

Abstract
GABAA receptors (GABAARs) composed of αβγ subunits are allosterically modulated by the benzodiazepines (BDZs). Agonists at the BDZ binding site potentiate submaximal GABA responses by increasing the apparent affinity of GABAARs for GABA. Although BDZs were initially thought to affect the binding of GABA agonists, recent studies suggest an effect on receptor gating; however, the involvement of preactivation steps in the modulation by BDZs has not been considered. Consequently, we examined whether BDZ agonists could exert their modulatory effect by displacing the equilibrium between resting and preactivated states of recombinant α1β2γ2 GABAARs expressed in Xenopus oocytes. For GABA and the partial agonists 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol and piperidine-4-sulfonic acid, we examined BDZ modulation using a simple three-step model incorporating agonist binding, receptor preactivation, and channel opening. The model accounted for diazepam modulation simply by increasing the preactivation constant by approximately fourfold. To assess whether BDZs preferentially affected a specific GABA binding site, pentameric concatamers were used. This demonstrated that single GABA-binding site mutant receptors were equally sensitive to modulation by BDZs compared with wild-type counterparts. Overall, our results suggest that BDZs affect the preactivation step to cause a global conformational rearrangement of GABAARs, thereby modulating receptor function.
Introduction
GABAA receptor (GABAARs) are members of the Cys-loop ligand-gated ion channel family, which also comprises nicotinic acetylcholine, glycine, and 5-HT3 receptors. They form pentameric heteromers whereby the extracellular domains of each subunit contribute toward a pseudo-symmetrical ring providing two binding sites for GABA located at the β/α interfaces (Ernst et al., 2003; Smart and Paoletti, 2011). GABAARs display considerable heterogeneity as a result of numerous subunit families (Korpi et al., 2002), but generally, the most prevalent subtypes found at inhibitory synapses throughout the brain are assembled from αβγ subunits (Whiting et al., 1995).
These αβγ receptors are functionally modulated by the benzodiazepines (BDZs) (Korpi et al., 2002), which bind to the α/γ extracellular interface (Sigel, 2002). At nonsaturating GABA concentrations, BDZ agonists potentiate GABA responses by increasing the apparent affinity of the receptor for GABA. Because this is unaccompanied by increased efficacy of channel gating, it had long been thought that BDZ agonists simply increased the binding affinity of GABA agonists (Study and Barker, 1981; Twyman et al., 1989; Rogers et al., 1994). However, this traditional view has been challenged because BDZ agonists not only potentiate responses elicited by saturating concentrations of partial agonists at the GABA binding site (Maksay et al., 2000; Downing et al., 2005; Rüsch and Forman, 2005) but also directly gate GABAARs that are spontaneously active (Bianchi and Macdonald, 2001; Campo-Soria et al., 2006). These results are incompatible with BDZs solely affecting GABA binding affinity, suggesting that an alternative allosteric mechanism needs to be considered.
For some members of the Cys-loop receptor family, agonist efficacy has been shown to depend on the equilibrium between a ligand-bound resting state and a ligand-bound preactivated (flipped) state that is still inactive, whereas the final step involving channel opening remains almost equally efficient for both full and partial agonists (Lape et al., 2008; Mukhtasimova et al., 2009). Our study provides the first evidence that BDZs can modulate GABAARs by simply shifting the equilibrium between the ligand-bound resting and preactivated states before channel opening, rather than by affecting affinity or efficacy.
Materials and Methods
Molecular biology.
GABAAR subunits (α1, β2, γ2S, and γ2L) were subcloned into pRK5. For constructing the concatamer, five subunits were assembled into a single open reading frame, separated by polyglutamine linkers in the order β2–α1–β2–α1–γ2S. The concatamer was cloned in pRK5 between ClaI and HindIII restriction sites after removing the endogenous HindIII sites from the β2 subunits. The five individual subunits were recloned after PCR to encode unique flanking restriction sites. Inverse PCR was then used to add 20 primer-encoded glutamines to the C-terminal end of the β2 subunits and 15 to the end of the α1 subunits. The initial β2 subunit was cloned into a low copy number vector pRK5, and the remaining subunits were sequentially added. The initial β2 subunit contained the signal peptide and a myc epitope tag, inserted between amino acids 4 and 5 of the mature β2 subunit protein. All the other subunits were composed only of their mature proteins. To enhance translation, a 37 bp 5′ UTR from Xenopus oocyte globin was added to the first subunit (β2), and the Kozak sequence was optimized. Between the ClaI and HindIII restriction sites, the final construct order is ClaI–Kozak–β2myc–20Q–AgeI–α1–15Q–SalI–β2–20Q–NheI–α1–15Q–XhoI–γ2S–Stop–HindIII. Concatamers containing selected point mutations in either of the two β2 subunits (SU1–Y157S and SU3–Y157S) were prepared by mutating the corresponding single subunits, which were then recloned into the concatamer.
Expressing GABAARs in Xenopus laevis oocytes.
Ovaries were removed from female African clawed frogs (Xenopus laevis) using procedures approved by the Animals (Scientific Procedures) Act 1986. Free oocytes were obtained by incubating segments of ovary in collagenase type 1 (Worthington) dissolved in a Ca2+-free OR2 solution, which contained the following (in mm): 85 NaCl, 5 HEPES, and 1 MgCl2, pH adjusted to 7.6 with KOH. After 2–4 h exposure to collagenase I, defolliculated oocytes were washed several times with OR2 and thereafter maintained in a Barth's solution containing the following (in mm): 88 NaCl, 1 KCl, 0.33 Ca(NO3)2, 0.41 CaCl2, 0.82 MgSO4, 2.4 NaHCO3, and 10 HEPES, pH adjusted to 7.6 with NaOH. Single oocytes were injected with 27.6 nl of GABAAR subunit cDNAs (nuclear injection), in a ratio for α1/β2/γ2 of 1:1:10 using a total cDNA concentration of 20 ng/μl for expressing GABAAR heteromers or 30 ng/μl for the concatamers. Oocytes were incubated at 17°C in Barth's solution devoid of serum or antibiotics.
Two-electrode voltage-clamp recording.
Oocytes expressing regular heteromeric receptors were used 1 d after injection, whereas the pentameric concatamers typically showed robust expression after 2–3 d (maximal currents ranged from 500 nA to 2 μA). They were superfused with a solution containing the following (in mm): 100 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, and 5 HEPES, pH adjusted to 7.4 with NaOH. Currents were recorded using an Axoclamp 2B amplifier, a Digidata 1322A interface, and pClamp 8 (Molecular Devices). Currents were digitized at 500 Hz and filtered at 50 Hz (10 Hz used for display purposes). Oocytes were voltage clamped at −60 mV, and experiments were conducted at room temperature.
Data analysis.
Concentration response data were fitted with the Hill equation (below) or one based on a three-step linear reaction scheme (see Results; Origin version 6; OriginLab):
For the Hill equation, I and Imax represent the current induced by various agonist concentrations and the maximal agonist-activated current, respectively, [A] is the agonist concentration, EC50 is the concentration producing a half-maximal response, and nH is the Hill coefficient. GABA data points were normalized to provide a maximal current of 1 in control and in the presence of diazepam. Although this is a constraint, it accords with the consensus view from the literature, and, after normalization, the diazepam-induced potentiation of responses to 1 mm GABA is predicted to be 13% for α1β2γ2S receptors, which falls within the experimentally measured range (7 ± 11% potentiation; n = 6; Fig. 1). Under control conditions, 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP) data points were normalized according to the ratio of currents for 1 mm GABA and 10 mm THIP, whereas the THIP data points in diazepam were normalized according to the potentiation of the responses to 10 mm THIP by diazepam. In control and in the presence of diazepam, piperidine-4-sulfonic acid (P4S) data points were normalized according to the current ratio between 1 mm GABA and 1 mm P4S. Because THIP was a hydrochloride salt, high concentrations will affect the driving force, resulting in an underestimation of the THIP-induced currents. The values of the measured currents induced by 1, 3, and 10 mm THIP were thus increased by 0.6, 2.2, and 7.4%, respectively, before analysis. All data values are means ± SD.
Figure 1.

Diazepam increases THIP and P4S potencies and efficacies at α1β2γ2S receptors. A, Left, Membrane currents activated by 1 mm P4S in the absence, presence, and after recovery from potentiation by 1 μm diazepam (DZ). Bars indicate durations of drug application. Right, Pooled data for the potentiation by 1 μm diazepam of currents activated by 1 mm GABA (n = 6), 1 mm P4S (n = 11), 10 mm P4S (n = 5), and 10 mm THIP (n = 4). Control current amplitudes = 1. B, Concentration–response curves for GABA (black), THIP (red), and P4S (blue) under control conditions (open symbols, solid lines) and in the presence of 1 μm diazepam (filled symbols, dotted lines). Curves were generated from the Hill equation for GABA in control (Imax normalized to 1; EC50 = 153 ± 22 μm; nH = 0.84 ± 0.02; n = 5) and in 1 μm diazepam (Imax normalized to 1, see Materials and Methods; EC50 = 35 ± 15 μm; nH = 0.85 ± 0.05; n = 4). For THIP in control (Imax = 0.77 ± 0.04; EC50 = 834 ± 154 μm; nH = 1.09 ± 0.03; n = 4) and in 1 μm diazepam (Imax = 0.87 ± 0.03; EC50 = 314 ± 85 μm; nH = 0.93 ± 0.11; n = 4); and for P4S in control (Imax = 0.27 ± 0.03; EC50 = 185 ± 30 μm; nH = 0.86 ± 0.02; n = 5) and in 1 μm diazepam (Imax = 0.51 ± 0.07; EC50 = 63 ± 13 μm; nH = 0.90 ± 0.01; n = 4).
Drugs and chemicals.
All compounds were purchased from Sigma, except diazepam (Roche) and THIP (Tocris Bioscience). GABA and P4S were prepared as 1 m stock solutions in recording solution. THIP hydrochloride was first prepared at a 30 mm concentration in recording solution; pH was then readjusted to 7.4 with NaOH before serial dilutions (Mortensen et al., 2010). Diazepam was prepared as a 10 mm stock solution in DMSO. Aliquots were stored at −20°C.
Results
Effect of diazepam on GABA agonist potency and efficacy
Using α1β2γ2S GABAARs expressed in Xenopus oocytes, we examined the potentiation by diazepam of responses to saturating concentrations of the partial agonists THIP and P4S. Diazepam (1 μm) increased the maximal currents elicited by 10 mm THIP and 1 mm P4S by 22 ± 8 and 76 ± 18%, respectively (Fig. 1A). In contrast, responses to saturating GABA (1 mm) were virtually unaffected by diazepam (Fig. 1A). The increased maximum response to 1 mm P4S was reproduced with a super-saturating 10 mm P4S, yielding a 61 ± 12% potentiation by diazepam (Fig. 1A). To investigate how diazepam affected GABA, THIP, and P4S responses, agonist concentration–response curves were constructed in the absence and presence of 1 μm diazepam. Although diazepam increased the potencies of GABA (approximately fourfold) and THIP and P4S (approximately threefold), it only increased the maximal current for THIP and P4S (Fig. 1B), which is likely to reflect a change in the macroscopic efficacy of these partial agonists. This was complemented by directly measuring the ratio of maximal currents (IP4S/IGABA) elicited by saturating (1 mm) concentrations of P4S and GABA (control, 26 ± 5%; with diazepam, 43 ± 7%; n = 6; data not shown).
BDZs and the preactivation step: a receptor model
Consistent with previous studies, these results clearly show that a BDZ agonist does not modulate GABAAR function simply by increasing the binding of GABA agonists but instead affects channel gating. However, other studies primarily discounted this possibility on the basis that BDZs do not affect the efficacy of GABA-induced single-channel openings (Rogers et al., 1994). To reconcile these views, we investigated whether BDZ modulation could be accounted for by a linear three-step model incorporating agonist binding, preactivation, and channel activation (model 1; Fig. 2A).
Figure 2.

Linear receptor model of diazepam modulation at GABAARs. A, Top, three-step model describing the activation of GABAARs (R) by GABA agonists (A), incorporating an agonist dissociation constant (K), a preactivation constant (F), and an efficacy constant for channel opening (E). AR represents the resting agonist-bound state, AF the preactivation state, and AR* the activated open channel state. Bottom, In the same model, diazepam is hypothesized to cause solely an increase in the preactivation constant (C > 1). B, Model-generated concentration–response curves for GABA (black), THIP (red), and P4S (blue) in the absence (solid lines) and presence (dotted lines) of diazepam. The experimental data points are replotted from Figure 1B to assess the adequacy of the model. Data are normalized to a GABA Po,max of 0.85 (see Results). These concentration–response curves predict Po,max and EC50: GABA control, 0.847 and 144 μm and with diazepam, 0.850 and 36.1 μm; THIP control, 0.636 and 909 μm and with diazepam, 0.737 and 263 μm; and P4S control, 0.218 and 141 μm and with diazepam, 0.420 and 68 μm. The values used to generate the model curves are tabulated.
In this model, we postulated that BDZ agonists can potentiate GABAAR function by just increasing the preactivation conformational constant, F, by a constant factor, C (Fig. 2A). To provide starting values for the model parameters, we used direct substitution with the following equations and values for the maximum open probabilities (Po,max) and half-maximally effective concentrations (EC50) obtained for GABA, THIP, and P4S under control conditions and in the presence of a BDZ.
For the three-step linear model 1, the open probability (PoCtrl) is given by the following:
 |
where K is the agonist dissociation constant, F is the preactivation conformation constant, [A] is the agonist concentration, and E is the efficacy of channel gating. Similarly, the open probability in the presence of BDZ (PoBDZ) is given by the following:
 |
By rearranging Equation 1,
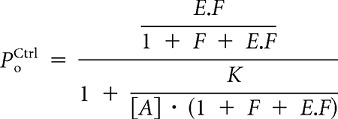 |
the following features are evident: (1) slope coefficient, nH = 1, (2) maximal open probability is given by
and (3) the half-maximally effective concentration is
Equivalent equations can be derived for Po,maxBDZ and EC50BDZ, replacing F by C.F.
Determining the ratio EC50/Po,max from Equations 3 and 4 yields the following equality:
 |
which in the presence of a BDZ equates to
 |
The ratio of Equations 5 and 6 provides a value for C obtained from the agonist EC50 values and the relative maximal current responses:
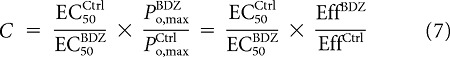 |
where Eff is the macroscopic efficacy of the agonist. Thus, determining C does not require knowledge of the absolute maximal open probabilities. Rearranging Equation 3 yields
with an equivalent expression in the presence of a BDZ:
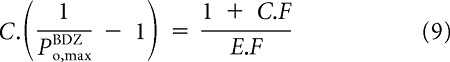 |
Subtracting Equation 8 from Equation 9 yields
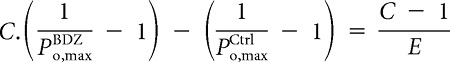 |
Rearranging in terms of E,
 |
According to Equation 4 and its equivalent in the presence of BDZs,
Thus,
Using Equation 7, this equates to
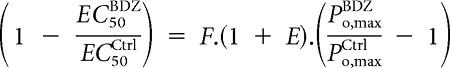 |
Providing an estimate of F,
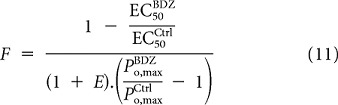 |
After calculating E and F, Equations 4 and 5 provide a value for K. According to Equation 11, although we require Po,maxCtrl ≠ Po,maxBDZ to provide accurate values for F and K, it should be noted that this is not needed for the determination of E, C, and the ratio K/F. Moreover, when Po,maxCtrl ∼ Po,maxBDZ, our model predicts high values of F and K.
To numerically determine the model parameters, we first estimated the values of EC50Ctrl and EC50BDZ (in the presence of diazepam) and Po,maxCtrl and Po,maxBDZ for GABA, THIP, and P4S. A good estimate of C can then be obtained by assuming the maximal responses to GABA are unchanged by diazepam, because the calculation of C only requires the macroscopic efficacy of the agonist (see Eq. 7). We then set Po,maxCtrl (GABA) to 0.85, which is consistent with previous single-channel recording estimates (Mortensen et al., 2004; Lema and Auerbach, 2006; Keramidas and Harrison, 2010). This allowed E to be approximated as Po,max for GABA is thought to be unchanged by diazepam, which concurs with previous studies (Rogers et al., 1994). However, F and K cannot be determined in this way, because our model strictly requires that diazepam does increase Po,max (GABA) (see Eq. 11). Considering that a significant change in Po,max (GABA) should have been experimentally observed, we arbitrarily postulated that the diazepam-induced change in Po,max (GABA) is <3%.
We then considered two scenarios in which
 |
is either ∼0.3% (scenario 1) or ∼3% (scenario 2). The value for Po,maxCtrl (THIP) was determined according to the ratio of maximal currents evoked by 10 mm THIP and 1 mm GABA in control, whereas the value for Po,maxBDZ (THIP) was inferred from the potentiation of THIP-induced currents by diazepam. The values for Po,maxCtrl (P4S) and Po,maxBDZ (P4S) were determined according to the ratio of maximal currents evoked by 1 mm P4S and 1 mm GABA, and notably, did not vary significantly regardless of the scenario. Thus, for P4S, all parameters were unaltered by the two scenarios (CP4S = 5.64 and 5.82, EP4S = 1.23 and 1.34, FP4S = 0.32 and 0.29, and KP4S = 317 and 309 μm for scenarios 1 and 2, respectively). For GABA, both C and E were also essentially unaffected (CGABA = 4.39 and 4.53, EGABA = 5.84 and 8.00, for scenarios 1 and 2, respectively); however, both FGABA and KGABA varied significantly (FGABA = 31.9 and 2.43; KGABA = 33.6 and 3.5 mm for scenarios 1 and 2, respectively), whereas the ratio K/F remained relatively unaltered. Our model therefore suggests that KGABA cannot be accurately determined in this way and that its value could lie anywhere above 3.5 mm, whereas the lower limit of FGABA is ∼2.4. By comparing the parameter values obtained for GABA, THIP, and P4S, it is notable that C is similar for all three agonists (Table 1). Considering the likelihood that small changes to C could just reflect variability in the fitting of the experimental data, we fixed C to an intermediate value of 4 and used the model to generate curve fits whereupon diazepam induced an increase in Po,maxCtrl (GABA) of ∼0.3%, providing an accurate fit to the data (Fig. 2B).
Table 1.
Parameters for the three-step model
| Po,maxCtrl | Po,maxBDZ | EC50Ctrl | EC50BDZ | C | E | F | K (mm) | |
|---|---|---|---|---|---|---|---|---|
| γ2S | ||||||||
| GABA | 0.850 | 0.853 | 153 | 35 | 4.4 | 5.84 | 31.9 | 33.6 |
| P4S | 0.230 | 0.441 | 140.4 | 54.1 | 5.6 | 1.23 | 0.32 | 0.32 |
| THIP | 0.655 | 0.735 | 834 | 314 | 3.0 | 3.61 | 1.11 | 5.09 |
| γ2L | ||||||||
| GABA | 0.850 | 0.853 | 53.9 | 13.6 | 4.0 | 5.85 | 30.9 | 11.5 |
| P4S | 0.075 | 0.185 | 138.8 | 64.2 | 5.3 | 0.388 | 0.264 | 0.190 |
Parameters for the three-step model in the scenario in which diazepam increases Po,max (GABA) by ∼0.3% (see Results).
Alternative models: agonist binding and channel gating
Although the potentiation of agonist activation of GABAARs by BDZs seemed adequately explained by changes to the preactivation step, we next considered alternative models in which diazepam would either increase the efficacy of channel opening by a factor CE (model 2; Fig. 3A) or decrease the agonist dissociation constant by a factor CK (model 3; Fig. 3B).
Figure 3.
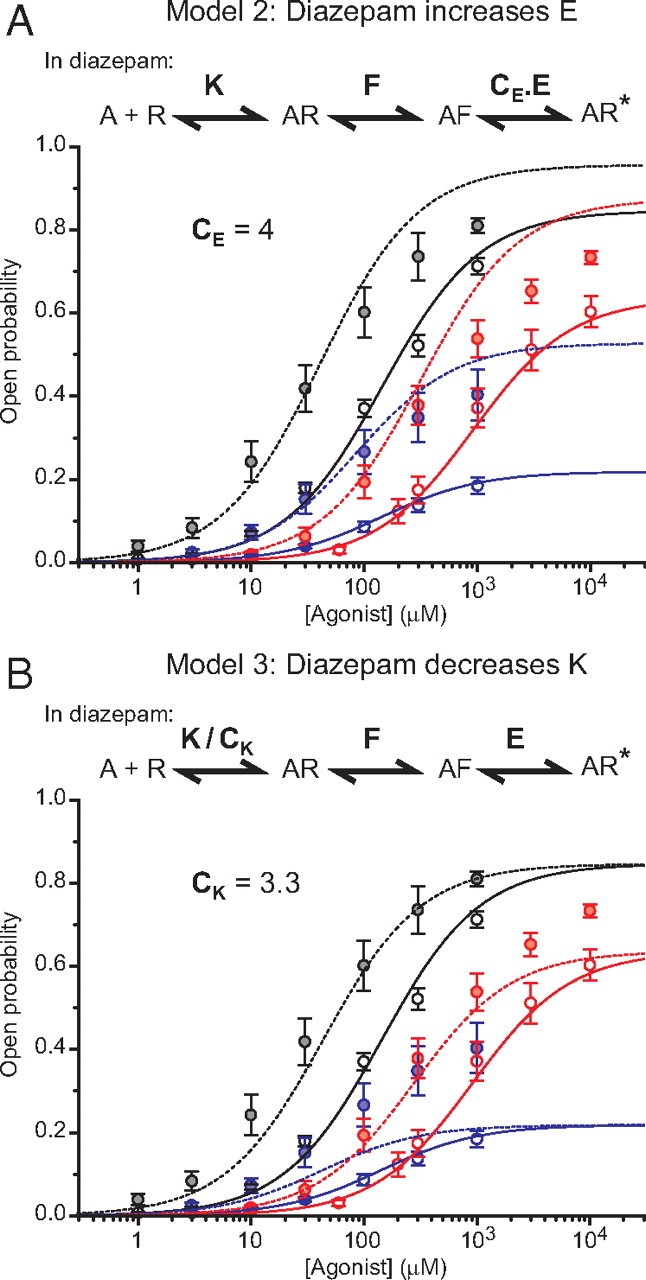
Alternative models fail to describe diazepam modulation at α1β2γ2S receptors. A, Top, Model 2 in which diazepam causes an increase in the efficacy of channel opening (CE > 1). Bottom, Concentration–response curves predicted by model 2 with CE = 4 and with the same values for K, F, and E as for model 1 in Figure 2. These concentration–response curves predict Po,max and EC50 in diazepam: GABA, 0.957 and 40.6 μm; THIP, 0.875 and 313 μm; and P4S, 0.528 and 85.1 μm. AR represents the resting agonist-bound state, AF the preactivation state, and AR* the activated open channel state. B, Top, Model 3 in which diazepam causes a reduction in the agonist dissociation constant (CK > 1). Bottom, Concentration–response curves predicted by model 3 with CK = 3.3 and with the same values for K, F, and E as in Figure 2. These concentration–response curves predict EC50 in diazepam: 43.6 μm for GABA; 275 μm for THIP; and 42.7 μm for P4S.
For model 2, because Equation 7 is still valid, we decided to use the same value for CE as that used for C in model 1 (CE = 4). The ratio of EC50 values gives
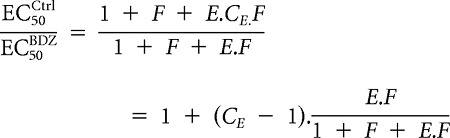 |
This equates to
 |
Rearranging for EC50BDZ,
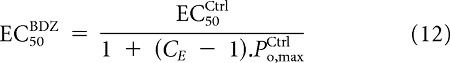 |
Moreover,
 |
Thus,
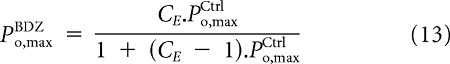 |
Equations 12 and 13 show that the prediction made by model 2 does not depend on the exact values of E, F, and K, because the diazepam-induced increase in agonist potency and open probability are expressed as a function of CE and the potency and open probability in control.
Similarly to our first model, model 2 predicts a potentiation of responses elicited by saturating agonist concentrations. However, the predicted potentiations considerably overestimate the experimental values (Fig. 3A). For instance, model 2 predicts a 13% increase in Po,max (GABA) and a 142% increase in Po,max (P4S), which do not accord with our experiments.
In the final alternative model (model 3), the ratio of the EC50 values in the absence and presence of a BDZ gives
 |
To model our data with the same value of CK regardless of the agonist, we used the mean value (3.3) obtained from the individual determinations of CK for GABA, THIP, and P4S.
Although this model, as expected, accounted for the diazepam-induced lateral shift of the agonist concentration–response curves to higher potency, it failed to account for the potentiation of responses to saturating concentrations of partial agonists (Fig. 3B). Together, our first model, in which diazepam affects just the preactivation step, is the only one able to reproduce the experimental results.
The preactivation step model applies to γ2L subunit-containing GABAARs
The γ2S subunit has been reported recently to produce anomalous results because of unusual interactions with other GABA receptor subunits (Boileau et al., 2010). To ensure that our results were unaffected by this potential anomaly, we expressed α1β2γ2L GABAARs and constructed GABA and P4S concentration–response curves in the absence and presence of 1 μm diazepam. In control, the GABA concentration–response curve indicated a higher apparent affinity and Hill coefficient (EC50 = 53.9 ± 8.5 μm; nH = 1.16 ± 0.10; n = 8) compared with that obtained with γ2S-containing receptors. The macroscopic efficacy of P4S was also lowered (Fig. 4A), possibly indicative of γ2S engaging in anomalous subunit interactions. Nevertheless, diazepam (1 μm) had similar effects on α1β2γ2L GABAARs compared with γ2S-containing receptors. Diazepam increased the potencies of both GABA (∼4-fold; EC50 = 13.6 ± 2.1 μm and nH = 1.18 ± 0.14 in diazepam; n = 5) and P4S (∼2.2-fold; in control and diazepam, respectively, EC50 = 139 ± 27 and 64.2 ± 1.3 μm; nH = 0.92 ± 0.06 and 1.02 ± 0.01; n = 6 and 5) and increased the relative efficacy of P4S (∼2.5-fold) without affecting the current induced by 1 mm GABA (potentiation by 6 ± 3%; n = 9).
Figure 4.
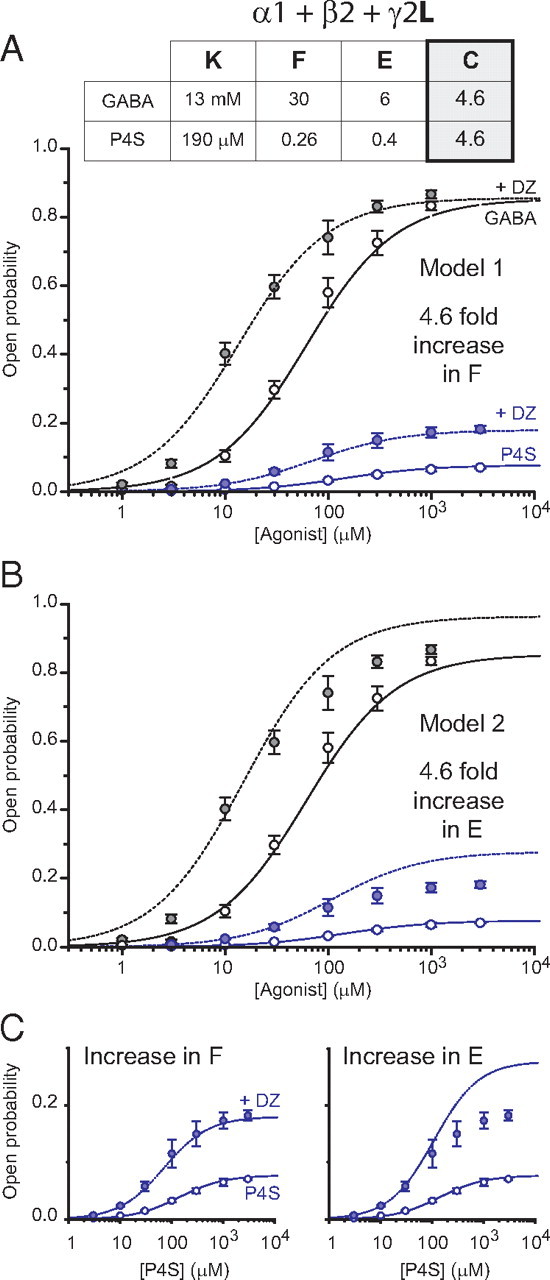
Diazepam modulation of α1β2γ2L receptors is best described by changes to the preactivation constant, F. A, Model 1-generated concentration–response curves for GABA (black) and P4S (blue) in the absence (solid lines) and presence (dotted lines) of diazepam. Diazepam is presumed to increase the preactivation constant by 4.6-fold. The experimental data points were normalized, after fitting by the Hill equation, to give a GABA Po,max of 0.85. These concentration–response curves predict Po,max and EC50: GABA control, 0.853 and 61.6 μm and with diazepam, 0.856 and 13.4 μm; P4S control, 0.077 and 139 μm and with diazepam, 0.180 and 70.4 μm. The values used to generate the model curves are tabulated. B, Alternative model 2, in which diazepam induces a 4.6-fold increase in the efficacy of channel opening. These agonist concentration–response curves predict Po,max and EC50 in diazepam: GABA, 0.964 and 15.1 μm; P4S, 0.278 and 109 μm. C, Magnified view of the predictions, from A and B, for the diazepam-induced modulation of P4S responses.
After normalizing Po,maxCtrl (GABA) to 0.85 and hypothesizing that diazepam potentiates the maximal GABA current by an undetectable value of 0.3% as proposed for γ2S-containing receptors (see above), we refitted our data with the three-step model 1 in which diazepam increases the preactivation constant (F) by 4.6-fold, determined as the mean of the values obtained for GABA and P4S (Table 1). Despite changing the γ2 subunit, the model still provides an accurate fit of the experimental data (Fig. 4A,C). Notably, the alternative model 2, in which diazepam increases the efficacy of gating (E), failed to fit the data and overestimated the macroscopic efficacies for both GABA and P4S in the presence of diazepam (Fig. 4B,C). For model 1, proposing that BDZs modulated the preactivation state, the increase in F predicts a diazepam-induced 2.5-fold potentiation of responses to 1 mm P4S, which is consistent with the experimental value (2.75 ± 0.18; n = 9). In contrast, from model 2, the increase in E predicts a much stronger potentiation of 3.7-fold (Fig. 4C). Model 3, in which diazepam decreases the agonist dissociation constant was discounted because it cannot account for the increased macroscopic efficacy of P4S in the presence of diazepam. Therefore, as for γ2S-containing receptors, the results obtained with α1β2γ2L GABAARs are entirely in accord with diazepam modulating the preactivation step in the GABAAR activation pathway.
Preactivation step and the agonist binding sites
The outcome of the fitting procedure for the experimental data using our model is independent of whether we incorporate one or two agonist binding steps. However, given that GABAARs require the binding of two agonist molecules for efficient gating (Miller and Smart, 2010), it is conceivable that the modulation by BDZ ligands might differentially affect one or other of the agonist binding sites. The increase in the preactivation constants by BDZs may therefore reflect a local conformational rearrangement at a specific neurotransmitter binding site or a global rearrangement equally affecting both binding sites.
A local effect would be plausible given the nonsymmetrical location of the two GABA binding sites relative to the BDZ binding site (Fig. 3B). A conformational rearrangement at just one GABA binding site should be resolved by selective mutation. However, limiting the disruption to a single specified GABA binding site is difficult when expressing α1β2γ2S heteromeric receptors using separate cDNAs. Previously, concatamers have been combined to constrain receptor subunit position and stoichiometry for GABAARs (Minier and Sigel, 2004; Baur et al., 2010; Bracamontes et al., 2011).
In a recent study to decipher the importance of each individual agonist binding site to the modulation by diazepam, a γ2–β2 dimer and α1–β2–α1 trimer, incorporating single GABA binding site mutations, were coinjected into Xenopus oocytes (Baur and Sigel, 2005). Using the β2Y205S mutation, which strongly disrupts GABA binding (Amin and Weiss, 1993), submaximal GABA responses were potentiated when either the dimer or trimer construct contained the binding site mutation. This suggests that BZDs affect the channel opening of GABAARs activated from either agonist binding site. However, the use of dimers and trimers can give rise to expression artifacts (Ericksen and Boileau, 2007; Sigel et al., 2009). This potential uncertainty in terms of GABAAR expression and stoichiometry is essentially removed by using pentameric concatamers (Baur et al., 2006).
To overcome those possible limitations, we generated a pentameric concatamer with a defined stoichiometry and arrangement of receptor subunits (Fig. 5B). This concatamer is a single protein with the C terminus of each subunit connected to the N terminus of the following subunit via a small inert linker (Fig. 5B). The β2Y205S mutation is so disruptive that it prevents a precise measure of GABA potency. The GABA concentration–response data for mutant concatamers containing β2Y205S in the second β2 subunit required fitting with a two-binding site equation with high affinity (570 μm; relative amplitude, ∼5%; n = 3) and dominant low-affinity components (160 mm; relative amplitude, ∼95%; n = 3), the latter presumably corresponding to the potency at the mutated binding site (data not shown). We therefore decided to mutate tyrosine 157 in the β2 subunit to serine (Y157S), an exchange that greatly disrupts GABA binding (Amin and Weiss, 1993) but still enables a reliable measurement of GABA potency. This allows a comparison of the diazepam modulation of various GABAARs when GABA occupancies are similar.
Figure 5.
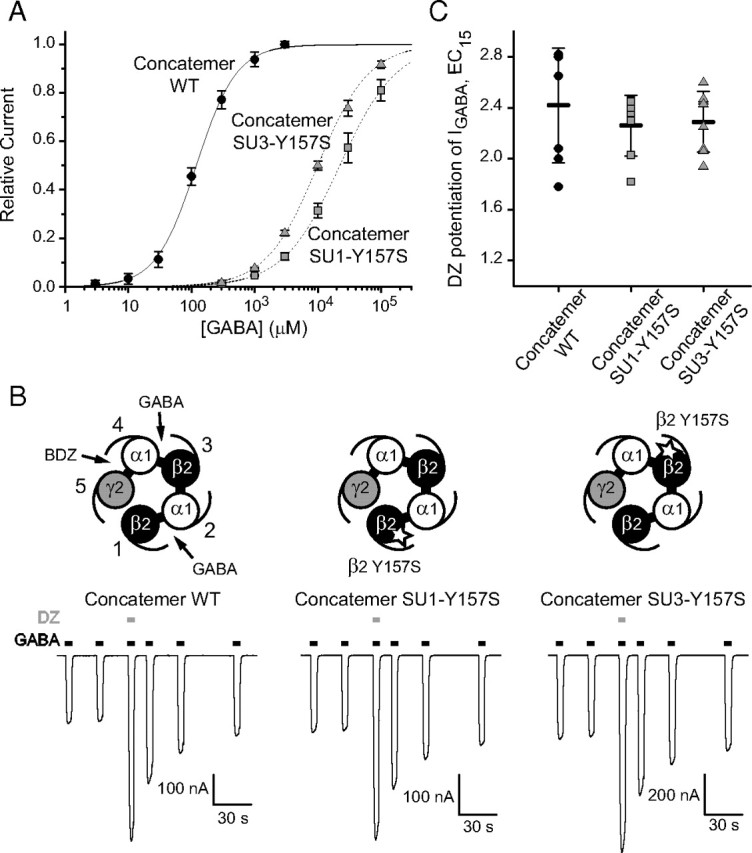
Effect of diazepam on pentameric concatamers incorporating single GABA binding site mutations. A, GABA concentration curves for wild-type (WT) β2–α1–β2–α1–γ2S concatamers (black circles; EC50 = 120 ± 15 μm; nH = 1.38 ± 0.17; n = 6), SU1–Y157S concatamers (gray squares; EC50 = 22.6 ± 5.1 mm; nH = 0.98 ± 0.07; n = 3), and SU3–Y157S concatamers (gray triangles; EC50 = 10.4 ± 0.9 mm; nH = 1.03 ± 0.06; n = 3). B, Top, Schematic plan view showing the subunit arrangement in the concatamer, including the location of the binding sites for GABA and BDZs and the location of the β2 subunit mutations (white stars). Bottom, Recordings showing the effect of 1 μm diazepam (DZ) on currents induced by GABA (EC15) for wild-type (left; 34 μm), SU1–Y157S (middle; 4 mm), and SU3–Y157S (right; 2 mm) concatamers. C, Pooled data for potentiation by 1 μm diazepam of currents induced by GABA (EC15; n = 7–8).
GABA concentration–response curves were constructed for wild-type and mutant concatamers containing β2Y157S in the first (SU1–Y157S) or second (SU3–Y157S; Fig. 5B) β2 subunit. GABA retained a similar potency at the wild-type concatamers to that measured for wild-type α1β2γ2S heteromers (EC50 = 120 μm). However, GABA potency was reduced by 100-fold for the SU1–Y157S and SU3–Y157S concatamers (EC50 = 22.6 and 10.4 mm, respectively; Fig. 5A). When the β2 subunit mutations were combined in SU1–Y157S/SU3–Y157S concatamers, GABA potency was further reduced (EC50 ∼75 mm, n = 4; data not shown). The non-additive nature of the two β2 subunit mutations suggests that, at singly mutated concatamers, the agonist potency corresponds mainly to that of GABA binding to the mutated site.
The potentiating effect of diazepam on GABA-activated currents was then examined using the wild-type and two single mutant concatamers. For each concatamer, 1 μm diazepam potentiated currents induced by GABA (EC15) to similar extents (potentiation: wild type, 2.42 ± 0.45-fold; SU1–Y157S, 2.26 ± 0.24; and SU3–Y157S, 2.29 ± 0.24; Fig. 5B,C). Of note, at their GABA EC15 (2–4 mm range), the two mutant concatamers should have their wild-type GABA binding site almost saturated with GABA. Together, these results suggest that BDZ agonists cause a global conformational rearrangement of the GABAAR to potentiate function rather than a local conformational change at a specific GABA binding site.
Discussion
The GABAAR is a widely recognized molecular target for BDZs to exert their anxiolytic and anticonvulsant effects. Although BDZs were considered to increase the binding affinity of the receptor for GABA, recent evidence was not in favor of such an explanation, suggesting an effect on receptor gating. Here, we provide a plausible explanation that links the previous observations by demonstrating that BDZs can increase the adoption of a transitional receptor state, the preactivated state. This state of the receptor is likely to encompass several distinct brief transitional states that are passed through after GABA binding, before proceeding to channel activation.
By using a simple receptor model, we present a plausible mechanism that accounts for the experimental observations. Of particular importance is the result that the BDZ-induced increase in the preactivation constant is similar regardless of whether the receptor is activated by GABA or partial agonists. This suggests that the putative BDZ-induced change in the preactivation constant is independent of the nature of the agonist occupying the GABA site, depending solely on the BDZ.
According to such a model, we can make several assertions. (1) If the value of (F + E.F) is already much more than 1, the limiting step to setting Po,max is the final step involving channel opening Po,max = E/(1 + E), which implies that BDZ agonists cannot significantly potentiate maximal agonist-induced responses. This situation pertains for the full agonist GABA. (2) Even if (F + E.F) > 1, an increase of the preactivation constant by BDZ agonists (i.e., C > 1) would increase the agonist potency (often interpreted as affinity), indicative of the many studies reporting the modulation of GABA responses by BDZ agonists. (3) Consistent with the actions of partial agonists at glycine and nACh receptors (Lape et al., 2008), the partial agonist nature of THIP and P4S stems mainly from a low preactivation constant. The diazepam-induced increase in this constant can entirely account for the increase in both THIP and P4S potency and efficacy. (4) We can now propose a unifying view of the mode of action of BDZ agonists (e.g., diazepam) and inverse agonists [e.g., methyl-6,7-dimethoxy- 4-ethyl-β-carboline-3-carboxylate (DMCM)]. Indeed, DMCM reduces the efficacy of P4S (Maksay et al., 2000), and DMCM is able to inhibit spontaneously active GABAARs (Campo-Soria et al., 2006). This indicates that this class of BDZ-site ligands acts on the gating of the receptor and not on the binding of GABA agonists. Our model therefore predicts that modulation by inverse agonists could be explained by a decrease in the preactivation constant (C < 1). In addition, BDZ antagonists such as flumazenil, which compete with BDZ-site ligands but do not modulate GABA-induced currents (Rudolph and Knoflach, 2011), would, in the context of the model, produce no change to the preactivation constant (i.e., C = 1). Importantly, when dwell time distributions are corrected for missed events, changes to the preactivation constant should not produce any alteration to the open time distribution of single-channel events but should influence the shut time distribution. This again is consistent with observations of GABA single channels modulated by diazepam or DMCM (Rogers et al., 1994).
The Monod–Wyman–Changeux (MWC) model has also been used to account for the effects of BDZs on GABA-activated and spontaneously open α1β2γ2 GABAARs (Downing et al., 2005; Rüsch and Forman, 2005; Campo-Soria et al., 2006). In all these models, however, no preactivated state is depicted (Fig. 6A). Diazepam was considered to increase the gating efficacy. This could account for the influence of saturating concentrations of diazepam on the GABA concentration–response curves (Downing et al., 2005; Campo-Soria et al., 2006). However, when studying the effect of high diazepam concentrations in these models, the GABA responses were normalized to give a maximal response of 1. Under such circumstances, diazepam, of course, cannot potentiate the maximal GABA response. In contrast, if the experimental data are normalized to the maximum open probability, which for GABAARs is <1, such a gating model would predict that diazepam potentiates the responses to saturating concentrations of GABA. Similarly, such an allosteric MWC model could account for the slight leftward shift of the GABA concentration–response curve induced by a low (10 nm) concentration of the BDZ midazolam (Rüsch and Forman, 2005), but in this latter study, the calculated maximum open probability is also expected to increase by 6.3% by 10 nm midazolam (from 0.862 to 0.916), and saturating concentrations of midazolam are expected to increase the maximal open probability by 10.2% (from 0.862 to 0.95), quite similar to what we predict when we use a model in which diazepam affects the gating efficacy. Hence, these MWC models cannot account for the effects of BZDs at saturating concentrations of GABA agonists. We therefore expanded this allosteric model to incorporate a preactivation step. In the resulting modified MWC model (Fig. 6B), diazepam is hypothesized to affect the equilibrium preactivation constant (L0) between the preactivated (F) and resting (R) states by a potency ratio d. In this model, diazepam does not affect L1, the equilibrium gating constant between the preactivated and the open (O) states. We also hypothesized the binding of the agonist to affect L0, rather than L1, by a potency ratio c. This is indeed more consistent with the partiality of agonists stemming mainly from differences in the preactivation step (Lape et al., 2008). The three-step preactivation model used in the present study corresponds to a subset of this modified MWC model, in which we consider there is no spontaneous activity of the receptor (i.e., L0 ≫ 1 ≫ c) and in which we consider only one single agonist binding site (Fig. 6C). In this analogy, it is worth noting that the reciprocal of the potency ratio d, in the MWC models, corresponds to the constant C from our three-step model. Our results, suggesting C = ∼4, are thus consistent with published values for d (∼0.3; Downing et al., 2005; Rüsch and Forman, 2005).
Figure 6.

Relationship of preactivation model to the MWC model. A, MWC model used by Rüsch and Forman (2005), which is similar to the models used by Downing et al. (2005) and Campo-Soria et al. (2006). R and O depict the resting and open states, respectively. The models in schemes A–C all use the same nomenclature in which K represents ligand dissociation constants for either BDZs (Bz; KBz) or the agonist, e.g., GABA (G; KG). Note the use of statistical factors (e.g., 2KG and KG/2) attributable to the provision of two agonist binding sites in these models. Bz and G affect L0, the equilibrium gating constant between resting (R) and open (O) states of the receptor, by the potency ratios d and c, respectively. Note L0 = [R]/[O]. B, The MWC model is expanded to incorporate a preactivated state, F. In this model, Bz and G affect L0, the equilibrium gating constant between R and F, and not L1, the equilibrium gating constant between states F and O (L1 = [F]/[O]). C, Highlighted pathways (black) are shown corresponding to the three-step model 1 used to describe the effect of diazepam in the current study (see Fig. 2A). Of note, 1/d corresponds to the constant C in model 1.
One potentially confounding issue not yet considered concerns a contribution from fast desensitization. It is conceivable that the macroscopic effects of BDZs, measured with whole-cell responses, might be subject to a dual increase in macroscopic efficacy and desensitization. In such a scheme, the maximal BDZ-induced potentiation would be reduced through desensitization, possibly accounting for our data. However, this is not in accord with the effect of BDZs on single GABA channel currents, because diazepam altered neither the mean open channel duration (for 3 μm GABA) nor the shortest shut time constants (Rogers et al., 1994). On the contrary, diazepam shortened the longest shut time constants and increased the frequency of bursts, both effects that are consistent with diazepam facilitating a preactivation step rather than promoting both gating and desensitization (Rogers et al., 1994; Lape et al., 2008).
A recent study on nACh receptors has reported the existence of at least two intermediate preactivation states (called “primed states”) that are transitional between resting and open states (Mukhtasimova et al., 2009). At the ACh binding site, priming was thought to correspond to the transition of the agonist binding loop C from an uncapped to capped conformation. The loop C capping during receptor priming is probably accompanied by a global conformational change in the extracellular domains, as proposed after structural analyses of “open” and “shut” states of the bacterial homologs of Cys-loop receptors (Hilf and Dutzler, 2008; Bocquet et al., 2009; Miller and Smart, 2010). Such a global conformational rearrangement would be consistent with our explanation of the effects of BDZs on GABAARs deduced using the concatamers. It is indeed plausible that BDZs promote, in addition to a C-loop capping, the rotation of the extracellular domains of subunits participating in the interfacial BDZ site. This rotation could in turn be communicated to the neighboring subunits reaching both GABA binding sites. If we hypothesize that preactivation involves conformational changes in the extracellular domain, it is plausible that ligands that bind to this domain act primarily through a change in the preactivation step, as suggested in our extended MWC model.
Together, our study demonstrates that the BDZs can allosterically modulate GABAARs by modifying the preactivation step, without affecting GABA binding or channel gating. The discovery that an allosteric modulator can act in this way at a Cys-loop receptor may facilitate our understanding of the molecular mechanisms underlying the elusive preactivation process.
Footnotes
This work was supported by the Medical Research Council and the EU-FP7 Consortium, Neurocypres, and a Long-Term Fellowship from the Human Frontier Science Program Organization (M.C.G.).
References
- Amin J, Weiss DS. GABAA receptor needs two homologous domains of the beta-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- Baur R, Sigel E. Benzodiazepines affect channel opening of GABAA receptors induced by either agonist binding site. Mol Pharmacol. 2005;67:1005–1008. doi: 10.1124/mol.104.008151. [DOI] [PubMed] [Google Scholar]
- Baur R, Minier F, Sigel E. A GABAA receptor of defined subunit composition and positioning: concatenation of five subunits. FEBS Lett. 2006;580:1616–1620. doi: 10.1016/j.febslet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Baur R, Kaur KH, Sigel E. Diversity of structure and function of α1α6β3δ GABAA receptors: comparison with α1β3δ and α6β3δ receptors. J Biol Chem. 2010;285:17398–17405. doi: 10.1074/jbc.M110.108670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Agonist trapping by GABAA receptor channels. J Neurosci. 2001;21:9083–9091. doi: 10.1523/JNEUROSCI.21-23-09083.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Boileau AJ, Pearce RA, Czajkowski C. The short splice variant of the γ2 subunit acts as an external modulator of GABAA receptor function. J Neurosci. 2010;30:4895–4903. doi: 10.1523/JNEUROSCI.5039-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracamontes J, McCollum M, Esch C, Li P, Ann J, Steinbach JH, Akk G. Occupation of either site for the neurosteroid allopregnanolone potentiates the opening of the GABAA receptor induced from either transmitter binding site. Mol Pharmacol. 2011;80:79–86. doi: 10.1124/mol.111.071662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo-Soria C, Chang Y, Weiss DS. Mechanism of action of benzodiazepines on GABAA receptors. Br J Pharmacol. 2006;148:984–990. doi: 10.1038/sj.bjp.0706796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing SS, Lee YT, Farb DH, Gibbs TT. Benzodiazepine modulation of partial agonist efficacy and spontaneously active GABAA receptors supports an allosteric model of modulation. Br J Pharmacol. 2005;145:894–906. doi: 10.1038/sj.bjp.0706251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericksen SS, Boileau AJ. Tandem couture: Cys-loop receptor concatamer insights and caveats. Mol Neurobiol. 2007;35:113–128. [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Brauchart D, Boresch S, Sieghart W. Comparative modeling of GABAA receptors: limits, insights, future developments. Neuroscience. 2003;119:933–943. doi: 10.1016/s0306-4522(03)00288-4. [DOI] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Keramidas A, Harrison NL. The activation mechanism of α1β2γ2S and α3β3γ2S GABAA receptors. J Gen Physiol. 2010;135:59–75. doi: 10.1085/jgp.200910317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Gründer G, Lüddens H. Drug interactions at GABAA receptors. Prog Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema GM, Auerbach A. Modes and models of GABAA receptor gating. J Physiol. 2006;572:183–200. doi: 10.1113/jphysiol.2005.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksay G, Thompson SA, Wafford KA. Allosteric modulators affect the efficacy of partial agonists for recombinant GABAA receptors. Br J Pharmacol. 2000;129:1794–1800. doi: 10.1038/sj.bjp.0703259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Minier F, Sigel E. Techniques: use of concatenated subunits for the study of ligand-gated ion channels. Trends Pharmacol Sci. 2004;25:499–503. doi: 10.1016/j.tips.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Kristiansen U, Ebert B, Frølund B, Krogsgaard-Larsen P, Smart TG. Activation of single heteromeric GABAA receptor ion channels by full and partial agonists. J Physiol. 2004;557:389–413. doi: 10.1113/jphysiol.2003.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Ebert B, Wafford K, Smart TG. Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABAA receptors. J Physiol. 2010;588:1251–1268. doi: 10.1113/jphysiol.2009.182444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459:451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers CJ, Twyman RE, Macdonald RL. Benzodiazepine and β-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J Physiol. 1994;475:69–82. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov. 2011;10:685–697. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüsch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–792. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]
- Sigel E. Mapping of the benzodiazepine recognition site on GABAA receptors. Curr Top Med Chem. 2002;2:833–839. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- Sigel E, Kaur KH, Lüscher BP, Baur R. Use of concatamers to study GABAA receptor architecture and function: application to δ-subunit-containing receptors and possible pitfalls. Biochem Soc Trans. 2009;37:1338–1342. doi: 10.1042/BST0371338. [DOI] [PubMed] [Google Scholar]
- Smart TG, Paoletti P. Synaptic neurotransmitter-gated receptors. In: Sheng M, Sabatini B, Südhof T, editors. The synapse. Cold Spring Harbor, NY: Cold Spring Harb Perspect Biol; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study RE, Barker JL. Diazepam and (−)-pentobarbital: fluctuation analysis reveals different mechanisms for potentiation of γ-aminobutyric acid responses in cultured central neurons. Proc Natl Acad Sci U S A. 1981;78:7180–7184. doi: 10.1073/pnas.78.11.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman RE, Rogers CJ, Macdonald RL. Differential regulation of γ-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann Neurol. 1989;25:213–220. doi: 10.1002/ana.410250302. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, McKernan RM, Wafford KA. Structure and pharmacology of vertebrate GABAA receptor subtypes. Int Rev Neurobiol. 1995;38:95–138. doi: 10.1016/s0074-7742(08)60525-5. [DOI] [PubMed] [Google Scholar]