

Abstract
In the developing mammalian cerebral cortex, excitatory neurons are generated in the ventricular zone (VZ) and subventricular zone; these neurons migrate toward the pial surface. The neurons generated in the VZ assume a multipolar morphology and remain in a narrow region called the multipolar cell accumulation zone (MAZ) for ∼24 h, in which they extend and retract multiple processes dynamically. They eventually extend an axon tangentially and begin radial migration using a migratory mode called locomotion. Despite the potential biological importance of the process movement of multipolar cells, the molecular mechanisms remain to be elucidated. Here, we observed that the processes of mouse multipolar cells were actin rich and morphologically resembled the filopodia and lamellipodia in growth cones; thus, we focused on the actin-remodeling proteins Lamellipodin (Lpd) and Ena/vasodilator-stimulated phosphoprotein (VASP). Lpd binds to phosphatidylinositol (3,4)-bisphosphate [PI(3,4)P2] and recruits Ena/VASP, which promotes the assembly of actin filaments, to the plasma membranes. In situ hybridization and immunohistochemistry revealed that Lpd is expressed in multipolar cells in the MAZ. The functional silencing of either Lpd or Ena/VASP decreased the number of primary processes. Immunostaining and a Förster resonance energy transfer analysis revealed the subcellular localization of PI(3,4)P2 at the tips of the processes. A knockdown experiment and treatment with an inhibitor for Src homology 2-containing inositol phosphatase-2, a 5-phosphatase that produces PI(3,4)P2 from phosphatidylinositol (3,4,5)-triphosphate, decreased the number of primary processes. Our observations suggest that PI(3,4)P2, Lpd, and Ena/VASP are involved in the process movement of multipolar migrating cells.
Introduction
In the developing mammalian cerebral cortex, projection neurons derive directly from radial glia in the ventricular zone (VZ) or indirectly from progenitors in the subventricular zone (SVZ). In both cases, they migrate radially through the intermediate zone (IZ) toward the pial surface and form the well-organized cortical plate (CP). We previously reported differences in the migratory behaviors between the two cell populations (Tabata et al., 2009, 2012). Mitotically active progenitors migrate from the VZ with a bipolar or monopolar morphology and become diffusely distributed throughout the SVZ–IZ, in which they undergo cell division. In contrast, postmitotic neurons derived directly from radial glia exit the VZ slowly and transform into multipolar cells. They remain for ∼24 h in the multipolar cell accumulation zone (MAZ), located just above the VZ and overlapping with the lower part of the SVZ. These cells extend and retract multiple processes dynamically as their somata move slowly (multipolar migration) (Tabata and Nakajima, 2003; Noctor et al., 2004). Eventually, the multipolar cells extend an axon tangentially, transform into bipolar cells, and quickly migrate using the radial fiber as a scaffold (locomotion mode of migration) (Rakic, 1972; Nadarajah et al., 2001; Tabata and Nakajima, 2003; Hatanaka and Yamauchi, 2012). These observations suggest that the process movement of multipolar cells is a biologically important, exploratory behavior resembling that of the growth cones, which sense the microenvironment. However, the molecular mechanisms responsible for the characteristic process movements of the multipolar cells are not fully understood.
Lamellipodin (Lpd) and the Ena/VASP family (Ena, vertebrate Mena, VASP, and EVL) are known to regulate growth cone dynamics. Ena/vasodilator-stimulated phosphoprotein (VASP) proteins antagonize capping proteins at actin filament-barbed ends and regulate filopodia and lamellipodia (Michael et al., 2010). Lpd is located in the phosphatidylinositol (3,4)-bisphosphate [PI(3,4)P2]-rich region on the plasma membrane and recruits Ena/VASP proteins (Krause et al., 2004). PI(3,4)P2 is generated mainly from phosphatidylinositol (3,4,5)-triphosphate [PI(3,4,5)P3] by 5-phosphatases, including SH2 (Src homology 2)-containing inositol phosphatase-2 (SHIP2).
Here, we focused on the process movement of multipolar cells in the MAZ and found that their behaviors were similar to those of lamellipodia and filopodia in the growth cones of growing axons. Furthermore, the processes of the multipolar cells were rich in actin, suggesting that the molecular mechanisms responsible for process movement rely on actin-associated proteins, as in growth cones. Although the roles of the Ena/VASP family in the radial migration and positioning of neurons (Goh et al., 2002; Kwiatkowski et al., 2007) and the knockdown phenotype of Lpd on locomoting neurons have been described previously (Pinheiro et al., 2011), the roles of these proteins in multipolar cells have scarcely been investigated. Here, we showed that the functional blocking of Lpd and the Ena/VASP family reduced the number of processes. We also showed that PI(3,4)P2 is located in the processes of multipolar cells. The knockdown and the pharmacological inhibition of SHIP2 also reduced the number of processes. Our observations provide evidence that PI(3,4)P2, Lpd, and Ena/VASP are involved in the process movement/formation of multipolar cells.
Materials and Methods
Animals.
Pregnant ICR mice were purchased from Japan SLC. The day of vaginal plug detection was considered embryonic day 0 (E0). All the animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals of Keio University School of Medicine.
Vector construction.
Pippi–PI(3,4)P2 (Yoshizaki et al., 2007) was a kind gift from Dr. M. Matsuda (Kyoto University, Kyoto, Japan). The pCALNL–DsRed vector (Matsuda and Cepko, 2007) was purchased from Addgene. Human Lpd–enhanced green fluorescent protein (EGFP) cDNA (a kind gift from Dr. F. Gertler, Massachusetts Institute of Technology, Cambridge, MA), was subcloned into a pCAGGS vector (a gift from J. Miyazaki, Osaka University, Osaka, Japan); this vector contained a chicken β-actin promotor, the CMV–IE enhancer, polycloning sites, and a rabbit β-Globin poly(A) signal. ΔPH–Lpd–EGFP is a deletion mutant of Lpd in which the Pleckstrin homology (PH) domain (397–505) is substituted with XhoI (Leu-Glu) sites in-frame. As for the pCAX–LacZ vector, a XbaI–BamHI fragment containing LacZ cDNA from pcDNA3.1Hygro (Invitrogen) was subcloned into the NheI–BamHI sites of a pCAX vector, a modified pCAGGS vector kindly provided by the late K. Umesono (last affiliated with Kyoto University, Kyoto, Japan). To generate VASP–HA/pCAGGS, the full-length coding sequence of mouse VASP (FANTOM III clone AK171715) and HA tag (YPYDVPDYAE) was inserted into a pCAGGS vector using the EcoRI–XhoI sites. A Cre recombinase gene modified according to the optimal codon usage for mammalian systems (Koresawa et al., 2000) was inserted into the XhoI site of the pCAGGS vector. To construct myr–SHIP2/pCAGGS, the FLAG tag of FLAG–mSHIP2–EGFP/pCAGGS (a gift from Dr. M. Matsuda) was replaced with synthesized oligonucleotides encoding the Src myristoylation signal (5′-AATTCATGGGTTCTTCTAAATCTAAACCTAAAGATCCTTCTCAACGTCGTCGTTCTTTAGAA-3′ and its complementary sequence). To construct shRNA-expressing vectors, oligonucleotides targeting the Lpd and SHIP2 coding sequences (shLpd, 5′- GATCCAGAAGTGCTGTTAGAGGAATGCTGTGAAGCCACAGATGGGCATTCCTCTAACAGCACTTCTTTTTTTGGAAA-3′; shSHIP2, 5′-GATCCGCTATCTGTGTTAAAGGACTTCTGTGAAGCCACAGATGGGAAGTCCTTTAACACAGATAGCTTTTTTGGAAA-3′) and their complementary sequences were inserted into the H1pSilencer 3.0 vector (Ambion).
To generate FPPPP (FP4)–mito/–CAAX constructs, a sequence containing five EVH1 binding sites (FP4 motifs) (866–1108) of Lpd was amplified from a mouse cDNA library using PCR and was subcloned into the EcoRI–KpnI site of the pEGFP-C2 (Clontech) multicloning site, together with synthetic oligonucleotides for the mitochondrial-targeting sequence of Listeria ActA (LILAMLAIGVFSLGAFIKIIQLRKNN) or the CAAX box (GKKKKKKSKTKCVIM) of human Ki-Ras in-frame. The coding sequence with the mitochondria-targeting signal was further subcloned into a pCAGGS vector. For the activator protein 4 (AP4) version, the phenylalanine residues in each EVH1 binding site, including four FP4 motifs and one FPSPP motif (1079–1083), were changed to alanine residues by mutagenesis.
All the constructs were verified using DNA sequencing, immunological detection, and/or restriction enzyme reactions.
In utero electroporation.
Pregnant mice were deeply anesthetized using barbiturate (Dainippon Sumitomo Pharma), and their intrauterine embryos were surgically manipulated as described previously (Nakajima et al., 1997). In utero electroporation was performed as described previously (Tabata and Nakajima, 2001). Briefly, vectors were purified using a Nucleobond Xtra Midi (Macherey-Nagel), followed by phenol/chloroform purification and ethanol precipitation. The concentration of the plasmids was adjusted to the desired concentrations using HEPES-buffered saline. Plasmids in 0.01% fast green solution were injected into the lateral ventricle of the intrauterine embryos, and electronic pulses (30 V, 50 ms, four times) were then applied using an electroporator (CUY-21; NEPA GENE) with a forceps-type electrode (CUY650P5). The embryos were allowed to live within the uterine horn until the desired time of observation.
Time-lapse analyses.
Time-lapse observations of slice cultures were performed using a previously described method (Tabata and Nakajima, 2003) with minor modification. Briefly, coronal brain slices (200 μm thick) from the anterior one-third of the cortex were placed on a glass-bottom dish and firmly fixed with collagen gel (70% collagen type I-A, 1× DMEM, and 0.22% NaHCO3) and a polycarbonate membrane (13 mm diameter; pore size, 10 μm; Neuroprobe) and were cultured in Neurobasal medium (NB) containing 2% B27 (Invitrogen). The dishes were then mounted in a CO2 incubator chamber (60% O2, 35% N2, and 5% CO2 at 37°C) fitted onto a confocal microscope FV1000 (Olympus Optical). The lateral region of the cortex was analyzed. Approximately 10–20 optical Z-sections were obtained automatically every 1.5–3 min, and 5–10 focal planes were merged to visualize the shape of the entire cell. A kymograph was generated using the ImageJ Kymograph (written by J. Rietdorf and A. Seitz, European Molecular Biology Laboratory, Heidelberg, Germany) Plugin on NIH ImageJ.
In vitro culture of multipolar cells.
Dissociated primary neuron cultures were prepared as described previously with some modifications (Ajioka and Nakajima, 2005). Briefly, 12-mm-diameter glass coverslips were pretreated with HNO3, 1 mg/ml poly-l-lysine (Sigma), and 20 μg/ml laminin (Sigma). In utero electroporation was performed at E14.5 or E15.0. At 1.5 days later, the cerebral cortices containing the electroporated cells were manually dissected under a dissecting fluorescent microscope. After the removal of the meninges and CP, the VZ–IZ tissue fragments were incubated in Ca2+/Mg2+-free PBS supplemented with DNaseI for 5 min at 37°C and dissociated by pipetting. Cells were incubated in NB + B27 supplemented with penicillin–streptomycin in a CO2 (5% CO2 at 37°C) incubator. For the experiments shown in Figure 6A, 100 ng/ml recombinant mouse Netrin-1 (R & D Systems) was added. Essentially the same results were obtained without Netrin-1 (data not shown). Cells were fixed at ∼12 h later unless otherwise noted. For pharmacological treatment, the cells were incubated in NB + B27 for ∼4 h so that most of the cells attached to the coverslips; a 10 μm final concentration of 3-[(4-chlorobenzyl)oxy]-N-[(1S)-1-phenylethyl]-2-thiophenecarboxamide (AS1949490) was then added. The cells were allowed to grow for another 10 h.
Figure 6.
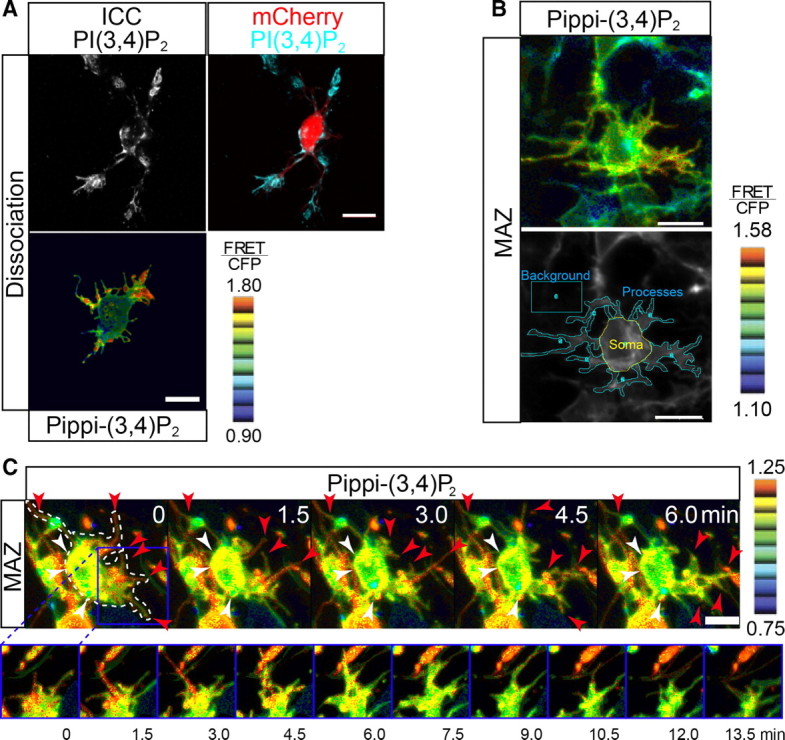
Subcellular localization of PI(3,4)P2 in multipolar cells. A, PI(3,4)P2 is localized at the growth cone-like structures and/or processes of cultured multipolar cells. To visualize the multipolar cells, the brains electroporated with mCherry/pCAGGS at E15.0 were dissociated and cultured at E16.5. The top is a representative image of immunocytochemistry (ICC) findings for an mCherry-positive multipolar cell stained with an anti-PI(3,4)P2 antibody. The bottom is a representative ratio image (FRET/CFP) of a cell transfected with Pippi-(3,4)P2. Eight colors from red to blue are used to represent the FRET/CFP ratio from 0.90 to 1.80. B, A representative ratio and Venus image of multipolar cells in the MAZ transfected with Pippi-(3,4)P2. The processes, soma, and backgrounds were defined as shown in the image. The FRET/CFP ratio was significantly higher in the processes compared with in the soma (FRETprocess/CFPprocess divided by FRETsoma/CFPsoma was 1.014 ± 0.006; n = 21 cells; Student's t test, p = 0.04), indicating the subcellular localization of PI(3,4)P2 in vivo. Eight colors from red to blue are used to represent the FRET/CFP ratio from 1.10 to 1.58. C, A high FRET/CFP ratio was observed in active processes (red arrowheads), suggesting the possible involvement of PI(3,4)P2 in the dynamic morphological changes of multipolar cells. High time resolution images of the boxed region shown in the first frame of the top panel are shown in the bottom panel. Eight colors from red to blue are used to represent the FRET/CFP ratio from 0.75 to 1.25.
Determination of knockdown efficiency.
The knockdown efficiency of the knockdown vector for Lpd (shLpd) on endogenous Lpd expression was examined. E15.5 mouse embryonic cerebral cortices were treated with 0.25% trypsin–EDTA for 15 min at 37°C and were dissociated into single cells by gentle trituration. Transfections into primary cultures of E15 cerebral cortices were performed using the Amaxa mouse neuron Nucleofector kit (Amaxa Biosystems) according to the instructions of the manufacturer, with some modifications. Briefly, 2.5 μg of shLpd or control plasmids together with 0.5 μg of pCAGGS vector carrying EGFP (for quality control) were prepared in 100 μl of Nucleofector reagent to make the plasmid solution. The cells were dissociated in the plasmid solution, and electroporation was performed. The electroporated cells were planted on 60 mm dishes coated with 1 mg/ml poly-l-lysine. After 2 d in vitro, the cells were washed with ice-cold PBS and harvested with a cell scraper. The harvested cells were centrifuged at 2300 rpm for 5 min and lysed in lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, and EDTA-free Complete protease inhibitor cocktail (Roche; 10 mm β-glycerophosphate, 50 mm sodium fluoride, and 1 mm sodium orthovanadate). After 1 h of incubation on ice, the lysates were sonicated and centrifuged at 5000 rpm for 5 min at 4°C to remove cell debris. The supernatants were mixed with SDS sample buffer [50 mm Tris-HCl, pH6.8, 2% SDS, 10% glycerol, 100 mm dithiothreitol (DTT), and bromophenol blue] and were subjected to blotting. The knockdown efficiency of the knockdown vector for SHIP2 (shSHIP2) was determined using a Western blot analysis of COS-7 cells transfected with pCAGGS–FLAG–mSHIP2–EGFP (a gift from Dr. M. Matsuda), together with shSHIP2 or a control knockdown vector (H1pSilencer).
Western blotting and coimmunoprecipitation.
Cell lysates or pallium lysates in SDS sample buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 100 mm DTT, and bromophenol blue) were separated using SDS-PAGE and were electrophoretically transferred onto polyvinylidene difluoride membranes using i-Blot (Invitrogen). The membranes were blocked with 5% skim milk in TBST (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20) for 1 h and probed with anti-Lpd rabbit IgG (1:1000; Sigma) in Can Get Signal reagents (TOYOBO), followed by treatment with horseradish peroxidase-conjugated secondary antibodies and ECL Plus Western blotting detection reagents (GE Healthcare). Signals were detected and measured with a cooled CCD camera (LAS-4000mini; Fujifilm). β-Tubulin was detected in the same membrane using anti-β-tubulin mouse monoclonal IgG (1:10,000; clone TUB2.1; Sigma) as the loading control after stripping with 0.2N NaOH for 20 min.
For the coimmunoprecipitation experiments, COS-7 cells cultured in DMEM/F-12 with 10% fetal calf serum under conditions of 37°C and 5% CO2 were transfected with HA–VASP and the indicated plasmids using GeneJuice (Merck). One day later, the transfected cell lysates were precleared with protein G–Sepharose (GE Healthcare), incubated with anti-GFP rabbit IgG (MBL International) or control rabbit IgG at 4°C for 90 min, followed by incubation with protein G–Sepharose beads overnight. The beads were washed eight times with PBS, and the bound proteins were analyzed using SDS-PAGE, followed by a Western blotting analysis using anti-HA (1:1000; Covance), as described above. Five percent skim milk in TBST was used instead of Can Get Signal.
Immunocytochemistry.
Cells were fixed with 4% PFA for 10 min, washed with PBS, blocked with 10% normal donkey serum (NDS), and then incubated overnight at 4°C with the primary antibody. The primary antibodies used were anti-β-tubulin mouse monoclonal IgG (1:200; Tuj1; Sigma), anti-Lpd rabbit IgG (1:100; Sigma), anti-NeuroD goat IgG (1:200; Santa Cruz Biotechnology), anti-GFP chick IgY (1:500; Abcam), anti-RFP rabbit IgG (1:200; Rockland Immunochemicals), and anti-VASP rabbit monoclonal IgG (1:100; 9A2; Cell Signaling Technology) in 5% NDS. Biotinylated anti-VASP antibody was used to double stain VASP and Lpd. To detect Lpd, VASP, and NeuroD, coverslips were incubated at 70°C in 10% HistoVT One (Nacalai) for 20 min before treatment with the primary antibody for antigen retrieval. PI(3,4)P2 staining using anti-PI(3,4)P2 mouse IgG (1:800 echelon; in 5% BSA/PBS) was performed as described previously (Bae et al., 2010). Briefly, the cells were fixed and permeabilized with 3.7% formaldehyde/0.1% glutaraldehyde in 0.15 mg/ml saponin solution for 1 h at 37°C, followed by overnight incubation with the primary antibody at 4°C. After washing with PBS, the cells were incubated for 1 h at room temperature with the secondary antibodies and, in some cases, together with phalloidin–Alexa Fluor 647 and/or 4′,6-diamidino-2-phenilindole (DAPI; Invitrogen).
Tissue preparation and immunohistochemistry.
The tissue samples were prepared as described previously (Tabata and Nakajima, 2001). For phalloidin staining (phalloidin–Alexa Fluor 647; Invitrogen) and Förster resonance energy transfer (FRET) experiments, fixed brains embedded in a 3% low-melting agarose gel were sectioned coronally using a vibratome. The polyclonal antibodies used in the present study were anti-SHIP2 rabbit IgG (1:200; Bethyl), anti-Lpd rabbit IgG (1:100; Sigma), anti-β-galactosidase (β-gal) rabbit IgG (1:100; Chappel), anti-GFP rabbit IgG (1:500; MBL International), and anti-GFP chick IgY (1:500; Abcam). The monoclonal antibodies that were used were anti-L1 rat IgG (1:200; Millipore) and anti-Mena mouse IgA (1:200; B D Biosciences). To detect Lpd, Mena, and SHIP2, sections were incubated at 85°C (Lpd) and 121°C (Mena and SHIP2) in 0.01 m citrate buffer, pH 6.0, for 10 min (Lpd) and 1 min (Mena and SHIP2), respectively, before treatment with the primary antibody for antigen retrieval. A tyramide signal amplification fluorescence system (PerkinElmer Life and Analytical Sciences) was used to detect Lpd and Mena. DAPI was used for nuclear staining. Images were acquired using confocal microscopes (Olympus FV1000).
To confirm the specificity of anti-Lpd, an absorption test with the antigen was performed. To produce the epitope peptide (QLSDEEIDHGAEEDSDKEDQDLDKMFGAWLGELDKLTQSLDSDKPMEPVKRSPLRQETNMANFSYRFSIYNLN) in Escherichia coli, the epitope sequence of the anti-Lpd antibody (Sigma) was amplified from human Lpd cDNA using PCR and was subcloned into the BamHI–NotI sites of the pGEX 4T-1 vector (GE Healthcare). BL21-codonPlus-competent cells were transformed with pGEX 4T-1 encoding the epitope sequence, and the expression was induced by isopropyl β-d-1-thiogalactopyranoside. The soluble fraction of the sonicated products was fixed with glutathione–Sepharose 4B (GE Healthcare). Anti-Lpd was incubated with the fraction overnight, and the supernatant was used as the primary antibody.
In situ hybridization.
PCR products amplified from AK133359 (RIKEN FANTOM III clone) and the E18.5 mouse cDNA library (forward, 5′-CGGAATTCTTGGGGCCTGGCTTGGAGAAC-3′; reverse, 5′-cgggatccagtaattggctgcccctggtgtg-3′) were used as templates for the in vitro transcription for SHIP2 and Lpd, respectively. Digoxigenin (DIG)-labeled RNAs were synthesized using the DIG RNA labeling kit (Roche). Embryonic mice were fixed by perfusion with 4% PFA, and their brains were postfixed in the same fixative overnight, incubated in 30% sucrose in diethylpyrocarbonate (DEPC)-treated PBS overnight, and embedded in OCT compound diluted with ⅓ volume of 30% sucrose in DEPC-treated PBS. Sections (14 μm) were digested with 1 μg/ml proteinase K for 5 min at 37°C. Hybridizations were performed with 2–4 ng/ml probes in 50% formamide, 10 mm Tris-HCl, pH 7.6, 600 mm NaCl, 0.25% SDS, 1 mm EDTA, 500 μg/ml yeast tRNA, 1× Denhardt's solution, and 10% dextran sulfate at 50°C for 16 h. After hybridization, the slides were washed with 50% formamide/2× SSC, 2× SSC, and 0.2× SSC at 50°C. The signal was detected using alkaline phosphatase-conjugated anti-DIG F(ab′)2 antibody and the 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (SHIP2) or 2-hydroxy-3-naphthoic acid-2′-phenylanilide phosphate fluorescent detection set (Roche) (Lpd). Nuclear fast red was used for nuclear staining of the SHIP2 samples. Immunohistochemistry for GFP and nuclear staining were performed using anti-GFP rabbit IgG (1:1000; MBL International) and DAPI to visualize the cellular morphology of the electroporated cells in Lpd samples.
FRET.
Primary cortical neurons, slice cultures and vibratome-fixed sections expressing FRET probes were imaged using an inverted microscope (IX81; Olympus) equipped with a cooled charge-coupled device camera (Cool SNAP-HQ; Roper Scientific), a laser-based auto-focusing system (IX2-ZDC; Olympus), and an automatically programmable x–y stage (MD-XY30100T-Meta; SIGMA KOKI). The filters used for the dual-emission imaging were a DM405–440 excitation filter, an SDM 510 dichroic mirror, and emission prism filters (460–500 nm for a CFP channel and 515–615 nm for the FRET channel). The cells were illuminated with a 440 nm diode laser at a power of 2.5% (living cells) or 5% (fixed specimens) and viewed through a 60× oil-immersion objective lens (UPLSAPO, 60×/1.35) with a 5× digital zoom. The exposure times for each pixel were 4 ms for live imaging and 20 ms for fixed samples. After performing a Gaussian blur (with a σ of 1.5 radius) and average projection (for slice samples) using NIH ImageJ software, the FRET/CFP ratio images were created in an intensity-modified display mode using MetaMorph software (Universal Imaging Corporation) to represent the subcellular localization of PI(3,4)P2.
Quantification of the number of primary processes.
The MAZ is a high-cell-density area in which the nuclei of the cells are randomly and tangentially oriented, the lower borders of which can be clearly identified using nuclear staining from the VZ, in which the nuclei of the cells are radially oriented (Fig. 1A). Thus, the MAZ was identified based on nuclear staining, and all the cells in the MAZ within two high power fields from the pallium–subpallium boundaries were subjected to quantification. Optical slices were obtained to cover all the cryosectioned brain (∼14 μm thick) by 1 μm, and image stacks were examined to count the number of all the primary processes or the processes that were derived directly from the somata. The immunohistochemical detection of β-gal was used for this purpose in most of the experiments. In Figure 5C and D, we used pCALNL–DsRed, which expresses DsRed in the presence of Cre recombinase. We introduced an FP4 or AP4 fusion protein-expressing vector together with pCALNL–DsRed and a low concentration of Cre expression vector to investigate the individual cell morphologies more easily.
Figure 1.
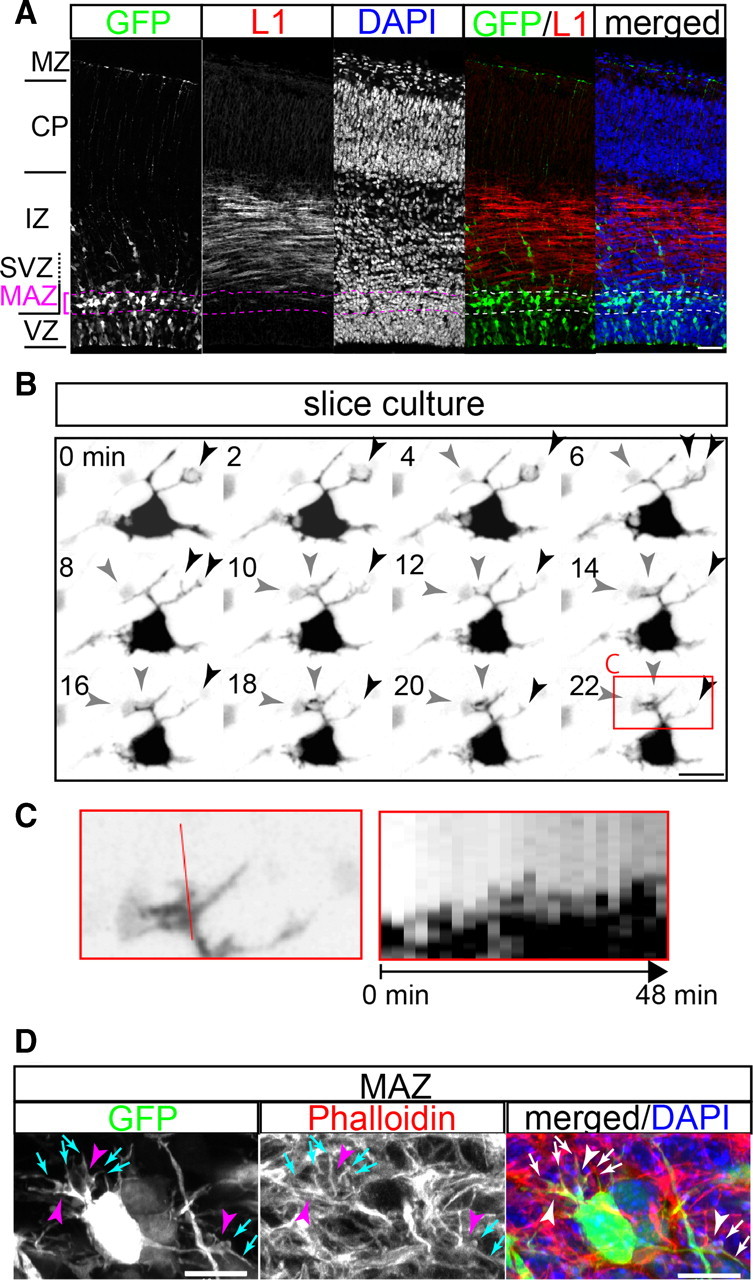
Multipolar cells in the MAZ have F-actin-rich processes with growth cone-like structures. A, Location of the MAZ in the developing cerebral cortex. E15.0 mouse cerebral cortex was electroporated with EGFP/pCAGGS and fixed at E16.5. The sections of the brains were subjected to immunohistochemistry for L1 (red), an axonal marker, and the cell nuclei were stained with DAPI (blue). The lateral pallium is shown. Many GFP-positive multipolar cells accumulated in the MAZ, in which the cell density was high and the nuclei of the cells were randomly oriented. The apical border of the MAZ (bottom broken lines) was identified by the orientation of the nuclei, which were radially oriented in the VZ. L1-positive thick axon bundles were present just above the MAZ. The cell density in the L1-positive region was low because of the presence of axon bundles, in which the cells are sandwiched obliquely in alignment. Scale bar, 50 μm. B, Growth cone-like dynamic “fan-shaped” structures were observed at the tip of some of the processes of the multipolar cells. EGFP/pCAGGS was electroporated at E15.0, slices were prepared at E16.5, and time-lapse imaging was performed. Images were acquired every 2 min. The black arrowheads point to a fan-shaped structure that turned, gradually arborized, and shrank. The gray arrowheads indicate another fan-shaped structure with protruding and retracting lamellipodia. Scale bar, 10 μm. C, Kymograph showing the dynamic process movements of the multipolar cells. A high-magnification view of a process in the boxed region shown in B is shown in the left. In the right, the GFP signals crossing the red line in the left during the course of time-lapse observation are aligned in a chronological order from 0 to 48 min. D, F-actin-positive growth cone-like structures (arrowheads) were observed at a higher magnification. Strong F-actin signals were observed especially at microspikes (arrows). Scale bar, 10 μm.
Figure 5.
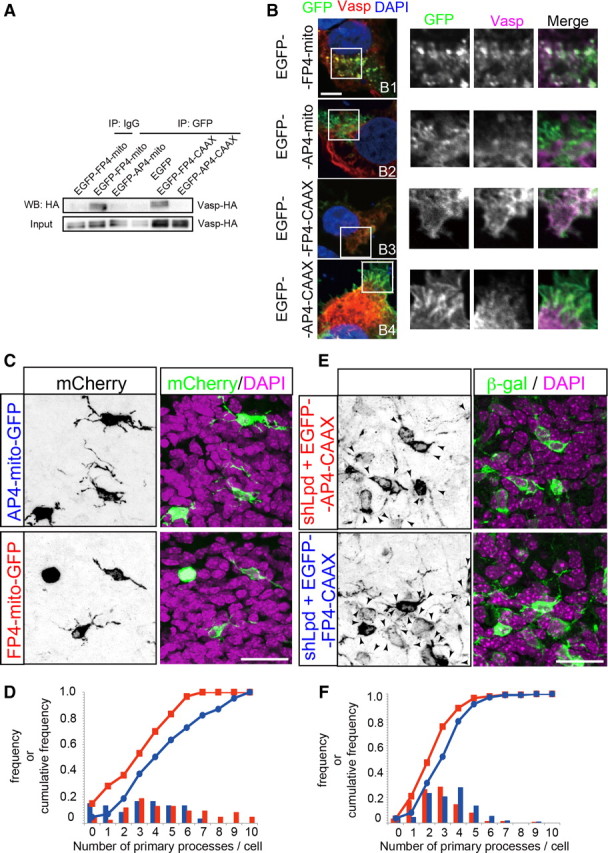
Ena/VASP family proteins interact with Lpd to form multipolar cell processes. A, B, Mitochondria-targeted and plasma-membrane-targeted FP4 proteins (from Lpd) bind to and colocalize with Ena/VASP proteins. A, EGFP–FP4–mito and EGFP–FP4–CAAX bound to VASP–HA, but mutated FP4 (AP4) fusion proteins did not. COS-7 cells were transfected with VASP–HA/pCAGGS and the indicated plasmids. The cell lysates were immunoprecipitated (IP) with an anti-GFP antibody and subjected to Western blotting (WB) analyses. B, FP4–mito–GFP sequestered endogenous VASP from the plasma membrane, whereas FP4–CAAX–GFP recruited it to the plasma membrane. COS-7 cells expressing FP4–mito–GFP, AP4–mito–GFP, FP4–CAAX–GFP, or AP4–CAAX–GFP were stained with an anti-VASP (9A2) antibody. Higher-magnification views are shown in the right. C, Either FP4–mito–GFP/pCAGGS (Ena/VASP depletion) or AP4–mito–GFP/pCAGGS (control) was electroporated at E15.0, and the brains were fixed at E16.5. Maximum projection images are shown. Scale bar, 20 μm. D, Frequency and cumulative frequency distributions of the number of primary processes per cell electroporated in utero with FP4–mito (blue circles) or AP4–mito (red squares). The number of primary processes was significantly reduced with the sequestration of Ena/VASP proteins from the plasma membrane (FP4–mito, 62 cells from 4 brains vs AP4–mito, 85 cells from 6 brains; Mann–Whitney U test, p = 0.001). E, FP4–CAAX can rescue the phenotype induced by shLpd. Either FP4–CAAX (Ena/VASP recruitment to the plasma membrane) or AP4–CAAX (control) was electroporated with shLpd at E15.0. Maximum projection images are shown. Scale bar, 20 μm. F, Quantification of E. The frequency and cumulative frequency distributions of the number of primary processes per cell (shLpd + AP4–CAAX, red squares; shLpd + FP4–CAAX, blue circles) are shown. Overexpression of FP4–CAAX rescued the knockdown phenotype of Lpd (FP4–CAAX, 154 cells from 4 brains vs 113 cells from 3 brains; Mann–Whitney U test, p < 0.001).
Data analyses.
Images collected using confocal microscopy were analyzed with Photoshop CS5 (Adobe Systems) or NIH ImageJ 1.45e. Linear changes in tone and background subtraction were made. Quantitative analyses of the cellular morphology and fluorescence signals were performed using NIH ImageJ. Maximum projection images of the optical slices were made to show the morphology of the whole cells. Statistical significance was determined using a two-tailed unpaired t test or Mann–Whitney U test, and p values <0.05 were considered significant.
Results
Multipolar cells have F-actin-rich processes with growth cone-like tips
Our previous time-lapse imaging with an interval of 30 min revealed that multipolar cells extend and retract multiple processes (Tabata and Nakajima, 2003). This observation raised the possibility that the mechanisms for the process movement of multipolar cells are similar to that of a growth cone of axons. A growth cone is a very dynamic structure formed at the distal end of a growing axon. It has rod-like projections called filopodia and flat sheet-like projections called lamellipodia. Both structures are F-actin-rich and move via actin fiber remodeling. The dynamic movement of filopodia and lamellipodia, however, happens within a few minutes, and the time-lapse observations of multipolar cells with 30 min intervals are not sufficient to examine their similarities with a growth cone. We therefore performed time-lapse imaging of multipolar cells in the MAZ in brain slices using a 1.5–3 min interval. We introduced a CAG-promoter-driven EGFP expression vector by in utero electroporation at E15.0 and prepared the slices at E16.5, when multipolar cells in the MAZ are most typically observed (Fig. 1A, see the figure legend for details); the time-lapse observations were initiated several hours later once the slices had settled. The processes of the multipolar cells moved very dynamically within a few minutes (Fig. 1B; see Fig. 6C). The processes sometimes assumed fan- or rod-like structures. The process movement within a few minutes was also demonstrated using a kymograph (Fig. 1C). These movement and structures strikingly resembled lamellipodia and filopodia in the growth cones of axons. Phalloidin staining in fixed brains confirmed that the processes of the multipolar cells in the MAZ were rich in F-actin (Fig. 1D), as shown previously (Kawauchi et al., 2006). Certain processes of multipolar cells had growth cone-like structures (Fig. 1D, arrowheads) that were rich in F-actin (arrows) in vivo. These results raised the possibility that molecules involved in the actin filament remodeling that occurs in the lamellipodia and filopodia of a growth cone are also involved in the movement of multipolar cell processes.
Lpd is expressed in multipolar cells at both the mRNA and protein levels
The movement of filopodia and lamellipodia is conducted by various actin-remodeling factors. Lpd and its binding partners, the Ena/VASP family, are known as such molecules (Krause et al., 2004). Lpd is localized at the leading edge of migrating cells and recruits Ena/VASP proteins. Ena/VASP proteins, in turn, promote the incorporation of globular actin to the barbed-end of actin filaments (Dent et al., 2011). These proteins are, therefore, good candidates for process formation factors in multipolar cells.
To investigate the expression of Lpd in multipolar cells, we performed double staining for Lpd mRNA and GFP protein in mouse brains electroporated with a GFP expression plasmid (CAG–EGFP) at E15 and fixed the brains at E16.5 (Fig. 2A1–A3). Lpd mRNA expression was detected mainly in the MAZ and CP, as well as slightly in the VZ and IZ. Almost no signals were detected in the marginal zone (MZ). These signals were not detected by the sense probe (Fig. 2A3). Observation at a single-cell level revealed that Lpd mRNA was expressed in the GFP-labeled multipolar cells in the MAZ (Fig. 2A2). Next, we tried to investigate the distribution of Lpd proteins using immunohistochemistry. We used an antibody that detected one single band with a predicted size of ∼200 kDa on a Western blot assay of E15.5 mouse pallium tissue (Fig. 2B). Immunohistochemistry using this antibody was performed on the brain sections that had been electroporated with CAG–EGFP and fixed 36 h later (Fig. 2C1–C3). Signals were broadly detected from the MAZ to the MZ, and these signals became barely detectable after the preabsorption of the antibody with epitope–GST fusion peptides (Fig. 2C4). In particular, strong signals were observed in the upper IZ and the upper CP to MZ, in which minimal mRNA was detected. These discrepancies may be attributed to the presence of Lpd in the growth cones of axons running in the IZ and the developing dendrites in the MZ. A high magnification revealed the existence of Lpd proteins in the multipolar cells in the MAZ (Fig. 2C3). The subcellular localization of Lpd proteins was difficult to observe, because the cell density in the MAZ was extremely high. To overcome this difficulty, we electroporated a human Lpd–GFP expression vector together with an mCherry expression vector and evaluated the relative intensities of the GFP signals compared with the mCherry signals in the multipolar cells in the MAZ. As a result, a relatively large proportion of the Lpd–GFP was detected in the processes of the multipolar cells (Fig. 2D). In this experiment, the punctate distribution of Lpd was not observed, unlike the situation shown in Figure 2C. This difference might be attributable to the high concentration of Lpd–GFP in the transfected cells.
Figure 2.

Lpd is expressed in multipolar cells at both the mRNA and protein level. A1–A3, Fluorescent in situ hybridization of Lpd. To visualize the multipolar cells, the brains were electroporated with EGFP/pCAGGS at E15.0 and fixed at E16.5. A1, Lpd mRNA expression was detected mainly in the MAZ and CP. A2, A higher magnification shows that the multipolar cells (enclosed by a dotted line) express Lpd. A z-axis reconstruction of the signals along the x-axis and y-axis is shown at the bottom and right side, respectively. Scale bar, 10 μm. A3, Fluorescent in situ hybridization with an Lpd sense probe. The image was obtained under the same conditions as A1. B, A Western blot analysis of Lpd in extracts of mouse E15.5 pallium using a polyclonal Lpd antibody revealed a single band at ∼200 kDa. C1–C4, Immunohistochemistry of Lpd. C1, C2, To visualize the multipolar cells, the brains were electroporated with EGFP/CAGGS at E15.0 and fixed at E16.5. The cell nuclei were stained with DAPI. Scale bar, 50 μm. C3, x–z and y–z planes as well as the Z-projection of the image stacks are shown to illustrate the colocalization of GFP and Lpd. A higher magnification shows that Lpd is expressed in multipolar cells (enclosed by a dotted line). Scale bar, 5 μm. C4, Preabsorption of the antibody with the epitope–GST fusion peptides (absorbed, left) reduced the immunohistochemistry signals (control, right). The top panels show the immunohistochemistry signals, and the bottom panels show the merged images with GFP (green) and nuclear (DAPI, blue) staining. Scale bar, 10 μm. D, Representative ratio image of a multipolar cell in the MAZ transfected with Lpd–GFP and mCherry. Eight colors from red to blue are used to represent the GFP/mCherry ratio from 0.7 to 2.2. The upper and lower limits of the ratio range are shown. Scale bar, 10 μm.
Lpd regulates the processes of multipolar cells
To investigate the role of Lpd in the multipolar cells, we generated a short hairpin RNA expression vector (shLpd), which can knockdown the expression of Lpd. The endogenous Lpd protein expression was reduced to 33% compared with the control vector when dissociated neurons were electroporated with the shLpd vector in vitro and cultured for 2 d (Fig. 3A). Using this shLpd, we investigated the effect of Lpd knockdown on the processes of multipolar cells in a dissociated culture system. We electroporated the cortices with the shLpd vector together with CAG–mCherry in utero at E15.0 and cultured the transfected cortical cells 1.5 d later (Fig. 3B). The cultured cells were fixed at 12 h in vitro (HIV), when the cells dynamically extended and retracted multiple processes as in the slice culture. The number of primary processes belonging to the mCherry-positive cells was significantly lower in the knockdown group than in the control [knockdown, mean 1.33 (157 cells) vs control, mean 2.49 (112 cells) from three independent experiments; Mann–Whitney U test, p < 0.001] (Fig. 3B,C). This phenotype was rescued by the overexpression of human Lpd–EGFP, which does not have the identical target sequence of the shLpd vector against mouse Lpd [knockdown, mean 1.33 (157 cells) vs knockdown + Lpd, mean 2.06 (110 cells) from three independent experiments; Mann–Whitney U test, p < 0.001] (Fig. 3B,C). To examine whether this effect is also observed in vivo, we electroporated the shLpd vector together with a CAG–β-gal at E15.0, fixed the brains 36 h later, and investigated the number of processes of β-gal-positive cells in the MAZ (Fig. 3D). The number of primary processes was also significantly lower in the knockdown group than in the control in vivo group [knockdown, mean 2.09 (161 cells from 3 brains) vs control, mean 3.71 (81 cells from 3 brains); the mean process number was different from that of Fig. 3C, probably because of the difference between the culture and in vivo situations; Mann–Whitney U test, p < 0.001] (Fig. 3D,E).
Figure 3.

Suppression of Lpd reduces the number of processes of multipolar cells both in vitro and in vivo. A, Evaluations of the knockdown efficiency of the shLpd plasmid. Lysates from primary cortical neurons (2 DIV) transfected in vitro with the shLpd plasmid or control silencer were subjected to immunoblot analyses with the indicated antibodies. The numbers indicate the Lpd/β-tubulin ratio standardized to the control (H1pSilencer) (0.33 ± 0.11). Three independent experiments were performed. B, C, Suppression of Lpd in dissociated multipolar cells significantly reduces the number of processes in vitro. B, Representative images of cells transfected with the shLpd vector + EGFP/pCAGGS (shLpd), control vector + EGFP/pCAGGS (control), and shLpd/H1pSilencer + human Lpd–EGFP/pCAGGS (shLpd + hLpd) in addition to mCherry/pCAGGS for labeling electroporated cells. Scale bar, 5 μm. C, Quantification of the effect of shLpd on multipolar cells cultured in vitro. Frequency and cumulative frequency distributions of the number of primary processes per cell (shLpd, red circles; control, blue squares; shLpd + hLpd, green triangles) is shown. The number of primary processes was significantly reduced with the suppression of Lpd (knockdown, 157 cells vs control, 112 cells; Mann–Whitney U test, p < 0.001), which was rescued by the overexpression of human Lpd (knockdown, 157 cells vs knockdown + Lpd, 110 cells; Mann–Whitney U test, p < 0.001). Three independent experiments were performed, with similar results. D, Silencing of Lpd resulted in a reduced number of processes in vivo. The shLpd or control plasmid, together with LacZ/pCAX, was electroporated at E15.0, and the brains were fixed at E16.5. To evaluate the morphology of each cell, sections were immunostained with an anti-β-gal polyclonal antibody. Maximum projection images are presented. Scale bar, 20 μm. E, Quantification of the effect of shLpd on multipolar cells in vivo. The frequency and cumulative frequency distributions for the number of primary processes per cell (shLpd, red circles; control, blue squares) are shown. The number of primary processes was significantly reduced by the suppression of Lpd (knockdown, 161 cells from 3 brains vs control, 81 cells from 3 brains; Mann–Whitney U test, p < 0.001).
Although we observed a lower number of primary processes of the transfected cells in the knockdown experiments, we scarcely observed cells with the typical locomotion cell morphology, that is, a thick leading process pointed to the brain surface and a thin trailing process extending in the opposite direction, indicating that the primary effect of Lpd knockdown may not be the premature transformation of multipolar cells into bipolar locomotion cells. These results suggest that Lpd is a positive regulator of process formation in multipolar cells.
Ena/VASP proteins regulate the morphology of multipolar cells
A previous study revealed that all three known mammalian Ena/VASP family members, Mena, VASP, and EVL, are highly expressed in the developing mouse neocortex (Goh et al., 2002). We performed immunohistochemistry on brain sections in which the multipolar cells were visualized using GFP and observed that multipolar cells in the MAZ expressed Mena (Fig. 4A,B). Moreover, the immunostaining of VASP and Lpd on dissociated multipolar cells revealed the colocalization of these proteins in the processes of the multipolar cells (Fig. 4C), suggesting that the Ena/VASP family proteins might be involved in the process formation of multipolar cells through their interactions with Lpd. Because the functions of three members of the Ena/VASP family essentially overlap, knockdown experiments using shRNA vectors are thought to be difficult. Considering that the Ena/VASP proteins specifically bind to the FP4 motifs of Lpd, we constructed an expression vector of a fusion protein of the FP4 motifs and a mitochondrial targeting signal (FP4–mito) to sequester the Ena/VASP family proteins into the mitochondria, which is basically the same approach as that used in previous reports (Bear et al., 2000; Krause et al., 2004). Conversely, targeting the FP4 motifs to plasma membranes by fusing them to a membrane localization signal (CAAX box) can mimic the active status of Ena/VASP proteins (Bear et al., 2000). As the negative control, we used an AP4–mito and an AP4–CAAX vector, in which a phenylalanine residue of each motif was changed to an alanine residue. An immunoprecipitation assay showed that FP4–mito and FP4–CAAX successfully bound to exogenous VASP–HA in COS-7 cells (Fig. 5A). The colocalizations of the endogenous VASP with FP4–mito in the intracellular space and with FP4–CAAX in the plasma membranes were observed in COS-7 cells, as expected (Fig. 5B), whereas such strong colocalizations were not observed in cells transfected with AP4–mito or AP4–CAAX. To investigate the effect of functional Ena/VASP depletion in multipolar cells in vivo, we electroporated the FP4–mito vector at E15.0 and fixed the brains at E16.5. The number of primary processes of the multipolar cells was significantly lower in the FP4–mito electroporated brains than in the control brains [FP4–mito, mean 3.17 (62 cells from 4 brains) vs AP4–mito, mean 4.80 (85 cells from 6 brains); Mann–Whitney U test, p = 0.001; the mean process number and the overall distribution of the process numbers differed from those in the other in vivo analyses because of the difference in the labeling methods, as described in Materials and Methods] (Fig. 5C,D).
Figure 4.
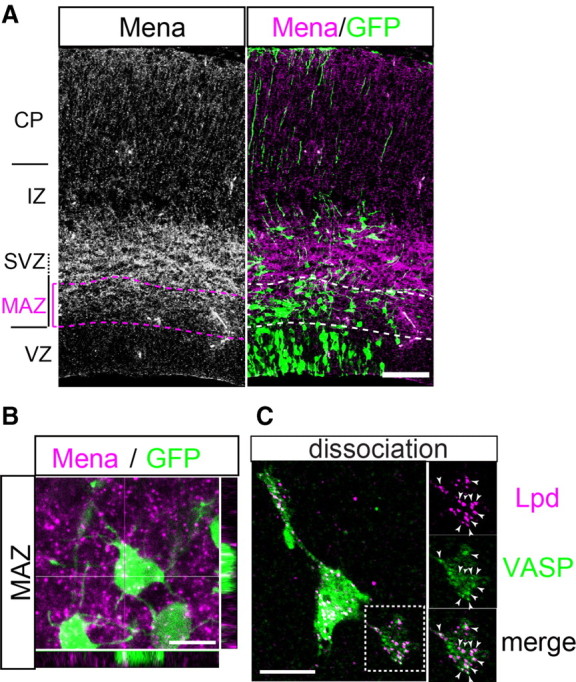
Localization of Ena/VASP family proteins in multipolar cells. A, Immunohistochemistry for Mena. Mena was expressed in the MAZ–IZ. Scale bar, 100 μm. B, Higher magnification of the multipolar cells revealing the expression of Mena. A Z-projection of the image stacks as well as the x–z and y–z planes are shown to illustrate the colocalization. Scale bar, 10 μm. C, Colocalization of VASP (green) and Lpd (magenta) proteins in the processes of the dissociated multipolar cells. High magnifications of the boxed region in the left are shown in the right. The arrowheads indicate the colocalization of VASP and Lpd. Scale bar, 10 μm.
To examine whether the Ena/VASP proteins are located downstream of Lpd in the multipolar cells, we attempted to rescue the knockdown phenotype of Lpd using FP4–CAAX. Again, the number of primary processes was slightly but significantly higher in the FP4–CAAX electroporated brains than in the control [AP4–CAAX, mean 2.72 (154 cells from 4 brains) vs FP4–CAAX, mean 3.40 (113 cells from 3 brains); Mann–Whitney U test, p < 0.001] (Fig. 5E,F). These results suggest that Ena/VASP proteins positively regulate the protrusion of the processes of the multipolar cells at a point downstream of Lpd.
Subcellular localization of PI(3,4)P2
Lpd is known to bind to PI(3,4)P2 in the plasma membrane. If Lpd is implicated in the process formation of multipolar cells, PI(3,4)P2 should be localized in their processes or process buds. To investigate the distribution of PI(3,4)P2 in multipolar cells, we labeled the multipolar cells in the MAZ with an mCherry expression vector, dissociated the cells, cultured them from E16.5 for 12 h, and then performed immunocytochemistry using a specific antibody for PI(3,4)P2 (Bae et al., 2010). As a result, immunoreactivity was found in the growth cone-like structures and limited parts of the soma and processes (Fig. 6A, top). Analyses using Pippi-(3,4)P2, a FRET probe that detects PI(3,4)P2 (Yoshizaki et al., 2007), also showed similar results (Fig. 6A, bottom). To investigate the localization of PI(3,4)P2 in multipolar cells in vivo, we electroporated Pippi-(3,4)P2 at E15.0 and fixed the brains at E16.5. Observations of the coronal slices using a confocal microscope (Fig. 6B) revealed that the FRET/CFP ratio was significantly higher in the processes than in the soma (FRETprocess/CFPprocess divided by FRETsoma/CFPsoma was 1.014 ± 0.006; n = 21 cells; Student's t test, p = 0.04). The differences between the total FRET/CFP ratios in the soma and processes were rather small, but punctate high-ratio domains were observed in the processes, suggesting that these local differences regulate the dynamic process movement. Time-lapse imaging of living slices from the electroporated brains revealed that PI(3,4)P2 accumulated in the actively moving processes (Fig. 6C).
PI(3,4)P2 is produced mainly from PI(3,4,5)P3 by the 5- phosphatases SHIP1 and SHIP2. In particular, SHIP2 is expressed in the developing brain, suggesting the possibility that SHIP2 may also be involved in process formation via PI(3,4)P2 production. In situ hybridization showed strong SHIP2 expression in the VZ and SVZ, as reported previously (Muraille et al., 2001), including the MAZ (Fig. 7A, left). SHIP2 protein was also detected in the VZ and SVZ, including the MAZ marked by Unc5D staining (Fig. 7A, right). A higher magnification revealed that the SHIP2 protein was expressed in the multipolar cells (Fig. 7B).
Figure 7.
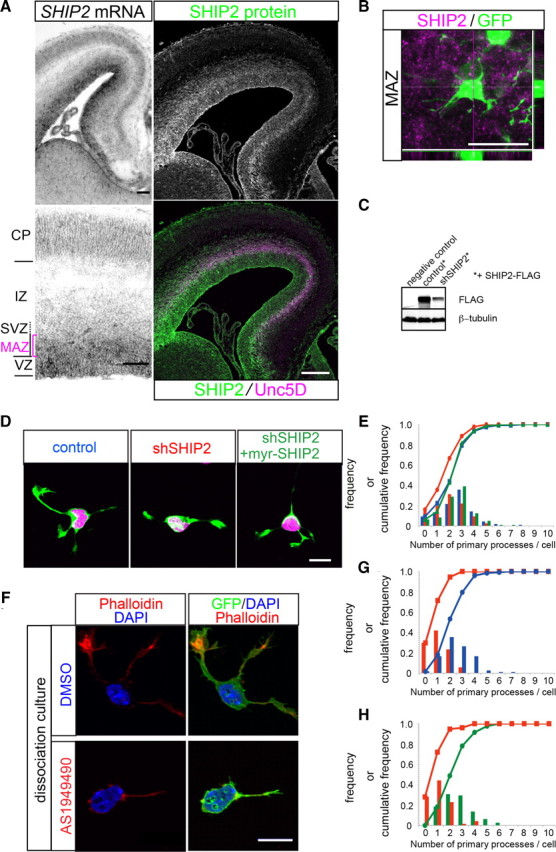
SHIP2 is involved in the process formation of multipolar cells. A, Left, Strong SHIP2 mRNA expression was detected in the VZ and MAZ. Scale bars, 100 μm. Right, Immunohistochemistry against SHIP2 (green). Immunoreactivity was detected in the VZ as well as the MAZ–SVZ, which is marked by Unc5D staining (Sasaki et al., 2008) (magenta). B, A higher magnification revealed that SHIP2 (magenta) is expressed in multipolar cells (green) at a single-cell level. To label the multipolar cells in the MAZ, brains were electroporated with GFP/pCAGGS at E15.0 and fixed 1.5 d later. A Z-projection of the image stacks as well as the x–z and y–z planes are shown to illustrate the colocalization. Scale bar, 20 μm. C, Knockdown efficiency of shSHIP2 on exogenous SHIP2–FLAG expression in COS-7 cells. D, SHIP2 knockdown reduced the number of processes in dissociated multipolar cells, and the reduction was rescued by the forced expression of myr–SHIP2. The cells were prepared from brains electroporated with the indicated combinations of plasmids in utero and were cultured for 20–24 h. E, Frequency and cumulative frequency distributions of the number of primary processes per cell that had been electroporated in utero with a control vector (blue squares), shSHIP2 (red circles), or shSHIP2 + myr–SHIP2/pCAGGS. The number of primary processes was significantly reduced after electroporation with shSHIP2 compared with the control (control, 121 cells from 2 brains vs shSHIP2, 87 cells from 2 brains; Mann–Whitney U test, p = 0.001); this reduction was rescued by CAG–myr–SHIP2 (shSHIP2 vs shSHIP2 + CAG–myr–SHIP2, 147 cells from 2 brains; Mann–Whitney U test, p < 0.001). F, Representative images of cells treated with either 10 μm AS1949490 or vehicle (DMSO). G, Frequency and cumulative frequency distributions of number of primary processes per cell treated with either AS1949490 (red squares) or vehicle (blue circles). The number of primary processes, which sprouted directly from the cell soma, was significantly reduced in the AS1949490 treatment group (AS1949490 treatment, 91 cells vs vehicle control, 122 cells; Mann–Whitney U test, p < 0.001). Three independent experiments were performed, with similar results. H, Withdrawal of AS1949490 from the culture medium restored the process number. After treatment with 10 μm AS1949490, as in A, the culture medium was replaced with conditioned medium with the vehicle control (green circles) or 10 μm AS1949490 (red squares), and the cells were cultured for another 4 h. Replacement with the AS1949490-free medium restored the phenotype (washout, 82 cells vs continued treatment, 75 cells; Mann–Whitney U test, p < 0.001). Two independent experiments were performed, with similar results. Scale bars, 10 μm.
To investigate the possible involvement of SHIP2 in the dynamic process movement of multipolar cells, we constructed a knockdown vector for SHIP2 (shSHIP2). The knockdown efficiency was confirmed using a Western blot analysis of COS-7 cells transfected with SHIP2–FLAG (Fig. 7C). We introduced an shSHIP2 vector together with an mCherry-expressing vector using in utero electroporation at E15.0, prepared the cells 24 h later, and cultured the cells for 24 h. The knockdown of SHIP2 resulted in a significantly reduced number of primary processes that were directly derived from the soma [control, mean 2.56 (121 cells) vs knockdown, mean 1.95 (87 cells) from two independent experiments; Mann–Whitney U test, p = 0.001]. Because the membrane localization of SHIP2 is reported to be important for its function (Ishihara, 2002), we performed rescue experiments using membrane-targeted SHIP2. As a result, the reduction was rescued using an expression vector for SHIP2 tagged with an Src myristoylation signal (myr–SHIP2/pCAGGS) [knockdown, mean 1.95 (87 cells) vs knockdown + myr–SHIP2/pCAGGS, mean 2.63 (147 cells) from two independent experiments; Mann–Whitney U test, p < 0.001] (Fig. 7D,E).
To investigate the role of the phosphatase activity of SHIP2 on the process formation of multipolar cells, we then used AS1949490, a specific competitive phosphatase inhibitor for SHIP2 (Suwa et al., 2009). We prepared a primary cortical culture, applied AS1949490 to the culture medium at 4 HIV (when most of the cells adhered to the coverslips with a round morphology), and fixed the cells at 12 HIV. AS1949490 treatment resulted in a significantly reduced number of primary processes [AS1949490 treatment, mean 1.04 (91 cells) vs vehicle control, mean 2.54 (122 cells) from three independent experiments; Mann–Whitney U test, p < 0.001] (Fig. 7F,G). To confirm this phenotype, the culture medium was replaced with inhibitor-free conditioned medium at 12 HIV, and the cells were cultured for another 4 h, resulting in a recovery in the number of processes [washout, mean 2.66 (82 cells) vs continued treatment, mean 1.09 (75 cells) from two independent experiments; Mann–Whitney U test, p < 0.001] (Fig. 7H). These results suggest that the phosphatase activity of SHIP2 is important for the process formation of multipolar cells, although other functions of SHIP2 may also be involved.
Because Lpd binds to PI(3,4)P2 with the PH domain, we next examined the importance of the PH domain to the process formation of multipolar cells. The knockdown phenotype of Lpd was, at least partially, rescued by the overexpression of human Lpd–EGFP [knockdown + a control plasmid, mean 2.30 (139 cells from 6 brains) vs knockdown + Lpd, mean 3.16 (232 cells from 7 brains); Mann–Whitney U test, p < 0.001] (Fig. 8A,B). However, the overexpression of a PH domain-deleted human Lpd–GFP [mean 1.98 (66 cells from 3 brains)] did not rescue the phenotype (Fig. 8A,B), supporting the hypothesis that Lpd regulates the process formation of multipolar cells in a PH-domain-dependent manner in vivo.
Figure 8.
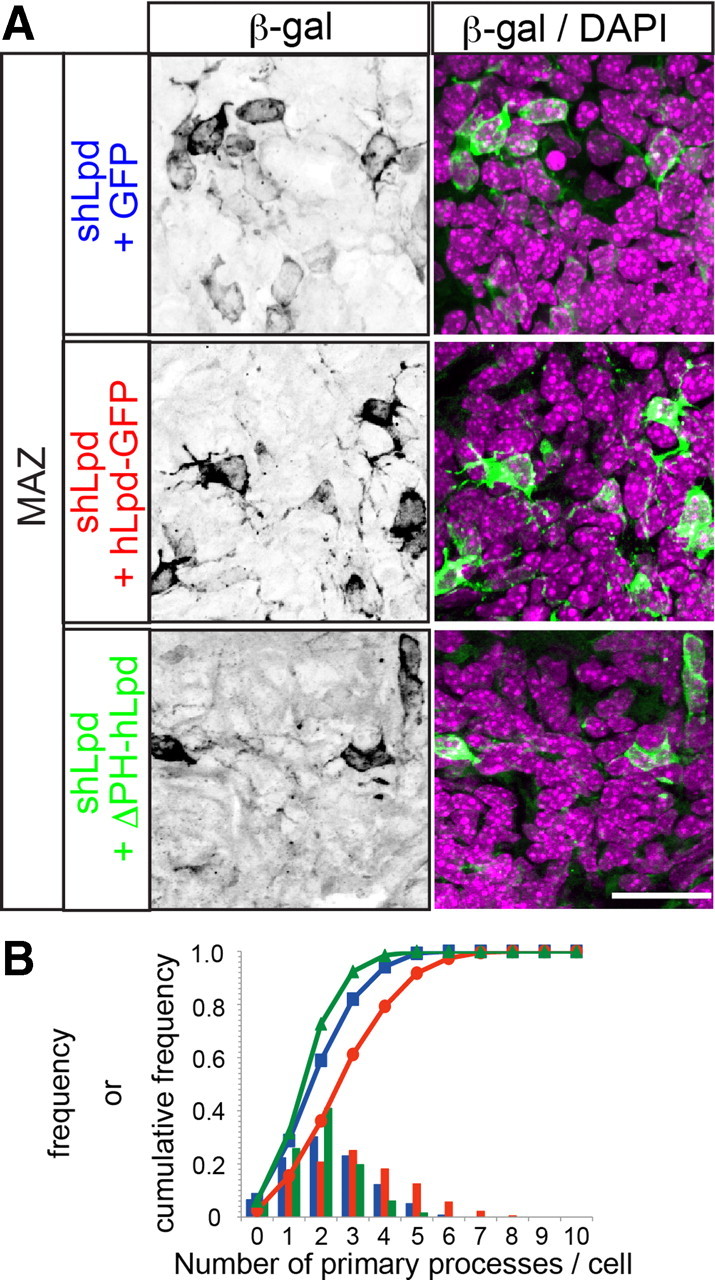
Knockdown phenotype of shLpd was not rescued by PH-domain-deleted Lpd. A, In utero electroporation of shLpd together with GFP, human Lpd–EGFP, or PH-domain-depleted human Lpd–EGFP was performed at E15.0, and the brains were fixed at E16.5. B, Frequency and cumulative frequency distributions of the number of primary processes per cell electroporated in utero with the indicated plasmids (shLpd + GFP, blue squares; shLpd + hLpd, red circles; shLpd + ΔPH–Lpd, green triangles). Overexpression of human Lpd rescued the phenotype that was characteristic of the suppression of Lpd (knockdown + a control plasmid, 139 cells from 6 brains vs knockdown + Lpd, 232 cells from 7 brains; Mann–Whitney U test, p < 0.001), whereas that of PH-depleted human Lpd did not (knockdown + a control plasmid, 139 cells from 6 brains vs knockdown + ΔPH Lpd, 66 cells from 3 brains; Mann–Whitney U test, p < 0.001). Scale bar, 20 μm.
Discussion
A considerable amount of research has been done on the mechanism responsible for the transformation from a multipolar migration mode to a locomotion mode, which is called a “point of vulnerability to disruption” (LoTurco and Bai, 2006). However, relatively few studies have examined the mechanisms of multipolar process movement. Here, we investigated the molecular mechanism of dynamic morphological changes of multipolar cells. We first observed the movement of the processes of multipolar cells at a high time resolution and found that they strikingly resembled the filopodia and lamellipodia in the growth cones of axons. We therefore focused on the actin remodeling proteins involved in growth cone dynamics. We found that the Lpd–Ena/VASP pathway positively regulates the number of primary processes of multipolar cells in the MAZ. We further observed that PI(3,4)P2, a phosphoinositide to which Lpd specifically binds, was localized in the moving processes of multipolar cells and that knockdown and an inhibitor of SHIP2, which produces PI(3,4)P2 from PI(3,4,5)P3, reduced the number of multipolar cell processes. These observations suggest that the SHIP2–PI(3,4)P2–Lpd–Ena/VASP pathway positively regulates the number of primary processes of multipolar cells.
Phosphoinositides are produced from phosphatidylinositol, a phospholipid in the plasma membrane, by phosphorylation at the 3, 4, and/or 5 positions of inositol. Depending on the phosphorylated sites, seven different phosphoinositides are currently known in mammalian cells, acting as second messengers for distinct functions. Therefore, their amounts are tightly regulated by specific kinases and phosphatases. Among them, PI(3,4,5)P3 is the most characterized phosphoinositide, recruiting enzymes such as PDK1 or PKB to regulate the proliferation and survival of cells. Hence, SHIP2, a 5-phosphatase that removes a phosphate from the 5 position of PI(3,4,5)P3 to produce PI(3,4)P2, has been studied as a negative regulator of PI(3,4,5)P3 (Aoki et al., 2007). However, it was shown that inositol polyphosphate 4-phosphatase-II, a 4-phosphatase that acts on PI(3,4)P2, is a tumor suppressor in epithelial carcinomas and is lost in some human basal-like breast cancers (Fedele et al., 2010). This illustrates the important role of PI(3,4)P2 as a second messenger that is regulated in a cell-context-dependent manner (Erneux et al., 2011). The importance of PI(3,4)P2 was also indicated by a mouse knock-in analysis showing that TAPP1 (tandem PH-domain-containing protein 1) and TAPP2 bind to PI(3,4)P2 and function as regulators of insulin and phosphatidylinositol 3-kinase (PI3K) signaling (Westerlund et al., 2011).
Previous reports have shown the roles of PI(3,4)P2 and Lpd in membrane dynamics. In HeLa cells, transfection of the catalytic domain-deleted SHIP2 and silencing of SHIP2 inhibited cell spreading and lamellipodia formation, respectively (Prasad et al., 2001; Prasad and Decker, 2005). In addition, Profilin1 inhibits MDA-MB-231 breast cancer cell motility by negatively regulating PI(3,4)P2 at the membrane, thereby limiting the recruitment of Lpd and Ena/VASP proteins to the leading edge (Bae et al., 2010). A genetic analysis of Caenorhabditis elegans suggested that age-1, a PI3K homolog, is one of the regulators of mig-10/Lpd in AVM axon guidance (Chang et al., 2006), probably because mig-10/Lpd specifically binds to PI(3,4)P2, and PI3K activity is indirectly required for PI(3,4)P2 production. In primary cortical neurons, Lpd promotes axonogenesis, but the relationship with PI(3,4)P2 has not been examined (Michael et al., 2010). Thus, the direct role of PI(3,4)P2 in the process formation of vertebrate neurons has yet to be explored.
Interestingly, PI(3,4,5)P3 elevation occurs locally in the leading edge of the growth cone and can serve as an early regulator during chemotactic guidance in Xenopus spinal neurons (Henle et al., 2011). In addition, the involvement of PI(3,4,5)P3 in neurite dynamics in cultured mouse hippocampal neurons was reported using PI3K inhibitors and Akt–PH–EGFP as a fluorescent probe (Ménager et al., 2004). Because the processes of multipolar cells are similar to these growth cones and neurites, these reports raise the possibility that the regulated conversion of PI(3,4,5)P3 to PI(3,4)P2 may extend the signaling network that originates from PI(3,4,5)P3 in the processes of multipolar cells.
It is generally accepted that growth cones probe for biochemical cues. Evidence obtained using primary cortical cultures suggests that Ena/VASP (Lebrand et al., 2004) and Lpd work downstream of Netrin-1 (Michael et al., 2010). The Netrin receptor Unc5d is expressed in multipolar cells (Sasaki et al., 2008). Whether the extent of the extension and retraction of the processes of multipolar cells may be modulated in response to ligands such as Netrins analogous to the growth cones of axons awaits additional investigation.
Ena/VASP proteins promote the assembly of long, sparsely branched actin filament networks, and the actin networks shaped by Ena/VASP give rise to filopodia or dynamic, protrusive lamellipodia (Dent et al., 2011), which were observed in the growth cone-like structures of multipolar cells in the present study. Growth cone motility is driven primarily by the cyclical polymerization and depolymerization of actin filaments, and the balance between the rate of polymerization and F-actin retrograde flow determines whether the growth cone extends or withdraws protrusions. Also, VASP is implicated in filopodial initiation (Svitkina et al., 2003), and most Ena/VASP-deficient neurons lack filopodia (Kwiatkowski et al., 2007). Thus, a reduced number of primary processes of multipolar cells is thought to reflect the withdrawal of protrusions (and thus growth cones and process shafts) and/or impaired process initiation.
Regarding the involvement of an actin cytoskeletal system in multipolar cells during cortical development, cells electroporated with an shRNA-based silencing vector for p27 or a dominant-negative form of Cdk5 exhibited a relatively round morphology with thin processes in the lower IZ, an MAZ equivalent, 3 d after electroporation (Kawauchi et al., 2006). The Cdk5–p27 pathway activates the actin-binding protein cofilin, which was also shown to be involved in cortical neuronal migration in vivo. Cofilin increases the rate constant for the dissociation of monomers from the pointed ends of actin filaments, leading to their depolymerization (Carlier et al., 1997), whereas Ena/VASP increases the filament length by interacting directly with the free barbed ends. Thus, although Kawauchi et al. (2006) and we observed a similar phenotype that could be explained by the disrupted assembly of the actin cytoskeleton, the underlying mechanism and point of action may differ.
Migrating neurons in the MAZ confront obstacles during their way to their final destination. In fact, just above the MAZ, there is a transverse layer of dense axon bundles, between which cells are sandwiched obliquely in alignment (Fig. 1A). A similar cell alignment was also found in the monkey inner fiber layer just above the inner SVZ (Smart et al., 2002), the putative primate counterpart of the MAZ (Tabata et al., 2012). This finding suggests that cells leave the MAZ when they successfully find their way among the thick axon bundles, in contrast to radial fiber-guided locomotion. Random migration with the extension and retraction of multiple processes would be a good strategy for finding a proper course among obstacles. In this study, multipolar cells expressing Lpd shRNA showed a reduced number of processes 1.5 d after electroporation. Pinheiro et al. (2011) demonstrated that, at 4 d after electroporation, cells expressing Lpd shRNA were mostly oriented tangentially in a bipolar morphology among and parallel to the axon bundles in the IZ–SVZ, whereas the control cells were oriented radially in the CP after passing the IZ successfully. They also showed that the migration speed of knockdown neurons that had successfully migrated using the locomotion mode in the CP was not affected, suggesting that the knockdown of Lpd mainly affected the migratory process during passage through the IZ. There is a possibility that cells expressing Lpd shRNA may become entangled in the obstacle-rich IZ–MAZ, probably because of an impaired multipolar stage as observed in our experiments.
In summary, we revealed that Lpd binds to PI(3,4)P2, which is produced by SHIP2, and recruits Ena/VASP, probably to the protruding membrane. This is one of the mechanistic bases of the dynamic morphology of multipolar cells. The suppression of this pathway may shed light on the biological significance of the unique dynamic movements of multipolar cells during cortical development.
Footnotes
This work was supported by the Strategic Research Program for Brain Sciences (“Understanding of Molecular and Environmental Bases for Brain Health”), the Grant-in-Aid for Scientific Research, the Global COE program of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Japan Society for the Promotion of Science, and Keio Gijuku Academic Development Funds. We thank F. B. Gurtler (Massachusetts Institute of Technology, Cambridge, MA) for Lpd–EGFP cDNA, J. Miyazaki (Osaka University, Osaka, Japan) for the CAG promoter, M. Matsuda (Kyoto University, Kyoto, Japan) for Pippi-(3,4)P2, T. Kawauchi (Keio University) for critical reading of this manuscript, and H. Arai for technical assistance and quantitation of processes in blind. We express our greatest gratitude to all the members of Nakajima laboratory for the valuable discussions, advice, expertise, and encouragement.
The authors declare no competing financial interests.
References
- Ajioka I, Nakajima K. Birth-date-dependent segregation of the mouse cerebral cortical neurons in reaggregation cultures. Eur J Neurosci. 2005;22:331–342. doi: 10.1111/j.1460-9568.2005.04214.x. [DOI] [PubMed] [Google Scholar]
- Aoki K, Nakamura T, Inoue T, Meyer T, Matsuda M. An essential role for the SHIP2-dependent negative feedback loop in neuritogenesis of nerve growth factor-stimulated PC12 cells. J Cell Biol. 2007;177:817–827. doi: 10.1083/jcb.200609017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YH, Ding Z, Das T, Wells A, Gertler F, Roy P. Profilin1 regulates PI(3,4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA-MB-231 cells. Proc Natl Acad Sci U S A. 2010;107:21547–21552. doi: 10.1073/pnas.1002309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Adler CE, Krause M, Clark SG, Gertler FB, Tessier-Lavigne M, Bargmann CI. MIG-10/lamellipodin and AGE-1/PI3K promote axon guidance and outgrowth in response to slit and netrin. Curr Biol. 2006;16:854–862. doi: 10.1016/j.cub.2006.03.083. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gupton SL, Gertler FB. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol. 2011;3:a001800. doi: 10.1101/cshperspect.a001800. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erneux C, Edimo WE, Deneubourg L, Pirson I. SHIP2 multiple functions: a balance between a negative control of PtdIns(3,4,5)P-3 level, a positive control of PtdIns(3,4)P-2 production, and intrinsic docking properties. J Cell Biochem. 2011;112:2203–2209. doi: 10.1002/jcb.23146. [DOI] [PubMed] [Google Scholar]
- Fedele CG, Ooms LM, Ho M, Vieusseux J, O'Toole SA, Millar EK, Lopez-Knowles E, Sriratana A, Gurung R, Baglietto L, Giles GG, Bailey CG, Rasko JE, Shields BJ, Price JT, Majerus PW, Sutherland RL, Tiganis T, McLean CA, Mitchell CA. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010;107:22231–22236. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh KL, Cai L, Cepko CL, Gertler FB. Ena/VASP proteins regulate cortical neuronal positioning. Curr Biol. 2002;12:565–569. doi: 10.1016/s0960-9822(02)00725-x. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Yamauchi K. Excitatory cortical neurons with multipolar shape establish neuronal polarity by forming a tangentially oriented axon in the intermediate zone. Cerebral Cortex. 2012 doi: 10.1093/cercor/bhr383. [DOI] [PubMed] [Google Scholar]
- Henle SJ, Wang G, Liang E, Wu M, Poo MM, Henley JR. Asymmetric PI(3,4,5)P3 and Akt signaling mediates chemotaxis of axonal growth cones. J Neurosci. 2011;31:7016–7027. doi: 10.1523/JNEUROSCI.0216-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Sasaoka T, Ishiki M, Wada T, Hori H, Kagawa S, Kobayashi M. Membrane localization of Src homology 2-containing inositol 5′-phosphatase 2 via Shc association is required for the negative regulation of insulin signaling in Rat1 fibroblasts overexpressing insulin receptors. Mol Endocrinol. 2002;16:2371–2381. doi: 10.1210/me.2002-0083. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol. 2006;8:17–26. doi: 10.1038/ncb1338. [DOI] [PubMed] [Google Scholar]
- Koresawa Y, Miyagawa S, Ikawa M, Matsunami K, Yamada M, Shirakura R, Okabe M. Synthesis of a new Cre recombinase gene based on optimal codon usage for mammalian systems. J Biochem. 2000;127:367–372. doi: 10.1093/oxfordjournals.jbchem.a022617. [DOI] [PubMed] [Google Scholar]
- Krause M, Leslie JD, Stewart M, Lafuente EM, Valderrama F, Jagannathan R, Strasser GA, Rubinson DA, Liu H, Way M, Yaffe MB, Boussiotis VA, Gertler FB. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev Cell. 2004;7:571–583. doi: 10.1016/j.devcel.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, Bronson RT, Mori S, Fässler R, Gertler FB. Ena/VASP is required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Dent EW, Strasser GA, Lanier LM, Krause M, Svitkina TM, Borisy GG, Gertler FB. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 2004;42:37–49. doi: 10.1016/s0896-6273(04)00108-4. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménager C, Arimura N, Fukata Y, Kaibuchi K. PIP3 is involved in neuronal polarization and axon formation. J Neurochem. 2004;89:109–118. doi: 10.1046/j.1471-4159.2004.02302.x. [DOI] [PubMed] [Google Scholar]
- Michael M, Vehlow A, Navarro C, Krause M. c-Abl, Lamellipodin, and Ena/VASP proteins cooperate in dorsal ruffling of fibroblasts and axonal morphogenesis. Curr Biol. 2010;20:783–791. doi: 10.1016/j.cub.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraille E, Dassesse D, Vanderwinden JM, Cremer H, Rogister B, Erneux C, Schiffmann SN. The SH2 domain-containing 5-phosphatase SHIP2 is expressed in the germinal layers of embryo and adult mouse brain: Increased expression in N-CAM-deficient mice. Neuroscience. 2001;105:1019–1030. doi: 10.1016/s0306-4522(01)00240-8. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci U S A. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Pinheiro EM, Xie Z, Norovich AL, Vidaki M, Tsai LH, Gertler FB. Lpd depletion reveals that SRF specifies radial versus tangential migration of pyramidal neurons. Nat Cell Biol. 2011;13:989–995. doi: 10.1038/ncb2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad NK, Decker SJ. SH2-containing 5′-inositol phosphatase, SHIP2, regulates cytoskeleton organization and ligand-dependent down-regulation of the epidermal growth factor receptor. J Biol Chem. 2005;280:13129–13136. doi: 10.1074/jbc.M410289200. [DOI] [PubMed] [Google Scholar]
- Prasad N, Topping RS, Decker SJ. SH2-containing inositol 5′-phosphatase SHIP2 associates with the p130(Cas) adapter protein and regulates cellular adhesion and spreading. Mol Cell Biol. 2001;21:1416–1428. doi: 10.1128/MCB.21.4.1416-1428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Tabata H, Tachikawa K, Nakajima K. The cortical subventricular zone-specific molecule Svet1 is part of the nuclear RNA coded by the putative Netrin receptor gene Unc5d and is expressed in multipolar migrating cells. Mol Cell Neurosci. 2008;38:474–483. doi: 10.1016/j.mcn.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Smart IH, Dehay C, Giroud P, Berland M, Kennedy H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex. 2002;12:37–53. doi: 10.1093/cercor/12.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa A, Yamamoto T, Sawada A, Minoura K, Hosogai N, Tahara A, Kurama T, Shimokawa T, Aramori I. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br J Pharmacol. 2009;158:879–887. doi: 10.1111/j.1476-5381.2009.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Kanatani S, Nakajima K. Differences of migratory behavior between direct progeny of apical progenitors and basal progenitors in the developing cerebral cortex. Cereb Cortex. 2009;19:2092–2105. doi: 10.1093/cercor/bhn227. [DOI] [PubMed] [Google Scholar]
- Tabata H, Yoshinaga S, Nakajima K. Cytoarchitecture of mouse and human subventricular zone in developing cerebral neocortex. Exp Brain Res. 2012;216:161–168. doi: 10.1007/s00221-011-2933-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund N, Zdrojewska J, Padzik A, Komulainen E, Björkblom B, Rannikko E, Tararuk T, Garcia-Frigola C, Sandholm J, Nguyen L, Kallunki T, Courtney MJ, Coffey ET. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat Neurosci. 2011;14:305–313. doi: 10.1038/nn.2755. [DOI] [PubMed] [Google Scholar]
- Yoshizaki H, Mochizuki N, Gotoh Y, Matsuda M. Akt-PDK1 complex mediates epidermal growth factor-induced membrane protrusion through Ral activation. Mol Biol Cell. 2007;18:119–128. doi: 10.1091/mbc.E06-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]