Fig. 8b). These findings could be explained by a reduction of the number of synapses per neuron or defective signal transmission by individual synapses. Presynaptic release depends on the size of the RRP and probability of vesicular release (Pvr). The pool size as measured by hypertonic sucrose application (or calcimycin treatment) (data not shown) was reduced to similar extents as EPSC amplitudes (Fig. 8b) such that the Pvr rendered unchanged (9.3 ± 0.6% in controls and 8.3 ± 0.8% in mutants). The number of synapses as determined by counting synaptophysin puncta per cell was reduced to 57 ± 7% (Fig. 8c,f). This reduction in synapses correlated with the smaller size of mutant neurons: The cell surface measured by membrane capacitance was 52 ± 4% of controls (Fig. 8c). The axonal area (Tau-positive area) was reduced to 61 ± 8%, and the area of the somatodendritic compartment (MAP2-positive area) was reduced to 62 ± 6% (Fig. 8g). Importantly, the density of synapses per dendrite length was unchanged (96 ± 14% of control). In separate experiments, the supplementation of low-level cholesterol to microisland cultures rescued the amplitude of evoked responses, the RRP size, cell size (capacitance), and synapse numbers close to control levels (Fig. 8d,e). In accordance with the above electrophysiological results, ultrastructural analysis of continental cultures of hippocampal neurons (after 14 DIV) revealed normal boutons in mutants and controls (Fig. 8h). These findings show that (in contrast to acutely cholesterol-depleted neurons) mutant neurons form fewer yet functionally normal synapses.
Figure 8.
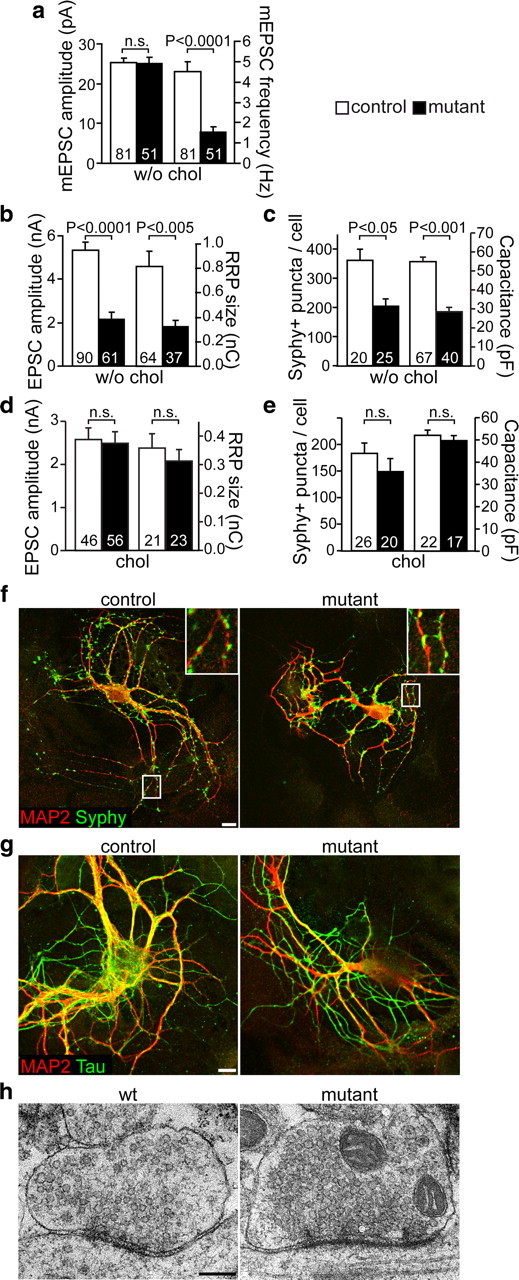
Formation of synapses. a, b, Electrophysiology on microisland cultures (without supplemented cholesterol) measuring mEPSC amplitude and mEPSC frequency (a), and EPSC amplitude and RRP size (b). The numbers indicate sample size. c, Number of synaptophysin-positive (Syphy+) puncta (apposed to MAP2-positive dendrites) per cell and the membrane capacitance as a measure of cell size were also reduced in mutants compared with control neurons (without supplemented cholesterol). d, In a different set of experiments, microisland cultures were cultivated in the presence of cholesterol, and EPSC amplitude and RRP size were measured. e, Supplementation of cholesterol almost rescues the number of synaptophysin-positive puncta per cell and the membrane capacitance in mutant neurons compared with controls. f, g, Microisland cultures (11 DIV) were immunostained for synaptophysin and MAP2 (d) or Tau and MAP2 (e). Scale bar, 10 μm. h, Representative synaptic boutons from wild-type (wt) and mutant hippocampal neurons grown in continental cultures for 14 DIV. Scale bar, 200 nm. Error bars indicate SEM.
Discussion
Cholesterol metabolism in the brain and horizontal cholesterol transfer
Using molecular and genetic strategies, we found a critical time window for cell-autonomous cholesterol biosynthesis in projection neurons of the mouse forebrain during development. Newly postmitotic projection neurons (this study) are especially susceptible to cholesterol deprivation in contrast to neural progenitors (Saito et al., 2009), mature projection neurons (this study), or postmitotic postmigratory cerebellar granule cells (Fünfschilling et al., 2007). While neural progenitors stimulate angiogenesis for cholesterol uptake from the circulation (Saito et al., 2009), neurons in the cortical plate depend on the cholesterol supply from within the brain. Our data suggest that astrocytes respond with enhanced cholesterol biosynthesis to the lack of cholesterol in neighboring neurons in postnatal brain. Because astrocytes are generated after neurogenesis has been practically completed (Vallejo, 2009), microglia likely provide cholesterol for newly committed mutant projection neurons in embryonic brain. Microglial lipoprotein particles containing ApoE ameliorate mevastatin-induced neuronal death in vitro (Xu et al., 2000), and lipoprotein particles can be used as a cholesterol source for axon growth (Hayashi et al., 2004). Similarly, microglia in SQS/Nex-cre mutants could locally provide ApoE-containing cholesterol-rich lipoprotein particles to neighboring mutant neurons.
We and others have determined the life span of neurons lacking cholesterol biosynthesis in vitro: Squalestatin induces neuronal death after 2 DIV (Michikawa and Yanagisawa, 1999) and only few mutant neurons survived 4 DIV in this study, when cholesterol biosynthesis was abolished genetically. A comparable survival period of 3 d (beginning of apoptosis after the onset of recombination) is found in vivo in SQS/Nestin-cre mutants targeting neuronal and glial progenitors (Saito et al., 2009). This is remarkable given the additional time required for SQS loss after Cre-mediated Fdft1 inactivation. In SQS/Nestin-cre mutants, no signs of astroglial or microglial support were evident (G. Saher and U. Fünfschilling, unpublished observation). Astonishingly, we found prolonged survival of mutant neurons in vivo in SQS/Nex-cre mice for at least 1 week (onset of recombination at E11.5 to evident neuronal degeneration at E18.5/P0). Hence, neurons must have received external support. SQS/Nex-cre mice demonstrate in vivo the neuroprotective function of microglia. This microglial support, however, does not suffice to meet the entire neuronal cholesterol demand leading to neuronal degeneration around birth. While numerous neuronal factors may alert microglia (Nakamura, 2002), the signals that specifically increase cholesterol biosynthesis in microglia and astrocytes are currently unknown.
Cholesterol-dependent neuronal integrity
Why are some neuronal subtypes more susceptible to cholesterol deprivation than others? Also in vitro, neuronal subpopulations react differently to statin-mediated cholesterol deprivation (Ko et al., 2005). Theoretically, cholesterol depletion could be critical in several independent ways. First, the time after inactivation of cell-autonomous cholesterol synthesis could be important. Indeed, lower cortical layers that contain the “oldest” neurons (born between E11.5 and E13.5) (Molyneaux et al., 2007) are more affected than upper cortical layers (born after E14.5) (Molyneaux et al., 2007). Although subplate neurons are reduced in number in SQS/Nex-cre mutants, the strongest reduction is found in the younger layer 5 subcerebral projection neurons. These neurons are born ∼2 d later, indicating that the time span without cholesterol biosynthesis is not the sole determinant of neuronal cell death.
Second, the reduction of cholesterol may affect neurosteroid synthesis and consequently CNS development. Mice that lack the rate-limiting enzyme of steroidogenesis that converts cholesterol to pregnenolone, P450scc (Cyp11a1), or the steroidogenic acute regulating protein (StAR; Star gene) that facilitates cholesterol import into mitochondria, do not exhibit obvious neurological phenotypes (Miller and Auchus, 2011). Hence, an involvement of neurosteroids in the pathology of SQS/Nex-cre mutant mice seems unlikely.
Third, inactivating SQS could lead to the accumulation of farnesyl-pyrophosphate (the substrate of SQS) and its metabolite farnesol. However, the stability of HMG-CoA reductase, the rate-limiting enzyme of the sterol biosynthesis pathway, is regulated by negative feedback of both isoprenoids (Correll et al., 1994). Although long-term accumulation of isoprenoids seems unlikely, we cannot formally exclude an impact on the isoprenoid pathway in SQS mutant neurons.
Fourth, subcerebral and subcortical projection neurons increase their membrane surface dramatically during peak axonal outgrowth (around E18–P1 in mice) (Polleux, 2005). These neurons would consequently run into cholesterol deprivation faster than neurons with short projections, which could explain the strong reduction of the pyramidal tract and the death specifically of subcerebral and subcortical projection neurons in our SQS/Nex-cre mutants. Consequently, loss of cholesterol synthesis in adult cortical neurons may be tolerated so well because of limited membrane growth after maturation.
In the case of layer 5 subcerebral projection neurons, cholesterol appears to be critical already during the period of final specification (approximately E13.5–E16.5) (Molyneaux et al., 2007). In that aspect, SQS/Nex-cre mutants phenocopy Ctip2 (Bcl11b gene) null mice and Fezf2 (a transcription factor potentially regulating Ctip2 expression) null mice: both are devoid of Ctip2-positive neurons, and corticospinal projections are virtually absent beyond the pons (Arlotta et al., 2005; Molyneaux et al., 2005). Although the relationship between cholesterol and these transcriptional regulators remains unknown, one could envision a scenario similar to myelin-forming glia. Myelinating glia need cholesterol for final differentiation marked by high expression of genes encoding myelin-specific proteins (Saher et al., 2005, 2009). Likewise, low cholesterol could prevent neurons from expressing the full set of genes specific for subcerebral projection neurons.
We and others have found that the lengths of axons and dendrites are affected by cholesterol depletion in vitro (Fig. 6d) (Fan et al., 2002; Pooler et al., 2006). In comparison, neurons from DHCR7 null mice (that lack the last step of cholesterol synthesis) accumulate the precursor 7-deydrocholesterol, which is predicted to show different membrane dynamics (Róg et al., 2008). Despite their altered membrane composition, these neurons actually form longer axons and dendrites in vitro (Jiang et al., 2010).
Normal physiology of synapses
Acute pharmacological depletion of cholesterol alters synaptic transmission with supposedly unchanged numbers of synapses (Wasser et al., 2007; Smith et al., 2010). In contrast, genetic cholesterol deprivation leads to electrophysiologically normal synapses, however reduced in number with concomitant reduction in synaptic transmission in vitro. The underlying wild-type astrocytes likely provide sufficient cholesterol for neuronal survival but not the saturating levels of cholesterol to mutant neurons as achieved in our cholesterol supplementation experiments that also restore neurite outgrowth (Fig. 6). This is further supported by the almost complete rescue of electrophysiological values (Fig. 8) when microisland cultures were supplemented with only 10% of the cholesterol concentration used in continental cultures. These findings are in agreement with a study that shows only negligible influence of glial factors on synapse formation by wild-type hippocampal neurons (Steinmetz et al., 2006).
Cholesterol or other glial-derived factors promote synaptogenesis in cultured retinal ganglion cells probably by enhancing dendrite growth (Mauch et al., 2001; Christopherson et al., 2005; Göritz et al., 2005; Eroglu and Barres, 2010). It is conceivable that the reduction of the cell size contributed to the reduction of the number of synapses in our mutant neurons.
Quality control
As expected, reduced availability of the major plasma membrane constituent, cholesterol, dramatically reduces cellular growth of mutant neurons both in vivo and in vitro. In contrast to pharmacological cholesterol deprivation (Fan et al., 2002; Sun et al., 2007; Sierra et al., 2011), genetic inactivation of cholesterol synthesis in SQS mutant neurons leads only to a minor decrease in cellular cholesterol content, and only to a slight increase in plasma membrane tension. It is well known that wild-type cells tightly regulate their cholesterol synthesis rate by elaborate feedback mechanisms to maintain their cellular cholesterol content (Sato, 2010). Our study shows that cells can also adjust their size to the available cholesterol and extends this concept to functional aspects. The quality control that tolerates only minimal changes in plasma membrane properties guarantees normal differentiation of mutant neurons: Neurites form functional growth cones, differentiate to axons and dendrites, and establish fully functional synapses.
We conclude that (1) the quality control system that maintains cholesterol content of plasma membranes in a physiological range restricts neurite outgrowth but enables differentiation. (2) Neurons can function and mature in the absence of cell-autonomous cholesterol synthesis provided sufficient external cholesterol supply. However, glial support during embryogenesis or in culture only partially rescues defective neurons. (3) In the adult brain, glial cholesterol can fully compensate for defective neuronal cholesterol biosynthesis.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant SA2114/1-1 (G.S.), European Community Marie Curie Fellowship 509506 (U.F.), and Swiss National Science Foundation Grant PA0033-117479 (U.F.). I.A.T.S. and S.L. were supported by the DFG Research Center for Molecular Physiology of the Brain/Excellence Cluster 171. We cordially thank I. Beulshausen, M. Dörre, G. Fricke-Bode, A. Fahrenholz, A. Galinski, S. Joo, and N. Eiselt for excellent technical help, and V. Tarabykin for generous sharing of antibodies. We also thank Judith Stegmüller for helpful comments on this manuscript.
The authors declare no competing financial interests.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Brinkmann BG, Agarwal A, Sereda MW, Garratt AN, Müller T, Wende H, Stassart RM, Nawaz S, Humml C, Velanac V, Radyushkin K, Goebbels S, Fischer TM, Franklin RJ, Lai C, Ehrenreich H, Birchmeier C, Schwab MH, Nave KA. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron. 2008;59:581–595. doi: 10.1016/j.neuron.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Connert S, Wienand S, Thiel C, Krikunova M, Glyvuk N, Tsytsyura Y, Hilfiker-Kleiner D, Bartsch JW, Klingauf J, Wienands J. SH3P7/mAbp1 deficiency leads to tissue and behavioral abnormalities and impaired vesicle transport. EMBO J. 2006;25:1611–1622. doi: 10.1038/sj.emboj.7601053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll CC, Ng L, Edwards PA. Identification of farnesol as the non-sterol derivative of mevalonic acid required for the accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 1994;269:17390–17393. [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Yu W, Gong JS, Zou K, Sawamura N, Senda T, Yanagisawa K, Michikawa M. Cholesterol-dependent modulation of dendrite outgrowth and microtubule stability in cultured neurons. J Neurochem. 2002;80:178–190. doi: 10.1046/j.0022-3042.2001.00686.x. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kawano H, Ohyama K, Li HP, Takeda Y, Oohira A, Kawamura K. Immunohistochemical localization of neurocan and L1 in the formation of thalamocortical pathway of developing rats. J Comp Neurol. 1997;382:141–152. [PubMed] [Google Scholar]
- Fünfschilling U, Saher G, Xiao L, Möbius W, Nave KA. Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. 2007;8:1. doi: 10.1186/1471-2202-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Göritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Guirland C, Suzuki S, Kojima M, Lu B, Zheng JQ. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 2004;42:51–62. doi: 10.1016/s0896-6273(04)00157-6. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Campenot RB, Vance DE, Vance JE. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J Biol Chem. 2004;279:14009–14015. doi: 10.1074/jbc.M313828200. [DOI] [PubMed] [Google Scholar]
- Jiang XS, Wassif CA, Backlund PS, Song L, Holtzclaw LA, Li Z, Yergey AL, Porter FD. Activation of Rho GTPases in Smith-Lemli-Opitz syndrome: pathophysiological and clinical implications. Hum Mol Genet. 2010;19:1347–1357. doi: 10.1093/hmg/ddq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sørensen JB, Varoqueaux F, Rhee JS, Brose N. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 2007;131:796–808. doi: 10.1016/j.cell.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Knöll B, Kretz O, Fiedler C, Alberti S, Schütz G, Frotscher M, Nordheim A. Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci. 2006;9:195–204. doi: 10.1038/nn1627. [DOI] [PubMed] [Google Scholar]
- Ko M, Zou K, Minagawa H, Yu W, Gong JS, Yanagisawa K, Michikawa M. Cholesterol-mediated neurite outgrowth is differently regulated between cortical and hippocampal neurons. J Biol Chem. 2005;280:42759–42765. doi: 10.1074/jbc.M509164200. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Liu Z, Conroy WG, Stawicki TM, Nai Q, Neff RA, Berg DK. EphB receptors co-distribute with a nicotinic receptor subtype and regulate nicotinic downstream signaling in neurons. Mol Cell Neurosci. 2008;38:236–244. doi: 10.1016/j.mcn.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Yanagisawa K. Inhibition of cholesterol production but not of nonsterol isoprenoid products induces neuronal cell death. J Neurochem. 1999;72:2278–2285. doi: 10.1046/j.1471-4159.1999.0722278.x. [DOI] [PubMed] [Google Scholar]
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Nakamura Y. Regulating factors for microglial activation. Biol Pharm Bull. 2002;25:945–953. doi: 10.1248/bpb.25.945. [DOI] [PubMed] [Google Scholar]
- Pereira DB, Chao MV. The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J Neurosci. 2007;27:4859–4869. doi: 10.1523/JNEUROSCI.4587-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Zhao B, Bedford F, Lamarche-Vane N. Compartmentalized DCC signalling is distinct from DCC localized to lipid rafts. Biol Cell. 2009;101:77–90. doi: 10.1042/BC20070108. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays. 2003;25:72–78. doi: 10.1002/bies.10195. [DOI] [PubMed] [Google Scholar]
- Polleux F. Genetic mechanisms specifying cortical connectivity: let's make some projections together. Neuron. 2005;46:395–400. doi: 10.1016/j.neuron.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Xi SC, Wurtman RJ. The 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitor pravastatin enhances neurite outgrowth in hippocampal neurons. J Neurochem. 2006;97:716–723. doi: 10.1111/j.1471-4159.2006.03763.x. [DOI] [PubMed] [Google Scholar]
- Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini LM. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler Thromb Vasc Biol. 2006;26:372–378. doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- Qin Q, Liao G, Baudry M, Bi X. Cholesterol perturbation in mice results in p53 degradation and axonal pathology through p38 MAPK and Mdm2 activation. PLoS One. 2010;5:e9999. doi: 10.1371/journal.pone.0009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JQ, Aika Y, Heizmann CW, Kosaka T. Quantitative analysis of neurons and glial cells in the rat somatosensory cortex, with special reference to GABAergic neurons and parvalbumin-containing neurons. Exp Brain Res. 1992;92:1–14. doi: 10.1007/BF00230378. [DOI] [PubMed] [Google Scholar]
- Róg T, Vattulainen I, Jansen M, Ikonen E, Karttunen M. Comparison of cholesterol and its direct precursors along the biosynthetic pathway: effects of cholesterol, desmosterol and 7-dehydrocholesterol on saturated and unsaturated lipid bilayers. J Chem Phys. 2008;129:154508. doi: 10.1063/1.2996296. [DOI] [PubMed] [Google Scholar]
- Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Saher G, Quintes S, Möbius W, Wehr MC, Krämer-Albers EM, Brügger B, Nave KA. Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J Neurosci. 2009;29:6094–6104. doi: 10.1523/JNEUROSCI.0686-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Dubreuil V, Arai Y, Wilsch-Bräuninger M, Schwudke D, Saher G, Miyata T, Breier G, Thiele C, Shevchenko A, Nave KA, Huttner WB. Ablation of cholesterol biosynthesis in neural stem cells increases their VEGF expression and angiogenesis but causes neuron apoptosis. Proc Natl Acad Sci U S A. 2009;106:8350–8355. doi: 10.1073/pnas.0903541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R. Sterol metabolism and SREBP activation. Arch Biochem Biophys. 2010;501:177–181. doi: 10.1016/j.abb.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Sheetz MP, Dai J. Modulation of membrane dynamics and cell motility by membrane tension. Trends Cell Biol. 1996;6:85–89. doi: 10.1016/0962-8924(96)80993-7. [DOI] [PubMed] [Google Scholar]
- Sierra S, Ramos MC, Molina P, Esteo C, Vázquez JA, Burgos JS. Statins as neuroprotectants: a comparative in vitro study of lipophilicity, blood-brain-barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J Alzheimers Dis. 2011;23:307–318. doi: 10.3233/JAD-2010-101179. [DOI] [PubMed] [Google Scholar]
- Skene JH, Shooter EM. Denervated sheath cells secrete a new protein after nerve injury. Proc Natl Acad Sci U S A. 1983;80:4169–4173. doi: 10.1073/pnas.80.13.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Sugita S, Charlton MP. Cholesterol-dependent kinase activity regulates transmitter release from cerebellar synapses. J Neurosci. 2010;30:6116–6121. doi: 10.1523/JNEUROSCI.0170-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz CC, Buard I, Claudepierre T, Nägler K, Pfrieger FW. Regional variations in the glial influence on synapse development in the mouse CNS. J Physiol. 2006;577:249–261. doi: 10.1113/jphysiol.2006.117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Graham JS, Hegedüs B, Marga F, Zhang Y, Forgacs G, Grandbois M. Multiple membrane tethers probed by atomic force microscopy. Biophys J. 2005;89:4320–4329. doi: 10.1529/biophysj.104.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Northup N, Marga F, Huber T, Byfield FJ, Levitan I, Forgacs G. The effect of cellular cholesterol on membrane-cytoskeleton adhesion. J Cell Sci. 2007;120:2223–2231. doi: 10.1242/jcs.001370. [DOI] [PubMed] [Google Scholar]
- Tashiro Y, Yamazaki T, Shimada Y, Ohno-Iwashita Y, Okamoto K. Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Niemann-Pick type C model cells. Eur J Neurosci. 2004;20:2015–2021. doi: 10.1111/j.1460-9568.2004.03677.x. [DOI] [PubMed] [Google Scholar]
- Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res. 2006;47:1535–1541. doi: 10.1194/jlr.M600141-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozawa R, Ishibashi S, Osuga J, Yagyu H, Oka T, Chen Z, Ohashi K, Perrey S, Shionoiri F, Yahagi N, Harada K, Gotoda T, Yazaki Y, Yamada N. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J Biol Chem. 1999;274:30843–30848. doi: 10.1074/jbc.274.43.30843. [DOI] [PubMed] [Google Scholar]
- Vallejo M. PACAP signaling to DREAM: a cAMP-dependent pathway that regulates cortical astrogliogenesis. Mol Neurobiol. 2009;39:90–100. doi: 10.1007/s12035-009-8055-2. [DOI] [PubMed] [Google Scholar]
- Wasser CR, Ertunc M, Liu X, Kavalali ET. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J Physiol. 2007;579:413–429. doi: 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR. Inherited disorders of cholesterol biosynthesis. Clin Genet. 2002;61:393–403. doi: 10.1034/j.1399-0004.2002.610601.x. [DOI] [PubMed] [Google Scholar]
- Xu Q, Li Y, Cyras C, Sanan DA, Cordell B. Isolation and characterization of apolipoproteins from murine microglia. Identification of a low density lipoprotein-like apolipoprotein J-rich but E-poor spherical particle. J Biol Chem. 2000;275:31770–31777. doi: 10.1074/jbc.M002796200. [DOI] [PubMed] [Google Scholar]