

Abstract
Parkinson's disease (PD) is the most common representative of a group of disorders known as synucleinopathies, in which misfolding and aggregation of α-synuclein (a-syn) in various brain regions is the major pathological hallmark. Indeed, the motor symptoms in PD are caused by a heterogeneous degeneration of brain neurons not only in substantia nigra pars compacta but also in other extrastriatal areas of the brain. In addition to the well known motor dysfunction in PD patients, cognitive deficits and memory impairment are also an important part of the disorder, probably due to disruption of synaptic transmission and plasticity in extrastriatal areas, including the hippocampus.
Here, we investigated the impact of a-syn aggregation on AMPA and NMDA receptor-mediated rat hippocampal (CA3-CA1) synaptic transmission and long-term potentiation (LTP), the neurophysiological basis for learning and memory. Our data show that prolonged exposure to a-syn oligomers, but not monomers or fibrils, increases basal synaptic transmission through NMDA receptor activation, triggering enhanced contribution of calcium-permeable AMPA receptors. Slices treated with a-syn oligomers were unable to respond with further potentiation to theta-burst stimulation, leading to impaired LTP. Prior delivery of a low-frequency train reinstated the ability to express LTP, implying that exposure to a-syn oligomers drives the increase of glutamatergic synaptic transmission, preventing further potentiation by physiological stimuli.
Our novel findings provide mechanistic insight on how a-syn oligomers may trigger neuronal dysfunction and toxicity in PD and other synucleinopathies.
Introduction
Parkinson′s disease (PD) is typically known for the loss of nigrostriatal dopaminergic neurons associated with motor impairment, of which muscle rigidity, resting tremor, bradykinesia, and postural instability are the cardinal symptoms (Jankovic, 2008). However, neuropsychiatric disturbances, cognitive deficits, and memory impairment also occur in PD (Turner et al., 2002), probably due to disruption of synaptic transmission and plasticity in extrastriatal areas. In fact, the accumulation of proteinaceous intracellular inclusions named Lewy bodies (LBs) (Spillantini et al., 1997), a pathological hallmark of diseases known as synucleinopathies, is not restricted to the substantia nigra but is extended to several areas including hippocampus and cortex (Spillantini et al., 1997; Mattila et al., 2000; Braak et al., 2004). These inclusions are mainly composed of α-synuclein (a-syn), a neuronal presynaptic protein implicated in vesicular trafficking, neurotransmitter release, and regulation of neurotransmission (Clayton and George, 1999; Auluck et al., 2002; Fujiwara et al., 2002). This protein is known to undergo misfolding and oligomerization, under specific conditions, forming dimers, oligomers, and fibrillar structures (Conway et al., 2000; Uversky et al., 2001; Lashuel et al., 2002). While the precise origin for the toxicity of a-syn is still unclear, existing data suggest that oligomeric forms, rather than the larger intracellular inclusions, might be more bioactive and, possibly, cytotoxic, leading not only to cell death but actually impacting neuronal function (Spillantini et al., 1997; Lansbury and Brice, 2002; El-Agnaf et al., 2003; Cookson, 2005; Outeiro et al., 2007). Recent studies support the possible transmission of a-syn pathology across different brain regions and the existence of extracellular forms of a-syn (Lee et al., 2005; Li et al., 2008; Hansen et al., 2011). Nevertheless, the mechanism by which extracellular a-syn impacts synaptic activity remains largely unknown. The purpose of this work was to investigate the effects of extracellular a-syn species on basic readouts of neuronal function, such as synaptic transmission and plasticity. For this, we used normal rodent hippocampus as a model, and determined the effect of the different a-syn species on the well established long-term potentiation (LTP) paradigm. Furthermore, to dissect the mechanisms involved, we explored the involvement of glutamate receptors on the impact of a-syn on synaptic function. We focused on NMDA and calcium-permeable AMPA receptors since they play a major role both in synaptic plasticity and glutamate-induced neuronal dysfunctions (Doble, 1999). Indeed, their overactivation is a prominent synaptic event leading to excitotoxicity (Besancon et al., 2008).
Our data show that a-syn oligomers, but not monomers or fibrils, impair LTP and increase basal synaptic transmission through a mechanism dependent on NMDA receptor activation. These effects cannot simply be attributed to cytotoxicity induced by a-syn oligomers. This is accompanied by an increase in the expression of GluR1-containing AMPA receptors and an NMDA receptor increased contribution. Importantly, prior delivery of a low-frequency train could rescue LTP impairments, consistent with the observation that LTP was saturated in slices treated with a-syn oligomers.
In summary, we provide novel insights into the mechanisms triggered by the accumulation of extracellular a-syn and how it may contribute to neuronal dysfunction in PD and other synucleinopathies.
Materials and Methods
Reagents and Materials
6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX; Ascent Scientific) and 1-naphthylacetyl spermine trihydrochloride (NASPM; Sigma-Aldrich) were prepared as a 10 mm stock solution in water. dl-2-Amino-5-phosphonopentanoic acid (APV; Ascent Scientific) was prepared at 50 mm stock solution in water and picrotoxin (Sigma-Aldrich) also as a 50 mm stock solution, in dimethylsulfoxide. All aliquots were kept frozen until use.
Oligomerization of a-syn and insulin
Purified a-syn was resuspended at 2 mg · ml−1 in water (Millipore Milli-Q system). Monomeric sample was readily used or aliquoted and stored at −80°C for further use. Oligomerization was induced by continuous shaking for 6 d at 37°C in a Thermomixer (Eppendorf) at 1400 rpm. Samples were ultracentrifuged and the resulting supernatant (oligomers) readily used or aliquoted and stored at −80°C. 4-Hydroxy-2-nonenal (HNE)-modified a-syn (a-syn HNE-oligomers) and a-syn fibrils were prepared as previously described (Näsström et al., 2011). Human insulin was prepared as previously (Ribero Morais et al., 2011) with the following modifications. Human insulin was prepared in Tris-HCl 50 mm at 3 mg.ml−1, pH 3, and incubated at 42°C for 24 h in a Thermomixer (Millipore). Fibrils were separated by ultracentrifugation at 40,000 × g for 1 h at 4°C and supernatant containing oligomers collected. Oligomer concentration was measured using BCA assay (Pierce).
Expression and purification of human a-syn
The expression and purification procedure of human a-syn was a modified version of a previously described method (Kessler et al., 2003). Briefly, cells of Escherichia coli strain BL-21 (GE Healthcare) were transformed by heat shock with a-syn PT7–7 construct, and expression was induced by the addition of isopropyl d-thiogalactopyranoside (IPTG) at a final concentration of 1 mm. Cells were harvested after 4 h incubation with IPTG and stored overnight at −20°C. E. coli was resuspended in 50 mm Tris, pH 8.5, 50 mm KCl, 5 mm MgAc, 0.1% (w/v) NaN3, and 300 μm phenylmethylsulfonyl fluoride, and lysed by three passages through a French cell press. The extract was centrifuged at 18,000 × g at 4°C for 30 min to eliminate cell debris. The supernatant was saved and boiled for 20 min. The boiled extract was centrifuged at 45,000 × g at 4°C for 40 min and the supernatant was filtered with a 0.22 mm filter to remove possible pellet contamination. The a-syn containing extract was loaded on to an ion-exchange chromatography Q SepharoseTM (GE Healthcare) fast-flow column equilibrated with 20 mm Tris/HCl, pH 8.0. Proteins were eluted with a linear NaCl gradient (0.12–0.5 m) at a flow rate of 1.5 ml.min−1 and the eluate was monitored at 280 nm. Protein-containing fractions were collected and probed by immunoblot analysis using Syn-1 anti-a-syn antibody (BD Transduction Laboratories). Fractions containing a-syn were collected, concentrated by centrifugation using Amicon filters (Millipore), and applied to a gel filtration Superdex 75 column (GE Healthcare), equilibrated with 50 mm Tris/HCl buffer, pH 7.5, containing 150 mm NaCl. Proteins were eluted with the same buffer at a flow rate of 1 ml · min−1. Again, fractions containing a-syn, probed by immunoblot, were collected and combined for dialysis against water and then lyophilized for future analysis.
SDS-PAGE
The composition of different a-syn samples, monomers, oligomers, HNE-oligomers, and fibrils, was evaluated by SDS-PAGE.
Total protein concentration was measured using BCA assay (Pierce). Five micrograms of each a-syn sample was separated by SDS-PAGE using a Tetra Cell (Bio-Rad) in a precast 4–15% polyacrylamide gel (Bio-Rad) using standard procedures. Proteins were transferred to a nitrocellulose membrane (Bio-Rad) using the Mini Tans-Blot system (Bio-Rad). Prestained standard proteins were also loaded on the gel. Membrane was blocked for 1 h at room temperature with blocking solution (5% bovine serum albumin in 50 mm Tris, 150 mm NaCl, 0.1% and Tween 20, pH 7.5. The membrane was incubated overnight at 4°C with the primary antibody anti-a-Syn (c-20) (BD Transduction Labs) using a dilution of 1:1000 in blocking solution. Membrane was washed and incubated for 1 h at room temperature with anti-mouse-horseradish peroxide (HRP)-conjugated secondary antibody (Invitrogen) using a dilution of 1:10,000 in blocking solution. Detection procedures were performed according to ECL system (Millipore) with 10 s of exposure time. Films were scanned and densitometry performed using ImageJ-Image Processing and Analysis in Java (Abramoff et al., 2004). Each immunoblot was repeated at least three times from independent experiments.
Thioflavin T binding
The formation of oligomers and fibrils was confirmed by Thioflavin T binding assay (Nilsson, 2004). Briefly, Thioflavin T was incubated at a final concentration of 20 μm with 0.7 μm a-syn and insulin species in 50 mm Tris-HCl, pH 7.4. Emission wavelength scan was performed with an excitation wavelength of 450 nm using a plate reader (Tecan Infinite 200).
Atomic force microscopy
Atomic force microscopy (AFM) was used to characterize the morphology of a-syn species. Samples for AFM were prepared as follows: 10 μl protein sample at a concentration of 30 μm was applied on a freshly cleaved mica substrate previously glued to a glass coverslip and allowed to adsorb for 5 min. Unbound protein was rinsed with 100 μl Milli-Q water (resistivity > 18 MW/cm) 0.22 mm filtered. The sample was then mounted on the AFM stage in 50 μl Mili-Q water.
AFM imaging was performed with uncoated silicon nitride cantilevers OMCL-TR800PSA-1 from Olympus with a typical stiffness of 0.57 N/m for intermittent contact mode. The scan rate was set to <1 Hz and the force applied on the sample was maintained at the lowest possible value by continuously adjusting the set point and gain during the imaging. The cantilever oscillation was turned to a frequency of 15–20 kHz. Height, deflection, and phase-shift signals were collected and images were line fitted as required. Tip artifacts were ruled out by image analysis. All AFM images have 512 × 512 pixels and are made at a typical scan speed of 0.8 mm/s.
Cell culture and species cytotoxicity assays
DMEM (DMEM + GlutaMAX II) and fetal bovine serum (FBS) were from Invitrogen. Cytotoxicity LDH Detection Kit was from Clontech.
SH-SY5Y neuroblastoma cells were maintained in Opti-MEM and DMEM, high glucose, and GlutaMAX II, respectively, supplemented with 10% FBS at 37°C. Cells were plated onto 24-well plates (1.9 cm2) at a density of 80,000 cells/cm2 24 h before species exposure. a-syn monomers, oligomers, HNE-oligomers, and fibrils were incubated at 500 nm for 90 min and conditioned media collected for cytotoxicity assays and lactate dehydrogenase (LDH) activity measured in a plate reader (Tecan Infinite 200), according to the manufacturer's protocol.
Slice preparation
The experiments were performed in acute transverse hippocampal slices taken from male Wistar rats (10–12 weeks old) from Harlan Interfauna Iberica, SL. The animals were handled according to European Community guidelines and Portuguese law concerning animal care and were anesthetized with halothane before decapitation.
For field extracellular EPSP (fEPSP) recordings the hippocampus was dissected free within ice-cold artificial CSF (aCSF) solution containing in (mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4, 2 CaCl2, and 10 d-glucose, previously gassed with 95% O2 and 5% CO2, pH 7.4. Slices (400 μm thick) were cut perpendicularly to the long axis of the hippocampus with a McIlwain tissue chopper and allowed to recover functionally and energetically for at least 60 min in a resting chamber, filled with the same solution, at room temperature (22–25°C). After incubation with or without a-syn (or insulin) species, slices were transferred to a recording chamber for submerged slices and continuously superfused at 3 ml.min−1 with gassed bathing solution at 32°C. Therefore, all synaptic plasticity recordings were obtained at 32°C.
For EPSC recordings transverse slices for patch-clamp recordings (300 μm thick) were cut on a vibratome (VT 1000 S; Leica) in ice-cold dissecting solution containing (in mm) 110 sucrose, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 7 d-glucose, oxygenated with 95% O2 and 5% CO2, pH 7.4. Slices were first incubated in aCSF solution for 30 min at 35°C and used after recovering for at least 60 min at room temperature. Individual slices were fixed on a grid in a recording chamber and continuously superfused at 2 ml · min−1 at room temperature.
Slice incubation with a-syn species
Incubation with the different species of a-syn (or insulin) was carried for 90 min at room temperature in gassed aCSF and compared with incubation with vehicle in the same conditions. The length of the incubation period relates to the main goal of the present work, which was to evaluate the impact of sustained exposure of neurons to extracellular synuclein upon synaptic transmission and plasticity. Pre-incubation with vehicle, a-syn, or insulin was performed at room temperature to avoid neuronal damage by hypoxic injury (Schiff and Somjen, 1985). The range of concentration chosen for a-syn incubation was based on previous data using amyloid-β (Aβ) oligomers ranging from 10 to 100 nm (Shankar et al., 2008). In a set of preliminary experiments, we tested 10–50 nm syn in fEPSPs but did not detect significant changes.
fEPSP recordings
Evoked fEPSPs were recorded as extracellularly through a microelectrode filled with 4 m NaCl (2–4 MΩ resistance) placed in the stratum radiatum of the CA1 area, as previously described (Diógenes et al., 2011). The pathway of Schaffer collateral fibers was stimulated (rectangular pulses of 0.1 ms duration) once every 15 s by a bipolar wire electrode placed on the Schaffer fibers in the CA3 area. The initial intensity of the stimulus (300 μA) was adjusted to obtain a submaximal fEPSP slope with a minimum population spike contamination, near one-third of the fEPSP slope obtained with supramaximal stimulation. The averages of eight consecutive fEPSPs from the Schaffer collateral CA1 pathway were obtained and quantified as the slope of the initial phase of the potential. Recordings were obtained with an Axoclamp 2B amplifier (Molecular Devices), digitized, and continuously stored on a personal computer with the WinLTP program (Anderson and Collingridge, 2001).
Input/output curves.
Input/output (I/O) curves were performed after a stable baseline of at least 15 min. The stimulus delivered to the slice was decreased until no fEPSPs evoked and subsequently increased by steps of 20 μA. Data from three consecutive fEPSPs were collected for each stimulation intensity. The range of all the input delivered to the slice was typically from 60 μA to a supramaximum stimulation amplitude of 360 μA. The I/O curve was plotted as the relationship of fEPSP slope versus stimulus intensity, which provides a measure of synaptic efficiency. The max slope values were obtained by extrapolation upon nonlinear fitting of the I/O curve and an F test was used to determine differences between the parameters.
LTP induction.
LTP was induced after obtaining a stable recording of fEPSP (each fEPSP is the average of eight individual fEPSPs) slope in the Schaffer collateral pathway for at least 30 min. LTP was induced by a theta-burst protocol (10 trains separated by 200 ms, four pulses each, 100 Hz) in the Schaffer collaterals/CA1 synapse. The intensity of the stimulus was kept constant throughout these induction protocols. LTP was quantified as the percentage of change in the average slope of the fEPSP taken from 46 to 60 min after LTP induction in relation to the average slope of the fEPSP measured during the 10 min that have preceded the induction of LTP.
For LTP saturation experiments, a second theta-burst protocol was delivered 60 min after LTP was first induced. LTP saturation was quantified by the LTP2/LTP1 ratio (LTP1: change in fEPSP slope at 46–60 min; LTP2: change in fEPSP slope at 106–120 min) induced by theta-burst stimulation in relation to baseline values.
Where indicated, LTP was induced 60 min following delivery of low-frequency stimulation (LFS; one train of 900 pulses, 1 Hz). In these experiments, LTP was quantified as the percentage of change in the average slope of the fEPSP taken from 46 to 60 min after LTP induction in relation to the average slope of fEPSPs measured during the 10 min that preceded theta-burst stimulation.
EPSC-whole-cell patch-clamp recordings
Glass micropipettes were pulled from a 1.0 mm outer diameter thin-walled capillary tube (Warner Instruments) obtaining a resistance of 4–7 MΩ when filled with an internal solution (125 mm K-gluconate, 11 mm KCl, 0.1 mm CaCl2, 2 mm MgCl2, 1 mm EGTA, 10 mm HEPES, 2 mm MgATP, 0.3 mm NaGTP, and 10 mm phosphocreatine, pH 7.3, 280–290 mOsm). For AMPA and NMDA current–voltage relationship a different intracellular solution was used [125 mm CsCl, 8 mm NaCl, 1 mm CaCl2, 10 mm EGTA, 10 mm HEPES, 10 mm d-glucose, 5 mm MgATP, and 0.4 mm NaGTP, pH 7.2, adjusted with CsOH (50 wt% in water), 280–290 mOsm; 0.1 mm spermine was added for EPSCAMPA recordings]. Mixed NMDA/AMPA EPSCs were recorded from CA1 pyramidal cells upon stimulation of the Schaffer collateral fibers (0.2 ms rectangular pulses delivered each 15 s), in the presence of glycine (50 μm) and picrotoxin (50 μm), as described previously (Dias et al., 2012). Whole-cell recordings were performed at room temperature in voltage-clamp mode (Vh = −80 mV, after liquid junction potential correction) with either an EPC-7 (List Biologic) or an Axopatch 200B (Molecular Devices) amplifier, under the control of pClamp10 software (Molecular Devices). CA1 pyramidal cells were identified by visualization with upright microscopes (Zeiss Axioskop 2FS) equipped with infrared video microscopy and differential interference contrast optics. Holding current and series resistance were continuously monitored throughout the experiment, and if any varied significantly, the experiment was discarded.
Biotinylation and immunoblot assay
Hippocampal slices were isolated and incubated with 500 nm a-syn or with the equivalent volume of vehicle. Fresh slices were submitted to a biotinylation technique as described previously (Dias et al., 2012) to isolate the membrane fraction from the total fraction. Slices were washed three times with ice-cold PBS/Ca2+/Mg2+ washing buffer (137 mm NaCl, 2.7 mm KCl, 4.3 mm NaH2PO4, 1 mm CaCl2, and 0.5 mm MgCl2) and then incubated with 1 mg · ml−1 of EZ-Link Sulfo-NHS-LC-biotin (Sigma) resuspended in a tetraethylammonium (TEA) buffer (10 mm TEA, 2 mm CaCl2, and 150 mm NaCl), for 60 min at 4°C using gentle rotation. Slices were washed with the same washing buffer and incubated with 100 mm glycine solution for 30 min at 4°C using gentle rotation to quench free biotin. Followed by another round of washes, slices were finally lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, 1 mm EDTA, 150 mm NaCl, and 0.1% SDS; 1% NP40, pH 8) supplemented with protease and phosphatase inhibitors (Roche) using a mechanical homogenization. Samples were centrifuged at 20,000 relative centrifugal force for 10 min, supernatants were saved, and total protein concentration was measured with BCA. Approximately 400 μg of total protein of each sample was incubated with 80 μl streptavidin beads conjugated (Sigma) overnight with gentle rotation. Samples were centrifuged again and pellets were saved as membrane fraction. Pellets were washed three times with RIPA buffer, resuspended in PBS, and heated 5 min at 95°C.
Samples were loaded onto a polyacrylamide gel (7.5%) and SDS-PAGE separation was done as previously mentioned. Proteins were then transferred to a nitrocellulose membrane (Bio-Rad) blocked with blocking solution for 60 min at room temperature. Membranes were incubated overnight at 4°C with anti-Glur1 (Millipore) primary antibody using a dilution of 1:1000 in blocking solution. Membranes were washed and incubated for 60 min at room temperature with the respective HRP-conjugated secondary antibody (Invitrogen) using a dilution of 1:10,000 in blocking solution. Membranes were developed as previously mentioned with 5 min of exposure time. Films were scanned and densitometry performed using ImageJ (Abramoff et al., 2004). Each immunoblot was repeated at least three times from independent experiments.
Statistical analyses
The significance of the differences between the mean values obtained in two different conditions in the same slice (e.g., absence vs presence of APV) was evaluated by Student's t test. The Emax values were obtained by extrapolation upon nonlinear fitting of the I/O curve and an F test was used to determine differences between the parameters.
For patch-clamp experiments, statistical significance was assessed by two-tailed Student's t test. Paired comparisons were made between absolute current values before and after application of a given drug; unpaired comparisons were applied to data from distinct experimental groups. A p value <0.05 was considered statistically significant. All values are expressed as mean ± SEM from n experiments.
Results
Preparation and characterization of a-syn oligomeric species
To investigate the effect of different a-syn species on neuronal function, monomers, two types of oligomeric species (unmodified and HNE-modified oligomers) (Näsström et al., 2011), and fibrils were prepared. Upon separation on a standard SDS-PAGE, we confirmed that monomers migrated with a typical molecular weight of ∼15 kDa (monomeric molecular weight of a-syn). Unmodified oligomeric a-syn samples displayed a molecular weight of >30 kDa and <180 kDa and, in contrast, a-syn HNE-oligomers displayed a high range of molecular weights, whereas fibrils displayed a very high molecular weight of >180 kDa (Fig. 1a). As expected, Thioflavin T interacted with a-syn oligomers, a-syn HNE-oligomers, and fibrils (Fig. 1b). To confirm the structural properties of the different species used in the study, we also performed AFM. While in the monomeric a-syn preparation no detectable species were identified, the a-syn oligomers and a-syn HNE-oligomers appeared as a heterogeneous population with globular and protofibrillar structures identical to those described in the literature. For the fibrillar sample, only large aggregates were identified (Fig. 1c). To assess putative toxic actions of the different a-syn species, we investigated their effect when applied exogenously to SH-SY5Y neuroblastoma cells. We detected no significant differences in the cytotoxicity induced by any of the species tested (Fig. 1d). Moreover, we controlled a possible overexposure to a-syn by comparing the amount of endogenous synuclein in rat brain extracts with the extracts after incubation with 500 nm a-syn oligomers for 90 min (Fig. 1e). Western blot analysis revealed an enrichment in a-syn within the same range of that found in the CSF of the A53T versus WT mice (Emmanouilidou et al., 2011). Therefore, for the complete study, we selected 500 nm a-syn.
Figure 1.

Characterization of a-syn species and biological effects. a, SDS-PAGE separation of the different a-syn species (monomers, oligomers, HNE-oligomers, and fibrils). Monomers migrate with monomeric molecular weight (15 kDa) whereas a-syn oligomers, a-syn HNE-oligomers, and fibrils display SDS-resistant high molecular weight species. b, Thioflavin T normalized fluorescence assay in the presence of the different a-syn species with excitation and emission wavelengths of 450 and 490 nm, respectively. The a-syn oligomers, a-syn HNE-oligomers, and fibrils interacted with Thioflavin T, suggesting that they display an increased β-sheet structure, typical of oligomeric and fibrillary species. c, AFM analysis of the different a-syn species. No detectable species are shown for monomers; a-syn oligomers and a-syn HNE-oligomers appeared as a heterogeneous population with globular and protofibrillar structures; fibrils displayed fibrillary structures. Scale bar, 500 nm. d, Cytotoxicity of a-syn species in SH-SY5Y cells. LDH activity of SH-SY5Y cells incubated with different a-syn species. LDH release was expressed as relative levels of nontreated (Ctrl) samples. Data are expressed as mean ± SD, n = 3. e, To control a possible overexposure to a-syn we performed immunoblot analysis comparing endogenous a-syn to that from hippocampal slices treated with a-syn oligomers (500 nm; 90 min). We detected an increment of ∼15% incorporation in the slices exposed to exogenous a-syn.
a-syn oligomeric species impair LTP
To evaluate the effect of a-syn oligomers on LTP we performed extracellular recordings of fEPSPs evoked by stimulation of Schaffer-collaterals/CA1 glutamatergic fibers in hippocampal slices pre-incubated with a-syn oligomers (500 nm for 90 min). LTP was significantly reduced when compared with that observed in slices treated with vehicle only (control slices) in which the theta-burst stimulation caused a robust potentiation (LTPCtr 31.5 ± 4.6%; LTPa-syn oligomers 2.7 ± 3.9%; n = 6 per group; p < 0.05, t test; Fig. 2a,b,f).
Figure 2.

a-syn oligomeric species impair LTP. a, Changes of fEPSP slope induced by theta-burst stimulation in control slices (○, n = 6) and in slices pre-incubated with a-syn oligomeric species (●, n = 6, 500 nm; 90 min). b, Traces from representative experiments obtained before (1) and 46–60 min after (2) LTP induction, composed of the stimulus artifact followed by the presynaptic volley and the fEPSP. c, Changes in fEPSP slope induced by theta-burst stimulation in control slices (○, n = 3) and in slices pre-incubated with a-syn monomeric species (●, n = 3, 500 nm; 90 min). d, Changes of fEPSP slope induced by theta-burst stimulation in control slices (○, n = 3) and in slices pre-incubated with a-syn HNE-oligomers (●; n = 5) (500 nm; 90 min). e, Changes of fEPSP slope induced by theta-burst stimulation in control slices (○, n = 3) and in slices pre-incubated with a-syn fibrils (●, n = 3, 500 nm; 90 min). f, LTP magnitude (change in fEPSP slope at 46–60 min) induced by theta-burst stimulation in relation to pre-theta-burst values (100%) in control slices and those that had been pre-incubated with a-syn oligomeric species (500 nm; 90 min), a-syn monomers (500 nm; 90 min), and a-syn fibrils (500 nm; 90 min) as indicated below each column. All values are mean ± SEM. g, Changes of fEPSP slope induced by theta-burst stimulation in control slices (○, n = 3) and in slices pre-incubated with insulin oligomeric species (●, n = 3, 500 nm; 90 min). h, Plot of the LTP magnitude (change in fEPSP slope at 46–60 min) induced by theta-burst stimulation in relation to pre-theta-burst values (100%) from experiments shown in g as indicated below each column. All values are mean ± SEM.
To assess the specificity of the effects observed with a-syn oligomers, we tested the same concentration of insulin oligomers, which caused no significant changes in LTP magnitude (LTPCtr 35.1 ± 14.9%, n = 3; LTPinsulin oligomers 41.9 ± 11.0%; p > 0.05; n = 3; Fig. 2g,h). There was a tendency for an increase in post-tetanic potentiation, which did not reach statistical significance (p = 0.19; n = 3). It was previously shown by others that insulin, by itself, does not affect LTP magnitude in animals of similar age (Zhao et al., 2010).
Stabilized a-syn HNE-oligomeric species, but not monomers or fibrils, impair LTP
In contrast to what we observed for oligomers, pre-incubation of hippocampal slices with a-syn monomers (500 nm for 90 min) did not significantly alter the magnitude of LTP when compared with control slices (LTPCtr 24.2 ± 3.6%; LTPmonomers 24.3 ± 5.6%, n = 3 per group; Fig. 2c,f). Similar results were obtained for slices incubated with fibrillar a-syn species (LTPfibrils 40.0 ± 13.0%; Fig. 2e,f). These data show that oligomeric but not monomeric or fibrillar a-syn species influence synaptic plasticity. Oligomeric a-syn species can undergo post-translational modifications like those prompted by oxidative stress, which give rise to a-syn HNE-oligomers, also detected in PD brains (Friguet et al., 1994). We then tested the effect of HNE-stabilized a-syn oligomers to determine whether they elicited similar effects to those obtained with unmodified oligomers. LTP recorded from slices pre-incubated with a-syn HNE-oligomers (500 nm for 90 min) had a similar magnitude to that recorded from slices treated with the unmodified oligomeric form, which was significantly smaller than that observed in control slices (LTPCtr 30.6 ± 1%, n = 3; LTPHNE-oligomers 8.8 ± 2.7%, n = 5; p < 0.05, t test; Fig. 2d,f).
a-syn oligomers enhance basal synaptic transmission through NMDA receptor activation
NMDA receptors are coincident detectors of high-frequency neuronal firing and postsynaptic depolarization and their function is critical for LTP induction and expression (Bliss and Collingridge, 1993; Collingridge et al., 2004). To investigate whether exacerbation of NMDA receptor function was involved in the effect of a-syn oligomeric species on LTP, slices were pre-incubated for 90 min with an NMDA receptor antagonist, APV (50 μm) (Morris et al., 1986), in the presence or in the absence of a-syn oligomers. APV was washed out together with a-syn oligomers before recording any synaptic events. This pretreatment with APV blocked the disruption of LTP induced by a-syn-oligomers and resulted in LTP levels similar to those obtained in the control (LTPa-syn oligomers+APV 27.9 ± 8.3%, n = 4). Pretreatment with APV alone did not affect LTP magnitude (LTPCtr+APV 29.8 ± 6.3%, n = 5; Fig. 3 a,b). Similarly, the decrease in LTP magnitude observed upon treatment with a-syn HNE-oligomers was fully recovered by pre-incubating slices with 50 μm APV (LTPHNE-oligomers+APV 22.7 ± 1.4%; n = 3; Fig. 3c,d). In contrast, coincubation of a-syn-treated slices with calcium-permeable AMPA receptor antagonist (NASPM, 100 μm) was unable to restore LTP. These data suggest that, during incubation with a-syn HNE-oligomers, activation of NMDA receptors (but not calcium-permeable AMPA receptors) is required for subsequent LTP impairments.
Figure 3.

The LTP impairment induced by unmodified and HNE a-syn oligomers is fully recovered by NMDA receptor blockade. a, Changes in fEPSP slope induced by theta-burst stimulation in slices pre-treated for 90 min with the NMDA receptor antagonist, APV (50 μm), with (n = 4) or without (n = 5) a-syn oligomeric species (*p < 0.05, t test). b, Plot of the LTP magnitude (change in fEPSP slope at 50–60 min) induced by theta-burst stimulation in relation to pre-theta-burst values (100%) from experiments shown in a as indicated below each column. c, Changes in fEPSP slope induced by theta-burst stimulation in slices pretreated for 90 min with the NMDA receptor antagonist, APV (50 μm), with (n = 4) or without (n = 3) HNE a-syn oligomers (*p < 0.05, t test). In a and c the ordinates represent normalized fEPSP slopes where 100% corresponds to the averaged slopes recorded for 10 min before theta-burst stimulation and the abscissae represent the time (min). d, Plot of the LTP magnitude (change in fEPSP slope at 46–60 min) induced by theta-burst stimulation in relation to pre-theta-burst values (100%) from experiments shown in c as indicated below each column. All values are mean ± SEM.
To evaluate whether the changes observed in LTP magnitude in the presence of either unmodified or HNE-modified a-syn oligomers could be due to changes in baseline synaptic efficiency, I/O curves were performed. a-syn-treated slices displayed higher Emax values when compared with vehicle (EmaxCtr = 1.2 ± 0.04 mV/ms, n = 5; Emaxa-syn oligomers = 2.0 ± 0.04 mV/ms, n = 6; p < 0.05, F test; Fig. 4a). As expected, similar results were obtained in the I/O curves from slices pre-incubated with HNE-modified a-syn oligomers.
Figure 4.

a-syn oligomers enhance basal synaptic transmission in the hippocampus. a, I/O curves corresponding to responses generated between fiber volley amplitude and fEPSP slope evoked by various stimulation intensities (60–300 μA) in control slices (○, n = 5), slices treated with a-syn oligomers (●, n = 6) (500 nm, 90 min) and slices treated with a-syn oligomers (■, n = 4) coincubated with 50 μm APV. The increase in synaptic transmission induced by a-syn oligomers was completely recovered by coincubation with APV (*p < 0.05, ANOVA F test). b, Averaged time course of changes in fEPSP slope after superfusion with APV for 30 min, in slices treated with (●, n = 4) or without (○, n = 4) a-syn oligomeric species (500 nm, 90 min) (*p < 0.05, t test). In slices treated with a-syn oligomers the acute application of APV induces a decrease in the EPSP slope. c, Averaged fEPSP (change in slope from the last 10 min of APV application) from acute APV superfusion experiments as indicated below each column. All values are mean ± SEM.
To assess the involvement of NMDA receptors in facilitation of baseline synaptic efficiency, APV (50 μm) was added to the incubation solution together with a-syn oligomers (unmodified or HNE) for 90 min. Again, APV was washed away together with a-syn oligomers before recording of synaptic events. This pre-incubation with APV completely prevented the shift observed in the I/O curve of a-syn oligomer-treated slices (Emaxa-syn oligomers+APV = 0.9 ± 0.02 mV/ms; n = 4 per group; Fig. 4a) as well as in a-syn HNE-oligomer-treated slices. This suggests that NMDA receptors might be contributing to basal synaptic transmission in a-syn oligomer-treated slices. Pre-incubation with APV alone had no effect on I/O curves in the absence of oligomers (n = 3). The acute influence of APV applied during recordings of fEPSPs was therefore evaluated. Adding APV to the recording chamber for 30 min induced a progressive and significant decrease (p < 0.05, t test) of fEPSP slope in a-syn oligomer-treated slices (unmodified: fEPSP 13.9 ± 2.8%, n = 4; Fig. 4b,c; HNE: fEPSP 13.3 ± 1.6%; n = 3; Fig. 4c), but not in control slices (Fig. 4c).
a-syn HNE-oligomers increase the NMDA component of synaptic transmission
To further dissect the mechanism through which a-syn oligomers affect synaptic transmission, whole-cell patch-clamp recordings of afferent-evoked EPSCs were performed from CA1 pyramidal neurons (Vh = −80 mV), in the presence of extracellular Mg2+ (1 mm), glycine (50 μm), and the GABAA receptor antagonist picrotoxin (50 μm; Fig. 5). In agreement with the fEPSP data, APV superfusion significantly inhibited EPSCs recorded from slices treated with 500 nm a-syn HNE-oligomers for 90 min (EPSCCtr 101 ± 1.1% of the baseline; EPSCHNE-oligomers 81.4 ± 2.9% of the baseline; n = 11 per group; p < 0.05, t test; Fig. 6a–c). Furthermore, superfusion of the AMPA/kainate receptor antagonist CNQX (10 μm; 20–25 min) revealed a residual EPSC component that was significantly larger in slices treated with a-syn HNE-oligomers (EPSCCtr 6.9 ± 1.1% of the baseline; EPSCHNE-oligomers 14 ± 2.1% of the baseline; n = 7 per group; p < 0.05, t test) and was blocked by APV (Fig. 6f–h). Similar modulatory effects were also observed when using oligomeric unmodified species both for APV (EPSCCtr 100 ± 1.7% of the baseline; EPSCa-syn oligomers 84 ± 2.9% of the baseline; n = 4 per group; p < 0.05, t test; Fig. 6d,e) and CNQX application (EPSCCtr 3.8 ± 0.7% of the baseline; n = 6; EPSCa-syn oligomers 9.7 ± 1.8% of the baseline; n = 4; p < 0.05, t test; Fig. 6i,j).
Figure 5.

Treatment with HNE-oligomers induces LTP saturation, which is reversed by prior LFS. a, Changes of fEPSP slope induced by two consecutive theta-burst stimulations in a representative control slice (○) and in a slice pre-incubated with a-syn oligomeric species (●, 500 nm; 90 min). b, LTP2/LTP1 ratio (LTP1: change in fEPSP slope at 46–60 min; LTP2: change in fEPSP slope at 106–120 min) induced by theta-burst stimulation in relation to baseline values (100%; −10 to 0 min) in control slices (n = 3) and those that had been pre-incubated with HNE a-syn oligomeric species (n = 3; 500 nm; 90 min) (*p < 0.05, t test). c, Changes of fEPSP slope induced by theta-burst stimulation in slices treated with HNE a-syn oligomers (500 nm; 90 min) with ●, n = 4) or without (○, n = 4) previous delivery of a LFS protocol, which decreased synaptic transmission by 8.3 ± 6.8 (n = 4). d, LTP magnitude (change in fEPSP slope at 46–60 min) induced by theta-burst stimulation in relation to pre-theta-burst values (100%) in slices pre-incubated with HNE a-syn oligomeric species (500 nm; 90 min) with (n = 4) or without (n = 4) previous delivery of a LFS protocol as indicated below each column. *p < 0.05, t test. All values are mean ± SEM.
Figure 6.
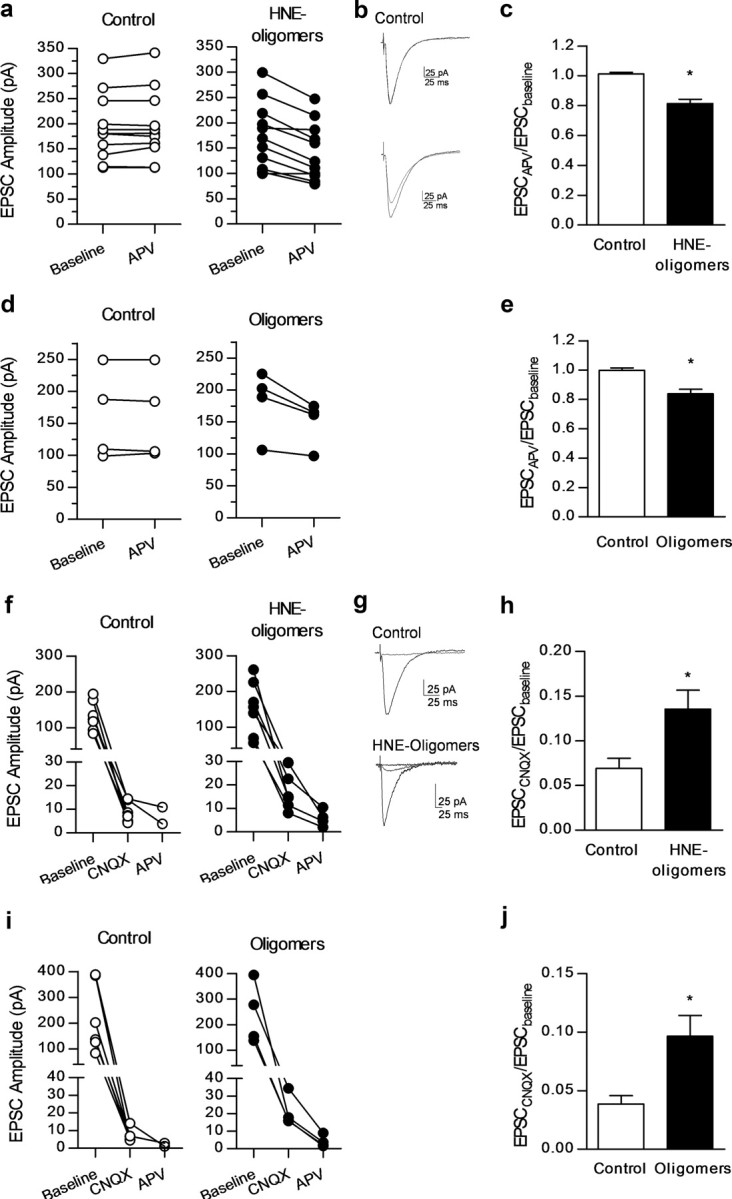
Treatment with a-syn oligomers is associated with an increased NMDA component of synaptic transmission. a, APV (50 μm, 20 min) significantly decreased EPSC amplitude in a-syn HNE-oligomer-treated neurons (●) but not in control ones (○), as evidenced by representative current traces shown in b. The effect of APV is illustrated as the evoked EPSC amplitude after APV application normalized to the baseline period (c). (*p < 0.05, t test, n = 11 per group). d, APV (50 μm) superfusion for 20 min decreased EPSC amplitude in neurons treated with a-syn oligomers (●) but not in control neurons (○). e, Average effects of APV superfusion are illustrated as the EPSC amplitude after drug application normalized to the baseline period. (p < 0.05, n = 4 per group). f, Blockade of EPSCs by CNQX (10 μm, 20–25 min) was virtually complete in control neurons (○) but not in those treated with a-syn HNE-oligomers (●), where small residual currents could subsequently be blocked by APV. Representative current tracings (g) are shown for each condition, corresponding to the baseline period and upon CNQX superfusion (control and treated neurons); tracings recorded after APV superfusion are additionally shown for HNE a-syn oligomer-treated neurons. h, Residual currents recorded in the presence of CNQX, normalized to the baseline period, were significantly larger in a-syn HNE-oligomer-treated neurons compared with control ones (n = 7 per group; *p < 0.05, t test). i, EPSC blockade by CNQX (10 μm, 20–25 min) was virtually complete in control neurons (○) but not in those treated with a-syn oligomers (●) where small residual currents were subsequently blocked by APV. j, Residual currents recorded in the presence of CNQX, normalized to the baseline period, were significantly larger in a-syn oligomer-treated neurons (n = 4) than in control ones (n = 6) (*p < 0.05, t test).
To investigate possible changes in the gating properties of NMDA receptors, current–voltage (I-V) relationships were performed in the presence of CNQX (10 μm) to isolate the NMDA component of the EPSCs. The voltage-dependency of NMDA receptor activation, including current rectification at negative holding potentials, was similar in control and a-syn HNE-oligomer-treated slices (EPSC−90 mV/EPSC+40 mv ratio: 0.7 ± 0.2 in control, n = 5; 0.6 ± 0.1 in a-syn HNE-oligomers, n = 7; Fig. 7a–c). No significant changes in resting membrane potential or in input resistance values were observed between conditions. The absence of changes in current–voltage relationships from control and treated neurons indicates that treatment with a-syn HNE-oligomers does not affect NMDA receptor properties. Importantly, higher stimulus intensities (near twofold increase) were required to elicit NMDA-mediated EPSCs of comparable size in control slices than in a-syn HNE-oligomer-treated slices, in agreement with an a-syn HNE-oligomer-induced increase in the contribution of NMDA receptors for basal synaptic transmission (Fig. 6). It should also be noted that NMDA-mediated EPSCs could be recorded even at hyperpolarized potentials, despite the presence of extracellular magnesium (Fig. 7). In fact, NMDA receptor voltage-dependent blockade by magnesium at resting potentials is not absolute (Espinosa and Kavalali, 2009) and both increased extracellular availability of glutamate as well as increased receptor levels could account for the observed gain of function. Together, these data suggest that increased contribution of NMDA receptors to synaptic transmission by treatment with a-syn HNE-oligomers does not involve alterations in NMDA receptor gating.
Figure 7.

a-syn HNE-oligomers induce changes in AMPA receptor subunit composition. a, Normalized EPSCNMDA current–voltage relationships recorded from neurons in control conditions (○, n = 5) and after incubation with a-syn HNE-oligomers (●, n = 7), in the presence of CNQX (10 μm). b, Representative EPSCNMDA traces recorded at +40 and −90 mV for each condition. c, Rectification index values (EPSC−90 mV/EPSC+40 mV) for NMDA receptor-mediated EPSCs calculated from control and a-syn HNE-oligomers. d, Plots of normalized EPSCAMPA current–voltage relationships recorded in the presence of APV (50 μm) from control (○, n = 11) and a-syn HNE-oligomer-treated (●, n = 9) treated slices. e, Representative EPSCAMPA traces recorded at +40 and −60 mV from control and a-syn HNE-oligomers. f, Rectification index values (EPSC+40 mV/EPSC−60 mV) for AMPA receptor-mediated EPSCs calculated from control and a-syn HNE-oligomers treated neurons (*p < 0.05, t test). g, h, i, Plots of individual EPSC amplitudes before and after NASPM (100 μm, 20 min) superfusion. NASPM significantly decreased EPSC amplitude in a-syn HNE-oligomer-treated neurons (●, n = 6) but not in control ones (○, n = 6) nor in those coincubated with HNE-oligomers and APV (50 μm) (●, n = 4). j, Effect of NASPM illustrated as the averaged EPSC amplitude after NASPM superfusion normalized to the baseline period (*p < 0.05, t test). All values are mean ± SEM. k, Immunoblot analysis of samples from rat hippocampal slices incubated with a-syn HNE-oligomers (500 nm; 90 min) and control, were submitted to biotinylation technique and immunoblotted with anti-GluR1 antibody. In samples incubated with a-syn HNE-oligomers it is possible to detect an increment in the membrane fraction of GluR1 subunit levels. l, Quantification of three independent experiments. Statistically significant values (*p < 0.05 t test) are normalized to those of total fraction.
An alternative mechanism that could explain the LTP impairment is the possibility that enhanced NMDA receptor-mediated signaling under basal conditions leads to LTP saturation. To directly address this hypothesis, we performed LTP saturation experiments in which a second induction of LTP (LTP2) was elicited 60 min after the first one (LTP1). In contrast to what was observed in control conditions, delivery of a second theta-burst protocol to slices treated for 90 min with a-syn HNE-oligomers produced no facilitation above baseline (pre-theta-burst stimulation) values (LTP2/LTP1CTR ratio: 1.8 ± 0.1 n = 3; LTP2/LTP1HNE-oligomers ratio: 0.87 ± 0.4, n = 3; p < 0.05, Fig. 5a,b). In a subsequent set of experiments, we further found that a-syn HNE-oligomer-induced LTP saturation could be reversed by delivery of LFS, 60 min before inducing LTP (LTPHNE-oligomers 12.4 ± 3.3%, n = 4; LTPHNE-post LFS 40.1 ± 8.9%, n = 4; p < 0.05; Fig. 5c,d). These findings are consistent with common substrates underlying bidirectional synaptic plasticity, namely with the ability for both theta-burst stimulation and LFS to affect the phosphorylation status and membrane levels of GluR1-containing AMPA receptors (Lee et al., 2000) and point toward involvement of AMPA receptors in the synaptic dysfunctions induced by a-syn HNE-oligomers. Since it is not necessary to activate AMPA receptors during incubation with a-syn HNE-oligomers (see above), they might be involved downstream in the cascade of injury induction.
a-syn HNE-oligomers alter AMPA receptor subunit composition
Changes in synaptic efficiency and plasticity also rely upon tuning of the synaptic AMPA receptor component (Bliss and Collingridge, 1993). GluR2-lacking AMPA receptors are characterized by a high permeability to calcium and strong inward current rectification due to voltage-dependent block by intracellular polyamines (Donevan and Rogawski, 1995), which allows their distinction from GluR2-containing AMPA receptors while performing current–voltage relationships in the presence of APV (to isolate the AMPA component of EPSCs). Current rectification at positive potentials was markedly increased in a-syn HNE-oligomer-treated neurons, when compared with control (EPSC+40 mV/EPSC−60 mV ratio: 0.47 ± 0.07 in control, n = 11; 0.18 ± 0.03 in a-syn HNE-oligomers, n = 9; p < 0.05, t test; Fig. 7d–f). The increased contribution of inwardly rectifying GluR2-lacking AMPA receptors for synaptic transmission in slices treated with a-syn HNE-oligomers was further confirmed by bath application of NASPM (100 μm), a selective blocker of AMPA receptors (McElligott et al., 2010). Indeed, NASPM superfusion for 20 min significantly decreased EPSC amplitude recorded from a-syn HNE-oligomer-treated neurons, but not from control neurons (EPSCCtr 100 ± 0.9% of the baseline; EPSCHNE-oligomers 80.9 ± 1.6% of the baseline; n = 6 per group; p < 0.05, t test; Fig. 7g–j). NASPM was also devoid of effect upon neurons that had been simultaneously incubated with a-syn HNE-oligomers and APV (50 μm; 99.4 ± 1.1%; n = 4; Fig. 7i,j), suggesting that changes in AMPA receptor subunit composition are downstream to NMDA receptor activation.
a-syn HNE-oligomers induce an increase in GluR1 AMPA receptors levels
In the mature hippocampus, most GluR2-lacking AMPA receptors are thought to correspond to homomeric assemblies of GluR1 subunits (Wenthold et al., 1996). To evaluate the surface membrane expression of GluR1 subunits, we combined biotinylation assays with immunoblot analysis. Our data showed that in slices incubated with a-syn HNE-oligomers there was a significant increase in GluR1 expression (>1.7) in the membrane fraction (n = 3; p < 0.05, t test, normalized to the total lysate values; Fig. 7k,i).
Discussion
Protein misfolding and aggregation are key events in several neurodegenerative disorders such as PD and Alzheimer's disease. Oligomeric species of the proteins implicated in these diseases, rather than the fibrillar forms, seem to be the more bioactive and, possibly, cytotoxic (Lansbury and Brice, 2002; El-Agnaf et al., 2003; Cookson, 2005; Outeiro et al., 2007; Martin et al., 2012). In addition, the levels of HNE-modified a-syn are increased in LBs in the brains of PD patients (Yoritaka et al., 1996; Castellani et al., 2002). Previous studies showed that monomeric species of Aβ peptide (Aβ1–42) are neuroprotective while oligomeric and fibrillary species are toxic and impair LTP (Townsend et al., 2006; Selkoe, 2008; Shankar et al., 2008; Giuffrida et al., 2009) suggesting similarities in the mechanism of action of these amyloidogenic proteins. Nevertheless, the mechanisms by which different a-syn species cause sustained changes in synaptic transmission had not been previously explored.
Our findings demonstrate that prolonged treatment with a-syn oligomers, but not monomers or fibrils, caused a long-lasting increase in hippocampal basal synaptic transmission through NMDA receptor activation, triggering enhanced contribution of calcium-permeable AMPA receptors for synaptic transmission and LTP impairment. Prior delivery of an LFS train rescued LTP in treated slices, consistent with our observation that LTP was saturated in slices incubated with HNE a-syn oligomers. Importantly, our data suggest that the effects are not simply due to increased cytotoxicity of the oligomeric species, or to acute actions of a-syn on synapses. Acute influences of a-syn have already been tested using autaptic hippocampal cultures (Hüls et al., 2011) and the major findings are consistent with an early increase in excitability.
We found that different species of a-syn have distinct effects on synaptic activity. Whereas fibrils and monomers did not affect synaptic function, pre-incubation of hippocampal slices with a-syn oligomeric species resulted in exacerbated synaptic transmission, reflected in the steeper I/O curve. On the other hand, synaptic plasticity was compromised since LTP induced by theta-burst stimulation was impaired. These effects were detected with two different preparations of a-syn oligomeric species, including HNE-stabilized oligomers, a post-translationally modified form detected in PD brains (Friguet et al., 1994). This effect seems to be related to the specific properties of a-syn, since oligomeric species of insulin, another fibrillization-prone protein, did not induce a similar effect.
The increase in synaptic transmission and abolishment of LTP induced by a-syn oligomeric species required NMDA receptor activation, since both effects were prevented by APV, a selective antagonist of this glutamate receptor subtype. This suggests that a-syn oligomers were exacerbating NMDA receptor activation, as already seen for Aβ oligomers (Kelly and Ferreira, 2006). To address this hypothesis, we studied the contribution of NMDA receptors for basal synaptic transmission, known to be minimal under physiological conditions. Indeed, NMDA receptor blockade had no noticeable effect in untreated slices, but it significantly inhibited synaptic transmission in a-syn-treated slices. Notably, this was even observed in neurons clamped at negative (−80 mV) membrane potentials, which physiologically restrains NMDA receptor activation. Although this increase in NMDA receptor-mediated transmission was not accompanied by a shift in the channel-gating properties, it does not exclude the occurrence of other modifications underlying the observed gain of function. Because these receptors play a crucial role in synaptic plasticity (Bliss and Collingridge, 1993; Collingridge et al., 2004), disrupted NMDA receptor function is expected to affect the ability of individual synapses to undergo reinforcement in response to high-frequency neuronal firing. This could occur through either an inability of the receptor to function as a coincidence detector or through anomalous NMDA receptor-mediated signaling upstream of transduction pathways leading to an enhancement of synaptic strength that prevents further facilitation. While the first hypothesis is discouraged by the absence of changes in the I-V plots of neurons from HNE a-syn oligomer-treated slices, the second hypothesis is favored by the observation that LTP was saturated in a-syn HNE-oligomer-treated slices and that prior delivery of a standard LFS train was able to rescue LTP back to control levels (Fig. 8).
Figure 8.

Model for LTP impairment induced by a-syn oligomers in CA1 pyramidal synapses. a, Under physiological conditions, theta-burst stimulation (TBS) induces Ca2+ influx through NMDA receptors into the postsynaptic cell and the activation of calmodulin (CaM). Through currently unknown mechanisms, Ca2+/CaM activates Ca2+/CaM-dependent protein kinase II (CaMKII), which modulates synaptic efficiency through phosphorylation of GluR1 subunits. In addition to prompting an enhancement in single-channel conductance, CaMKII promotes the trafficking and transient insertion of calcium-permeable AMPA receptors into the postsynaptic membrane, thought to be critical for the induction and/or stabilization of LTP. b, When exposed to a-syn oligomers, synapses lose the ability to respond to theta-burst stimulation. In resting synapses, the oligomers are able to activate, either directly or indirectly, the NMDA receptors, leading to an increase of NMDA channel contribution to transmission. This promotes a basal increase in the intracellular calcium and modulation of AMPA receptor trafficking, favoring the insertion of higher conductance calcium-permeable versus GluR2-containing AMPA receptors that facilitates basal synaptic transmission. Whenever activated by a theta-burst, the synapse is saturated and unable to recruit extra AMPA receptors, which translates into impaired long-term potentiation.
NMDA receptor activation is critical for determining the expression level and composition of AMPA receptors at the postsynaptic membrane (Bliss and Collingridge, 1993; Martin et al., 2000; Collingridge et al., 2004), through which significant alterations of synaptic transmission and plasticity are achieved (Plant et al., 2006; Guire et al., 2008). For instance, in neurons overexpressing the GluR1 subunit, prompting the formation of calcium-permeable GluR1 homomeric receptors, LTP rapidly decays to the baseline within 20 min while cells expressing GluR2-containing AMPA receptors exhibit large and sustained LTP (Shi et al., 1999; Collingridge et al., 2004). Interestingly, our results revealed an increase in the expression of GluR1-containing receptors upon treatment with a-syn HNE-oligomers, accompanied by an increase in the contribution of calcium-permeable AMPA receptors for basal synaptic transmission. This was further corroborated by the EPSC inhibition caused by NASPM, a blocker of GluR2-lacking AMPA receptors. These changes might contribute to LTP impairment since a proper balance of GluR1 and GluR2 subunits is critical for LTP induction and maintenance. Indeed, calcium-permeable GluR2-lacking AMPA receptors are required to trigger their replacement with GluR2-containing AMPA receptors, which underlie synaptic stabilization (Pozo and Goda, 2010). It is therefore plausible that, by increasing the synaptic expression of GluR2-lacking receptors, a-syn oligomers compromise AMPA receptor remodeling necessary for synaptic reinforcement (Fig. 8). This effect seems to be dependent on activation of NMDA receptors, since acute EPSC inhibition by NASPM was lost when slices were coincubated with a-syn HNE-oligomers and APV. Importantly, calcium entry through NMDA and GluR2-lacking AMPA receptors plays a prominent role not only in plasticity, but also in the excitotoxicity phenomena known to be involved in the pathogenesis of several conditions such as stroke, epilepsy, and several neurodegenerative diseases (Lipton and Rosenberg, 1994; Doble, 1999). Hence, the present results may help shed light into the mechanisms by which extracellular a-syn oligomers may trigger neuronal dysfunction and death. The toxicity induced by a-syn oligomers might be a consequence of enhanced Ca2+ influx through NMDA receptors (Kelly and Ferreira, 2006; Martin et al., 2012). Recently, it was shown that soluble Aβ oligomers affect GluR1 phosphorylation and AMPA receptor trafficking through increased calcium influx and activation of calcium-dependent phosphatase calcineurin (Miñano-Molina et al., 2011). Our data fully support a similar mechanism for a-syn oligomers.
Moreover, since ionotropic glutamate receptors also play a key role in regulating the strength of corticostriatal excitatory synapses, their abnormal activation might be also involved in the alteration of neuronal firing patterns in certain regions of the basal ganglia and significantly contribute to the motor symptoms associated with PD (Wenthold et al., 1996; Hallett and Standaert, 2004; Mullasseril et al., 2010).
Altogether, our findings provide important insight into the mechanisms associated with chronic accumulation of extracellular a-syn oligomeric species and might contribute to the understanding of the neuronal dysfunction observed in PD and other synucleinopathies, opening novel avenues for therapeutic intervention.
Footnotes
This research is supported by a Marie Curie International Reintegration Grant (Neurofold; T.F.O.), an EMBO Installation Grant, Fundação para a Ciência e Tecnologia (FCT), PTDC/QUI/73430/2006 Grant from FCT (A.Q), PDCT/SAU-NMC/099853/2008 from FCT (L.V.L.), PDCT/SAU-NMC/110838/2009 from FCT (A.M.S.), FCT fellowships SFRH/BD/60386/2009 (D.M.R.) and SFRH/BD/27761/2006 (R.B.D.), FCT fellowship SFRH/BPD/64702/2009 (H.V.M.), and by the University of Rome Sapienza (F.M.). We also thank Alex Kasrayan and Monica Ekberg for the production of recombinant a-syn.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Anderson WW, Collingridge GL. The LTP program: a data acquisition program for on-line analysis of long-term potentiation and other synaptic events. J Neurosci Methods. 2001;108:71–83. doi: 10.1016/s0165-0270(01)00374-0. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, Smith MA. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–129. [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Dias RB, Ribeiro JA, Sebastião AM. Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. Hippocampus. 2012;22:276–291. doi: 10.1002/hipo.20894. [DOI] [PubMed] [Google Scholar]
- Diógenes MJ, Costenla AR, Lopes LV, Jerónimo-Santos A, Sousa VC, Fontinha BM, Ribeiro JA, Sebastião AM. Enhancement of LTP in aged rats is dependent on endogenous BDNF. Neuropsychopharmacology. 2011;36:1823–1836. doi: 10.1038/npp.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Intracellular polyamines mediate inward rectification of Ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci U S A. 1995;92:9298–9302. doi: 10.1073/pnas.92.20.9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D. Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17:1945–1947. doi: 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K. Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PLoS One. 2011;6:e22225. doi: 10.1371/journal.pone.0022225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101:2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friguet B, Szweda LI, Stadtman ER. Susceptibility of glucose-6-phosphate dehydrogenase modified by 4-hydroxy-2-nonenal and metal-catalyzed oxidation to proteolysis by the multicatalytic protease. Arch Biochem Biophys. 1994;311:168–173. doi: 10.1006/abbi.1994.1222. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüls S, Högen T, Vassallo N, Danzer KM, Hengerer B, Giese A, Herms J. AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J Neurochem. 2011;117:868–878. doi: 10.1111/j.1471-4159.2011.07254.x. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Kessler JC, Rochet JC, Lansbury PT., Jr The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–678. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- Lansbury PT, Jr, Brice A. Genetics of Parkinson's disease and biochemical studies of implicated gene products. Curr Opin Cell Biol. 2002;14:653–660. doi: 10.1016/s0955-0674(02)00377-0. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G. alpha-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J Neurochem. 2012;120:440–452. doi: 10.1111/j.1471-4159.2011.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Röytt ä M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol. 2000;100:285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL, Winder DG. Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci U S A. 2010;107:2271–2276. doi: 10.1073/pnas.0905568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miñano-Molina AJ, España J, Martín E, Barneda-Zahonero B, Fad ó R, Solé M, Trullás R, Saura CA, Rodríguez-Alvarez J. Soluble oligomers of amyloid-beta peptide disrupt membrane trafficking of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J Biol Chem. 2011;286:27311–27321. doi: 10.1074/jbc.M111.227504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat Commun. 2010;1:90. doi: 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsström T, Fagerqvist T, Barbu M, Karlsson M, Nikolajeff F, Kasrayan A, Ekberg M, Lannfelt L, Ingelsson M, Bergström J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of alpha-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic Biol Med. 2011;50:428–437. doi: 10.1016/j.freeradbiomed.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34:151–160. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro Morais G, Vicente Miranda H, Santos IC, Santos I, Outeiro TF, Paulo A. Synthesis and in vitro evaluation of fluorinated styril benzazoles as amyloid-probes. Bioorg Med Chem. 2011;19:7698–7710. doi: 10.1016/j.bmc.2011.09.065. [DOI] [PubMed] [Google Scholar]
- Schiff SJ, Somjen GG. The effects of temperature on synaptic transmission in hippocampal tissue slices. Brain Res. 1985;345:279–284. doi: 10.1016/0006-8993(85)91004-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MA, Moran NF, Kopelman MD. Subcortical dementia. Br J Psychiatry. 2002;180:148–151. doi: 10.1192/bjp.180.2.148. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wu X, Xie H, Ke Y, Yung WH. Permissive role of insulin in the expression of long-term potentiation in the hippocampus of immature rats. Neurosignals. 2010;18:236–245. doi: 10.1159/000324040. [DOI] [PubMed] [Google Scholar]