

Abstract
Proteins are versatile macromolecules that can perform a variety of functions. In the past three decades, they have been commonly used as building blocks to generate a range of biomaterials. Owing to their flexibility, proteins can either be used alone or in combination with other functional molecules. Advances in synthetic and chemical biology have enabled new protein fusions as well as the integration of new functional groups leading to biomaterials with emergent properties. In this review, we discuss protein-engineered materials from the perspectives of domain-based designs as well as physical and chemical approaches for crosslinked materials, with special emphasis on the creation of hydrogels. We also review engineered proteins that organize or template metal ions, bear non-canonical amino acids (NCAAs), and their potential applications.
Keywords: Engineered proteins, Coiled-Coils, Biomaterials, Functional materials, Recombinant protein
Graphical Abstract
ToC figure Illustration of protein engineered materials from the perspective of domain-based protein materials, crosslinked materials, metal templating protein materials and non-canonical amino acid (NCAA) bearing protein materials.
This paper provides a comprehensive review of state of art protein-engineered functional materials. The work is divided into five different sections with representative examples: single and multi-domain based protein materials, crosslinked materials, metal templating protein materials and non-canonical amino acid (NCAA) bearing protein materials. At the end, insights on the advantages of functional protein-engineered materials are discussed.
1. Introduction
Proteins are multifunctional macromolecules that regulate a number of biological processes and pathways.[1,2] With the recent advances in protein engineering including synthetic and chemical biology, new variants have been designed with improved or novel functionalities. The diverse properties of proteins make them excellent candidates for building new biomaterials. Over the past few decades, continuous efforts have been made to develop protein engineered materials, capable of replacing synthetic polymers, owing to their biocompatible and biodegradable properties.[1]
There are several advantages associated with the use of protein-based materials. The proteins can undergo conformational changes based on external stimuli, such as changes in temperature, pH, or ionic strength. These triggers can further modulate the design and synthesis of novel biomaterials with increasingly complex functions. Because of their versatile nature, proteins can be combined with a variety of other materials to create materials with novel functionalities. Additionally, the inherent property of proteins to self-assemble has paved the ways to generate new protein assemblies. By precisely controlling the self-assembly of proteins, novel architectures with improved functional properties can be designed.[3]
The increasing interest in self-assembly of proteins has contributed significantly to the development of biomaterials. These self-assembled protein based materials have a wide range of biomedical applications in tissue engineering, biosensors, drug delivery, medical imaging, gene therapies and protein therapeutics.[4–10] The properties of proteins can be dynamically regulated by the organization or templation of inorganic metal ions and more recently by introducing non-canonical amino acids (NCAAs). [11,12]
In this review, we highlight various recombinant proteins with a particular focus on their ability to assemble into various structures. In the following sections, we discuss functional materials from the perspectives of domain-based designs as well as physically- and chemically- crosslinked materials, with special emphasis on the design of hydrogels. In addition, we review protein engineered materials bearing sequences for metal crystallization and non-canonical amino acids, and their potential applications.
2. Domain-Based Protein Engineered Materials
2.1. Single Domain Protein Engineered Materials
Single domain protein engineered materials are comprised of sequences that are derived from a single protein conformation. While the sequences of single domain protein may vary with similar or different repeats, the conformation of the domain remains singular. In this section, we describe such single domains that can interact with each other to form hydrogel networks.
2.1.1. Elastin
Elastin is a major component of the extracellular matrix (ECM) that directly impacts the elasticity of blood vessels and overall movement of joints and limbs.[13] Inspired by the natural protein, tailor-made elastin-like polypeptides (ELPs) consisting of pentapeptide sequences have been investigated for their remarkable elasticity and self-assembling properties (Figure 2.1-1).[14] These properties render ELPs useful for applications in tissue engineering,[15,16] as drug delivery systems,[17–19] and as probes for bioimaging applications.[16,20,21]
Figure 2.1-1.
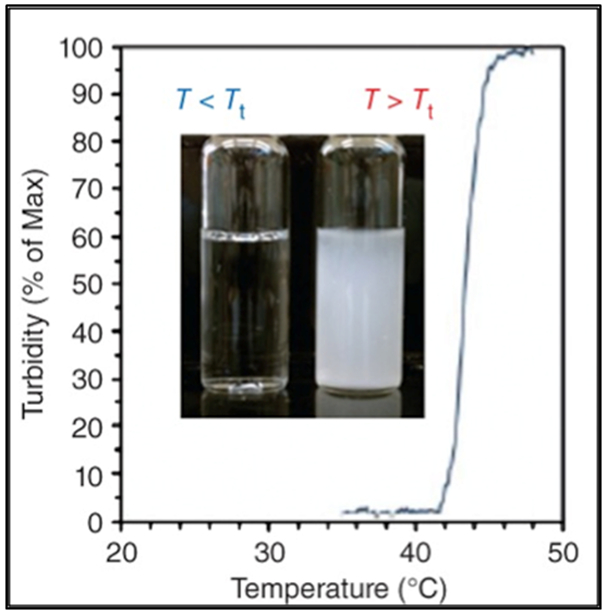
Graph shows the inverse phase transition properties for ELPs. Reproduced with permission from Ref [14].Copyright 2014, John Wiley and Sons.
ELPs are comprised of a pentapeptide sequence, Val-Pro-Gly-Xaa-Gly or (VPGXG)n, where ‘Xaa’ is an interchangeable amino acid (except proline) and ‘n’ is the number of repeating units; the properties of ELPs can be tuned by varying Xaa or n (Table 1).[16,22] The glycine and proline residues of ELPs maintain the structure and function of elastin. ELPs exhibit a unique lower critical solution temperature (LCST), which is also referred to as the inverse transition temperature (Tt); below their Tt, ELPs are soluble in aqueous solution and as the temperature increases beyond Tt, ELPs undergo phase separation and form aggregates.[23] Introduction of hydrophobic residues at the ‘Xaa” position decreases Tt while ionic and polar residues increase Tt. By varying the guest residue, it is possible to tune the ELP transition temperature from 0 to 60 °C.[24,25]
Table 1.
Sequences of single domain proteins
| Type of protein |
Name of protein |
Sequence | Reference |
|---|---|---|---|
| Elastin Protein | Elastin Like Proteins (ELPs) | (VPGVG)n (VPGAG)n[(VPGIG)2(VPGKG)(VPGIG)2] |
Urry D.W. et al. (1978) Le D.H.T. et al. (2013) Welsh E.R. et al. (2000) |
| [MSKGPG-(XGVPG)n-Y or WP] | McDaniel J.R. et al. (2013) | ||
| ELP[V5A2G3]
-n ELP[V1A8G7] -n ELP[V5]-n *n= length of the pentapeptide.. |
Meyer D.E. et al. (2004) | ||
| BAB Thermoplastic Elastomer Hydrogels | Two BAB
sequences: VPAVG[(IPAVG)4(VPAVG)]16IPAVG}-[VPGVG[(VPGVG)2VPGEG(VPGVG)2]30VPGVG]{VPAVG[(IPAVG)4(VPAVG)]16IPAVG} {VPAVG[(IPAVG)4(VPAVG)]16IPAVG}-[VPGVGVPGVG]-VPGVG{VPAVG[(IPAVG)4(VPAVG)]16IPAVG} *A block is Bold Hydrophobic B block : VPAVG[(IPAVG)4(VPAVG)]16IPAVG}-[A]-{VPAVG[(IPAVG)4(VPAVG)]16IPAVG} Hydrophilic elastomeric A blocks: [VPGVG[(VPGVG)2VPGEG(VPGVG)2]30VPGVG] [VPGVGVPGVG] |
Wright E.R. et
al. (2002) Nagapudi K. et al. (2005) |
|
| Resilin Protein | Drosophila melanogaster, CG15920 gene |
MVRPEPPVNS YLPPSDSYGA PGQSGPGGRP
SDSYGAPGGG NGGRPSDSYG APGQGQGQGQ GQGGYAGKPS DTYGAPGGGN GNGGRPSSSY GAPGGGNGGR PSDTYGAPGG GNGGRPSDTY GAPGGGGNGN GGRPSSSYGA PGQGQGNGNG GRSSSSYGAO GGGNGGRPSD TYGAPGGGNG GRPSDTYGAP GGGNNGGRPS SSYGAPGGGN GGRPSDTYGA PGGGNGNGSG GRPSSSYGAP GQGQGGFGGR PSDSTGAPGQ NQKPSDSYGA PGSGNGNGGR PSSSYGAPGS GPGGRPSDSY GPPASGSGAG GAGGSGPGGA DYDNDEPAKY EFNYQVEDAP SGLSFGHSEM RDGDFTTGQY NVLLPDGRKQ IVEYEADQQG YRPQIRYEGD ANDGSGPSGP GGPGGQNLGA DHYSSHRPGN GNGNGNGGYS GGRPGGQDLG PSGYSGGRPG GQDLGAGGYS NGKPGGQDLG PGGYSGGRPG GQDLGRDGYS GGRPGGQDLG ASGYGNGRPG GNGNGGSDGG RVIIGGRVIG GQDGGDQGYS GGRPGGQDLG RDGYSSGRPG GRPGGNGQDS QDGQGYSSGR PGQGGRNGFG PGGQNGDNDG SGYRY Exon 1 sequence in italic, Exon 2 sequence in Bold and Exon 3 sequence underlined |
Qin G. et al. (2011) |
| Putative Resilin Sequence from the Drosophila melanogaster CG15290 gene | 26 342-403 Signal peptide-(GGRPSDSYGAPGGGN)18-Exon 2-(GYSGGRPGGQDLG)11 |
Su R.S. et al. (2014) | |
| Rec1-resilin | (GGRPSDSYGAPGGGN)17 |
Elvin C.M. et
al. (2005) Mayavan S. et al. (2011) |
|
| Dros16 | (GYSGGRPGGQDLG)16 | Lin C.Y. et al. (2016) | |
| Two Exons (1 and 3) | (GGRPSDSYGAPGGGN)18-(GYSGGRPGGQDLG)11 | Qin G. et al. (2011) | |
|
Anopheles gambiae
AN16 |
(AQTPSSQYGAP) (AQTPSSQYGAP)16 |
Nairn K.M. et
al. (2008) Lyons R.E. et al. (2007) |
|
| Silk Proteins | Silkworm Silks from B. mori | repeated motifs [GAGSGA]n and
[GAGXGA]n
X= tyrosine or valine |
Asakura T. et
al. (2003) Kim U.J. et al. (2004) |
| Spider Silks from N. clavipes | (GPGGYGPGQQGPGGYGPGQQGPSGPGS(A)n) | Vepari C. et al. (2007) | |
| Major Ampullate spidroin 1 (MaSp1) Protein Analog | SGRGGLGGQAAGAAAAAGGAGQYGGLGSQG)n |
Kluge J.A. et
al. (2008) Prince J.T. et al. (1995) |
|
| Helices and Coiled Coil Proteins | COMPccs C Q CSP |
MGRSH6GSGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGKLN MGRSH6-GSIEGRAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTSKL MGRSH6-GSIEGRVKEITFLKNTAPQMLRELQETNAALQDVRELLRQQSKL MGRSH6GSGRLRPQMLRELQRTNAALRDVRELLRQQVKEITRLKNTVRRSRASGKLN |
Gunasekar S.K.
et al. (2009) Gunasekar S.K. et al. (2012) Hume J. et al. (2014) Hume J. et al. (2015) More H.T. et.at. (2014) |
| GCN4-pAA 7HSAP1 |
VKQLAMKVEELADANYHLASAVARLANAVGERAKA VEKLAQAVEKLARA NEKLAYAVEKLARAVEKLAQA |
Xu C. et al. (2013) | |
| Consensus TPR Protein | Consensus TPR Protein | WLGYAFAP | Lamb J.R. et al. (1995) |
The transition temperature of ELPs is also influenced by their amino acid sequence, chain length, buffer concentration, and polypeptide concentration.[26] Chilkoti and coworkers have designed ELPs that form injectable depots capable of providing sustained release of peptide therapeutics.[22] Additionally, ELP solution when mixed with chondrocytes, result in the formation of coacervates that can maintain the cell viability (Figure 2.1-2-a).[27] The Chilkoti group has precisely tuned the Tt by varying the guest residue and ELP chain length (Figure 2.1-2-b) using recombinant DNA techniques.[28] They have proposed a model that can predict the Tt of ELPs (Table 1) with the sequence repeats of VPGVG and VPGAG across a range of molecular weights and chain lengths.[28] These studies have allowed the design of new ELPs for applications in medicine and biotechnology. [22,26]
Figure 2.1-2.
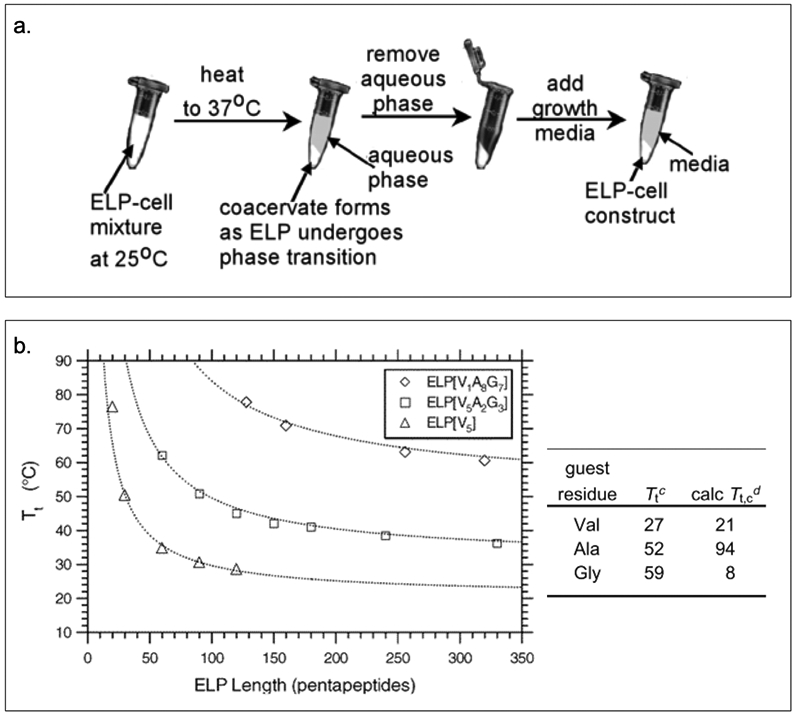
a. The process shows how ELP forms coacervates under temperature stimulation and how chondrocytes are captured within the ELP coacervates. b. Graph shows inverse phase transition properties for ELPs by varying length and guest residues. Reproduced with permission from Ref [27][28]. Copyright 2002, 2004, American Chemical Society.
The Conticello group has generated different types of BAB (Table 1) elastin-based triblock protein polymers, capable of forming thermoplastic elastomer hydrogels (Figure 2.1-3).[29,30] The protein copolymer contains a hydrophobic B block derived from the pentapeptide sequence [(V/I)PAVG] and a hydrophilic elastomeric A block derived from different pentapeptide or tetrapeptide sequences comprised of [VPXYG], where X is glycine and Y can be changed into glutamic acid that display change in lower critical transition temperature (Tt) (Table 1).[29–31] While the B block governs the Tt ; mechanical properties can be tuned by modifying block A.[30] Remarkably, the mechanical behavior of the copolymer changes from elastomeric to plastic, when glycine at the X position is replaced with alanine.[30] The unique properties of the thermoplastic hydrogel can be utilized for a variety of tissue engineering applications.[32]
Figure 2.1-3.
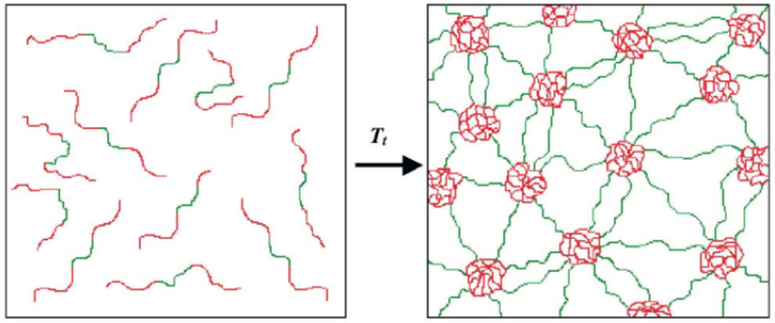
Schematic of formation of water-swollen network through micellization of hydrophobic end-block domains (red) of the BAB triblock copolymer. Unassociated unimers in aqueous solution below phase transition temperature (left) and virtually crosslinked structure between the central hydrophilic domains (green) and hydrophobic end-block domains (right). Reproduced with permission from Ref. [31]. Copyright 2002, Elsevier Science B.V.
2.1.2. Resilin
Resilin, an intrinsically disordered protein found in cuticles of winged insects, is known for its elastic properties.[33] In general, tyrosine residues within the protein sequence react with each other to form dityrosine and trityrosine, which can further crosslink to form a network (Figure 2.1-4-a).[34,35] Owing to its crosslinked network, natural resilin exhibits high resiliency and a very high fatigue lifetime.[34,36] It is heat stable[33] and demonstrates phase transition behavior, pH-responsiveness and autofluorescence properties (Figure 2.1-4-b).[36,37] These properties make resilin a unique biomaterial often referred to as protein rubber.[34,36,37] Resilin-based materials benefit from its unique LCST phase transition behavior, allowing them to be soluble below their Tt and becoming insoluble with properties similar to natural rubbers above the Tt.[36] These features allow the use of resilin in biomedical and tissue engineering fields for the design of scaffolds, biosensors, and environmentally responsive materials.[38]
Figure 2.1-4.
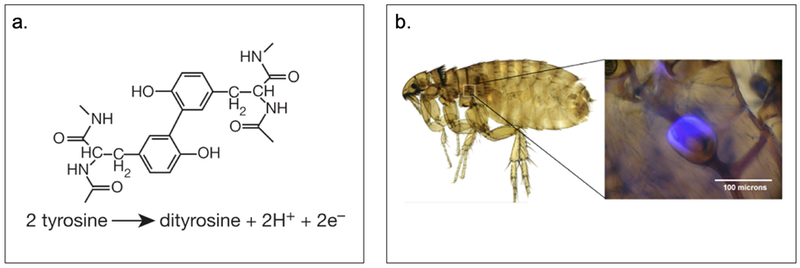
a. Structure and reaction of dityrosine forming in resilin. b. Auto fluorescence property of resilin pad of the flea in the pleural arch at the top of the hind legs under ultraviolet illumination. Reproduced with permission from Ref. [34] Copyright 2005, Nature Publishing Group
Since the discovery of resilin gene in Drosophila melanogaster, a flurry of research focused on the construction of resilin-like polypeptides has been conducted.[34,39,40] The gene precursor of resilin, gene product CG15920 (Table 1), is comprised of 620 amino acids with a signal peptide sequence at the N-terminus and three exon domains (Figure 2.1-5).[36] The three exon domains include: the N-terminal elastin domain (exon 1), the chitin-binding domain (exon 2) and the C-terminal elastic domain (exon 3).[36] Different copies of the repetitive sequences derived from CG15920 gene, particularly from the exon 1 (GGRPSDSYGAPGGGN)n and exon 3 (GYSGGRPGGQDLG)n, have been reported.[36] Rec1-resilin[34,41] with 17 copies of the 15 repeats of exon 1, Dros16[42] with 16 copies of the 15 repeats from exon 1 and two exons (1 and 3) are all excellent examples of recombinant resilin protein derived from the D. melanogaster exon domains (Table 1).[43,44] These repeat motifs are responsible for the elasticity of resilin. In addition, researchers have engineered recombinant resilin-like polypeptides (RLPs) containing a number of repeating units based on different species (such as fruit fly, African malaria, flea and mosquitos) and have also combined them with other functional domains.[36,39]
Figure 2.1-5.
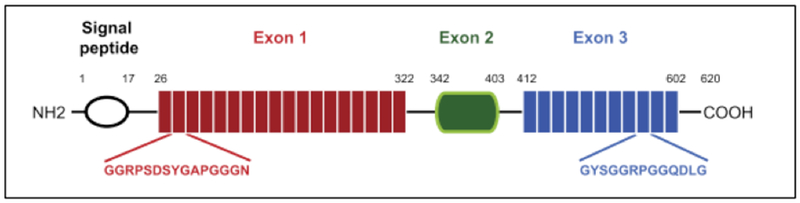
The putative resilin sequence from the Drosophila melanogaster CG15290 gene product. It has 620 amino acids and consists of a signal peptide sequence and three main regions. Exons 1 with 18 repeat motifs; exon 2 involved in binding of chitin and exons 3 with 11 repeat motifs. Reproduced with permission from Ref. [36] Copyright 2013, Elsevier Ltd
Rec1-resilin[34,39,40] (Table 1), derived from the first exon of the fruit fly gene, exhibit resilience comparable to natural resilin.[45] It exhibits dual upper critical solution temperature (UCST) and LCST behavior and can be crosslinked into a rubber like material.[37,44] Similar to Rec1-resilin, AN16 (Table 1), derived from African malaria mosquito gene Anopheles gambiae (Table 1), is comprised of 16 copies of proresilin repeats.[36,46] It has similar secondary structure and mechanical properties as Rec1-resilin.[45] AN16 having higher resilience and elasticity, displays UCST at a higher temperature when compared to Rec1-resilin (Figure 2.1-6).[40] Although these RLPs are derived from different sources, they exhibit similar properties as that of natural resilin and can be used as scaffolds for applications including tissue engineering and biosensors.[36]
Figure 2.1-6.

3% AN16 solution observed over a range of temperature for UCST behavior. Reproduced with permission from Ref.[40] Copyright 2014, Acta Materialia Inc. Published by Elsevier Ltd.
2.1.3. Silk and Silk-Like Proteins
Silk is a fibrous protein spun by Lepidoptera larvae such as silkworms, spiders, scorpions, mites and flies.[47,48] Among these different species, silkworm (Bombyx mori) and spider (Nephila clavipes and Araneus diadematus) silks are the most extensively characterized with remarkable mechanical properties.[49]
Silkworm silks have been first investigated as biomaterials centuries ago and used as suture materials.[47,48] As recombinant DNA techniques have matured, allowing facile cloning and expression through a variety of hosts, rapid development of silk-like proteins ensued.[50] Despite the differences in functionality of silk proteins due to variations in primary amino acid sequence and processing procedures, they represent a unique family of structural proteins that are biocompatible, biodegradable, mechanically superior, and can be chemically modified to suit a wide range of biomedical applications.[47,51]
Silkworm silk from B. mori contains fibroin, the structural protein that forms antiparallel beta-sheets, composed of repeated motifs [GAGSGA]n and [GAGXGA]n where X can be tyrosine or valine residues (Table 1).[52,53] Similar to silkworm silk, spider silk from N. clavipes contains glycine and alanine (Table 1). It also includes a high proportion of glutamic acid, proline and arginine.[48] Overall, silk fibroin possesses a higher amino acid composition of glycine and alanine (43% and 30%) than that of spider silk (30% and 20%).[48,54] Both spider and silkworm silks are semicrystalline and selectively incorporate specific repetitive motifs into β-sheet crystallites.[55] The beta-sheet conformation help silk proteins maintain their solubility in strong salts and acids during storage and after being spun.[55,56]
Kaplan and colleagues have reported that recombinant silkworm silk protein demonstrates relatively weak mechanical properties and brittle behavior when compared to recombinant spider silk.[57] The spider silk exhibits exceptionally high tensile strength and remarkable mechanical properties. More specifically, recombinant spider silk protein derived from dragline spider silk (Table 1) is soluble in water, organic solvents, and ionic liquids and has been used to produce various novel biomaterials including fibers and textiles, biofilms, and hydrogels (Figure 2.1-7). [48,57] Major ampullate spidroin 1 (MaSp1, Table 1) protein analog from the N. clavipes spider dragline silk has been electrospun to form spider silk fibers with diameter ranging from 10–60 μm.[57,58] A porous 3D scaffold generated from spider silk fibroin has been used to promote chondrocyte cell growth, with applications in articular cartilage engineering.[57,59]
Figure 2.1-7.
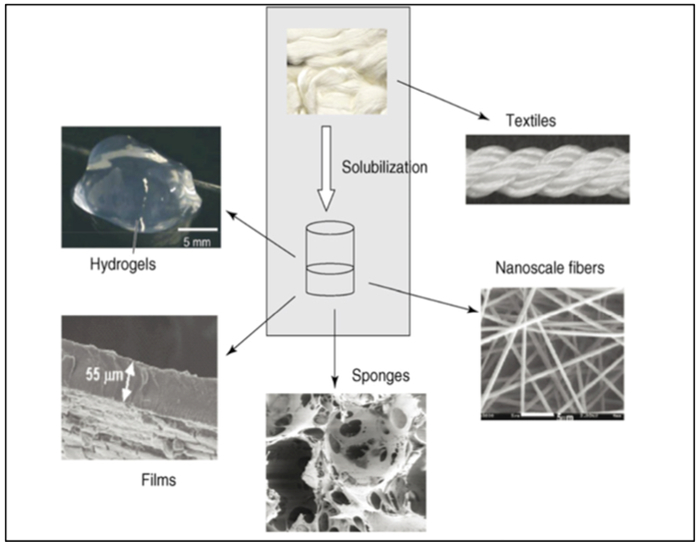
Schematic of recombinant silk processed into new biomaterials. The silk proteins can be solubilized with a variety of solvents, including formic acid, hexafluoroisopropanol, calcium nitrate, lithium salts, or ionic liquids. Once solubilized, the silk protein solutions can be processed into the range of different structures shown. Reproduced with permission from Ref.[57] Copyright 2008, Elsevier Ltd.
2.1.4. Engineered Helices and Coiled-Coils
The α-helix is the most common and highly favored stereochemical secondary structure present within proteins.[60] Crick and coworkers suggest that the coiled-coil is the predominant structure of the α-class fibrous proteins (keratin, myosin, epidermin and fibrinogen, also referrred as k-m-e-f class).[61] α-helical coiled-coils are ubiquitous in protein-protein interaction domains,[62] found in over 200 natural proteins. These proteins are involved in many biological activities, such as muscle contraction, transcription, metabolism, membrane channel, molecular chaperons, and immobilization of proteins and enzymes.[60,63]
Typically, right-handed α-helices tend to self-assemble into a coiled-coil structure with heptad repeats (abcdefg)n.[64] The e and g positions are often occupied by charged residues (lysine and glutamic acid), while the c, b, and f positions usually contain polar residues. The a and d positions are usually located at the hydrophobic interface of two helices and are mostly non-polar residues (leucine, isoleucine and valine). Both a and d residue positions determine whether the interhelical interactions between the two coiled-coils would be anti-parallel or parallel (Figure 2.1-8).[60,62,65] The helices can form a number of oligomers (dimers, tetramers, pentamers, etc.) and attain different topologies to direct a wide variety of protein assemblies.[65] Coiled-coil systems are being studied as models for stability and structural specificity and for their potential as drug delivery vehicles.[64]
Figure 2.1-8.
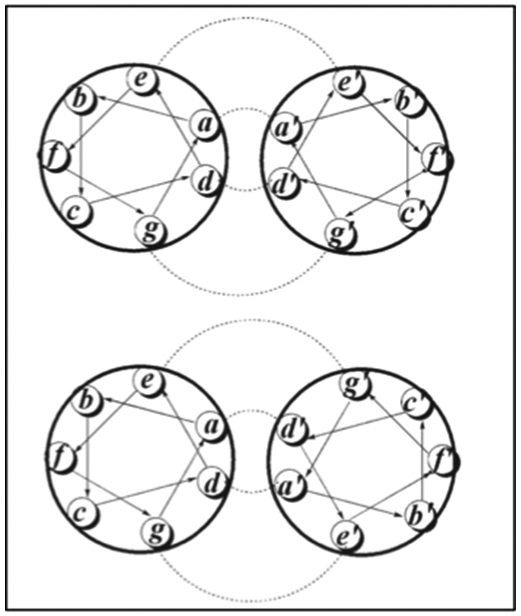
Anti-parallel helical wheel, where residues a and d lines up with a’ and d’, respectively (Top). Parallel helical wheel where residues a and d line up with d’ and a’, respectively (Bottom). Hydrophobic and ionic interactions are highlighted. Reproduced with permission from Ref.[65] Copyright 2008, John Wiley & sons, ltd
Montclare and coworkers have investigated α-helical assemblies based on the coiled-coil domain of cartilage oligomeric matrix protein (COMPcc) (Figure 2.1-9-a, Table 1).[19,66–68] COMPcc self-assembles into a pentameric unit with a hydrophobic pore that can bind small hydrophobic molecules like vitamin D, curcumin and all-trans retinol.[66] They have demonstrated that a variant of COMPcc, bearing two serines (COMPccs, Table 1) self-assemble into nanofibers; binding to zinc metal ions stabilizes the fibers while binding to nickel ions causes aggregation.[69] Another variant Q (Table 1), engineered by truncating the COMPccs by one heptad to form C (Table 1) and swapping regions with respect to glutamine 54, self-assembles into nanofibers and upon binding to a small molecule, curcumin, forms microfibers[70,71] (Figure 2.1-9-b). Introduction of positively charged residues on the surface of COMPccs to form a supercharged protein, CSP endows it the ability to effectively deliver genes (Table 1).[72] CSP exhibits greater helicity compared to the parent protein and can bind with nucleic acids and cationic lipids to form spherical particles termed “lipoproteoplexes”. These assemblies are capable of condensing and delivering nucleic acids (Figure 2.1-9-c).[72] CSP has been used as a dual delivery system for delivering siRNA and doxorubicin, a potent anticancer agent to breast carcinoma MCF7 cells.[73] It has also been used to accelerate wound healing in diabetic mice models.[73].
Figure 2.1-9.
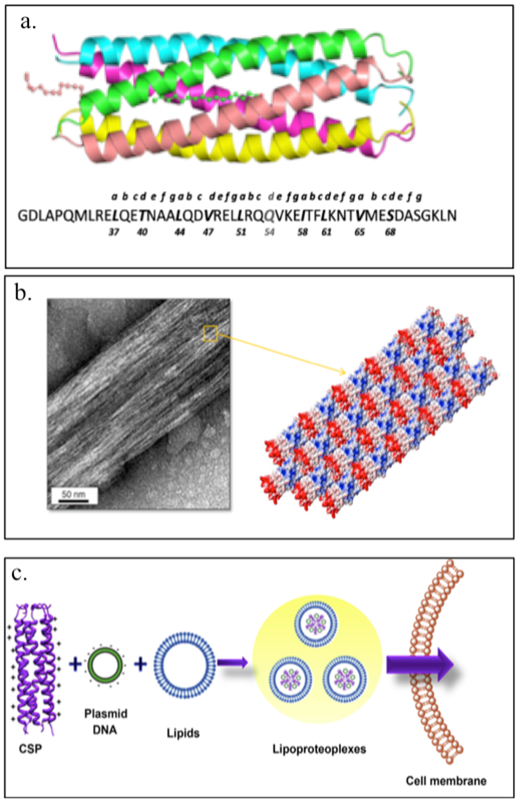
a. Structure and sequence of COMPcc. b. Transmission electron micrograph of Q fiber and schematic representation of Q fiber assembly with staggered positive (red) and negative (blue) regions of the pentamer. c. Schematic of CSP complexation with plasmid DNA and a tertiary complex with cationic lipids to form lipoproteoplexes for gene delivery. Reproduced with permission from Refs.[70–72] Copyright 2014,2015, American Chemical Society. Copyright 2014, Elsevier Ltd.
The Conticello group has developed a coiled-coil derived from the leucine zipper region of the Saccharomyces cerevisiae transcription factor GCN4 to create GCN4-pAA (Table 1).[74] GCN4-pAA self-assembles into a 7-helix (helices A-G) bundle and resembles a supramolecular lock-washer as the A and G helices interfaces provided extra surface area for complementary interaction between coiled-coil promoters (Figure 2.1-10-a). Structurally informed mutagenesis of GCN4-pAA has produced 7HSAP1 (Table 1), which retains the 7-helix bundle and forms nanotubes via noncovalent interactions between the complementary interfaces of the coiled-coil lock-washer structures (Figure 2.1-10-b, 2.1-10-c).[74] These rationally designed helical nanotubes can encapsulate shape-appropriate small molecules such as cyclodextrins with high binding affinity.[74] These examples demonstrate the tremendous technical advantage of coiled-coils as functional nanoporous biomaterials for tissue engineering and drug delivery purposes.[70]
Figure 2.1-10.
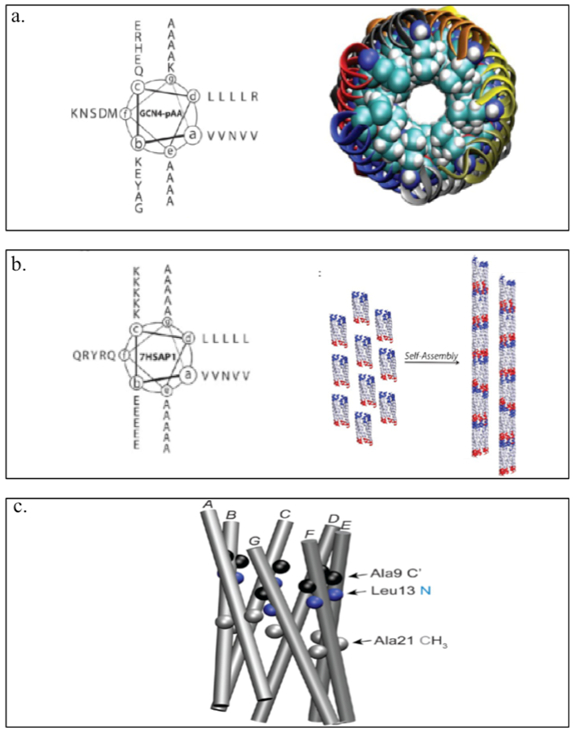
a. Helical wheel of GCN4-pAA (left) and 7-helix bundle with helices A-G represented by the different colors. Coiled-coil lock washer is represented by the displaced edge of the structure occurs at the interface between the first (A, blue) and the seventh (G, gray) helices. b. Helical wheel of the sequence of peptide 7HSAP1(left) and schematic representation of the proposed model for self-assembly of lock-washer structures derived from the 7-helix bundle of peptide 7HSAP1 into helical nanotubes. c. The 7-helix bundle of GCN4-pAA that occupies structural homologous positions with respect to the isotopically labeled sites in 7HSAP1*: carbonyl carbons (C’) of Ala9 (black), amide nitrogen of Leu13 (blue), and methyl carbons of Ala21 (gray). Reproduced with permission from Ref. [74] Copyright 2013, American Chemical Society
2.1.5. Consensus Tetratricopeptide Repeats (TPRs)
In 1990, both Hirano and Sikorski groups introduced the tetratricopeptide repeat (TPR) protein.[75,76] TPR, comprised of 34 amino acids as a basic repeat unit, was originally identified as a protein interaction module in the cell division cycle proteins (Table 1).[75,76] The TPR unit could be found in unrelated functional proteins in a range of organisms from a simple bacteria to human beings.[77,78] TPR mediates a variety of protein-protein interactions, thereby bringing together different proteins in complex biological machines.[78,79]
The amphipathic nature of the TPR protein suggests that it might form a coiled-coil or helical bundle structure. However, the three-TPR domain of phosphatase 5 as the earliest solved TPR structure, showed that the structure was helical but the folding presented a novel helical array.[77] Due to the consensus TPR motif being involved in protein-protein interactions, redesign of the superhelix structure has been explored.[77] TPR domains can be used as a “switch” for either binding specificity or to disrupt protein-protein interactions, resulting in a unique scaffold for protein engineering.[77]
Regan and coworkers have designed recombinant consensus TPR proteins, manipulating the structure and stability in a rational way.[79] These TPR modules have been engineered into supramolecular arrays with practical applications including: (1) affinity purification capture reagent and (2) nanostructured functional films as novel anticancer agents.[79] affiTRAPs, designed as TPR affinity proteins (TRAPs), are more cost efficient than antibody-based affinity purification as it is easier to produce high yields of these proteins. affiTRAP can specifically bind a target short peptide having a sequence MEEVF.[73] This interaction is resistant to washing but can be disrupted under mild conditions for purification purposes (Figure 2.1-11-a).[80] A designed TPR module, CTPR390+ binds the anticancer chaperone Hsp90 with high affinity and greater specificity than the endogenous co-chaperones, enabling effective inhibition of Hsp90 functions that are essential to the folding of many oncogenic proteins (Figure 2.1-11-b).[81]
Figure 2.1-11.
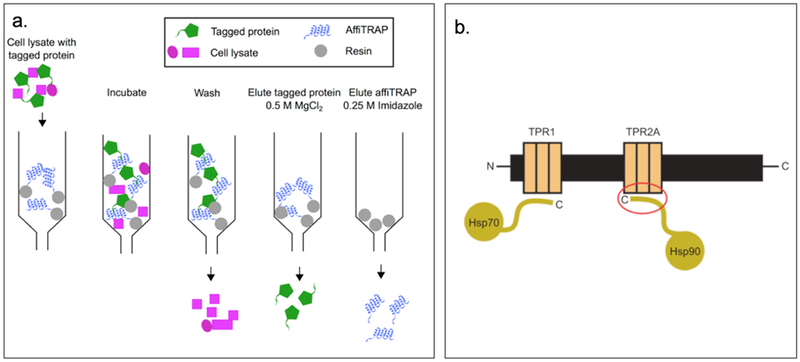
a. Schematic representation of all the components of the protein purification system. AffiTRAP is represented in blue and target protein in green. Through washing, the cell lysate in pink can be easily washed out. By adding magnesium salt, protein can be eluted. While upon addition of imidazole, affiTRAP is eluted. b. Schematic representation of Hsp organizing protein indicating the two independent TPR domains, TPR1 and TPR2A, which interact with the C-terminal tails of Hsp70 and Hsp90 anticancer target. Red circle highlighted the TPR2A-Hsp90 interaction. Reproduced with permission from Ref. [80,81] Copyright 2015, Portland Press Limited. Copyright 2008, American Chemical Society
2.2. Multi-Domain Protein Engineered Materials
Multi-domain protein engineered materials are comprised of more than one domain within a polymeric chain.[82] Many of the single domains discussed in the previous section are employed to generate multi-domain systems. Theoretically, each domain or block of the protein material can self-assemble, preserving each domain’s function, leading to an overall enhanced functionality of the multi-domain system.
2.2.1. α-Helix (H)-Random Coil (RC)-α-Helix (H) Protein Polymers
α-Helix-random coil-α-helix proteins (H-RC-H) represent a class of triblock proteins consisting of a random coil domain flanked by either the same or different coiled-coil domain at each end.[23] A combination of α-helix coiled-coil and random coil can help tune the physical properties of the protein for a desired application. As previously discussed, right-handed α-helices found in nature tend to self-assemble into coiled-coil structures with heptad repeats (abcdefg)n. These helices can form a number of oligomers (dimers, tetramers, pentamers, hexamers and octamers) having different topologies in the presence of random coil domain.[65]
Tirrell and coworkers have constructed a triblock copolymer, AC10A (Figure 2.2-1-a, Table 2), consisting of a central polyelectrolyte random coil domain ((AG)3PEG)10 flanked by terminal leucine zipper coiled-coil domains A, where a and d positions of the coiled-coil are based on Jun oncogene product .[21,83] The coiled-coil domain of AC10A, which self-assembles into a tetramer, is responsible for pH controlled gelation and viscoelastic properties, while the random coil assists in the formation of a highly swollen hydrogel.[21,83] The design of such reversible hydrogels is valuable for understanding and controlling physical and biological properties such as the strength, porosity and sensitivity that impact drug delivery and cell encapsulation applications.[83] Recently, they have developed a protein triblock copolymer in which the end coiled-coil block of AC10A is replaced with the coiled-coil P derived from the N-terminal helical domain of cartilage oligomeric matric protein (COMP) that self-assembles into a pentameric structure, to yield PC10P (Figure 2.2-1-b, Table 2)[17]. This protein hydrogel demonstrates a transition from a linear elastic gel to sol at large strains, exhibiting shear thinning by three orders of magnitude.[17] These unique properties allows the hydrogel to be injected through narrow gauge needles with moderate pressure, facilitating its use in tissue engineering.[17]
Figure 2.2-1.
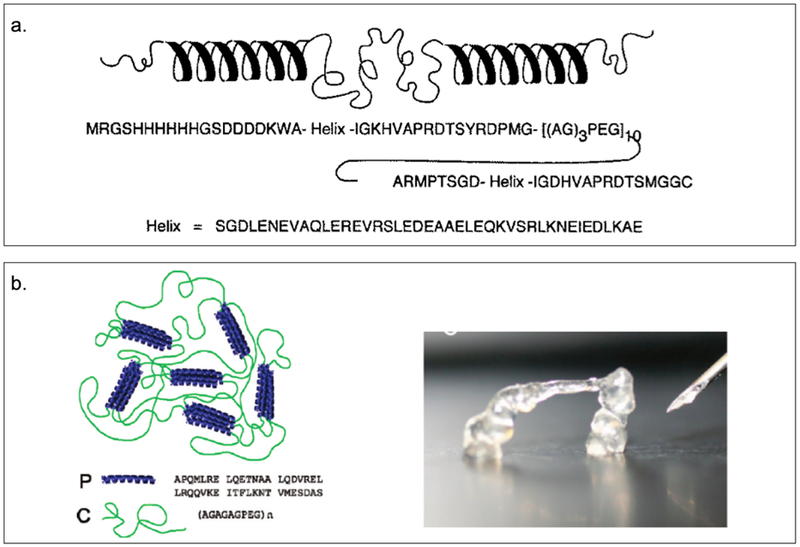
a. Amino acid sequence and representation of secondary structure of a triblock polymer (AC10A) with a central polyelectrolyte domain, a random coil (AG3PEG)x, flanked by terminal leucine zipper domains. b. The fast recovery of injectable protein hydrogel PC10P allows self-supporting structures to be produced. Reproduced with permission from Ref.[17,83] Copyright 1998, The American Association for the Advancement of Science. Copyright 2010, American Chemical Society.
Table 2.
Sequences of multi-domain proteins
| Type of protein |
Name of protein |
Sequence | Reference |
|---|---|---|---|
| H- RC- H Protein Polymers | AC10A | MRGSHHHHHHGSDDDDKWA-SGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKA-EIGDHVAPRDTSYRDPMG-[AGAGAGPEG]10-ARMPTSGD-SGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKA-IGDHVAPRDTSMGGC *central polyelectrolyte block is Bold ; Coiled-Coil domain is italic |
Petka W.A. et al. (1998) |
| Telechelic PC10P | MRGSHHHHHHGSGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGTSYRDPMG[AGAGAGPEG]10ARMPTSGSGDLAPQMLRE
LQETNAA LQDVRELLRQQVKE ITFLKNT VMESDASGKLN * P block is italic; C10 is Bold |
Olsen B.D et al. (2010) | |
| Diblock and Triblock Copolymer Protein | Block A:
(VSSLESK)6 Block B: (AG)3PEG10 Block C:(VSSLESK)2-VSKLESK-KSKLESK-VSKLESK-VSSLESK |
Xu C. et al. (2008) (2005) | |
| ABA | H6VNADP[(VSSLESK)6]ASYRDPMG[(AG)2PEG]10ARMPTSADP[(VSSLESK)6] | Xu C. et al. (2005) | |
| CBA | H6VNADP[(VSSLESK)2-(VSKLESK)-(KSKLESK)-(VSKLESK)-(VSSLESK)]-ASYRDPMG[(AG)2PEG]10ARMPTSADP[(VSSLESK)6] | ||
| ABC | H6VNADP[(VSSLESK)6] ASYRDPMG[(AG)2PEG]10 ARMPTSADP[(VSSLESK)2-(VSKLESK)-(KSKLESK)-(VSKLESK)-(VSSLESK)] | ||
| CBC | H6VNADP[(VSSLESK)2-(VSKLESK)-(KSKLESK)-(VSKLESK)-(VSSLESK)]- ASYRDPMG[(AG)2PEG]10 ARMPTSADP[(VSSLESK)2-(VSKLESK)-(KSKLESK)-(VSKLESK)-(VSSLESK)] | ||
| ZR–C10–ZR | MGLEIRAAALRRRNTALRTRVAELRQRVQRLRNEVSQYETRYGPLKLRDWMG [(AG)3PEG]10ALMPVDLEIRAAALRRRNTALRTRVAELRQRVQRLRNEVSQYET RYGPLR *C10 is Bold ; ZR is italic |
Park W.M et al. (2016) | |
| H-RC-Protein Polymers | H-S-H | MRGS HHHHHH GSDDDDKA SGDLENE VAQLERE
VRSLEDE AAELEQK VSRLKNE IEDLKAEIGDHVAPRDTSYRDPMG
AGAGAGPEG AGAGAGPEG AGAGAGPEG AGAGAGPEG AGAGAGPEGAGAGAGPEG
AGAGAGPEG AGAGAGPEG AGAGAGPEG AGAGAGPEG ARMPT SGDLENE
VAQLERE VRSLEDE AAELEQK VSRLKNE IEDLKAE
IGDHVAPRDTSW *S is Bold ; H is italic |
Shen W. et al. (2004) Wheeldon I.R. et al. (2008) |
| H-S-AdhD-H | MRGSHHHHHHGSDDDDKWASGDLENEVAQL
EREVRSLEDE AAELEQKVSR
LKNEIEDLKA EIGDHVAPRDTSYRDPMGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAG PEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGARMPHGMAKRVN AFNDLKRIGDDKVTAIGMGTWGIGGRETPDYSRDKESIEAIRYGLELGMNLIDTAEFYGAGHAEEIVGEAIKEFEREDIFIVSKVWPTHFGYEEAKKAARASAKRLGTYIDLYLLHWPVD DFKKIEETLHALEDLVDEGVIRYIGVSNFN LELLQRSQEVMRKYEIVANQVKYSVKDRWP ETTGLLDYMKREGIALMAYTPLEKGTLARNECLAKIGEKYGKTAAQVALNYLIWEENVVA IPKASNKEHLKENFGAMGWRLSEEDREMARRCVGMPTSGDLENEVAQLEREVRSLEDEAA ELEQKVSRLKNEIEDLKAEIGDHVAPRDTSMGGC *S is Bold ; H is italic ; AdhD is underlined |
Wheeldon I.R. et al. (2009) | |
| β-roll HS-Leuβ HS-DLeuβ |
GSARDDVLIGDAGANVLNGLAGNDVLSGGAGDDVLLGDEGSDLLSGDAGNDD LFGGQGDDTYLFGVGYGHDTIYESGGGHDTIRINAGADQLWFARQGNDLEIR ILGTDDALTVHDWYRDADHRVEIIHAANQAVDQAGIEKLVEAMAQYPD WASGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKAEIGDHVAPRDTSYRDPMGAGAGAGPEGAGAGA GPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGARMLGSARDDVLIGDAGANLLLGLAGNDVLSGGAGDDLLLGDEGSDLLSGDAGNDLLLGGQGDDTYLFGVGYGHDLILESGGGHDTIRINAGADQLWFARQGNDLEIRILGTDDALTVHDWYRDADHRVEIIHAANQAVDQAGIEKLVEAMAQYPD WASGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKAEIGDHVAPRDTSYRDPMGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGAGAGAGPEGARMLGSARDDLLLGDAGANLLLGLAGNDLLLGGAGDDLLLGDEGSDLLLGDAGNDLLLGGQGDDLYLFGVGYGHDLILESGGGHDTIRINAGADQLWFARQGNDLEIRILGTDDALTVHDWYRDADHRVEIIHAANQAVDQAGIEKLVEAMAQYPD *mutated β-roll is Bold |
Dooley K. et.al. (2014) | |
| Alpha Helical-Elastin Protein Polymers | E | [(VPGVG)2VPGFG(VPGVG)2]5VP |
Haghpanah J.S. et al. (2009) Haghpanah J.S. et al. (2010) Olsen et al. (2018) |
| C | DLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASG | ||
| EC | MRGSHHHHHHGSKPIAASA-E-LEGSELA(AT)6AACG-C-LQA(AT)6AVDLQPS | ||
| CE | MRGSHHHHHHGSACELA(AT)6AACG-C-LQA(AT)6AVDKPIAASA-E-LEGSGTGAKLN | ||
| ECE | MRGSHHHHHHGSKPIAASA-E-LEGSELA(AT)6AACG-C-LQA(AT)6AVDKPIAASA-E-LEGSGTGAKLN | ||
| CEC | MRGSHHHHHHGSACELA(AT)6AAC-C-LQA(AT)6AVDKPIAASA-E-LEGSGT- C-LQALSI | ||
| EPE | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGELGSGLGSAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASKLNTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM | Tirrell D.A. et.al. (2016) | |
| T40A | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGV GVPGVGVPGVGVPGVGVPGEGVPGVGVP GVGELGSGLGSAPQMLRELQEANAALQDVRELLRQQVKEITFLKNTVMESDASKLNTSVPGVGVPGVGVP GEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| Q54A | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGV GVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVG ELGSGLGSAPQMLRELQETNAALQDVRELLRQAVKEITFLKNTVMESDASKLNTSVPGVGVPGVGVPGEGVPGVGVPGV GVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| I58A | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGE GVPGVGVPGVGVPGVGVPGVGVPGEGVPGV GVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGELGSGLGSAPQMLRELQETNAALQDVRELLRQQVKEATFLKNTVMESDASKLNTSVPGVGVPGVGVPGEGVPGVGVPGV GVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| L37A | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPG VGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVG ELGSGLGSAPQMLREAQETNAAAQDVRELLRQQVKEITFLKNTVMESD ASKLNTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEG VPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| L37V | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPG
VGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVG
ELGSGLGSAPQMLREVQETNAAAQDVRELLRQQVKEITFLKNTVMESD
ASKLNTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEG
VPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| L37I | MRCSSHHHHHHVDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPG
VGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVG
ELGSGLGSAPQMLREIQETNAAAQDVRELLRQQVKEITFLKNTVMESDA
SKLNTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVP
GVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLECM *mutation is Bold |
||
| Silk-Elastin Protein Polymers | SELP-47K SELP-415K SELP-815K |
MDPVVLQRRDWENPGVTQLVRLAAHPPFASDPMGAGSGAGS[(GVGVP)4GKGVP(GVGVP)3(GAGAGS)4]12(GVGVP)4GKGVP(GVGVP)2(GAGAGS)2GAGAMDPGRYQDLRSHHHHHH MDPVVLQRRDWENPGVTQLVRLAAHPPFASDPMGAGSGAGS[(GVGVP)4GKGVP(GVGVP)11(GAGAGS)4]7(GVGVP)11GKGVP(GVGVP)4(GAGAGS)2GAGAMDPGRYQDLRSHHHHHH MDPVVLQRRDWENPGVTQLVRLAAHPPFASDPM[GAGS(GAGAGS)2(GVGVP)4GKGVP(GVGVP)11(GAGAGS)5GAGA]6MDPGRYQDLRSHHHHHH |
Gustafson J. et al. (2009) |
| Silk-Collagen Protein Polymers | CSSC and SCCS/ CSESEC and SECCSE | S:
[(GA)3GE]24 C:LEKREAEAGPPGEPGNPGSPGNQGQPGNKGSPGNPGQPGNEGQPGQPGQNGQPGEPGSNGPQGSQGNPGKNGQPGSPGSQGSPGNQGSPGQPGNPGQPGEQGKPGNQGPAGG |
Werten M.W.T. et al. (2001) Martens A.A. et al. (2009) |
| Two-Componentα-Helix-Elastin Polymers andα-Helix-Globular Protein | Leucine Zipper Coiled Coils
ZE-ZR
Pairs mCherry–ZE EGFP–ZE |
LEIEAAALEQENTALETEVAELEQ
EVQRLENIVSQYRTRYGPL-LEIRAAALRRRNTALRTRVAELRQRVQRLRNEVSQYETRYGPL MGGSRSMVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEG TQTAKLKVTKGGPLPFAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGFKW ERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGW EASSERMYPEDGALKGEIKQRLKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVN IKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYKSKLRGSGSLEIEAAALEQEN TALETEVAELEQEVQRLENIVSQYRTRYGPLRSHHHHHH MGGSRSMASKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKF ICTTGKLPVPWPTLVTTLCYGVQCFSRYPDHMKRHDFFKSAMPEGYVQERTIF FKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIM ADKQKNGIKVNFKTRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQS ALSKDPNEKRDHMVLLEFVTAAGITHGMDELYNLRGSGSLEIEAAALEQENTA LETEVAELEQEVQRLENIVSQYRTRYGPLRSHHHHHH *ZE and ZR is Bold |
Park W.M et al. (2014) |
| MITCH | CC43 WW Domain Proline-Rich PPxY Peptide C7 P9 |
RLPAGWEQRMDVKGRPYFVDHVTKSTTWEDPRPE EYPPYPPPPYPSG MGSSHHHHHHSSGLVPRGSSSGHIDDDDKV DGT[RLPAGWEQRMDVKGRPYFVDHVTKSTTWEDPRPE]GTLDEL[AGAGAGPEG]2RGDSAGPEG[AGAGAGPEG]2ELLDGT([RLPAGWEQRMDVKGRPYFVDHVTKSTTWEDPRPE]GTLDEL[AGAGAGPEG]2[RGDSAGPEG][AGAGAGPEG]2 ELLDGT)5[RLPAGWEQRMDVKGRPYFVDHVTKSTTWEDPRPE]GTLE MGSSHHHHHHSSGLVPRGSSSGHIDDDDKVDGT[EYPPYPPPPYPSG]GTLDEL[AGAGAGPEG]2ELLDGT([EYPPYPPPPYPSG]GTLDEL[AGAGAGPEG]2ELLDGT)7[EYPPYPPPPYPSG]GTLE |
Mulyasasmita W. et al. (2011) |
| P1 | EYPPYPPPPYPSGC | Foster A.A. et.al. (2018) | |
| Reversible Ca2+-Sensitive Two-Component Hydrogel | CaM-(8)-Zip PGD-(n)-PGD PGD-(n)-eNOS |
MHHHHHHAADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAEL QDMINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISA AELRHVMTNLGEKLTDEEVDEMIREADIDGDGQVNYEEFVQMMTAK(AGAGAGPEG)8AGSGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKAE MHHHHHHAGHKKTDSEVQLEMITAWKKFVEEKKKK(AGAGAGPEG)nAGHKKTDSEVQLEMITAWKKFVEEKKKK MHHHHHHAGHKKTDSEVQLE MITAWKKFVEEKKKK(AGAGAGPEG)nAGRKKTFKEVANAVKISASLMGAERLI |
Topp S. et al. (2006) |
Kopecěk and colleagues have generated a series of stimuli-responsive reversible protein block polymers, ABA, CBA, ABC, CBC (Table 2) that are capable of forming hydrogels (Figure 2.2-2).[84,85] While block B is similar to the polyelectrolyte domain ((AG)3PEG)10 discussed above, block A has a sequence of (VSSLESK)6 exhibiting a transition temperature of 95 °C. Introducing lysine at key positions within block A results in block C (Table 2), lowering its transition temperature. The self-assembly of hydrogels are accompanied by intermolecular association of coiled-coil domain forming physically crosslinked network.[85,86] Tailoring the amino acid sequence of the coiled-coil domain allows these triblock hydrogels to exhibit temperature- and pH-responsiveness.[85] The hydrogel assembly has been also shown to be influenced by the overall length and structure of the coiled-coil domain.[84] The proteins containing block A (ABA, CBA and ABC) are more stable at neutral conditions rather than either acidic or basic pH, while the CBC protein polymer reveals the structure to be more stable from acidic to basic condition.[85] CBC forms reversible gel, which is strongly elastic at room temperature and predominantly viscous at 55°C. These engineered protein materials are useful in biomedical applications where stimuli responsiveness is required.[87]
Figure 2.2-2.

Representation of the four different morphologies of triblock polymer protein hydrogels (Polymer I II III IV refers to ABA, CBA, ABC, CBC respectively) characterized by SEM. Reproduced with permission from Ref.[85] Copyright 2005, American Chemical Society
Park et. al have designed hybrid flower-shaped (FS) nanoparticles using an artificial recombinant protein ZR–C10–ZR (Table 2, Figure 2.2-3).[63] ZR represents the helical domain derived from a b-ZIP protein and C10, is a random coil block composed of [(AG)3PEG]10. Addition of calcium chloride (CaCl2) to the triblock copolymer in the presence of phosphate buffered saline (PBS), results in hybrid FS nanoparticles with co-precipitation of calcium phosphate (CaP). These FS nanoparticles assemble into clusters that form porous higher order supraparticles at the air–water interface.[63] This colloidal system can be adapted by other similar systems for the production of complex suprastructures.[63,87] Such colloidal systems can contribute greatly to the study of biomineralization, biocatalysis and biosensors since the hybrid supraparticles provide high-affinity binding sites for protein immobilization applications.[63]
Figure 2.2-3.
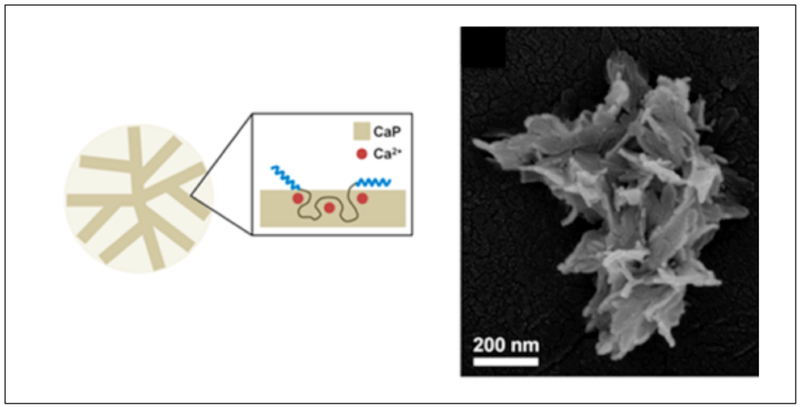
Co-precipitation of CaP and ZR–C10–ZR (left). SEM image of Single FS nanoparticle composed of nanoplate petals (right). Reproduced with permission from Ref.[63] Copyright 2016, American Chemical Society.
2.2.2. α-Helix (H)-Random Coil (RC) Fused to Globular Protein Domains
H-RC-globular protein copolymers represent a class of multi-domain materials that contains an α-helix coiled-coil domain and a random coil domain along with a functionalized protein segment.[88,89] A wide variety of such polymers self-assemble into protein hydrogels, resulting in multifunctional protein materials with enzymatic,[90,91] optically active,[88] analyte-binding,[92] or signaling abilities.[93]
Banta and coworkers have generated a series of successful bifunctional fusion proteins by creating H-S diblocks[94] fused with other proteins including green fluorescent protein (GFP) and the tetrameric discosoma red fluorescent protein (DSRED),[95,96] or enzymes like small laccase (SLAC),[89,97] a polyphenol oxidase from Streptomyces coelicolor, and AdhD,[90,98] an aldo-keto reductase from Pyrococcus furiosus (Table 2).[89–91] The H represents an α-helical coiled-coil derived from leucine zipper domain (Table 2) while S is comprised of [AGAGAGPEG]10.[94] The bifunctional fusions combine self-assembly properties with the bioactivity profile of fluorescent proteins or enzymes. The resulting H-S-GFP-H, H-S-DSRED, and H-S-GFP constructs are expressed and purified to form hydrogels via physical crosslinking (Table 2, Figure 2.2-4-a). All constructs assemble into elastic hydrogels having lower erosion rates when compared to the parent H-S-H hydrogel.[89] More recently, they have developed a fusion protein containing a thermostable aldo-keto reductase (HS-AdhD-H) that self-assembles to form a thermostable enzymatic hydrogel (Table 2, Figure 2.2-4-c).[90] The hydrogel retains the enzymatic activity of the enzyme even at elevated temperatures.[90] Such bi-functional fusion proteins have tremendous potential for use in biofuel cells, and as tissue engineering scaffolds.[88–91]
Figure 2.2-4.
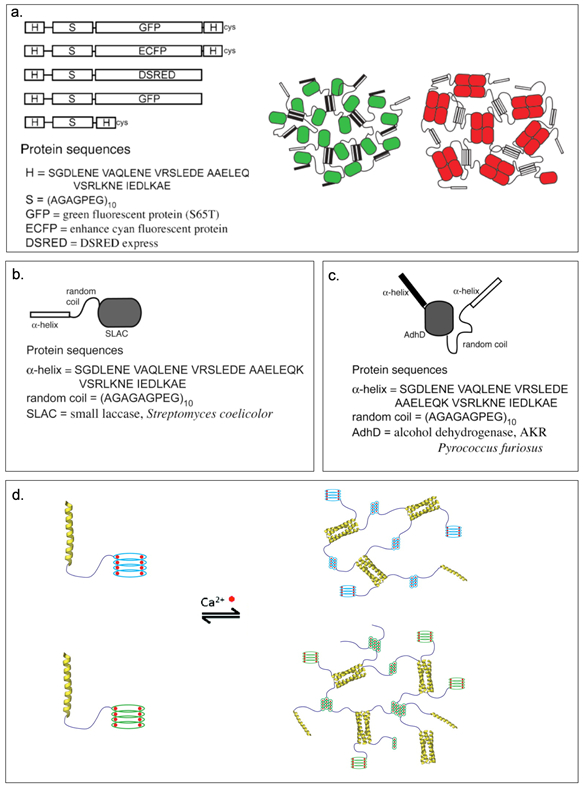
a. Representation of H-S-H with fluorescence protein and partial sequences (left). Schematic representation of H-S-GFP-H and H-S-DSRED physically crosslinked fluorescent protein hydrogels (right). Green and red ovals are fluorescence protein GFP and DSRED respectively, bars are H-domains. b. H-S-SLAC constructs with partial sequences. c. HS-AdhD-H constructs with partial sequences. d. Cartoon represents hydrogel formation of HS-Leuβ and HS-DLeuβ. Calcium ions are shown in red. The monomeric units consists an α-helical leucine zipper (yellow), randomly coiled linker (purple), and the mutant Leuβ (light blue) and DLeuβ mutant (green). Reproduced with permission from Ref. [89,90,100] Copyright 2007, American Chemical Society. Copyright 2009, Elsevier Ltd. Copyright 2014, American Chemical Society.
Banta and coworkers have also created calcium-dependent hydrogels comprising of an α-helical leucine zipper domain (H) and a peptide motif derived from repeats-in-toxin (RTX) domain of adenylate cyclase linked by a hydrophilic linker (S).[99] Introduction of Ca2+ ions changes the peptide motif of RTX from intrinsically disordered to a folded beta roll (β-roll) structure.[100] The β-roll structure is divided into two short parallel β-sheet faces by turns. Hydrophobic surface(s) can result from mutating the residues on one (Leuβ) or both (DLeuβ) β-sheet faces to leucines, leading to self-assembly of HS-Leuβ and HS-DLeuβ hydrogels (Table 2, Figure 2.2-4-d).[99,100] Only mutated HS-Leuβ and HS-DLeuβ can form hydrogels in calcium rich environment, with HS-Leuβ needing extra crosslinking by the H domain and HS-DLeuβ forming hydrogel with lower protein concentration and without extra crosslinking.[99,100] These scaffolds can be potentially used for creating calcium dependent stimuli-responsive protein based hydrogels and other biomaterials.[99]
2.2.3. α-Helix-Elastin Protein Polymers
While H-RC-H protein polymers use α-helical coiled-coils and random coil domains to form multifunctional hydrogels, coiled-coils have also been combined with ELPs. Montclare and coworkers have designed artificial protein block polymers bearing two self-assembling domains (SADs), consisting of the coiled-coil region of cartilage oligomeric matrix protein (COMPcc or C) and elastin domain (E).[13,66] Three different constructs, EC, CE, and ECE have been assessed for their overall conformation, thermoresponsive behavior, and physico-chemical properties (Table 2). Interestingly, the number of repeats and orientation of C and E domains impact the mechanical properties with EC exhibiting elastic character, CE being viscous and ECE demonstrating a viscoelastic behavior (Figure 2.2-5).[67] Of the three protein polymers, the CE diblock exhibited stronger affinity to curcumin followed by ECE and EC, making CE as an excellent drug delivery carrier capable of providing sustained release effects. Furthermore, EnC and CEn (n=1-5) libraries have been generated to study the effects of orientation of the two domains and the effect of the length of the E domain on the physicochemical properties.[101] The EnC library show elastic behavior (above the transition temperature) that allow them to form soft gels, while the CEn library exhibit viscous behavior.[101] Overall, the orientation of the C and E blocks influences the hydrogel formation and binding/release of small molecules.[19,67] Recently, a triblock protein, CEC has been created for forming protein hydrogels (Table 2).[102] The addition of an extra C domain to CE improves the stability with CEC exhibiting enhanced elasticity and increased small molecule binding ability.[102]
Figure 2.2-5.
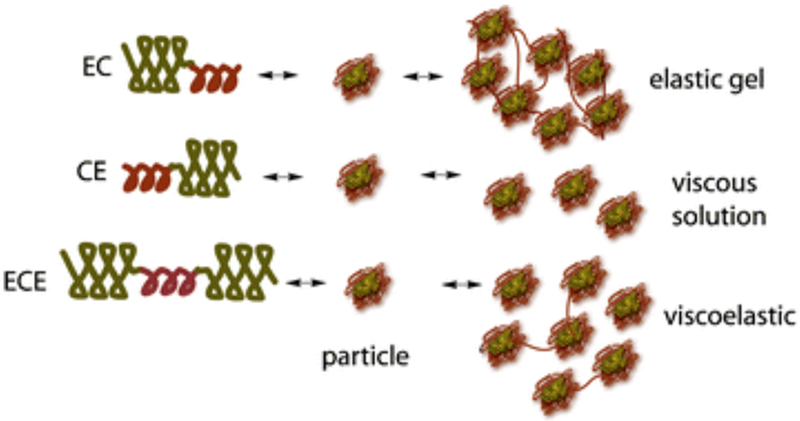
Cartoons of block polymers CE, EC, and ECE structure and how they self-assemble forming networked soft gels. Reproduced with permission from Ref. [152] Copyright 2012, American Chemical Society.
The Tirrell group have produced the EPE protein (Table 2), which consists of a central domain derived from COMPcc (P) flanked by elastin-like domains (E). The tri-block consisits of N- and C-terminal cysteine residues. Under denaturing conditions, EPE can covalently react with four-arm PEG vinyl sulfone (PEG-4VS) via free thiol groups.[103] Upon removal of the denaturant, the P domain associates to form physical crosslinks. In order to study the mechanical properties of the EPE hydrogels, six variants (T40A, Q54A, I58A, L37A, L37V, and L37I, Table 2 ) are created by mutating a single amino acid within the P domain at a and d positions based on the work of Montclare et.al. [66,104] The variants Q54A and T40A exhibit a higher folded fraction of the P domain than I58A mutant, leading to stiffer gels. Further studies mutating leucine 37 which is known to decrease the helicity of the P domain is explored. The variants L37A, L37V and L37I demonstrate different relaxation behaviour with L37A revealing longer relaxation time compared to L37V and L37I. Differences of the sequence within the coiled-coil domain of each variant changes the energy necessary for mechanical deformation at the macroscopic level. Hydrogels with complex relaxation dynamics have been also prepared by mixing block polymers with multiple relaxation times. Thus, programmed molecular genetics play an important role in controlling the dynamic relaxation behaivor in engineered coiled-coil protein based hydrogels. [104]
2.2.4. Silk-Elastin Protein Polymers
Silk-elastin like polypeptides (SELPs) consist of repeating domains of silk and elastin.[50,105] These recombinant proteins combine the high strength of silk domain with the elastic properties of the elastin domain.[50] For example, recombinant SELP47K has been fabricated into nanofibrous scaffolds using the electrospinning technique (Table 2).[106] Three SELPs (SE8Y, S2E8Y, S4E8Y) with silk-to-elastin ratios of 1:8, 1:4, and 1:2 have been expressed in E. coli (Figure 2.2-6). The silk-to-elastin ratio governs the particle size and self-assembly of SELPs with the formation of micelle-like particles being predominantly affected by the amount of silk present.[14] By precisely tuning the silk-to-elastin ratio, different nanostructures such as nanofibers, hydrogels, and nanoparticles have been generated with potential applications in biosensors, drug delivery, and tissue engineering.[105,107]
Figure 2.2-6.
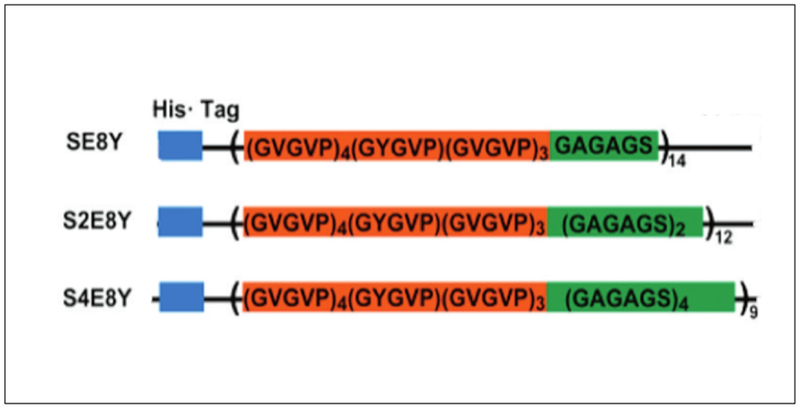
Constructs of silk–elastin-like proteins that contain varying ratios of the silk-to-elastin blocks in each monomer repeat. SE8Y (1:8), S2E8Y (1:4) and S4E8Y (1:2). Reproduced with permission from Ref. [107] Copyright 2011, American Chemical Society.
In addition to the above listed applications, SELPs have been used for delivering nucleic acids.[108–113] The Ghandehari group has created SELP47K and SELP415K (Table 2) having repeats of silk like domain (GAGAGS) with different numbers of elastin units (GVGVP). In addition to GVGVP, modified elastin includes lysine at the second position (GKGVP) (Figure 2.2-7-a, b).[113] The molecular structure and composition of the SELPs greatly affect the physicochemical and mechanical properties including the time required for gelation, degree of swelling and shear modulus.[7,113,114] The gelation time and swelling ratio are influenced by the number of elastin repeats, temperature and pH conditions. The higher the number of elastin repeats, the faster the gelation time and higher the swelling ratio; lower shear modulus indicates lower mechanical strength enabling the gel to spread around and deform.[114] These SELP hydrogels undergoes an irreversible sol-to-gel transition allowing DNA or viral particles to be loaded under aqueous conditions.[111] SELPs (SELP47K, SELP415K and SEL815K) have also been employed as matrices for controlled delivery of gene, targeting head and neck tumors.[7,8,113–117] SELP815K (Table 2) hydrogels, designed to contain silk and elastin repeats at a ratio of 8:16, has been encapsulated with reporter genes. Amongst the different variants, SELP815K shows the highest transfection efficiency in in vivo tumor model.[108] These hydrogels serve as a liquid chemoembolic, capable of co-delivering (Figure 2.2-7-c) chemotherapeutic drugs doxorubicin and sorafenib in an in vitro system.[109]
Figure 2.2-7.
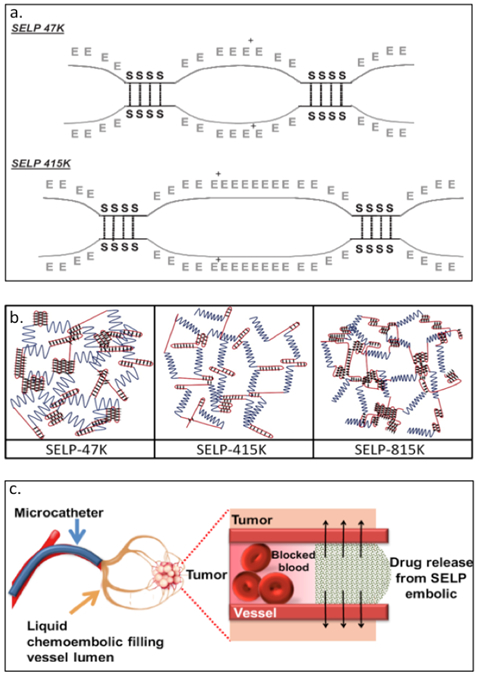
a. Schematic of SELP47K and SELP415K showing hydrogen bonds between silk-like blocks as points of crosslinking between polymer chains. b. Schematic of hydrogel structures formed by SELP47K, SELP415K, and SELP815. Blue lines represent elastin blocks, red line represents silk blocks, and the black diamond represents covalent crosslinks between silk blocks. c. Schematic of liquid chemo-embolic process for localized release of doxorubicin and sorafenib. Reproduced with permission from Ref.[109,114] Copyright 2016, American Chemical Society. Copyright 2010, Elsevier B.V
2.2.5. Silk-Collagen Protein Polymers
The Cohen Stuart group has generated self-assembling silk-collagen like block polymers, CSSC and SCCS (Table 2), where S is a pH–sensitive silk-like domain comprising of 24 repeats of (GA)3GE octapeptide and C is asparagine, serine and glutamine rich hydrophilic collagen-like domain.[118–120] Both CSSC and SCCS are able to form nanotapes that self-assemble to form transparent gels when uncharged.[119] Interestingly, the order of the blocks did not affect the properties of the gel except the gelation time. In a separate study, they used the same block polymers, which they referred as CSESEC and SECCSE (Figure 2.2-8), to test the mechanical properties of the transparent gels. At low pH, both CSESEC and SECCSE can self-assemble to form similar nanotapes, ultimately forming the gels.[118] Both the variants exhibit comparable elastic modulus with SECCSE gelating much faster than CSESEC. These pH responsive gels have high potential biocompatibility for blood and tissue engineering applications. [119]
Figure 2.2-8.
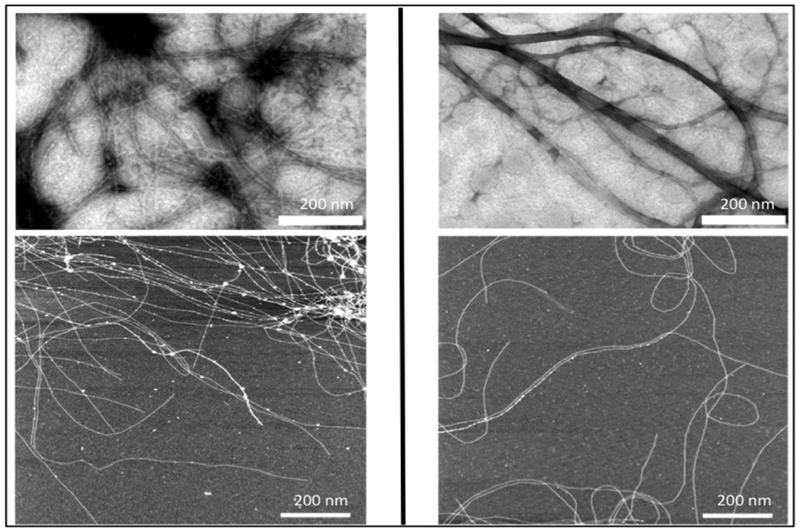
TEM images (top) and AFM images (bottom) of CSSC (left) and SCCS (right) supramolecular structures. The white bar indicates 200 nm. It can be clearly seen from both images that CSSC and SCCS form nanotape microstructures and self-assembly into fibrils. Reproduced with permission from Ref. [119] Copyright 2009, American Chemical Society.
2.3. Two-Component Protein Engineered Materials
The above mentioned examples are focused on single component materials, where a single polymer chain can self-assemble to form particles, fibers, colloids and nanotapes.[57,63,69,119] While these assemblies possess interacting domains in the single polymer chain that leads to self-assembly, an alternative approach is to combine two different protein chains that are able to interact with each other. In this section, we will discuss two component protein engineered materials resulting in more stable, controlled and functional systems.
2.3.1. Biocataytic Hydrogels from Two-Components
Expanding on the H-RC-globuar protein motif, the Banta group has generated a bioelectrocatalytic hydrogel by mixing osmium bis-bipyridine ([Os(bpy)2Cl2]Cl) complexed H-S-H (referred to as metallopolypeptide, Table 2) with H-S-SLAC.[91] The [Os(bpy)2Cl2]Cl moiety is used as redox-mediator while SLAC, an oxidoreductase, catalyzes the reduction of dioxygen to water. The helical domains within the metallopolypeptide and H-S-SLAC can self-assemble into a tetramer coiled-coil structure forming a supramolecuar hydrogel (Figure 2.3-1).[91] The mixed hydrogel demonstrates the ability to generate a catalytic current, thus enabling bioelectrocatalysis.[91] These types of bioelectrocatalytic hydrogels can be used as biosensors and in tissue engineering and drug delivery applications.[91]
Figure 2.3-1.
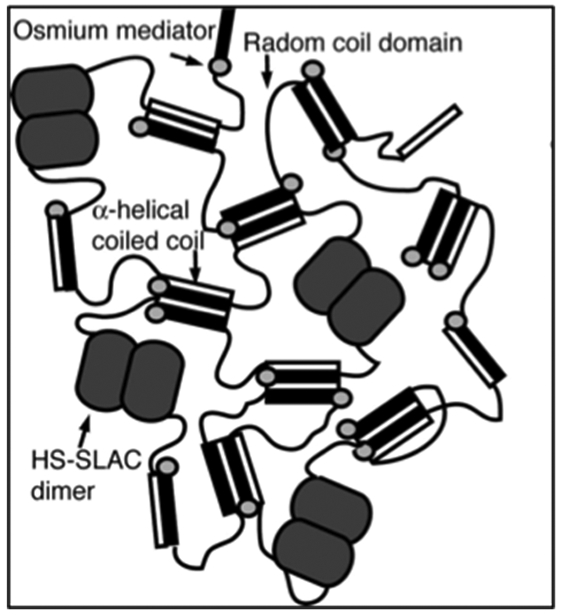
A bi-functionalized hydrogel with two components. Schematic showing crosslinking of the supramolecular hydrogel through: (1) formation of coiled coil, (2) dimerization of H-S-SLAC motif, and (3) crosslinking of metallopolypeptide motif mediated by osmium bis-bipyridine. Reproduced with permission from Ref. [91] Copyright 2008, The National Academy of Sciences of the USA
2.3.2. Two-Componentα-Helix-Elastin Polymers and α-Helix-Globular Protein
The Champion group has used leucine zipper coiled-coils ZE-ZR pairs in which they fuse the ZR domain with an ELP (Table 2) to yield ZR-ELP and the ZE domain with either mCherry or EGFP, resulting in mCherry–ZE or EGFP–ZE (Table 2, Figure 2.3-2-a).[121,122] As the ELP domain exhibits a phase transition behavior, the ZR-ELP forms a “rod–coil” and “globule-rod–coil” protein complex when mixed with either mCherry–ZE or EGFP–ZE.[122] These complexes self-assemble into thermally responsive coiled-coil protein vesicles capable of encapsulating fluorescein and small molecules for drug delivery (Figure 2.3-2-b).[122] These biocompatible protein vesicles can be used to incorporate enzymes or receptor ligands into vesicle membranes for various practical applications and have potential as “carrier-free” protein delivery system capable of self-assembly within the extracellular matrix.[123]
Figure 2.3-2.
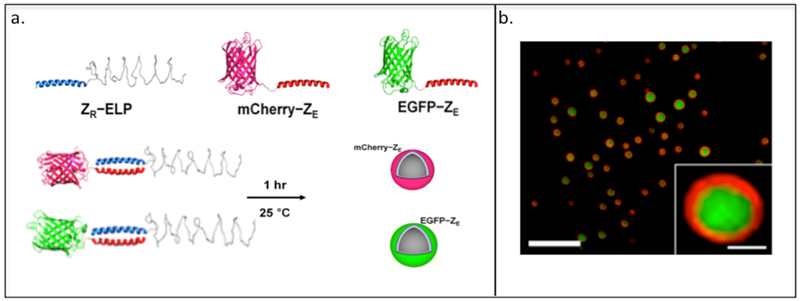
a. Representation of “rod-coil” and “globule-rod-coil” protein complexes and protein vehicles formed through the mixing of ZR that contains elastin-like protein (ELP, shown as blue coiled-coil structure) with its ZE counterpart that contains two different types of fluorescence protein (red globular protein: mCherry, green globular protein: EGFP). b. Confocal microscopy image of vesicle encapsulating fluorescein. Green fluorescence indicates the fluorescein and red fluorescence indicates the mCherry-ZE. Reproduced with permission from Ref. [122] Copyright 2014, American Chemical Society
2.3.3. Mixing-Induced Two-Component Hydrogel (MITCH)
Heilshorn and coworkers have developed mixing-induced two-component hydrogels (MITCH) comprising of seven repeats of the CC43 WW domain (C7) and nine repeats of the proline-rich PPxY peptide (P9) (Table 2, Figure 2.3-3-a).[124] The C7 and P9 domains are selected since they possess multiple repeats with short amino acid sequences, exhibit specific interactions and do not interfere with the cell signaling pathways.[124] MITCH demonstrates tunable mechanical properties capable of encapsulating cells under physiological conditions.[23,124,125] In addition, C7 has been mixed with P1 that contains an 8-arm polyethylene glycol (PEG) tethered to poly(N-isopropylacrylamide) (PNIPAM) to create a shear-thinning hydrogel for injectable encapsulation and long-term delivery (SHIELD) (Figure 2.3-3-b).[126] SHIELD minimizes destruction to the cell membrane during injection and improves viability of human induced pluripotent stem cell-derived endothelial cells (iPSC-ECs) in vivo. The physically crosslinked hydrogels of the SHIELD family demonstrates potential in cell transplantation for treating peripheral arterial disease.[126]
Figure 2.3-3.
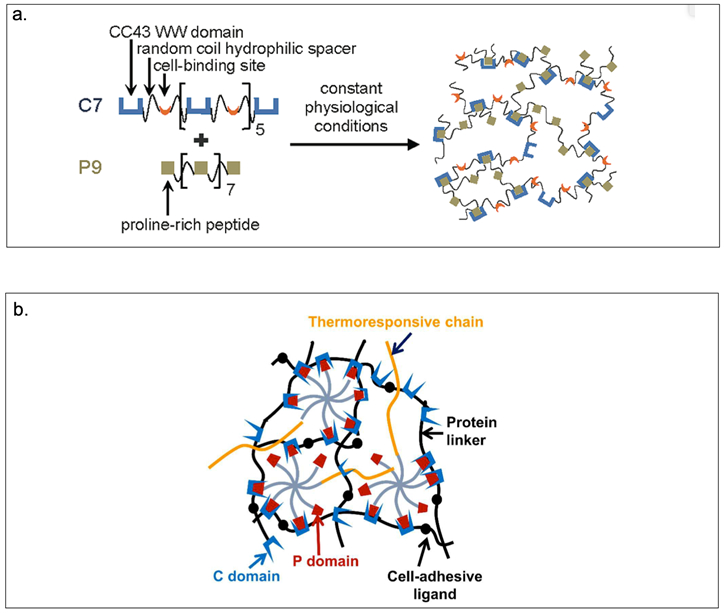
a.Schematic of MITCH. Protein polymers contain repeats of the CC43 WW domain and proline-rich peptide mixed together at constant physiological conditions results in hydrogel formation. b. Schematic of SHIELD prepared by mixing together two components: C7 engineered protein and an 8-arm PEG-P1 with or without thermoresponsive PNIPAM. Reproduced with permission from Ref.[125,126] Copyright 2011, American Chemical Society. Copyright 2018, Royal Society of Chemistry
2.3.4. Reversible Ca2+-Sensitive Two-Component Hydrogel
Calmodulin, a calcium modulated protein, regulates many calcium dependent processes found in eukaryotic cells.[127] Gallivan and colleagues have explored self-assembling materials comprised of calmodulin and calmodulin binding domains, exhibiting calcium dependent changes in microrheological properties.[128] Three different triblock proteins are generated: one consisting of calmodulin and tetrameric leucine zipper domain connected via a hydrophilic protein sequence ((AG)3-PEG)8, referred as CaM-(8)-Zip and the other two triblock proteins comprising of petunia glutamate decarboxylase (PGD) connected with either the human endothelial NO synthase (eNOS) or another copy of PGD connected via the hydrophilic linker ((AG)3PEG)n with n=8,40 (Table 2).[128] The CaM-(8)-Zip is mixed with either of the PGD containing block proteins to form a two-component system leading to a calcium sensitive network (Figure 2.3-4-a).[128] An increase in viscosity (about 5000-fold increase) is observed when calcium is added to the mixture of CaM-(8)-Zip and PGD-(8)-PGD, indicating the formation of a hydrogel (Figure 2.3-4-b).[128] These engineered proteins can be used to generate novel calcium sensitive materials useful in tissue engineering and drug delivery applications.
Figure 2.3-4.
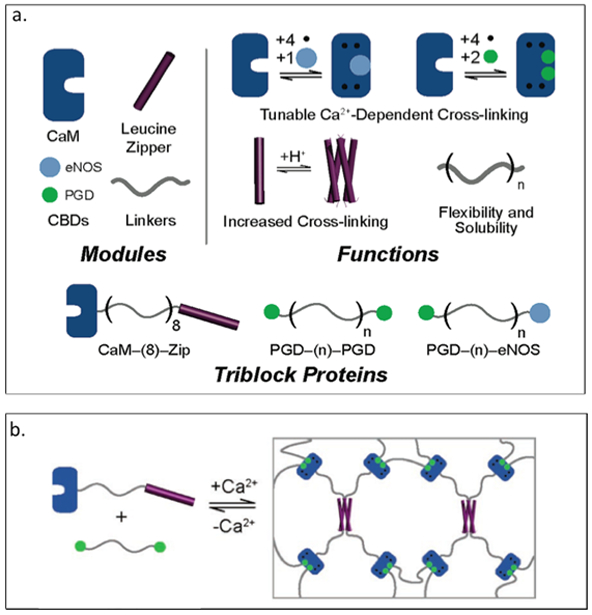
a. Representation of each module with structure and the specific functions for each module. Modules can be combined together to produce different functionalized materials. b. Schematic of PGD- (8)-PGD and CaM- (8)-Zip mixed with equal molar-formed network. Reproduced with permission from Ref.[128] Copyright 2006, American Chemical Society.
3. Chemically Crosslinked Protein Materials
While the aforementioned examples focus on physically crosslinked protein materials, in this section, we explore chemically crosslinked protein-engineered materials, which prevent gel dissolution through covalent networks.[129,130] Chemically crosslinked protein-engineered materials have the ability to form hydrophilic networks that absorb large amounts of solution, yet remain insoluble and maintain their specific structure.[23,131] Crosslinked protein hydrogels are classified as promising biomaterials[129] and have been of great interest in a variety of biomedical[129,132,133] and pharmaceutical[131,134] applications.
3.1. Chemically Crosslinking Through Side Chains
Chilkoti and coworkers have explored several chemical crosslinkers and enzymatic crosslinkers of ELPs (Figure 3.1-1) for its applications as potential injectable materials and scaffolds in tissue engineering.[27,135–137] The lysines within ELP[KV6] or ELP[KV16] (Table 3) are crosslinked with tris-succinimidyl aminotriacetate (TSAT) to form a networked gel.[135] The difference in molecular weight, concentration, and lysine content significantly impacts the swelling and mechanical properties of the chemically linked hydrogel.[135] An increase in mechanical integrity is observed for these crosslinked ELPs, making them useful in cartilage tissue repair.[135] In addition, ELP[KV6-112] and ELP[QV6-112] (Table 3) with X=lysine (K) or glutamine (Q) within the VPGXG repeating sequence, have been crosslinked using an enzyme, transglutaminase (tTG)) to form networked hydrogels (Figure 3.1-1).[136] These gels not only exhibit stabilization of the ELP matrix in situ, but can also promote cell viability and cartilage matrix synthesis and accumulation.[136] These findings promote the use of ELP gels as injectable materials and functional cartilage repair.[135,136] Furthermore, ELP[KV7F] and ELP[KV2F] can be crosslinked using β-[tris(hydroxymethyl)phosphino]propionic acid (THPP) to form hydrogels (Figure 3.1-1).[137] The reaction takes place under mild aqueous conditions. The resulting gels maintain cell viability, and their mechanical strength is comparable to the tissues found in cartilage.[137] Overall, these crosslinked gels have a tremendous potential in tissue engineering.
Figure 3.1-1.
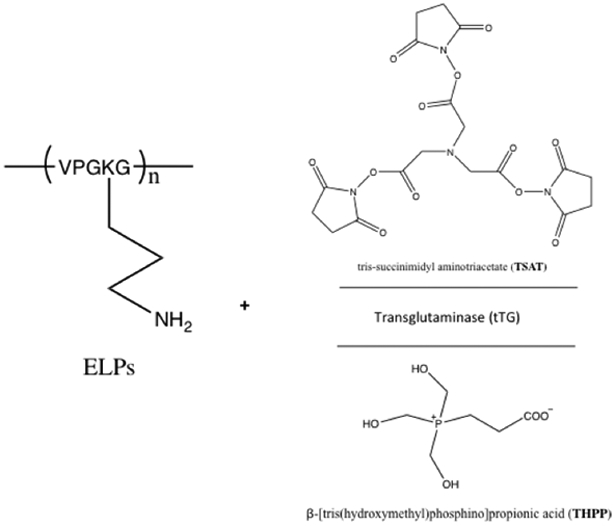
Reaction scheme for ELPs with chemical tris-succinimidyl aminotriacetate (TSAT) and β-[tris(hydroxymethyl)phosphino]propionic acid (THPP) and enzymatic (tTG) crosslinker to crosslink ELPs into networked hydrogels. Modified with permission from Ref.[137]
Table 3.
Sequences of chemically crosslinking proteins
| Type of Protein Materials |
Name of protein |
Sequence | Reference |
|---|---|---|---|
| Chemically Crosslinking Protein Hydrogel | ELP | MSKGPG(VGVPGVGVPGGGVPGAGVPGVGVPGVGVPGVGVPGGGVPGAGVPGGGVPG)9WPTer | Betre H. et al. (2002) |
| ELP[KV6] ELP[KV16] | VPGKG(VPGVG)6 VPGKG(VPGVG)16 |
Trabbic-Carlson K. et al. (2003) | |
| ELP[KV6-112] ELP[QV6-112] | MSKGPG[VGVPGKGVPGVGVPGVGVPGVGVPGVGVPGVGVPG]16WPTerTer MSKGPG[VGVPGQGVPGVGVPGVGVPGVGVPGVGVPGVGVPG]16WPTerTer |
McHale M.K. et al. (2005) | |
| ELPs with lysine at X position | (VPGKG)n | Di Zio K. et al. (2003) | |
| Two ELPs X position replaced with both lysine and isoleucine | Sequence
1: M-MASMTGGQQMG-HHHHHHH-DDDDK(LD-GEEIQGHIPREDVYHLYP-G((VPGIG)2VPGKG(VPGIG)2)4VP)3-KLE Sequence 2: M-MASMTGGQQMG-HHHHHHH-DDDDK(LD-GEEIQGHIPREVDYHLYP-G((VPGIG)2VPGKG(VPGIG)2)4VP)3-KLE |
Di Zio K. et al. (2003) | |
| Poly(Lys-25) | [(VPGVG)4(VPGKG)] | McMillan R.A. et al. (1999) | |
| LysB10 |
[VPAVGKVPAVG(IPAVG)4][(IPAVG)5]33[IPAVGKAAKVPGAG][(VPGAG)2VPGEG(VPGAG)2]28
[VPAVG-
KAAKVPGAGVPAVG(IPAVG)4][(IPAVG)5]33
[IPAVGKAAKA] *elastic-like central block is Bold ; plastic-like end block is italic; crosslinking block is underlined |
Sallach R. et al. (2009) | |
| ELP-Based Protein | MASMTGGQQMG-HHHHH-DDDDK-LQ[LDAS-bioactive site-SA((VPGIG)2VPGKG(VPGIG)2)3]4LE | Raphel J. et al. (2012), (2016) | |
| RGD | TVYAVTGRGDSPASSAA | Straley K.S et al. (2008) | |
| u1 u2 u3 |
YAVTGGTARSASPASSA YAVTGTSHRSASPASSA YAVTGDRIRSASPASSA |
||
| ELP-RGD with Crosslinking Site | MASMTGGQQMG-HHHHH-DDDDK-LQ[LDAS-
(TVYAVTGRGDSPASSAA)-[(VPGIG)2VPGKG(VPGIG)2]3]4
LE Crosslinking site is bold |
Chang D. et al. (2015) | |
|
A. gambiae fruit fly gene
RZ10 |
AQTPSSQYGAP (AQTPSSKYGAP -AQTPSSKYGAP)5 |
Renner J.N. et al. (2012) | |
| RLP12 | (GGRPSDSFGAPGGGN)12 |
Li L. et al.
(2011) Charati M.B. et al. (2009) |
|
| SpyTag SpyCatcher AAA BB A-LIF-A |
AHIVMVDAYKPTK AMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGK TISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHID MKGSSHHHHHHVDAHIVMVDAYKPTKLDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGV PGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGELAHIVMVDAYKPTKTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEG VPGVGVPGVG VPGGLLDAHIVMVDAYKPTKLEWKK MKGSSHHHHHHVDIPTTENLYFQGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSS GKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIDGPQGIWGQLDGHGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGV GVPGVGELYAVTGRGDSPASSAPIATSVPGVGVPGV GVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGGLLDIPTTENLYFQGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGKTISTW ISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIDGPQGIWGQLEWKK MKGSSHHHHHHVDAHIVMVDAYKPTKLDGHGVGVPGVGVPGVGVPGVGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVP GVGVPGVGVPGVGVPGEGVPGVGVPGVGELSPLPITPVNATCAIRHPCHGNLMNQIKNQLAQLNGSANALFISYYTAQGEPFPNNLDKLCGPNVTDFPPFHANGTEKAKLV ELYRMVAYLSASLTNITRDQKVLNPSAVSLHSKLNATIDVMRGLLSNVLCRLCNKYRVGHVDVPPVPDHSDKEVFRKKKLGCQLLGTYKQVISVVVQAFTSVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGVGVPGEGVPGVGVPGVGVPGVGVPGGLLDAHIVMVDAYKPTKLEWKK *SpyTag is Bold ; SpyCatcher is italic: LIF is underlined |
Zhang W. et
al. (2013) Sun F. et al. (2014) |
|
| GB1-Sc GB1-St (GB1-St)2 (GB1-Sc)3 TNfn3-(GB1-Sc)3 (GB1-St)4 |
MRGSHHHHHHGSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGKTISTWISDGQ VKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHI MRGSHHHHHHGSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTEAHIVMVDAYKPTK MRGSHHHHHHGSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTKRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDN GVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTK MRGSHHHHHHGSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATM ELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGA MELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDED GKELAGATMELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETA APDGYEVATAITFTVNEQGQVTVNGKATKGDAHI MRGSHHHHHHGSRLDAPSQIEVKDVTDTTALITWFKPLAEIDGIELTYGIKDVPGDRTTIDLTEDENQYSIGNLKPDTEYEVSLISRRGDMSSNPAKETFTT RSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIRSMDTYKLILNGKTLKGETTTEAVDAATAEKV FKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHIRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSGAMVDTLSGLSSEQGQSGDMTIEEDSATHIKFSKRDEDGKELAGATMELRDSSGKTIST WISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQ GQVTVNGKATKGDAHI MRGSHHHHHHGSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTKRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTKRSMDTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTKRSMDTYKLILNGKTLKGETTTEAVDA ATAEKVFKQYANDNGVDGEWTYDDATKTFTVTERSAHIVMVDAYKPTK *SpyTag is Bold ; SpyCatcher is italic: GB1 and TNfn3 is underlined |
Gao X. et al. (2016) | |
| ELP | [TVYAVYGRGDSPASSA-((VPGIG)2VPGKG(VPGIG)2)3]4 | Mald C.M. et al. (2016) |
Tirrell et. al have replaced X within (VPGXG)n of ELPs with the charged residue lysine, generating (VPGKG)n with pH-responsive properties and crosslinkable sites (Table 3). [16] The lysine residues have been crosslinked using bifunctional electrophiles.[16,138] Two different sequences of ELPs with both lysine and isoleucine as the guest residues are generated (Table 3). Lysine residues are introduced as crosslinking sites whereas isoleucine residue decreases the Tt to below ambient temperature for facile processing.[138,139] Bis(sulfosuccinimidyl) suberate (BS3) can react with ELPs via primary amine groups to generate protein films (Figure 3.1-2). By varying the ratio between the crosslinker and the ELP, the tensile properties and moduli of the crosslinked elastin protein can be tuned. These protein films mimic the extracellular matrix and thus can be used as grafts for small blood vessels.[138]
Figure 3.1-2.
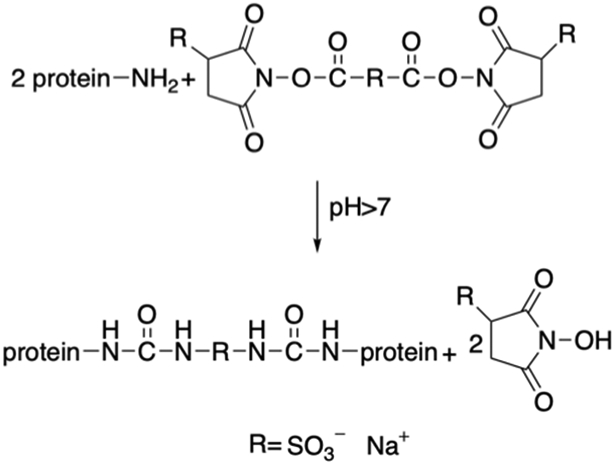
Reaction scheme of ELP proteins chemically crosslinked with bis (sulfosuccinimidyl) suberate (BS3). Modified with permission from Ref.[138]
Focusing on ELPs, Conticello et al. have genetically engineered and expressed poly(Lys-25) based on the elastin-mimetic repeat sequence [(VPGVG)4(VPGKG)], which is crosslinked with an equivalent amount of bifunctional N-hydroxysuccinimidyl (NHS) ester resulting in a well-defined microstructured hydrogel (Table 3).[140] A comparison of crosslinked protein hydrogel and uncrosslinked poly(Lys-25) reveals the hydrogel undergoes structural rearrangement (macroscopic and microscopic) during phase transition (Figure 3.1.3-a).[140] Its precisely defined structure permits a more rational evaluation of the effects of reaction conditions on its microstructure and materials properties.[18,21] In addition, they have generated a recombinant elastin-mimetic triblock copolymer, LysB10, comprised of hydrophobic sequences (IPAVG) that display plasticity separated by a central block that is both hydrophilic and elastomeric (VPGEG) with crosslinking sequence (KAAK) inserted between the plastic-like and elastic-like domain (Table 3, Figure 3.1.3-b); enabling both physical and chemical crosslinking.[141] Vapor phase crosslinking can be introduced by reacting the lysine residues with glutaraldehyde after LysB10 forms a protein film by physical crosslinking. Chemical crosslinking maintains the material’s integrity upon loading, thus enhancing the capacity of LysB10 films.[141] This elastin-mimetic protein polymer with plastic-like mechanical responses allows for its applications in vascular and non-vascular biomedical studies.[141]
Figure 3.1-3.
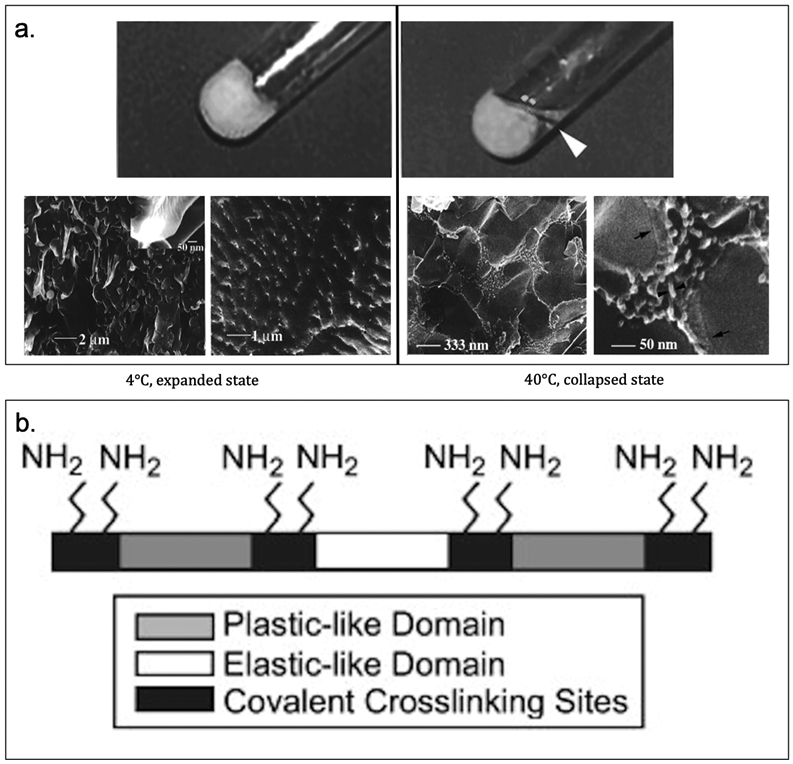
a. Photo (top) and cryo-HRSEM (bottom) images of poly(Lys-25) hydrogel to show macroscopic and microscopic gel behavior. At low temperature (4°C), the gel is in expanded state (left). At high temperature (40°C), the gel is in collapsed state (right). b. Schematic of LysB10 showing crosslinking primary amine sites and hydrophobic plastic-like endblock and hydrophilic elastic-like central block. Reproduced with permission from Ref. [140,141] Copyright 1999, American Chemical Society. Copyright 2008, Elsevier Ltd.
Heilshorn and colleagues have developed ELP-based hydrogels crosslinked with the chemical crosslinkers N-hydroxysuccinimide (NHS) ester (Figure 3.1.4-a) or tetrakis(hydroxymethyl) phosphonium chloride (THPC) (Figure 3.1.4-b).[142–144] Both these chemicals react with primary amines (lysine residues) to form a crosslinked network. These hydrogels have been used for neural regeneration,[142] as dental and orthopaedic implants,[145] as well as for maintaining 3-D culture of murine cochlea[143] and neural progenitor cell (NPC) stemness.[144]The recombinant proteins (Table 3) designed for neural regeneration consists of lysine bearing ELP and bioactive sites of either RGD containing sequence (Table 3) for cell adhesion or u1, u2 and u3 (Table 3) to be cleaved by urokinase plasminogen activator (uPA), an enzyme secreted by neurites.[142,143] The lysine within the proteins are chemically crosslinked with NHS esters to form hydrogels, which are capable of supporting cell adhesion and neural growth.[142] In another example, ELP-RGD with crosslinking sites (Table 3) is chemically crosslinked with THPC resulting in hydrogels capable of encapsulating cochlear cultures.[143] These protein hydrogels maintain the structural integrity of the cochlea compared to the non-encapsulated cochlea.[143] The THPC crosslinked ELP-RGD hydrogel matrix have also been used to maintain the NPC stemness.[144] These gels have been prepared with varied stiffness and degradability. While hydrogel degradation is essential for NPC stemness, gel stiffness has no specific correlation in maintaining the stemness.[144] The lysine bearing ELP-RGD with crosslinking sites is also conjugated using a photoactive covalent crosslinker, NHS–diazirine (NHS–diazirine, succinimidyl 4,40-azipentanoate) (Figure 3.1-4-c).[145,146] The development of a modular photoactive protein gel is useful for fabricating biomaterial coatings, films, and scaffolds.[146] The ELP-based protein (hydrogel) with NHS–diazirine, has been spin or dip coated on Ti6Al4V, a commercially available dental and orthopaedic titanium-based implant material.[145] UV exposure of ELP gel containing the photocrosslinker, allows the gel to be chemically conjugated with Ti6Al4V, which can improve the speed of osseointegration of titanium implants by increasing the bone-implant contact (Figure 3.1-4-d).[145]
Figure 3.1-4.
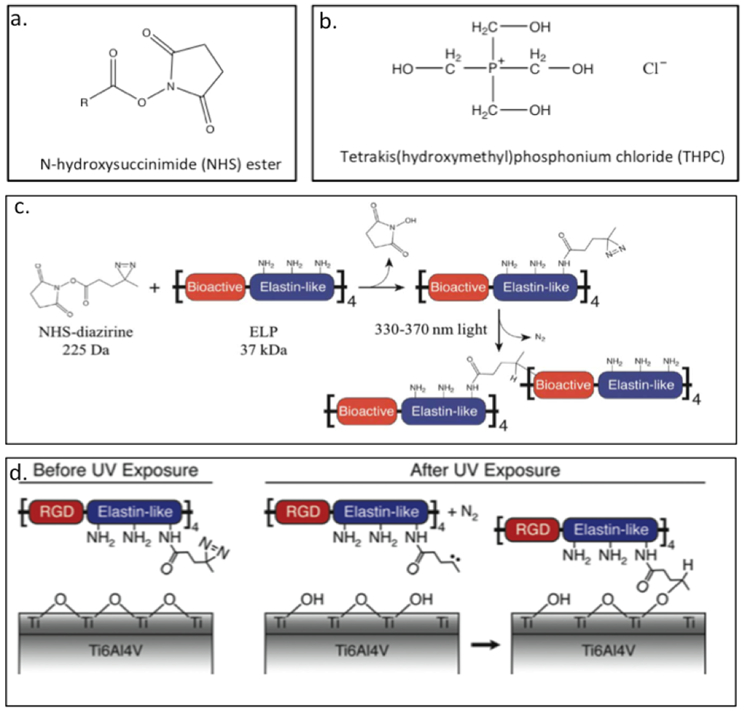
Chemical structure of a. N-hydroxysuccinimide (NHS) ester and b. tetrakis(hydroxymethyl)phosphonium chloride (THPC). c. Schematic of crosslinker conjugation chemistry. Lysine residues in ELPs protein react with the NHS–diazirine crosslinker to form an amide bond, thereby synthesizing a diazirine-modified ELP (ELP–D). Upon exposure to ultraviolet light, the diazirine group forms a highly reactive carbene intermediate, which can insert into a neighboring ELP chain. d. Schematic of proposed ELPs protein gel conjugation to titanium substrates upon exposure to UV light. Reproduced with permission from Ref. [146] Copyright 2012, Royal Society of Chemistry.
The Liu group has created the resilin mimic RZ10 (Table 3) derived from the fruit fly gene (Table 3).[38] By replacing tyrosine with phenylalanine and adding lysine as chemical cross-linking sites, RZ10 can be readily crosslinked with tris(hydroxymethyl)phosphine (THP) to form hydrogel.[38] Furthermore, they have attached different cell-binding domains, such as the RGD motif and vascular endothelial growth factor (VEGF), to exploit the cloning scheme containing bioactive domains. These gels exhibit a high compressive modulus, can maintain the viability of mesenchymal stem cells (MSCs) and has potential use in cartilage tissue engineering (Figure 3.1-5-a).[36,38,44] In a separate study, Kiick and coworkers have created RLP12 (Figure 3.1-5-b, Table 3), a modified exon 1 repeat motif, having similar mutations as RZ10. The additional lysine residues can react with [tris(hydroxymethyl)phosphino]propionic acid (THPP) to form a crosslinked hydrogel network that possess high elasticity with great resilience, and can be used in tissue regeneration.[147,148]
Figure 3.1-5.
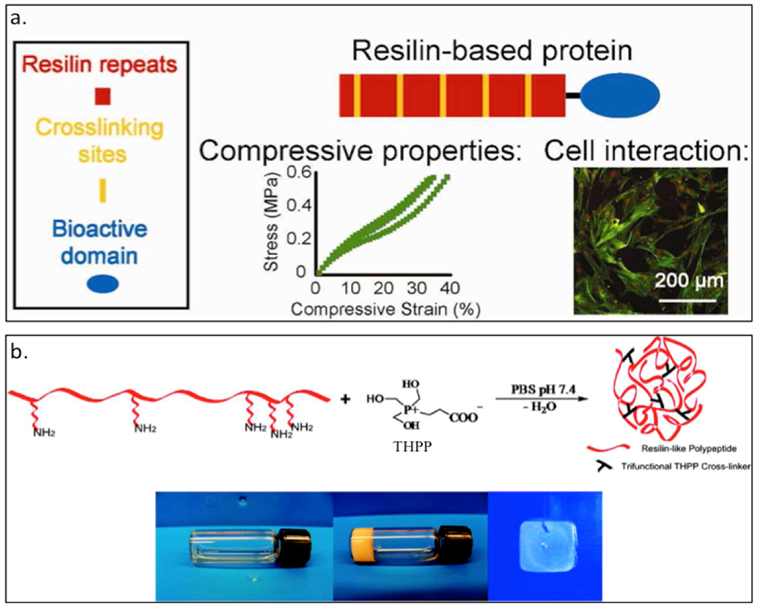
a. Scheme showing RZ10 resilin-based protein bearing crosslinking sites and bioactive domains. Characterization of cell interaction and compressive properties are studied for tissue engineering applications. b. RZP12 reslilin-like protein able to form a hydrogel by crosslinking with [tris(hydroxymethyl)phosphino]propionic acid (THPP). The gel formation can happen under mild conditions and tuning crosslinker ratio can generate RLP12 hydrogels and films. Reproduced with permission from Ref. [38][148] Copyright 2012, American Chemical Society. Copyright 2011, American Chemical Society.
Azide-alkyne cycloaddition or click chemistry represents a bioorthogonal reaction that enables specific reaction without cross reacting with common biological functional groups.[149] In the context of protein crosslinking via click chemistry, side chains bearing azides and alkynes are required.[150] Heilshorn and coworkers have created ELP hydrogels that crosslink via strain-promoted azide-alkyne cycloaddition (SPAAC) or Staudinger ligation for the application of cell encapsulation (Figure 3.1-6).[151] SPAAC mediated gelation happens instantaneously when ELP with azide moieties (N3-ELP) are mixed with ELP with bicyclo[6.1.0]nonyne (BCN) groups (BCN-ELP) (Table 3) to form hydrogel network.[151] On the contrary, Staudinger ligation mediated gelation happens at a much slower rate when N3-ELP is mixed with triarylphosphine functionalized ELP. While the azide groups are introduced in the side chains of lysine residues (of ELP) via diazo transfer protocol, BCN and triarylphosphine ELPs are obtained by reacting the lysine residues with activated esters of BCN and the triarylphosphine reagent.[151] ELP hydrogels formed via SPAAC can encapsulate human mesenchymal stem cells (hMSCs), human umbilical vein endothelial cells (HUVECs), and murine neural progenitor cells (mNPCs) with improved cell viability.[151]
Figure 3.1-6.
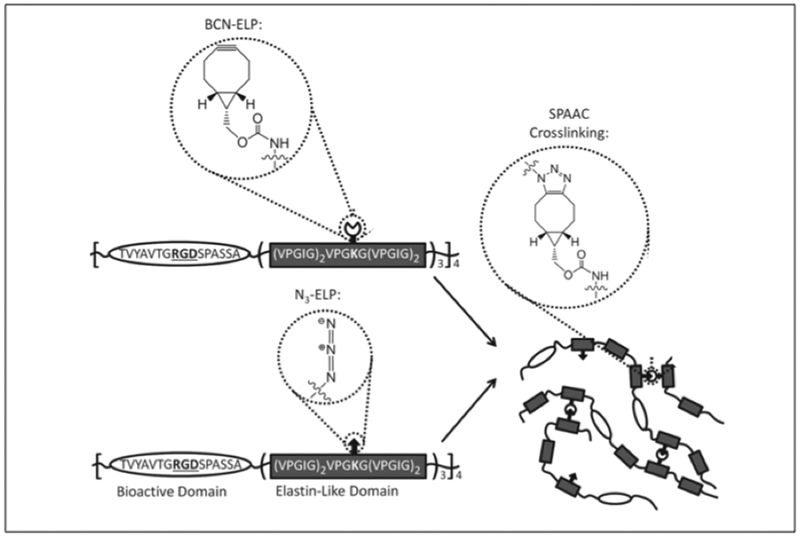
Crosslinking chemistry of engineered ELP hydrogel via strain-promoted azide-alkyne cycloaddition (SPAAC) for the application of cell encapsulation. ELPs are functionalized with azides (N3‐ELP) and strained cyclooctynes (BCN‐ELP). Upon mixing, the functionalized ELPs crosslink by SPAAC to form hydrogels.. Reproduced with permission from Ref. [151] Copyright 2016, John Wiley and Sons.
Recently, several groups have employed SpyCatcher-SpyTag chemistry to generate protein hydrogels.[152,153] First introduced by Howarth and coworkers, SpyCatcher-SpyTag chemistry is comprised of two peptide tags (SpyCatcher and SpyTag, Table 3) that can rapidly form an isopeptide bond between the Lys-31 and Asp-117 of SpyCatcher and SpyTag (Figure 3.1-7).[152,154] SpyCatcher and SpyTag are split from the second immunoglobulin-like collagen adhesion domain (CnaB2) from the fibronectin binding protein (FbaB) of Streptococcus pyogenes.[154] Tirrell and Arnold have used elastin-like proteins to explore the potential of using SpyCatcher-SpyTag chemistry to create chemically crosslink hydrogels.[152] They designate SpyTag as A and SpyCatcher as B. The AAA protein (Table 3) bearing three SpyTags at the head, center, and tail of the elastin-like protein and BB protein (Table 3) bearing two SpyCatchers at the end of each N- and C-terminal of the elastin-like protein with an integrin-binding site (RGD) and a matrix metalloproteinase-1 (MMP-1) cleavage site are biosynthesized .[152] AAA can react with BB to form a protein hydrogel through the Spy network covalent crosslinking. When mouse 3T3 fibroblasts are suspended in BB and reacted with AAA, the AAA+BB hydrogel successfully encapsulates fibroblasts showing no signs of cytotoxicity. Furthermore, they design the SpyTag protein, A-LIF-A (Table 3), with a chimeric leukemia inhibitory factor (LIF) variant that can be recognized by mouse embryonic stem cells (mESCs); when A-LIF-A is dissolved with BB, a hydrogel is formed, resulting in a 3D cell culture while maintaining the pluripotency of stem cells.[152]
Figure 3.1-7.
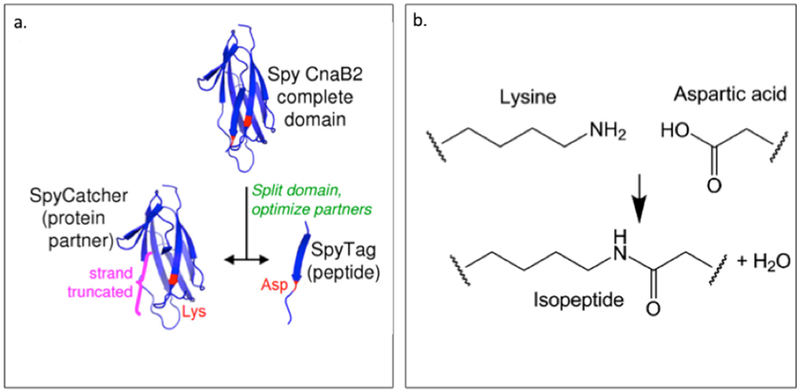
a. Cartoon of SpyCatcher-SpyTag split from CnaB2 b. Mechanism of rapid isopeptide bond reaction between the Lys-31 and Asp-117 of SpyCatcher and SpyTag. Reproduced with permission from Ref. [183] Copyright 2012, National Academy of Sciences.
Li and coworkers have utilized the SpyCatcher-SpyTag chemistry on tandem modular proteins to engineer them into hydrogels.[153] They select a small globular protein, GB1, and the third fibronectin type III (FnIII) from the human extracellular matrix protein tenascin (TNfn3) to construct the Spy network. They design GB1-SpyCatcher (GSc), GB1-SpyTag (GSt), GB1-SpyTag dimer ((GSt)2), GB1-SpyCatcher trimer ((GSc)3), GB1-SpyTag tetramer ((GSt)4), and TNfn3-(GB1-SpyCatcher trimer) (TNfn3-(GSc)3) (Table 3).[153] The (GSc)3 and (GSt)4 result in soft chemically crosslinked hydrogels.[153] They are able to suspend human lung fibroblasts (HFL1) in TNfn3-(GSc)3 and added (GSt)4 to form soft crosslinked hydrogel that can encapsulate HFL1 within its 3D network.[153] Also, they investigate the cumulative release of the (GSc)3/(GSt)4 hydrogel loaded with cyan fluorescence protein (CFP) to explore the capability of the hydrogel as a drug carrier.[153] Chemical crosslinking protein into hydrogels using the SpyCatcher-SpyTag chemistry have great potential applications in tissue engineering and developing scaffolds for biomedical usage and drug delivery.[152,153]
Protein crosslinking can be achieved via enzyme catalysis such as using sortase A.[155] Sortase family has the ability to carry out site-specific transpeptidation reactions.[155,156] Liu and Xiao employ Staphylococcus aureus sortase A (SrtA) as a molecular stapler enabling the crosslinking of (S)-carbonyl reductase II (SCRII) from Candida parapsilosis into dimers and trimers (Figure 3.1-8, Table 3),[157] SCRII is a short-chain alcohol dehydrogenase/reductase with higher efficiency in biotransformation and increased thermostability upon crosslinking.[157] SrtA is an attractive tool for protein engineering since it not only can mediate crosslinking between proteins but also can engineer bacterial surfaces by ligating protein to surfaces of gram-positive bacteria as well as covalently attach proteins to solid supports including PEG hydrogels and crosslinked polymer beads.[156,158,159]
Figure 3.1-8.
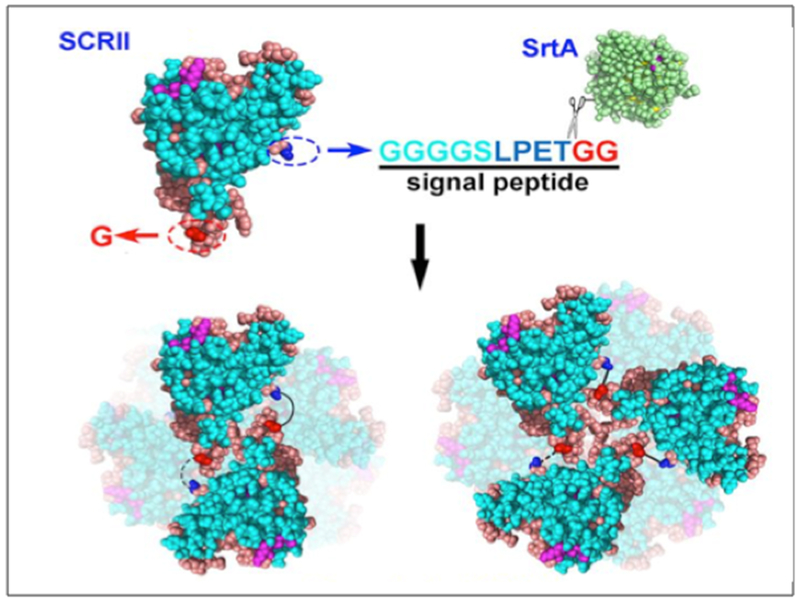
Schematic illustration showing the SrtA-mediated crosslinking of SCRII into dimers and trimers. Reproduced with permission from Ref. [157] Copyright 2017, Nature.
4. Metal Templating Protein Materials
Nanocomposites comprised of engineered proteins that can template inorganic metal ions, possess promising applications as efficient biosensors, contrast agents, delivery vehicles and therapeutics.[71,160,161] In this section, different protein engineered materials will be discussed to illustrate how templation of metal nanoparticles enhance/alter properties including structure, stability, self-assembly, chemical reactivity and stimuli-responsiveness.[162]
4.1. Gold Nanoparticles and Nanocomposites
Kiick and colleagues have generated a recombinant polypeptide 17H6 that assembles into irreversible β-sheet that allows it to form fibers at pH 2.3 and at a temperature of 80°C (Table 4). The polypeptide consists of repeats of alanine rich motifs with positively charged histidine patches that complex with negatively charged gold nanoparticles (GNPs) (Figure 4.1-1). The electrostatic interaction between 17H6 fibers and GNPs (approximately 2–3 nm size) results in formation of one-dimensional arrays (Figure 4.1-1). These nanoparticle arrays have potential applications in constructing optoelectronic devices.[68]
Table 4.
Sequences of metal templating proteins
| Type of Protein Materials |
Name of protein |
Sequence | Reference |
|---|---|---|---|
| Gold Nanoparticles and Nanocomposites | 17H6 | MGH10SSGHIHM (AAAQEAAAAQAAAQAEAAQAAQ)6AGGYGGMG | Sharma N. et al. (2009) |
| Cx Qx |
APQMLRE LQETNAA LQDVREL LRQQVKE ITFLKNT
SKL VKE ITFLKNT APQMLRE LQETNAA LQDVREL LRQQSKL |
Hume J. et al. (2015) | |
| Recombinant Clathrin Hub-His6 | MGSSHHHHHHSSGLVPRGSSHMLD-1074Clathrin heavy chain1675 | Hom N. et al. (2012) |
Figure 4.1-1.
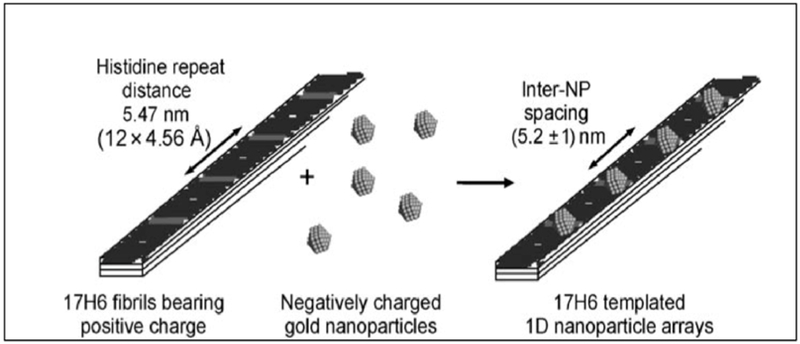
Schematic of 17H6 bearing positively charged patches assembly with negatively charged gold nanoparticles through electrostatic interactions. Reproduced with permission from Ref. [68] Copyright 2009, John Wiley and Sons.
Using the engineered coiled-coils C and Q, Hume et. al have templated gold nanoparticles exploiting the N-terminal His-tag. The nanocomposites C-GNP and Q-GNP exhibit distinct morphologies when compared to His-tag cleaved Cx and Qx and are further studied for their electronic properties (Table 4, Figure 4.1-2-a).[71] The His-tagged C and Q nanocomposites form large aggregates that adsorb onto the glassy carbon electrode surface, thereby decreasing the peak current. In contrast, the His-cleaved protein-GNPs remain soluble and exhibit an increase in current.[71] These assemblies aggregate upon the addition of trifluoroethanol, thus providing a strategy that can tune the supramolecular assemblies of these nanocomposites.
Figure 4.1-2.
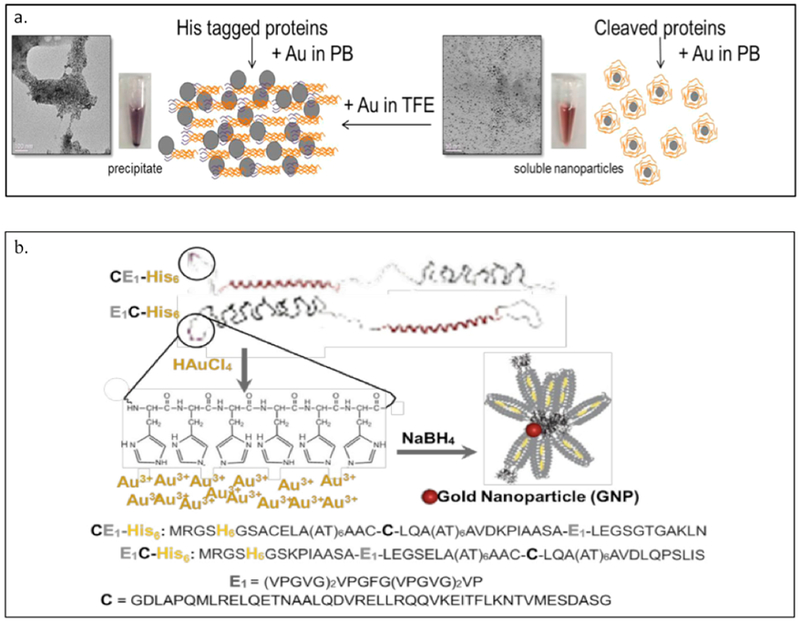
a. Representation of His tagged C and Q protein (left) comparing to cleaved His tagged Cx Qx protein (right) to template GNP. TEM images shown for both nanocomposites demonstrated very distinct assembly morphologies. b. Gold nanoparticle (GNP) templated-synthesis strategy and P-GNP sequences. Reproduced with permission from Ref.[148] [160] Copyright 2015 American Chemical Society. Copyright 2016, OMICS international.
The α-helix–elastin diblock copolymers comprised of C and E domains (as mentioned in section 2.2) has been employed for templation of GNPs.[160] The presence of hexahistidine tag at the N-terminus of E1C and CE1 assists in crystallization of GNPs leading to the production of protein-GNP nanocomposites (P-GNPs) (Table 4, Figure 4.1-2-b).[160] The CE1-His6–GNP and E1C-His6-GNP nanocomposites demonstrate improved delivery of curcumin with an increased uptake by breast cancer MCF-7 cells. [160]
5. Protein Materials Bearing Non-Canonical Amino Acids
Incorporation of non-canonical amino acids (NCAAs) within the proteins offers unique chemical and biological functionalities, paving the way to novel materials with extraordinary properties and functions.[12,163] Although NCAA incorporation can be technically challenging, requiring arduous optimization efforts in increasing protein yields or incorporating multiple/different NCAA within the same protein, recent advances and successful approaches in NCAA incorporation have expanded the genetic code, resulting in biomaterials with extraordinary and unique capabilities.[164] NCAAs can be introduced into recombinant proteins through either residue-specific incorporation or site-specific incorporation.[164–166] Residue-specific incorporation globally replaces a particular canonical amino acid with its NCAA analogue, whereas site-specific incorporation targets a specific residue within the protein sequence.[164,165]
5.1. Fluorination
Although fluorine atoms are not found in any of the twenty amino acids, incorporation of fluorine analogues into the protein have been shown to enhance their stability with improvements in physical and mechanical properties.[167] With the help of modern protein engineering incorporation techniques, fluorine can be readily introduced into a protein structure. Furthermore, fluorination can be used to study the folding mechanism of proteins and activity of biological molecules.[168,169]
Montclare and coworkers have studied the impact of fluorination on fiber assembly, supramolecular assemblies and mechanical properties.[167,170] Using the α-helical coiled–coils C and Q mentioned previously, incorporation of trifluoroleucine (TFL) is achieved via residue-specific incorporation (Table 5, Figure 5.1-1-a). The resulting C-TFL and Q-TFL exhibits an increase in α-helicity, resulting in enhanced drug binding ability, improved thermal stability and enhanced fiber assembly at pH 8.0 compared to their non-fluorinated variants.[170] Fluorination of the α-helix-elastin protein polymer through the residue-specific incorporation of p-fluorophenylalanine (pFF) results in pFF-EC, pFF-CE, and pFF-ECE (Table 5, Figure 5.1-1-b).[167] As fluorination affects the properties and stability of the target proteins, it has been used to tune the mechanical properties of the proteins. The fluorinated block polymers exhibit more elastic character with superior mechanical properties when compared to their non-fluorinated variants.[167] Incorporation of TFL and pFF improves overall stability, mechanical properties and allows self-assembly of proteins into higher ordered structures.[167,170]
Table 5.
Sequences of NCAA bearing proteins
| Type of Protein Materials |
Name of protein |
Sequence | Reference |
|---|---|---|---|
| Coiled-Coil Protein | C-TFL | MRGSHHHHHHGSIEGR APQMTFLRE TFLQETNAA TFLQDVRETFL TFLRQQVKE ITETFLKNT SKL | More H. et al. (2015) |
| Q-TFL | MRGSHHHHHHGSIEGR VKE ITFTFLKNT APQMTFLRE TFLQETNAA TFLQDVRETFL TFLRQQSKTFL | ||
| Diblock or Triblock Copolymer Protein with Two SADs | EpFF | [(VPGVG)2VPGFG(VPGVG)2]5VP | Yuvienco C. et al. (2012) |
| CpFF | DLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASG | ||
| pFF-EC | MRGSHHHHHHGSKPIAASA-EpFF-LEGSELA(AT)6AACG-CpFF-LQA(AT)6AVDLQPS | ||
| pFF-CE | MRGSHHHHHHGSACELA(AT)6AACG-CpFF-LQA(AT)6AVDKPIAASA-EpFF-LEGSGTGAKLN | ||
| pFF-ECE | MRGSHHHHHHGSKPIAASA-EpFF-LEGSELA(AT)6AACG-CpFF-LQA(AT)6AVDKPIAASA-EpFF-LEGSGTGAKLN | ||
| Elastin Mimic Protein with NCAA | Elastin Repetitive Protein Polymers | (Val-Pro-Gly-Val-Gly)2 (Val-Pro-Gly-(Xaa)-Gly)(Val-Pro-Gly-Val-Gly)2 | Wu I.L. et al. (2013) |
| Elastin with Globular Protein | ZE ZR Linker ELF ZE-ELP |
LEI EAAALE RTRYG ALEVQ ENTALET EVAELEQ EVQRLEN
IVSQY PL LEI RAA RR RNTALRT RVAELRQ RVQRLRN SQYET RYGPL G(GS)6G [(VPGVG)2-VPGFG(VPGVG)2]5VPGC LEI EAAALE RTRYG ALEVQ ENTALET EVAELEQ EVQRLEN IVSQY PL-G(GS)6G-[(VPGVG)2-VPGFG(VPGVG)2]5VPGC *p-azidophenelalanine is bold |
Zhang K. et al. (2005) |
Figure 5.1-1.
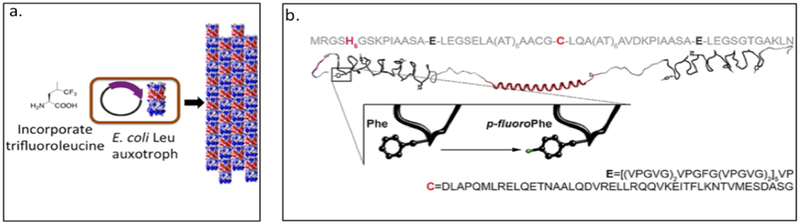
a. Schematic of residue-specific incorporation of TFL using leucine auxotroph E. coli cells to produce fluorinated protein that allow protein fiber formation. b. The schematic shows the sequence of pFF-ECE constructs containing six histidine tags near the N terminal and E domain highlight in black and C domain highlighted in red. Residue-specific incorporation of pFF happens at the residue site of phenylalanine (Phe). Reproduced with permission from Ref. [167,170] Copyright 2012, American Chemical Society. Copyright 2015, American Chemical Society.
5.2. Photocrosslinking
The approach of photocrosslinking proteins has been commonly employed to study protein-protein and protein and cell interactions.[171,172] Recently, there has been a growing interest in using photocrosslinking as a strategy to generate protein-based materials. Compared to chemical crosslinking, photocrosslinking can be performed under milder conditions to generate novel biomaterials.[173]
Conticello and coworkers have introduced new chemical functionalities into elastin mimetic protein polymers by incorporating NCAAs at the guest residue position (Table 5).[173] The NCAAs para-benzoyl-L-phenylalanine (Bpa) and para-azidophenylalanine (AzF) (Figure 5.2-1) have been introduced into elastin-mimetic protein polymers for up to twenty-two repeats. E. coli MRA30, a release factor −1 (RF-1) attenuated strain, is used to support multi-site suppression at selected amber codon positions, thereby introducing NCAA at multiple sites with the elastin sequence.[173] Bpa and AzF have been used to test how each NCAA can efficiently incorporate at designated locations. They have shown that the ability to recognize the substrate and charge the tRNA differs; the AzF incorporation into elastin mimetic protein polymer exhibits higher yield than Bpa.[173] Through this study, they have introduced a way to incorporate multiple photocrosslinkable residues at desired positions for enhancing protein functions.
Figure 5.2-1.
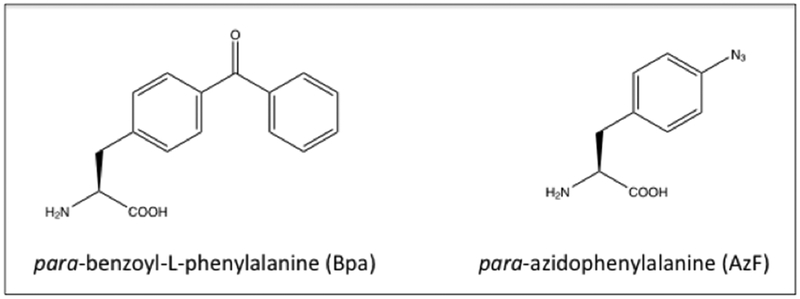
Structures of the NCAAs with photocrosslinking properties.
Tirrell et. al have developed a fusion protein, ZE-ELF comprising of: (1) a surface anchor domain that consists of an ELP bearing p-azidophenylalanines (ELF) and has the ability to provide strong adhesion to hydrophobic surfaces;[121][174] and (2) a protein capture domain composed of an acidic leucine zipper motif ZE (glutamic acid-rich leucine zipper motif).[174,175] A second polypeptide, ZR-target protein comprised of a target protein fused to a basic motif ZR (arginine-rich leucine zipper motif) is employed (Table 5, Figure 5.2-2).[121] The ZE-ELF immobilized and photocrosslinked to covalently attach on a solid support enable the ZR-target protein (green fluorescent protein (GFP) and glutathione-S-transferase (GST)) to be non-covalently captured on the solid surface through the paring of the ZE and ZR.[121] Compared to traditional methods such as physical adsorption or covalent conjugation through cysteine and lysine residues, this leucine zipper-elastin protein engineered material presents high specificity and stability due to its heterodimeric leucine zipper association.
Figure 5.2-2.
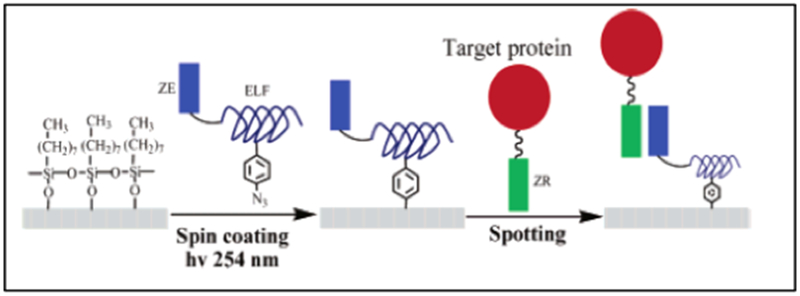
Schematic process of surface functionalization and coiled coil mediated immobilization of recombinant proteins on substrates. The phenylalanine residues in the ELF domain are partially replaced by a photo-reactive non-natural amino acid, para-azidophenylalanine. This moiety can be used to generate covalent linkages to substrates upon UV irradiation. ZE is incorporated as the protein capture domain, and the basic portion ZR is fused to target proteins as an affinity tag. Reproduced with permission from Ref. [121] Copyright 2005, American Chemical Society.
6. Conclusions
Protein engineered biomaterials have many advantages compared to natural or synthetic materials. In this review, we provide an overview of the different techniques used for protein functionalization including crosslinking, metal templation, and incorporation of non-canonical amino acids. Protein materials are discussed form the perspectives of domain designs that can self-assemble or can be mixed together to form protein-networks. Since chemical crosslinking is known to enhance the mechanical properties of the gels[130], we have discussed various examples that corroborate the use of crosslinkers for various protein-based gels. Furthermore, we have described the examples highlighting the assembly of gold nanoparticles-templated proteins and their applications in drug delivery. Lastly, we have briefly discussed functionalization via NCAA incorporation.
Overall, protein engineered functional materials are advantageous because: (1) they are able to combine properties from both naturally found materials and chemically synthesized materials;[176] (2) they can better simulate and interface with cells and tissue, which may effectively minimize foreign immune responses;[177] and (3) they can surpass the limitations of existing biotechnological natural/synthetic materials.[178] Single domain protein materials such as elastin, silk or coiled-coil proteins can result in hydrogels with tunable properties including temperature-dependent phase transition, tensile strength and drug storage cavities.[23,57,66] However, different applications may require the protein materials to be able to adapt and possess multi-properties. Multi-domain protein materials provide a solution by combining various functionalities. While many of the described single and multi-domain protein materials can self-assemble into a physically crosslinked network forming reversible hydrogels, the strength of such hydrogels may not be sufficient. Therefore strategies to develop stronger and stiffer crosslinked protein materials are crucial, demanding the use of chemical crosslinkers. Furthermore, templation of engineered proteins with inorganic metal ions enable the construction of various biological assemblies and composites with diverse functions. These assemblies have been typically used as biosensors, contrast agents and delivery vehicles.[71,160,161] NCAAs are also incorporated within the proteins to pave the way to novel materials with extraordinary properties and functions..[179]
The use of functionalized proteins holds great promise to generate novel biomaterials. Different strategies can be used to modify or decorate these materials with a range of functional groups. For example, protein-materials templated with iron oxide particles are being used for biomedical imaging.[180] Other approaches include the use of lithography techniques to pattern protein-based materials.[181] These strategies have been used to create biomimetics that are endowed with properties that are similar, or better than originally found in nature. Overall, the use of protein-based functionalized materials provides an attractive approach for generating advanced materials and devices.
7. Acknowledgements
This work was supported by NSF-DMREF under Award Number DMR 1728858, NSF-MRSEC Program under Award Number DMR 1420073, NSF BMAT under Award Number DMR 1505214, ARO W911NF-15-1-0304, the NYU Shiffrin-Myers Breast Cancer Discovery Fund, and the NYU CTSA grant UL1 TR000038 from the National Center for Advancing Translational Sciences, National Institutes of Health.
Biography

Yao Wang was born in Beijing China and holds a bachelor in Polymer materials. In 2015, she finished her M.S. in materials science and engineer at University of Florida. The same year September, she joined New York University Tandon School of engineering where she is currently pursuing her Ph.D. in materials chemistry. Her work focus on patternable environmentally responsive hydrogels derived from protein tri-block copolymers for biomedical applications.

Priya Katyal completed her Ph.D. in Pharmaceutical Sciences from University of Connecticut, where she investigated protein-protein and protein-polymer interactions using biophysical and biochemical approaches. She is a postdoctoral fellow in Professor Montclare’s lab at New York University, Tandon School of Engineering. Her research is focused on developing self-assembled injectable hydrogels for post-traumatic osteoarthritis.

Dr. Jin Kim Montclare is a Professor in the Department of Chemical and Biomolecular Engineering at NYU Tandon School of Engineering. She has appointments in Biochemistry at SUNY Downstate Medical Center, Chemistry at NYU, Radiology at NYU School of Medicine and Biomaterials at NYU College of Dentistry. Professor Montclare is performing groundbreaking research in engineering proteins to mimic nature and, in some cases, work better than nature. She exploits nature’s biosynthetic machinery and evolutionary mechanisms to design new artificial proteins. Her lab focuses on two research areas: (1) developing protein biomaterials capable of self-assembling into supramolecular structures and (2) engineering functional proteins/enzymes for particular substrates with the aim of targeting human disorders, drug delivery and tissue regeneration.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- [1].Silva NHCS, Vilela C, Marrucho IM, Freire CSR, Neto CP, Silvestre AJD, J. Mater. Chem. B. 2014, 2, 3715. [DOI] [PubMed] [Google Scholar]
- [2].Kaplan D, McGrath K, Eds. , in Protein-Based Mater, Birkhäuser Boston, 1996, pp. xiii–xx. [Google Scholar]
- [3].Sun H, Luo Q, Hou C, Liu J, Nano Today 2017, 14, 16. [Google Scholar]
- [4].Kim W, Chaikof EL, Adv. Drug Deliv. Rev 2010, 62, 1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Frandsen JL, Ghandehari H, Chem. Soc. Rev 2012, 41, 2696. [DOI] [PubMed] [Google Scholar]
- [6].Ghandehari H, Hatefi A, Adv. Drug Deliv. Rev 2010, 62, 1403. [DOI] [PubMed] [Google Scholar]
- [7].Haider M, Hatefi A, Ghandehari H, J. Control. Release 2005, 109, 108. [DOI] [PubMed] [Google Scholar]
- [8].Hatefi A, Cappello J, Ghandehari H, Pharm. Res 2007, 24, 773. [DOI] [PubMed] [Google Scholar]
- [9].Hu X, Cebe P, Weiss AS, Omenetto F, Kaplan DL, materialstoday 2012, 15, 208. [Google Scholar]
- [10].Yin L, Yuvienco C, Montclare JK, Biomaterials 2017, 134, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dickerson MB, Sandhage KH, Naik RR, Chem. Rev 2008, 108, 4935. [DOI] [PubMed] [Google Scholar]
- [12].Liu CC, Schultz PG, Annu. Rev. Biochem 2010, 79, 413. [DOI] [PubMed] [Google Scholar]
- [13].Baker P, Haghpanah J, Montclare JK, in Polym. Biocatal. Biomater. II (Eds: Cheng HN, Gross R), 2008, pp. 37–51. [Google Scholar]
- [14].Desai MS, Lee SW, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2015, 7, 69. [DOI] [PubMed] [Google Scholar]
- [15].Langer R, a Tirrell D, Nature 2004, 428, 487. [DOI] [PubMed] [Google Scholar]
- [16].Le DHT, Hanamura R, Pham DH, Kato M, Tirrell DA, Okubo T, Sugawara-Narutaki A, Biomacromolecules 2013, 14, 1028. [DOI] [PubMed] [Google Scholar]
- [17].Olsen BD, Kornfield JA, Tirrell DA, Macromolecules 2010, 43, 9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McMillan RA, Conticello VP, Macromolecules 2000, 33, 4809. [Google Scholar]
- [19].Haghpanah JS, Yuvienco C, Civay DE, Barra H, Baker PJ, Khapli S, Voloshchuk N, Gunasekar SK, Muthukumar M, Montclare JK, ChemBioChem 2009, 10, 2733. [DOI] [PubMed] [Google Scholar]
- [20].Rosano GL, Ceccarelli EA, Front. Microbiol 2014, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].van Hest JCM, Tirrell DA, Chem. Commun 2001, 1897. [DOI] [PubMed] [Google Scholar]
- [22].Qi Y, Chilkoti A, Curr. Opin. Chem. Biol 2015, 28, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dooling LJ, a Tirrell D, in Polym. Self Assem. Hydrogels From Fundam. Underst. to Appl, 2013, pp. 93–124. [Google Scholar]
- [24].Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A, J. Am. Chem. Soc. 1991, 113, 4346. [Google Scholar]
- [25].MacEwan SR, Chilkoti A, Biopolymers 2010, 94, 60. [DOI] [PubMed] [Google Scholar]
- [26].McDaniel JR, Radford DC, Chilkoti A, Biomacromolecules 2013, 14, 2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Betre H, Setton LA, Meyer DE, Chilkoti A, Biomacromolecules 2002, 3, 910. [DOI] [PubMed] [Google Scholar]
- [28].Meyer DE, Chilkoti A, Biomacromolecules 2004, 5, 846. [DOI] [PubMed] [Google Scholar]
- [29].Nagapudi K, Brinkman WT, Leisen J, Thomas BS, Wright ER, Haller C, Wu X, Apkarian RP, Conticello VP, Chaikof EL, Macromolecules 2005, 38, 345. [Google Scholar]
- [30].Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP, Adv. Funct. Mater. 2002, 12, 149. [Google Scholar]
- [31].Wright ER, Conticello VP, Adv. Drug Deliv. Rev 2002, 54, 1057. [DOI] [PubMed] [Google Scholar]
- [32].Wu X, Sallach R, Haller CA, Caves JA, Nagapudi K, Conticello VP, Levenston ME, Chaikof EL, Biomacromolecules 2005, 6, 3037. [DOI] [PubMed] [Google Scholar]
- [33].Weis-Forg T, J. Exp. Biol 1960, 37, 889. [Google Scholar]
- [34].Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DCC, Merritt DJ, Dixon NE, Nature 2005, 437, 999. [DOI] [PubMed] [Google Scholar]
- [35].Andersen SO, Biochim. Biophys. Acta - Gen. Subj 1964, 93, 213. [DOI] [PubMed] [Google Scholar]
- [36].Su RSC, Kim Y, Liu JC, Acta Biomater. 2014, 10, 1601. [DOI] [PubMed] [Google Scholar]
- [37].Dutta NK, Truong MY, Mayavan S, Roy Choudhury N, Elvin CM, Kim M, Knott R, Nairn KM, Hill AJ, Angew. Chemie - Int. Ed 2011, 50, 4428. [DOI] [PubMed] [Google Scholar]
- [38].Renner JN, Cherry KM, Su RSC, Liu JC, Biomacromolecules 2012, 13, 3678. [DOI] [PubMed] [Google Scholar]
- [39].Nairn KM, Lyons RE, Mulder RJ, Mudie ST, Cookson DJ, Lesieur E, Kim M, Lau D, Scholes FH, Elvin CM, Biophys. J. 2008, 95, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Balu R, Dutta NK, Choudhury NR, Elvin CM, Lyons RE, Knott R, Hill AJ, Acta Biomater. 2014, 10, 4768. [DOI] [PubMed] [Google Scholar]
- [41].Mayavan S, Dutta NK, Choudhury NR, Kim M, Elvin CM, Hill AJ, Biomaterials 2011, 32, 2786. [DOI] [PubMed] [Google Scholar]
- [42].Lin CY, Liu JC, Curr. Opin. Biotechnol 2016, 40, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lyons RE, Lesieur E, Kim M, Wong DCC, Huson MG, Nairn KM, Brownlee AG, Pearson RD, Elvin CM, Protein Eng. Des. Sel 2007, 20, 25. [DOI] [PubMed] [Google Scholar]
- [44].Renner JN, Kim Y, Cherry KM, Liu JC, Protein Expr. Purif 2012, 82, 90. [DOI] [PubMed] [Google Scholar]
- [45].Lyons RE, Nairn KM, Huson MG, Kim M, Dumsday G, Elvin CM, Biomacromolecules 2009, 10, 3009. [DOI] [PubMed] [Google Scholar]
- [46].Qin G, Rivkin A, Lapidot S, Hu X, Preis I, Arinus SB, Dgany O, Shoseyov O, Kaplan DL, Biomaterials 2011, 32, 9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen J, Lu H, Richmond J, Kaplan DL, Biomaterials 2003, 24, 401. [DOI] [PubMed] [Google Scholar]
- [48].Vepari C, Kaplan DL, Prog. Polym. Sci 2007, 32, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kapoor S, Kundu SC, Acta Biomater. 2016, 31, 17. [DOI] [PubMed] [Google Scholar]
- [50].Dinjaski N, Kaplan DL, Curr. Opin. Biotechnol 2016, 39, 1. [DOI] [PubMed] [Google Scholar]
- [51].Kaplan DL, Nat. Biotechnol 2002, 20, 239. [DOI] [PubMed] [Google Scholar]
- [52].Asakura T, Nitta K, Yang M, Yao J, Nakazawa Y, Kaplan DL, Biomacromolecules 2003, 4, 815. [DOI] [PubMed] [Google Scholar]
- [53].Kim UJ, Park J, Li C, Jin HJ, Valluzzi R, Kaplan DL, Biomacromolecules 2004, 5, 786. [DOI] [PubMed] [Google Scholar]
- [54].Lombardi SJ, Kaplan DL, The Amino Acid Composition of Major Ampullate Gland Silk (Dragline) of Nephila Clavipes, 1990. [Google Scholar]
- [55].Szela S, Avtges P, Valluzzi R, Winkler S, Wilson D, Kirschner D, Kaplan DL, Biomacromolecules 2000, 1, 534. [DOI] [PubMed] [Google Scholar]
- [56].Zhang X, Reagan MR, Kaplan DL, Adv. Drug Deliv. Rev 2009, 61, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kluge JA, Rabotyagova O, Leisk GG, Kaplan DL, Trends Biotechnol. 2008, 26, 244. [DOI] [PubMed] [Google Scholar]
- [58].Prince JT, McGrath KP, DiGirolamo CM, Kaplan DL, Biochemistry 1995, 34, 10879. [DOI] [PubMed] [Google Scholar]
- [59].Gellynck K, Verdonk P, Almqvist KF, van Nimmen E, Gheysens T, Mertens J, van Langenhove L, Kiekens P, Verbruggen A, Eur. Cells Mater 2005, 10, 45. [Google Scholar]
- [60].Yu YB, Adv. Drug Deliv. Rev 2002, 54, 1113. [DOI] [PubMed] [Google Scholar]
- [61].Crick FHC, Acta Crystallogr. 1953, 6, 689. [Google Scholar]
- [62].Woolfson DN, Bartlett GJ, Bruning M, Thomson AR, Curr. Opin. Struct. Biol 2012, 22, 432. [DOI] [PubMed] [Google Scholar]
- [63].Park WM, Champion JA, ACS Nano 2016, 10, 8271. [DOI] [PubMed] [Google Scholar]
- [64].McFarlane AA, Orriss GL, Stetefeld J, Eur. J. Pharmacol 2009, 625, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gunasekar SK, Haghpanah JS, Montclare JK, Polym. Adv. Technol 2008, 19, 454. [Google Scholar]
- [66].Gunasekar SK, Asnani M, Limbad C, Haghpanah JS, Hom W, Barra H, Nanda S, Lu M, Montclare JK, Biochemistry 2009, 48, 8559. [DOI] [PubMed] [Google Scholar]
- [67].Haghpanah JS, Yuvienco C, Roth EW, Liang A, Tu RS, Montclare JK, Mol. Biosyst. 2010, 6, 1662. [DOI] [PubMed] [Google Scholar]
- [68].Sharma N, Top A, Kiick KL, Pochan DJ, Angew. Chemie - Int. Ed 2009, 48, 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gunasekar SK, Anjia L, Matsui H, Montclare JK, Adv. Funct. Mater 2012, 22, 2154. [Google Scholar]
- [70].Hume J, Sun J, Jacquet R, Renfrew PD, Martin JA, Bonneau R, Gilchrist ML, Montclare JK, Biomacromolecules 2014, 15, 3503. [DOI] [PubMed] [Google Scholar]
- [71].Hume J, Chen R, Jacquet R, Yang M, Montclare JK, Biomacromolecules 2015, 16, 1706. [DOI] [PubMed] [Google Scholar]
- [72].More HT, Frezzo JA, Dai J, Yamano S, Montclare JK, Biomaterials 2014, 35, 7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu CF, Chen R, Frezzo JA, Katyal P, Hill LK, Yin L, Srivastava N, More HT, Renfrew PD, Bonneau R, et al. , Biomacromolecules 2017, 18, 2688. [DOI] [PubMed] [Google Scholar]
- [74].Xu C, Liu R, Mehta AK, Guerrero-Ferreira RC, Wright ER, Dunin-Horkawicz S, Morris K, Serpell LC, Zuo X, Wall JS, et al. , J. Am. Chem. Soc 2013, 135, 15565. [DOI] [PubMed] [Google Scholar]
- [75].Hirano T, Kinoshita N, Morikawa K, Yanagida M, Cell 1990, 60, 319. [DOI] [PubMed] [Google Scholar]
- [76].Sikorski RS, Boguski MS, Goebl M, Hieter P, Cell 1990, 60, 307. [DOI] [PubMed] [Google Scholar]
- [77].D’Andrea LD, Regan L, Trends Biochem. Sci 2003, 28, 655. [DOI] [PubMed] [Google Scholar]
- [78].Blatch GL, Lässle M, BioEssays 1999, 21, 932. [DOI] [PubMed] [Google Scholar]
- [79].Main ERG, Xiong Y, Cocco MJ, D’Andrea L, Regan L, Structure 2003, 11, 497. [DOI] [PubMed] [Google Scholar]
- [80].Speltz EB, Brown RSH, Hajare HS, Schlieker C, Regan L, Biochem. Soc. Trans 2015, 43, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cortajarena AL, Yi F, Regan L, ACS Chem. Biol 2008, 3, 161. [DOI] [PubMed] [Google Scholar]
- [82].Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA, Janin J, Simonson T, Curr. Opin. Struct. Biol 2004, 14, 208. [DOI] [PubMed] [Google Scholar]
- [83].Petka WA, Harden JL, Mcgrath KP, Wirtz D, Tirrell DA, Science (80-. ). 1998, 281, 389. [DOI] [PubMed] [Google Scholar]
- [84].Xu C, Kopeček J, Pharm. Res 2008, 25, 674. [DOI] [PubMed] [Google Scholar]
- [85].Xu C, Breedveld V, Kopeček J, Biomacromolecules 2005, 6, 1739. [DOI] [PubMed] [Google Scholar]
- [86].Kopeček J, Yang J, Acta Biomater. 2009, 5, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jang Y, Champion JA, Acc. Chem. Res 2016, 49, 57. [DOI] [PubMed] [Google Scholar]
- [88].Banta S, Wheeldon IR, Blenner M, Annu. Rev. Biomed. Eng 2010, 12, 167. [DOI] [PubMed] [Google Scholar]
- [89].Wheeldon IR, Barton SC, Banta S, Biomacromolecules 2007, 8, 2990. [DOI] [PubMed] [Google Scholar]
- [90].Wheeldon IR, Campbell E, Banta S, J. Mol. Biol 2009, 392, 129. [DOI] [PubMed] [Google Scholar]
- [91].Wheeldon IR, Gallaway JW, Barton SC, Banta S, Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wu J, Park JP, Dooley K, Cropek DM, West AC, Banta S, PLoS One 2011, 6, e24948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Banta S, Dooley K, Shur O, Annu. Rev. Biomed. Eng 2013, 15, 93. [DOI] [PubMed] [Google Scholar]
- [94].Shen W, Lammertink RGH, Sakata JK, Kornfield JA, Tirrell DA, Macromolecules 2005, 38, 3909. [Google Scholar]
- [95].Glykys DJ, Szilvay GR, Tortosa P, Suárez Diez M, Jaramillo A, Banta S, Syst. Synth. Biol 2011, 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Garcia KE, Babanova S, Scheffler W, Hans M, Baker D, Atanassov P, Banta S, Biotechnol. Bioeng 2016, 113, 2321. [DOI] [PubMed] [Google Scholar]
- [97].Campbell E, Wheeldon IR, Banta S, Biotechnol. Bioeng 2010, 107, 763. [DOI] [PubMed] [Google Scholar]
- [98].Szilvay GR, Brocato S, Ivnitski D, Li C, De La Iglesia P, Lau C, Chi E, Werner-Washburne M, Banta S, Atanassov P, Chem. Commun. (Camb) 2011, 47, 7464. [DOI] [PubMed] [Google Scholar]
- [99].Dooley K, Kim YH, Lu HD, Tu R, Banta S, Biomacromolecules 2012, 13, 1758. [DOI] [PubMed] [Google Scholar]
- [100].Dooley K, Bulutoglu B, Banta S, Biomacromolecules 2014, 15, 3517. [DOI] [PubMed] [Google Scholar]
- [101].Dai M, Haghpanah J, Singh N, Roth EW, Liang A, Tu RS, Montclare JK, Biomacromolecules 2011, 12, 4240. [DOI] [PubMed] [Google Scholar]
- [102].Olsen AJ, Katyal P, Haghpanah JS, Kubilius MB, Li R, Schnabel NL, Neill SCO, Wang Y, Dai M, Singh N, et al. , Biomacromolecules 2018, 19, 1552. [DOI] [PubMed] [Google Scholar]
- [103].Dooling LJ, Buck ME, Bin Zhang W, Tirrell DA, Adv. Mater 2016, 28, 4651. [DOI] [PubMed] [Google Scholar]
- [104].Dooling LJ, Tirrell DA, ACS Cent. Sci 2016, 2, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yigit S, Dinjaski N, Kaplan DL, Biotechnol. Bioeng 2016, 113, 913. [DOI] [PubMed] [Google Scholar]
- [106].Qiu W, Huang Y, Teng W, Cohn CM, Cappello J, Biomacromolecules 2010, 11, 3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Xia X, Xu Q, Hu X, Qin G, Kaplan DL, Biomacromolecules 2011, 12, 3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Gustafson J, Greish K, Frandsen J, Cappello J, Ghandehari H, J. Control. Release 2009, 140, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Poursaid A, Jensen MM, Nourbakhsh I, Weisenberger M, Hellgeth JW, Sampath S, Cappello J, Ghandehari H, Mol. Pharm 2016, 13, 2736. [DOI] [PubMed] [Google Scholar]
- [110].Hatefi A, Megeed Z, Ghandehari H, J. Gene Med 2006, 8, 468. [DOI] [PubMed] [Google Scholar]
- [111].Megeed Z, Haider M, Li D, O’Malley BW, Cappello J, Ghandehari H, J. Control. Release 2004, 94, 433. [DOI] [PubMed] [Google Scholar]
- [112].Megeed Z, Cappello J, Ghandehari H, Adv. Drug Deliv. Rev 2002, 54, 1075. [DOI] [PubMed] [Google Scholar]
- [113].Haider M, Leung V, Ferrari F, Crissman J, Powell J, Cappello J, Ghandehari H, Mol. Pharm 2005, 2, 139. [DOI] [PubMed] [Google Scholar]
- [114].Gustafson JA, Ghandehari H, Adv. Drug Deliv. Rev 2010, 62, 1509. [DOI] [PubMed] [Google Scholar]
- [115].Nagarsekar A, Crissman J, Crissman M, Ferrari F, Cappello J, Ghandehari H, J. Biomed. Mater. Res 2002, 62, 195. [DOI] [PubMed] [Google Scholar]
- [116].Greish K, Araki K, Li D, O’Malley BW, Dandu R, Frandsen J, Cappello J, Ghandehari H, Biomacromolecules 2009, 10, 2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Megeed Z, Cappello J, Ghandehari H, Pharm. Res 2002, 19, 954. [DOI] [PubMed] [Google Scholar]
- [118].Martens AA, Van Der Gucht J, Eggink G, De Wolf FA, Cohen Stuart MA, Soft Matter 2009, 5, 4149. [Google Scholar]
- [119].Martens AA, Portale G, Werten MWT, De Vries RJ, Eggink G, Cohen Stuart MA, De Wolf FA, Macromolecules 2009, 42, 1002. [Google Scholar]
- [120].Schor M, Martens AA, Dewolf FA, Cohen Stuart MA, Bolhuis PG, Soft Matter 2009, 5, 2658. [Google Scholar]
- [121].Zhang K, Diehl MR, Tirrell DA, J. Am. Chem. Soc 2005, 127, 10136. [DOI] [PubMed] [Google Scholar]
- [122].Park WM, Champion JA, J. Am. Chem. Soc 2014, 136, 17906. [DOI] [PubMed] [Google Scholar]
- [123].Park WM, Champion JA, Angew. Chemie - Int. Ed 2013, 52, 8098. [DOI] [PubMed] [Google Scholar]
- [124].Mulyasasmita W, Lee JS, Heilshorn SC, Biomacromolecules 2011, 12, 3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wong CTS, Foo P, Seok J, Mulyasasmita W, Parisi-amon A, Heilshorn SC, PNAS 2009, 106, 22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Foster AA, Dewi RE, Cai L, Hou L, Strassberg Z, Alcazar CA, Heilshorn SC, Huang NF, Biomater. Sci 2018, 6, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Crivici A, Ikura M, Annu. Rev. Biophys. Biomol. Struct 1995, 24, 85. [DOI] [PubMed] [Google Scholar]
- [128].Topp S, Prasad V, Cianci GC, Weeks ER, Gallivan JP, J. Am. Chem. Soc 2006, 128, 13994. [DOI] [PubMed] [Google Scholar]
- [129].Rosiak JM, Yoshii F, Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. with Mater. Atoms 1999, 151, 56. [Google Scholar]
- [130].Hennink WE, van Nostrum CF, Adv. Drug Deliv. Rev 2012, 64, 223. [DOI] [PubMed] [Google Scholar]
- [131].Censi R, Di P, Vermonden T, Hennink WE, J. Control. Release 2012, 161, 680. [DOI] [PubMed] [Google Scholar]
- [132].Hoffman AS, Adv. Drug Deliv. Rev 2002, 54, 3. [DOI] [PubMed] [Google Scholar]
- [133].Huglin MB, Br. Polym. J 1989, 21, 184. [Google Scholar]
- [134].Peppas NA, Bures P, Leobandung W, Ichikawa H, Eur. J. Pharm. Biopharm 2000, 50, 27. [DOI] [PubMed] [Google Scholar]
- [135].Trabbic-Carlson K, Setton LA, Chilkoti A, Biomacromolecules 2003, 4, 572. [DOI] [PubMed] [Google Scholar]
- [136].McHale MK, Setton LA, Chilkoti A, Tissue Eng. 2005, 11, 1768. [DOI] [PubMed] [Google Scholar]
- [137].Lim DW, Nettles DL, Setton LA, Chilkoti A, Biomacromolecules 2007, 8, 1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Di Zio K, Tirrell DA, Macromolecules 2003, 36, 1553. [Google Scholar]
- [139].Welsh ER, a Tirrell D, Biomacromolecules 2000, 1, 23. [DOI] [PubMed] [Google Scholar]
- [140].McMillan RA, Caran KL, Apkarian RP, Conticello VP, Macromolecules 1999, 32, 9067. [Google Scholar]
- [141].Sallach RE, Cui W, Wen J, Martinez A, Conticello VP, Chaikof EL, Biomaterials 2009, 30, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Straley KS, Heilshorn SC, Soft Matter 2009, 5, 114. [Google Scholar]
- [143].Chang DT, Chai R, Dimarco R, Heilshorn SC, Cheng AG, Otol. Neurotol. 2015, 36, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Madl CM, Lesavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M, Lampe KJ, Nguyen D, Chaudhuri O, Enejder A, et al. , Nat. Mater 2017, 16, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Raphel J, Karlsson J, Galli S, Wennerberg A, Lindsay C, Haugh MG, Pajarinen J, Goodman SB, Jimbo R, Andersson M, et al. , Biomaterials 2016, 83, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Raphel J, Parisi-Amon A, Heilshorn SC, J. Mater. Chem 2012, 22, 19429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Charati MB, Ifkovits JL, Burdick JA, Linhardt JG, Kiick KL, Soft Matter 2009, 5, 3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Li L, Teller S, Clifton RJ, Jia X, Kiick KL, Biomacromolecules 2011, 12, 2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Best MD, pubs.acs.org/Biochemistry Biochem. 2009, 48, 6571. [Google Scholar]
- [150].Nwe K, Brechbiel MW, Cancer Biother. Radiopharm 2009, 24, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Madl CM, Katz LM, Heilshorn SC, Adv. Funct. Mater 2016, 26, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Sun F, Zhang W-B, Mahdavi A, Arnold FH, Tirrell DA, PNAS 2014, 111, 11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gao X, Fang J, Xue B, Fu L, Li H, Biomacromolecules 2016, 17, 2812. [DOI] [PubMed] [Google Scholar]
- [154].Zhang W-B, Sun F, Tirrell DA, Arnold FH, J. AM. CHEM. SOC 2013, 135, 13988. [DOI] [PubMed] [Google Scholar]
- [155].Heck T, Faccio G, Richter M, Thöny-Meyer L, Appl Microbiol Biotechnol 2013, 97, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Popp MWL, Ploegh HL, Angew. Chemie - Int. Ed 2011, 50, 5024. [DOI] [PubMed] [Google Scholar]
- [157].Li K, Zhang R, Xu Y, Wu Z, Li J, Zhou X, Jiang J, Liu H, Xiao R, Sci. Rep 2017, 7, 3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Parthasarathy R, Subramanian S, Boder ET, Bioconjug. Chem 2007, 18, 469. [DOI] [PubMed] [Google Scholar]
- [159].Chan L, Cross HF, She JK, Cavalli G, Martins HFP, Neylon C, PLoS One 2007, 2, e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Dai M, Frezzo J. a., Sharma N, Chen R, Singh N, Yuvienco C, Caglar E, Xiao S, Saxena A, Montclare JK, J. Nanomed. Nanotechnol 2016, 07, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Saha K, Bajaj A, Duncan B, Rotello VM, Small 2011, 7, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Sontz PA, Song WJ, Tezcan FA, Curr. Opin. Chem. Biol 2014, 19, 42. [DOI] [PubMed] [Google Scholar]
- [163].Voloshchuk N, Montclare JK, Mol. Biosyst. 2010, 6, 65. [DOI] [PubMed] [Google Scholar]
- [164].Johnson JA, Lu YY, Van Deventer JA, Tirrell DA, Curr. Opin. Chem. Biol 2010, 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Link AJ, Mock ML, Tirrell DA, Curr. Opin. Biotechnol 2003, 14, 603. [DOI] [PubMed] [Google Scholar]
- [166].Ngo JT, Tirrell DA, Acc. Chem. Res 2011, 44, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Yuvienco C, More HT, Haghpanah JS, Tu RS, Montclare JK, Biomacromolecules 2012, 13, 2273. [DOI] [PubMed] [Google Scholar]
- [168].Neil E, Marsh G, Acc. Chem. Res 2014, 47, 2878. [DOI] [PubMed] [Google Scholar]
- [169].Yoo TH, Link a J., Tirrell D. a, Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].More HT, Zhang KS, Srivastava N, Frezzo JA, Montclare JK, Biomacromolecules 2015, 16, 1210. [DOI] [PubMed] [Google Scholar]
- [171].Mitra N, Mater. Methods 2013, 3, 204. [Google Scholar]
- [172].Chin JW, Martin AB, King DS, Wang L, Schultz PG, PNAS 2002, 99, 11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Wu IL, Patterson MA, Carpenter Desai HE, Mehl RA, Giorgi G, Conticello VP, ChemBioChem 2013, 14, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Zhang K, Sugawara A, Tirrell DA, ChemBioChem 2009, 10, 2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Diehl MR, Zhang K, Lee HJ, Tirrell DA, Science (80-. ). 2006, 311, 1468. [DOI] [PubMed] [Google Scholar]
- [176].Dimarco RL, Heilshorn SC, Adv. Mater 2012, 24, 3923. [DOI] [PubMed] [Google Scholar]
- [177].Gomes S, Leonor IB, Mano JF, Reis RL, Kaplan DL, Prog Polym Sci 2012, 37, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Van Hest JCM, Tirrell DA, Chem. Commun 2001, 1897. [DOI] [PubMed] [Google Scholar]
- [179].Brustad EM, Arnold FH, Curr. Opin. Chem. Biol 2011, 15, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Bulte JWM, Kraitchman DL, NMR Biomed. 2004, 17, 484. [DOI] [PubMed] [Google Scholar]
- [181].Carrico IS, Maskarinec SA, Heilshorn SC, Mock ML, Liu JC, Nowatzki PJ, Franck C, Ravichandran G, Tirrell DA, J. Am. Chem. Soc 2007, 129, 4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Haghpanah JS, Tu R, Da Silva S, Yan D, Mueller S, Weder C, Foster EJ, Sacui I, Gilman W, Montclare JK, Biomacromolecules 2013, 14, 4360. [DOI] [PubMed] [Google Scholar]
- [183].Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M, PNAS 2012, 109, E690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Hom N, Mehta KR, Chou T, Foraker AB, Brodsky FM, Kirshenbaum K, Montclare JK, J. Mater. Chem 2012, 22, 23335. [DOI] [PMC free article] [PubMed] [Google Scholar]