

An engineered viral vector of bacteriophage T4 linked to AAV delivers large payloads of genes and proteins into human cells.
Abstract
Development of safe and efficient nanoscale vehicles that can deliver large molecular cargos into human cells could transform future human therapies and personalized medicine. Here, we design a hybrid viral vector composed of a prokaryotic virus (bacteriophage T4) and a eukaryotic virus [adeno-associated virus (AAV)]. The small 25-nm AAV is attached to the large 120 nm × 86 nm T4 head through avidin-biotin cross-bridges using the phage decoration proteins Soc and Hoc. AAV “piggy-backed” on T4 capsid, by virtue of its natural ability to enter human cells acted as an efficient “driver,” delivering the largest payloads of foreign DNA (up to 170 kb) and protein (up to 1025 molecules) reported to date, and elicited robust immune responses in mice against flu and plague pathogens and conferred complete protection against lethal pneumonic plague challenge. The T4-AAV represents a unique platform for assembly of natural building blocks into potential therapeutics against genetic and infectious diseases.
INTRODUCTION
Therapeutics that can deliver cargos of genes and proteins into human cells safely and efficiently could lead to a transformative impact on the prevention and treatment of many human diseases, ranging from correction of complex genetic defects to gene/protein augmentation for chronic disease and to vaccines against deadly pathogens or cancer. However, the formulation and delivery of these vehicles remain a major challenge (1, 2).
Eukaryotic viral vectors such as adenoviruses, adeno-associated viruses (AAVs), retroviruses, and/or lentiviruses have naturally evolved to deliver their genomes efficiently into mammalian cells. This property has been extensively exploited by using these viruses as therapeutic vehicles to deliver genes of interest into human cells (3). However, their capacity is very limited, generally to one or two genes and 4 to 8 kb in size, and they are not suitable for delivering proteins. Furthermore, their tropism to human cells, preexisting immunity, and safety concerns restrict their broad application (2). New delivery vehicles based on novel concepts are desperately needed to develop future human therapies.
As one of the most abundant and ubiquitous life forms on Earth, bacteriophages (phages) provide a broad range of platforms to incorporate cargo molecules (4, 5). Phage T4 is an excellent platform for several reasons (table S1). Molecular and genetic analyses led to the engineering of an empty protein shell (head or capsid) lacking the outer capsid proteins Soc (small outer capsid protein) and Hoc (highly antigenic outer capsid protein), neck, or tail as a delivery vehicle (6). The shell has a very large capacity and can be filled in vitro with up to ~170 kb of foreign genes using a powerful DNA packaging motor (7), far exceeding the capacity of any eukaryotic viral vector. The phage T4 packaging initiation does not require a sequence-specific packaging signal. The packaging adenosine triphosphatase (ATPase) attaches to the end of any linear DNA molecule and initiates DNA packaging. After one molecule is packaged, it attaches to the end of another DNA molecule and the process is repeated until the head is full (6, 7). Furthermore, the same head can also be used to deliver up to ∼1025 molecules of foreign proteins displayed on its surface as fusions of Soc and Hoc (8). The whole packaging process is reduced to only two components, head and motor protein (gp17), and the entire process including packaging and display is reduced to about 3 hours. All the components are pure and stable and can be stored for years at −80°C (6, 8). Last, the phage T4 head has no tropism to human cells, has no known toxicity or pathogenicity, exhibits no preexisting immunity, and can be inexpensively produced on a large scale (9–13).
However, phages are poor delivery vehicles because they lack natural mechanisms to enter mammalian cells or reach appropriate intracellular compartments following entry. Here, we addressed this critical barrier by creating a novel prokaryotic-eukaryotic hybrid viral vector. This hybrid vector combines the large capacity, easy genetic manipulation, and engineering flexibility of phage T4 (14) with the superior delivery and persistent expression of the AAV virus (15). The phage T4 head is decorated with 870 molecules of Soc and 155 molecules of Hoc, neither of which is essential. We took advantage of this feature by attaching the 25-nm AAV to the 120 nm × 86 nm T4 head through avidin-biotin cross-bridges to generate the T4-AAV hybrid nanoparticle. Consequently, the small AAV virus is “piggy-backed” on the large T4 head and functions as an efficient “driver” to deliver the cargos associated with each into mammalian cells.
The ability of the T4-AAV hybrid vector to deliver large payloads into mammalian cells was assessed in comparison with the unconjugated T4 vector. A variety of T4-AAV nanoparticles carrying different cargos of reporter genes including luciferase, green fluorescent protein (GFP), and mCherry packaged in either the T4 capsid or the AAV capsid or in both the capsids were engineered and tested. In addition, the external lattice of T4 was decorated with β-galactosidase, and the copy number of each component was precisely quantified. For instance, about 10 molecules of ~6-kb plasmid DNAs, 250 molecules of 116-kDa β-galactosidase, and two to four AAV particles carrying 1 molecule each of ~4.7-kb single-stranded DNA were incorporated into each T4-AAV nanoparticle. Our results show that the entire cargo, far exceeding the capacity of any vector, was efficiently delivered into the same cell and the respective functions were expressed. Furthermore, when injected into mice, the T4-AAV particles expressed strong luminescence and the signal persisted for at least 60 days. Last, the T4-AAV particles packaged with influenza virus hemagglutinin (HA) DNA and displayed with plague F1mutV antigen elicited durable immune responses against both flu and plague pathogens, responses typically observed in natural infections. In addition, these responses provided complete protection to mice against pneumonic plague evoked by challenging with a high dose of lethal Yersinia pestis CO92 bacteria. These data demonstrate the versatility of the hybrid T4-AAV vector approach to deliver complex and large cargos into mammalian cells that could, in the future, lead to the development of powerful nanoparticle therapies against genetic and infectious diseases.
RESULTS
Delivery principles of the T4-AAV hybrid vector
The T4 head is a prolate icosahedron with a length of 120 nm and a width of 86 nm (Fig. 1A). At approximately 25 nm in diameter, the non-enveloped AAV is one of the smallest viruses known (Fig. 1B). The T4 head is assembled with hexamers of a single major capsid protein, gp23* (“*” represents the cleaved mature form of gp23). Two minor capsid proteins, gp24* and gp20, occupy the vertices. Gp24* forms pentamers at 11 of the 12 pentameric vertices, whereas the dodecameric gp20 is at the unique vertex known as the portal vertex that allows entry and exit of DNA during DNA packaging and phage infection (16). In addition, the capsid lattice is decorated with two nonessential outer capsid proteins, Soc and Hoc. The 10-kDa Soc, on the one hand, assembles as trimers at the quasi-threefold axes of the capsid to form a molecular cage around the shell. The 40-kDa Hoc, on the other hand, is at the center of each capsomer as a monomer. Hoc is a linear fiber consisting of a string of four domains, with the C-terminal domain attached to the head and the N-terminal domain projected ~170 Å away from the shell (Fig. 1A) (17). Our previous studies demonstrated that both Soc and Hoc can be used for the display of a variety of foreign proteins, including short polypeptides, large domains, full-length proteins, or multimeric complexes (8, 10).
Fig. 1. Strategy for construction of T4-AAV hybrid vectors and schematic for delivery into mammalian cells.
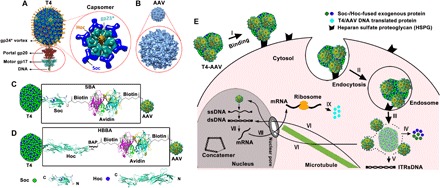
(A) T4 DNA packaging and protein display machine. The zoomed oval view below indicates the DNA packaging motor gp17 (pentamer) and portal gp20 (dodecamer). Enlarged capsomer (hexamer) shows the arrangement of the major capsid protein gp23* (cyan), Soc trimers (blue), and Hoc fiber (yellow). (B) Sixty AAV DJ subunits assemble to form the icosahedrally symmetric virus capsid [Protein Data Bank (PDB) 3J1Q]. The T4 (120 nm × 86 nm) and AAV (25 nm) capsids are shown approximately to scale. (C and D) Design principles of the T4-AAV vector. Soc-biotin-avidin (SBA) (C) and Hoc–biotin acceptor peptide (BAP)–biotin–avidin (HBBA) (D) are applied to conjugate the T4 head with biotin-labeled AAV via the avidin-biotin interaction. The protein ribbon structures and biotin structure in the rectangle depict the detailed SBA and HBBA linkers. The ribbon diagrams at the bottom show Hoc and Soc structures with exposed N and C termini (PDB 3SHS and PDB 3IG9). (E) Schematic representation of protein and gene delivery into mammalian cells by the T4-AAV vector. I, binding to receptors on the cell membrane; II, endocytosis; III, endosomal/lysosomal escape; IV, uncoating to release protein cargo; V, T4-encapsidated DNA release; VI, transport of DNAs into the nucleus along microtubules; VII, transcription of delivered genes; VIII and IX, mRNA transport into cytosol and protein production. ssDNA, single-stranded DNA; dsDNA, double-stranded DNA; ITRs, inverted terminal repeats.
To attach AAV to the T4 head, we constructed two bridge molecules, Soc-biotin-avidin (SBA) and Hoc–biotin acceptor peptide (BAP)–biotin–avidin (HBBA) (Fig. 1, C and D). Soc and Hoc attach to the T4 head, and avidin attaches to biotinylated AAV (bioAAV) on the other side. We reasoned that this T4-AAV nanoparticle would bind to the mammalian host cell surface through the ubiquitously present AAV receptor, such as the heparin sulfate proteoglycan, which would then trigger internalization of the nanoparticle by endocytosis (Fig. 1E) (15, 18). Moreover, AAV is known to have a strong endosome-rupturing capacity upon endosome maturation (19). This would then lead to efficient escape of the T4-AAV cargo into the cytoplasm. Mechanisms for further intracellular trafficking and processing of T4-AAV cargo are unknown. Current evidence suggests that uncoating of T4 capsid occurs in cytosol, thereby leading to the release of cargos in the cytosol. Proteins exert their function in the cytosol, whereas the DNAs migrate to the nucleus, possibly assisted by microtubules, and trigger recombinant gene (transgene) expression under the control of the strong cytomegalovirus (CMV) promoter (Fig. 1E; see below).
Designing the bridging molecules attached to phage T4 head nanoparticles
To design Soc bridges, the purified Soc protein was first biotinylated by activation with biotin–N-hydroxysuccinimide-ester (NHS) (Fig. 2A). Soc has five lysines, which provide primary amine groups for efficient biotinylation, as confirmed by Western blotting using streptavidin–horseradish peroxidase (HRP) conjugate (Fig. 2B). To design Hoc bridges, we recombinantly attached a BAP (1.8 kDa) to the N terminus of Hoc. The Hoc-BAP was then biotinylated using biotin ligase (BirA, 35 kDa), which specifically recognizes a single lysine residue in BAP (Fig. 2C and fig. S1A) (20). Biotinylation was confirmed by the ability of the modified Hoc to bind avidin (fig. S1B), and the resulting Hoc-avidin complex was purified by size exclusion chromatography (SEC) (fig. S1, C and D). Intermolecular bridging was minimized by adding an excess of avidin molecules to saturate the Soc-biotin or Hoc-biotin ligands, and any oligomeric complexes formed by Soc-Soc or Hoc-Hoc bridging were separated by SEC.
Fig. 2. Synthesis and characterization of SBA and HBBA cross-bridges.
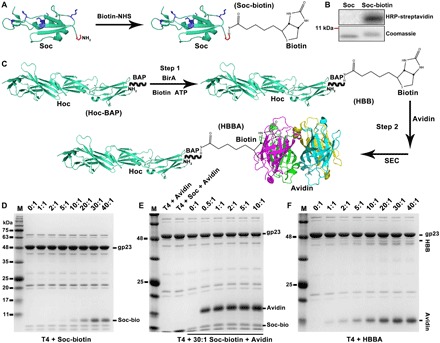
(A) Schematic of Soc modification with biotin. (B) HRP-streptavidin Western blot and Coomassie staining analyses of Soc and Soc-biotin. (C) Schematic representation of the HBBA assembly process. (D) Display of Soc-biotin on the T4 surface at increasing ratios of Soc-biotin molecules to capsid Soc binding sites (0:1 to 40:1). (E) Binding of avidin to the T4-Soc-biotin. (F) Display of HBBA on the T4 capsid at increasing ratios of HBBA molecules to capsid Hoc binding sites (0:1 to 40:1). The copy numbers of Soc- or Hoc-fused proteins displayed on T4 heads were determined by co-sedimentation assay (D to F). See Materials and Methods for details.
Next, T4-Soc-avidin (TSA) and T4-Hoc-avidin (THA) bridges were prepared. TSA was prepared by displaying biotinylated Soc on the T4 head (Fig. 2D) and then attaching avidin to the Soc molecules (Fig. 2E). Binding of biotinylated Soc increased with an increasing ratio of Soc-biotin molecules to capsid binding sites, reaching a maximum at 30:1, similar to that reported for the unlabeled Soc (Fig. 2D and fig. S1E). Subsequent binding of avidin to T4-Soc-biotin was very efficient, reaching saturation at a ratio of only 2:1 (Fig. 2E and fig. S1F). A similar approach was used to prepare THA bridges by displaying the HBBA complex on T4, which was also saturated at a ratio of ~30:1, similar to that reported for the unlabeled Hoc (Fig. 2F and fig. S1G). The above data show that modifications of Soc and Hoc by biotinylation and avidin binding do not substantially affect their ability to bind to the T4 capsid. The Soc and Hoc bridges expose the unliganded biotin binding sites of the tetravalent avidin molecules on the surface of the T4 nanoparticle.
To determine the functionality of the exposed avidin to bind additional biotinylated ligand molecules, the modified T4 heads were mixed with a high–molecular weight protein, the 83-kDa biotinylated anthrax protective antigen (PA) (fig. S2A) (12). The PA-biotin molecules bound very efficiently to the TSA and THA bridges, whereas the control unbiotinylated PA showed no detectable binding (fig. S2, B and C). The copy numbers of bound PA were 550 and 90 for Soc and Hoc bridges, respectively. The above results demonstrate that the T4 heads were functionalized by displaying Soc- and Hoc-avidin bridge molecules at high density, and the exposed avidin molecules can efficiently capture externally added biotinylated proteins.
Attachment of AAV to T4 through Soc/Hoc cross-bridges
To attach AAV to T4, we first biotinylated AAV-DJ virus particles by activation with biotin-NHS and purified the particles by dialysis (fig. S3A). AAV-DJ is a chimera of AAV types 2, 8, and 9 and combines the best features of AAV-2 with those of AAV-8 and AAV-9 (21). Western blotting showed that all three AAV capsid proteins—VP1, VP2, and VP3—were biotinylated, whereas no signal was detected in the unlabeled control (fig. S3B). The biotinylated AAV-DJ retained a similar level of infectivity as the unlabeled virus, as measured by the efficiency of transduction of the GFP reporter gene into human embryonic kidney (HEK) 293 cells (fig. S3, C and D) and as was reported previously (22).
The biotinylated AAV-DJ particles were then attached to the TSA bridges by passing the purified virus particles through a column of nickel beads on which the T4 heads decorated with Soc-avidin bridge molecules were immobilized through the hexahistidine tag present at the N terminus of Soc (Fig. 3A). This column thus allowed efficient capture of AAV by the TSA bridge molecules and simultaneous removal of unbound AAV in the flow-through. The T4-AAV nanoparticles were then eluted with imidazole. The capture efficiency was followed by packaging the AAV particles with the GFP reporter gene, and the specificity of cross-bridging was ascertained by numerous controls. For instance, the same protocol but lacking the hexahistidine tag and/or bound avidin failed to capture the biotin-AAV particles (Fig. 3, B and C). Second, very little unbound AAV was detected in the flow-through when the ratio of AAV to T4 was less than 4:1, suggesting that, on average, up to four AAV particles were conjugated to one T4 capsid (Fig. 3D and fig. S3E). Further addition of AAV resulted in the elution of increasing amounts of the virus in the flow-through (fig. S3E). In addition, transmission electron microscopy confirmed the presence of conjugated AAV particles on the T4 capsid (Fig. 3E).
Fig. 3. Attachment of AAV to T4 capsids through biotin-avidin cross-bridges.
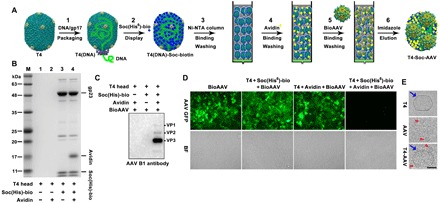
(A) Workflow for assembly of the T4-Soc-AAV vector on Ni beads. (B) SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis of elution fractions from Ni beads loaded with different combinations of the T4 head, Soc(His6)-Biotin, and avidin, as indicated at the bottom of the panel. No head proteins (gp23) are present in lanes 1 and 2 because the control heads lacking the His-tagged Soc molecules were not captured by the Ni beads. Because the beads were eluted with imidazole and analyzed for the captured heads by SDS-PAGE, gp23 was present only in the positive test lanes (lanes 3 and 4). (C) Western blot using an AAV B1 monoclonal antibody demonstrates that only TSA interacts with bioAAV. (D) Capturing efficiency of bioAAV was assessed by GFP expression in cells transduced with flow-through fractions containing unbound bioAAV(GFP). Equal amounts of bioAAV(GFP) vectors were loaded and bound to TSA on Ni beads. (E) Transmission electron micrograph of negatively stained T4, AAV, and T4-Soc-AAV particles. Blue arrows and red arrowheads point to T4 head and AAV particles, respectively. Scale bar, 50 nm.
A similar protocol and additional controls were used to prepare T4-AAV conjugates through Hoc-avidin bridges (fig. S3F). The main difference compared with Soc, other than the copy number of Hoc, was that in the T4-Hoc-AAV conjugate, the attached AAV was ∼170 Å from the capsid, providing more flexibility to interact with the cell surface receptor molecules. The capture efficiency assay showed that most of the bioAAV bound to the THA bridges when less than twofold the amount of AAV was loaded (fig. S3E).
T4-AAV nanoparticles efficiently delivered genes and proteins into mammalian (human) cells
Gene delivery through T4
Three different plasmids containing luciferase (Luci), GFP, or mCherry genes under the control of the CMV promoter were packaged into the T4 capsid using the DNA packaging motor (fig. S4A). At a 20:1 ratio of plasmid DNA molecules to T4 head particles, an average of ~9 to 10 molecules of luciferase, GFP, or mCherry plasmid DNA were packaged into each head (fig. S4B). In agreement with our previous data, transduction into mammalian cells (HEK293) by T4 heads alone is very inefficient. However, attachment to AAV resulted in a remarkable increase in transduction efficiency. As shown in Fig. 4A, a 40,000-fold increase in luciferase activity was observed for T4-AAV compared to control T4 particles lacking AAV. In addition, the amount of luciferase activity was proportional to the number of Soc-avidin bridges (fig. S5A), hence the number of AAVs displayed per T4 capsid, and also to the number of T4-AAV particles added per cell, which reached a maximum at 105 particles per cell (Fig. 4B).
Fig. 4. T4-AAV nanoparticles efficiently deliver combinations of genes and proteins into mammalian cells.
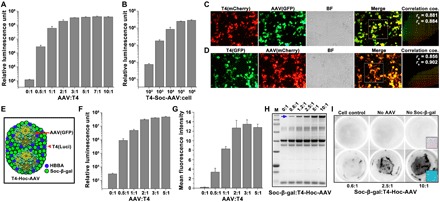
(A) Increasing the amount of displayed AAV increased the delivery of luciferase DNA into cells using T4(luci)-Soc-AAV. (B) Dose-dependent delivery of luciferase DNA into cells by T4(luci)-Soc-AAV. (C and D) Colocalization of AAV- and T4-packaged gene expression in HEK293 cells treated with T4(mCherry)-Soc-AAV(GFP) (C) and T4(GFP)-Soc-AAV(mCherry) (D). The merge channel represents the overlay of the corresponding green and red channels. The mixed fluorescence signals from the white hatched boxes are described as scatterplots and correlation coefficients (coe.). The rp and rs values suggest the levels of colocalization ranging from −1 for negative correlation to +1 for perfect colocalization. (E) Schematic of T4-Hoc-AAV. (F) Luciferase gene delivery by T4(luci)-Hoc-AAV(GFP) increases as the binding ratio of AAV to T4 increases. (G) Quantitative analysis of GFP expression in cells transduced with T4(luci)-Hoc-AAV(GFP). The mean fluorescence pixel intensity in arbitrary units (a.u.) was analyzed. (H) Display of Soc-β-gal on THA. The T4 head was incubated with HBBA (20:1; to hoc sites) and Soc-β-gal at increasing ratios (0:1 to 10:1; to Soc sites). The blue arrow indicates displayed Soc-β-gal. (I) Intact and functional Soc-β-gal transduction into cells by T4-Hoc-AAV. No enzymatic activity was shown in the absence of AAV or Soc-β-gal. The black hatched box in the right corner indicates the magnified image of stained cells.
Parallel experiments were performed with the Hoc-bridged T4-AAV particles, which also showed greatly increased transduction efficiency when compared to T4 alone (fig. S5, B to D). However, the maximum luciferase activity was approximately sixfold lower compared with T4-Soc-AAV particles (fig. S5B), probably because there were 5.6 times fewer Hoc bridges than Soc bridges per T4 capsid. These transduction experiments were repeated with a second reporter gene, GFP, and the results were similar to those observed with the luciferase reporter.
Gene delivery through both T4 and AAV
An advantage of the T4-AAV hybrid vector is that both T4 and AAV can be simultaneously used to deliver genes. T4 delivers genes in the form of double-stranded DNA (23), whereas AAV delivers genes in the form of single-stranded DNA, the natural state of the packaged AAV genome (15). To test this possibility, mCherry DNA was packaged into T4 capsid by in vitro packaging and GFP DNA was packaged into AAV by in vivo packaging, and both were conjugated through the Soc bridges. When added to HEK293 cells, these T4(mCherry)-AAV(GFP) nanoparticles efficiently delivered and expressed both green and red fluorescence, and the fluorescence signals were evenly distributed throughout the cells (Fig. 4C). The signals highly merged, demonstrating the expression of both signals in the same cell as a result of co-delivery of both the T4-associated mCherry and AAV-associated GFP by the T4-AAV hybrid vector. Furthermore, quantification of linear Pearson (rp) and nonlinear Spearman’s rank (rs) correlation coefficients for pixels representing the fluorescence signals in green and red channels showed high rp and rs values, demonstrating high coexpression of the fluorescence genes delivered by T4 and AAV (Fig. 4C). We repeated this experiment by reversing the encapsidated reporter plasmids, GFP into T4 and mCherry into AAV, and the results showed very similar values as the T4(mCherry)-AAV(GFP) particles (Fig. 4D). However, we observed weaker signal for T4-delivered transgene expression than for the AAV-delivered transgenes (Fig. 4, C and D). This might be due to inefficient transport of T4-delivered DNAs to nucleus when compared to the AAV DNAs (see below).
Simultaneous delivery of genes and proteins
Another advantage of the T4-AAV vector is that it can deliver foreign proteins in addition to foreign genes, further expanding the therapeutic potential of the hybrid nanoparticle. To demonstrate simultaneous gene and protein delivery, we chose the 116-kDa β-galactosidase from Escherichia coli fused to Soc (Soc-β-gal) and displayed on the T4 head. Approximately 250 copies of Soc-β-gal were displayed per head particle, in which about nine molecules of ~6.2-kb luciferase plasmid were also packaged. At the same time, AAV was packaged with GFP and attached to T4 through Hoc bridges (Fig. 4E). The resultant β-gal-Soc-T4(luci)-AAV(GFP) particles were transduced into HEK293 cells, and gene expression and protein activity were analyzed. The expression of T4-associated luciferase and AAV-associated GFP showed a clear dose dependence on the ratio of AAV to T4 (Fig. 4, F and G). The T4-AAV particles carried the surface-displayed β-galactosidase molecules in addition to the encapsidated luciferase and GFP transgenes (Fig. 4H). Furthermore, the appearance of the blue color of the cleaved X-galactosidase (X-Gal) substrate showed the successful formation of the functional tetrameric β-galactosidase enzyme (only the tetramer has cleavage activity). The intensity of the color was proportional to the copy number of the displayed Soc-β-gal protein (Fig. 4I). These data demonstrated that the entire cargo of luciferase plasmids, GFP DNA, and β-galactosidase proteins was efficiently delivered into the cells.
To further demonstrate that the delivered proteins exerted function following their release from the T4 nanoparticle, the T4 capsid was displayed with either GFP or GFP fused to the nuclear localization signal (NLS) and packaged with mCherry plasmid DNA. These T4-AAV nanoparticles were then transduced into HEK293 cells and imaged for green fluorescence. In the GFP-T4(mCherry)-AAV–transduced cells, the green fluorescence signal was evenly distributed throughout the cell, whereas in the NLS-GFP-T4(mCherry)-AAV–transduced cells, most of the fluorescence signal was localized in the nucleus (fig. S5E). The mCherry signal, as expected, was distributed throughout the cell in both the cases. These data indicate that the T4 capsid has undergone some sort of uncoating, releasing the displayed proteins (and the packaged nucleic acids) in the cytosol, which then traffic to the respective destinations. In addition, Alexa Fluor–labeled T4 capsid remained in the cytosol and did not get trapped in the nucleus after delivery by the hybrid nanoparticle (fig. S5F). Consistent with the above hypothesis, treatment with tubastatin A, a microtubule stabilization drug, enhanced transgene expression in a dose-dependent manner, suggesting that the microtubule tracking system might be involved in the translocation of T4-delivered DNAs into the nucleus (fig. S5G).
The above series of experiments demonstrated that the T4-AAV nanoparticles robustly co-delivered large molecular cargos containing multiple DNA molecules and foreign proteins into cultured human cells. Furthermore, no measurable impact on cell viability was observed following transduction by T4-AAV particles, whether Soc-bridged or Hoc-bridged, even when added to a level of 106 T4-AAV vectors per cell (fig. S5, H and I).
AAV facilitates endosomal escape
Endosomal escape appears to be a significant bottleneck for T4-associated cargo delivery and transgene expression. Compounds such as chloroquine that enhance endosomal escape and acidification by nonspecifically disrupting endosomes (24) greatly enhanced T4 delivery (fig. S6, A and B). Because endosomal escape by AAV has been well documented to be very efficient, it is possible that the enhanced delivery by T4-AAV hybrid vector, at least in part, might be due to linkage of T4 to AAV. To test this hypothesis, we mutated a phospholipase A2 (PLA2) of AAV VP1 capsid protein, which mediates endosomal escape by lipolytic pore formation. Mutation at these two critical amino acids in the catalytic region of PLA2, H75A, and D76N abrogated virus infectivity (fig. S6C) but not virus attachment, entry, and endocytosis (25). The T4-AAV nanoparticles prepared using the AAV-PLA2 mutant either through SBA bridges or through HBBA bridges failed to enhance T4(luci)-AAV transduction (fig. S6D), suggesting that association with endosomal escape-competent wild-type AAV is essential for efficient transduction of T4-AAV nanoparticles. Furthermore, T4 and AAV must be physically linked because simple mixing of T4 or T4-Hoc-Tat peptide nanoparticles with AAV did not lead to enhanced transduction (fig. S6E).
Efficient in vivo gene delivery by T4-AAV nanoparticles
We next studied whether the T4-AAV particles could deliver and express transgenes in vivo and how long this expression persisted, using a mouse model. To enhance persistence, we have engineered AAV inverted terminal repeats (ITRs) to flank the transgene of our packaged DNAs. This is to mimic the double-stranded AAV DNA produced by replication of the single-stranded AAV genome upon its entry into the nucleus. The double-stranded DNA is thought to be converted to head-to-tail concatemers via intra- or intermolecular recombination at the ITRs and persists as episomes for long-term transgene expression. Studies on various AAV-transduced tissues including liver and muscle have shown that the concatemers persist for a long period of time, up to 22 months (15, 26). Our previous studies have shown that the ITRs notably enhanced the in vivo persistence of T4-packaged DNAs (10). Hence, ITRs were engineered to endow this persistence character to the T4-AAV hybrid vector (fig. S7A). However, note that although ITRs allow integration of the genome, it should not be a significant issue here because integration requires AAV rep gene, which is absent in the T4-AAV vector. In the new constructs, the ITRs were inserted into the luciferase plasmid such that cleavage with Pst I restriction endonuclease would result in a linear DNA molecule containing the ITRs at the termini flanking the luciferase expression cassette, a structure similar to the duplex AAV genome (fig. S7A). In addition, the presence of ITRs did not affect DNA packaging efficiency (fig. S7, A and B).
The T4 heads were packaged with approximately nine ITR-Luci DNA molecules and attached to AAV through either Soc or Hoc cross-bridges. To determine whether the in vivo delivery efficiency is enhanced in the T4-AAV conjugate, we injected ~2 × 1011 T4(ITRs-Luci)-Soc/Hoc-AAV(GFP) particles into mice intramuscularly and monitored them for the expression of luciferase by whole-body imaging (Fig. 5A). T4(ITRs-Luci) alone and phosphate-buffered saline (PBS) were used as controls, and the signal was quantified by measuring photon flux (Fig. 5B). Although T4 heads lack natural mechanisms to enter mammalian cells, they are taken up by myocytes at the site of intramuscular injection presumably by nonspecific endocytosis. In addition, the data showed that a single injection of T4-AAV nanoparticles resulted in efficient and prolonged expression of luciferase in mice. The luciferase signal was detectable only at the local injection site. Because the skeletal muscle tissue is composed of a large number of myocytes and very few immune system cells (27), most of the nanoparticles were likely taken up by myocytes. In a time course analysis, T4-Soc-AAV particles resulted in ~25-fold enhancement of the luciferase signal when compared with control T4 particles lacking AAV (Fig. 5). The signal reached a peak at day 30 and retained at a high level even on day 60. By contrast, the signal reached a peak at day 15 and nearly disappeared by day 60 in mice that received T4 particles lacking AAV (Fig. 5, A and B). The Hoc-bridged T4-AAV particles generated more pronounced luciferase signals, ~5-fold higher than the Soc-bridged T4-AAV and ~130-fold higher than T4 lacking AAV, although the copy number of Hoc is 5.6 times less than that of Soc. This result is in contrast to the in vitro transduction results, where the signal was proportional to the copy number. This is probably because, as reported previously, the T4 Hoc protein has a tendency to bind to mammalian cells and tissues, presumably through its immunoglobulin (Ig)–like domains, which enhances the efficiency of uptake of phage T4 (17, 28). In addition, the enhanced expression efficiency by T4-AAV was further confirmed by its administration at a fourfold less molar dose (fig. S7, C and D). These data indicated that the conjugated T4-AAV facilitated more efficient delivery of T4 cargo into myocytes, resulting in enhanced gene expression for a longer period of time when compared to T4 alone. The T4-AAV hybrid vector might therefore provide an attractive strategy for therapeutic applications.
Fig. 5. In vivo gene delivery by the T4-AAV nanoparticles.
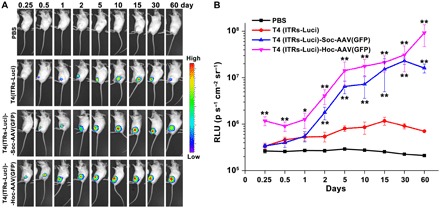
(A) Luciferase signal dynamics at the site of injection of mice. The bioluminescence scale is provided on the right of the panel and ranges from the most intense (red) to the least intense (blue) on the basis of the radiance intensity. (B) The luminescence signal intensities of PBS, T4(ITRs-Luci), T4(ITRs-Luci)-Soc-AAV(GFP), and T4(ITRs-Luci)-Hoc-AAV(GFP) were quantified as photons per second per centimeter squared per steradian (p s−1 cm−2 sr−1). The values represent means ± SD of three independent replicates. *P < 0.05 and **P < 0.01 compared with T4(ITRs-Luci) alone. Statistical significance was determined using two-tailed Student’s t tests.
Multivalent DNA vaccine and protein antigen delivery by T4-AAV
DNA vaccines are attractive candidates for vaccine development due to their overall safety and facile production. Formulating DNA vaccines to protect the DNA from degradation and developing efficient delivery technology play a key role in the final efficacy of DNA vaccines (29). In addition, the immune responses elicited by DNA vaccine were markedly enhanced by a homologous protein boost (30). Our T4-AAV hybrid vector, which can efficiently deliver both DNA and proteins and can also sustain the expression of the delivered genes up to 60 days, would lend itself as a multifunctional genetic and protein (prime-boost) vaccine delivery platform.
We tested this possibility by incorporating DNA and protein from two deadly pathogens, one being the influenza virus and the second being the plague bacterium Y. pestis. The HA “stem” region of the influenza virus envelope protein is a very attractive “universal” vaccine candidate because it encodes highly conserved epitopes and can elicit broadly neutralizing antibodies that can block infection by diverse influenza A virus strains (31). We constructed a T4 delivery vector containing the HA stem (HA4900) (31) DNA flanked by AAV ITRs under the control of the strong CMV promoter. Cleavage by Pst I resulted in the linearization of the HA4900 expression cassette with ITRs at the termini (fig. S8A). This DNA was packaged into T4 heads at ~10 molecules per capsid at a packaging ratio of 30:1 (fig. S8B). Transduction of these particles into HEK293 cells resulted in efficient expression and secretion of the HA4900 antigen into the culture medium (fig. S8C). Because the Hoc-bridged T4-AAV was the most efficient from the above in vivo delivery experiments (Fig. 5), it was used to deliver the HA4900 DNA cargo. Mice were immunized intramuscularly with ~2 × 1011 T4(HA4900)-AAV(VRCO1) particles using a prime-boost scheme without any external adjuvant (Fig. 6, A and B). Serum titers of HA-specific total IgG and their subclass specificity, IgG1 or IgG2a, were determined by enzyme-linked immunosorbent assay (ELISA). In mice, the IgG2a titers reflect T helper 1 (TH1) immune responses, whereas the IgG1 titers reflect TH2 immune responses.
Fig. 6. Multivalent DNA vaccine and protein antigen delivery by T4-AAV nanoparticles.
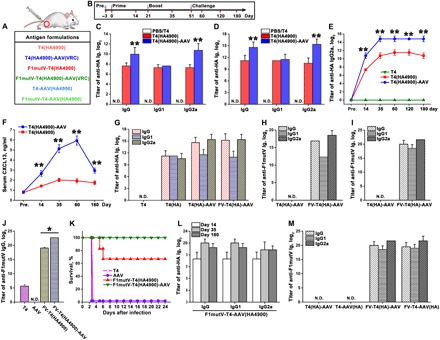
(A) Formulations used for mice vaccinations. (B) Prime-boost immunization scheme. The days of prebleeding (pre.), bleeding, prime, boost, and challenge with Y. pestis are indicated. (C and D) Sera from the PBS, T4, T4(HA4900), and T4(HA4900)-AAV(VRCO1) groups were assessed for specific anti-HA IgG/G1/G2a antibody titers by ELISA on day 14 (prime) (C) and day 35 (boost) (D). (E) Measurement of anti-HA IgG2a antibody titers in sera from groups T4, T4(HA4900), and T4(HA4900)-AAV(VRCO1) at serial time points (on prebleeding day and days 14, 35, 60, 120, and 180). (F) Serum CXCL13 concentration (ng/ml) in mice immunized with T4(HA4900) or T4(HA4900)-AAV(VRCO1) at prebleeding day and days 14, 35, 60, and 180 after immunization. (G) Comparison of anti-HA antibody titers in sera from groups T4, T4(HA4900), T4(HA4900)-AAV(VRCO1), and F1mutV-T4(HA4900)-AAV(VRCO1) on day 35. (H and I) Anti-F1mutV–specific antibody (IgG/G1/G2a) titers were determined by ELISA using sera from mice vaccinated with T4(HA4900)-AAV(VRCO1) and F1mutV-T4(HA4900)-AAV(VRCO1) on days 14 (H) and 35 (I). (J) Comparison of anti-F1mutV IgG antibody titers in sera from groups T4, AAV, F1mutV-T4(HA4900), and F1mutV-T4(HA4900)-AAV on day 35. (K) Kaplan-Meier survival curves of mice against intranasal pneumonic plague challenge with 295 LD50 (median lethal dose) of Y. pestis CO92 (1 LD50 = 100 colony-forming units in BALB/c mice). (L) Anti-HA–specific antibody titers in immunized sera from the F1mutV-T4-AAV(HA4900) group at various time points. (M) Comparison of anti-F1mutV antibody titers in sera from groups T4(HA4900)-AAV, T4-AAV(HA4900), F1mutV-T4(HA4900)-AAV, and F1mutV-T4-AAV(HA4900) on day 35. The values represent means ± SD of three independent replicates. **P < 0.01 compared with the T4(HA4900) vaccination. *P < 0.05 compared with the F1mutV-T4 vaccination. Statistical significance was determined using two-tailed Student’s t tests. N.D., not detected. See Materials and Methods for additional details.
A single injection of T4(HA4900) elicited anti-HA IgG titers in mice (day 14) (Fig. 6C), which were further boosted with a second injection (day 35) (Fig. 6D). No such antibodies were detectable in the naïve control group (PBS or empty T4). Considerably higher anti-HA IgG titers were elicited by T4(HA4900)-AAV immunization than by T4(HA4900) immunization (Fig. 6, C and D), indicating that conjugation to AAV significantly improved T4(HA4900) delivery that, in turn, resulted in increased antigen expression and antibody elicitation, which have greater potential to clear an infectious agent (32). Notably, however, the T4(HA4900)-AAV sera contained ~25-fold higher subclass IgG2a titers, whereas the IgG1 titers were similar in both groups (Fig. 6D). Even more notable was the durability of the antibodies: The anti-HA IgG2a titers remained stable even at 6 months after immunization (Fig. 6E). These data suggest that the T4-AAV nanoparticles elicit TH1-biased immune responses when compared to the unconjugated T4 particles.
Strong and durable humoral immune activities are usually characterized by germinal center response and long-lived plasma and memory B cells and are highly dependent on the help of CD4+ follicular helper T (Tfh) cells (33). CXCL13 [chemokine (C-X-C motif) ligand 13] chemokine is a recognized plasma biomarker for the activities of germinal center and Tfh cells in lymphoid tissue (34). To explore the possible mechanism for elicitation of the remarkably durable humoral responses by T4(HA4900)-AAV, we analyzed serum CXCL13 levels. As shown in Fig. 6F, T4(HA4900)-AAV–immunized mice had much higher serum concentrations of CXCL13 when compared to T4(HA4900)-immunized mice, indicating enhanced activities of germinal center and Tfh cells elicited by the hybrid vector.
Another advantage of the T4-AAV hybrid platform is that it can co-deliver cargo containing both DNA and protein vaccines. To evaluate this capability, we displayed the plague antigen F1mutV on the exterior of the T4(HA4900)-AAV nanoparticles as a Soc fusion protein (fig. S8D). Each particle was decorated with ~590 molecules of F1mutV-Soc protein outside and packaged with ~10 molecules of HA4900 DNA inside. The plasmid DNA encapsidation in T4 has no effect on the anti-F1mutV antibody responses induced by the displayed F1mutV protein. These particles elicited robust anti-F1mutV antibody responses, in addition to the anti-HA antibodies (Fig. 6, G to I). Additionally, consistent with the HA DNA vaccine, F1mutV-T4(HA4900)-AAV also stimulated higher total anti-F1mutV IgG titers than F1mutV-T4(HA4900) (Fig. 6J). We then tested the protective efficacy of the T4(HA4900)-AAV–displayed F1mutV antigen in a mouse pneumonic challenge model. We used greater than three times higher challenge dose in this study than in our previous study (12) to more rigorously evaluate the efficacy of the T4-AAV hybrid vector in comparison with the unconjugated T4. At the challenge dose of 295 LD50 (median lethal dose) (1 LD50 = 100 colony-forming units in BALB/c mice) of the most lethal Y. pestis CO92, the F1mutV-T4(HA4900)-AAV vector showed complete protection, whereas the unconjugated F1mutV-T4(HA4900) vector showed partial 67% protection and all the control animals died (Fig. 6K), demonstrating the beneficial effect of AAV conjugation on antigen delivery by the hybrid vector.
We have also switched the cargo carrier by packaging HA4900 DNA in AAV and displaying F1mutV on T4, which also elicited durable and high anti-F1mutV and anti-HA antibody titers. However, curiously, the anti-HA IgG2a bias observed when the HA4900 DNA was packaged in the T4 capsid was not seen when the HA4900 DNA was delivered through AAV (Fig. 6, L and M, and fig. S8, E to G). This might be due to different processing pathways (35) and/or due to the different forms of the packaged DNAs. The prokaryotic T4-packaged double-stranded DNA containing unmethylated CpG motifs might stimulate innate immune responses by interacting with Toll-like receptors (TLRs) such as TLR9 expressed on the endosomal surface of the antigen-presenting cells (36). Endosomal TLR signaling polarizes TH1-biased adaptive immune responses (37). On the other hand, the human cell–derived AAV single-stranded DNA might generate more balanced (equivalent) TH1 and TH2 responses, as has been reported previously (38).
Notably, no signs of adverse or toxicity-related effects such as changes in serum alkaline phosphatase (ALP), weight loss, or temperature change were evident in any of the mice immunized with T4-AAV nanoparticles (fig. S9).
DISCUSSION
We report here a unique prokaryotic-eukaryotic hybrid vector that can deliver complex and large cargos of genes and/or proteins into mammalian (human) cells. Future human therapies, such as genome editing by CRISPR-Cas, would require delivery of multiple genes encoding guide RNA(s), Cas proteins(s), donor DNA molecule(s), and/or regulatory molecules. Currently, very few vector systems can accomplish these complex deliveries. In this study, we addressed the question of whether combining different natural viral building blocks, a phage and a virus, could lead to new delivery platforms with superior properties. The T4-AAV vector developed here incorporates the useful properties of two key viruses: high-capacity, multifunctional T4 phage coupled with efficient entry and long-term gene expression by AAV. The current study focuses on the design and testing of this hybrid vector for delivery of a variety of reporter genes and proteins as well as vaccine molecules from two different pathogens, plague and flu, both in vitro and in vivo.
The T4-AAV vector design exploits the multivalent and high-affinity avidin-biotin interactions to create bridging molecules that attach to T4 Soc or Hoc on one side and to AAV on the other side. Because Soc and Hoc also have nanomolar affinity and the packaging motor of T4 is the fastest and most powerful (23), varieties of T4-AAV nanoparticles could be prepared by simple mixing of various components. Furthermore, the copy number of each component could be precisely quantified and tuned as desired, by varying the ratios of the interacting components.
Customized T4-AAV nanoparticles containing various combinations of genes and proteins were produced. For instance, nanoparticles—each carrying ~9 molecules of double-stranded luciferase plasmid (packed in T4 capsid), ~2 to 4 molecules of single-stranded GFP DNA (packaged in AAV capsid), and ~250 molecules of β-galactosidase (displayed on T4 capsid)—delivered the entire cargo into the same cell in vitro. The T4-AAV delivery led to ~40,000-fold increase in luciferase activity when compared to control T4 particles lacking AAV.
Entry of nanoparticles into mammalian cells is a complex process requiring the routing of particles through several cellular compartments. This includes binding of the vector to cell surface molecules, internalization by mechanisms such as endocytosis, endosomal escape and release, dissociation of displayed proteins into the cytosol, and transport of nucleic acid into the nucleus (39). Because AAV has naturally evolved mechanisms to facilitate some of these processes, attachment to AAV allowed T4 to co-opt these mechanisms for its own delivery. Notably, our studies indicate that endosomal escape is a significant bottleneck for T4 delivery because compounds such as chloroquine that enhance endosomal escape by nonspecific disruption of endosomes (24) greatly enhanced T4 delivery. Attachment to AAV has a similar effect. Neither simple mixing of T4 and AAV nanoparticles (without avidin bridging) nor a PLA2 mutant in the AAV capsid that disrupted the endosomal escape function, but not virus attachment and entry, facilitated efficient transduction. These results thus provide good evidence that endosomal escape facilitated by AAV is at least one factor for efficient transduction of T4-AAV hybrid nanoparticles.
The T4-AAV nanoparticles also delivered the cargos efficiently in vivo. When injected into mice, the T4-AAV nanoparticles showed ~130 times stronger luminescence when compared to T4 alone. The signal persisted for at least 60 days, whereas without AAV, the signal was nearly lost by 60 days. Furthermore, immune responses were elicited against both the protein expressed from the delivered gene and the protein directly delivered. Consequently, immune responses against two deadly pathogens, one being a virus (flu) and another being a bacterium (plague), were produced by a single immunization with the nanoparticles without any adjuvant. Notably, the antibody titers remained stable even at 6 months following immunization, demonstrating the durability of the T4-AAV–stimulated response.
Our results show that the hybrid T4-AAV vector can function both as a gene delivery vehicle, eliciting transgene expression, and as a vaccine, eliciting robust immune responses. This might be because the nanoparticles are taken up by different types of cells in vivo. Presumably, while some of the particles are taken up by myocytes present at the site of the intramuscular injection and exhibited luciferase reporter gene expression locally, other particles were captured by immune system cells (dendritic cells, B cells, and T cells) and led to stimulation of immune responses. In addition, the myocyte-expressed proteins are likely able to be cross-primed or cross-presented to the local immune cells for the induction of immune responses.
The T4-AAV hybrid nanoparticles stimulated both the TH1- and TH2-mediated responses of the immune system, as evidenced by the elicitation of both IgG2a and IgG1 subtype antibodies, respectively. Significantly higher IgG2a titers against both the flu and plague antigens were elicited by the T4-AAV hybrid particles. These antibodies are considered to be essential for the clearance of infectious pathogens. Plasma levels of CXCL13 correlate with germinal center Tfh cell frequencies and high-affinity, durable antibody production in mice, macaques, and humans (34). The T4-AAV–immunized mice showed elevated levels of the CXCL13 chemokine, which indicate high germinal center activity and frequency of draining lymph nodes that likely lead to a durable humoral response. Furthermore, the T4-AAV–immunized mice were completely protected against pneumonic plague, whereas the unconjugated T4 showed partial protection against a challenge dose of 295 LD50 Y. pestis CO92 (greater than three times higher than the previous study) (12). Together, these data point to high-quality immune responses elicited by the T4-AAV–delivered vaccines. This might be because the T4-AAV vector could use diverse antigen presentation and processing pathways, leading to stimulation of both innate and adaptive immune responses. In addition, by mimicking the pathogen-associated molecular patterns, the T4-AAV nanoparticles might be recognized by the host’s TLRs that allow cross-presentation of the antigens, resulting in broad and protective immune responses. Last, note that no significant adverse effects due to T4-AAV immunizations have been observed, as evaluated by serum ALP, temperature, and weight changes.
Our hybrid vector approach combined the useful features of two different types of viruses while not compromising the capabilities of either. The T4-AAV hybrid vector has a very high capacity, and it is safe and scalable and efficiently delivers large cargos of genes and proteins, features not available in any other systems. It elicits robust and high-quality immune responses that are durable. Hence, the T4-AAV hybrid vector might provide a desirable platform for various therapeutic applications that require the delivery of a complex set of molecules. However, more detailed studies are needed to understand the mechanisms of delivery and maximize delivery efficiency and signal generation, vector distribution in vivo, and safety assessment before it can be transitioned to human therapies. Although integrating the complementary properties of T4 and AAV is ideal for many therapeutic applications, this hybrid vector approach could be broadly applied to create novel combinations of prokaryotic and eukaryotic viruses, bacteria, or synthetic nanoparticles for different applications in the future. These hybrid vectors with collaborative and superior delivery attributes will be potentially useful for creating future gene and protein therapeutics against genetic and infectious diseases.
MATERIALS AND METHODS
Plasmid construction
The plasmids pET-28b-Soc, pET-28b-Soc-β-gal, and pET-28b-F1mutV-Soc were constructed as previously described (10, 12). For the construction of pET-28b-BAP-Hoc, two rounds of polymerase chain reaction (PCR) were performed to amplify BAP-Hoc using three primers: FW1, ATCGAGTGGCACGAGGGTCTTTCGATGACTTTTACAGTTGATATAACTCC; FW2, TTCTAGCTAGCGGTCTTAACGACATCTTCGAGGCACAGAAGATCGAGTGGCACGAGGGTCTTTCG; and RW, ATAAAGCTTTTATGGATAGGTATAGATGATACCAGTTTC. The first round of PCR was performed by fusing Hoc to part of the BAP sequence using the FW1 and RW primers. Full-length BAP-Hoc was obtained by the second round of PCR using the FW2 and RW primers and then digested with Nhe I and Hind III. The digested BAP-Hoc fragment was subcloned into the pET-28b vector. The BirA-expressing plasmid pET-21a-BirA was purchased from Addgene (plasmid no. 20857). Plasmids pAAV-DJ, pAAV-Helper, pAAV-GFP, pAAV-luciferase, and pAAV-mCherry were purchased from Cell Biolabs. To clone DJ-H75A-D76N mutant, upstream fragment was amplified using the primers AN-F (CGCGGAAGCTTCGATCAACTACGCAGACAG) and HD-AN-R (GTTTGCCTCGAGGGCCGCGGCGTCTGCCTC) and the template pAAV-DJ, and downstream fragment was amplified using the primers HD-AN-F (CCGCGGCCCTCGAGGCAAACAAAGCCTACGACCGGCAGCTCGACA) and AN-R (GAGAACGTACGGCAGCTGGTACTCCGAGTC). Then, the two fragments were mixed in equal amounts and used as a template to amplify DJ-H75A-D76N using the primers AN-F and AN-R. The PCR products were subsequently cloned into the pAAV-DJ vector at the Hind III/Bsi WI restriction sites to construct pAAV-DJ-H75A-D76N. Influenza HA4900 DNA fragment was synthesized by Invitrogen according to a recent report (31) and then subcloned into the pAAV vector after digestion with Eco RI and Hind III. All the constructed plasmids were sequenced to confirm correct fragment insertion (Retrogen, CA).
Protein expression and purification
The recombinant proteins expressed in E. coli BL21 (DE3) RIPL cells were purified according to previously described protocols (10, 12). Influenza HA4900 was purified from HEK293F suspension cells (Thermo Fisher Scientific, MA) according to previous reports (31). Briefly, the pAAV-ITRs-HA4900 plasmid was transiently transduced into HEK293F cells maintained in FreeStyle 293 expression medium (Thermo Fisher Scientific, MA) using the 293fectin Transfection Reagent (Thermo Fisher Scientific, MA). The cells were then incubated at 37°C and 8% CO2 while shaking at 130 rpm overnight. After 12 hours, an equal volume of fresh medium supplemented with sodium butyrate solution (enhancing protein expression; 2 nM final concentration) (Sigma-Aldrich, MO) was added to the cells. On day 7, the culture supernatants were harvested by centrifugation and filtered over 0.22-μm filters (Sartorius Stedim Biotech, Germany). Proteins were purified using HisTrap HP and gel filtration columns (GE Healthcare, IL).
Production of the T4 head and AAV
The 10-amber 13-amber hoc-del soc-del T4 heads were purified according to previously described protocols (6). To produce rAAV-DJ, the triple-plasmid transfection method (Cell Biolabs, CA) was used according to the manufacturer’s instructions. Briefly, plasmids, including an adenovirus helper plasmid (pHelper), a rep/cap plasmid expressing rep and cap (pAAV-DJ or pAAV-DJ-H75A-D76N), and a transgene plasmid carrying the AAV transgene cassette (AAV genome) (pAAV-GFP, pAAV-mCherry, pAAV-luciferase, pAAV-HA4900, or pAAV-VRCO1) (1:1:1) were cotransfected into HEK293 cells expressing adenovirus E1a and E1b proteins using polyethyleneimine (Polysciences, PA). After 72 hours, cells were harvested by centrifugation at 1140g for 10 min. The cell pellet was resuspended in lysis buffer [50 mM tris-HCl (pH 8.5) and 0.15 M NaCl] and lysed by three freeze/thaw cycles in dry ice–ethanol and a 37°C water bath. Benzonase was added to the mixture (50 U/ml final concentration), and the lysate was incubated for 30 min at 37°C. The lysate was clarified by centrifugation at 3700g for 20 min, and the virus-containing supernatant was considered to be the crude lysate. Viruses were then purified by discontinuous iodixanol density gradient centrifugation at 500,000 rpm for 16 hours at 4°C (type 55 Ti rotor; Beckman, CA) and using HiTrap AVB Sepharose HP according to the manufacturer’s instructions (GE Healthcare, IL). The peak fractions were dialyzed against PBS-MK buffer [PBS (pH 7.4), 2.5 mM KCl, and 1 mM MgCl2] in Slide-A-Lyzer dialysis cassettes (Thermo Fisher Scientific, MA). The dialyzed AAV particles were concentrated and stored at −80°C. AAV titers were determined using a QuickTiter AAV Quantitation kit (Cell Biolabs, CA).
In vitro DNA packaging and protein display of the T4 head
For in vitro DNA packaging assays, each 20 μl of reaction mixture contained purified T4 heads (∼2 × 1010 particles), purified full-length gp17 (∼3 μM), and linearized DNA in packaging buffer [30 mM tris-HCl (pH 7.5), 100 mM NaCl, 3 mM MgCl2, and 1 mM adenosine 5′-triphosphate (ATP)]. The mixture was incubated at 37°C for 30 min, followed by benzonase nuclease addition and incubation at 37°C for 20 min to remove excess unpackaged DNA. The encapsidated nuclease-resistant DNA was released by treatment with 50 mM EDTA, proteinase K (0.5 μg/μl; Thermo Fisher Scientific, MA), and 0.2% SDS for 30 min at 65°C. The packaged DNA was analyzed by 1% (w/v) agarose gel electrophoresis followed by staining with ethidium bromide, and the amount of packaged DNA was quantified using Quantity One software (Bio-Rad, CA). The packaging efficiency was defined as the number of DNA molecules packaged per T4.
In vitro protein display on the T4 head was assessed by the co-sedimentation described previously (8). Briefly, after encapsidating linearized DNA as described above, T4 heads were incubated with Soc- and/or Hoc-fused proteins at 4°C for 45 min. The mixtures were sedimented by centrifugation at 30,000g for 45 min, and unbound proteins in the supernatants were removed. After washing twice with PBS, the pellets were incubated at 4°C overnight and then resuspended in PBS for SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis or Opti-MEM for transduction. After Coomassie Blue R-250 (Bio-Rad, CA) staining and destaining, the protein bands on SDS-PAGE gels were scanned and quantified by laser densitometry (Personal Densitometer SI; GE Healthcare, IL). The densities of the Hoc, Soc, and gp23 bands were determined for each lane separately, and the copy numbers of bound Hoc or Soc fusion molecules per capsid were calculated using gp23 as the internal control (930 copies per capsid).
Biotinylation of AAV vectors and Soc proteins
AAV vectors and Soc proteins were biotinylated using EZ-Link Sulfo-NHS-LC-biotin according to the manufacturer’s instructions (Thermo Fisher Scientific, MA). Purified AAV vectors (1 × 1012 particles) were incubated with 500 nmol of Sulfo-NHS-LC-biotin at 37°C for 2 hours in PBS buffer. For Soc protein biotinylation, a 20-fold molar excess of the biotin reagent was used to label 10 mg of Soc in PBS buffer. To remove free biotin, the reaction mixture was dialyzed against PBS-MK buffer in Slide-A-Lyzer dialysis cassettes (Thermo Fisher Scientific, MA). Dialyzed bioAAV and bioSoc were concentrated and stored at −80°C.
HBBA production
A 15–amino acid BAP (GLNDIFEAQKIEWHE) was fused to the N terminus of the Hoc protein. The E. coli enzyme biotin ligase (BirA) sequence specifically ligates biotin to BAP. Briefly, His6-tagged BirA and Hoc-BAP were induced in BL21 RIPL cells and purified via a HisTrap HP column and SEC. For Hoc-BAP biotinylation, 1 μM recombinant BirA ligase was added to 30 μM Hoc-BAP monomer [in PBS-MgCl2 buffer (pH 7.4)] in the presence of 0.3 mM biotin and 5 mM ATP. The reaction proceeded at room temperature (RT) for 3 hours. Biotinylation was confirmed by streptavidin-HRP Western blot analysis. Free biotin was removed using Zeba Desalt spin columns (Thermo Fisher Scientific, MA). The biotinylation reaction mixture was loaded onto the center of the compact resin bed and centrifuged at 1000g for 2 min to collect the desalted sample. Purified Hoc-BAP-biotin was incubated with avidin at a 1:3 ratio for 1 hour at 4°C. HBBA was purified by SEC. The peak fractions were concentrated and stored at −80°C.
T4-Soc-AAV and T4-Hoc-AAV vector production
For assembly of T4-Soc-AAV vectors on nickel beads, T4-Soc(His6)-biotin vectors were loaded onto Ni2+-NTA (nitrilotriacetic acid) agarose beads (Qiagen, The Netherlands). After incubation for 1 hour at 4°C, the mixture was centrifuged at 100g for 30 s and washed five times with binding buffer. Avidin dissolved in binding buffer was then added to the beads. After 20 min of incubation at 4°C, free avidin was removed by washing and centrifugation. BioAAV vectors were then added to the T4-Soc-SBA immobilized beads and incubated for 30 min. After washing five times with binding buffer, bound T4-Soc-AAV vectors were eluted with elution buffer [50 mM tris-HCl (pH 8.0), 300 mM NaCl, and 300 mM imidazole]. The protocol for assembling T4-Hoc-AAV on nickel beads was similar to that used for T4-Soc-AAV. Last, the eluted vectors were exchanged into PBS-MK buffer.
Western blot analysis
Briefly, AAV or T4-AAV particles were boiled in loading buffer for 10 min, separated by 12% SDS-PAGE, and then transferred to nitrocellulose membranes (Bio-Rad, CA). Blocking was performed in 5% bovine serum albumin (BSA)/PBS-T buffer [PBS (pH 7.4) and 0.05% Tween 20] at RT for 1 hour with gentle shaking. Blots were then washed three times with PBS-T. Primary antibodies were added to the blots and incubated overnight at 4°C in PBS with 5% BSA. AAV VP proteins were detected using the AAV capsid protein–specific antibody B1 (diluted 1:50; American Research Products, MA), which recognizes VP1, VP2, and VP3 on the basis of their identical C-terminal regions. After washing with PBS-T three times, a secondary goat anti-mouse HRP-conjugated antibody (Thermo Fisher Scientific, MA) was applied at a 1:2000 dilution in 5% BSA/PBS-T for 1 hour at RT, followed by rinsing three times with PBS-T. The bioAAV, bioSoc, and Hoc-BAP-biotin were directly detected by HRP-conjugated streptavidin (Abcam, UK). Signals were visualized with an enhanced chemiluminescence substrate (Bio-Rad, CA) using the Bio-Rad Gel Doc XR+ System and Image Lab software according to the manufacturer’s instructions (Bio-Rad, CA).
Labeling of T4 capsid
To label the T4 capsid with amine-reactive Alexa Fluor 594 (Thermo Fisher Scientific, MA), the pelleted T4 heads were resuspended in 0.1 M carbonate buffer (pH 9.0). Alexa Fluor 594 was added to a final concentration of 0.2 mg/ml. After incubation for 1 hour at RT with rotation in the dark, unbound dye was removed via buffer exchange into PBS (pH 7.4).
Transmission electron microscopy
The T4-AAV complex was applied to the carbon grid for 5 min at RT. The grid was then frozen in liquid nitrogen using Gatan CP3 cryo-plunger. The cryo–electron microscopy images of T4, AAV, and T4-AAV complex were collected by Q. Fang at Purdue University using a Titan Krios microscope equipped with a charge-coupled device camera. AAV particles for this experiment were provided by M. Agbandje-McKenna, University of Florida.
Cell culture
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, MA), 1× Hepes (Thermo Fisher Scientific, MA), and 1% antibiotics (Thermo Fisher Scientific, MA) (complete DMEM). Cells were passaged with 0.05% trypsin/EDTA at a subcultivation ratio of 1:5 at 80 to 90% confluence and incubated in a humidified atmosphere at 37°C and 5% CO2.
Cell transduction and the detection of gene and protein delivery
HEK293 cells were seeded in 24-well plates at 2.0 × 105 cells per well in complete DMEM. After 24 hours, the cells were incubated with the AAV, bioAAV, T4, or T4-AAV vectors at different multiplicities of infection in antibiotic-free Opti-MEM for 6 hours. Thereafter, Opti-MEM was removed and replaced with complete DMEM. The cells were further incubated at 37°C for an additional 48 hours. GFP/mCherry transgene expression was observed by fluorescence microscopy (Carl Zeiss, Germany) at 48 hours after transduction, and the mean fluorescence intensities were quantified using ImageJ software. The nucleus was stained using Hoechst 33342 (Thermo Fisher Scientific, MA). To analyze luciferase gene delivery into cells by T4 or T4-AAV, we measured luciferase activity with the Luciferase Assay System (Promega, WI) according to the manufacturer’s instructions. Briefly, growth medium was removed, and cells were rinsed with PBS buffer. After removing the wash buffer, 150 μl of passive lysis buffer was added to each well, followed by gentle shaking at RT for 20 min. Twenty microliters of the cell lysate was then transferred to a 96-well white opaque plate and mixed with 80 μl of Luciferase Assay Reagent, and the luminescence signal was recorded using the GloMax-Multi Detection System (Promega, WI). The activity of the Soc-β-gal enzyme displayed on the T4 head in cells was determined by staining with X-Gal using the β-Galactosidase Staining Kit (Sigma-Aldrich, MO). Triplicate measurements were applied to each group.
Colocalization analysis
Images were processed using Zen (Carl Zeiss, Germany) and ImageJ software. To quantify the extent of colocalization for a two-color comparison, the linear Pearson (rp) and nonlinear Spearman’s rank (rs) correlation coefficients for pixels that represented the fluorescence signals in the green and red channels were calculated using the ImageJ PSC plugin.
Cell viability assay
After transfection for 48 hours, cell viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, WI) following the manufacturer’s instructions. Briefly, an equal volume of CellTiter-Glo Reagent was added to the cell culture in each well. The mixture was placed on an orbital shaker for 2 min to induce cell lysis and then incubated at RT for 10 min to stabilize the luminescence signal, which was recorded using the GloMax-Multi Detection System (Promega, WI). The viability of the untreated cell group was arbitrarily set to 100%, and triplicate measurements were applied to each group.
In vivo bioluminescence imaging
As previously described (10), 5 × 1010 and 2 × 1011 T4 or T4-AAV particles were intramuscularly injected into BALB/c mice. At 0.25, 0.5, 1, 2, 5, 10, 15, 30, and 60 days after administration, 30 μg of RediJect D-Luciferin Ultra (PerkinElmer, MA), a luciferase substrate dissolved in 0.9% saline, was injected intraperitoneally. After 5 min, the mice were lightly anesthetized with 2% isoflurane and placed on an IVIS 200 bioluminescence whole-body imaging workstation (Caliper). The bioluminescence emission signal was quantified using the camera control program, Living Image software, and displayed in physical units of surface radiance, photons per second per centimeter squared per steradian (photons s−1 cm−2 sr−1).
Mouse immunizations
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Catholic University of America (Washington, DC) and the University of Texas Medical Branch (Galveston, TX). Mice (BALB/c, female, 6 to 8 weeks old; The Jackson Laboratory, ME) were randomly grouped and allowed to acclimate for 7 days, followed by intramuscular immunizations into their hind legs with vectors (priming on day 0 and boosting on day 21). A group of mock-immunized mice (PBS only) was included as negative control. Blood was drawn from each animal on days 0 (prebleed), 14, 21, 35, 60, 120, and 180, and the isolated sera were stored at −80°C.
Enzyme-linked immunosorbent assay
ELISA plates (Evergreen Scientific, CA) were coated with 0.1 μg of protein per well in coating buffer [0.05 M sodium carbonate–sodium bicarbonate (pH 9.6)] overnight at 4°C. After washing three times with PBS-T buffer, the plates were blocked with PBS–3% BSA buffer for 1 hour at 37°C. The concentrations of antigen-specific IgG/G1/G2a in sera were monitored using a 5-fold dilution series beginning with an initial 100-fold dilution in PBS–1% BSA. The diluted serum samples were added to each well, and the plates were incubated at 37°C for 1 hour and washed five times with PBS-T buffer. The secondary goat anti-mouse IgG-HRP antibody (HRP-conjugated goat anti-mouse IgG1 or IgG2a secondary antibodies were used for IgG1/IgG2a subtypes; Invitrogen) was then added to each well at a 1:5000 dilution and incubated for 1 hour at 37°C, followed by washing five times with PBS-T buffer. Next, the TMB (3,3′,5,5′-tetramethylbenzidine) Microwell Peroxidase Substrate System (KPL) was applied in the dark for color development. After 10 min, the enzymatic reaction was quenched by adding TMB BlueSTOP (KPL) solution, and plates were read within 30 min at 650 nm using an ELISA reader (VersaMax; Molecular Devices). Endpoint titers were presented as the sample dilution resulting in an OD650 (optical density at 650 nm) equal to twice the mean background (negative serum) of the assay. For CXCL13 quantification in sera, the Mouse CXCL13 ELISA Kit (Boster Biological Technology, CA) was used by following the manufacturer’s instructions.
ALP assay
The ALP biochemical parameter was determined in sera of mice immunized with T4, AAV, or T4-AAV. Naïve mice were used as control. ALP level was measured using a commercial ALP assay kit (Elabscience, MD) following the manufacturer’s instructions.
Mouse challenges
The acclimated mice were intramuscularly immunized into their hind legs with T4, AAV, F1mutV-T4(HA4900), or F1mutV-T4(HA4900)-AAV (priming on day 0 and boosting on day 21). Sera were collected from each animal on days 0 (prebleeds) and 35 for immunological analyses. On day 42, mice were intranasally challenged with 295 LD50 of Y. pestis CO92 bacteria (40). Animals were monitored and recorded for mortality, body weight, and body temperature.
Statistics
All quantified data were presented as means ± SD. Statistical analyses were performed by two-tailed Student’s t tests. Significant differences between two groups were indicated by *P < 0.05 or **P < 0.01.
Supplementary Material
Acknowledgments
We thank A. Raafat at NCI, NIH, for critical review and editing of the manuscript. Funding: This work was supported by NIAID/NIH grant AI111538 to V.R. Author contributions: J.Z. and V.R. designed research. J.Z., P.T., and M.M. planned and performed the experiments. J.S., P.K., and A.K.C. performed mouse in vivo imaging and plague challenge experiments. J.Z. and V.R. analyzed and interpreted the data. V.R. and J.Z. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax0064/DC1
Fig. S1. Characterization of HBBA and quantification of the SBA and HBBA displays.
Fig. S2. TSA and THA efficiently bind biotinylated molecules in vitro.
Fig. S3. Recombinant AAV (serotype DJ) efficiently attaches to T4 capsid through biotin-avidin cross-bridges.
Fig. S4. Characterization of T4 DNA packaging.
Fig. S5. The T4-AAV vector efficiently delivers packaged DNAs and displayed proteins into mammalian cells without affecting cell viability.
Fig. S6. Endosomal escape is critical for T4-AAV intracellular delivery.
Fig. S7. Representative in vivo bioluminescence images of mice at various time points.
Fig. S8. DNA packaging, protein expression, and antigen delivery by the T4-AAV vector.
Fig. S9. In vivo toxicity measurement of T4-AAV particles.
Table S1. Comparison of T4 nanoparticle delivery with other viral delivery platforms.
REFERENCES AND NOTES
- 1.Yoo J. W., Irvine D. J., Discher D. E., Mitragotri S., Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat. Rev. Drug Discov. 10, 521–535 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Stewart M. P., Sharei A., Ding X., Sahay G., Langer R., Jensen K. F., In vitro and ex vivo strategies for intracellular delivery. Nature 538, 183–192 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Giacca M., Zacchigna S., Virus-mediated gene delivery for human gene therapy. J. Control. Release 161, 377–388 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Cao B., Yang M., Mao C., Phage as a genetically modifiable supramacromolecule in chemistry, materials and medicine. Acc. Chem. Res. 49, 1111–1120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karimi M., Mirshekari H., Moosavi Basri S. M., Bahrami S., Moghoofei M., Hamblin M. R., Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 106, 45–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z., Kottadiel V. I., Vafabakhsh R., Dai L., Chemla Y. R., Ha T., Rao V. B., A promiscuous DNA packaging machine from bacteriophage T4. PLOS Biol. 9, e1000592 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leffers G., Rao V. B., A discontinuous headful packaging model for packaging less than headful length DNA molecules by bacteriophage T4. J. Mol. Biol. 258, 839–850 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Li Q., Shivachandra S. B., Zhang Z., Rao V. B., Assembly of the small outer capsid protein, Soc, on bacteriophage T4: A novel system for high density display of multiple large anthrax toxins and foreign proteins on phage capsid. J. Mol. Biol. 370, 1006–1019 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruttin A., Brüssow H., Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 49, 2874–2878 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao P., Mahalingam M., Marasa B. S., Zhang Z., Chopra A. K., Rao V. B., In vitro and in vivo delivery of genes and proteins using the bacteriophage T4 DNA packaging machine. Proc. Natl. Acad. Sci. U.S.A. 110, 5846–5851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao P., Mahalingam M., Zhu J., Moayeri M., Sha J., Lawrence W. S., Leppla S. H., Chopra A. K., Rao V. B., A bacteriophage T4 nanoparticle-based dual vaccine against anthrax and plague. MBio 9, e01926-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tao P., Mahalingam M., Kirtley M. L., van Lier C. J., Sha J., Yeager L. A., Chopra A. K., Rao V. B., Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLOS Pathog. 9, e1003495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao P., Wu X., Rao V., Unexpected evolutionary benefit to phages imparted by bacterial CRISPR-Cas9. Sci. Adv. 4, eaar4134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tao P., Wu X., Tang W. C., Zhu J., Rao V., Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth. Biol. 6, 1952–1961 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samulski R. J., Muzyczka N., AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 1, 427–451 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Chen Z., Sun L., Zhang Z., Fokine A., Padilla-Sanchez V., Hanein D., Jiang W., Rossmann M. G., Rao V. B., Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl. Acad. Sci. U.S.A. 114, E8184–E8193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fokine A., Islam M. Z., Zhang Z., Bowman V. D., Rao V. B., Rossmann M. G., Structure of the three N-terminal immunoglobulin domains of the highly immunogenic outer capsid protein from a T4-like bacteriophage. J. Virol. 85, 8141–8148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Summerford C., Samulski R. J., Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72, 1438–1445 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farr G. A., Zhang L. G., Tattersall P., Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. U.S.A. 102, 17148–17153 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schatz P. J., Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: A 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology 11, 1138–1143 (1993). [DOI] [PubMed] [Google Scholar]
- 21.Grimm D., Lee J. S., Wang L., Desai T., Akache B., Storm T. A., Kay M. A., In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 82, 5887–5911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold G. S., Sasser A. K., Stachler M. D., Bartlett J. S., Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol. Ther. 14, 97–106 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Rao V. B., Feiss M., Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu. Rev. Virol. 2, 351–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seglen P. O., Grinde B., Solheim A. E., Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur. J. Biochem. 95, 215–225 (1979). [DOI] [PubMed] [Google Scholar]
- 25.Zadori Z., Szelei J., Lacoste M. C., Li Y., Gariepy S., Raymond P., Allaire M., Nabi I. R., Tijssen P., A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 1, 291–302 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Penaud-Budloo M., Le Guiner C., Nowrouzi A., Toromanoff A., Cherel Y., Chenuaud P., Schmidt M., von Kalle C., Rolling F., Moullier P., Snyder R. O., Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 82, 7875–7885 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gause K. T., Wheatley A. K., Cui J., Yan Y., Kent S. J., Caruso F., Immunological principles guiding the rational design of particles for vaccine delivery. ACS Nano 11, 54–68 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Barr J. J., Auro R., Furlan M., Whiteson K. L., Erb M. L., Pogliano J., Stotland A., Wolkowicz R., Cutting A. S., Doran K. S., Salamon P., Youle M., Rohwer F., Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M. A., Immunologic basis of vaccine vectors. Immunity 33, 504–515 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Flotte T. R., Lu S., DNA vaccination in 2018: An update. Hum. Gene Ther. 29, 963–965 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Impagliazzo A., Milder F., Kuipers H., Wagner M. V., Zhu X., Hoffman R. M., van Meersbergen R., Huizingh J., Wanningen P., Verspuij J., de Man M., Ding Z., Apetri A., Kukrer B., Sneekes-Vriese E., Tomkiewicz D., Laursen N. S., Lee P. S., Zakrzewska A., Dekking L., Tolboom J., Tettero L., van Meerten S., Yu W., Koudstaal W., Goudsmit J., Ward A. B., Meijberg W., Wilson I. A., Radosevic K., A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 349, 1301–1306 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Tao P., Zhu J., Mahalingam M., Batra H., Rao V. B., Bacteriophage T4 nanoparticles for vaccine delivery against infectious diseases. Adv. Drug Deliv. Rev. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitfeld D., Ohl L., Kremmer E., Ellwart J., Sallusto F., Lipp M., Forster R., Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192, 1545–1552 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Havenar-Daughton C., Lindqvist M., Heit A., Wu J. E., Reiss S. M., Kendric K., Belanger S., Kasturi S. P., Landais E., Akondy R. S., McGuire H. M., Bothwell M., Vagefi P. A., Scully E., Investigators I. P. C. P., Tomaras G. D., Davis M. M., Poignard P., Ahmed R., Walker B. D., Pulendran B., McElrath M. J., Kaufmann D. E., Crotty S., CXCL13 is a plasma biomarker of germinal center activity. Proc. Natl. Acad. Sci. U.S.A. 113, 2702–2707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon J. J., Huang B., Irvine D. J., Engineering nano- and microparticles to tune immunity. Adv. Mater. 24, 3724–3746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwasaki A., Medzhitov R., Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Schwarz K., Storni T., Manolova V., Didierlaurent A., Sirard J. C., Rothlisberger P., Bachmann M. F., Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur. J. Immunol. 33, 1465–1470 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Johnson P. R., Schnepp B. C., Connell M. J., Rohne D., Robinson S., Krivulka G. R., Lord C. I., Zinn R., Montefiori D. C., Letvin N. L., Clark K. R., Novel adeno-associated virus vector vaccine restricts replication of simian immunodeficiency virus in macaques. J. Virol. 79, 955–965 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ni R., Zhou J., Hossain N., Chau Y., Virus-inspired nucleic acid delivery system: Linking virus and viral mimicry. Adv. Drug Deliv. Rev. 106, 3–26 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Tao P., Mahalingam M., Zhu J., Moayeri M., Kirtley M. L., Fitts E. C., Andersson J. A., Lawrence W. S., Leppla S. H., Chopra A. K., Rao V. B., A bivalent anthrax-plague vaccine that can protect against two tier-1 bioterror pathogens, Bacillus anthracis and Yersinia pestis. Front. Immunol. 8, 687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax0064/DC1
Fig. S1. Characterization of HBBA and quantification of the SBA and HBBA displays.
Fig. S2. TSA and THA efficiently bind biotinylated molecules in vitro.
Fig. S3. Recombinant AAV (serotype DJ) efficiently attaches to T4 capsid through biotin-avidin cross-bridges.
Fig. S4. Characterization of T4 DNA packaging.
Fig. S5. The T4-AAV vector efficiently delivers packaged DNAs and displayed proteins into mammalian cells without affecting cell viability.
Fig. S6. Endosomal escape is critical for T4-AAV intracellular delivery.
Fig. S7. Representative in vivo bioluminescence images of mice at various time points.
Fig. S8. DNA packaging, protein expression, and antigen delivery by the T4-AAV vector.
Fig. S9. In vivo toxicity measurement of T4-AAV particles.
Table S1. Comparison of T4 nanoparticle delivery with other viral delivery platforms.