

Abstract
Growing evidence suggests that galectin-3 is involved in fine tuning of the inflammatory responses at the periphery, however, its role in injured brain is far less clear. Our previous work demonstrated upregulation and coexpression of galectin-3 and IGF-1 in a subset of activated/proliferating microglial cells after stroke. Here, we tested the hypothesis that galectin-3 plays a pivotal role in mediating injury-induced microglial activation and proliferation. By using a galectin-3 knock-out mouse (Gal-3KO), we demonstrated that targeted disruption of the galectin-3 gene significantly alters microglia activation and induces ∼4-fold decrease in microglia proliferation. Defective microglia activation/proliferation was further associated with significant increase in the size of ischemic lesion, ∼2-fold increase in the number of apoptotic neurons, and a marked deregulation of the IGF-1 levels. Next, our results revealed that contrary to WT cells, the Gal3-KO microglia failed to proliferate in response to IGF-1. Moreover, the IGF-1-mediated mitogenic microglia response was reduced by N-glycosylation inhibitor tunicamycine while coimmunoprecipitation experiments revealed galectin-3 binding to IGF-receptor 1 (R1), thus suggesting that interaction of galectin-3 with the N-linked glycans of receptors for growth factors is involved in IGF-R1 signaling. While the canonical IGF-1 signaling pathways were not affected, we observed an overexpression of IL-6 and SOCS3, suggesting an overactivation of JAK/STAT3, a shared signaling pathway for IGF-1/IL-6. Together, our findings suggest that galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury.
Introduction
Microglial cells are the principal immune cells of the brain and they react to modifications in the cellular environment through a graded response termed activation. Activation includes phenomena such as changes in morphology, proliferation, and migration of the resident cell population (Hansson and Rönnbäck, 2003; Raivich, 2005; Hanisch and Kettenmann, 2007), ultimately leading to an inflammatory cascade promoting the synthesis and the secretion of inflammatory cytokines and trophic factors. It has been now well established that activated microglial cells can act as a double edge sword. Indeed, microglial cells can produce reactive oxygen and nitrogen species, cytokines such as IL-1β and TNF-α, which promote neuronal death (Allan and Rothwell, 2001; Alexandrova and Bochev, 2005; Allan et al., 2005; Lai and Todd, 2006). On the other hand, many groups, including ours, demonstrated beneficial role of microglial cells by their capacities to produce trophic factors (O'Donnell et al., 2002; Nakajima and Kohsaka, 2004; Lalancette-Hébert et al., 2007) or play a role in phagocytosis (Neumann et al., 2008).
One of the characteristic features of the injury-induced microglial activation is a massive proliferation of the resident cells peaking 48–72 h after injury (Ladeby et al., 2005). By using a CD11bTK mouse model in which we selectively ablated the population of proliferating microglial cells by ganciclovir, we observed that a great majority of the proliferating resident microglial cells coexpress Mac-2/galectin-3 and insulin-like growth factor-1 (IGF-1) (Gowing et al., 2006; Lalancette-Hébert et al., 2007). The fact that in the first 72 h after stroke, galectin-3 was indeed expressed by the resident microglial cells rather than infiltrating monocytes, was further confirmed in studies using chimeric mice in which the peripheral bone-marrow derived cells of myeloid origin were labeled with GFP (Lalancette-Hébert et al., 2007). While IGF-1 is a known autocrine/paracrine mitogen for microglia/macrophages after hypoxic/ischemic injury, the role of galectin-3 in microglial activation/proliferation is less clear. Galectin-3 belongs to a 15-member galectin family (β-galactosidase binding proteins), and growing evidence suggests that galectins may be involved in fine tuning of the inflammatory response (for review, see Rabinovich and Toscano, 2009). While recently galectin-3 has been increasingly used as a marker of microglial activation, its biological role and the molecular mechanisms of its action in the injured brain remain unclear. Based on our findings, we hypothesized that galectin-3 may serve as endogenous mediator of injury-induced microglial activation/proliferation.
To test this possibility, we took advantage of the galectin-3 knock-out (Gal-3KO) mouse. Here, we report that the genetic depletion of galectin-3 induces aberrant microglial activation followed by a deficient microglial proliferation and a decrease in the total number of microglial cells after ischemia. This was further associated with significant increase in the size of ischemic lesion, the number of apoptotic cells, predominantly neurons, and by aberrant responses to IGF-1-mitogenic signals. Together, our findings suggest that galectin-3 plays a pivotal role in mediating injury-induced microglial activation and proliferation.
Materials and Methods
Mouse models
Galectin-3 null mutant mice (C57BL/6 background) were obtained from NIH-supported functional glycomics consortium (www.functionalglycomics.org). The mice were generated by standard gene targeting techniques as described previously (Colnot et al., 1998). Wild-type C57BL/6 (WT) and galectin-3 null mutant mice (C57bl/6 background) were used for our experiments. The galectin-3 null mice (Gal-3KO) were genotyped by PCR with Thermus aquaticus DNA polymerase (GE Healthcare) in 1.7 mm MgCl2 PCR buffer with the following primers: WT, 5′AGGGCCAGTCCCTTAACAAC and 5′GGGCACATCCTCTCATTTGT; Gal3-KO, 5′ CTTGGGTGGAGAGGCTATTC and 5′AGGTGAGATGACAGGAGATC. The PCR conditions were as follows: 95°C, 5 min, 30 cycles (94°C, 30 s; 62°C, 30 s; 72°C, 3 min); 72°C, 10 min. All experimental procedures were approved by the Laval University animal care ethics committee and are in accordance with The Guide to the Care and Use of Experimental Animals of the Canadian Council on Animal Care.
Surgical procedures
Unilateral transient focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO) during 1 h followed by 24 or 72 h reperfusion periods (Belayev et al., 1999; Beaulieu et al., 2002; Lalancette-Hébert et al., 2007). The MCAO was performed on 2–3-month-old male Gal-3KO mice and WT C57bl/6 mice (20–25 g). The animals were anesthetized with 2% isoflurane in 100% oxygen at a flow rate of 2 L/min. To avoid cooling, the body temperature was regularly checked and maintained at 37°C with a heating pad. Under an operating microscope, the left common carotid artery and ipsilateral external carotid artery (ECA) were exposed through a midline neck incision and were carefully isolated from surrounding tissue. The internal carotid artery (ICA) was isolated and carefully separated from the adjacent tissue and a 12-mm-long 6–0 silicon-coated monofilament suture was inserted via the proximal ECA into the ICA and then into the circle of Willis, thus, occluding the MCA. The size of the infarct was estimated using 2% solution of 2,3,5 triphenyltetrazolium chloride (TTC) (Sigma) vital die, dissolved in saline, and stained for 20 min at 37°C in the dark. The relative size of the infarction was measured by using the Scion image-processing and analysis program, calculated in arbitrary units (pixels), and expressed as a percentage of the control, nonstroked area in the contralateral nonischemic hemisphere (100%) for each section (Weng and Kriz, 2007). All animals were allowed ad libitum access to water and food before and after surgery.
Tissue collection
The animals were anesthetized via an intraperitoneal injection of chloral hydrate (10 mg/ml) and transcardially perfused with 30 ml of 0.9% NaCl, followed by PBS-buffered 4% paraformaldehyde (PFA) at pH 7.4. Tissue samples were then postfixed overnight in 4% PFA and equilibrated in phosphate-buffered 20% sucrose for 48 h. Brains were embedded into Tissue-Tek (O.C.T. compound; Sakura), frozen at −80°C overnight, and cut into coronal sections (35 μm thick) with a Cryostat and stored at −20°C.
BrdU labeling
To visualize dividing cells, mice were injected intraperitoneally two times with bromodeoxyuridine (BrdU; 50 μg/g mouse weight in saline) 24 h before being killed. Mice were anesthetized and transcardially perfused with PBS and 4% PFA. The brains were removed and postfixed overnight in 4% PFA. All brain samples were prepared for immunofluorescence manipulation.
Immunohistochemistry
Immunohistochemistry was performed according to the following procedures. Brain sections were blocked in PBS containing 5% goat serum and 0.25% Triton X-100. Attenuation of endogenous peroxidases was done by incubation in 0.6% hydrogen peroxide. Using the same buffer solution, the sections were then incubated overnight at room temperature in primary antibody rabbit polyclonal anti-Iba1 (ionized calcium-binding adapter molecule 1; Wako) and rat anti-Mac-2 (American Type Culture Collection). Incubation for 2 h at room temperature in corresponding biotinylated goat secondary antibody (1:500; Jackson ImmunoResearch) was performed. For the amplification of the positive signal, incubation in a complex of avidin-biotine solution (Vectastain ABC Kit; Vector Laboratories) was performed. Staining was developed for in nickel-DAB solution (0.3%) or in peroxidase substrate kit Vector SG (Vector Laboratories). Each of the above steps was followed by four 5 min rinses in TBS-0.25% Triton X-100. The sections were then dehydrated and coverslipped with DPX (a mixture of distyrene, tricresyl phosphate, and xylene; Electron Microscopy Sciences).
Immunofluorescence
As previously described (Lalancette-Hébert et al., 2007), the sections were then incubated overnight at room temperature using primary antibodies, 1:750 rabbit polyclonal anti-Iba1 (Wako), 1:1000 anti-Mac2 (ATCC), 1:500 anti-cleaved caspase 3 (New England Biolabs), and 1:1000 rat polyclonal anti-BrdU (Axyll). After wash in PBS, the sections were incubated in corresponding fluorescent goat secondary antiserum (Invitrogen).
Protein analysis
Cytokine and growth arrays.
The protein expression analysis of inflammatory cytokine was performed with a mouse antibody array (RaybioMouse Inflammation Antibody Array 1.1, Ray Biotech, #AAM-INF-1L). Protein lysates were obtained by homogenization of brain of control and transgenic mice in 1× cell lysis buffer (included in the Ray Biotech kit) with protease inhibitor cocktail (Sigma #P8340). The protein concentration was determined for each sample and diluted at 500 μg in 1× blocking buffer. Samples for each group (3 mice/group) were pooled and incubated with the array membrane overnight at 4°C. After washes, the membranes were incubated with the biotin-conjugated antibodies overnight at 4°C. The membranes were then processed according to Raybiotech protocol. Membrane were exposed to x-ray film (Kodak film Biomax MR1, #8701302) and analyzed by Agfa Arcus II system and ImageJ software.
Protein analysis–immunoblot.
Immunoblot analyses of phospho-proteins were performed as described previously (Beaulieu et al., 2004, 2008). Briefly, mice were killed by decapitation, after which the heads of the animals were immediately cooled by immersion in liquid nitrogen for 6 s. The stroked and control brains were rapidly dissected out on an ice-cold surface and frozen in liquid nitrogen before protein extraction. Tissue samples were homogenized in boiling 1% SDS solution and boiled for 5 min, and protein concentration was measured using a DC-protein assay (Bio-Rad). Protein extracts (50 μg) were separated on 10% SDS/PAGE Tris-glycine-gels (Invitrogen) and transferred to nitrocellulose membranes. Blots were immunostained overnight at 4°C with primary antibodies. Immune complexes were revealed using appropriate IR dye labeled secondary antibodies from Licor Biotechnology. Quantitative analyses of fluorescent IR dye signal were performed using an Odyssey Imager (Licor Biotechnology). For quantification, total (phospho-independent) protein signals were used as loading controls for each phospho-protein signal. Results were further normalized to respective control conditions to allow for comparison between separate experiments. The gels shown in the figures correspond to representative experiments, where each lane corresponds to a separate animal. The anti-phospho-GSK3β Ser9 polyclonal antibodies, as well as anti-phospho-ERK1/2 (Thr202/Tyr204), anti-total ERK1/2 were purchased from Cell Signaling Technology. Anti-GSK3α/β clone 0011-A monoclonal antibodies were from Santa Cruz Biotechnology. Anti-SOCS3 antibody was purchased from Cell Signaling Technology. Anti-GSK3α/β clone 0011-A monoclonal antibodies were from Santa Cruz Biotechnology. Anti-total-Akt monoclonal antibodies were obtained from Biosources.
Immunoprecipitation.
The immunoprecipitation experiments were done by using Dynabeads standard protocol from Invitrogen. Galectin-3 antibody was first preincubated for 1 h with Dynabeads coupled with protein G. Then, 300 μg of proteins from stroked brain were incubated overnight with the Dynabeads at 4°C. After several washes, 2× SDS was added to the samples and incubated to 95°C for 10 min. Following an addition of DTT, samples were finally incubated at 95°C for 10 min. The eluate was collected using the magnetic device. As previously described, the eluate was run on a SDS-PAGE gel following a regular Western blot using the IGFR1 antibody (Cell Signaling Technology).
Cell cultures
Neonatal microglia.
Primary cell cultures were obtained from the brains of PN1-PN4 WT and Gal3-KO pups. After death, the brain was removed and placed in ice-cold PBS. After mechanical dissociation, 5–6 brains are incubated in a 0.25% Trypsin-EDTA solution (Sigma) containing 250K U/ml of DNase I (Sigma). After centrifugation, the cells pellet was plated in T-75 cm2 flask (Sarstedt) for at least 7 d at 37°C 5% CO2 in DMEM high-glucose media with 10% fetal bovine serum and 1× antibiotic antimycotic solution (Sigma). At confluence, the cells were plated in 24 wells plate at concentration of 10 000 cells/well in media. After 24 h, to allow adhesion, cells were incubated in a control serum free medium, an IGF-1 (50 ng/ml) (Cedarlane) supplemented serum free medium or in a mixture of IGF-1 (50 ng/ml) and tunicamycin (5 μg/ml) (Sigma) serum-free medium for a period of 24 h.
Adult microglia.
Adult C57BL/6 wild-type (WT) and Gal-3KO mice, age 5–6 weeks old, were used for these experiments. After death, the whole brain was extracted and placed in ice-cold Hibernate A medium (BrainBits) supplemented with B-27 (1×) and 0.5 mm GlutaMAX-I, l-alanyl-l-glutamine (Invitrogen). Following mechanical dissociation, the digested tissues were softly triturated by up and down pipetting and the supernatant was collected and filtered through 40 μm cell strainer (BD Biosciences). Then cells were separated using a discontinuous Percoll density gradient centrifugation. A stock isotonic Percoll (100%) was prepared by mixing nine parts of Percoll (GE Healthcare) with one part of 10× HBSS. The gradient concentrations were prepared by diluting with Hibernate A medium containing EDTA (US Biological) and supplemented with B-27 and 0.5 mm GlutaMAX-I. Resultant cell suspension was centrifuged at 300 × g for 10 min at room temperature, and the pellet was resuspended in 2 ml of 37% Percoll that was overlaid 2 ml of 70% Percoll in a 5 ml centrifuge tube and centrifuged at 600 × g for 40 min at room temperature with slow acceleration and no stop-brake. Cells were collected from the interphase and washed in DMEM-GlutaMAX-I medium supplemented with 15% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin. The cell suspension from one mouse brain was plated in eight chambers on 8-chamber culture-treated glass slide with a culture area of 0.7 cm2/well (Becton Dickinson Biosciences) as 150,000 cells per well and incubated at 37°C in 95% air/5% CO2 humidified culture incubator. After 24 h of plating, the cells were treated with 50 ng/ml IGF-1 (Cedarlane) in serum-free DMEM-GlutaMAX-I medium, and incubation was continued for another 24 h period.
Glutamate treatment of cells.
Before separating, the crude cell homogenate was treated with 100 μm glutamate (l-glutamic acid sodium salt—Sigma) in DMEM-GlutaMax-I culture medium at 37°C for 10 min. Glutamate-containing medium was aspirated and cells were washed three times with fresh culture medium and analyzed 24 h after acute exposure to glutamate.
Cell proliferation-imaging protocols.
Cell proliferation was determined with the CyQUANT NF cell proliferation assay kit (Invitrogen) in accordance with manufacturer recommendations. After 1 h incubation, the 24 wells plates were place in an IVIS 200 Imaging System (Xenogen-Caliper). Images were collected using high-sensitivity CCD camera with wavelengths ranging from 300–500 nm. The exposition time for imaging was 1 s using different field of views and f/1 lens aperture. The light output from specified region of interest was quantified by determining the total number of photons emitted per second using the Living Image 2.5 acquisition and imaging software (Xenogen-Caliper). Region-of-interest (ROI) measurements on the images were used to convert surface radiance (photons/s/cm2/sr) to source flux or total flux of photons expressed in photons/seconds. The data were represented as pseudo-color images indicating light intensity (red and yellow, most intense), which were superimposed over grayscale reference photographs.
In vivo bioluminescence/biophotonic imaging.
To obtained innate immune signals from the brains of living mice we crossed the toll-like receptor 2 (TLR2)-luc/gfp transgenic mice with the Gal-3KO mice and obtained the TLR2 reporter colonies in Gal-3KO background. As previously described in detail (Lalancette-Hébert et al., 2009; Cordeau and Kriz, 2012), the images were gathered using IVIS 200 Imaging System (CaliperLS-Xenogen); 25 min before imaging session, the mice received intraperitoneal injection of d-luciferine (150 mg/kg; CaliperLS-Xenogen). ROI measurements on the images were used to convert surface radiance (photons/s/cm2/sr) to source flux or total flux of photons expressed in photons/seconds (p/s). The data were represented as pseudo-color images indicating light intensity (red and yellow, most intense), which were superimposed over greyscale reference photographs.
Flow cytometric studies.
WT and Gal-3KO mice were anesthetized using 10% chloral hydrate and transcardially perfused with ice-cold 1× HBSS to remove all blood from the nervous tissue. Brain samples were surgically removed and tissue was then mechanically dissociated and filtered through a 70 μm cell strainer to obtain a single cell suspension [Becton Dickinson (BD)]. To isolate mononuclear cell fraction, the single cell suspensions were loaded on a 30–37-70% Percoll tri-gradient (GE Healthcare) and centrifuged for 40 min at 200–300 × g. After centrifugation, the mononuclear cells from the 37–70% interface were recovered and stained with CD11b conjugated with allophycocyanin (APC) [Becton Dickinson (BD)]. Samples were then analyzed on a flow cytometer FACStar Plus or FACS Canto Cytometer (BD). Cells were gated using side and forward scatter to eliminate unviable cells and selected for viable cells. For proliferation analysis, mice were injected with BrdU (10 mg/kg, i.p.) twice daily 24 h after MCAO and killed 72 h following stroke. Mononuclear cells were isolated as described above and stained for CD11b-APC and BrdU-FITC according to BrdU flow kit protocol [Becton Dickinson (BD)]. Non-BrdU treated animals were used as negative controls.
Statistical analysis.
All data are presented as mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by post hoc comparison test (Bonferroni test). p ≤ 0.05 was considered statistically significant.
Results
Galectin-3 is expressed in activated microglial cells localized in the ischemic lesion
In physiological conditions, ramified microglial cells exert resting/quiescent and/or surveillance phenotypes (Hanisch and Kettenmann, 2007). In response to injuries, including cerebral ischemia, microglial cells become activated, proliferate, and change morphology. While the ramified/resting microglial cells are positive for the Iba-1 marker (general marker of microglia), in normal, physiological conditions quiescent microglial cells are devoid of any galectin-3 immunoreactivity (Fig. 1A,B). After stroke, Iba-1-positive microglial cells were found in both hemispheres with a more pronounced expression in the stroke area (Fig. 1D,F). Seventy-two hours after stroke, immunohistological analysis revealed a strong increase in galectin-3 immunoreactivity (E). Interestingly, the expression of galectin-3 was restricted to microglial cells that acquired amoeboid morphology and were almost exclusively present within and/or closely surrounding the site of ischemic lesion (Fig. 1E,G). As shown in Figure 1H–J, the high-magnification confocal microscopy images reveled that galectin-3 immunostaining indeed colocalized with Iba1, thus further suggesting rather selective induction of galectin-3 in microglial cells.
Figure 1.

Galectin-3 expression is silent in normal condition and it is restricted to the stroke area region after MCAO. A, Iba-1, a common microglial marker, is normally expressed in the resting microglia in the whole brain and in baseline condition. B, Resting microglial cells in normal conditions are completely devoid of any galectin-3 immunoreactivity. C, Schematic representation of the brain sections used for immunohistochemistry experiments. Box represents the cortex area magnified in D–G. The dash line represents the stroke area region. D–G, Immunohistochemistry of different microglial markers showed an Iba-1 expression in the stroke area region but also in noninfarct area (D, F) contrary to the galectin-3 expression, which is restricted to the stroke area region (E, G). H–J, High-magnification confocal images of the activated microglial cells from the stroked brain show colocalization of Iba-1 staining with galectin-3. Scale bars: A, D, 100 μm; F, 50 μm; H, 20 μm.
Galectin-3 is required for microglia activation
Galectin-3 has been proposed to have multifaceted functions in many pathological conditions, including modulation of innate and adaptive immune responses in peripheral immune cells (Sato et al., 2002; Sato and Nieminen, 2004; Nieminen et al., 2005; Lalancette-Hébert et al., 2007; Rotshenker, 2009), however, its role in brain immune response remains unclear. Because the above presented results suggest that galectin-3 is abundantly expressed in the injury-activated but not in the resting microglial cells, we next examined whether galectin-3 is involved in the injury-induced microglial activation. The effects of galectin-3 were first evaluated in in vitro conditions, by using purified microglia cell culture systems. As described in detail in Materials and Methods, the adult primary microglial cells were derived from the cortexes of the 2–3-month-old WT and Gal-3KO mice. To examine the effects of galectin-3 on microglial activation in an acute injury model, the primary microglial cells from WT and Gal-3KO mice were treated with 100 μm glutamate for 10 min. The microglia activation profiles were analyzed 24 h following initial glutamate stimuli. As shown in Figure 2A–D, the adult, Iba1-positive microglial cells in normal culture conditions express low levels of galectin-3 and the TLR2. Analysis of the WT microglia activation profiles revealed that 10 min exposure to 100 μm glutamate induced a robust upregulation of microglia activation markers such as TLR2, galectin-3, as well as Iba1 (Fig 2E–G). Further analysis revealed that the majority of WT adult primary microglial cells in response to acute glutamate treatment upregulate and coexpress all three glial activation markers (Fig 2H). The analysis of the glutamate-induced activation profiles in Gal-3KO primary microglial cells revealed, however, rather intriguing results. While Iba1 expression levels were comparable between WT and Gal-3KO microglia (Fig 2I,M), contrary to WT cells, the adult microglial cells derived from the brains of Gal-3KO mice did not upregulate TLR2 (Fig. 2K,O,P) following acute glutamate treatment. Here, it is important to mention that we also analyzed additional markers of microglial activation such as CD68. In both WT and Gal-3KO primary microglia, the baseline expression levels and induction patterns for CD68 followed and were almost identical with the TLR2 expression patterns. Thus, due to the space limitations we did not show photomicrographs. However, as demonstrated in Figure 2Q, statistical analysis revealed that contrary to WT cells the Gal-3KO microglia did not upregulate CD68 after glutamate-induced injury. Together, these data suggest that in in vitro model of an acute injury galectin-3 is instrumental for early induction of microglial activation. Because defective microglial activation may significantly affect the evolution of the brain damage following stroke, we next examined whether galectin-3 is also required for injury-induced microglial activation in more complex in vivo conditions. Namely, we recently generated a transgenic mouse model (the TLR2 reporter mouse) (Lalancette-Hébert et al., 2009) in which reporter genes luciferase and GFP are coexpressed under transcriptional control of the murine TLR2 gene promoter. Importantly, in this mouse model, by using biophotonic/bioluminescence imaging and high-resolution CCD camera, we can visualize microglial activation/TLR2 response longitudinally from the brains of living reporter mice (Lalancette-Hébert et al., 2009). To validate whether defective microglial activation/TLR2 response occurs in Gal-3KO mice in vivo, we generated colonies of double transgenic Gal-3KO/TLR2 reporter mice and analyzed in real time TLR2 biohotonic signal induction in stroked TLR2/WT and TRL2/Gal-3KO reporter mice. We previously demonstrated that the imaged TLR2 biophotonic signals represent a valid measure of the TLR2 induction in microglial cells after stroke (Lalancette-Hébert et al., 2009, 2011). As shown in Figure 3A–H, the WT and Gal-3KO reporter mice were subjected to MCAO and imaged for 7 d after initial stroke. Importantly, in concordance with our in vitro data, in vivo analysis of the TLR2 signal after stroke revealed a significant decrease in the signal intensity, a total photon emission after stroke in TLR2/Gal-3KO mice when compared with signals obtained from the stroked WT littermates (Fig. 3I). Together, our results suggest that presence of galectin-3 is required for an adequate injury-induced microglia activation and induction of the TLR2 response.
Figure 2.

Galectin-3-deficient primary adult microglial cells in culture do not upregulate TLR2 signals in response to excitotoxic injury. A–D, In basal conditions the adult WT microglial cells derived from the cortexes of the 2–3-month-old mice express general Iba1 microglial cells marker (red) and low levels of microglia activation markers including galectin-3 (blue) and TLR2 (green). E–H, Acute 10 min exposure to 100 μm glutamate induced marked increase in Iba-1, glaeticn-3, and TLR2 immunoreactivities in WT microglial cells. Majority of the activated microglial cells in culture coexpressed all three markers (H). I–L, In basal conditions, the Gal-3KO primary microglia cells express similar levels of Iba-1 immunoreactivity as WT cells (red); however, we did not detect any TLR2 immunostaining (green). M–P, While short-term glutamate treatment induced a marked increase in Iba-1 staining, unlike WT microglia, the Gal-3KO microglial cells did not upregulate TLR2. (K, O). Nuclear Dapi staining (blue) confirmed the presence of the viable cells (J, N). Q, Densitometry quantification revealed significant increase in Iba-1 immunoreactivities 24 h after 10 min glutamate (100 μm) treatment in both WT and Gal-3KO primary adult microglial cells. Contrary to WT microglia, the Gal-3KO microglial cells were completely devoid of any TLR2 and CD68 immunoreactivities in basal conditions and after glutamate treatment. Data are expressed as mean ± SEM as average of at least 4 independent experiments. p values are presented in the graph. Scale bar, 50 μm.
Figure 3.

Real-time visualization of the biophotonic/bioluminescent TLR2 signals obtained from the brains of live WT and Gal-3KO/TLR2-luc/gfp reporter mice. A–H, Representative images of the WT male (A–D) and Gal-3KO male 2–3-month-old mice (E–H) imaged before and then 2, 5, and 7 d after MCAO. The images were longitudinally recorded from the same animal and represent the spatial and temporal dynamics of the TLR2 response/microglial activation over a 7 d time period. Scales on the right are color maps of the photon counts. I, Quantification of the luciferase signals obtained by LivingImage 4.1 software (CaliperLS) revealed significantly higher TLR2 signals induction in the brains of the WT reporter mice (n = 8–10, *p = 0.02, **p = 0.007).
Galectin-3 deficiency is associated with defective microglial proliferation
One of the characteristic features of microglial activation after stroke is a marked increase in proliferation peaking 48–72 h after initial ischemic injury. Therefore, to further decipher the role of galectin-3 in injury-induced microglial activation, we next asked whether defective microglial responses observed in previous experiments would also affect a potential of stroke-activated microglial cells to proliferate. Namely, we previously reported that the majority of the activated microglial cells in proliferation coexpress galectin-3 and IGF-1. The analysis was performed in WT and Gal-3KO mice. Importantly, the morphology of microglial cells as determined by Iba1 immunofluorescence was similar in WT and Gal-3KO brain sections (Fig. 4A,B), and the analysis of the flow cytometry staining (CD11b-APC) from the brain samples before stroke (naive brains) revealed no differences in the numbers as determined by FACS count for CD11b+ cells (WT: 33.2 ± 2.2% cells, Gal-3KO: 30.6 ± 3% cells, n = 3, p = 0.82) (Fig. 4E,F). Evidence suggests that the brain response to ischemic injury is associated with a several fold increase in number of microglial cells due to a marked increase in the injury-induced proliferation. In accordance with previous studies, the flow cytometry analysis of the stroked brains in WT mice revealed a 2.4-fold increase in the number of CD11b+ cells after stroke (72 h after MCAO). However, the FACS counts showed a significant decrease in numbers of CD11b+ microglial cells in Gal-3KO mice compared with the ischemic brain sections from WT littermates (Fig. 4C,D). Namely, the analysis revealed >6-fold decrease in numbers of CD11b+ microglial cells after stroke in Gal-3KO when compared with WT brains (CD11b+ cells WT: 14.5 ± 2.1% cells, Gal-3KO: 2.1 ± 0.52% cells, n = 5, p < 0.0001) (Fig. 4M). Because at present the exact functional role of galectin-3 in microglial response to brain ischemia is unclear, we next investigated whether observed decrease in the numbers of microglial cells is due to a defective proliferative response or whether it is a consequence of an increase in glial cell death/apoptosis.
Figure 4.
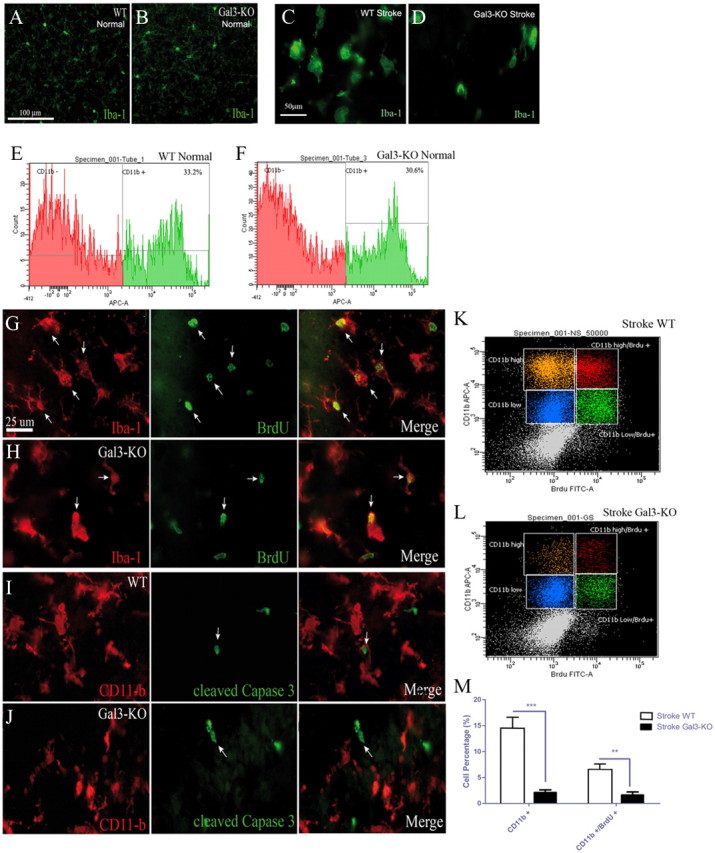
Reduction in microglial cell numbers and proliferation after MCAO in Gal-3KO mice. A, B, Iba-1, a general marker of microglial cell (green), is expressed in normal brain in WT (A) and Gal-3KO (B) mice. E, F, In the basal control conditions, no difference in the number of microglial cells is found between WT and Gal-3KO brain samples as seen by CD11b-APC flow cytometry staining (WT: 33.2 ± 2.2% cells, Gal-3KO: 30.6 ± 3% cells, n = 3, p = 0.82) (E, F). C, D, Seventy-two hours after MCAO, Iba-1-positive microglia change in morphology and appear to be in less numbers Gal-3KO compared with WT mice. G, H, Representative photomicrographs of Iba-1 (in red) and BrdU (in green) staining in WT (G) and Gal-3KO (H) mice show colocalization between both markers in greater amount in WT mice. I, J, Double immunofluorescence staining revealed no colocalization between the microglial marker CD11b and the cleaved caspase-3 staining. K–M, Isolated brain mononuclear cells were analyzed using 2-color flow cytometry. Cells were gated using side and forward scatter to include viable cells and were analyzed for the of CD11b and BrdU expression in WT (K) and Gal-3KO (L) tissues 72 h after MCAO. Flow cytometry analysis shows that there is a significant decrease in the number of CD11b+ cells in Gal-3KO as compared with WT after stroke (CD11b+ cells WT: 14.5 ± 2.1% cells, Gal-3KO: 2.1 ± 0.52% cells, n = 5, ***p < 0.0001). The number of proliferating microglia (CD11b-high/BrdU+ population) in Gal-3KO mice is also significantly reduced as compared with WT (WT: 6.4 ± 1.2% cells, Gal-3KO: 1.6 ± 0.3% cells, n = 5, **p < 0.001). Scale bars: A, 100 μm; C, 50 μm; G, 25 μm.
We first analyzed the rate of microglial proliferation after stroke. BrdU was injected to WT and Gal-3KO mice and the number of proliferating CD11b+ microglial cells was quantified 72 h after initial injury using BrdU-FITC Proliferation kit, BD Biosciences. We have previously demonstrated that 72 h represent an optimal time-window to analyze microglial proliferation after stroke. The Iba-1:BrdU double immunofluorescence analysis confirmed that most of the BrdU+ cells were also Iba-1+ microglial cells in WT and Gal-3KO brains, suggesting that the majority of proliferating cells in the stroke lesion were indeed microglial cells (Fig. 4G,H). The two-color flow cytometry analysis revealed that there was a significant decrease in numbers of CD11b+/BrdU+ cells in Gal-3KO mice compared with the ischemic brain from WT littermates (WT: 6.4 ± 1.2% cells, Gal-3KO: 1.6 ± 0.3% cells, n = 5, p < 0.001) (Fig. 4K–M). Next, we analyzed whether galectin-3 deficiency is associated with an increase in number of dying/apoptotic microglial cells. The double immunofluorescence analysis of the brain sections from Gal-3KO and WT mice at 72 h after stroke revealed no colocalization between the cleaved caspase-3 and the CD11b immunoreactivities (Fig. 4I,J), thus suggesting that marked reduction in the number of microglial cells observed in Gal-3 KO mice is rather due to a defective proliferation than apoptotic cell death.
Absence of galectin-3 exacerbates ischemic damage and neuronal apoptosis after cerebral ischemia
Proliferating and IGF-1/galectin-3 expressing microglial cells have been associated with neuroprotective phenotypes. To determine whether defective microglial proliferation and galectin-3 deficiency exacerbate ischemic injury, we next analyzed the size of ischemic lesions in WT and Gal-3KO mice. As shown in Figure 5, A and B, the analysis of the stroke region by TTC staining, 72 h after MCAO, revealed a 25% increase in the size of the ischemic lesion in Gal-3KO mice when compared with WT control mice (WT: 42.6 ± 4.13% n = 24, Gal-3KO: 67.6 ± 8.13, n = 6, *p = 0.0166). Furthermore, the analysis of the cleaved caspase-3 immunoreactivity revealed a 2.2-fold increase in the number of apoptotic cells (Fig. 5I) (WT: 3077 ± 433.7 cells/mm3, n = 4, Gal-3KO: 6736 ± 546.9 cells/mm3, n = 4, **p = 0.0019). Double immunofluorescence analysis revealed a strong colocalization between cleaved caspase-3 and NeuN, a marker of neurons, in WT (Fig. 5C–E) and in Gal-3KO (Fig. 5F–H), thus suggesting that the majority of apoptotic cells were neurons. Intriguingly, the response to brain ischemia observed in Gal-3KO mice carried a strong resemblance with the brain response to ischemic injury previously observed in the CD11TK mouse model with ablated resident microglia proliferation (Lalancette-Hébert et al., 2007).
Figure 5.

Increase in stroke area and neuronal apoptosis in Gal-3KO mice. A, A 25% increase in the stroke area is observed in Gal-3KO mice compared with WT mice 72 h after MCAO (*p = 0.0166). B, TTC staining was performed to calculate the stroke area region (dotted line). C–H, Immunofluorescence of NeuN (neuronal marker, in red) (C, F) and cleaved caspase-3 (apoptosis marker, in green) (D, G) demonstrated a colocalization between both markers (E, H) 72 h after MCAO in WT and Gal-3KO mice. I, Quantification of the number of cleaved caspase-3-positive cells reveals a twofold increase in the number of apoptotic cells in Gal-3KO mice compared with WT (**p = 0.0019). Scale bar, 250 μm.
Gal-3KO microglial cells are not responsive to IGF-induced mitogenic signals
IGF-1 is a potent mitotic factor for resident microglial cells (O'Donnell et al., 2002). Based on the results of our previous study, where we demonstrated that proliferating microglial cells coexpress galectin-3 and IGF-1 and consequently the ablation of proliferating microglia was associated with significant decrease in IGF-1 and galectin-3 levels, we hypothesized that defective microglial proliferation observed in stroked Gal-3KO mice may be caused by alterations in IGF-1 levels (O'Donnell et al., 2002, Lalancette-Hébert et al., 2007; Ohtaki et al., 2008). Therefore, by using the RayBiotech mouse antibody arrays designed for detection of IGF-1, IGF-2, and the distinct members of the IGF-binding protein family (IGFBP), we analyzed changes in levels of the IGF protein family after stroke in WT and Gal-3KO mice. Here it is important to mention that IGFBP-3 is the most abundant protein and accounts for more than 80% of all IGF binding. Contrary to our expectation, when compared with controls, the protein levels of IGF-1, IGF-2 as well as IGFBP-2,3 were significantly increased after stroke in Gal-3KO mice (Fig. 6A–D), suggesting an early deregulation of the IGF-1 system axes in galectin-3 deficient mice in response to ischemic injury. The observed differences in the expression profiles of IGF-1 and related proteins found in ischemic Gal-3KO brains led us to investigate in more details the underlying mechanisms of the IGF-1/galectin-3 interaction in injury-induced microglial proliferation. As previously described, the mitotic effects of the IGF-1 were tested on primary microglia cell derived from the brains of P1–P4 and 2–3-month-old Gal-3KO and WTs. The galectin-3 deficient and WT primary microglia cultures were exposed to IGF-1 stimuli, and the subsequent proliferative response was quantified in vivo using fluorescence imaging (Fig. 6E–H). As shown in Figure 6E, addition of recombinant IGF-1 in cell culture medium induced proliferation of WT cells with a significant 13% increase in the total number of cells (***p < 0.0001). In contrast, the Gal-3KO cells were not responsive to IGF-1 (see representative images, Fig. 6F,H). The IGF-1-mediated microglial proliferation was also blocked by a selective N-glycosylation inhibitor, tunicamycine (Fig. 6E). Namely, it has been established that N-glycosylation is required for proper expression of IGF-R1 on the cell surface (Carlberg et al., 1996). The adequate surface expression of the IGF-R1 may represent a first sequence of events in the IGF-1-mediated and injury-induced microglial proliferation. Because evidence suggests that neonatal microglial cells have different functional properties and may exert different responses to distinct stimuli, we also tested the mitotic potential of IGF-1 on primary microglial cells derived from 2–3-month-old WT and Gal-3KO mice. Importantly, as revealed in Figure 6, G and H, microglia derived from the adult 2–3-month-old WT and Gal-3KO brains exerted a similar response to IGF-1/galectin-3 mediated proliferation as microglial cells derived from P1–P4 brains. Namely, the quantitative analysis of in vivo fluorescence signal showed that contrary to WT microglia, the adult Gal-3KO microglial cells derived from 2–3-month-old mice were not responsive to IGF-1 (Fig. 6G,H). Further, we investigated how IGF-1 signaling and function of IGF-R1 may be affected in the context of galectin-3 deficiency. Recent work by Partridge et al. (2004) suggests that the interaction of galectin-3 with N-linked glycans attached to various growth factor (GF) receptors enhances GF-induced signal transduction. The persistent retention of ligand-receptor complexes on the surface by galectin-3 alters the receptor dynamics, thereby promoting GF-initiated signaling. Indeed, when the lysates of poststroked brains of WT mice were immunoprecipitated with anti-galectin-3 antibody, IGF-R1 was coimmunoprecipitated with galectin-3 (Fig. 6I,J) and detected using anti-IGFR1 monoclonal antibody, suggesting that galectin-3 interacts with IGF-R1 in vivo. We next investigated whether the downstream of IGF-1 signaling transduction pathways and synthesis of cytokines were directly affected by galectin-3 deficiency and thus involved in modulation of the ischemia-induced microglial proliferation.
Figure 6.

Gal-3KO microglial cells are not responsive to IGF-induced mitogenic signals. A–D, Protein analysis of brain homogenate reveals different expression profile in the level of IGF-1 and the IGF-1 protein member family in WT compared with Gal-3KO mice after stroke. The levels of IGF-1 and IGF-2 (A, B) and IGFBP-2 and -3 (C, D), major IGF-1 binding proteins, were significantly increased in the stroked brains of Gal-3KO mice at 6 and 24 h (*p = 0.05, **p = 0.001). E, G, Primary cells culture experiments show a 10–13% increase in the number of cells after the exogenous addition of IGF-1 (50 μg/ml) in the WT culture. No significant difference was obtained in Gal-3KO culture in the presence of IGF-1. A similar effect was observed with the addition of the N-glycosylation inhibitor tunicamycine (5 μg/ml). Striped bar represents a baseline fluorescence levels obtained from in vivo imaging (F). F, H, Representative images of the cell proliferation quantitative assay using a fluorescent marker, CyQuant NF. The images were obtained with an IVIS 200 Imaging System 24 h after stimulation with IGF-1 and/or tunicamycin in neonatal primary microglial cultures (F) and in adult primary microglial cell cultures derived from the brains (cortex) of 2–3-month-old WT and Gal-3KO mice (H). I, J, Coimmunoprecipitation experiments using mouse monoclonal anti-galectin-3 antibody preincubated with Protein G magnetic beads revealed interaction between IGF-1 receptor and galectin-3.
Galectin-3 deficiency leads to overexpression of IL-6 and induction of suppressor of cytokines signaling 3
To determine whether galectin-3 deficiency leads to changes in profiles of proinflammatory cytokines, we analyzed protein levels of IL-1β, IL-6, and TNF-α at 24 and 72 h after stroke. We were in particular interested in potential alteration in the IL-6 levels since recent evidence suggests that galectin-3 exerts its functions primarily through the JAK-STAT pathway (Jeon et al., 2010). As shown in Figure 7, analysis of protein signals at 24 h following MCAO revealed a small decrease in the levels of IL-1β and the small increase in TNF-α protein level in Gal-3KO mice as compared with WT controls. However, contrary to relatively subtle changes in the levels of IL-1β and TNF-α, we observed more than a twofold increase in the levels of IL-6 in the ischemic brains of Gal-3KO mice. Moreover, while at 72 h after stroke levels of IL-1β and TNF-α were corresponding to controls, IL-6 protein levels stayed significantly upregulated in the ischemic brains of Gal-3KO mice (Fig. 7A–C) (***p < 0.0001). Since growing evidence suggests a crosstalk and synergy between IL-6 and IGF-R1, we further analyzed downstream IGFR1-mediated signaling events (Abroun et al., 2004; Sprynski et al., 2009).
Figure 7.

Galectin-3 deficiency is associated with overexpression of IL-6 and induction of SOCS3. Increase in protein levels of proinflammatory cytokines 24 and 72 h after MCAO. A–C, Brain lysates were generated and the presence of key inflammatory cytokines was determined using a cytokine array (RayBiotech). While IL 1-β was downregulated, galectin-3 deficiency was associated with significant increase in the levels of IL-6 24 and 72 h after MCAO and the early increase in TNFα at 24 h. D, Overexpression of IL-6 was accompanied by an increase in the levels of SOCS3. Analysis of the end effectors of the canonical IGF-1 downstream targets revealed no changes in the phosphorylation of the end effectors of the canonical IGF-1 signaling pathways. E, F, There were no significant changes in the levels of phospho-ERK and pGSK3β levels between WT and galectin-3 deficient mice 72 h after stroke. G, H, Analysis of the brain lysates using cytokine arrays further revealed that galectin-3 deficiency was associated with an increase in M-CSF levels 24 h after stroke while GM-CSF levels were increased 24 and 72 h after stroke (*p < 0.01; **p < 0.001; ***p < 0.0001).
Here, it is important to mention that in addition to the canonical IGF-R1 signaling through mitogen activated protein kinase (MAP kinase) and phopshatidylinositol-3 kinase (PI3K) pathways, IGF-1 receptor can also activate JAK /STAT pathway (strongly induced in stroke and shared by cytokines such as IL-6). Activation of JAK/STAT pathway leads to induction of suppressors of cytokines signaling molecules (SOCSs), in particular SOCS3 (Tan and Rabkin, 2005). The functional role of SOCS3 is to act as negative feedback inhibitor of the JAK/STAT pathway by avoiding STAT3 phosphorylation (Krebs and Hilton, 2001; Himpe and Kooijman, 2009). As shown in Figure 7D, the analysis of the total protein brain extracts revealed significant increase in the levels SOCS3 in the stroked brains of Gal-3KO mice. The SOCS1 levels were not changed (data not shown). We next investigated how/whether galectin-3 deficiency affected activation patterns of the two canonical IGF-1 signaling pathways the Raf/MEK/ERK (MAP kinase) and the PI3K/Akt/GSK3 (PI3K) pathways. Contrary to observed overactivation of the JAK/STAT pathway in Gal-3KO mice, the analysis of the total protein brain extracts from the stroked brains revealed no difference in Gal-3KO and WT mice in the phosphorylation levels of ERK1/2 and GSK3β, the end effectors of the MAP kinase and PI3K pathway, respectively (Fig. 7E,F), suggesting that the two canonical IGF-R1 signaling pathways were not affected by galectin-3 deficiency. Together, our results suggest that galectin-3 deficiency leads to a defective IGF-R1-mediated mitogenic response, an overexpression of IL-6, and an overactivation of JAK/STAT pathway followed by upregulation of a feedback inhibitor SOCS3, which in terms may block the STAT3-mediated prosurvival effects of IGF-1 and may negatively regulate IGF-1-mediated microglial proliferation. Finally, as revealed in Figure 7, G and H, defective IGF-1/Gal-3-mediated proliferative response of microglial cells observed in Gal-3KO mice after stroke was accompanied by a significant compensatory increase in M-CSF and GM-CSF levels. M-CSF and GMCSF are the two major colony stimulating factors and chemokines involved in the regulation of proliferation of peripheral monocytes and resident microglia (Raivich et al., 1991, 1994; Lee et al., 1994). Importantly, the mice deficient in M-CSF and GM-CSF signaling also exert defects in microglia colony proliferation (Raivich et al., 1991, 1994). Hence, based on our results we may conclude that following acute ischemic injury, more than 50% of microglial activation/proliferation is mediated via galectin-3/IGF-1 axis, while the remaining proliferative response observed in Gal-3KO mice is likely modulated by a compensatory increase in the levels of promitotic colony stimulating factors M-CSF and GM-CSF.
Discussion
From the data presented in this study we propose that following brain ischemia galectin-3 acts as an important modulator of the injury-induced microglia activation and proliferation. We report here that galectin-3 deficiency is associated with (1) defective early microglia activation and innate immune response; (2) significant decrease in the number of proliferating microglial cells followed by a sixfold decrease in the total number of microglial cells after stroke; (3) significant increase in the size of ischemic lesion and an increase in the number of apoptotic cells, predominately neurons; (4) aberrant responses to IGF-1-induced mitogenic signals and inability of galectin-3 KO cells to proliferate in presence of IGF-1; and (5) overexpression of IL-6 and induction of suppressors of cytokines signaling 3 (SOCS3). Furthermore, our results suggest that the presence of galectin-3/IGF-R1 interaction appears to be necessary for IGF-mediated microglial proliferation in response to ischemic injury. Together, our findings indicate that galectin-3, expressed in activated microglial cells, is an important intrinsic modulator of microglial activation and IGF-1-mediated mitogenic signaling in the brain responses to ischemic injury.
Galectins are defined by conserved peptide sequence elements in the carbohydrate recognition domain (CRD) that mediates glycan binding. Galectin-3 is composed of the C-terminal CRD and the N-terminal nonlectin domain, which is involved in the oligomerization of galectin-3 upon binding to its glycan ligands on cell surfaces (Rabinovich and Toscano, 2009; Sato et al., 2009). During innate and adaptive immune response, galectin-3 can be expressed by variety of immune cells at the periphery (Leffler et al., 2004; Rabinovich et al., 2007; Marth and Grewal, 2008; Yang et al., 2008; Rabinovich and Toscano, 2009). Galectin-3 binds to different surface glycosylated proteins, inducing immune cell activation, differentiation, and homeostatic programs (Rabinovich and Toscano, 2009). To date, however, most of the galectin-3 functions have been found and characterized in the peripheral immune system. Originally identified as a cell surface marker of inflammatory macrophages, galectin-3 is expressed and secreted by peripheral macrophages (Sato et al., 1993; Sato and Hughes, 1994). Both adaptive and innate immunity are modulated by galectin-3. For example, through the formation of galectin-3 lattice with T cell receptor, galectin-3 increases threshold of antigen-inducing T cell activation (Demetriou et al., 2001). Galectin-3 also modulates cytokine responses to pathogen (Rabinovich and Toscano, 2009). For innate immune cells, galectin-3 is suggested to be involved in neutrophil migration in streptococcal pneumonia (Sato et al., 2002; Nieminen et al., 2008). Once migrated, neutrophils are also activated by galectin-3, which results in degranulation of various layers of microbicidal factors (Yamaoka et al., 1995; Karlsson et al., 1998; Nieminen et al., 2005, 2007). For macrophages, galectin-3 is reported to induce chemoattraction of monocyte/macrophages (Sano et al., 2000) and participate in the alternative activation of macrophages (MacKinnon et al., 2008). For both stromal and immune cells, various GF receptors clustering by galectin-3 on the surface alter the dynamics of GF receptors, leading to prolonged signaling because of delay in the ligand-induced receptor endocytosis that arrests the signal transduction (Dennis et al., 2009).
While the role of galectin-3 is relatively well defined in peripheral immune cells, little is known about its role in the brain. Recent evidence obtained from Gal-3KO suggests that galectin-3 may have a role in the oligodendrocyte differentiation and the control of myelin integrity (Pasquini et al., 2011). Furthermore, studies from Compte et al. (2011) reported potential role of galectin-3 in the modulation of cell motility in the rostral migratory stream. While galectin-3 is abundant activated microglia (Kriz et al., 2003; Lalancette-Hébert et al., 2007; Ohtaki et al., 2008; Comte et al., 2011; Satoh et al., 2011), resting cells do not express galectin-3 and consequently there is no overt microglial phenotype in galectin-3-deficient mice in normal conditions (Figs. 1, 2). However, after ischemic injury (within the first 72 h), there is a robust upregulation of the expression of galectin-3 in the subpopulation of activated resident microglial cells, suggesting its role in microglial cells activation in CNS injury and/or degeneration (Lalancette-Hébert et al., 2007; Rotshenker, 2009; Yan et al., 2009). Recent study on adult rat brains revealed that postischemic induction of galectin-3 may affect angiogenesis and neurogenesis (Yan et al., 2009), and studies in neonates revealed that galectin-3 may contribute to hypoxic/ischemic injury (Doverhag et al., 2010). While at present we cannot exclude the possibility that the absence of galectin-3 during early postnatal period may have affected susceptibility of the adult brain to ischemic injury (this particular issue may have been addressed by development of a novel conditional knock-out model), here it is important to mention that the results described by Doverhag et al. (2010) are in line and supportive of the current views that immature microglial cells have distinct functional properties in response to injuries and different pathologies. Studies by our group and others, on the other hand, have demonstrated that after ischemia galectin-3 is induced in the subpopulation of neuroprotective, proliferating microglial cells that also express IGF-1 (Lalancette-Hébert et al., 2007; Ohtaki et al., 2008). In addition, results of our recent work have strongly suggested that galectin-3 may have a role in modulation of microglial cell proliferation in response ischemic injury (Lalancette-Hébert et al., 2007). This possibility has been further supported in the present study. Namely, our results show >3-fold reduction of proliferating microglial cells in ischemic brains of Gal-3KO mice. Moreover, the results from our present study revealed that in addition to microglial cell proliferation, the presence of galectin-3 may be instrumental for an early induction of the injury-induced innate immune response/TLR2 signaling. While previous studies reported that galectin-3 is required for the recognition of pathogen-associated molecular patterns (PAMPs) to TLRs (Debierre-Grockiego et al., 2010), our results for the first time showed that galectin-3 may also be required for proper induction of the innate immune response after ischemic injury. Although at this time the effects of galectin-3 on other cells types should not be neglected and/or excluded (Pasquini et al., 2011), our results suggest that early after stroke galectin-3 may represent a major immunomodulatory molecule that coordinates processes of injury-induced microglial activation and proliferation. Namely, the results of our present study revealed that galectin-3 deficiency was associated with a marked deregulation of the IGF-1 system responses to injury as well as the inability of Gal-3 KO microglial cells to proliferate in response to IGF-1-mediated mitogenic signals (Figs. 6, 7). Previous works suggest that upon binding to glycans attached to GF receptors, oligomerized galectin-3 molecules crosslink GF receptors at the surface and delays their removal by endocytosis, resulting in prolongation of GF signaling (phosphorylation of Erk and nuclear translocation in cancer cells) (Partridge et al., 2004; Mendelsohn et al., 2007). Here, it is important to mention that our in vitro and in vivo results further support this view, suggesting that similar molecular mechanisms (galectin-3 interaction with IGF-R1) may be involved in IGF-1-mediated and injury-induced proliferation of microglia cell after ischemic injury.
Although within the first 48–72 h after stroke, the analysis of downstream events have not revealed changes in the phosphorylation of the end effectors of the canonical IGF-1 MAP kinase and PI3K signaling pathways, we observed an overexpression of IL-6 and significant increase in the level of SOCS3, suggesting an overactivation of JAK/STAT3, a shared signaling pathway for IGF-1 and IL-6. These data are in agreement with previous work by Jeon et al. (2010), who demonstrated that galectin-3 may exert its functions primarily through the JAK-STAT pathway. Together, our results indicate that galectin-3 deficiency leads to a defective microglia activation and defective IGF-R1 signaling/mitogenic response in microglial cells, and likely compensatory overexpression of IL-6 (Abroun et al., 2004). This may lead to an overactivation of JAK/STAT pathway and upregulation of a feedback inhibitor SOCS3 (Wormald et al., 2006). Early after stroke, the observed upregulation of SOCS3 may block the STAT3-mediated prosurvival effects of IGF-1 and negatively regulate IGF-1-mediated microglial proliferation. Our results also suggest that remaining injury-induced microglial proliferation is likely regulated by the M-CSF and GM-CSF shown to be upregulated in the context of galectin-3 deficiency (Fig. 7).
In conclusion, microglial activation and proliferation are a hallmark of many CNS injuries, including stroke and brain trauma. However, the underlying molecular mechanisms that modulate microglial activation and proliferative response after injuries remain unclear. Based on our results, we propose that galectin-3 plays an important role in mediating early microglial activation/TLR2 induction. Moreover, our results suggest that in addition to established mitogenic signals, the colony stimulating factors such as GM-CSF and M-CSF (Raivich et al., 1991, 1994; Lee et al., 1994), galectin-3, in concert with IGF-1, may act as an important endogenous modulator of injury-induced microglial proliferative response after stroke.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR) and Heart and Stroke Foundation of Canada (HSFC). J.K. is recipient of the Career Award from the R&D Health Research Foundation and CIHR. J.M.B. is recipient of Canada Chair in Molecular Psychiatry (Tier 2). S.S. is recipient of a Scholarship Award (senior level) from the Fonds de la Recherche en Santé du Québec. I.B. visit was sponsored by Unity for Knowledge Fund (UKF/35/08).
The authors declare no competing financial interests.
References
- Abroun S, Ishikawa H, Tsuyama N, Liu S, Li FJ, Otsuyama K, Zheng X, Obata M, Kawano MM. Receptor synergy of interleukin-6 (IL-6) and insulin-like growth factor-I in myeloma cells that highly express IL-6 receptor alpha [corrected] Blood. 2004;103:2291–2298. doi: 10.1182/blood-2003-07-2187. [DOI] [PubMed] [Google Scholar]
- Alexandrova ML, Bochev PG. Oxidative stress during the chronic phase after stroke. Free Radic Biol Med. 2005;39:297–316. doi: 10.1016/j.freeradbiomed.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Kriz J, Julien JP. Induction of peripherin expression in subsets of brain neurons after lesion injury or cerebral ischemia. Brain Res. 2002;946:153–161. doi: 10.1016/s0006-8993(02)02830-5. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR, Caron MG. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105:1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Fernandez G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-l-lysine: neurological and histological validation. Brain Res. 1999;833:181–190. doi: 10.1016/s0006-8993(99)01528-0. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Dricu A, Blegen H, Wang M, Hjertman M, Zickert P, Höög A, Larsson O. Mevalonic acid is limiting for N-linked glycosylation and translocation of the insulin-like growth factor-1 receptor to the cell surface. Evidence for a new link between 3-hydroxy-3-methylglutaryl-coenzyme a reductase and cell growth. J Biol Chem. 1996;271:17453–17462. doi: 10.1074/jbc.271.29.17453. [DOI] [PubMed] [Google Scholar]
- Colnot C, Fowlis D, Ripoche MA, Bouchaert I, Poirier F. Embryonic implantation in galectin 1/galectin 3 double mutant mice. Dev Dyn. 1998;211:306–313. doi: 10.1002/(SICI)1097-0177(199804)211:4<306::AID-AJA2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Comte I, Kim Y, Young CC, van der Harg JM, Hockberger P, Bolam PJ, Poirier F, Szele FG. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J Cell Sci. 2011;124:2438–2447. doi: 10.1242/jcs.079954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeau P, Kriz J. Real-time imaging after cerebral ischemia: model systems for visualization of inflammation and neuronal repair. Methods Enzymol. 2012;506:117–133. doi: 10.1016/B978-0-12-391856-7.00031-7. [DOI] [PubMed] [Google Scholar]
- Debierre-Grockiego F, Niehus S, Coddeville B, Elass E, Poirier F, Weingart R, Schmidt RR, Mazurier J, Guérardel Y, Schwarz RT. Binding of Toxoplasma gondii glycosylphosphatidylinositols to galectin-3 is required for their recognition by macrophages. J Biol Chem. 2010;285:32744–32750. doi: 10.1074/jbc.M110.137588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Dennis JW, Lau KS, Demetriou M, Nabi IR. Adaptive regulation at the cell surface by N-glycosylation. Traffic. 2009;10:1569–1578. doi: 10.1111/j.1600-0854.2009.00981.x. [DOI] [PubMed] [Google Scholar]
- Doverhag C, Hedtjärn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Sävman K. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2010;38:36–46. doi: 10.1016/j.nbd.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Gowing G, Vallières L, Julien JP. Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia. 2006;53:331–337. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hansson E, Rönnbäck L. Glial neuronal signaling in the central nervous system. FASEB J. 2003;17:341–348. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- Himpe E, Kooijman R. Insulin-like growth factor-I receptor signal transduction and the Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT) pathway. Biofactors. 2009;35:76–81. doi: 10.1002/biof.20. [DOI] [PubMed] [Google Scholar]
- Jeon SB, Yoon HJ, Chang CY, Koh HS, Jeon SH, Park EJ. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J Immunol. 2010;185:7037–7046. doi: 10.4049/jimmunol.1000154. [DOI] [PubMed] [Google Scholar]
- Karlsson A, Follin P, Leffler H, Dahlgren C. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood. 1998;91:3430–3438. [PubMed] [Google Scholar]
- Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- Kriz J, Gowing G, Julien JP. Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol. 2003;53:429–436. doi: 10.1002/ana.10500. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Phaneuf D, Soucy G, Weng YC, Kriz J. Live imaging of Toll-like receptor 2 response in cerebral ischaemia reveals a role of olfactory bulb microglia as modulators of inflammation. Brain. 2009;132:940–954. doi: 10.1093/brain/awn345. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Julien C, Cordeau P, Bohacek I, Weng YC, Calon F, Kriz J. Accumulation of dietary docosahexaenoic acid in the brain attenuates acute immune response and development of postischemic neuronal damage. Stroke. 2011;42:2903–2909. doi: 10.1161/STROKEAHA.111.620856. [DOI] [PubMed] [Google Scholar]
- Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia. 1994;12:309–318. doi: 10.1002/glia.440120407. [DOI] [PubMed] [Google Scholar]
- Leffler H, Carlsson S, Hedlund M, Qian Y, Poirier F. Introduction to galectins. Glycoconj J. 2004;19:433–440. doi: 10.1023/B:GLYC.0000014072.34840.04. [DOI] [PubMed] [Google Scholar]
- MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180:2650–2658. doi: 10.4049/jimmunol.180.4.2650. [DOI] [PubMed] [Google Scholar]
- Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn R, Cheung P, Berger L, Partridge E, Lau K, Datti A, Pawling J, Dennis JW. Complex N-glycan and metabolic control in tumor cells. Cancer Res. 2007;67:9771–9780. doi: 10.1158/0008-5472.CAN-06-4580. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O, Gunzer M, Reymann KG. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. 2008;28:5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen J, St-Pierre C, Sato S. Galectin-3 interacts with naive and primed neutrophils, inducing innate immune responses. J Leukoc Biol. 2005;78:1127–1135. doi: 10.1189/jlb.1204702. [DOI] [PubMed] [Google Scholar]
- Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. 2007;282:1374–1383. doi: 10.1074/jbc.M604506200. [DOI] [PubMed] [Google Scholar]
- Nieminen J, St-Pierre C, Bhaumik P, Poirier F, Sato S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J Immunol. 2008;180:2466–2473. doi: 10.4049/jimmunol.180.4.2466. [DOI] [PubMed] [Google Scholar]
- O'Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia. 2002;39:85–97. doi: 10.1002/glia.10081. [DOI] [PubMed] [Google Scholar]
- Ohtaki H, Ylostalo JH, Foraker JE, Robinson AP, Reger RL, Shioda S, Prockop DJ. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc Natl Acad Sci U S A. 2008;105:14638–14643. doi: 10.1073/pnas.0803670105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Millet V, Hoyos HC, Giannoni JP, Croci DO, Marder M, Liu FT, Rabinovich GA, Pasquini JM. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011;18:1746–1756. doi: 10.1038/cdd.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–352. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol. 2007;17:513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Raivich G, Gehrmann J, Kreutzberg GW. Increase of macrophage colony-stimulating factor and granulocyte-macrophage colony-stimulating factor receptors in the regenerating rat facial nucleus. J Neurosci Res. 1991;30:682–686. doi: 10.1002/jnr.490300412. [DOI] [PubMed] [Google Scholar]
- Raivich G, Moreno-Flores MT, Müller JC, Kreutzberg GW. Regulation of microglial proliferation: colony-stimulating factors and their receptors. Neuropathol Appl Neurobiol. 1994;20:209–211. [PubMed] [Google Scholar]
- Rotshenker S. The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J Mol Neurosci. 2009;39:99–103. doi: 10.1007/s12031-009-9186-7. [DOI] [PubMed] [Google Scholar]
- Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–2164. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- Sato S, Hughes RC. Regulation of secretion and surface expression of Mac-2, a galactoside-binding protein of macrophages. J Biol Chem. 1994;269:4424–4430. [PubMed] [Google Scholar]
- Sato S, Nieminen J. Seeing strangers or announcing “danger”: galectin-3 in two models of innate immunity. Glycoconj J. 2004;19:583–591. doi: 10.1023/B:GLYC.0000014089.17121.cc. [DOI] [PubMed] [Google Scholar]
- Sato S, Burdett I, Hughes RC. Secretion of the baby hamster kidney 30-kDa galactose-binding lectin from polarized and nonpolarized cells: a pathway independent of the endoplasmic reticulum-Golgi complex. Exp Cell Res. 1993;207:8–18. doi: 10.1006/excr.1993.1157. [DOI] [PubMed] [Google Scholar]
- Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. 2002;168:1813–1822. doi: 10.4049/jimmunol.168.4.1813. [DOI] [PubMed] [Google Scholar]
- Sato S, St-Pierre C, Bhaumik P, Nieminen J. Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs) Immunol Rev. 2009;230:172–187. doi: 10.1111/j.1600-065X.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- Satoh K, Niwa M, Binh NH, Nakashima M, Kobayashi K, Takamatsu M, Hara A. Increase of galectin-3 expression in microglia by hyperthermia in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia. Neurosci Lett. 2011;504:199–203. doi: 10.1016/j.neulet.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Sprynski AC, Hose D, Caillot L, Réme T, Shaughnessy JD, Jr, Barlogie B, Seckinger A, Moreaux J, Hundemer M, Jourdan M, Meissner T, Jauch A, Mahtouk K, Kassambara A, Bertsch U, Rossi JF, Goldschmidt H, Klein B. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood. 2009;113:4614–4626. doi: 10.1182/blood-2008-07-170464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JC, Rabkin R. Suppressors of cytokine signaling in health and disease. Pediatr Nephrol. 2005;20:567–575. doi: 10.1007/s00467-004-1766-8. [DOI] [PubMed] [Google Scholar]
- Weng YC, Kriz J. Differential neuroprotective effects of a minocycline-based drug cocktail in transient and permanent focal cerebral ischemia. Exp Neurol. 2007;204:433–442. doi: 10.1016/j.expneurol.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, Speed TP, Nicola NA, Hilton DJ. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J Biol Chem. 2006;281:11135–11143. doi: 10.1074/jbc.M509595200. [DOI] [PubMed] [Google Scholar]
- Yamaoka A, Kuwabara I, Frigeri LG, Liu FT. A human lectin, galectin-3 (epsilon bp/Mac-2), stimulates superoxide production by neutrophils. J Immunol. 1995;154:3479–3487. [PubMed] [Google Scholar]
- Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009;1288:116–124. doi: 10.1016/j.brainres.2009.06.073. [DOI] [PubMed] [Google Scholar]
- Yang RY, Rabinovich GA, Liu FT. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med. 2008;10:e17. doi: 10.1017/S1462399408000719. [DOI] [PubMed] [Google Scholar]