

Population screening and double-blind phenotyping confirm unique clinical profile in FMR1 premutation carriers.
Abstract
The impact of the FMR1 premutation on human health is the subject of considerable controversy. A fundamental unanswered question is whether carrying the premutation allele is directly correlated with clinical phenotypes. A challenging problem in past genotype-phenotype studies of the FMR1 premutation is ascertainment bias, which could lead to invalid research conclusions and negatively affect clinical practice. Here, we created the first population-based FMR1-informed biobank to find the pattern of health characteristics in premutation carriers. Our extensive phenotyping shows that premutation carriers experience a clinical profile that is significantly different from controls and is evident throughout adulthood. Comprehensive understanding of the clinical risk associated with this genetic variant is critical for premutation carriers, their families, and clinicians and has important implications for public health.
INTRODUCTION
The phenotypic features of human disease are initially defined by studies of selected patient groups that may not represent the full genetically affected population. Resulting genotype-phenotype correlations may be biased, which could affect the outcomes of research and the health and well-being of millions of people worldwide (1). In this study, our focus is on the gene known as fragile X mental retardation 1 (FMR1). The conditions related to the FMR1 gene are a prime example of genetic variants for which phenotypes were defined from clinically ascertained data and possibly not representative of the full genetically affected population.
The FMR1 gene is responsible for the production of a protein called fragile X mental retardation protein (FMRP). This protein regulates the translation of ~30% of all transcripts in the synaptic proteome, predicting a key and widespread role in the functioning of the nervous system (2). In the 5′ untranslated region of the FMR1 mRNA, there are varying numbers of cytosine-guanine-guanine (CGG) trinucleotide repeats. The modal number of CGG repeats in the human population is around 30. Large expansions in the number of CGG repeats can disrupt the functioning of the FMR1 gene (2–4). Expansion of CGG repeats beyond 200 leads to hypermethylation and at least partial silencing of FMR1, which results in the fragile X syndrome (FXS) (5–7). This genetic condition is the most common inherited cause of intellectual disability and autism (5–7). However, the phenotypic effect of shorter repeat expansions, below 200 CGGs, remains a subject of considerable controversy (6).
The FMR1 premutation (55 to 200 CGG repeats) is carried by over 1 million individuals in the United States, and carriers originally were believed to be clinically unaffected except for their risk of having a child with FXS (8, 9). However, two known disorders have now been well documented to cause clinical symptoms in a subset of individuals carrying the premutation: fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI). FXTAS is a neurodegenerative disorder that emerges after age 50 and affects 30 to 40% of male premutation carriers (10) and 8 to 16% of female premutation carriers (11). FXPOI is defined as menopause before age 40 and other reproductive symptoms and affects 20 to 25% of female premutation carriers (12). The penetrance of these conditions is highly correlated with the number of CGG repeats, and they are more prevalent in individuals with larger than 70 CGGs (7, 10, 13).
In addition, some clinical and community-based reports have suggested that premutation carriers are at higher risk for a range of other health conditions and symptoms, including autoimmune diseases, migraines, fibromyalgia, neuropathy, infertility, depression, anxiety, and cognitive dysfunction, all with variable frequency and emerging at different stages of the life course (6, 8, 14–17). These two characteristics—differential age of symptom onset and variability in frequency of symptoms within the premutation population—make it a significant challenge to determine the genotype-phenotype association of the FMR1 premutation.
There is controversy regarding whether these symptoms are the direct result of the premutation because nearly all studies describing these phenotypes have been based on individuals who were ascertained through a family member with FXS who was diagnosed in a clinical setting. Following diagnosis of FXS, family members are offered cascade genetic testing, leading to the identification of relatives with the premutation. Family members, thus, become aware of their own genetic status, as do their clinicians, which may artifactually elevate prevalence estimates of symptoms. An additional limitation of these studies is that the biological significance of the premutation may be confounded with parenting/family stress due to the challenges posed by a family member with FXS (18). Thus, the published literature may be skewed toward larger CGG repeats, reports of more serious symptoms, and substantial ascertainment bias. To date, genotype-phenotype associations have not been evaluated systematically for all potential symptoms in the absence of familial FXS or studied in a population-based unbiased context.
The purpose of the present research was to use double-blind methods where both clinicians and patients were blind to genotype to evaluate whether premutation carriers differ significantly from those with normal numbers of CGGs in the pattern of clinical diagnoses recorded in their electronic health records (EHRs). We further determined whether these double-blind methods replicate the clinical literature. Incorporation of EHRs into genetic research provides an unprecedented opportunity to refine the definition of the genotype-phenotype association in human diseases. Mining the EHR is a rich and powerful tool to rapidly ascertain a comprehensive and diverse collection of clinical phenotypes (19). By linking population-based EHRs to genomic data, we investigated associations of the FMR1 premutation with a wide range of possible health conditions. We analyzed females and males separately, as the premutation phenotypes associated in the clinical literature differed by sex.
The present study, using a discovery-oriented approach, is the first research to investigate the health characteristics of FMR1 premutation carriers in a population setting incorporating the entire spectrum of available health diagnoses. Using machine learning approaches, we mined the EHRs of nearly 20,000 participants (all of whom were served by a single health care system), with the goal of identifying potential phenotypes associated with the FMR1 premutation (Fig. 1). In addition, we studied the EHRs of participants of various ages to investigate age-related differences in premutation phenotypes. The FMR1 premutation carriers in our study are unaware of their CGG repeat length, and thus, unlikely to have been familiar with the medical literature identifying symptoms associated with the premutation. Therefore, the results of this study offer an opportunity to evaluate the extent of primary phenotypes in premutation carriers unconfounded by knowledge or concerns about one’s own genetic status, the effects of stressful parenting, or clinical ascertainment bias. Understanding the clinical risks associated with the FMR1 premutation potentially will result in more effective preventive care and personalized treatment that could improve the quality of life of millions of people around the globe and have a positive impact on public health. The study has the potential to inform a long-standing research controversy within the FMR1 field. More generally, our approach provides a window into how screening for genetic variants can inform health care prior to receiving a diagnosis.
Fig. 1. Workflow overview of creating and mining the FMR1-informed biobank.
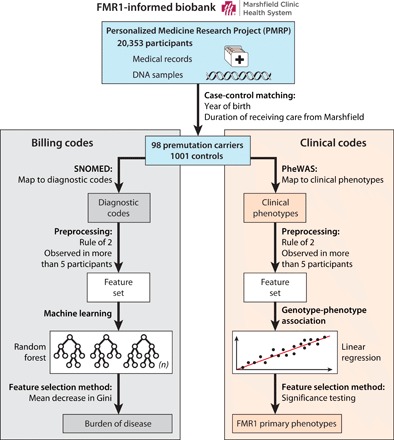
Starting with recruiting 20,353 PMRP participants, 19,996 individuals were genotyped. We identified premutation carriers and controls and matched them on year of birth and duration of receiving care from Marshfield Clinic. The diagnostic codes were used to examine whether the overall health profile of premutation carriers is different than controls. To filter possible noise and error in the EHR data, we applied the rule of 2 and limited our dataset to health conditions that were observed in more than five participants. We applied random forest to create a model representing the health conditions differentiating the two groups. Further examination of the model showed that premutation carriers suffer from a higher burden of disease throughout the life span compared with the controls for differentiating conditions. In a separate set of analysis, we used PheWAS to identify individual clinical conditions that are primary phenotypes of premutation. The resulting phenotypes are unconfounded by concerns about one’s own genetic status, stressful parenting, or clinical ascertainment bias.
RESULTS
Here, we report on the first U.S. population–based FMR1-informed biobank that we created by connecting EHRs of 19,996 adults with their FMR1 genetic information. All participants received their health care from the Marshfield (Wisconsin) Clinic health care system. As members of the Personalized Medicine Research Project (PMRP), these participants consented to contribute their deidentified EHRs, DNA, and other biosamples to be used in research. The phenotypic data for this study include almost 40 years of detailed EHR data (1979 to 2018) in the form of International Classification of Disease, Ninth and Tenth Revisions (ICD-9 and ICD-10 codes) harmonized as SNOMED (systematized nomenclature of medicine–clinical data) codes.
We screened 19,996 PMRP participants to identify all those who had premutation-range CGG repeats (i.e., 55 to 200 repeats) (20). In total, 98 premutation carriers were identified (72 females and 26 males), who are the focus of the present study. All individuals who had a CGG repeat in the premutation range were included in the analysis. CGGs of female premutation carriers ranged from 55 to 125 (mean, 66.9) on longer allele and 7 to 45 (mean, 28.8) on shorter allele; CGGs of male premutation carriers ranged from 55 to 96 (mean, 63.81) (Fig. 2). Thus, this is a study of genotype-phenotype associations among individuals who were primarily in the lower half of the premutation range. Most studies based on ascertainment through family diagnosis are skewed toward larger number of CGGs, and thus, here we have a unique opportunity to investigate premutation carriers beyond the scope of previous studies.
Fig. 2. Distributions of CGG repeats in female and male participants.
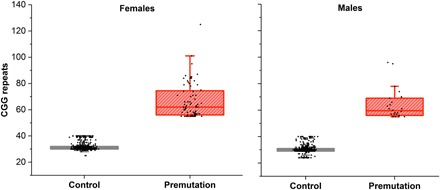
Premutation-range CGG repeats are defined as 55 to 200 repeats, and normal range is considered to be 24 to 40 CGG repeats. In total, 98 premutation carriers were identified (72 females and 26 males), and we selected 1001 age-matched controls (494 male and 507 female controls). CGG repeats in female premutation carriers ranged between 55 and 125, and in male premutation carriers, the range was between 55 and 96 CGGs.
We selected 1001 controls with CGGs in the normal range (i.e., 24 to 40 CGG repeats)—494 male controls and 507 female controls (Fig. 2). The female controls had both FMR1 alleles in the normal range. As the presence (or absence) of certain health phenotypes in the EHR is a function of the participant’s age and observation window, we matched premutation carriers and controls on year of birth and duration of receiving care from the Marshfield Clinic (Table 1). The participants did not differ in many parameters that otherwise could have confounded the interpretation of the data. As shown in Table 1, these included total number of medical encounters to providers in the Marshfield health care system, total number of SNOMED codes in the EHR, number of unique SNOMED codes in the EHR, or number of medical encounters or codes per year of receiving care from the Marshfield system. Hence, phenotypic characteristics that are found to differentiate premutation carriers from controls would not be attributable to differences between the two groups in age, health care utilization from the Marshfield Clinic, or total diagnoses received. None of the participants—case or control—had a diagnosis of FXS, FXTAS, or FXPOI in their EHR. Our data include deceased cases (8 premutation carriers and 108 controls), but EHRs from all cases were included in this study regardless of mortality status. Individuals with 41 to 54 CGG repeats (the “gray zone”) were excluded from the control group, as there is some evidence suggesting possible phenotypic associations with gray zone CGG repeats (21). In addition, those with low numbers of CGG repeats (<2 SDs below the mean; 7 to 23 CGGs in the present population) were similarly excluded from the control group because recent research has suggested that having low numbers of CGGs might possibly be associated with clinical symptoms (22, 23).
Table 1. Participants’ characteristics.
Participants included 72 female premutation carriers with 55 to 125 CGGs, 507 female controls with 24 to 40 CGGs, 26 male premutation carriers with 55 to 96 CGGs, and 494 male controls with 24 to 40 CGGs. Female deceased: premutation carriers = 5 and control group = 40. Male deceased: premutation carriers = 3 and control group = 68.
| Premutation carriers | Controls | |||||
| Variable | Range | Mean | Range | Mean | P value | |
| Females | Year of birth* | 1918–1987 | 1957.74 | 1911–1988 | 1956 | 0.62 |
|
Duration of receiving care from Marshfield* |
3–40 | 32.47 | 1–40 | 31.96 | 0.67 | |
|
Number of medical encounters |
12–1114 | 268.55 | 3–1652 | 300.28 | 0.26 | |
|
Number of SNOMED codes |
21–2985 | 748.77 | 10–6130 | 859.54 | 0.24 | |
|
Unique SNOMED codes |
10–369 | 151.80 | 5–482 | 159.45 | 0.47 | |
|
SNOMED codes/ years in the system |
1.17–218 | 28.44 | 0.59–234.9 | 27.79 | 0.85 | |
|
Number of medical encounters/years in the system |
0.67–54.11 | 9.57 | 0.30–64.64 | 9.63 | 0.95 | |
| Males | Year of birth* | 1912–1988 | 1956 | 1911–1988 | 1956.06 | 0.99 |
|
Duration of receiving care from Marshfield* |
2–40 | 28.5 | 1–40 | 31.26 | 0.12 | |
|
Number of medical encounters |
16–703 | 197.92 | 4–1617 | 246.94 | 0.29 | |
|
Number of SNOMED codes |
20–2335 | 608.85 | 7–6416 | 745.89 | 0.43 | |
|
Unique SNOMED codes |
9–306 | 115.65 | 5–495 | 132.20 | 0.35 | |
|
SNOMED codes/ years in the system |
0.67–59.87 | 21.10 | 0.48–195.46 | 24.14 | 0.57 | |
|
Number of medical encounters/years in the system |
0.53–18.02 | 7.12 | 0.38–44.08 | 8.02 | 0.53 | |
*Cases and controls were matched on year of birth and duration of receiving care from Marshfield.
Differentiating premutation carriers from controls
We used a machine learning approach, random forest, to differentiate premutation carriers from individuals with normal alleles using EHR data. The input vector for each participant represents the frequency of appearance of each feature (SNOMED concept identifiers) in the EHRs. Although the use of EHR data provides a unique opportunity to examine a broad spectrum of phenotypes in a population setting, this approach also has multiple challenges. EHRs contain noise, other errors, and missing data, in part because their primary role is billing and also because patients choose whether and when to come into the clinic. We attempted to minimize these limitations by restricting the analyses to codes that appeared at least twice for a given participant (rule of 2) and that were observed in at least five individuals (24, 25).
To investigate the possible effect of age, we conducted the analyses at various age thresholds including all of the codes that were recorded in the EHRs before the ages of 40, 60, and 80, as well as lifetime diagnoses. We include cumulative lifetime diagnoses because 186 participants received care beyond the age of 80. 10-fold cross-validation was used to train and test the models. To measure the success of classification, the area under the receiver operating characteristic curve (AUROC) is reported. If the AUROC is significantly greater than 0.5, as determined by a Mann-Whitney-Wilcoxon test (Mann-Whitney U test), we conclude that the premutation carriers are significantly different from controls (26).
Given the variability in both age of onset and frequency of symptoms, we expected a significant but not perfect classification of the two groups. Using this approach, we were able to differentiate premutation carriers from controls based on diagnostic codes in their EHRs. Our random forest classifiers predicted the premutation status of participants before the ages of 40, 60, 80, or lifetime diagnoses with AUROC = 0.63, P = .0000; AUROC = 0.65, P = .0000; AUROC = 0.65, P = .0000; and AUROC = 0.6, P = .007, respectively, for females, and AUROC = 0.61, P = 0.039; AUROC = 0.63, P = .0000; AUROC = 0.64, P = .0000; and AUROC = 0.66, P = .0000, respectively, for males (Fig. 3). The statistically significant values of AUROCs for these models indicate the importance of the target genotype (FMR1 premutation) and are consistent with the variability in frequency and age-related symptom manifestation of the FMR1 premutation.
Fig. 3. Classifier performances using different sets of inputs in premutation carriers.
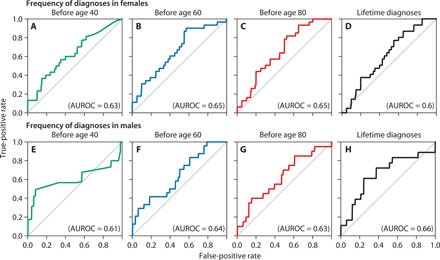
Frequency of diagnoses received (A) before age 40 in females (AUROC = 0.63), (B) before age 60 in females (AUROC = 0.65), (C) before age 80 in females (AUROC = 0.65), and (D) in the entire lifetime in females (AUROC = 0.6). Frequency of diagnoses received (E) before age 40 in males (AUROC = 0.61), (F) before age 60 in males (AUROC = 0.64), (G) before age 80 in males (AUROC = 0.63), and (H) in the entire lifetime in males (AUROC = 0.66).
To identify the specific clinical diagnoses that differentiate premutation carriers from controls, we used a measure called mean decrease in impurity based on Gini score (MDG), which is defined as the total decrease in node impurities from splitting on the variable, averaged over all trees in a trained random forest. Variables with higher MDGs are more influential in creating decision trees for prediction (27). After identifying these variables, we examined the participants’ EHR data in terms of three indicators of burden of disease (28): (i) the percentage of cases and controls who received each diagnosis, (ii) the number of medical encounters for each condition for cases versus controls, and (iii) the age of cases versus controls when they first received each diagnosis (data S1 and S2). Figure 4 shows the distribution of three indicators of the burden of disease for the 100 variables with the highest MDG at age 40. Similar distributions based on ages 60 and 80 and lifetime are shown in figs. S1 to S3. Results indicated that for all of these variables, female premutation carriers had either a higher likelihood of receiving the diagnosis, had a higher frequency of medical encounters for this diagnosis, or were first diagnosed at a younger age compared with controls.
Fig. 4. The distribution of the target diagnoses in premutation carriers and controls for codes received before age 40.
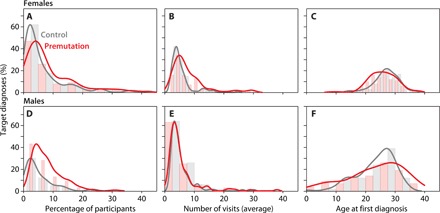
(A) Prevalence of target diagnosis codes in females; the percentage of premutation carriers who received the target codes was higher than controls. (B) Average frequency of medical encounters for target diagnoses in females; premutation carriers had a higher number of medical encounters for the differentiating diagnoses than controls. (C) Average age when participants received the target codes for the first time in females, with premutation carriers experiencing symptoms at younger ages than controls. (D) Prevalence of target diagnosis codes in males; the percentage of premutation carriers who received the target codes was higher than controls. (E) Average frequency of medical encounters for target diagnoses in males; the distributions did not differ for male premutation carriers from controls. (F) Average age when participants received the target codes for the first time in males; premutation carriers and controls had similar age of onset for target conditions.
Focusing on diagnostic codes that were received before age 40 in females (Fig. 4, figs. S1 to S3, and data S1), we observed that the percentage of receiving the code was higher in premutation carriers for 75% of the 100 target conditions. A greater number of medical encounters for female premutation carriers than for controls was observed in 70% of the target conditions, and in 64% of conditions, premutation carriers were diagnosed at a younger average age than controls. For 32% of conditions, female premutation carriers had worse health outcomes on all three indicators of disease than the controls—higher frequency of receiving the diagnosis, greater number of medical encounters for the diagnosis, and younger age at receiving the diagnosis. We did not observe any condition in which all criteria were higher in controls than in premutation carriers (data S1).
For males, focusing on diagnostic codes that were received before age 40 (Fig. 4 and data S2), we observed that the percentage of receiving the code was higher in premutation carriers for 78% of the 100 target conditions. However, unlike for females, the frequency of medical encounters for target conditions did not differ for male premutation carriers than for controls, and the two groups had similar distribution of ages when first receiving these diagnoses. For 14% of conditions, male premutation carriers had worse health outcomes on all three indicators of disease than the controls—higher frequency of receiving the diagnosis, greater number of medical encounters for the diagnosis, and younger age at receiving the diagnosis. We did not observe any condition in which all criteria were higher in male controls than in male premutation carriers. These patterns at age 40 are less definitive than those observed in females. This observation could be the result of older age of onset for FMR1-related health conditions in males (i.e., the onset of FXTAS symptoms is at age 50 or older). As shown in figs. S1 to S3, the burden of disease in males appears to be greater at older ages.
Participants were not aware of their CGG repeat length, so their higher burden of disease was not an artifact of anxiety emanating from knowledge of their premutation status. On the basis of available data, premutation carriers and controls had an equivalent likelihood of parenting a child with a disability, meaning that stressful parenting does not confound the health outcomes in these individuals.
Phenotypic profiles of premutation carriers
To further examine the phenotypic association of clinical diagnoses and the FMR1 premutation, we used the Phenome-Wide Association Study (PheWAS) methodology, separately for females and for males (29). We converted all diagnostic codes to ICD-9 codes and then created a map connecting those codes to clinical phenotypes (phecodes) (30). To take into account the effect of both X chromosomes for females, the number of CGGs on the shorter allele was included as a covariate in all analyses. Here, we report the health profile of participants based on clinical phenotypes that were received before age 40. In the Supplementary Materials, we report similar analyses for codes received before ages 60 and 80 and for lifetime codes received (figs. S4 to S6). We declared the level of statistical significance at a P value of 0.05 without adjusting for multiple comparisons. In addition, we report the statistical significance at adjusted P value of 0.1 for the false discovery rate (FDR). Although the current research is one of the largest studies examining premutation carriers, more data will be needed to generate substantial individual findings with a lower FDR. In addition, using this significance threshold reduces the risk of underreporting medical conditions that could possibly affect the well-being of individuals with the premutation (31).
Applying linear regression on phecodes that represent diagnoses received before age 40 in females, we identified 37 significant associations with FMR1 premutation status (Fig. 5). Among these conditions, fracture of upper limb survived adjustment for multiple comparisons. The results suggest that three phenotypic categories were strongly related to premutation status in females: mental disorders, genitourinary problems, and injuries (P < 0.01), all previously suggested in the clinical literature (6). With respect to mental disorders, findings of agoraphobia, social phobia, and panic disorder confirm the higher risk of anxiety disorder in female premutation carriers that has been reported in the clinical literature (6). With respect to genitourinary problems, specific codes that were identified include infertility, menstrual-related symptoms, and dysmenorrhea; premature reproductive aging has been previously reported in the clinical literature in premutation carriers (32). With respect to injuries, specific codes that we identified for females in the present analysis include fractures and sprains, perhaps reflective of ataxia and falls, or estrogen insufficiency, both of which have been reported in the clinical literature as characteristic of females with the premutation. Thus, patterns of conditions reported in the clinical literature were largely replicated by this double-blind approach (6).
Fig. 5. Manhattan plots of unadjusted −log10 (P values) for phecodes observed before age 40.
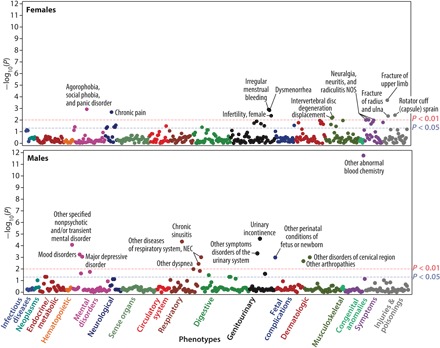
Each point shows one phecode. All associations with P < 0.01 are annotated. For females, the conditions with P values between 0.05 and 0.01 include alteration of consciousness, abnormality of gait, convulsions, obstructive sleep apnea, abnormal movement, sleep apnea, chronic venous insufficiency, other diseases of the teeth and supporting structures, endometriosis, inflammatory diseases of the uterus (except cervix), noninflammatory disorders of the vulva and perineum, disorders of menstruation and other abnormal bleeding from the female genital tract, complications of labor and delivery, hyperhidrosis, acne, cellulitis and abscess of the leg (except the foot), diseases of the sebaceous glands, chronic ulcer of the skin, synovitis and tenosynovitis, intervertebral disc disorders, disorders of the coccyx, thoracic or lumbosacral neuritis or radiculitis, symptoms of the muscles, swelling of limbs, malaise and fatigue, and myalgia and myositis. For males, the conditions with P values between 0.05 and 0.01 include other symptoms of the respiratory system, other diseases of the respiratory system, personality disorders, depression, orchitis and epididymitis, gingival and periodontal diseases, disorders of carbohydrate transport and metabolism, intestinal disaccharidase deficiencies and disaccharide malabsorption, disorders of the function of the stomach, dyspepsia, and other specified disorders of the function of the stomach.
In addition, at the .05 level, the results indicate significant associations of the premutation with other conditions and symptoms for females: mental disorders (alteration of consciousness); neurological (abnormality of gait, convulsions, obstructive sleep apnea, abnormal movement, and sleep apnea); circulatory system (chronic venous insufficiency); digestive (other diseases of the teeth and supporting structures); genitourinary [endometriosis, inflammatory diseases of the uterus (except cervix), noninflammatory disorders of the vulva and perineum, disorders of menstruation, and other abnormal bleeding from the female genital tract]; fetal complications (complications of labor and delivery); dermatologic [hyperhidrosis, acne, cellulitis and abscess of leg (except foot), diseases of sebaceous glands, and chronic ulcer of skin]; musculoskeletal (synovitis and tenosynovitis, intervertebral disc disorders, and disorders of coccyx); and symptoms and ill-defined conditions (thoracic or lumbosacral neuritis or radiculitis, symptoms of the muscles, swelling of limb, malaise and fatigue, and myalgia and myositis).
Focusing on the health profile of male participants based on diagnostic codes that were received before age 40, we identified a total of 22 significant associations (see Fig. 5). All of the conditions that are annotated (P < 0.01) in Fig. 5 survived adjustment for multiple comparisons. Abnormal blood chemistry has the highest association with the FMR1 premutation. The categories that differentiated male premutation carriers from male controls include mental disorders, respiratory conditions, and genitourinary disorders, all of which were elevated in premutation carriers (P < 0.01). Specific codes reflective of mental disorders were major depressive disorder, mood disorder, and other nonpsychotic and/or transient mental disorders. Specific codes reflective of respiratory problems include chronic sinusitis, other disease of the respiratory system (not elsewhere classified), and dyspnea. Specific codes reflective of genitourinary disorders include urinary incontinence and other symptoms and conditions of the urinary system. Mood disorder, incontinence, and arthropathies may suggest early possible signs of FXTAS (33). In addition, evidence (P < 0.05) of higher rates of respiratory disorders, digestive problems, and endocrine/metabolic conditions were observed in male premutation carriers, which have not been reported previously in the clinical literature (Fig. 5).
Automated mining of the research literature for phenotype confirmation
To verify our results, in addition to using cross-validation in conjunction with random forest learning, we incorporated the published literature as a positive control. Using the text mining tool KinderMiner (see Materials and Methods and table S1) (34), we processed 26 million articles in PubMed to identify the reported associations of FMR1 premutation with health conditions. The results of this validation task showed that our research is able to confirm in an unbiased population-based sample many of the conditions previously reported only in clinical research (e.g., anxiety, phobia, depression, falls, injuries, infertility and menstrual-related issues, pain, thyroid-related conditions, and sleep apnea).
The KinderMiner analysis also confirmed that our study resulted in the discovery of conditions that were not previously reported in the literature (e.g., acne and skin problems, bacterial infection, and adverse drug reactions). These previously unidentified discoveries not only are impactful for premutation carriers and their families but also can improve current clinical practice. For example, further examination of EHRs of the 21 premutation carriers who had an adverse drug reaction showed that 57% of them had a reaction to antibiotics, 43% to opioid (narcotic) analgesics, 29% to antihypertensive drugs, 24% to anti-inflammatory drugs, and 48% to other types of drugs. Clinicians, genetic counselors, and pharmacogeneticists can use this information to improve personalized treatment plans for premutation carriers. In addition, these new proposed phenotypes will create an opportunity to reevaluate the risks associated with FMR1 premutation.
DISCUSSION
The present study is the first to investigate the health characteristics of FMR1 premutation carriers in a representative population sample selected on the basis of CGG repeat length rather than being ascertained after a family member is diagnosed with FXS. The approach we used was double blind, as neither the patient nor the provider was aware of the individual’s FMR1 status, and so expectancy of associated conditions cannot account for the patterns.
Understanding how this genetic variant affects disease risk could be potentially used in developing personalized health plans and preventive care. This research informs a long-standing debate regarding the health implications of carrying the FMR1 premutation and provides new insights about the health and well-being of premutation carriers.
In our discovery approach, we examined EHR data to systematically assess phenotypic associations with FMR1 status. In addition to identifying the conditions that are more prevalent in FMR1 premutation carriers, our research revealed that premutation carriers had an elevated number of medical encounters for these conditions compared with controls, and the onset of symptoms was at a younger age. Together, these patterns indicate that premutation carriers suffer from a greater burden of disease for these differentiating conditions. Notably, premutation carriers, both female and male, were not sicker overall, as when all conditions in the health record were considered, there were no differences in number of conditions or number of medical encounters, whether considering the entire EHR or the averages of these indicators per year. It was specifically with respect to the conditions that differentiated premutation carriers from controls that the greater burden of disease was observed. In other words, the premutation is associated with a unique pattern of health conditions.
What could account for these findings? Past discussions implicated a number of alternative hypotheses, all of which were considered in the present research design. For example, participants were not aware of their CGG repeat length, so their higher burden of disease was not an artifact of knowledge of their premutation status. In addition, on the basis of the available data, these premutation carriers and controls had an equivalent likelihood of parenting a child with a disability, and thus, stressful parenting does not confound the difference in health outcomes in these individuals. The premutation carriers in the present sample had CGG counts in the lower portion of the premutation range, and even so, the clinical phenotype evident in their health records mirrored those reported for clinically ascertained carriers with higher numbers of CGG repeats. Thus, the most parsimonious explanation is that there are health consequences of the premutation, and these cannot be attributed to ascertainment bias or spurious self-report.
A unique aspect of the present study in examining whether those who had the premutation differed from the population in the normal CGG range is that we omitted from the control group both those with gray zone CGG repeats and those with particularly low numbers of CGG repeats, as both of these “zones” have been associated with an elevated risk of health conditions (21, 22). It is possible that having a control population for whom CGGs clustered more closely around the population mode of 30 CGG repeats sharpened the present study’s ability to detect a premutation phenotype.
The PMRP population is relatively homogenous, with a majority of the participants reporting themselves to be white Caucasian. Initial testing and discovery in a genetically relatively homogeneous population is a logical first step in a continuum of investigations because other genetic factors and population structure (admixture) are naturally well controlled. The advantage of studying a population with low genetic variation has been clearly demonstrated by the success of the Iceland genetics project (35).
However, the lack of diversity is a limitation of the current study. The number of FMR1 premutation carriers in this population is similar to the reported prevalence in other U.S.-based studies on white Caucasians (9, 20). However, it is higher than other ethnic groups, and additional studies are required to examine the presence of identified phenotypes in these populations (9).
Although the sample was drawn from a representative population cohort of 20,000 adults, the number of premutation carriers was small, particularly for the male premutation carriers. Thus, larger samples are needed to more fully investigate the genotype-phenotype associations reported here. Larger samples would make it possible to more robustly correct for multiple comparisons. The balance between types I and II error must be considered in every study, and in our discovery-oriented approach, we sought to identify associated phenotypes rather than to overlook these in an effort to reduce type II error. The results of the present study largely replicated the phenotypes reported in studies based on data ascertained through family diagnoses cases, providing some justification for this decision, although in future research with larger samples, correction for false discovery should be prioritized.
Knowledge of the clinical risk associated with the premutation will be critical for clinicians who diagnose and counsel families. Vulnerability of premutation carriers to falls and higher rates of injuries and fractures could be a part of genetic counseling for these individuals, with specific instructions about avoiding certain risky behaviors. Our results also showed that psychiatric features such as anxiety, depression, and panic disorder are more common and severe in premutation carriers. Psychiatric counseling, therapeutic intervention, and proper medication will be helpful in improving these conditions. It is possible that for women, hormonal insufficiency is a factor in both the injuries (bone thinning) and psychiatric phenotypes. Females identified as FMR1 premutation carriers via population screening should receive proper genetic counseling and be informed about the possibility of early menopause and infertility. Early detection of reproductive difficulties will help in avoiding the long and emotionally painful process of diagnosis and fertility treatments. Our machine learning approach can serve as a framework for discovery and evaluation of primary phenotypes in other genetic variants in which knowledge about the fundamental phenotype may be compromised by ascertainment bias.
MATERIALS AND METHODS
Study population
Participants in this study were all members of the Marshfield Clinic’s PMRP. The PMRP is one site of the National Institutes of Health (NIH)–funded eMERGE (Electronic Medical Records and Genomics) Network, a consortium of medical centers with EHR-linked banked DNA (36). The PMRP includes 20,353 individuals (40% of the eligible population of the Marshfield Epidemiologic Study Area, a 19–zip code region centered geographically around Marshfield, Wisconsin, and an additional 9–zip code area in northern Wisconsin) who consented to share their EHRs, DNA, and other biosamples for research. Recruitment into the PMRP began in 2002. We assayed CGG repeat length in all PMRP participants for whom DNA samples were available (n = 19,996) using the procedures described previously (8, 20). The exact size of the CGG repeat is available for 3998 participants (2114 females and 1884 males), from which 620 males and 650 females qualified as possible controls for this study; after matching, 494 males and 507 females were included in the data analysis. The PMRP is the only eMERGE site with FMR1 CGG data. The participants came from a very stable/stationary rural population for which the average participant has approximately 40 years of continuous and virtually comprehensive data within the Marshfield Clinic EHR system linked to both stored and assayed biospecimens (e.g., DNA).
Institutional review board (IRB) approval for this research was obtained by the Marshfield Clinic and the University of Wisconsin-Madison. According to the approved protocol, participants consented to contribution of their deidentified EHRs and DNA to be used in research without expectation of return of research results (37). The ethical considerations raised by this protocol have been discussed extensively in previous reports (38).
Random forest classifier
Random forest is a robust, accurate, and reliable classification method with low generalization error and high predictive performance (39). The algorithm repeatedly draws a bootstrap sample (random sampling with replacement) and trains an ensemble of decision trees, one tree per sample. To further ensure diversity among the trees in the forest, during training only, a random subset of variables is considered for use at any node in a decision tree. At prediction time (testing time), for a given test, the forest aggregates the predictions from all of the trees and identifies the most popular class as the final prediction (39). Although the current study is based on the largest FMR1-informed biobank derived from population data, the number of cases is still relatively small compared with the number of features in our analyses. This difference elevates the risk of overfitting the training data, a risk that is also raised by use of a nonlinear model, such as the decision trees in a random forest. Nevertheless, such nonlinear models provide an opportunity to find important multivariate interactions in the data. They enabled us to find predictive combinations of diagnostic codes that differentiate the two groups. The ensemble nature of random forests in practice reduces the risk of overfitting. The random forest method can be validly applied to studies in which the number of cases is much smaller than number of input features (39, 40). To measure the success of classification, the AUROC was reported. The ROC curve displays the false-positive rate versus the true-positive rate. AUROC of 1.00 shows 100% success in classification, and AUROC of 0.5 represents random classification (26). To ensure that the ROC curve is not overly optimistic, it was constructed by stratified 10-fold cross-validation, the form of hold-out testing widely used throughout machine learning for this same purpose. In stratified 10-fold cross-validation, cases and controls were each randomly partitioned into 10 parts, and on each fold, a different single part of the cases and of the controls were held aside for testing. A predictive model (in our case, random forest) was trained on the remaining of the data, tested on the held-out , and an ROC curve was constructed from the test set. The 10 final resulting ROC curves were vertically averaged to avoid any assumption of calibration between folds. Because a single ROC curve was returned by the overall method, no adjustment for multiple comparisons was necessary for the curve or the P value resulting from the Mann-Whitney U test based on it.
Replication via KinderMiner
The strength of machine learning is its ability to find previously unrecognized phenotypes, as well as to “rediscover” previously known phenotypes, simultaneously moving research beyond published literature and clinical studies. Therefore, the previous studies can serve as a positive control to determine whether machine learning is able to accurately identify the known phenotypes and how well it is able to actively expand the boundary of our knowledge. To verify our results, we used a text-mining tool called KinderMiner, which enabled us to screen the entire published literature on fragile X premutation available on PubMed and evaluate our list of phecodes (34). KinderMiner uses keyword matching and document counting to identify correlations of FMR1 premutation and target clinical phenotypes and ranks them by their co-occurrence proportion. For each target phenotype, KinderMiner returns the number of articles that contain both, either, and neither the target phenotype and FMR1 premutation. The one-sided Fisher’s exact test was performed to identify the significance level of each correlation. We processed 26 million article abstracts and identified 2070 published articles related to FMR1 premutation. Table S1 shows the list of target phenotypes and their association level with FMR1 premutation in the published literature.
Supplementary Material
Acknowledgments
We thank all of the volunteers for participation in the research. We are grateful to E. Allen, D. Bailey, D. Hall, J. Hunter, M. Raspa, S. Sherman, P. Todd, and A. Wheeler for technical advice and consultation. We also thank T. Kitchner and E. Kunze for assistance in the data preparation. We are also grateful to H. A. Steinberg for creating the figures and workflow. Funding: The National Institute of Child Health and Human Development (grant numbers R01 HD082110 and U54 HD090256) supported this project. The PMRP is supported by NHGRI UO1HG8701 (eMERGE) and NCATS UL1TR000427 (ICTR). We are also grateful for the support received from the Wisconsin Alumni Research Foundation (WARF). This project was funded in part by the Centers for Disease Control and Prevention (CDC), National Center on Birth Defects and Developmental Disabilities (NCBDDD) under Cooperative Agreement U01DD000231 to the Association of University Centers on Disabilities (AUCD). Ethics statement: All work with human subjects were carried out in accordance with institutional, national, and international guidelines and approved by the IRB at the University of Wisconsin-Madison and Marshfield Clinic Research Institute. Written informed consent was obtained before PMRP data collection. Author contributions: A.M., M.R.M., and D.P. designed the study and interpreted the results. A.M. performed the data analysis. M.R.M. and D.P. supervised the analysis. M.B. and M.R.M. coordinated and managed the genotype and phenotype data ascertainment. E.B.-K. and M.W.B. genotyped and processed the DNA samples. J.H. served as the data manager and ensured quality control over the data. F.K. and R.S. performed text mining. L.S.D., J.G., and K.S. reviewed the manuscript. A.M., M.R.M., and D.P. wrote the manuscript. All authors read, revised, and approved the manuscript. Competing interests: M.R.M. serves as the chair of the Scientific Advisory Board of the John Merck Fund. E.B.-K. is a consultant to Asuragen, the company that produces the assay to measure FMR1 CGG repeats. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw7195/DC1
Supplementary Text
Fig. S1. The distribution of the target diagnoses in premutation carriers and controls for codes received before age 60.
Fig. S2. The distribution of the target diagnoses in premutation carriers and controls for codes received before age 80.
Fig. S3. The distribution of the target diagnoses in premutation carriers and controls based on lifetime diagnoses.
Fig. S4. Manhattan plots of unadjusted −log10 (P values) for phecodes observed before age 60.
Fig. S5. Manhattan plots of unadjusted −log10 (P values) for phecodes observed before age 80.
Fig. S6. Manhattan plots of unadjusted −log10 (P values) for lifetime phecodes.
Table S1. Phenotypic association with FMR1 premutation reported in published literature, identified by KinderMiner.
Data S1. Lists of the first 100 variables that contributed in the classification of female premutation carriers (n = 72) versus control (n = 507).
Data S2. Lists of the first 100 variables that contributed in the classification of male premutation carriers (n = 26) versus control (n = 494).
Data S3. Linear regression models based on PheWAS phenotypes of female participants.
Data S4. Linear regression models based on PheWAS phenotypes of male participants.
REFERENCES AND NOTES
- 1.Minikel E. V., Zerr I., Collins S. J., Ponto C., Boyd A., Klug G., Karch A., Kenny J., Collinge J., Takada L. T., Forner S., Fong J. C., Mead S., Geschwind M. D., Ascertainment bias causes false signal of anticipation in genetic prion disease. Am. J. Hum. Genet. 95, 371–382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darnell J. C., Van Driesche S. J., Zhang C., Hung K. Y. S., Mele A., Fraser C. E., Stone E. F., Chen C., Fak J. J., Chi S. W., Licatalosi D. D., Richter J. D., Darnell R. B., FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santoro M. R., Bray S. M., Warren S. T., Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. Mech. Dis. 7, 219–245 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Eichler E. E., Holden J. J. A., Popovich B. W., Reiss A. L., Snow K., Thibodeau S. N., Richards C. S., Ward P. A., Nelson D. L., Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 8, 88–94 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Crawford D. C., Acuña J. M., Sherman S. L., FMR1 and the fragile X syndrome: Human genome epidemiology review. Genet. Med. 3, 359–371 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wheeler A. C., Bailey D. B. Jr., Berry-Kravis E., Greenberg J., Losh M., Mailick M., Milà M., Olichney J. M., Rodriguez-Revenga L., Sherman S., Smith L., Summers S., Yang J. C., Hagerman R., Associated features in females with an FMR1 premutation. J. Neurodev. Disord. 6, 30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagerman R. J., Leavitt B. R., Farzin F., Jacquemont S., Greco C. M., Brunberg J. A., Tassone F., Hessl D., Harris S. W., Zhang L., Jardini T., Gane L. W., Ferranti J., Ruiz L., Leehey M. A., Grigsby J., Hagerman P. J., Fragile-X–associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am. J. Hum. Genet. 74, 1051–1056 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seltzer M. M., Baker M. W., Hong J., Maenner M., Greenberg J., Mandel D., Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 589–597 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tassone F., Iong K., Tong T.-H., Lo J., Gane L. W., Berry-Kravis E., Nguyen D., Mu L. Y., Laffin J., Bailey D. B., Hagerman R. J., FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 4, 100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacquemont S., Hagerman R. J., Leehey M. A., Hall D. A., Levine R. A., Brunberg J. A., Zhang L., Jardini T., Gane L. W., Harris S. W., Herman K., Grigsby J., Greco C. M., Berry-Kravis E., Tassone F., Hagerman P. J., Penetrance of the fragile X–associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291, 460–469 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Adams P. E., Adams J. S., Nguyen D. V., Hessl D., Brunberg J. A., Tassone F., Zhang W., Koldewyn K., Rivera S. M., Grigsby J., Zhang L., DeCarli C., Hagerman P. J., Hagerman R. J., Psychological symptoms correlate with reduced hippocampal volume in fragile X premutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 775–785 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherman S. L., Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 97, 189–194 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Tassone F., Adams J., Berry-Kravis E. M., Cohen S. S., Brusco A., Leehey M. A., Li L., Hagerman R. J., Hagerman P. J., CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 566–569 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Bourgeois J. A., Coffey S. M., Rivera S. M., Hessl D., Gane L. W., Tassone F., Greco C., Finucane B., Nelson L., Berry-Kravis E., Grigsby J., Hagerman P. J., Hagerman R. J., A review of fragile X premutation disorders: Expanding the psychiatric perspective. J. Clin. Psychiatry 70, 852–862 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagerman R. J., Protic D., Rajaratnam A., Salcedo-Arellano M. J., Aydin E. Y., Schneider A., Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychol. 9, 564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gossett A., Sansone S., Schneider A., Johnston C., Hagerman R., Tassone F., Rivera S. M., Seritan A. L., Hessl D., Psychiatric disorders among women with the fragile X premutation without children affected by fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 171, 1139–1147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Movaghar A., Mailick M., Sterling A., Greenberg J., Saha K., Automated screening for Fragile X premutation carriers based on linguistic and cognitive computational phenotypes. Sci. Rep. 7, 2674 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartley S. L., Seltzer M. M., Hong J., Greenberg J. S., Smith L., Almeida D., Coe C., Abbeduto L., Cortisol response to behavior problems in FMR1 premutation mothers of adolescents and adults with fragile X syndrome: A diathesis-stress model. Int. J. Behav. Dev. 36, 53–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohane I. S., Using electronic health records to drive discovery in disease genomics. Nat. Rev. Genet. 12, 417–428 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Maenner M. J., Baker M. W., Broman K. W., Tian J., Barnes J. K., Atkins A., McPherson E., Hong J., Brilliant M. H., Mailick M. R., FMR1 CGG expansions: Prevalence and sex ratios. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162, 466–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall D. A., Berry-Kravis E., Zhang W., Tassone F., Spector E., Zerbe G., Hagerman P. J., Ouyang B., Leehey M. A., FMR1 gray-zone alleles: Association with Parkinson’s disease in women? Mov. Disord. 26, 1900–1906 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mailick M. R., Hong J., Rathouz P., Baker M. W., Greenberg J. S., Smith L., Maenner M., Low-normal FMR1 CGG repeat length: Phenotypic associations. Front. Genet. 5, 309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gleicher N., Weghofer A., Oktay K., Barad D., Relevance of triple CGG repeats in the FMR1 gene to ovarian reserve. Reprod. BioMed. Online 19, 385–390 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Rasmussen L. V., Thompson W. K., Pacheco J. A., Kho A. N., Carrell D. S., Pathak J., Peissig P. L., Tromp G., Denny J. C., Starren J. B., Design patterns for the development of electronic health record-driven phenotype extraction algorithms. J. Biomed. Inform. 51, 280–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheurwegs E., Cule B., Luyckx K., Luyten L., Daelemans W., Selecting relevant features from the electronic health record for clinical code prediction. J. Biomed. Inform. 74, 92–103 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Fawcett T., ROC graphs: Notes and practical considerations for researchers. Mach. Learn. 31, 1–38 (2004). [Google Scholar]
- 27.G. Louppe, L. Wehenkel, A. Sutera, P. Geurts, in Advances in Neural Information Processing Systems (2013), pp. 431–439. [Google Scholar]
- 28.McGuire T., Wells K. B., Bruce M. L., Miranda J., Scheffler R., Durham M., Ford D. E., Lewis L., Burden of illness. Ment. Health Serv. Res. 4, 179–185 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Denny J. C., Ritchie M. D., Basford M. A., Pulley J. M., Bastarache L., Brown-Gentry K., Wang D., Masys D. R., Roden D. M., Crawford D. C., PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Israel R. A., The International Classification of Disease. Two hundred years of development. Public Health Rep. 93, 150–152 (1978). [PMC free article] [PubMed] [Google Scholar]
- 31.Amrhein V., Greenland S., McShane B., Scientists rise up against statistical significance. Nature 567, 305–307 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Spath M. A., Feuth T. B., Smits A. P. T., Yntema H. G., Braat D. D. M., Thomas C. M. G., van Kessel A. G., Sherman S. L., Allen E. G., Predictors and risk model development for menopausal age in fragile X premutation carriers. Genet. Med. 13, 643–650 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brouwer J. R., Willemsen R., Oostra B. A., The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 782–798 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuusisto F., Steill J., Kuang Z., Thomson J., Page D., Stewart R., A Simple Text Mining Approach for Ranking Pairwise Associations in Biomedical Applications. AMIA Jt Summits Transl. Sci. Proc. 2017, 166–174 (2017). [PMC free article] [PubMed] [Google Scholar]
- 35.Palotie A., Widén E., Ripatti S., From genetic discovery to future personalized health research. N. Biotechnol. 30, 291–295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottesman O., Kuivaniemi H., Tromp G., Faucett W. A., Li R., Manolio T. A., Sanderson S. C., Kannry J., Zinberg R., Basford M. A., Brilliant M., Carey D. J., Chisholm R. L., Chute C. G., Connolly J. J., Crosslin D., Denny J. C., Gallego C. J., Haines J. L., Hakonarson H., Harley J., Jarvik G. P., Kohane I., Kullo I. J., Larson E. B., McCarty C., Ritchie M. D., Roden D. M., Smith M. E., Böttinger E. P., Williams M. S.; eMERGE Network , The electronic medical records and genomics (eMERGE) network: Past, present and future. Genet. Med. 15, 761–771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarty C. A., Wilke R. A., Giampietro P. F., Wesbrook S. D., Caldwell M. D., Marshfield Clinic Personalized Medicine Research Project (PMRP): design, methods and recruitment for a large population-based biobank. Pers. Med. 2, 49–79 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ossorio P., Mailick M., Genotype-driven recruitment without deception. Am. J. Bioeth. 17, 60–61 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Breiman L., Random forests. Mach. Learn. 45, 5–32 (2001). [Google Scholar]
- 40.Liaw A., Wiener M., Classification and Regression by randomForest. R News 2, 18–22 (2002). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw7195/DC1
Supplementary Text
Fig. S1. The distribution of the target diagnoses in premutation carriers and controls for codes received before age 60.
Fig. S2. The distribution of the target diagnoses in premutation carriers and controls for codes received before age 80.
Fig. S3. The distribution of the target diagnoses in premutation carriers and controls based on lifetime diagnoses.
Fig. S4. Manhattan plots of unadjusted −log10 (P values) for phecodes observed before age 60.
Fig. S5. Manhattan plots of unadjusted −log10 (P values) for phecodes observed before age 80.
Fig. S6. Manhattan plots of unadjusted −log10 (P values) for lifetime phecodes.
Table S1. Phenotypic association with FMR1 premutation reported in published literature, identified by KinderMiner.
Data S1. Lists of the first 100 variables that contributed in the classification of female premutation carriers (n = 72) versus control (n = 507).
Data S2. Lists of the first 100 variables that contributed in the classification of male premutation carriers (n = 26) versus control (n = 494).
Data S3. Linear regression models based on PheWAS phenotypes of female participants.
Data S4. Linear regression models based on PheWAS phenotypes of male participants.