

Abstract
Microglia are known to invade the mammalian spinal cord (SC) at an early embryonic stage. While the mechanisms underlying this early colonization of the nervous system are still unknown, we recently found that it is associated, at least partially, with the ability of microglia to proliferate at the onset of motoneuron developmental cell death and of synaptogenesis in mouse embryo (E13.5). In vitro studies have shown that the proliferation and activation of adult microglia can be influenced by the purinergic ionotropic receptor P2X7 via a coupling with Pannexin-1. By performing patch-clamp recordings in situ using a whole-mouse embryonic SC preparation, we show here that embryonic microglia already express functional P2X7R. P2X7R activation evoked a biphasic current in embryonic microglia, which is supposed to reflect large plasma membrane pore opening. However, although embryonic microglia express pannexin-1, this biphasic current was still recorded in microglia of pannexin-1 knock-out embryos, indicating that it rather reflected P2X7R intrinsic pore dilatation. More important, we found that proliferation of embryonic SC microglia, but not their activation state, depends almost entirely on P2X7R by comparing wild-type and P2X7R−/− embryos. Absence of P2X7R led also to a decrease in microglia density. Pannexin-1−/− embryos did not exhibit any difference in microglial proliferation, showing that the control of embryonic microglial proliferation by P2X7R does not depend on pannexin-1 expression. These results reveal a developmental role of P2X7R by controlling embryonic SC microglia proliferation at a critical developmental state in the SC of mouse embryos.
Introduction
Microglia are the resident immune cells of the CNS, which begin to colonize at early embryonic developmental stages (Marin-Teva et al., 1998; Dalmau et al., 2003; Billiards et al., 2006; Rigato et al., 2011). In the mouse embryo, the colonization of the spinal cord (SC) by microglia involves several processes, including microglia proliferation at the periphery of the parenchyma (Rigato et al., 2011). Remarkably, microglia accumulate in the motoneuron (MN) area at 13.5 d of embryonic age (E13.5), a time at which they reach an activated state, proliferate, and phagocyte dying MNs (Rigato et al., 2011). However, the molecular mechanisms underlying microglia proliferation during SC invasion in the embryo remain unknown. In addition it is unclear whether the proliferation and the activation state of microglia are controlled by the same signals and mechanisms at this early developmental stage. In the adult, in vitro studies have shown that a number of factors, among which purinergic receptors, such as P2X7R (Monif et al., 2009), evoke both microglia activation and proliferation (Kettenmann et al., 2011). While P2X7R, in addition to P2X4R and P2X1R, is also expressed by embryonic microglia (Xiang and Burnstock, 2005), its functions remain unknown at these developmental stages. In the adult, P2X7R is involved, in addition to microglia activation and proliferation, in a variety of important effector functions of microglia (Kettenmann et al., 2011), including the release of interleukin (IL)-1β, IL-1α, and TNF-α (Ferrari et al., 1997; Hide et al., 2000; Brough et al., 2002; Mingam et al., 2008). A remarkable feature of P2X7R is that it promotes the formation of a large pore permeable to hydrophilic moieties up to 900 Da (Surprenant et al., 1996), which has been proposed to evoke IL-1β release, to mediate P2X7R effects on the control of microglia activation and proliferation and to mediate the detrimental effect of continuous P2X7R activation (Surprenant et al., 1996; Di Virgilio et al., 1998; Le Feuvre et al., 2002; Pelegrin and Surprenant, 2006). Initially, it was proposed that dilatation of the integral pore of the P2X7R channel accounts for this large pore formation (Surprenant et al., 1996), but, later, experimental evidence indicated that large molecule uptake can occur through the formation of an independent plasma membrane pore (Jiang et al., 2005), which might be related to a functional coupling of P2X7R to the hemichannel pannexin-1 (Panx1) (Pelegrin and Surprenant, 2006).
Using multiple approaches, we determined to which extent P2X7R influences embryonic microglia proliferation during their colonization of the embryonic SC at E13.5 (Rigato et al., 2011) and whether this needs an association to Panx1. We show that, already at E13.5, microglia express P2X7R and that P2X7R activation evokes a biphasic current, even in Panx1 knock-out (KO) (−/−) mice. Microglia proliferation is strongly reduced in P2X7R−/− mice, but not in Panx1−/− mice. Our results provide direct evidence that P2X7Rs strictly control embryonic microglia proliferation independently of their activation state and without involving a coupling to Panx1. Our study discloses the function of P2X7R during embryonic SC colonization by microglia.
Materials and Methods
Animals.
The experiments were performed in accordance with the European Community guiding principles on the care and use of animals (86/609/CEE; Official Journal of the European Communities no. L358, December 18, 1986), French Decree no. 97/748 of October 19, 1987 (Journal Officiel de la République Française, October 20, 1987), and the recommendations of the Centre National de la Recherche Scientifique (CNRS). We used transgenic CX3CR1eGFP mice from CNRS/CDTA. P2X7R−/− mice were derived from Pfizer and from GlaxoSmithKline. Given the questionable specificity of the available P2X7R antibodies (Sim et al., 2004), we performed β-galactosidase immunostainings on the SC of GlaxoSmithKline P2X7R−/− mice as an indirect assay for P2X7R protein expression and cellular localization. Panx1−/− C57B1/6N mice were from the Knockout Mouse Project (KOMP). Panx1 immunostaining was no longer observed in Panx1−/− C57B1/6N mice (see Fig. 7B). Mice were coupled overnight and females were examined for vaginal plugs in the morning. Mice in which a vaginal plug was observed, were considered as E 0.5. The developmental stage of the embryos was confirmed using the e-Mouse Atlas from EMAP (http://www.emouseatlas.org/emap/ema/home.html). At the intended embryonic age (E13.5), SCs of mouse embryos were isolated as previously described (Rigato et al., 2011). Briefly, pregnant mice were anesthetized by intramuscular injection of a mix of ketamine and xylazine. Pregnant mice were then killed using CO2 and the embryos were removed immediately and the SC was isolated. Isolated SCs were then maintained for 1 h before recording in an artificial CSF (ACSF) containing (in mm): 130 NaCl, 25 NaHCO3, 1 NaH2PO4, 4.5 KCl, 10 glucose, 2 CaCl2, and 1 MgCl2, at 32°C, continuously bubbled with a 95% O2-5% CO2 gas mixture.
Figure 7.

Panx1 immunostaining was not observed in the SC of Panx1−/− mouse embryos. A, Confocal image of immunostaining against Panx1 (red; A2, A3, A5, and A6) in the ventral SC of CX3CR1eGFP mouse embryo. A3, B3, Superposition of IBA1 and Panx1 immunostainings. A4, A6, Single confocal sections showing microglia located in the ventrolateral area of the SC. A2, A5, Panx1 immunostaining in the ventral SC of CX3CR1eGFP mouse embryo. A3, Superimposed images (A1 and A2) showing Panx1 staining in microglia. A6, Superimposed images showing Panx1 staining in microglia localized in the ventrolateral region. B, Confocal image of immunostaining against Panx1 (red; B2, B3, B5. and B6) in the ventral SC of Panx1−/− mouse embryo. B1, B4, Microglia were stained using IBA1 antibody. B1, B2, B3, Panx1 immunostaining in the SC of E13.5 Panx1−/− mouse embryos. B1, B3, B4, B6, Microglia were revealed using IBA1 antibody (green). Note that Panx1 immunostaining was no longer observed in the SC of E13.5 Panx1−/− mouse, indicating that this antibody specifically recognizes Panx1. A1, A3, A4, A6, B1, B3, B4, B6, Cell nuclei are visualized with DAPI staining.
Pharmacological experiments with P2XR agonists and antagonists and hemichannel blockers were performed on transgenic C57BL/6 CX3CR1eGFP mice (Jung et al., 2000; Rigato et al., 2011) to record microglia in the SC of wild-type mouse embryos. CX3CR1eGFP mouse embryos were also used to analyze the passive electrophysiological properties of microglia in the SC of wild-type mouse embryos. Homozygous C57BL/6 CX3CR1eGFP mice were obtained from the CNRS/CDTA via the European Mouse Mutant Archive. The CX3CR1eGFP−/+ embryos used throughout this study were obtained by crossing CX3CR1eGFP+/+ male mice with C57BL/6 wild-type female mice. Two lines of transgenic C57BL/6 P2X7R KO mice were also used: P2X7R−/− mice derived from Pfizer and from GlaxoSmithKline (Sim et al., 2004). P2X7R−/− mouse lines were kindly provided by E. Audinat (INSERM U603, CNRS UMR 8154, Université Paris Descartes, France) and by Micaela Galante (IBBMC, CNRS UMR 8619, France). Pfizer mice were preferentially used instead of GlaxoSmithKline mice for patch-clamp recordings and for studying microglia cell activation and proliferation, since a P2X7R variant can still be expressed in GlaxoSmithKline homozygous P2X7R−/− mice (Nicke et al., 2009). P2X7R+/− mice were obtained by crossing C57BL/6 P2X7R−/− Pfizer mice with C57BL/6+/+ mice. Briefly, Pfizer mice were constructed by a deletion in the region containing amino acids 506–532, followed by insertion of a neomycin cassette in a 3′ to 5′ direction. The GlaxoSmithKline P2X7R−/− mice have a lacZ gene inserted at the beginning of the exon 1 region of the P2X7R gene. We also used Pannexin-1-targeted homozygous C57BL/6N mice from KOMP (Guan et al., 2010). Disruption of Panx1 was generated by insertion of a trapping cassette splice acceptor-β-geo-polyA flanked by Flp-recombinase target sites within intron 2 upstream of the critical exon 3, where it tags the gene with the lacZ reporter. This creates a constitutive null mutation in the target gene through efficient splicing to the reporter cassette, resulting in truncation of the endogenous transcript (pannexin1tm1a(KOMP)Wtsi). Reverse transcription-PCR (RT-PCR) of mouse ear punches and Western blot analysis of the brain confirmed the absence of Panx1 expression in this Panx1−/− mouse (Seminario-Vidal et al., 2011). Our immunohistochemical experiments showed a lack of Panx1 expression in the SC of Panx1−/− mouse embryos (Fig. 7B). Primers used for PCR analysis for genotyping P2X7R−/− and Panx1−/− mice are listed in Table 1. PCR results for Panx1−/− and P2X7R −/− mice are shown in Figures 6A and 8A, respectively.
Table 1.
Primers used for PCR analysis for genotyping P2X7 and Panx1 KO mice
| Primer | Sequence |
|---|---|
| Primers P2X7 | |
| P2X7 wt-F | 5′-GCAGCCCAGCCCTGATACAGACATT-3′ |
| P2X7 wt-R | 5′-TCGGGACAGCACGAGCTTATGGA-3′ |
| NEO-F | 5′-GCAGGATCTCCTGTCATCTCACC-3′ |
| NEO-R | 5′-GATGCTCTTCGTCCAGATCATCC-3′ |
| Primers Panx1 | |
| CSD-comLoxP-F | 5′-GAGATGGCGCAACGCAATTAAT-3′ |
| CSD-panx1-R | 5′-CTGGCTCTCATAATTCTTGCCCTG-3′ |
| CSD-panx1-wtF | 5′-CTGTATCACACAACCACTTCAGAGAAGG-3′ |
| CSD-panx1-wtR | 5′-GAGCTGACCCCTTTCCATTCAATAG-3′ |
Figure 6.

ATP-evoked biphasic currents were not observed in SC microglia of P2X7R−/− mouse embryos. A, PCR results for wild-type (+/+) mice, heterozygote mice (+/−), and P2X7R KO mice (−/−). B, Voltage-current relationship of microglia recorded in the SC of E13.5 P2X7R−/− mouse embryos. A voltage ramp protocol (−100 mV +100 mV; 2 s) was used to investigate voltage-activated currents. C1, Application of 3 mm ATP on microglia recorded in the SC of P2X7R −/− mouse E13.5 embryo failed to evoked large biphasic inward currents. C2, Examples of currents evoked by 3 mm ATP on microglia of a P2X7R +/− mouse E13.5 embryo. C3, Histogram showing differences in current density of 3 mm ATP-evoked responses in SC microglia of P2X7R −/− (0.93 ± 0.43 pA/pF; N = 5) and P2X7R +/− (16.5 ± 2.12 pA/pF; N = 7) mouse embryos. (Vh = −60 mV). Input capacitance of recorded microglia in P2X7R−/− embryos (11.9 ± 1.4 pF; N = 5) and in P2X7R +/− embryos (10.3 ± 1 pF; N = 7) were not significantly different (p > 0.1). Error bars indicate SEM.
Figure 8.

ATP-evoked biphasic currents did not result from Panx1 activation. A, PCR results for wild-type (+/+) mice, heterozygote mice (+/−), and Panx1 KO mice (−/−). B, Voltage-current relationship of currents recorded in microglia obtained from the SC of E13.5 Panx1−/− mouse embryos. A voltage ramp protocol (−100 mV +100 mV; 2 s) was used to investigate voltage-activated currents. C1, C2, Effect of high concentrations of the hemichannel blockers DIDS and CBX on ATP-evoked currents (3 mm ATP; Vh = −60 mV) in CXCR1eGFP mice. C3, 100 μm DIDS significantly (p < 0.01) inhibits 28.3 ± 2.9% (N = 11) of ATP responses while 100 μm CBX had no significant effect (p > 0.1) (9.9 ± 6.6% increase; N = 7). The percentage change in response amplitude observed during the application of the blockers was compensated according to the averaged decrease (rundown: 6.8%) in the corresponding responses observed on different cells in the absence of the blockers (Normal [Ca2+]o and [Mg2+]o ACSF: see Materials and Methods and Results). Error bars indicate SEM. D, Currents evoked by 3 mm ATP on microglia recorded from the SC of E13.5 Panx1 −/− mouse embryos. Note that ATP application evoked a biphasic current as observed in CX3CR1eGFP and in P2X7+/− mouse embryos (Figs. 3, 6).
Whole-cell recordings and analysis.
The isolated SC was placed in a recording chamber and continuously perfused at room temperature (20−24°C) with oxygenated ACSF (2 ml/min). Standard whole-cell patch-clamp and voltage-clamp recordings of fluorescent microglia were made under direct visualization using an infrared-sensitive CCD video camera. In CX3CR1eGFP mice embryo, all eGFP cells localized in the SC are microglial cells (Rigato et al., 2011). We recorded microglia localized in the ventrolateral region of the spinal since at E13.5. At this age activated microglial cells (Mac-2 staining) accumulated in the ventrolateral region close to motor columns where they proliferated at the onset of the developmental cell death of MNs (Rigato et al., 2011). Recordings of microglia in the ventrolateral part of the SC fiber zone of Pfizer P2X7R−/−, of Pfizer P2X7R+/−, and of Panx1−/− mouse embryos were performed using infrared microscopy. Microglia in this SC area can be easily recognized by their morphology. Their electrophysiological profile was identical to those of CX3CR1eGFP microglia (Figs. 2, 6B, 8B).
Figure 2.

Voltage-activated current profile in microglia of the developing SC. Voltage-activated current was analyzed using voltage ramps ranging from −100 to +100 mV over 2 s. Two different voltage-activated current patterns could be distinguished: an IrC, observed at membrane potentials lower than ≈−70 mV (A), and an OrC (B). C, The absence or the presence of IrC was significantly correlated (p < 0.05) to the morphology of the recorded microglia (Fisher's exact test, p < 0.05). Confocal images (Z-stack) showing amoeboid microglia (left) and branched microglia (right) in the ventral SC area of CXCR1 EGFP mice. D, The gap junction/hemichannel blocker CBX (100 μm) had no effect on the OrC (p > 0.1). This current was 8.4 ± 4.8% inhibited in the presence of 100 μm CBX (N = 8). CTR, Control.
Whole-cell patch-clamp electrodes were pulled from thick-wall borosilicate glass using a Brown-Flaming puller (Sutter Instrument). The tip of the electrode was fire polished using a microforge (Narishige). Patch-clamp electrodes had resistances of 5–7 MΩ. For voltage-clamp experiments, the electrode was filled with a solution containing (in mm): 130 KCl, 4 MgCl2, 4 Na2ATP, 10 EGTA, and 10 HEPES (pH adjusted to 7.2 with KOH, osmolarity 290 mosmol/kg-H2O, equilibrium potential for chloride ions ECl ≈ 0 mV). In some experiments with low [Cl−]i, the electrode contained (in mm): 113 potassium methyl sulfate, 17 KCl, 4 MgCl2, 4 Na2ATP 4, 10 HEPES, and 1 EGTA (pH adjusted to 7.2 with KOH, osmolarity 290 mosmol/kg-H2O, equilibrium potential for chloride ions ECl ≈ −40 mV).
Current fluctuations were recorded and lowpass filtered (2 kHz) using an Axopatch 1D amplifier, digitized (20 kHz) on-line using a Digidata 1322A interface card and analyzed off-line with the PClamp10 software package or the Axograph X.1.3.5 software (Axograph).
Because embryonic microglia are rapidly damaged by voltage steps, voltage-dependent currents were analyzed using 2 s voltage ramps from −100 mV to +100 mV from a holding potential of −60 mV. An initial voltage step (100 ms) to −100 mV was applied before the onset of the ramp. To control the stability of the recordings, three ramps were applied at 2 s intervals. The resulting evoked currents were then averaged for analysis using the Axograph X software. In the large majority of the recordings, outward voltage-gated currents evoked by voltage ramps slowly increased with time (runup), reaching a steady state and then decreasing (rundown) as previously described in cultures of murine microglia (Eder et al., 1998). The steady-state period lasted for 10–15 min, being the period during which the pharmacological experiments were performed. To estimate the stability of the recordings, voltage ramp-evoked currents in the presence of carbenoxolone (CBX) were compared with currents evoked by voltage ramps applied 1 min before and 3 min after application of the drug. A set of three ramps (2 s time interval) was applied before, during, and after drug applications. The resulting evoked currents for each voltage ramp set were then averaged for analysis. Recordings without evidence of recovery after drug treatment were discarded.
To analyze concentration–response relationships in these conditions, different ATP concentrations were applied alternatively with 1 mm ATP to control changes in response amplitudes due to rundown (see below). The responses evoked by different ATP concentrations were compared with the responses evoked by 1 mm ATP applied before and after each test application. The amplitude of the test response was normalized to the value obtained by the sum of the amplitudes of the response evoked by 1 mm ATP before and after the test response divided by two. Normalized concentration–response curves were fitted using the Hill equation I/Imax = 1/(1 + (EC50/[ATP])∧nH), where I/Imax is the normalized response amplitude, EC50 is the ATP concentration producing half of the maximal response, and nH is the Hill coefficient. For each concentration tested, the amplitude of the current, I, was measured at the peak of the response.
For pharmacological experiments with P2XR antagonists, P2XR activation was evoked through application of 500 μm ATP in [Ca2+]o and [Mg2+]o free ACSF (Vh = −60 mV). All experiments were performed using the first three responses elicited by ATP after inducing the enlarged inward (biphasic) current (see Fig. 3C). The first response was used as the control, the second response was the test response, and the third response corresponded to the recovery of antagonist effects. Application of the antagonist started between the control and the test responses and ended between the test response and the third ATP application (recovery) (3 min interval between ATP applications). To determine antagonist efficiency, the amplitude of the test response was compared with the amplitude of the control response (100%). Current amplitude was measured at the peak of the responses. Recovery from antagonist block was evaluated by comparing the amplitude of the third response to the amplitude of the test response. However, when microglia were exposed to successive ATP applications after inducing a biphasic inward current, the amplitude of the following responses showed a progressive decline with time, the extent of which was similar in all microglial cells (see Results). Accordingly, such a rundown can give rise to an overestimation of the efficiency of the antagonist tested. To overcome this problem, the percentage change in response amplitude observed during the application of the antagonist was compensated according to the averaged decrease in the corresponding responses observed on different cells in the absence of the antagonist in [Ca2+]o- and [Mg2+]o-free ACSF (13%; see Results).
Figure 3.

ATP evokes inward currents in embryonic microglia. A, Example of responses evoked by 0.5, 1, and 3 mm ATP in the presence of 1.3 mm [Ca2+]o and 3 mm [Mg2+]o. Note the biphasic activation phase (arrow) of the response evoked by 3 mm ATP. B, Currents evoked by 0.05, 0.1, and 0.3 mm ATP in [Ca2+ + Mg2+]o = 0. C, Currents evoked by repetitive applications of 0.5 mm ATP in [Ca2+ + Mg2+]o = 0. D, Current evoked by long application of 3 mm ATP in the presence (D1) and of 0.5 mm ATP in the absence (D2) of [Ca2+ + Mg2+]o. This response slowly desensitized (half-amplitude decay time = 26.9 s in the presence of [Ca2+ + Mg2+]o and 41.9 s in free [Ca2+ + Mg2+]o solution). Large inward currents evoked by 140 s application of ATP after eliciting a biphasic response slowly decreased with time in both normal [Ca2+, Mg2+]out (3 mm ATP) and free [Ca2+, Mg2+]out (0.5 mm ATP) solutions and reached a plateau representing 50.5 ± 3.1% (N = 5) of the peak current in normal [Ca2+, Mg2+]out solution and 64.7 ± 5.2% (N = 8) in free [Ca2+, Mg2+]out. The half-desensitization time was 25.9 ± 3.3 s (N = 5) in normal [Ca2+, Mg2+]out and 36.9 ± 6.5 s (N = 8) in free [Ca2+, Mg2+]out. E1, Example of currents evoked by increasing ATP concentrations after having obtained a biphasic response ([Ca2+ + Mg2+]o = 0). E2, Dose–response curve of ATP-evoked currents as shown in E1 (EC50 = 467 μm; Hill coefficient = 2.6). Each point represents the average of 5–10 measurements. The amplitude of the ATP-evoked responses has been normalized to the current amplitude of the responses evoked by 1 mm ATP (see Materials and Methods). (Vh =−60 mV). Error bars indicate SEM.
Drug application and pharmacological agents.
All drugs were applied using a 0.5 mm diameter quartz tubing positioned 1 mm away from the recording area (Scain et al., 2010). The quartz tubing was connected using a manifold to reservoirs filled with a control solution or with P2X receptor agonists and/or channel blockers or P2X receptor antagonists.
Adenosine triphosphate disodium salt (Na2ATP), 2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate triethylammonium salt (bzATP), α,β-methylene ATP (α,β-Me-ATP), Brilliant Blue G (BBG), 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt hydrate (DIDS), and the hemichannel blocker CBX (Gomes et al., 2005; Ma et al., 2009) were purchased from Sigma-Aldrich. CuCl2 and ZnCl2 (0.1 m stock solution) were also purchased from Sigma-Aldrich. 2′,3′-O-(2,4,6-Trinitrophenyl)adenosine-5′-triphosphate tetra(triethylammonium) (TNP-ATP), suramin, pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS), and 3-[[5-(2,3-dichlorophenyl)-1H-tetrazol-1-yl]methyl]pyridine hydrochloride (A438079), N-[1-[[(cyanoamino)(5-quinolinylamino)methylene]amino]-2,2-dimethylpropyl]-3,4-dimethoxybenzeneacetamide (A740003) were all purchased from Tocris Bioscience. Na2ATP, bzATP, α,β-Me-ATP, BBG, CuCl2, ZnCl2, TNP-ATP, suramin, PPADS, A438079, and A740003 were dissolved in the bathing solution. DIDS and CBX were dissolved in dimethylsulfoxide (0.1% final concentration). ATP solutions were prepared the day of the experiment.
Immunohistochemistry and confocal microscopy.
All primary antibodies used and their dilutions are listed in Table 2. To visualize P2X4R, we used rabbit polyclonal P2X4R antibodies (kindly provided by Francois Rassendren; CNRS UMR 5203/INSERM U661, Montpellier, France). P2X4R staining was abolished in P2X4R−/− mice indicating that this antibody specifically recognizes P2X4R (Sim et al., 2006). To perform multiple staining in the SC of mouse embryos other than CXCR1eGFP mouse embryos, microglia were marked using anti-IBA1 (Wako), anti-CD11b (Serotec), or anti-CD68 (Serotec) antibodies (Rigato et al., 2011). Activated caspase-3 Asp175 antibody (Cell Signaling Technology) was used to detect apoptotic cells. The Asp175 antibody recognizes two bands of 17 and 19 kDa on Western blot corresponding to large fragments of activated caspase-3. This antibody does not recognize full-length caspase-3 or other cleaved caspases (Cheong et al., 2003). We used a purified mouse anti-human Ki-67 antibody to determine the proliferation state of microglial cells (BD Pharmingen) (Charrier et al., 2006). To determine to what extent microglial cells can be activated, we used a monoclonal antibody directed against Mac-2 (Avignone et al., 2008). This antibody (hybridomas TIB-166; American Type Culture Collection-LGC Standards) was originally used to isolate the protein Galectin3/Mac-2 (Ho and Springer, 1982). In GlaxoSmithKline P2X7R+/− mice, we used anti-β-galactosidase antibody (MP Biomedical) as an indirect probe to visualize cells expressing P2X7Rs. This antibody was produced by hyperimmunizing rabbits with the enzyme β-galactosidase from Escherichia coli. To detect Panx1 in microglia, we used a polyclonal chicken anti-mouse Panx1 antibody from Diatheva (ANT0027) (Huang et al., 2007). Panx1 staining was no longer observed in Panx1 KO mice (Fig. 7B).
Table 2.
Primary antibodies
| Primary antibody | Company | Reference | Host/isotype | Dilution |
|---|---|---|---|---|
| Anti-KI67 | BD Pharmingen | 556003, clone B56 | Monoclonal mouse IgG1 | 1:200 |
| Anti-caspase3 | Cell Signaling Technology | 9661 | Rabbit polyclonal | 1:500 |
| Anti-pannexin-1 | Diatheva | ANT0027 | Chicken polyclonal | 1:100 |
| Anit-P2X4 | Provided by F. Rassendren, INSERM U661, Montpellier, France | Sim et al. (2006) | Rabbit polyclonal | 1:500 |
| Anti-Mac-2 | American Type Culture Collection | Clone M3/38.10.2.8 HL0.2 | Monoclonal rat IgG2a | 1:1000 |
| Anti-CD68 | Serotec | MCA1957, clone FA-11 | Monoclonal rat IgG2a | 1:400 |
| Anti-IBA1 | Wako | 019-19741 | Rabbit polyclonal | 1:500 |
| Anti-CD11b | Serotec | MCA74GA, clone M1/70.15 | Monoclonal rat IgG2b | 1:400 |
| Anti-β-galactosidase | MP Biomedicals | 55976 | Rabbit polyclonal | 1:500 |
Whole embryonic SCs were fixed in 4% paraformaldehyde for 1 h at 4°C and subsequently incubated in 30% sucrose overnight at 4°C for cryoprotection. Sucrose was dissolved in phosphate buffer. After overnight incubation, the tissue was embedded in optimal cutting temperature compound (VWR) and quickly frozen to −50°C in isopentane (Sigma-Aldrich). The embedded tissue was then stored at −80°C. Fifty micrometer transverse cryosections were made using a Leica Cryostat (Leica Microsystems). Tissue sections were washed three times in PBS for 5 min, in PBS-NH4Cl for 20 min, and again three times in PBS for 5 min. SC sections were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) and 0.25% fish gelatin (PBS-G-T) for 1 h at room temperature, before incubation with primary antibodies (18–20 h) at 4°C. After three washes in PBS-G-T for 5 min, sections were incubated with the secondary antibodies and with DAPI nuclear stain (Sigma-Aldrich, 1/1000) for 2 h at room temperature. Sections were washed three times in PBS for 5 min and rinsed in distilled water before addition of Mowiol 4–88 (475904; Calbiochem). Confocal images were taken on a Leica SP5 platform (Leica Microsystems). Preparations were analyzed using a Leica SP5 confocal microscope. For quantitative analysis of microglial cells on isolated SC or in total embryo transverse sections, serial optical sections were obtained with a Z-step of l μm and observed using a 20× oil-immersion objective with a numerical aperture (NA) of 0.7. A Z-step of 0.3 μm was performed when using a 63× oil-immersion objective (NA 1.32), plus a 3× digital zoom magnification. Images (1024 × 1024; 8-bit grayscale) were stored using Leica software LAS-AF. Images were analyzed using ImageJ 1.41 (National Institutes of Health [NIH]; http//rsb.info.nih.gov/ij/) and Photoshop CS3 (Adobe Software). Quantification was performed on 50-μm-thick transverse sections (N = 10 slices per SC) of the embryonic lumbar SC (N = 3–5 SCs). Only eGFP cell bodies were taken into account. Microglial cells immunoreactive (ir) to specific antibodies were counted manually in 10 optical sections per embryo. The threshold used to determine whether a microglial cell was coimmunolabeled was set at twice the intensity of background noise measured between eGFP-positive cells.
As cleaved caspase-3 is not only detectable in the cytoplasm of dying cells but also in apoptotic bodies, as a result of cell dismantling, it was not possible to quantify the exact number of cells that were undergoing apoptosis. For this reason, we performed a statistical analysis by measuring the area fraction of anti-caspase-3 fluorescence, setting the ventral region area as constant at all SC analyzed (347 × 260 μm). Thirty micrometer stack images for each SC slice were made using ImageJ 1.41 (NIH; http//rsb.info.nih.gov/ij/), and then transformed into binary images on a black background, by adjusting the threshold. If necessary, the option fill holes was selected and the area fraction checkbox was checked. The percentage of white area, corresponding to anti-caspase-3 fluorescence, was calculated, permitting comparisons of area fraction of anti-caspase-3 fluorescence between wild-type and mutant P2X7R SCs. Background noise for activated caspase-3-ir was measured around the central canal.
Microglia isolation.
Embryonic SCs were isolated and transferred into 2 ml Neurobasal media (Invitrogen), supplemented with N2 (100×), B27 (50×), l-glutamine (100×), and penicillin/streptomycin (200×), containing papain at a concentration of 48 U/ml (Sigma; 31 U/mg protein). After being cut into small pieces, they were incubated at 37°C for 30 min. Afterward, the tissue was allowed to settle for 5 min, the media was removed, and 1 ml of media was added. By making use of a 1 ml pipette, the tissue sections were quickly triturated 10 times. The larger pieces were allowed to settle for 2 min. Afterward, the supernatant was transferred to a fresh Falcon tube. These steps were repeated twice and the supernatants were combined. The obtained cell suspension was centrifuged at 400 g for 5 min at room temperature. The supernatant was discarded at the pellet resuspended in 1 ml fresh media. Large pieces were filtered away using a 70 μm cell strainer (BD Falcon-BD Biosciences). The cell strainer was prewet with 1 ml medium; afterward another 2 ml of medium was used to rinse the cell strainer. This cell suspension was centrifuged at 350 × g for 10 min at room temperature, supernatant was discarded, and the pellet resuspended at 10 × 106 cells/ml.
The microglial cell suspension was sorted out by means of their eGFP expression. The emitted fluorescence was detected using a fluorescence-activated cell-sorting (FACS) Aria flow cytometer (BD Biosciences) equipped with a 488 nm argon-ion laser. The eGFP-positive cells were sorted out and collected. Afterward, they were centrifuged at 350 × g for 10 min at room temperature and the cell pellet was washed with PBS and re-pelleted. The cell pellet was kept at −80°C until RNA isolation was performed. Then 3500–5500 microglia were obtained per SC upon FACS sorting. Thirty-two SC were used to perform quantitative PCR (qPCR) experiments.
RNA isolation, RT-PCR, and qPCR.
Total RNA was isolated using the PicoPure RNA isolation kit (Arcturus) according to the manufacturer's protocol. The RNA was reverse transcribed into cDNA with oligo(dT)15 primers in a 20 μl reaction using the Promega Reverse Transcription System. Quantitative real-time PCR (qRT-PCR) was performed in duplicate using four independent RNA samples. The cDNA was quantified using the SYBR green mix (Applied Biosystems) and Quantitect Primer Assay kit (Qiagen). Primer references are listed in Table 3. The PCR was run in 10 μl on 10 ng cDNA generated from total RNA using a 7900 HT Fast Real-Time PCR System (Applied Biosystems). Reaction conditions for the PCR amplification were 20 s at 95°C, followed by 40 cycles, each consisting of 1 s at 95°C and 20 s at 60°C. For the analysis, we used the comparative Ct method, and normalized the cycle threshold (Ct) value of each sample to the endogenous housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT), which gives the ΔCt value of the sample. This estimates the fold change in expression of the other genes relative to the housekeeping gene. The results of the qPCR were run on a 2% agarose gel at the end of the PCR process to verify the product sizes.
Table 3.
Primers used for RT-PCR analysis
| Gene | Product size | Company (ref.) | Acc. number |
|---|---|---|---|
| HPRT | 168 bp | Qiagen (QT00166768) | NM_013556 |
| P2X1 | 100 bp | Qiagen (QT00120694) | NM_008771 |
| P2X4 | 73 bp | Qiagen (QT00163149) | NM_011026 |
| P2X7 | 80 bp | Qiagen (QT00130900) | NM_011027, NM_001038839, NM_001038845 |
| Panx1 | 108 bp | Qiagen (QT00138845) | NM_019482 |
| Panx2 | 133 bp | Qiagen (QT01773737) | NM_001002005 |
Statistics.
Data are presented as mean ± SEM; N expresses the number of recorded cells or the number of independent experiments in each of which multiple cells were measured. Mean effects of drugs applied on the same cell or on the same group of cells were compared using nonparametric Wilcoxon rank sign test for paired data. Means for each group of cells treated in the same way were compared using the nonparametric Mann–Whitney U test for continuous and unpaired variables. Differences were considered significant at p < 0.05. Statistics were performed using GraphPad Prism 4 (2003) and Kaleidagraph 4.04 (2006; Synergy Software). Graphics were prepared using Kaleidagraph software.
Results
P2X1R, P2X4R, and P2X7R expression in embryonic microglia
Microglia are known to express ionotropic P2X1R, P2X4R, and P2X7R in the adult (Kettenmann et al., 2011) and during substantia nigra pars compacta development (Xiang and Burnstock, 2005). We first determined the expression of these P2XRs using qPCR in FACS-sorted microglia of E13.5 CX3CR1eGFP mouse embryonic SCs. We also investigated the expression of the mRNAs coding for Panx1 and pannexin-2 (Panx2) since P2X7R can interact with Panx hemichannels (Pelegrin and Surprenant, 2009). P2X1R mRNAs were poorly expressed when compared with P2X4R and P2X7R mRNAs. We observed a significant (p < 0.01) 19- and 7-fold larger expression of the mRNAs coding for P2X4R and for P2X7R, respectively (Fig. 1A), when compared with the expression of mRNAs coding for P2X1R. We observed a significant (p < 0.01) 2.25-fold larger expression of the mRNAs coding for Panx1 (Fig. 1A), when compared with the expression of mRNAs coding for P2X1R. To the contrary, Panx2 mRNAs were poorly expressed in microglia. We observed a significant (p < 0.01) 12.9-fold lower expression of the mRNAs coding for Panx2 (Fig. 1A), when compared with the expression of mRNAs coding for Panx1.
Figure 1.
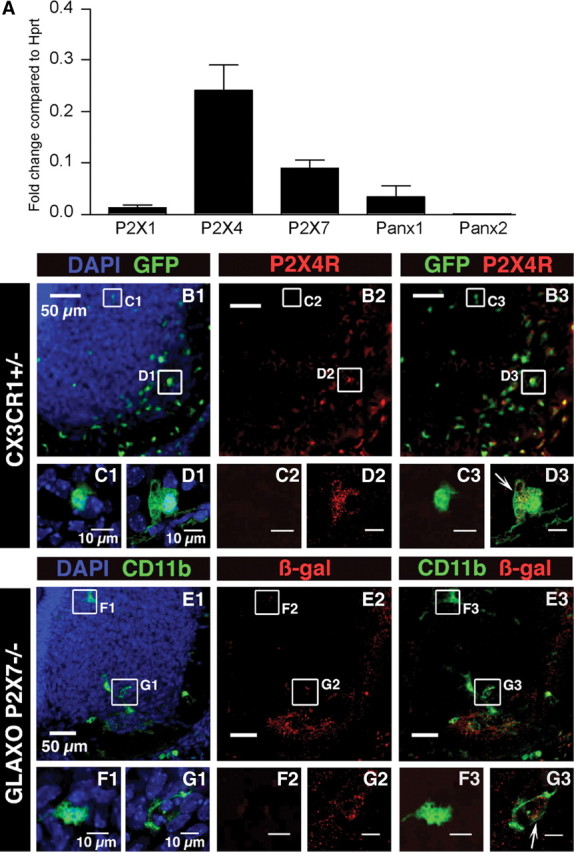
P2X1R, P2X4R, P2X7R, Panx1, and Panx2 expression in microglia. A, P2X1R, P2X4R, P2X7R, Panx1, and Panx2 expressions were analyzed on FACS-sorted SC microglia using qPCR. For each gene, the expression level corresponds to the x-fold change relative to the housekeeping gene Hprt. Note that P2X4R and P2X7R are the main P2XR transcripts expressed. The P2X1R transcript expression level is significantly (p < 0.01) lower than those of P2X4R and P2X7R. Note that low levels of Panx2 mRNAs were detected when compared with Panx1 mRNAs (≈13-fold significantly lower; p < 0.01). Error bars indicate SEM. B–D, P2X4R immunostaining in the ventral region of the SC of E13.5 CX3CR1eGFP mouse embryos. B1, Representative pictures of the SC ventral region. Note the accumulation of microglia (green) in the ventrolateral part of the SC. B2, P2X4R immunostaining. B3, Superimposed images shown in B1 and B2. C1, Enlarged image showing an example of eGFP microglia localized in the dorsomedial region of the ventral SC. C2, Note the lack of P2X4R immunostaining in the area shown in C1. C3, Superimposed images (C1 and C2) showing lack of P2X4R immunostaining within eGFP-positive microglia localized in the dorsomedial region. D1, Enlarged image showing an example of eGFP microglia localized in the ventrolateral region of the ventral SC. D2, P2X4R immunostaining in the area shown in D1 (red). D3, Superimposed images (D1 and D2) showing P2X4R immunostaining within eGFP-positive microglia localized in the ventrolateral region (arrow). Note that the staining is mainly located within the cytoplasm (single confocal section). E–G, β-Galactosidase immunostaining in the ventral region of the SC of E13.5 GlaxoSmithKline P2X7R−/− mouse embryos where the LacZ reporter gene has been inserted into the P2X7R gene. E1, Representative pictures of the ventral SC in E13.5 GlaxoSmithKline P2X7R−/− mouse embryos. Microglia were stained using CD11b antibody. E2, β-Galactosidase staining in the ventral SC. E3, Superimposed images shown in E1 and E2. F1, Enlarged image showing an example of microglia localized in the dorsomedial region of the ventral SC. F2, β-Galactosidase immunostaining in the area of the microglia shown in F1. Note the lack of galactosidase immunostaining. F3, Superimposed images shown in F1 and F2. G1, Enlarged image showing an example of microglia localized in the ventrolateral region of the ventral SC. G2, Galactosidase immunostaining in the area shown in G1. G3, Superimposed images (G1 and G2) showing galactosidase immunostaining within CD11b-positive microglia localized in the ventrolateral region (arrow; single confocal section), which indicates P2X7R gene expression in microglia. B1, C1, D1, E1, F1, G1, Cell nuclei are visualized with DAPI staining.
When analyzing P2X4R expression in the ventral part of the SC using immunostaining (Fig. 1B2,C2,C3,D2) we observed that 63.7 ± 2.2% (N = 5 SCs) of microglia localized in the ventral SC expressed P2X4R. However, at E13.5, 67.8 ± 1.9% (N = 5 SCs) of the ventral microglia were localized in the ventrolateral region (the MN area) of the SC (Fig. 1B1,B3) (Rigato et al., 2011), and a significant higher proportion (p < 0.01) of these microglia expressed P2X4R (81.3 ± 2.9%; N = 5 SCs) when compared with the other areas of the ventral SC (27.1 ± 5.1%; N = 5 SCs) (Fig. 1B2,D2). The MN area (12,000 μm2) was set as constant in all SCs analyzed as previously described (Rigato et al., 2011). P2X4R immunostaining was mainly localized in the cytoplasm (Fig. 1D3), as also observed in peritoneal macrophages (Qureshi et al., 2007). Due to the lack of a specific antibody for P2X7R, we used GlaxoP2X7R+/− mice, in which the LacZ gene has been inserted into the P2X7 gene, to analyze P2X7R cellular expression (Sim et al., 2004). Double immunostaining using antibodies directed against CD11b (Fig. 1E1,F1.G1) to stain microglia (Rigato et al., 2011) and against β-galactosidase encoded by the LacZ gene (Fig. 1E2,E3,F2,F3,G2,G3), showed that 77.1 ± 1.6% (N = 5 SCs) of microglia expressed P2X7R in the ventral part of the embryonic SC at E13.5. But as mentioned above most microglia accumulate within the ventrolateral region at E13.5 (72.2 ± 1.7% of ventral microglia in this set of experiments; N = 5 SCs) (Fig. 1E1). Moreover, 88.1 ± 1.4% (N = 5 SCs) of microglia localized in the ventrolateral region of the SC-expressed galactosidase while only 47.7 ± 4.1% (N = 5 SCs) of microglia localized in other areas of the ventral SC were immunoreactive to galactosidase antibody (Fig. 1E2,F2,F3,G2,G3), indicating that P2X7R was significantly (p < 0.01) more likely to be expressed by microglia localized in the MN area compared with other parts of the ventral SC.
ATP evokes biphasic membrane currents in embryonic microglia that result from P2X7R activation
To evaluate the functionality of the purinergic receptors expressed at the cell membrane of microglia, we performed an electrophysiological study of embryonic microglia ex vivo using whole-cell patch-clamp. We first investigated the passive electrophysiological properties of microglia in the embryonic SC. At E13.5, microglia had an irregular cell body shape and short processes (branched microglia) or an amoeboid morphology (Fig. 2C). The input resistances of the branched cells and of the amoeboid cells were 3.6 ± 0.3 GΩ and 4.3 ± 0.5 GΩ, respectively, being not significantly different (p > 0.1). The input capacitances of the branched cells (12 ± 1 pF; N = 56) and of the amoeboid cells (11 ± 1 pF; N = 26) were not significantly different either (p > 0.1). Almost all cells with an amoeboid morphology (96.2%; 24 of 26 recorded cells) displayed inward rectifying current (IrC), while only 57.1% (32 of 56 recorded cells) of the branched cells exhibited IrC (Fig. 2A,C). In contrast, all cells, regardless of their morphology, displayed outward rectifying current (OrC; Fig. 2A,B). Because outward rectification in the I/V relationship obtained on embryonic microglia (Figs. 2A,B, 6B) was reminiscent of that described for a Panx1 voltage-dependent current (Ma et al., 2009), we tested the effect of the hemichannel blocker CBX (Ma et al., 2009) on currents evoked by voltage ramps from −100 mV to +100 mV; 100 μm CBX had no significant effect (p > 0.1) on the outward rectification (Fig. 2D). It inhibited only 8.4 ± 4.8% of the current (N = 8). This clearly indicates that the voltage-activated current in embryonic microglia does not reflect Panx1 activation.
Next, we assessed the presence of functional P2XRs. The threshold concentration of ATP to elicit a response was 0.5 mm suggesting that P2XR involved in this current had a low affinity for ATP as previously described for P2X7R (Pelegrin and Surprenant, 2009). Applications of 0.5 and 1 mm ATP evoked currents of 0.3 +/− 0.1 pA/pF (N =9), and of 1.21 +/− 0.41 pA/pF (N =5), respectively, while 3 mm ATP evoked a biphasic current of 30.7 +/− 4.4 pA/pF (N = 7) (Fig. 3A). Such a biphasic response may reflect P2XR channel pore dilatation (Gudipaty et al., 2001; Shinozaki et al., 2009) and/or the formation of a large plasma membrane pore (Pelegrin and Surprenant, 2009). Since calcium and/or magnesium are known to modulate P2X7R and P2X4R activation (Khakh et al., 1999), we determined to which extent lowering their extracellular concentration could modify ATP-evoked responses. Responses evoked by ATP in the absence and in the presence of [Ca2+, Mg2+]out were analyzed in different cells. In free [Ca2+, Mg2+]out solution, the minimal ATP concentration necessary to evoke a current was 10 times lower (50 μm) than in normal [Ca2+, Mg2+]out (Fig. 3B). Similarly, a biphasic current was evoked by 0.3 mm ATP in free [Ca2+, Mg2+]out (Fig. 3B). ATP (0.3 mm) elicited a current of 25.97 ± 3.17 pA/pF (N = 5), being not significantly different (p > 0.05) from the one measured with 3 mm ATP in normal [Ca2+, Mg2+]out (30.7 +/− 4.4 pA/pF; N = 7). In free [Ca2+, Mg2+]out, 0.05 mm ATP (N = 9) and 0.1 mm ATP (N = 9) evoked currents of 0.42 ± 0.6 pA/pF and of 0.65 ± 0.14 pA/pF, respectively. Input capacitance of recorded microglia in normal [Ca2+, Mg2+]out (13.3 ± 1 pF; N = 9) and in free [Ca2+, Mg2+]out (13.8 ± 1.4 pF; N = 5) were not significantly different (p > 0.1).
ATP-induced biphasic responses occurred only at the first application, while subsequent responses did not display a biphasic activation phase anymore (Fig. 3C). These subsequent responses had a significantly (p < 0.01) faster 20–80% rise time (RT) (20–80% RT = 1.9 ± 0.4 s; N = 8) than the first response (20–80% RT = 10.1 ± 1.4 s; N =8). In free [Ca2+, Mg2+]out, these values were not significantly modified (p > 0.05): they were 13.7 ± 1.3 s and 3 ± 0.4 s (N = 16) for the biphasic current and the subsequent responses, respectively. We also observed a small rundown of the current amplitude with time of the responses after evoking a biphasic current (Fig. 3C). We compared the amplitude of the currents evoked after obtaining a biphasic response. When the responses were evoked on the 3 min (Fig. 3C), the amplitude of the second current was decreased by 6.8 ± 4% (N = 7) in normal [Ca2+, Mg2+]out, and by 13 ± 8% (N = 12) in free [Ca2+, Mg2+]out, being not significantly different (p > 0.1). The amplitude of the third current was decreased by 26.6 ± 3.2% (N = 7) in normal [Ca2+, Mg2+]out, and by 15.4 ± 3.8% (N =12) in free [Ca2+, Mg2+]out being significantly different (p < 0.05). Large inward currents evoked by long application of ATP (140 s) slowly decreased with time, indicating that these P2XRs have slow desensitization kinetics (Fig. 3D1,D2). In addition, responses evoked by application of different concentrations of ATP after evoking a biphasic current could be fitted with the Hill equation, yielding an EC50 of 0.47 mm and a Hill coefficient of 2.6 (Fig. 3E). Finally, ATP-induced inward current versus voltage relationship yielded a reversal potential of 3 ± 3 mV (N = 7) (ECl = −40 mV), indicating that ATP application caused an increase in cationic conductance (Fig. 4A).
Figure 4.

Whole-cell currents evoked by ATP and P2XR agonists in embryonic microglia in situ. A, A voltage ramp protocol (−100 mV +100 mV; 2 s) was used to determine the reversal potential of ATP-evoked currents (inset lower right). A set of three voltage ramps from −100 to 100 mV (interval) was applied before and during the response to 3 mm ATP (inset upper left) and the resulting currents were averaged. The averaged currents evoked before ATP application were subtracted from the averaged currents evoked during ATP application to obtain the current–voltage relationship of the current evoked by ATP. In this example, 3 mm ATP evoked inward currents with a reversal potential of −3.2 mV. B, Application of α,β-Me-ATP (300 μm), a P2X1R and P2X3R agonist, failed to evoke a response in embryonic SC microglia (Vh = −60 mV). C, In contrast, bzATP (100 μm), a compound acting as an agonist on P2X7Rs and also effective on P2X1Rs and P2X2Rs, induced inward currents (Vh = −60 mV). bzATP (100 μm)evoked a current of 9 ± 2 pA/pF (N = 6).
These functional characteristics of ATP-evoked currents are compatible with the activation of P2X4R and/or of P2X7R, but not of P2X1R (Coddou et al., 2002). To have an estimation of the pharmacological profile of these microglia P2XRs, we then analyzed the effect of two agonists and eight antagonists on the response evoked by repetitive applications of 500 μm ATP in free [Ca2+, Mg2+]out (see Materials and Methods). Agonist α,β-Me-ATP (300 μm), a P2X1R and P2X3R agonist (North and Surprenant, 2000), did not induce any current (N = 4) (Fig. 4B) unlike 100 μm bzATP (N = 6), a strong agonist on P2X7Rs that can also be effective on P2X1Rs and P2X2Rs, but not on P2X5Rs (North and Surprenant, 2000) (Fig. 4C). TNP-ATP (10 μm), a potent antagonist at concentrations ≥2 μm on P2X1R, P2X2R, P2X3R, P2X4R (North and Surprenant, 2000), and on P2X4/P2X7R (Guo et al., 2007), (Fig. 5A,L) and 10 μm PPADS (Fig. 5B,L) being known to antagonize P2X1R, P2X2R, P2X3R, and P2X5R (North and Surprenant, 2000), and to weakly antagonize mouse P2X7R (Hibell et al., 2001), had no significant effect (p > 0.1) on the ATP-evoked response, while 50 μm PPADS significantly (p < 0.01) blocked (≈70%) the response evoked by ATP (Fig. 5C,L). Similarly, 20 μm suramin, being known to fully antagonize all P2XR except P2X4R and P2X7R (North and Surprenant, 2000), had no significant inhibitory effect (p > 0.05) on the ATP-evoked current (Fig. 5D,J), while 200 μm suramin evoked a significant (p < 0.01) ≈66% inhibition (Fig. 5E,L). We then tested the effect of Cu2+ and Zn2+ on ATP-evoked currents. Cu2+ inhibits both P2X7Rs and P2X4Rs whereas Zn2+ increases the activity of P2X4R and is virtually without effect on rat P2X7R expressed in oocytes (Coddou et al., 2002) or fully blocks the activity of these two receptors expressed in transfected human embryonic kidney (HEK) cells (Liu et al., 2008). Then 50 μm Cu2+ evoked a significant (p < 0.01) ≈77% inhibition (Fig. 5F,L), while ATP-evoked responses were insensitive to 50 μm Zn2+ (Fig. 5G,L). Although this pharmacological profile was apparently consistent with P2X7R expression, 1 μm BBG, known to fully antagonize P2X7R (Jiang et al., 2000), had no significant effect (p > 0.1) (Fig. 5H,L), whereas 10 μm BBG, known to also inhibit P2X4R (Jiang et al., 2000), evoked a significant (p < 0.01) ≈79% inhibition (Fig. 5I,L). Accordingly, this pharmacological profile of ATP-evoked current did not allow discriminating clearly between P2X4R and P2X7R. To further address this issue we analyzed the effects of A438079 and A740003, two novel highly selective P2X7R antagonists (Donnelly-Roberts et al., 2009). Three (data not shown) and 10 μm (Fig. 5J) A438079 resulted in significant (p < 0.01) ≈87 and ≈100% inhibition, respectively (Fig. 5L), while 3 (data not shown) and 10 μm (Fig. 5K) A740003 resulted in significant (p < 0.01) ≈76 and ≈97% inhibition, respectively (Fig. 5L). These results are consistent with the hypothesis that ATP-evoked currents were due to the activation of P2X7R on embryonic SC microglia. To confirm this hypothesis, we then compared the effect of ATP on microglia in the isolated SC of E13.5 P2X7R−/− and P2X7R+/− mouse embryos. In microglia of P2X7R−/− embryos, the current evoked by a voltage ramp in microglia curve displayed both inward and outward rectifications as observed in wild-type (Fig. 6B), but the application of 3 mm ATP evoked a very small transient inward current (≤1 pA/pF; N = 5) only (Fig. 6C1), whereas it was larger and showed a biphasic activation time course in the SC of P2X7R+/− embryos (N = 7) (Fig. 6C2) as observed in C57BL/6 CX3CR1eGFP+/− mouse embryos. The current density in P2X7R−/− was significantly (≈16 times) smaller (p < 0.01) than the current measured in P2X7R+/− (N =7) (Fig. 6C3).Together, these results demonstrate that ATP-evoked biphasic current in embryonic microglia requires the expression of P2X7R.
Figure 5.
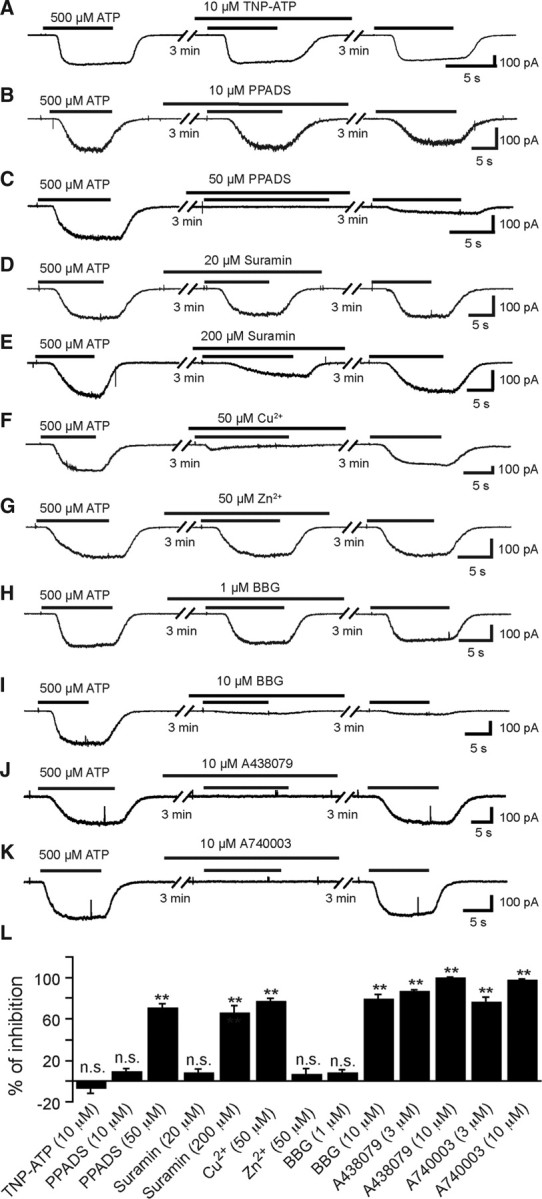
Effects of TNP-ATP, PPADS, suramin, copper, zinc, BBG, A438079, and A740003 on ATP-evoked currents in SC microglia. All compounds were applied before and during ATP application (500 μm; free [Ca2+, Mg2+]o; Vh = −60 mV). A, ATP-evoked current was poorly sensitive to TNP-ATP (10 μm), a selective antagonist of P2X1R, P2X3R, and P2X4R (p > 0.05). B, PPADS (10 μm) did not prevent ATP-evoked currents. C, PPADS (50 μm) fully blocked ATP-evoked currents. Note that ATP-evoked responses were poorly reversible 3 min after the end of the PPADS application. D, Suramin (20 μm) did not inhibit ATP-evoked responses as expected for P2X7Rs. E, Suramin (200 μm) partially blocked ATP-evoked currents. F, Copper (50 μm), known to block P2X7R activity, reversibly inhibited ATP-evoked currents. G, Zn2+ (50 μm) was poorly effective on ATP-evoked currents. H, BBG (1 μm) at this concentration was poorly effective on ATP-evoked currents. I, BBG (10 μm), known at this concentration to inhibit both P2X7R and P2X4R, irreversibly inhibited ATP-evoked currents. J, A438079 (10 μm) known at this concentration to inhibit selectively murine P2X7R fully inhibited ATP-evoked currents. K, A740003 (10 μm) also known to inhibit selectively murine P2X7R fully blocked ATP-evoked currents. L, Quantification of the inhibitory effects of the antagonists on ATP-evoked responses. Inhibitions of ATP responses were as follows: −6.8 ± 5.5% (N = 6) for 10 μm TNP-ATP, 8.7 ± 3% (N = 5) for 10 μm PPADS, 70.3 ± 4.4% (N = 6) for 50 μm PPADS, 7.2 ± 3.9% (N = 6) for 20 μm suramin, 65.7 ± 7.4% (N = 5) for 200 μm suramin, 76.8 ± 3.4% (N = 13) for 50 μm Cu2+, 5.9 ± 5.6% (N = 10) for 50 μm Zn2+, 7.2 ± 3.4% (N = 10) for 1 μm BBG, 79.3 ± 4.5% (N = 5) for 10 μm BBG, 86.8 ± 1.5% (N = 5) for 3 μm A438079, 76 ± 5% (N = 5) for 3 μm A740003, 99.8 ± 0.8% (N = 5) for 10 μm A438079, and 97.4 ± 1.6% (N = 5) for 10 μm A740003. (Statistical significance: **p < 0.01, * p < 0.05). Error bars indicate SEM.
P2X7R activation does not need Panx1 expression to generate a biphasic current
Large biphasic currents evoked in response to ATP might result from the interaction between P2X7R and Panx1 (Pelegrin and Surprenant, 2009). Embryonic microglia express both Panx1 mRNAs (Fig. 1A) and proteins, as shown by immunohistochemistry (Fig. 7A); 61.2 ± 3% (N = 5 SCs; 249–282 cells tested per SC) of the microglia located in the ventral area of the SC were immunoreactive to the Panx1 antibody. Panx1 staining was not observed in SC microglia of E13.5 Panx1−/− mouse embryos (Fig. 7B). Microglia obtained from Panx1−/− still express the voltage-dependent current (N = 5) observed in wild-type (Fig. 2A,B,D), further supporting its independence on Panx1 expression (Fig. 8B). To determine to what extent the biphasic currents evoked by ATP applications might reflect Panx1 opening, we first tested the effect of two putative hemichannel blockers, DIDS and CBX (Gomes et al., 2005; Ma et al., 2009), on successive responses evoked by 3 mm ATP in normal [Ca2+, Mg2+]out on SC microglia of CX3CR1eGFP mouse embryos. High concentration (100 μm) of DIDS had little but significant (p < 0.01) effect (20–30% inhibition) on ATP-evoked current whereas 100 μm CBX had no significant effect (p < 0.1) on ATP-evoked current (Fig. 8C1,C2,C3). Because the pharmacological profile of Panx1 could have been modified by its interaction with P2X7Rs (Ma et al., 2009), we then determined whether ATP could evoke a biphasic response on microglia of Panx1−/− embryos (Fig. 8D), although 3 mm ATP still evoked biphasic currents (27.7 ± 3.5 pA/pF; N =10), indicating that the biphasic response to ATP did not reflect functional coupling of P2X7R to Panx1.
Embryonic microglia proliferation requires the expression of P2X7R but not of Panx1
P2XRs have been proposed to promote both microglia activation and proliferation (Monif et al., 2009). At E13.5, microglia in the ventral area of the embryonic SC proliferate while they are activated when interacting with apoptotic MNs (Rigato et al., 2011). To determine whether P2X7R is involved in these processes, we compared KI67 staining (a proliferation marker) and Mac-2 staining (an activation marker for microglia) on microglia at E13.5 in the ventral SC area of P2X7R−/−, P2X7R+/−, and wild-type mouse embryos (Figs. 9A–C, 10A,B). Because microglia activation within the lateral motor column was associated with MN developmental cell death at E13.5 (Rigato et al., 2011), we also determine to what extent MN developmental cell death was altered in P2X7R−/− mouse embryos using activated caspase-3 immunostaining (Fig. 10C). The proportion of proliferating microglia (KI67 staining) was dramatically decreased in P2X7R−/− mouse embryos (2.1 ± 0.4%; N =5) when compared with P2X7R+/− mouse embryos (25.4 ± 1.5%; N = 5) (p < 0.01) or to P2X7R+/+ mouse embryos (26.9 ± 0.8%; N = 5) (Fig. 6A–C). The difference between P2X7R+/− and P2X7R+/+ was not significant (p > 0.1). Accordingly, the number of microglial cells was significantly decreased (by 12.5%; p < 0.05) in the ventral area of the lumbar SC of E13.5 P2X7R−/− mouse embryos (225 ± 3 cells; N = 5) when compared with P2X7R+/+ (257 ± 5 cells; N = 5). Similarly the density of microglia aggregating in the lateral motor column (LMC) at the onset of MN developmental cell death was also significantly decreased by 13.5% (p < 0.01) in E13.5 P2X7R−/− mouse embryos (4.7 ± 0.1 × 10−4 cells/μm2, N = 5) when compared with wild-type (5.4 ± 0. 2 × 10−4 cells/μm2, N = 5). We did not find any difference (p > 0.1) in microglia proliferation between wild-type and Panx1 −/− mice (24.8 ± 0.9%; N = 5) (Fig. 9C), indicating that the control of embryonic microglia proliferation by P2X7Rs did not involve coupling of P2X7Rs to Panx1. Microglia localized in the LMC have been previously shown to be in an activated state (Mac-2 staining) (Rigato et al., 2011), but the proportion of activated microglia in the LMC was not significantly different (p > 0.1) between P2X7R−/− (20 ± 0.5%; N =5) and wild-type (20.5 ± 0.6%; N = 5) (Fig. 5A,B). Accordingly, P2X7R was not involved in embryonic microglia activation at E13.5. Finally, MN developmental cell death at E13.5 was not affected in P2X7R−/− mouse embryo. No difference (p > 0.1) in activated caspase-3 staining in the ventral region of the SC was found between wild-type (3.18 ± 0.12% fluorescence/area; N = 4) and P2X7R−/− (3.24 ± 0.15% fluorescence/area; 40 slices, N = 4) (Fig. 10C,D).
Figure 9.
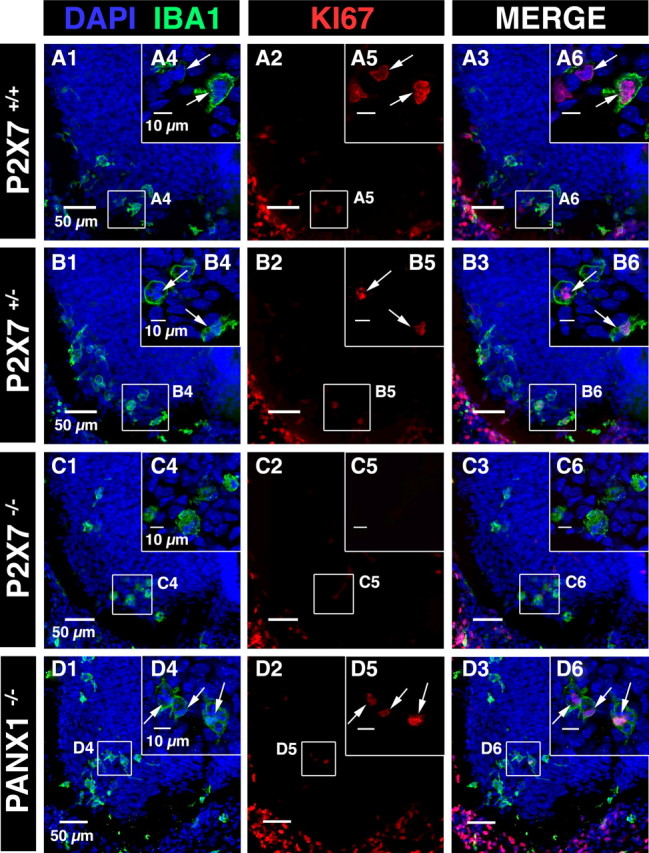
Microglia proliferation was dramatically reduced in the SC of P2X7R−/− mouse embryos but was unchanged in the SC of Panx1−/− mouse embryos. A1, B1, C1, D1, DAPI staining (blue) and microglia accumulation (IBA1 immunostaining, green) in the ventral region of P2X7R+/+, P2X7R+/−, P2X7R−/−, and Panx1−/− E13.5 SCs. A2, B2, C2, D2, Nuclear staining of KI67-expressing cells (red). A3, B3, C3, D3, Superposition of IBA1 and KI67 immunostainings. A4, B4, C4, Examples of microglia localized in the ventrolateral region of the SCs. A5, B5, C5, Example of KI67 staining in the ventrolateral region of the SC. A6, B6, White arrows indicate KI67 staining (single confocal section). Contrary to P2X7 +/+ SC (A6) and P2X7+/− SC (B6), LMC microglia are not stained for KI67 in P2X7R−/− SC (C6, single confocal section). D1, Example of microglia localized in the ventral SC of E13.5 Panx1−/− mouse embryos. D2, Immunostaining for KI67 in the ventral SC of E13.5 Panx1−/− mouse embryos. D3, superimposed images shown in D1 and D2. D4, Example of microglia localized in the ventrolateral region of the SC of E13.5 Panx1−/− mouse embryos. D5, KI67 staining in the ventrolateral region of the SC of E13.5 Panx1−/− mouse embryos. Note that KI67 immunostaining (arrows; D5) colocalized with IBA1 immunostaining (arrows; D6), indicating that microglia proliferated in the ventrolateral region of the SC of E13.5 Panx1−/− mouse embryos. D4, D5, D6, Single confocal section.
Figure 10.
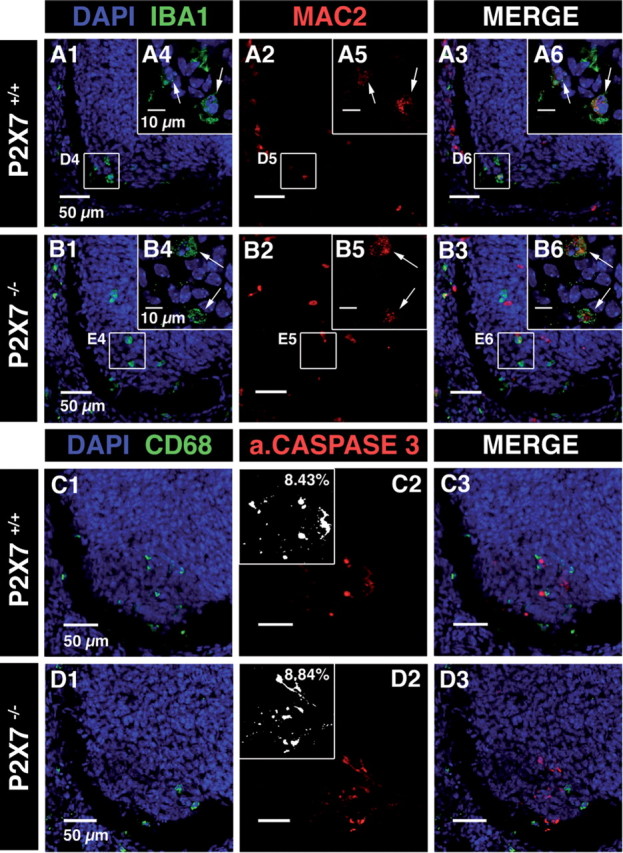
Microglia activation and MN developmental cell death were not altered in the SC of E13.5 P2X7R−/− mouse embryos. A, B, Confocal image of immunostainings against IBA1 (A1 and B1) and Mac-2 (red; A2 and B2) in the SC of wild-type (A) and P2X7R−/− (B) mouse embryos. A1, B1, DAPI staining (blue) and microglia staining (IBA1 immunostaining, green) in the ventrolateral region of the SC. A3, B3, Superposition of IBA1 and Mac-2 immunostainings. Note that Mac-2 immunostaining colocalized with IBA1 immunostaining, indicating that microglia were activated in the LMC of the SC of E13.5 P2X7R+/+ mouse embryos (arrows; A4–A6) and of E13.5 P2X7R−/− mouse embryos (arrows; B4–B6). A4, B4, Single confocal section showing microglia located in the ventrolateral region of the SC. C, D, Confocal image of immunostainings against CD68 (C1 and D1) and activated caspase-3 (red; C2 and D2) in the ventral SC of wild-type (C) and of P2X7R−/− (D) mouse embryos. C1, D1, DAPI staining (blue) and microglia staining (CD68 immunostaining, green) in the ventrolateral region of the SC. C3, D3, Superposition of CD68 and activated caspase-3 immunostainings. Insets in C2 and D2 are measurements of the percentage of fluorescence in the ventrolateral region of the SC (see Materials and Methods).
Discussion
Our results demonstrate a new physiological role of P2X7R during SC embryonic development. Microglia in the SC of the mouse embryo already express functional P2X7R at the onset of MN developmental cell death and synaptogenesis (Scain et al., 2010; Rigato et al., 2011). P2X7R activation by high concentration of ATP evokes a biphasic current in embryonic microglia, but our results indicate that although these cells express Panx1, there is no functional coupling of P2X7R to Panx1 in the embryo. Our results also demonstrate that P2X7R expression is required for the proliferation, but not for the activation of embryonic spinal microglia at the onset of MN developmental cell death. Since proliferation of nonactivated microglia is also affected in the SC of P2X7R−/− mouse embryos, our results also suggest that embryonic microglia proliferation and activation are controlled by two independent mechanisms.
Functional P2XRs expressed by embryonic microglia in situ
It is now well accepted that microglia express P2X1R, P2X4R, and P2X7R, both in vitro and in vivo in the adult (Kettenmann et al., 2011). Their expression in prenatal microglia (Xiang and Burnstock, 2005), let alone their function, was, however, unknown. Although we found low P2X1R mRNA expression in embryonic microglia, we cannot exclude the possibility that functional P2X1Rs are expressed on the cell membrane. P2X1Rs have fast desensitization kinetics (Parker, 1998), which render their detection difficult due to the relatively slow drug exchange in our ex vivo preparations (0.5–1 s). In embryonic SC microglia, P2X4R transcripts were the most abundantly expressed, as shown by qPCR, but our immunohistochemical results indicate that most P2X4Rs are rather located within the cytoplasm. This can result from a fast turnover of this receptor at the cell membrane (Royle et al., 2005). Our results contrast with recent data showing that P2X4Rs are functional in BV2 microglia cell lines and in cultured postnatal microglia (Bernier et al., 2012). This was unlikely to be the case in embryonic SC microglia of E13.5 mouse embryo, according to the pharmacological profile of ATP-evoked currents and to experiments performed on microglia of P2X7R−/− mouse embryos. Such an apparent discrepancy between our results obtained in situ and those recently described in culture could be explained by the activation state of cultured microglia. Cultured microglia adopt an activation state that can be variable depending on the culture conditions (Kettenmann et al., 2011), which could favor the membrane expression of P2X4R. Effectively, surface expression of P2X4R is increased by exposure of microglia to proinflammatory bacterial lipopolysaccharides, which mimic microglial activation observed in pathological conditions (Raouf et al., 2007).
Although our data demonstrate that P2X7R expression is required for ATP-evoked biphasic responses, the pharmacological profile of P2X7R is somewhat different from that classically described, especially with regard to the BBG potency in inhibiting ATP-evoked currents in transfected cells (Jiang et al., 2000). However, BBG efficiency on P2X7R largely depends on the method used to measure P2X7R activation and on animal species. (Donnelly-Roberts et al., 2009). We cannot completely rule out the possibility that BBG concentrations reaching the receptors were lower in situ than expected, but BBG has been applied 3 min before ATP, which is >40 times longer than necessary to reach equilibrium for other molecules in our preparation. Alternatively, a low BBG efficiency might indicate the expression of heteromeric P2X7/P2X4Rs, which is also consistent with the low efficiency of Zn2+ on ATP-evoked current (Guo et al., 2007). However, this hypothesis is not supported by our experiments (Fig. 5A) showing a lack of inhibition of ATP-evoked current by TNP-ATP (Guo et al., 2007). Another possibility might be that homomeric P2X7Rs and homomeric P2X4Rs directly interact, as previously proposed in cultured microglia (Boumechache et al., 2009), but the pharmacological consequences of such an interaction remain unknown. A lack of effect of Zn2+ on ATP-evoked current was surprising (Fig. 5G), but the Zn2+ effect on P2X7R remains controversial and depends on the preparation used. Indeed, Zn2+ has a low potency on rat P2X7R expressed in oocytes (Coddou et al., 2002; Acuña-Castillo et al., 2007), when compared with rat P2X7R expressed in transfected HEK cells (Liu et al., 2008).
P2X7R is not coupled to Panx1 in embryonic microglia
Our results indicate that P2X7R is not coupled to Panx1 in embryonic microglia in situ, which contrasts with results previously obtained in the adult and in vitro. Accordingly, the biphasic ATP response reported in our experiments rather reflects integral pore dilatation of P2X7Rs (Yan et al., 2008). However, out data do not rule out the possibility that P2X7R activation can evoke the formation of Panx1 plasma membrane pore in more mature microglia, as proposed in BV2 microglial cell line and in cultured microglia (Bernier et al., 2012). It could be envisioned that the difference in P2X7R/Panx1 interaction between embryonic microglia and adult microglia reflects a developmental process. Another consequence of the interaction of P2X7R with Panx1 is their induction of cell death (Locovei et al., 2007). In that respect, immature T-cells are less sensitive to cell death evoked by P2X7R activation when compared with mature T-cells (Tsukimoto et al., 2006). A lack of coupling between P2X7R and Panx1 in embryonic microglia may prevent this detrimental effect without affecting other P2X7R functions. Nevertheless, recent data also suggest that there is no functional coupling of P2X7R to Panx1 in mature macrophages. Effectively, the activation of P2X7R by ATP increases bone marrow-derived macrophages membrane permeability independently of Panx1, indicating that Panx1 is not essential for P2X7R channel formation (Qu et al., 2011). Panx1 activation independently of P2X7R signaling (Qu et al., 2011) was proposed to release a nucleotide “find me” signal from apoptotic cells (Chekeni et al., 2010; Qu et al., 2011) allowing purinergic receptor activation on phagocytes. This nucleotide release pathway is unlikely to be involved in the embryonic SC, at least at the onset of MN developmental cell death. Indeed, we did not observe any Panx1 expression in MNs at E13.5. The activation of P2X7R by an autocrine release of ATP by microglia through Panx1 activation seems also unlikely, since we did not find evidence for spontaneous activation of Panx1 expressed by embryonic microglia.
P2X7Rs control microglia proliferation independently of microglia activation
It has been previously proposed that microglia proliferation is an important mechanism that allows an increase in activated microglia density in response to brain injury (Kettenmann et al., 2011). By showing that P2X7R can control microglia proliferation during their early embryonic SC colonization, our results reveal a new physiological role of P2X7R on microglia in vivo that does not require pathological events leading to microglia activation. Although experiment performed in cultured microglia cell line and primary microglia have suggested that P2X7R activation promotes both microglia proliferation and activation (Bianco et al., 2006; Monif et al., 2009), our results clearly indicate that P2X7R is not involved in embryonic microglia activation. The reduction in microglia proliferation marker (KI67) observed in P2X7R−/− embryonic SC microglia was not restricted to Mac-2-positive cells, but was also observed in nonactivated cells, which lends further support to the conclusion that in vivo P2X7R controls microglia proliferation, either independently of its activation, or through a different mechanism. We determined the microglia activation state using Mac-2 staining (Rigato et al., 2011). Because Mac-2 (also named galectin-3) marks a unique state of activation in microglia that correlates to phagocytosis behavior (Dumic et al., 2006; Rotshenker, 2009) and promotes phagocytosis by macrophages (Sano et al., 2003), we can ascertain that P2X7R was not required to engage embryonic microglia in a phagocytic phenotype. An increase in cytosolic calcium level is known to induce proliferation of primary microglia cultures (Schwab et al., 2001) and of cultured N9 murine microglia cell line in response to P2X7R activation (Bianco et al., 2006). Accordingly, it is possible that embryonic microglia proliferation requires an increase in cytosolic calcium level provided by P2X7R activation (Egan and Khakh, 2004). The regional distribution of microglia within the parenchyma and their aggregation close to dying MN was not affected in P2X7R−/− embryonic mice, demonstrating that P2X7R is not involved in embryonic microglia migration and attraction by dying cells. At least in the adult, microglia migration and aggregation at damaged sites are regulated by several chemokine receptors (Kettenmann et al., 2011). In addition, microglia localized in the MN area showed an amoeboid morphology at the onset of MN developmental cell death. This morphological change is unlikely to result from P2X7R expression by microglia as amoeboid cells were still observed in P2XR−/− embryonic mice SC. Such a morphological change of activated microglia in the embryonic SC could result from the activation of adenosine A2 receptors by adenosine as recently shown in culture and in the adult brain (Orr et al., 2009).
In conclusion, our experiments revealed that embryonic microglia activation and proliferation can be controlled independently and that embryonic microglia proliferation is mainly under the control of P2X7Rs. Our data thus reveal a new role of this receptor during the SNC development by regulating embryonic microglia proliferation without the need of its interaction to Panx1 during their initial SC colonization.
Footnotes
This work was supported by AFM Grant n°15436, INSERM, Centre National de la Recherche Scientifique, the Belgian Charcot Foundation, and the Belgian Multiple Sclerosis Study Group. The P2X4 antibody and P2X2 plasmids were supplied by Dr. F. Rassendren, Institut National de la Santé et de la Recherche Médicale (INSERM U661 Montpellier, France). Mac-2 antibody was supplied by Dr. E. Audinat (INSERM U603, Paris, France). Pfizer P2X7R−/− mice and Glaxo P2X7R−/− mice were provided by Dr. E. Audinat (INSERM U603, Paris, France) and by Dr. M. Galante (BBMC, Orsay, France).
References
- Acuña-Castillo C, Coddou C, Bull P, Brito J, Huidobro-Toro JP. Differential role of extracellular histidines in copper, zinc, magnesium and proton modulation of the P2X7 purinergic receptor. J Neurochem. 2007;101:17–26. doi: 10.1111/j.1471-4159.2006.04343.x. [DOI] [PubMed] [Google Scholar]
- Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J Neurosci. 2008;28:9133–9144. doi: 10.1523/JNEUROSCI.1820-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier LP, Ase AR, Boué-Grabot E, Séguéla P. P2X4 receptor channels form large noncytolytic pores in resting and activated microglia. Glia. 2012;60:728–737. doi: 10.1002/glia.22301. [DOI] [PubMed] [Google Scholar]
- Bianco F, Ceruti S, Colombo A, Fumagalli M, Ferrari D, Pizzirani C, Matteoli M, Di Virgilio F, Abbracchio MP, Verderio C. A role for P2X7 in microglial proliferation. J Neurochem. 2006;99:745–758. doi: 10.1111/j.1471-4159.2006.04101.x. [DOI] [PubMed] [Google Scholar]
- Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, Kinney HC. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- Boumechache M, Masin M, Edwardson JM, Górecki DC, Murrell-Lagnado R. Analysis of assembly and trafficking of native P2X4 and P2X7 receptor complexes in rodent immune cells. J Biol Chem. 2009;284:13446–13454. doi: 10.1074/jbc.M901255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough D, Le Feuvre RA, Iwakura Y, Rothwell NJ. Purinergic (P2X7) receptor activation of microglia induces cell death via an interleukin-1-independent mechanism. Mol Cell Neurosci. 2002;19:272–280. doi: 10.1006/mcne.2001.1054. [DOI] [PubMed] [Google Scholar]
- Charrier C, Coronas V, Fombonne J, Roger M, Jean A, Krantic S, Moyse E. Characterization of neural stem cells in the dorsal vagal complex of adult rat by in vivo proliferation labeling and in vitro neurosphere assay. Neuroscience. 2006;138:5–16. doi: 10.1016/j.neuroscience.2005.10.046. [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JW, Chong SY, Kim JY, Eom JI, Jeung HK, Maeng HY, Lee ST, Min YH. Induction of apoptosis by apicidin, a histone deacetylase inhibitor, via the activation of mitochondria-dependent caspase cascades in human Bcr-Abl-positive leukemia cells. Clin Cancer Res. 2003;9:5018–5027. [PubMed] [Google Scholar]
- Coddou C, Villalobos C, González J, Acuña-Castillo C, Loeb B, Huidobro-Toro JP. Formation of carnosine-Cu(II) complexes prevents and reverts the inhibitory action of copper in P2X4 and P2X7 receptors. J Neurochem. 2002;80:626–633. doi: 10.1046/j.0022-3042.2001.00732.x. [DOI] [PubMed] [Google Scholar]
- Dalmau I, Vela JM, González B, Finsen B, Castellano B. Dynamics of microglia in the developing rat brain. J Comp Neurol. 2003;458:144–157. doi: 10.1002/cne.10572. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Chiozzi P, Falzoni S, Ferrari D, Sanz JM, Venketaraman V, Baricordi OR. Cytolytic P2X purinoceptors. Cell Death Differ. 1998;5:191–199. doi: 10.1038/sj.cdd.4400341. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br J Pharmacol. 2009;157:1203–1214. doi: 10.1111/j.1476-5381.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl- channels in ramification of murine microglia. J Neurosci. 1998;18:7127–7137. doi: 10.1523/JNEUROSCI.18-18-07127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Khakh BS. Contribution of calcium ions to P2X channel responses. J Neurosci. 2004;24:3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med. 1997;185:579–582. doi: 10.1084/jem.185.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:1208–1218. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- Guan C, Ye C, Yang X, Gao J. A review of current large-scale mouse knockout efforts. Genesis. 2010;48:73–85. doi: 10.1002/dvg.20594. [DOI] [PubMed] [Google Scholar]
- Gudipaty L, Humphreys BD, Buell G, Dubyak GR. Regulation of P2X(7) nucleotide receptor function in human monocytes by extracellular ions and receptor density. Am J Physiol Cell Physiol. 2001;280:C943–C953. doi: 10.1152/ajpcell.2001.280.4.C943. [DOI] [PubMed] [Google Scholar]
- Guo C, Masin M, Qureshi OS, Murrell-Lagnado RD. Evidence for functional P2X4/P2X7 heteromeric receptors. Mol Pharmacol. 2007;72:1447–1456. doi: 10.1124/mol.107.035980. [DOI] [PubMed] [Google Scholar]
- Hibell AD, Thompson KM, Xing M, Humphrey PP, Michel AD. Complexities of measuring antagonist potency at P2X(7) receptor orthologs. J Pharmacol Exp Ther. 2001;296:947–957. [PubMed] [Google Scholar]
- Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. 2000;75:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- Ho MK, Springer TA. Mac-2, a novel 32,000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J Immunol. 1982;128:1221–1228. [PubMed] [Google Scholar]
- Huang YJ, Maruyama Y, Dvoryanchikov G, Pereira E, Chaudhari N, Roper SD. The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc Natl Acad Sci U S A. 2007;104:6436–6441. doi: 10.1073/pnas.0611280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- Jiang LH, Rassendren F, Mackenzie A, Zhang YH, Surprenant A, North RA. N-methyl-D-glucamine and propidium dyes utilize different permeation pathways at rat P2X(7) receptors. Am J Physiol Cell Physiol. 2005;289:C1295–C1302. doi: 10.1152/ajpcell.00253.2005. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Bao XR, Labarca C, Lester HA. Neuronal P2X transmitter-gated cation channels change their ion selectivity in seconds. Nat Neurosci. 1999;2:322–330. doi: 10.1038/7233. [DOI] [PubMed] [Google Scholar]
- Le Feuvre R, Brough D, Rothwell N. Extracellular ATP and P2X7 receptors in neurodegeneration. Eur J Pharmacol. 2002;447:261–269. doi: 10.1016/s0014-2999(02)01848-4. [DOI] [PubMed] [Google Scholar]
- Liu X, Surprenant A, Mao HJ, Roger S, Xia R, Bradley H, Jiang LH. Identification of key residues coordinating functional inhibition of P2X7 receptors by zinc and copper. Mol Pharmacol. 2008;73:252–259. doi: 10.1124/mol.107.039651. [DOI] [PubMed] [Google Scholar]
- Locovei S, Scemes E, Qiu F, Spray DC, Dahl G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007;581:483–488. doi: 10.1016/j.febslet.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Hui H, Pelegrin P, Surprenant A. Pharmacological characterization of pannexin-1 currents expressed in mammalian cells. J Pharmacol Exp Ther. 2009;328:409–418. doi: 10.1124/jpet.108.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Teva JL, Almendros A, Calvente R, Cuadros MA, Navascués J. Tangential migration of ameboid microglia in the developing quail retina: mechanism of migration and migratory behavior. Glia. 1998;22:31–52. doi: 10.1002/(sici)1098-1136(199801)22:1<31::aid-glia4>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Mingam R, De Smedt V, Amédée T, Bluthé RM, Kelley KW, Dantzer R, Layé S. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1 beta in the murine brain. Brain Behav Immun. 2008;22:234–244. doi: 10.1016/j.bbi.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29:3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicke A, Kuan YH, Masin M, Rettinger J, Marquez-Klaka B, Bender O, Górecki DC, Murrell-Lagnado RD, Soto F. A functional P2X7 splice variant with an alternative transmembrane domain 1 escapes gene inactivation in P2X7 knock-out mice. J Biol Chem. 2009;284:25813–25822. doi: 10.1074/jbc.M109.033134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. 2009;12:872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KE. Modulation of ATP-gated non-selective cation channel (P2X1 receptor) activation and desensitization by the actin cytoskeleton. J Physiol. 1998;510:19–25. doi: 10.1111/j.1469-7793.1998.019bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. The P2X(7) receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal. 2009;5:129–137. doi: 10.1007/s11302-009-9141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol. 2011;186:6553–6561. doi: 10.4049/jimmunol.1100478. [DOI] [PubMed] [Google Scholar]
- Qureshi OS, Paramasivam A, Yu JC, Murrell-Lagnado RD. Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J Cell Sci. 2007;120:3838–3849. doi: 10.1242/jcs.010348. [DOI] [PubMed] [Google Scholar]
- Raouf R, Chabot-Doré AJ, Ase AR, Blais D, Séguéla P. Differential regulation of microglial P2X4 and P2X7 ATP receptors following LPS-induced activation. Neuropharmacology. 2007;53:496–504. doi: 10.1016/j.neuropharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Rigato C, Buckinx R, Le-Corronc H, Rigo JM, Legendre P. Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. Glia. 2011;59:675–695. doi: 10.1002/glia.21140. [DOI] [PubMed] [Google Scholar]
- Rotshenker S. The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J Mol Neurosci. 2009;39:99–103. doi: 10.1007/s12031-009-9186-7. [DOI] [PubMed] [Google Scholar]
- Royle SJ, Qureshi OS, Bobanović LK, Evans PR, Owen DJ, Murrell-Lagnado RD. Non-canonical YXXGPhi endocytic motifs: recognition by AP2 and preferential utilization in P2X4 receptors. J Cell Sci. 2005;118:3073–3080. doi: 10.1242/jcs.02451. [DOI] [PubMed] [Google Scholar]
- Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, Izui S, Liu FT. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scain AL, Le Corronc H, Allain AE, Muller E, Rigo JM, Meyrand P, Branchereau P, Legendre P. Glycine release from radial cells modulates the spontaneous activity and its propagation during early spinal cord development. J Neurosci. 2010;30:390–403. doi: 10.1523/JNEUROSCI.2115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Frei E, Klusman I, Schnell L, Schwab ME, Schluesener HJ. AIF-1 expression defines a proliferating and alert microglial/macrophage phenotype following spinal cord injury in rats. J Neuroimmunol. 2001;119:214–222. doi: 10.1016/s0165-5728(01)00375-7. [DOI] [PubMed] [Google Scholar]
- Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, Jones LC, O'Neal WK, Penuela S, Laird DW, Boucher RC, Lazarowski ER. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem. 2011;286:26277–26286. doi: 10.1074/jbc.M111.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Sumitomo K, Tsuda M, Koizumi S, Inoue K, Torimitsu K. Direct observation of ATP-induced conformational changes in single P2X(4) receptors. PLoS Biol. 2009;7:e1000103. doi: 10.1371/journal.pbio.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Young MT, Sung HY, North RA, Surprenant A. Reanalysis of P2X7 receptor expression in rodent brain. J Neurosci. 2004;24:6307–6314. doi: 10.1523/JNEUROSCI.1469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Chaumont S, Jo J, Ulmann L, Young MT, Cho K, Buell G, North RA, Rassendren F. Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci. 2006;26:9006–9009. doi: 10.1523/JNEUROSCI.2370-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- Tsukimoto M, Maehata M, Harada H, Ikari A, Takagi K, Degawa M. P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J Immunol. 2006;177:2842–2850. doi: 10.4049/jimmunol.177.5.2842. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Burnstock G. Expression of P2X receptors on rat microglial cells during early development. Glia. 2005;52:119–126. doi: 10.1002/glia.20227. [DOI] [PubMed] [Google Scholar]
- Yan Z, Li S, Liang Z, Tomić M, Stojilkovic SS. The P2X7 receptor channel pore dilates under physiological ion conditions. J Gen Physiol. 2008;132:563–573. doi: 10.1085/jgp.200810059. [DOI] [PMC free article] [PubMed] [Google Scholar]