

Abstract
The number and subunit composition of postsynaptic AMPA receptors (AMPARs) is a key determinant of synaptic transmission. The vast majority of AMPARs contain GluA2 subunit, which renders the channel impermeable to calcium. However, a small proportion are GluA2 lacking and therefore calcium permeable (CP-AMPARs). It has been proposed recently that long-term potentiation (LTP) involves not only an increase in the total number of AMPARs at the synapse but also a transient switch to CP-AMPARs in the first few minutes after LTP induction. The molecular mechanisms that underlie this switch to CP-AMPARs and the subsequent switch back to calcium-impermeable AMPARs are unknown. Here, we show that endogenous GluA1 is rapidly inserted at the synaptic plasma membrane of rat hippocampal neurons immediately after stimulation with elevated glycine, a treatment known to induce LTP. In contrast, GluA2 is restricted from trafficking to the cell surface by a glycine-induced increase in PICK1–GluA2 binding on endosomal compartments. Between 5 and 20 min after stimulus, activation of CP-AMPARs triggers a release of GluA2 from PICK1, allowing GluA2-containing AMPARs to traffic to the synaptic plasma membrane. These results define a PICK1-dependent mechanism that underlies transient alterations in the subunit composition and calcium permeability of synaptic AMPARs that is important during the early phase after stimulation with glycine and therefore is likely to be important during the expression of LTP.
Introduction
Long-term synaptic plasticity is thought to underlie learning and memory and the fine-tuning of neural circuitry during development. AMPA receptors (AMPARs) mediate the majority of fast excitatory synaptic transmission in the brain, and most forms of plasticity at excitatory synapses require alterations in AMPAR number brought about by regulated trafficking. A decrease in synaptic strength involves AMPAR removal from synapses in long-term depression (LTD), whereas an increase in synaptic AMPARs leads to enhanced synaptic strength during long-term potentiation (LTP) (Shepherd and Huganir, 2007; Collingridge et al., 2010; Henley et al., 2011).
It has been suggested that, as well as changes in AMPAR number, alterations in subunit composition are important during early stages of hippocampal CA3–CA1 LTP (Plant et al., 2006; Lu et al., 2007; Yang et al., 2010; but see Adesnik and Nicoll, 2007). A transient incorporation of GluA2-lacking AMPARs has been recorded electrophysiologically during the first 10 min after LTP induction but has not been observed using imaging techniques. GluA2-lacking AMPARs are calcium permeable, and it has been suggested that the initial insertion of calcium-permeable AMPARs (CP-AMPARs) provides additional signaling to contribute to calcium-dependent processes required for long-lasting synaptic changes, including the reincorporation of GluA2-containing AMPARs (Plant et al., 2006; Isaac et al., 2007). However, the molecular processes behind this switch in AMPAR subunit composition during LTP are unclear.
Most imaging studies that have investigated subunit-specific trafficking during LTP have used overexpressed AMPAR subunits tagged with GFP or pHluorin, which form homomers in neurons (Shi et al., 1999; Yudowski et al., 2007; Makino and Malinow, 2009; Kennedy et al., 2010). These studies have provided important information about a GluA1-driven mechanism for LTP. The specific trafficking behavior of GluA2 during LTP is much less clear, and data from similar studies have been inconsistent. It has been suggested that GluA2 trafficking is not regulated during LTP (Shi et al., 2001; Makino and Malinow, 2009), whereas another study reported similar trafficking behavior as GluA1 (Kopec et al., 2006). It is important to clarify these discrepancies and define the subunit-specific mechanisms that regulate trafficking of endogenous AMPARs during LTP.
PICK1 is a PDZ and BAR domain protein that binds calcium and has a central role in AMPAR internalization and LTD (Hanley and Henley, 2005; Hanley, 2008; Terashima et al., 2008; Citri et al., 2010). The effect of calcium concentration on GluA2–PICK1 binding is biphasic, with the maximal effect seen with small increases in calcium (Hanley and Henley, 2005). PICK1 has also been shown to be involved in LTP (Terashima et al., 2008), but the molecular mechanisms involved are unknown.
Here, we define the localization and trafficking properties of endogenous AMPAR subunits in response to stimulation with elevated glycine, a treatment known to induce LTP (Lu et al., 2001). During the 3 min stimulus, GluA1 is rapidly inserted into the synaptic plasma membrane, whereas GluA2 is restricted from trafficking to the surface by increased PICK1 binding on endosomes. Activation of GluA2-lacking CP-AMPARs later disrupts GluA2–PICK1 interactions, allowing GluA2-containing AMPARs to traffic to the synapse.
Materials and Methods
DNA constructs and viral vectors.
PICK1 shRNA, WT–PICK1, DD4,6AA–PICK1 and Δ380–390–PICK1 (previously referred to as ΔCT–PICK1) have been reported previously (Hanley and Henley, 2005; Nakamura et al., 2011). cDNA encoding mCherry–PICK1 was ligated into pCDNA3.1. GFP–Rab11 was a kind gift from M. Ehlers (Duke University, Durham, NC).
Primary neuronal culture.
Rat embryonic hippocampal neuronal cultures were prepared from E18 Wistar rats (of either sex) using standard procedures. The culture medium was Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 2 mm glutamine. Neurons were transfected at 12–13 DIV using Lipofectamine 2000 and used for experiments 5–6 d later.
Antibodies.
The following antibodies were used: N-terminal anti-GluA1, anti-GluA2, and anti-GluA3 (Millipore); anti-PICK1 (NeuroMab); anti-GRIP1 and anti-Rab11 (BD Biosciences); and anti-early endosomal antigen 1 (EEA1), anti-PSD95, and anti-tubulin (all from Sigma).
Glycine stimulation (chemical LTP).
Chemical LTP was induced as described previously (Lu et al., 2001; Park et al., 2004, 2006). Briefly, neuronal cultures were transferred from Neurobasal growth medium to extracellular solution (ECS) containing the following (in mm): 150 NaCl, 2 CaCl2, 5 KCl, 10 HEPES, pH 7.4, 30 glucose, 0.0005 TTX, 0.001 strychnine, and 0.02 bicuculline methiodide. After 5 min in ECS, neuronal cultures were treated with glycine (200 μm) for 3 min in ECS and then incubated in ECS without glycine for a range of time points before fixation.
For 1-naphthyl acetyl spermine (NASPM) treatment, cells were incubated with NASPM (30 μm; Tocris Bioscience) immediately after glycine treatment for the duration of the experiment.
Immunocytochemistry.
For endogenous surface and intracellular staining of AMPARs, after glycine treatment, neurons were first fixed for 5 min with 4% paraformaldehyde (PFA) in PBS. Surface AMPARs were labeled with primary antibody against the N-terminal extracellular domain of rat GluA1, GluA2, or GluA3, followed by a Cy3-conjugated secondary antibody (1:500) under nonpermeabilized conditions. Neurons were then fixed for 12 min with 4% PFA, permeabilized using 0.1% Triton X-100 including 10% horse serum for 10 min, and incubated in the same primary antibody, followed by Cy5-conjugated secondary antibodies (1:500; The Jackson Laboratory) to visualize intracellular receptors.
For double immunolabeling of AMPARs and PSD95, after glycine treatment, surface AMPARs were labeled as described above. After permeabilization of the cells with 0.1% Triton X-100 in 10% horse serum for 10 min, cells were then stained with anti-PSD95, followed by Cy5-conjugated secondary antibody (1:500). In some experiments, early endosomal (EEA1) and recycling endosomal (Rab11) markers were used in conjunction with PICK1 antibody.
Transferrin recycling assay.
Dissociated hippocampal neurons were incubated with Alexa Fluor 594–transferrin (50 μg/ml; Sigma) in normal growth medium for 60 min at 37°C to reach equilibrium. Neurons were then treated with or without glycine (200 μm, 3 min) in ECS as described above and then incubated in the same solutions without any added glycine for various time points. After washing, neurons were fixed and immunostained with specific primary antibodies against GluA2 and PICK1.
Image analysis and quantification.
Images were acquired using a Carl Zeiss LSM510 confocal microscope. Z-stacks of 5–10 images were taken at 1024 × 1024 resolution with Z-step of 0.5 μm. Images for all conditions in a particular experiment were obtained using identical acquisition parameters and were analyzed using NIH ImageJ software. Neurons were selected blind based on EGFP fluorescence. Quantification of surface AMPARs was performed by thresholding the EGFP fluorescence signal in NNIH ImageJ to define outlines of neurons, and the ratio of surface over total (surface + internal) fluorescence within this area was calculated. These values were then normalized to the average surface fluorescence of untreated control cells. For all experiments, except when noted, n values represent individual experiments in which 10–15 cells were imaged in each. Data are expressed as mean ± SEM, and significance was determined using two-tailed t tests, with the Bonferroni's post hoc correction for multiple comparisons. For presentation, images were processed using Adobe Photoshop software (Adobe Systems) by adjusting brightness and contrast levels to the same degree for all conditions illustrated in each experiment. Colocalization of different markers was determined using NIH ImageJ colocalization plugin (JaCoP plugin). The degree of colocalization between immunolabels was assessed on the original data in dendritic segments by calculating the overlap coefficient according to Manders. Automated thresholding to correct the background was used to avoid user bias in setting analysis parameters. Values were then normalized to untreated control cells. For each condition, n values represent individual experiments in which 10–15 cells were analyzed in each and three to five regions of interest per cell. Data are expressed as mean ± SEM, and significance was determined using two-tailed t tests, with the Bonferroni's post hoc correction for multiple comparisons. To control for random colocalization in the same set of images, one channel was rotated by 180 ° and then analyzed in the same way.
Live imaging.
Dissociated hippocampal neurons were transfected with GFP–Rab11 to label recycling endosomes and mCherry–PICK1. Cultures were transferred to ECS, and images were acquired at a rate of one per minute at a resolution of 512 × 512. For subsequent analysis, the GFP–Rab11 fluorescence was used to define a region of interest, and the mCherry–PICK1 fluorescence in this compartment was measured over time.
Surface biotinylation.
After glycine stimulation, neurons were chilled on ice, washed twice with ice-cold PBS, and incubated with 0.15 mg/ml Sulfo-NHS-SS-Biotin (Pierce) in PBS for 10 min on ice. Cells were then washed two times with cold PBS, incubated for 5 min in 50 mm NH4Cl in PBS, washed three times with PBS, and lysed in lysis buffer (150 mm NaCl, 25 mm HEPES, 1% Triton X-100, and 0.1% SDS). After centrifugation, lysate was incubated with streptavidin–agarose beads for 3 h at 4°C and washed four times in lysis buffer, and bound proteins were detected by Western blotting.
Coimmunoprecipitations.
Cultured neurons (15–19 DIV) were subjected to the glycine stimulation protocol (see above). Cultures were chilled on ice and then lysed in 150 mm NaCl, 25 mm HEPES, pH 7.5, 1% Triton X-100, plus protease inhibitors. Lysates were cleared by centrifugation and then incubated with 2 μg of anti-PICK1 or GRIP1 antibody for 1 h at 4°C. Protein G–Sepharose beads were added and rotated at 4°C for 1 h, followed by four to five washes in lysis buffer. Bound proteins were then detected by Western blotting.
Quantification of immunoblots.
Immunoblots from at least five independent experiments were scanned and analyzed using NIH Image J. Error bars represent SEM, and two-tailed t tests were performed to determine significant differences.
Results
Temporally distinct cell surface insertion of endogenous AMPAR subunits in response to glycine stimulation
We used a chemical stimulation protocol involving bath application of glycine to activate synaptic NMDARs in response to spontaneously released glutamate in dissociated hippocampal neurons. This protocol has been used previously to drive AMPAR insertion and dendritic spine enlargement and to potentiate synaptic transmission (Lu et al., 2001; Park et al., 2004, 2006; Yudowski et al., 2007). We used a 3 min glycine stimulus, and the time points described here are taken from the end of this stimulus. Changes in the localization of endogenous AMPAR subunits were assessed using fixed cell immunocytochemistry.
Immediately after glycine stimulation (0 min), GluA1 surface levels are significantly increased (20 ± 4% increase, p < 0.0125) compared with untreated controls and continue to increase until 5 min after stimulus (55 ± 8% increase compared with controls, p < 0.005; Fig. 1A). Interestingly, surface levels of GluA2 are unchanged immediately after glycine stimulus and also at 5 min (Fig. 1B). However, 10 min after stimulus, GluA2 surface expression begins to increase (30 ± 6% increase compared with control, p < 0.0125) and increases further until 20 min (70 ± 9% increase compared with control, p < 0.001; Fig. 1B). GluA1 surface levels remain elevated at 20 min after stimulus (30 ± 7% increase compared with control, p < 0.0125; Fig. 1A). Surface GluA3 increases by 16 ± 4% (p = 0.046) at 0 min and 26 ± 9% (p = 0.037) at 5 min after stimulation (Fig. 1C), which are nonsignificant after the Bonferroni's post hoc correction for multiple comparisons. These results indicate that, during the first 5 min after glycine exposure, GluA2-lacking AMPARs are inserted into the neuronal plasma membrane, whereas between 10 and 20 min after stimulus, AMPARs containing GluA2 are trafficked to the cell surface.
Figure 1.

Transient insertion of endogenous GluA2-lacking AMPARs at the plasma membrane after glycine stimulation (immunocytochemistry). Dissociated hippocampal neurons expressing EGFP were exposed to elevated glycine for 3 min, returned to normal medium for the specified time, and fixed at 0, 5, 10, or 20 min after stimulus. Controls did not receive the stimulus. Surface and internal GluA1 (A), GluA2 (B), and GluA3 (C) were sequentially labeled under nonpermeabilized conditions and permeabilized conditions with different colored secondary antibodies. Representative images are shown for all conditions, with zoomed sections of dendrites taken from the boxes marked. Graphs show surface/internal ratio relative to unstimulated controls. *p < 0.0125, **p < 0.005, ***p < 0.001. n = 4 independent experiments, with 12–15 cells in each. Scale bars, 20 μm.
We next used biotinylation assays to determine surface levels of AMPAR subunits, which gave similar results as the immunocytochemistry. At 5 min after glycine stimulation, GluA1 surface expression shows a 150 ± 15% increase (p < 0.005), GluA2 is unchanged, and surface GluA3 increases by 65 ± 21% (p < 0.05; Fig. 2A). At 20 min after stimulus, GluA2 surface expression is enhanced by 97 ± 19% (p < 0.01). GluA1 surface levels remain elevated at 20 min (81 ± 20% increase compared with control, p < 0.01), and GluA3 returns to control levels (Fig. 2B).
Figure 2.

Transient insertion of endogenous GluA2-lacking AMPARs at the plasma membrane after glycine stimulation (surface biotinylation). Dissociated hippocampal neurons were exposed to elevated glycine for 3 min and then returned to normal medium. Controls did not receive the stimulus. After 5 min (A) or 20 min (B), biotinylation was used to quantify surface levels of AMPAR subunits. Representative Western blots are shown of GluA1–GluA3 present in total lysates (T) and surface (S, biotinylated) GluA1–GluA3 after glycine stimulus or under control conditions. Graphs show surface/total ratio relative to control. *p < 0.05, ***p < 0.005. n = 6 independent experiments.
Internalization of GluA2 from the cell surface could be an alternative or complementary mechanism to GluA1 plasma membrane insertion for increasing the proportion of GluA2-lacking AMPARs. However, we observed no significant loss of surface GluA2 immediately after glycine stimulation, suggesting that this does not account for our results (Figs. 1B, 2A). Together, our results indicate that endogenous GluA2-lacking AMPARs are inserted into the neuronal plasma membrane immediately after glycine stimulation. These receptors are likely to be GluA1 homomers with a possible small contribution from GluA1/GluA3 heteromeric receptors. There is then a delayed insertion of GluA2-containing AMPARs at the cell surface between 10 and 20 min after stimulus.
Temporally distinct synaptic targeting of endogenous AMPAR subunits in response to glycine stimulation
To investigate whether the observed increases in AMPAR subunit surface levels are localized to synaptic sites, we costained cultured neurons for surface AMPAR subunits and intracellular PSD95 as a synaptic marker. At 5 min after glycine treatment, colocalization between surface GluA1 and PSD95 increases by 19 ± 5% compared with untreated controls (p < 0.025; Fig. 3A), whereas GluA2 is unchanged (Fig. 3B). Twenty minutes after the glycine stimulus, surface GluA1 shows an additional increase in PSD95 colocalization (41 ± 6% increase compared with control, p < 0.001; Fig. 3A), and GluA2 also shows a glycine-induced increase in colocalization with PSD95 (30 ± 4% increase compared with control, p < 0.001; Fig. 3B). Colocalization between surface GluA3 and PSD95 shows a small, transient increase at 5 min after stimulus (15 ± 2% increase compared with controls, p < 0.025). These experiments demonstrate that GluA2-lacking AMPARs are transiently inserted at synaptic sites in response to glycine stimulus. The smaller increase in synaptic incorporation of GluA1 compared with the increase in cell surface levels at 5 min is consistent with a model in which a proportion of the GluA2-lacking AMPARs inserted at the plasma membrane in response to LTP are located at extrasynaptic sites.
Figure 3.

Transient incorporation of GluA2-lacking AMPARs at synaptic sites after glycine stimulation. Dissociated hippocampal neurons were exposed to elevated glycine for 3 min, returned to normal medium for the specified time, and fixed at 5 or 20 min after stimulus. Controls did not receive the stimulus. Neurons were sequentially stained for surface AMPAR subunits GluA1 (A), GluA2 (B), GluA3 (C), (green channel), and PSD95 (red channel) under nonpermeabilized and permeabilized conditions, respectively. Representative images are shown for all conditions. Graphs show Manders' colocalization coefficients for the fraction of AMPAR subunit colocalized with PSD95 normalized to control. *p < 0.025, **p < 0.005. n = 3 independent experiments, with 10–15 cells in each. Scale bars, 5 μm.
Delayed insertion of GluA2 after glycine stimulation is PICK1 dependent
It has been demonstrated previously that PICK1 mediates certain GluA2-dependent trafficking events (Terashima et al., 2004; Ho et al., 2007; Dixon et al., 2009). We therefore investigated the possibility that PICK1 is involved in the delayed forward traffic of GluA2 during LTP, by manipulating PICK1 expression levels using overexpression and RNAi-mediated knockdown.
We first used shRNA to reduce endogenous PICK1 expression (Nakamura et al., 2011) and analyzed GluA2 surface expression at a range of time points after glycine treatment. As presented in Figure 1B, GluA2 trafficking to the cell surface in control neurons expressing EGFP is delayed until 10 min after glycine stimulus and continues to increase until 20 min after stimulus. However, in neurons expressing EGFP and PICK1 shRNA, surface GluA2 increases much sooner, with an increase of 34 ± 5% (p < 0.001) at 5 min after stimulus, which is maintained for at least 20 min (Fig. 4A). This demonstrates that PICK1 restricts GluA2 trafficking to the plasma membrane immediately after glycine stimulation. In the converse experiment, we overexpressed PICK1 and performed the same analysis. Surprisingly, in PICK1-overexpressing neurons, glycine treatment induces a loss of surface GluA2 at 5 min after stimulation (54 ± 4% decrease compared with control, p < 0.001; Fig. 4B). This indicates that the level of PICK1 expression determines the extent of GluA2 retention in response to glycine stimulation. To test this further and to confirm specificity of the shRNA, we knocked down endogenous PICK1 with shRNA and simultaneously expressed shRNA-resistant PICK1. Exogenously expressed PICK1 over-rescues the shRNA knockdown phenotype, resulting in reduced surface GluA2 at 5 min after glycine treatment (31 ± 4% decrease compared with control, p < 0.0125; Fig. 4C). Together, these data suggest that PICK1 plays a central role in controlling GluA2 traffic to the cell surface during the first few minutes after LTP induction.
Figure 4.
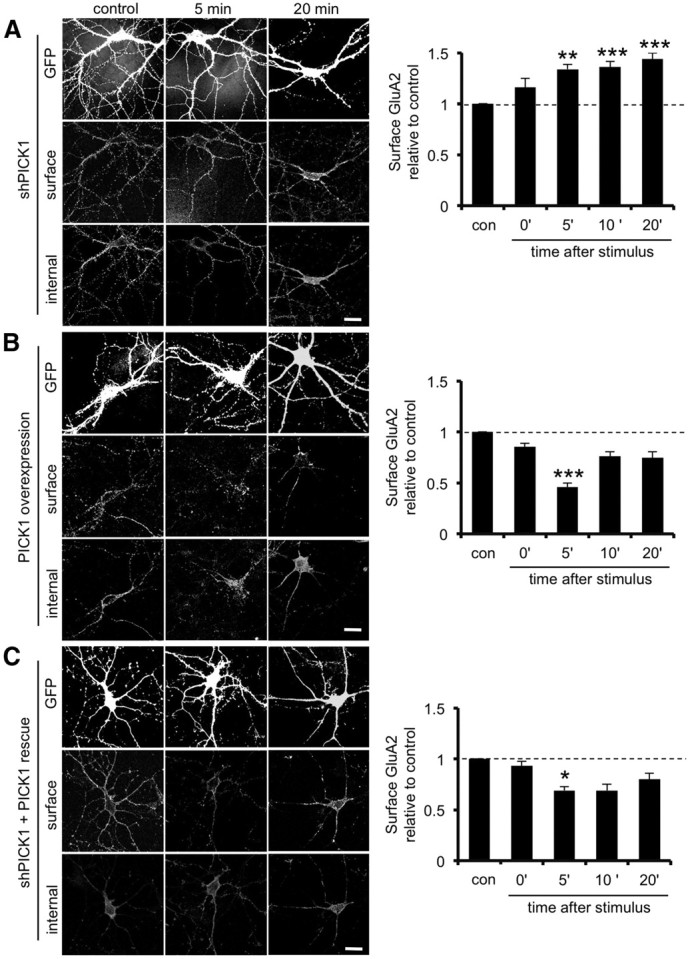
PICK1 restricts GluA2 trafficking to the cell surface after glycine stimulation. A, PICK1 knockdown allows the traffic of GluA2 to the plasma membrane within 5 min after glycine stimulation. Dissociated hippocampal neurons transfected with plasmids expressing EGFP alone or EGFP plus PICK1 shRNA were exposed to elevated glycine for 3 min, returned to normal medium for the specified time, and fixed 0, 5, 10, or 20 min after stimulation. Controls did not receive the stimulus. Cells were processed for immunocytochemistry as described for Figure 1. Graph shows surface/internal ratio relative to unstimulated controls. **p < 0.005, ***p < 0.001. n = 4 independent experiments, with 12–15 cells in each. Scale bar, 20 μm. B, PICK1 overexpression further restricts GluA2 trafficking to the plasma membrane in response to glycine stimulation. Dissociated hippocampal neurons transfected with plasmids expressing WT–PICK1–IRES–EGFP were treated as in A. Graph shows surface/internal ratio relative to unstimulated controls. ***p < 0.001. n = 4 independent experiments, with 12–15 cells in each. Scale bar, 20 μm. C, Coexpression of sh-resistant WT–PICK1 rescues the shRNA phenotype. Dissociated hippocampal neurons transfected with plasmids expressing shRNA against PICK1 plus sh-resistant WT–PICK1–IRES–EGFP were treated as in A. Graph shows surface/internal ratio relative to unstimulated controls. *p < 0.0125. n = 4 independent experiments, with 12–15 cells in each. Scale bar, 20 μm.
Colocalization of PICK1 and GluA2 on endosomal compartments is transiently enhanced after glycine stimulation
Because our data suggest a role for PICK1 in transiently restricting GluA2 trafficking to the synaptic plasma membrane during LTP and PICK1 has been shown to colocalize with both early and recycling endosomes (Sossa et al., 2006; Madsen et al., 2008), we investigated whether the association of PICK1 with GluA2 on endosomal compartments is regulated after glycine stimulation. We used Alexa Fluor-conjugated transferrin to label functional endosomes and analyzed endogenous GluA2–PICK1 colocalization in these compartments. Figure 5A shows that the proportion of GluA2 that colocalizes with PICK1 on transferrin-positive compartments increases at 5 min after the glycine stimulus (21 ± 4% increase compared with control, p < 0.0125), reaching a peak at 10 min (33 ± 4% increase compared with control, p < 0.001). At 20 min after stimulus, the proportion of GluA2 colocalizing with PICK1 on endosomes decreases dramatically to a level not significantly different from control.
Figure 5.

Transient increase in PICK1 localization at endosomal compartments after glycine stimulation. A, Colocalization between GluA2 and PICK1 is transiently increased at endosomal compartments after stimulation with glycine. Dissociated hippocampal neurons were preloaded with Alexa Fluor–transferrin to label endosomes (red channel). Neurons were exposed to elevated glycine for 3 min, returned to normal medium for the specified time, fixed at 0, 5, 10, or 20 min after stimulus, and stained using anti-GluA2 (green channel) and anti-PICK1 (blue channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graph shows Manders' colocalization coefficients for the fraction of GluA2 colocalized with PICK1 within the transferrin compartment normalized to control. *p < 0.0125, ***p < 0.001. n = 3 independent experiments, with 10–15 cells in each. Scale bar, 5 μm. B, Colocalization between PICK1 and EEA1 is transiently enhanced in dendrites at 5 min after glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in A and stained using anti-EEA1 (green channel) and anti-PICK1 (red channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graph shows Manders' colocalization coefficients for the fraction of PICK1 colocalized with EEA1 normalized to control. **p < 0.01. n = 4 independent experiments, with 10–12 cells in each. Scale bar, 5 μm. C, Colocalization between PICK1 and the recycling endosomal marker Rab11 is transiently enhanced in dendrites at 5 min after glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in A and stained using anti-Rab11 (green channel) and anti-PICK1 (red channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graph shows Manders' colocalization coefficients for the fraction of PICK1 colocalized with Rab11 normalized to control condition. **p < 0.01, ***p < 0.001. n = 4 independent experiments, with 10–12 cells in each. Scale bar, 5 μm. D, Colocalization between PICK1 and the synaptic marker PSD95 is unchanged at 5 min after glycine stimulation but increases at 20 min. Dissociated hippocampal neurons were glycine stimulated as in A and returned to normal medium for the specified time, fixed at 5 or 20 min after stimulus, and stained using anti-PSD95 (green channel) and anti-PICK1 (red channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graph shows Manders' colocalization coefficients for the fraction of PICK1 colocalized with PSD95 normalized to control condition. *p < 0.025. n = 3 independent experiments, with 10–12 cells in each. Scale bar, 5 μm. E, Association between PICK1 and Rab11 analyzed by live imaging. Dissociated hippocampal neurons expressing GFP–Rab11 and mCherry–PICK1 were exposed to elevated glycine for 3 min, followed by washout with normal medium. Images were collected every minute, and fluorescence intensity for mCherry–PICK1 was measured within a mask generated from the GFP–Rab11 signal. Images show representative images from selected time points, and graph shows fluorescence normalized to baseline (before glycine application). n = 7. Scale bar, 2 μm.
These results suggest that PICK1 may translocate to endosomal compartments to regulate GluA2 trafficking during LTP. To test this hypothesis, we costained cultured neurons for PICK1 and the early endosomal antigen marker EEA1 or the recycling endosome marker Rab11 at 5 and 20 min after glycine stimulation. At 5 min after stimulus, the colocalization of PICK1 with both endosomal markers is significantly increased [EAA1, 21 ± 3% increase compared with control, p < 0.01 (Fig. 5B); Rab11, 49 ± 5% increase compared with control, p < 0.001 (Fig. 5C)]. In both cases, these increases are transient and decrease at 20 min after stimulus. However, the colocalization of PICK1 with Rab11 remains higher than controls at the 20 min time point (18 ± 4% increase compared with control, p < 0.01; Fig. 5C). As a control for this experiment, we also analyzed the colocalization of PICK1 with the synaptic marker PSD95. Glycine stimulation has no effect on the synaptic localization of PICK1 at 5 min but does result in an increase at 20 min after stimulus (21 ± 4% increase compared with control, p < 0.025; Fig. 5D).
To explore further the association of PICK1 with endosomal compartments, we performed live imaging experiments using mCherry–PICK1 in conjunction with GFP–Rab11 to label recycling endosomes. We used the GFP (Rab11) fluorescence signal to define endosomes in sections of dendrite and measured the mCherry (PICK1) fluorescence in these endosomal compartments over time. Stimulation with glycine results in a rapid increase in PICK1 association with recycling endosomes, reaching a peak at 4 min after stimulus (102 ± 12% increase compared with baseline, p < 0.001; Fig. 5E). This association remains elevated for several minutes and then gradually decreases toward baseline levels, with a significant decrease at 20 min compared with 5 min after glycine stimulation (34 ± 8.9% decrease, p < 0.025). This demonstrates that PICK1 translocates to recycling endosomes in response to glycine stimulation, after a time course that is consistent with a role in restricting GluA2 trafficking through endosomal compartments during the early stages of LTP.
GluA2–PICK1 interaction is transiently enhanced after glycine stimulation
Our data so far suggest that LTP induction may cause a transient increase in GluA2–PICK1 interactions that restrict GluA2 trafficking to the synaptic plasma membrane, delaying the insertion of GluA2-containing receptors. Alternatively, it could be that basal levels of PICK1 binding to GluA2 are sufficient to retain GluA2 inside the cell at 5 min after LTP induction and that the PICK1–GluA2 interaction is actively inhibited between 5 and 20 min. To distinguish between these hypotheses, we performed coimmunoprecipitations (co-IPs) from hippocampal cultures at 5 and 20 min after glycine stimulation to investigate GluA2–PICK1 interactions. Control IPs were performed using normal IgG to demonstrate specificity of the interaction (data not shown). Figure 6A demonstrates that GluA2–PICK1 binding increases dramatically at 5 min after stimulus (187 ± 45% increase compared with control, p < 0.01) and returns to baseline at 20 min, consistent with a role for PICK1 in regulating GluA2 surface expression during LTP. GRIP1 is another PDZ domain protein that is involved in AMPAR trafficking and synaptic plasticity (Takamiya et al., 2008; Hanley and Henley, 2010) and can bind directly to PICK1 (Lu and Ziff, 2005). We therefore investigated GluA2–GRIP1 and PICK1–GRIP1 binding using the same approach. The association of GRIP1 with GluA2 also increases at 5 min after glycine treatment (100 ± 55% increase compared with control, p < 0.05; Fig. 6B). In contrast to the PICK1 interaction, GluA2–GRIP binding is maintained at 20 min after stimulus. The PICK1–GRIP1 interaction follows a similar pattern as GluA2–GRIP1, with a 105 ± 30% increase at 5 min compared with control (p < 0.05; Fig. 6C). No significant reduction in PICK1–GRIP1 binding is seen at 20 min compared with 5 min after stimulus, suggesting that this interaction is also maintained at this later time point.
Figure 6.

Interaction between GluA2 and PICK1 is transiently enhanced by glycine stimulation and is subsequently disrupted as a result of CP-AMPAR activation. A, GluA2–PICK1 interaction is transiently increased at 5 min after stimulus. Dissociated hippocampal neurons were stimulated with glycine for 3 min and returned to normal medium for the specified time. Lysates were prepared and IPs performed using anti-PICK1 antibody. Bound proteins were detected by Western blotting using anti-GluA2 and anti-PICK1 antibodies. Controls did not receive the stimulus. Left shows representative blots, and the graph shows PICK1–GluA2 binding relative to control. **p < 0.01. n = 7. B, GluA2–GRIP1 interaction is increased at 5 and 20 min after stimulus. Dissociated hippocampal neurons were glycine stimulated as in A. Lysates were prepared and IPs performed using anti-GRIP1 antibody. Bound proteins were detected by Western blotting using anti-GluA2 and anti-GRIP1 antibodies. Controls did not receive the stimulus. Left shows representative blots, and the graph shows GRIP1–GluA2 binding relative to control. *p < 0.05; n = 7. C, PICK1–GRIP1 interaction is increased at 5 min after stimulus. Dissociated hippocampal neurons were glycine stimulated as in A. Lysates were prepared and IPs performed using anti-PICK1 antibody. Bound proteins were detected by Western blotting using anti-PICK1 and anti-GRIP1 antibodies. Controls did not receive the stimulus. Left shows representative blots, and the graph shows GRIP1–PICK1 binding relative to control. *p < 0.05; n = 9. D, Blockade of GluA2-lacking AMPARs with NASPM completely blocks the release of GluA2 from PICK1 between 5 and 20 min after glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in A, but 30 μm NASPM was present throughout. Lysates were prepared and IPs performed using anti-PICK1 antibody. Bound proteins were detected by Western blotting using anti-GluA2 and anti-PICK1 antibodies. Controls did not receive the stimulus. Left shows representative blots, and the graph shows PICK1–GluA2 binding relative to control. *p < 0.05; n = 6. E, Blockade of GluA2-lacking AMPARs with NASPM has no effect on GluA1 trafficking in response to glycine stimulation and blocks the increase in GluA2 surface expression at 20 min after stimulus. Dissociated hippocampal neurons were stimulated with glycine for 3 min and returned to normal medium for the specified time. NASPM (30 μm) was present throughout. Controls did not receive the stimulus. Cells were fixed at 5 or 20 min after stimulus and processed for immunocytochemistry as described for Figure 1. **p < 0.01; n = 3 independent experiments, with 10–12 cells in each. Scale bar, 5 μm.
These data demonstrate that a PICK1–GRIP1–GluA2 complex is involved in the regulation of GluA2 trafficking after glycine stimulation and therefore suggest that this complex is involved in these processes during LTP expression. The tripartite complex associates soon after stimulation, and PICK1 later dissociates from GluA2. GRIP1 remains associated with both GluA2 and PICK1, suggesting that two distinct pools of GRIP1 are involved during this process.
Calcium influx via GluA2-lacking AMPARs triggers the release of GluA2 from PICK1
The delayed synaptic plasma membrane insertion of GluA2 suggests that this event is not an immediate consequence of calcium influx through NMDARs activated during LTP induction. We therefore investigated the possibility that the disruption of GluA2–PICK1 interactions, and plasma membrane insertion of GluA2 between 5 and 20 min after stimulus, is triggered by the activation of GluA2–lacking CP-AMPARs. We performed co-IPs after stimulation with glycine in neurons treated with NASPM to block CP-AMPARs (Koike et al., 1997; Dixon et al., 2009). Interestingly, NASPM completely blocks the reduction in PICK1–GluA2 interactions between 5 and 20 min after glycine stimulation (Fig. 6C), suggesting that calcium influx through CP-AMPARs inhibits the interaction of PICK1 with GluA2. To explore this further, we analyzed the plasma membrane expression levels of GluA1 and GluA2 after stimulation in the presence of NASPM. Although NASPM has no effect on GluA1 trafficking, it completely blocks the increase in surface GluA2 at 20 min after glycine treatment (Fig. 6D). This strongly suggests that the resetting of synaptic AMPARs to GluA2-containing subtypes depends on the activation of GluA2-lacking receptors that are transiently expressed at the synapse immediately after LTP induction. NASPM has no effect on the increase in PICK1–GluA2 binding between 0 and 5 min after stimulus or on GluA2 trafficking at this earlier time point, which is consistent with the notion that there is negligible synaptic expression of GluA2-lacking AMPARs under basal conditions (Lu et al., 2009).
PICK1 is a calcium sensor, and direct calcium binding alters the conformation of the protein, modulating its interaction with GluA2 during NMDAR-dependent LTD (Hanley and Henley, 2005; Citri et al., 2010). Two regions of acidic amino acids at the N terminus (residues D4–D12) and C terminus (D380–D390) of PICK1 are involved in calcium binding. The N-terminal region is crucial for sensing calcium during LTD, whereas the cellular function of the C-terminal region is unknown (Hanley and Henley, 2005; Citri et al., 2010). Because our data show that the PICK1–GluA2 interaction is regulated after glycine stimulation via the activity of CP-AMPARs, we asked whether PICK1 acts as a calcium sensor during the trafficking events triggered by this stimulus. To investigate this question, we analyzed GluA2 trafficking in hippocampal neurons expressing two different mutant forms of PICK1 targeting the acidic regions. A two amino acid substitution in the N-terminal acidic region (DD4,6AA–PICK1), which is known to reduce calcium binding to the same extent as deleting the entire acidic domain (Hanley and Henley, 2005), and a mutant lacking the C-terminal acidic region (Δ380–390–PICK1; Fig. 7A). Although overexpression of WT–PICK1flag leads to a 54% glycine-induced loss of surface GluA2 at 5 min after stimulus (Fig. 4B), this effect is completely abolished in neurons expressing Δ380–390–PICK1flag or DD4,6AA–PICK1flag (Fig. 7B). This suggests that calcium binding to PICK1 is required for the PICK1-mediated restriction of GluA2 trafficking to the plasma membrane during the first 5 min after glycine treatment. At 20 min after stimulus in WT–PICK1flag-overexpressing cells, surface GluA2 increases relative to the 5 min time point, suggesting that GluA2 is released from WT–PICK1 and allowed to traffic to the cell surface (Fig. 4B). In contrast, DD4,6AA–PICK1flag and Δ380–390–PICK1flag restrict GluA2 traffic at this later time point, suggesting that calcium binding is also required for the release of GluA2 from PICK1.
Figure 7.
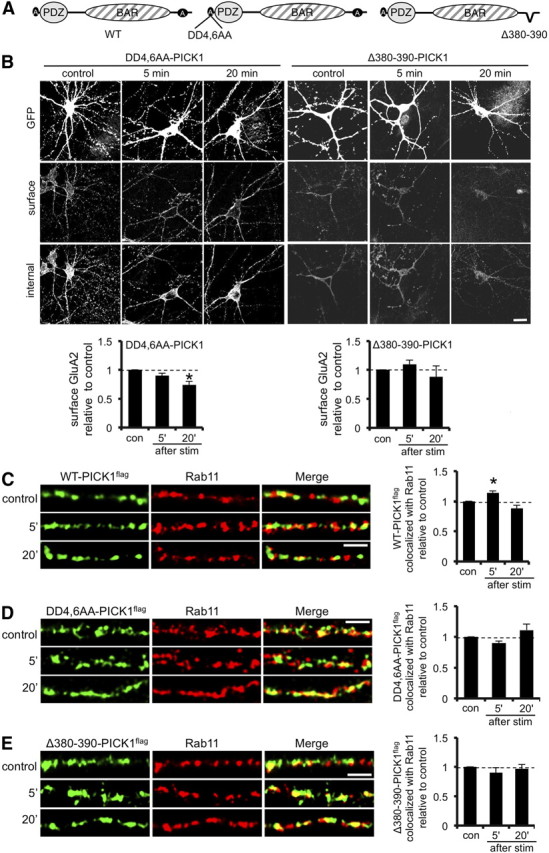
Calcium-sensing function of PICK1 is required for the regulation of GluA2 trafficking to the plasma membrane after glycine stimulation. A, Schematic diagram showing PICK1 mutants used. B, Calcium-sensing regions of PICK1 are involved in GluA2 trafficking to the plasma membrane after glycine stimulation. Dissociated hippocampal neurons expressing DD4,6AA–PICK1flag–IRES–EGFP or Δ380–390–PICK1flag–IRES–EGFP were stimulated with glycine for 3 min, returned to normal medium for the specified time, and fixed 5 or 20 min after stimulus. Controls did not receive the stimulus. Cells were processed for immunocytochemistry as described for Figure 1. *p < 0.025; n = 3 independent experiments, with 12–15 cells in each. Scale bar, 20 μm. C, Colocalization between WT–PICK1flag and the recycling endosomal marker Rab11 is transiently enhanced in dendrites at 5 min after glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in B and stained using anti-Rab11 (red channel) and anti-flag (green channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graphs show Manders' colocalization coefficients for the fraction of WT–PICK1flag colocalized with Rab11 normalized to control condition. *p < 0.05, n = 3 independent experiments, with 10–12 cells in each. Scale bar, 5 μm. D, Colocalization between DD4,6AA–PICK1flag and the recycling endosomal marker Rab11 is unaffected by glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in B and stained using anti-Rab11 (red channel) and anti-flag (green channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graphs show Manders' colocalization coefficients for the fraction of DD4,6AA–PICK1flag colocalized with Rab11 normalized to control condition. n = 3 independent experiments, with 10–12 cells in each. Scale bar, 5 μm. E, Colocalization between Δ380–390–PICK1flag and the recycling endosomal marker Rab11 is unaffected by glycine stimulation. Dissociated hippocampal neurons were glycine stimulated as in B and stained using anti-Rab11 (red channel) and anti-flag (green channel). Controls did not receive the stimulus. Representative images are shown for all conditions. Graphs show Manders' colocalization coefficients for the fraction of Δ380–390–PICK1flag colocalized with Rab11 normalized to control condition. n = 3 independent experiments, with 10–12 cells in each. Scale bar, 5 μm.
We also analyzed the association of these mutant forms of PICK1 with Rab11-positive recycling endosomal compartments after stimulation with glycine. Exogenous WT–PICK1flag shows a similar pattern as endogenous PICK1, with a transient increase in colocalization with Rab11 at 5 min after stimulus (14 ± 3% increase compared with control, p < 0.025). In contrast, neither DD4,6AA–PICK1flag nor Δ380–390–PICK1flag shows increased association with the Rab11 compartment at 5 min after glycine stimulation. These experiments suggest that the calcium-sensing properties of PICK1 are crucial for regulating GluA2 trafficking during LTP, for both the initial restriction of trafficking through the endosomal system and the delayed release of GluA2 for insertion at the cell surface.
Discussion
We have defined a mechanism for the switch in AMPAR subunit composition in hippocampal neurons that occurs after stimulation with elevated glycine, a treatment known to induce LTP. This stimulus rapidly activates PICK1 to increase its binding to GluA2 and also its association with endosomal compartments. Although allowing the trafficking of GluA2-lacking AMPARs to the plasma membrane within the first 5 min after glycine stimulation, calcium-dependent PICK1 interactions restrict the forward trafficking of GluA2-containing AMPARs, resulting in synaptic expression of GluA2-lacking CP-AMPARs. Subsequent activation of these CP-AMPARs results in the dissociation of PICK1 from GluA2, allowing GluA2-containing receptors to insert into the synapse between 5 and 20 min after stimulation (Fig. 8). Thus, increased PICK1–GluA2 binding is responsible for the appearance of CP-AMPARs, and CP-AMPAR activation is responsible for disrupting PICK1–GluA2. This elegant feedback loop would allow transient, self-limiting increases in synaptic calcium immediately after LTP induction.
Figure 8.

Model for subunit-specific AMPAR trafficking during LTP. A, Under basal conditions, the vast majority of synaptic AMPARs are GluA1/2 and GluA2/3 heteromers. These receptors continually exchange with endosomal compartments in rounds of recycling. It is assumed that a small proportion of intracellular AMPARs are GluA2 lacking (GluA1/3 heteromers or GluA1 homomers). A proportion of PICK1 associates with the recycling endosome system. B, Immediately after LTP induction, PICK1–GluA2 interactions are increased, and a larger proportion of PICK1 associates with endosomes, leading to retention of GluA2-containing AMPARs in endosomes. GluA2-lacking AMPARs are inserted at the plasma membrane and move laterally to the synapse. C, At 5 min after LTP, a proportion of synaptic AMPARs are GluA2 lacking, which are calcium permeable. D, Between 5 and 20 min after LTP induction, activation of GluA2-lacking CP-AMPARs leads to disruption of the PICK1–GluA2 interaction, allowing GluA2-containing AMPARs to be inserted into the plasma membrane. During this time, GluA2-lacking AMPARs are removed from the synaptic plasma membrane. E, This process leads to a net increase in synaptic AMPARs, the vast majority of which contain GluA2.
Subunit-specific trafficking
Our data indicate that the transiently surface-expressed GluA2-lacking AMPARs are composed primarily of GluA1 homomers, with a possible contribution from either GluA3 homomers or, more likely, GluA1/GluA3 heteromeric receptors. Interestingly, GluA3 surface levels return to baseline, and GluA1 shows a slight decrease at 20 min after glycine stimulation compared with 5 min, suggesting that the GluA2-lacking AMPARs may be internalized between 5 and 20 min and replaced by GluA1/GluA2.
The increase in GluA1 surface expression (55% increase) at 5 min after stimulation is considerably greater than the increase in GluA1 synaptic localization (19% increase) at the same time point. Furthermore, GluA1 synaptic localization increases further at 20 min compared with 5 min, compared with a small decrease in GluA1 surface expression during the same time period. This suggests that a proportion of the newly inserted GluA2-lacking AMPARs are initially expressed at extrasynaptic sites and then move by lateral diffusion to synapses. This is consistent with a recent study reporting the insertion of overexpressed pHluorin-tagged GluA1 homomers at extrasynaptic sites before lateral diffusion to synapses during LTP (Makino and Malinow, 2009). Our data are also in agreement with an emerging universal model for AMPAR trafficking whereby receptors are inserted extrasynaptically, diffuse in the plasma membrane, and are eventually trapped at the synapse by TARP/PSD95 complexes (Opazo and Choquet, 2011). The role of these extrasynaptic GluA2-lacking AMPARs is unclear, but a recent report suggested that they may be important in controlling the reversibility of LTP (Yang et al., 2010). A relatively small increase in synaptic GluA1 at 5 min after LTP fits well with a theoretical model put forward by Guire et al. (2008), suggesting that incorporation of GluA2-lacking receptors to just 5% of the total synaptic population would be sufficient to produce the increase in synaptic strength observed electrophysiologically.
Role of PICK1
PICK1 is required for the expression of both LTP and LTD (Terashima et al., 2008; Volk et al., 2010). Numerous studies have defined molecular mechanisms that underlie the role of PICK1 in LTD, which involve regulating AMPAR internalization at the level of endocytosis from the plasma membrane or endosomal compartments or both (Xu and Xia, 2006; Hanley, 2008). However this is, to our knowledge, the first to report the mechanistic details of PICK1-dependent mechanisms during a stimulation protocol that induces LTP. Interestingly, our data suggest that the role of PICK1 in early LTP has similarities to a proposed role in LTD, namely it acts in a calcium-dependent manner to constrain GluA2 trafficking from endosomal compartments to the plasma membrane. The difference in expression between LTD and early LTP presumably lies in the forward traffic of GluA2-lacking AMPARs. Either this process is specifically stimulated during LTP, or alternatively it may be inhibited during LTD.
PICK1 expression level has a profound effect on the trafficking of GluA2-containing AMPARs in response to glycine stimulus, demonstrating the importance of PICK1 in AMPAR trafficking during LTP. Endogenous PICK1 is sufficient to restrict GluA2 trafficking so that no change in surface expression is observed at 5 min after stimulation. Increasing PICK1 levels by overexpression results in a correspondingly stronger restriction of GluA2 trafficking to the cell surface in response to glycine stimulus. Conversely, reducing PICK1 expression by RNAi effectively removes this restriction of GluA2 trafficking to the cell surface, resulting in a very rapid increase in GluA2 surface expression similar to that of GluA1. GluA1 trafficking to the plasma membrane is initiated very rapidly in response to glycine treatment, with significantly increased surface levels observed immediately after stimulation (0 min time point). Under conditions of reduced PICK1 expression, surface levels of GluA2 show a trend toward increasing at 0 min, which is statistically significant at 5 min after stimulus. This slight discrepancy between GluA2 trafficking under conditions of PICK1 knockdown and GluA1 trafficking may reflect a contribution from other proteins, e.g., GRIP1, in modulating GluA2 traffic through recycling endosomes.
In addition to regulation of GluA2–PICK1 binding, our data indicate that GRIP1–GluA2 as well as GRIP1–PICK1 interactions are enhanced at 5 min after glycine stimulation. GRIP1 has been suggested to restrict surface trafficking of GluA2 during LTD (Daw et al., 2000) and also to function as part of a complex with PICK1 (Lu and Ziff, 2005). At 20 min after glycine stimulation, the enhanced GRIP1–GluA2 and GRIP1–PICK1 interactions persist, in contrast to PICK1–GluA2, which returns to baseline levels. This suggests that distinct pools of GRIP1 are involved; some is bound to GluA2 as it traffics to the cell surface, whereas some remains associated with PICK1. Differential palmitoylation of GRIP1 isoforms may occur during LTP, since it has been reported that GRIP1 palmitoylation regulates endosomal recycling of AMPARs (Hanley and Henley, 2010; Thomas et al., 2012).
The subsequent release of GluA2 from PICK1 and the trafficking of GluA2 to the plasma membrane is triggered by the activation of CP-AMPARs and depends on regions of PICK1 shown previously to be involved in binding calcium. Together, this suggests a mechanism in which CP-AMPARs mediate an influx of calcium that inhibits PICK1–GluA2 interactions, allowing the reincorporation of GluA2-containing AMPARs at the synapse. The calcium sensitivity of the PICK1–GluA2 interaction is biphasic, such that a moderate increase in calcium enhances the interaction and higher calcium concentrations attenuate the interaction (Hanley and Henley, 2005). Our results suggest that the PICK1–GluA2 complex formed at 5 min after stimulus is exposed to a relatively high concentration of calcium via CP-AMPARs to free GluA2 from PICK1.
PICK1-dependent switching of AMPAR subunit composition has been shown previously to be important in in vitro models for pathological situations, such as ischemia (Dixon et al., 2009) and mild traumatic brain injury (Bell et al., 2009) that ultimately lead to neuronal dysfunction or death. It has been suggested that the expression of GluA2-lacking AMPARs leads to excitotoxicity in these models. Glycine stimulation does not result in excitoxicity, which is probably attributable to the transient nature of the subunit switch, compared with a more sustained switch in pathological models (Liu et al., 2006; Dixon et al., 2009). In addition, a reduction in surface GluA2 is observed after these insults, whereas we see no loss of GluA2 from the plasma membrane in our experiments in this study. Perhaps this subtle difference is important in determining neuronal fate in response to the surface expression of CP-AMPARs. Additional studies will be needed to define the mechanisms that determine whether GluA2 is actively internalized from the plasma membrane or restricted from recycling back to the cell surface, depending on the stimulus.
PICK1 has been shown previously to associate with both early and recycling endosomal compartments (Sossa et al., 2006; Madsen et al., 2008). We show here that stimulation with elevated glycine, a treatment known to induce LTP, causes an increase in colocalization of PICK1 with EEA1-positive (early) and Rab11-positive (recycling) endosomal compartments at early time points after stimulus. This suggests a role for PICK1 in regulating trafficking through these compartments and is consistent with a model in which PICK1 restricts the recycling of GluA2-containing receptors and hence prevents their insertion at the plasma membrane. A reduction in GluA2–PICK1 binding and an increase in GluA2 insertion into the synaptic plasma membrane occur while PICK1 remains associated with the Rab11-positive recycling compartment. This suggests that PICK1 does not influence the trafficking of AMPAR-containing endosomes per se but instead is recruited to these compartments to regulate the sorting of GluA2-containing AMPARs from recycling endosomes to the plasma membrane. The translocation of PICK1 to both early and recycling endosomal compartments in response to glycine stimulation suggests that PICK1 may be involved in controlling trafficking at multiple stages in the recycling pathway during LTP.
In conclusion, we have defined a novel negative feedback mechanism that results in the transient incorporation of GluA2-lacking CP-AMPARs at synaptic sites for several minutes during the initial phase of glycine stimulation, which is known to induce LTP in hippocampal neurons. This PICK1-dependent mechanism may contribute to calcium-dependent signaling processes necessary for the long-term maintenance of LTP.
Footnotes
This work was funded by the Medical Research Council. We thank P. Rubin and P. Tidball for technical assistance, I. Gonzales-Gonzales, F. Konopacki, and S. Kantamneni for discussions, and D. Rocca and K. Wilkinson for critical reading of this manuscript.
References
- Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009;16:1665–1680. doi: 10.1038/cdd.2009.106. [DOI] [PubMed] [Google Scholar]
- Citri A, Bhattacharyya S, Ma C, Morishita W, Fang S, Rizo J, Malenka RC. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J Neurosci. 2010;30:16437–16452. doi: 10.1523/JNEUROSCI.4478-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- Dixon RM, Mellor JR, Hanley JG. PICK1-mediated glutamate receptor subunit 2 (GluR2) trafficking contributes to cell death in oxygen/glucose-deprived hippocampal neurons. J Biol Chem. 2009;284:14230–14235. doi: 10.1074/jbc.M901203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley LJ, Henley JM. Differential roles of GRIP1a and GRIP1b in AMPA receptor trafficking. Neurosci Lett. 2010;485:167–172. doi: 10.1016/j.neulet.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen KL, Eriksen J, Milan-Lobo L, Han DS, Niv MY, Ammendrup-Johnsen I, Henriksen U, Bhatia VK, Stamou D, Sitte HH, McMahon HT, Weinstein H, Gether U. Membrane localization is critical for activation of the PICK1 BAR domain. Traffic. 2008;9:1327–1343. doi: 10.1111/j.1600-0854.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Wood CL, Patton AP, Jaafari N, Henley JM, Mellor JR, Hanley JG. PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 2011;30:719–730. doi: 10.1038/emboj.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Sossa KG, Court BL, Carroll RC. NMDA receptors mediate calcium-dependent, bidirectional changes in dendritic PICK1 clustering. Mol Cell Neurosci. 2006;31:574–585. doi: 10.1016/j.mcn.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Mao L, Huganir RL, Linden DJ. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28:5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24:5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Chiu SL, Chen CM, Huganir RL. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012;73:482–496. doi: 10.1016/j.neuron.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk L, Kim CH, Takamiya K, Yu Y, Huganir RL. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci U S A. 2010;107:21784–21789. doi: 10.1073/pnas.1016103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xia J. Structure and function of PICK1. Neurosignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Zhou Q. Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad Sci U S A. 2010;107:11999–12004. doi: 10.1073/pnas.0913004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]