

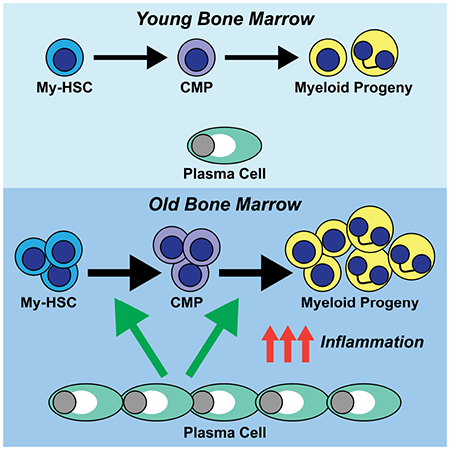
Summary
Aging results in increased myelopoiesis, which is linked to the increased incidence of myeloid leukemias and production of myeloid-derived suppressor cells. Here we examined the contribution of plasma cells (PCs) to age-related increases in myelopoiesis, as PCs exhibit immune regulatory function and sequester in bone marrow (BM). PC number was increased in old BM and they exhibited high expression of genes encoding inflammatory cytokines and pathogen sensors. Antibody-mediated depletion of PCs from old mice reduced the number of myeloid biased hematopoietic stem cells and mature myeloid cells to levels in young animals, but lymphopoiesis was not rejuvenated, indicating that redundant mechanisms inhibit that process. PCs also regulated the production of inflammatory factors from BM stromal cells, and disruption of the PC-stromal cell circuitry with inhibitors of the cytokines IL-1 and TNF-a attenuated myelopoiesis in old mice. Thus, the age-related increase in myelopoiesis is driven by an inflammatory network orchestrated by PCs.
Keywords: Hematopoiesis, Lymphopoiesis, Myelopoiesis, Aging, Plasmablast, Plasma Cell, Inflammaging, Inflammation, Interleukin-1, Tumor Necrosis Factor
Graphical Abstract
eTOC Blurb
Pioli et al. demonstrate that bone marrow plasma cells (PCs) increase in number with age and play an obligate role in the increased production of myeloid cells during aging. They further demonstrate that old PCs are a source of myelopoietic cytokines and regulate their production from BM stroma.
Introduction
Aging in both mice (Geiger et al., 2013; Montecino-Rodriguez et al., 2013) and humans (Pang et al., 2011; Rundberg Nilsson et al., 2016) results in increased myelopoiesis (Dykstra et al., 2011; Min et al., 2006; Rossi et al., 2005) that includes a significant increase in the frequency and number of hematopoietic stem cells (HSCs) (Dykstra and de Haan, 2008; Geiger and Van Zant, 2002), common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs) and mature myeloid cells (Dykstra et al., 2011; Tang et al., 2013). Enhanced myelopoiesis correlates with an increased incidence of myeloid leukemias (Rossi et al., 2007; Signer et al., 2007) and production of myeloid-derived suppressor cells (Flores et al., 2017). Thus, understanding why age-associated myeloid skewing occurs has translational relevance.
Age-related myeloid bias reflects population shifts within the HSC compartment (Beerman et al., 2010; Benz et al., 2012; Challen et al., 2010; Muller-Sieburg et al., 2004). HSCs include lymphoid biased (Ly-HSCs) and myeloid biased (My-HSCs) subsets, and the ability to resolve them phenotypically (Beerman et al., 2010; Benz et al., 2012; Challen et al., 2010) led to the realization that the number of My-HSCs increases over time and they become the predominant stem cell population in bone marrow (BM) of old mice (Beerman et al., 2010; Challen et al., 2010). Thus, the myeloid biased pattern of reconstitution in young recipients reconstituted with old total HSCs is due in part to the fact that the majority of transplanted cells are My-HSCs. An understanding of what triggers the increase in the number of My-HSCs in old BM would provide insights into why age-related increases in myelopoiesis occur.
The production of inflammatory cytokines and chemokines increases systemically (Zhang et al., 2004) and locally in the BM (Ergen et al., 2012; Henry et al., 2015; Kennedy and Knight, 2017; Zhang et al., 2004) with age and is referred to as inflammaging (Franceschi et al., 2000). Hematopoietic stem and progenitor cells (HSPCs) express receptors for various inflammatory factors, including interleukin (IL)-1, IL-6 and tumor necrosis factor-alpha (TNF-α) as well as various Toll-like receptors (TLRs) (King and Goodell, 2011; Mirantes et al., 2014; Nagai et al., 2006). The activation of these cytokine or TLR signaling pathways can stimulate myelopoiesis and inhibit lymphopoiesis (Baratono et al., 2015; Nagai et al., 2006; Ueda et al., 2005). The fact that old HSCs express genes indicative of increased pro-inflammatory signaling is consistent with the conclusion that residence in an inflammatory environment contributes to age-related changes in hematopoiesis (Chambers et al., 2007). Thus, extrinsic events may trigger population shifts within the HSC compartment and in downstream myeloid progenitors. This view is supported by our recent report that Ly-HSCs and My-HSCs exhibit normal developmental potential when removed from the old environment (Montecino-Rodriguez et al., 2019). Consequently, there is considerable interest in identifying the source of inflammation in aging BM.
Numerous non-hematopoietic populations that include osteolineage, perivascular, endothelial, neuronal and mesenchymal stromal cells play a role in the regulation of hematopoiesis (Boulais and Frenette, 2015; Crane et al., 2017; Lo Celso and Scadden, 2011). The effects of aging on these populations are poorly understood compared to the understanding of changes that occur in HSPCs (Geiger et al., 2013; Guidi and Geiger, 2017). Nevertheless, recent studies have demonstrated that aging affects the ability of these microenvironmental elements to support hematopoiesis. For example, old endothelial cells exhibit compromised support of hematopoietic recovery following irradiation (Poulos et al., 2017), stromal cells from old telomerase deficient mice have a decreased ability to support lymphopoiesis (Ju et al., 2007) and the reduced expression of osteopontin by osteoblasts can compromise stem cell function (Guidi et al., 2017). Despite these advances, the major cellular alterations in the aging hematopoietic microenvironment that contribute to myeloid skewing remain to be defined.
In addition to the above non-hematopoietic cells, hematopoietic populations such as megakaryocytes, T cells, NK cells and B cells (Hirata et al., 2018; King et al., 2009; Nakamura-Ishizu et al., 2014; Ratliff et al., 2013) can be considered as components of the hematopoietic microenvironment in view of their ability to regulate blood cell development. A regulatory role for B cells beyond their traditional function as a source of immunoglobulins is a relatively recent concept (Dang et al., 2014; Rauch et al., 2012), and the regulatory B cell pool includes cytokine secreting plasma cells (PCs) (Dang et al., 2014). PCs produce both pro- and anti-inflammatory cytokines such as IL-10 (Suzuki-Yamazaki et al., 2017), IL-17 (Bermejo et al., 2013), IL-35 (Shen et al., 2014) and TNF-α (Fritz et al., 2011) and they can modify immune responses in extramedullary tissues such as the intestine (Fritz et al., 2011; Kim and Kim, 2014). We considered the possibility that PCs contribute to age-related changes in hematopoiesis because, in addition to being a source of myelopoietic cytokines, they sequester in BM following their generation in secondary lymphoid tissues (Chu and Berek, 2013; Fairfax et al., 2008).
Here we examined the contribution of PCs to the increased myelopoiesis associated with aging. We found that the number of PCs significantly increased in aging BM and that PC depletion with CD138 antibodies resulted in a reduction in the numbers of My-HSCs and myeloid progenitors as well as an attenuation of myelopoiesis. Whole transcriptome profiling revealed that old PCs acquired a TLR responsive gene signature that distinguished these cells from their young counterparts and that TLR4 signaling promoted the production of inflammatory cytokines known to stimulate myelopoiesis. In addition, PCs regulated inflammatory cytokine gene expression in BM stroma, indicating the existence of a cross-talk between these two populations. Thus, the accumulation of plasma cells in the BM environment is a major contributor to enhanced myelopoiesis observed during aging.
Results
PCs accumulate in aging BM
Using cell surface expression of CD138, CD45R(B220) and IgD, we quantified the number of CD138high CD45R(B220)+ IgD™ short-lived plasmablasts (PBs) and CD138high CD45R(B220)™ IgD™ mature plasma cells (PCs) in the BM of young (2-3 months old), middle-aged (9-10 months old) and old (17-19 months old) mice (Figure 1A) (Chernova et al., 2014). While PBs were unchanged with age, mature PC frequency and number significantly increased in BM of middle-aged and old mice (Figure 1B). The PCs identified in Figure 1A exhibited heterogeneous expression of CD19 (Figure 1C) (Chernova et al., 2014) but not CD11b (Kunisawa et al., 2013) or Gr-1 (Figure 1C). PCs from young and old mice were labeled with anti-IgM, IgG and IgA (Figure 1D). In agreement with previous reports (Pinto et al., 2013), some PCs expressed surface immunoglobulin (Ig) (Figure 1D), and almost all cells expressed Ig following permeabilization (Figure 1D). Both young and old PCs also expressed Blimp-1 (Shapiro-Shelef et al., 2003; Taubenheim et al., 2012) (Figure 1E).
Figure 1. PCs Accumulate in old BM.
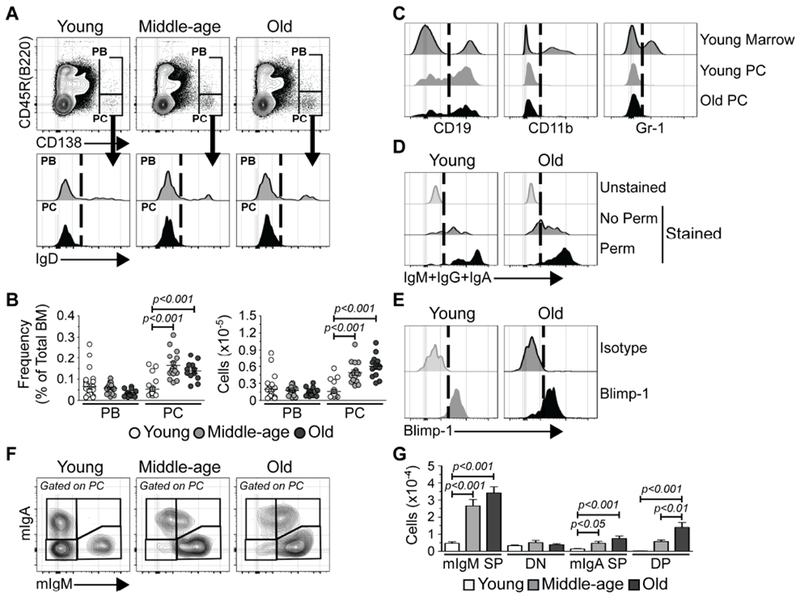
(A) Representative FACS plots depicting immunostaining of PBs and PCs in the bone marrow (BM) of young, middle-aged and old mice. (B) Frequencies and numbers of PBs and PCs in BM of young (n = 25), middle-aged (n = 16) and old (n = 13) mice. Each symbol represents an individual mouse. (C) Representative FACS plots depicting CD19, CD11b and Gr-1 immunostaining of PCs from young and old mice. Young total BM is shown for comparison. (D) Representative FACS plots depicting IgM+IgA+IgG immunostaining of PCs from young and old mice. Unstained samples are shown for comparison. No Perm = unpermeabilized; Perm = permeabilized. (E) Representative FACS plots depicting isotype control and Blimp-1 immunostaining of PCs in BM of young and old mice. (F) Representative FACS plots depicting mIgM and mIgA immunostaining of PCs in BM of young, middle-aged and old mice. (G) Numbers of SP, DN or DP mIgM+ and/or mIgA+ PCs in BM of young (n = 18), middle-aged (n = 12) and old (n = 13) mice. Bars represent mean ± SEM. (A, C-E): Vertical dashed lines on histograms depict cut-offs for positive staining. Statistics: One-way ANOVA with Bonferroni’s correction.
See also Figure S1.
PCs arise in disparate sites and secrete Igs of different classes (Lemke et al., 2016; Reynolds et al., 2015). We examined membrane-bound IgM (mIgM) and IgA (mIgA) expression (Pinto et al., 2013) and identified 1) mIgM+ mIgA™ single positive (mIgM SP); 2) mIgM™ mIgA™ double negative (DN); 3) mIgM™ mIgA+ single positive (mIgA SP); and 4) mIgM+ mIgA+ double positive (DP) (Figure 1F) PCs. Their quantification revealed significant increases in the numbers of IgM and IgA expressing PCs evident by middle-age and maintained in old mice (Figure 1G). This included a subset of IgM and IgA double positive cells absent in young animals but present in middle aged and old mice. IgM+ IgA+ B cells are present in patients with IgA deficiency (Conley and Cooper, 1981) and may arise due to defective class switch recombination (Yazdani et al., 2017). It is relevant in this regard that old B cells exhibit impaired class switching (Frasca et al., 2008).
We also analyzed PB and PC populations in the spleen (SPL) of young, middle-aged and old mice (Figures S1A–S1G). In contrast to BM, both SPL PBs and PCs exhibited an age-associated accumulation that was evident by middle-age (Figures S1A and S1B). Similar to BM PCs, SPL PCs displayed variable CD19 expression whereas these cells were negative for both CD11b and Gr-1 staining (Figure S1C). PCs in the SPL expressed intracellular Ig, in particular IgM, and Blimp-1 (Figures S1D–G).
PC depletion in old mice abrogates increased myelopoiesis
We investigated whether PC accumulation had functional relevance by depleting them in old mice with a mouse anti-mouse CD138 antibody (muCD138). This IgG2a heavy chain and κ light chain antibody originates from the 281–2 hybridoma (Jalkanen et al., 1985). Cohorts of old mice were treated in parallel with an isotype matched mouse anti-human CD20 (huCD20) antibody (Figure 2A). After two weeks, the status of lymphopoiesis and myelopoiesis was analyzed. Young, untreated mice were assessed in parallel to provide a reference for any effects seen upon PC depletion. muCD138 treated mice had a significant reduction in the frequency (Figure S2B) and number of PCs (Figure 2B) whereas PC numbers in the huCD20 treated mice were comparable to those in untreated old mice (Figures 1B and 2B).
Figure 2. PC Depletion Reverses the Age-associated Enhancement of Myelopoiesis.
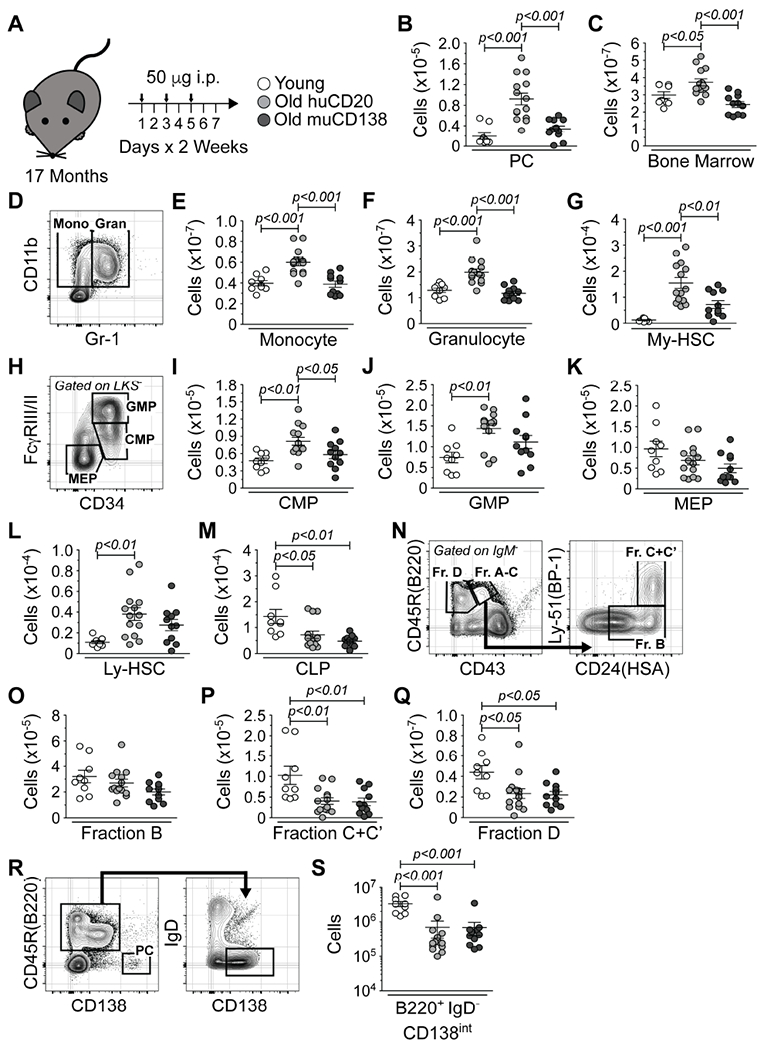
(A) Schematic of antibody-mediated depletion of PCs in old mice. Arrows indicate days of injection. (B) Number of PCs in young and huCD20 and muCD138 treated old mice. (C) Total BM cellularity in young, huCD20 and muCD138 treated old mice. (D) Representative FACS plot depicting immunostaining of monocytes and granulocytes in young mice. Numbers of (E) monocytes, (F) granulocytes and (G) My-HSCs in young, huCD20 and muCD138 treated old mice. (H) Representative FACS plot from young mice depicting immunostaining of CMPs, GMPs and MEPs. Numbers of (I) CMPs, (J) GMPs and (K) MEPs in young, huCD20 and muCD138 treated old mice. (L) Numbers of Ly-HSCs and (M) CLPs in young and huCD20 and muCD138 treated old mice. (N) Representative FACS plots from young mice depicting immunostaining of pro-B and pre-B cells. Numbers of (O) Fraction B early pro-B, (P) Fraction C+C’ late pro-B/large pre-B and (Q) Fraction D small pre-B cells in young, huCD20 and muCD138 treated old mice. (R) Representative FACS plots from young mice depicting immunostaining of B220+ IgD™ CD138int immature B lineage cells. The PC containing gated region is provided as a reference point for CD138 staining intensity. (S) Numbers of B220+ IgD™ CD138int immature B lineage cells in young, huCD20 and muCD138 treated old mice. (B, C, E-G, I-M, O-Q, S) Each symbol represents an individual mouse. Animals used: Young = 9; huCD20 = 14; muCD138 = 11. Statistics: One-way ANOVA with Bonferroni’s correction.
See also Figures S2 and S3.
Total BM cellularity in muCD138, but not huCD20, treated mice was reduced to levels observed in young animals (Figure 2C). This reduction resulted from a significant decline in the numbers of monocytes and granulocytes to levels observed in young mice (Figures 2D–2F). muCD138 treated mice also had a significant reduction in the numbers of My-HSCs (Figure 2G and Figure S3) and CMPs (Figures 2H and 2I) which increase with age (Min et al., 2006; Signer et al., 2007). While the numbers of GMPs and MEPs were also reduced, these changes were not significant (Figures 2J and 2K). These observations demonstrated that PC depletion results in a decrease in My-HSCs and myeloid progenitors and a significant reduction in myelopoiesis in the BM. Despite the fact that the number of myeloid lineage cells was reduced in PC depleted mice, only subtle changes in the frequency of these populations were observed (Figure S2). This is due to the fact that, while myelopoiesis was reduced in muCD138 treated mice, B lymphopoiesis did not rebound.
A reduction in BM lymphopoiesis occurs with age (Alter-Wolf et al., 2009; Miller and Allman, 2003; Min et al., 2006). muCD138 treated old mice exhibited no significant change in the numbers of Ly-HSCs (Figure 2L) while CLPs (Figure 2M) and committed B lymphoid progenitors (Hardy et al., 1991; Hardy and Hayakawa, 1995) (Figures 2N–2Q) remained depressed. Immature B220+ IgD™ B lineage cells in the BM express intermediate levels of CD138 (Tung et al., 2006) (Figure 2R), leading to the possibility that B cell development did not rebound in muCD138 treated mice due to their depletion. However, CLPs do not express CD138, and their numbers, which are decreased with age, remained depressed in muCD138 treated mice (Figure 2M). In addition, the muCD138 treatment did not reduce the number of B220+ IgD™ CD138int cells below levels observed in huCD20 treated old mice (Figure 2S).
PC depletion in young mice does not alter myelopoiesis
The above data demonstrated that old PCs are key effectors of the age-associated increase in myelopoiesis. However, it is unclear what, if any, regulatory role PCs play in the BM of young mice. To investigate this, we treated young (2 months old) mice with PBS and huCD20 or muCD138 antibodies (Figure 3A) and assessed hematopoiesis as described above. muCD138 treatment of young mice resulted in the depletion of PCs (Figure 3B) but had no other effect on overall BM cellularity (Figure 3C), the number of lymphoid and myeloid progenitors or the number of mature myeloid cells (Figures 3D–3N). These data indicated that the ability of PCs to regulate myelopoiesis is age-dependent and demonstrated that the CD138 antibody does not non-specifically deplete hematopoietic cells.
Figure 3. PC Depletion in Young Mice Does Not Alter Hematopoiesis.
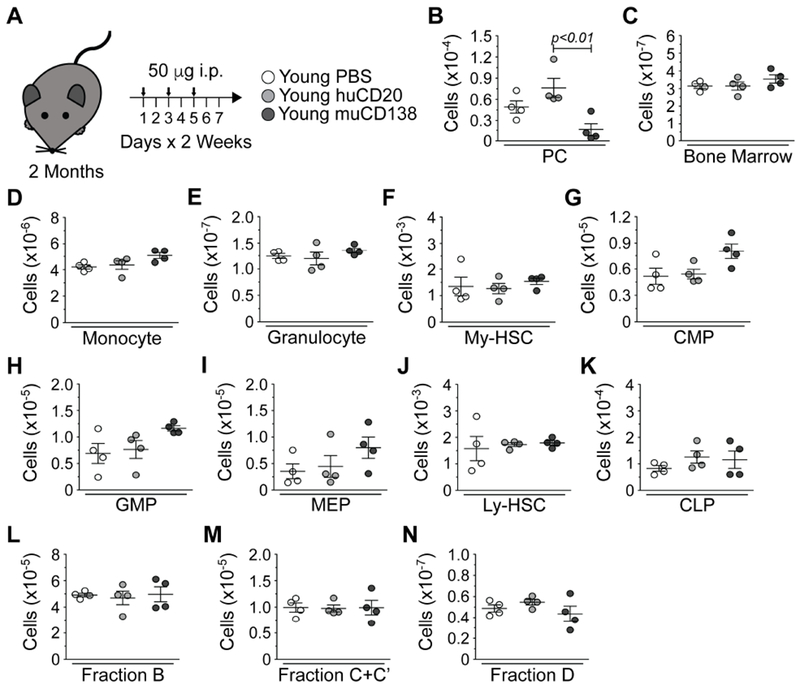
(A) Schematic of antibody-mediated depletion of PCs in young mice. Arrows indicate days of injection. (B) Numbers of PCs in PBS, huCD20 and muCD138 treated young mice. (C) Total BM cellularity in PBS, huCD20 and muCD138 treated young mice. Numbers of (D) monocytes, (E) granulocytes, (F) My-HSCs, (G) CMPs, (H) GMPs, (I) MEPs, (J) Ly-HSCs, (K) CLPs, (L) Fraction B early pro-B, (M) Fraction C+C’ late pro-B/large pre-B and (N) Fraction D small pre-B cells in PBS, huCD20 and muCD138 treated young mice. Each symbol represents an individual mouse. Young = 4; huCD20 = 4; muCD138 = 4. Statistics: One-way ANOVA with Bonferroni’s correction.
Old PCs stimulate myelopoiesis and inhibit lymphopoiesis in vitro
An in vitro culture system was used to identify the stages of hematopoiesis sensitive to PC derived signals. PCs were purified from young and old mice (Figure S3A) and pre-incubated at a 10:1 ratio with young Ly-HSCs, My-HSCs, LKS™ myeloid progenitors (MyPros) and CLPs for 15 hours before being transferred onto green fluorescent protein (GFP)-expressing OP9 stromal layers (Figures 4A and S3). This PC:progenitor ratio was chosen based on the numbers of PCs (Figure 1) and total HSCs found in young mice.
Figure 4. Old PCs Enhance Myelopoiesis and Suppress Lymphopoiesis In Vitro.
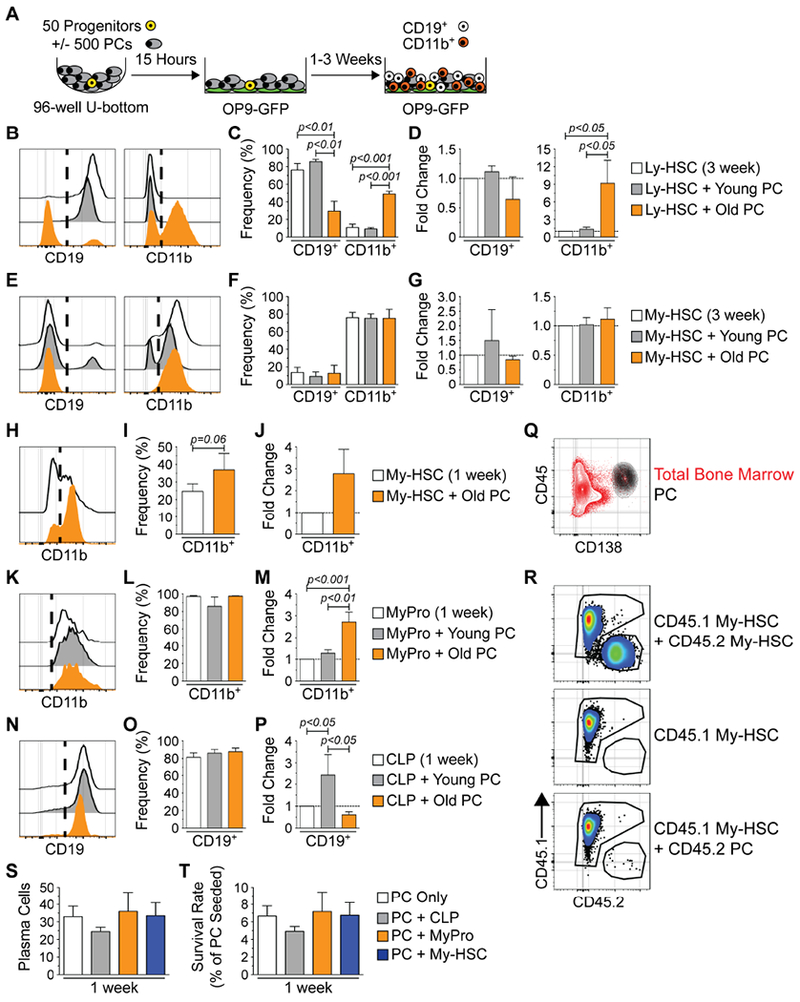
(A) Schematic depicting co-culture experiments utilizing PCs from young or old mice and hematopoietic progenitors from young mice. (B) Representative histograms of CD19 and CD11b expression on cells from 3 week cultures initiated with Ly-HSCs. (C) Frequencies and (D) fold change of CD19+ and CD11b+ cell production in 3 week cultures initiated with Ly-HSCs. (E) Representative histograms of CD19 and CD11b expression on cells from 3 week cultures initiated with My-HSCs. (F) Frequencies and (G) fold change of CD19+ and CD11b+ cell production in 3 week cultures initiated with My-HSCs. (H) Representative FACS histograms of CD11b expression in on cells from 1 week cultures initiated with My-HSCs. (I) Frequency and (J) fold change of CD11b+ cell production in 1 week cultures initiated with My-HSCs. (K) Representative FACS histograms of CD11b expression on cells from 1 week cultures initiated with MyPros. (L) Frequency and (M) fold change of CD11b+ cell production in 1 week cultures initiated with MyPros. (N) Representative FACS histograms of CD19 expression on cells from 1 week cultures initiated with CLPSs. (O) Frequency and (P) fold change of CD19+ cell production in 1 week cultures initiated with CLPs. (Q) Representative FACS contour plot depicting CD45 and CD138 expression in old BM. Gated PCs (black) are overlaid upon total bone marrow (red). (R) Representative FACS plots of CD45.1 and CD45.2 expression on cells harvested from 1 week cultures containing young CD45.1 My-HSCs and old CD45.2 My-HSCs (top panel), young CD45.1 My-HSCs only (middle panel) or young CD45.1 My-HSCs and old CD45.2 PCs (bottom panel). (S) Numbers of PCs remaining after 1 week in the indicated culture conditions. (T) Survival rate of PCs remaining after 1 week in the indicated culture conditions. Survival rate = (number of PCs detected / initial number of PCs seeded) × 100. (B, E, H, K, N) Vertical dashed lines on CD19 and CD11b histograms indicate cut-offs for positive staining. (C, D, F, G, I, J, L, M, O, P) Results are from a minimum of 3 experiments with 3-5 technical replicates per culture. Bars represent mean ± SEM. Statistics: For 3 groups, one-way ANOVA with Bonferroni’s correction. For 2 groups, Wilcoxon matched-pairs signed rank test. (D, G, J, M, P) Fold change was calculated using cell numbers derived from each culture condition (see Figure S4) as follows: (Progenitor + PC) / (Progenitor Only) = relative fold change. (S, T) Results are from 2 experiments with 3-4 technical replicates per culture. Bars represent mean ± SEM.
See also Figures S3 and S4.
Consistent with their lymphoid bias, Ly-HSCs predominantly produced CD19 expressing cells after 3 weeks in culture (Figure 4B) and addition of young PCs did not change this (Figures 4C and 4D). In contrast, the addition of old PCs to this system resulted in a reduced frequency of CD19+ and an increased proportion of CD11b+ cells (Figure 4C). As shown in Figures 4D and S4A, the production of CD11b+ cells from Ly-HSCs was stimulated approximately 9-fold. My-HSCs primarily produced CD11b+ myeloid cells in this culture system (Figures 4E and 4F), and PCs also enhanced their production but with different kinetics compared to Ly-HSCs. Unlike Ly-HSCs, PCs had little impact on My-HSC-derived cultures at 3 weeks post initiation (Figures 4F, 4G and S4B). However, old PCs increased myeloid output from My-HSCs at 1 week following culture initiation (Figures 4H–J and S4C).
We also investigated the ability of PCs to affect cell production from committed progenitors following 7 days in culture. The vast majority of cells produced by myeloid progenitors (MyPros), which include CMPs and GMPs, were CD11b+ (Figures 4K and 4L). CD11b+ cell production in old PC containing cultures increased approximately 3-fold, while young PCs did not affect production (Figures 4M and S4D). We cultured PCs with B lineage specified CLPs, which predominantly generate CD19+ committed B lineage cells after 1 week of culture (Figures 4N and 4O). The addition of old PCs to the cultures inhibited CD19+ cell production by approximately 40% (Figures 4P and S4E). Young PCs stimulated the production of CD19+ cells in this system (Figures 4P and S4E).
It was possible that PCs acquired expression of CD11b and expanded in the above cultures (Figures 4B–M), leading to an overestimation of myeloid cell production. To exclude this possibility, co-cultures were initiated with BM PCs, which expressed high levels of CD45 (Figure 4Q), from CD45.2 old mice and stem and progenitor cells were isolated from young CD45.1 mice. To validate our detection strategy, we also performed co-cultures with young CD45.1+ My-HSCs and old CD45.2+ My-HSCs. As expected, both congenic markers are readily detectable in co-cultures containing both CD45.1+ and CD45.2+ My-HSCs (Figure 4R, top panel), and cultures containing only CD45.1+ My-HSCs were negative for CD45.2 staining (Figure 4R, middle panel).
The cultures initiated with old CD45.2+ PCs and young CD45.1+ My-HSCs primarily contained CD45.1+ cells and only a few CD45.2+ PCs (Figure 4R, bottom panel). That result is in accord with a previous study showing that mature PCs are non-proliferative (Chernova et al., 2014). On average, 33 of the originally 500 seeded PCs were recovered after 1 week of culture on OP9 stroma (Figure 4S), and we observed an approximately 6.4% survival rate (Figure 4T) after 1 week that is consistent with previous studies of in vitro PC survival (Minges Wols et al., 2002). Thus, the vast majority of cells recovered in our culture system are derived from the CD45.1+ hematopoietic progenitors that were seeded. Furthermore, old PCs maintained their ability to suppress lymphopoiesis and enhance myelopoiesis in these culture conditions (Figures S4F–H).
Old PCs are primed for Toll-like receptor signaling
RNA sequencing (RNA-seq) was performed to identify differences between young and old PCs (>99% pure; Figure S3). We used SaVanT (Lopez et al., 2017) to compare our PC transcriptional signatures against those contained within the Immunological Genome Project (ImmGen) and Haemopedia databases (Figure S5). Our PC-derived transcriptional footprints were most enriched for B cell gene expression signatures from both databases. We also compared our data against already published RNA-seq analyses of in vitro-derived PCs as well as FACS purified PCs from the SPL and BM (Lam et al., 2018; Shi et al., 2015) (Figure S5). Again, this comparison demonstrated that our datasets strongly resembled the PC signatures obtained in those studies.
1413 protein-coding (non-Ig) genes were differentially expressed between young and old PCs (adjusted p-value < 0.05, Log2 fold change > |1.0|) (Figures 5A and 5B and Table S1), which represented ~10% of the total number of detectable genes (13,638). Of these, the expression of 939 genes increased with age whereas 474 genes exhibited decreased expression in old PCs (Figures 5A and 5B and Table S1). Metascape analysis (Tripathi et al., 2015) revealed that processes related to pathogen responses were enriched for in genes with increased expression in old PCs (Table S2 and Figure 5C). Within these networks, we found increased expression of transcripts for multiple pathogen sensors that included Naip2, Naip6, Nod2 and Toll-like receptors (TLRs) such as Tlr4, Tlr6 and Tlr7 (Figure 5D). We also observed that old PCs had enhanced expression of various effector molecules, such as Mapk3 (Erk1), Elk1 and Tbk1, involved in propagating responses to pathogen-associated molecular patterns (PAMPs) and host-derived damage-associated molecular patterns (DAMPs) (Table S1) (Kawai and Akira, 2010; Takeuchi et al., 1999). In addition, expression of the gene encoding IKKα (Chuk) was increased in old PCs (Table S1). IKKα complexes with IKKβ and NEMO to induce the degradation of IκB resulting in NF-κB activation (Kawai and Akira, 2010). Tnfaip3 (A20), which prevents excessive TLR signaling and subsequent inflammation (O’Reilly and Moynagh, 2003) also exhibited reduced expression in old PCs (Table S1).
Figure 5. Old PCs Possess a Toll-like Receptor Responsive Gene Signature.
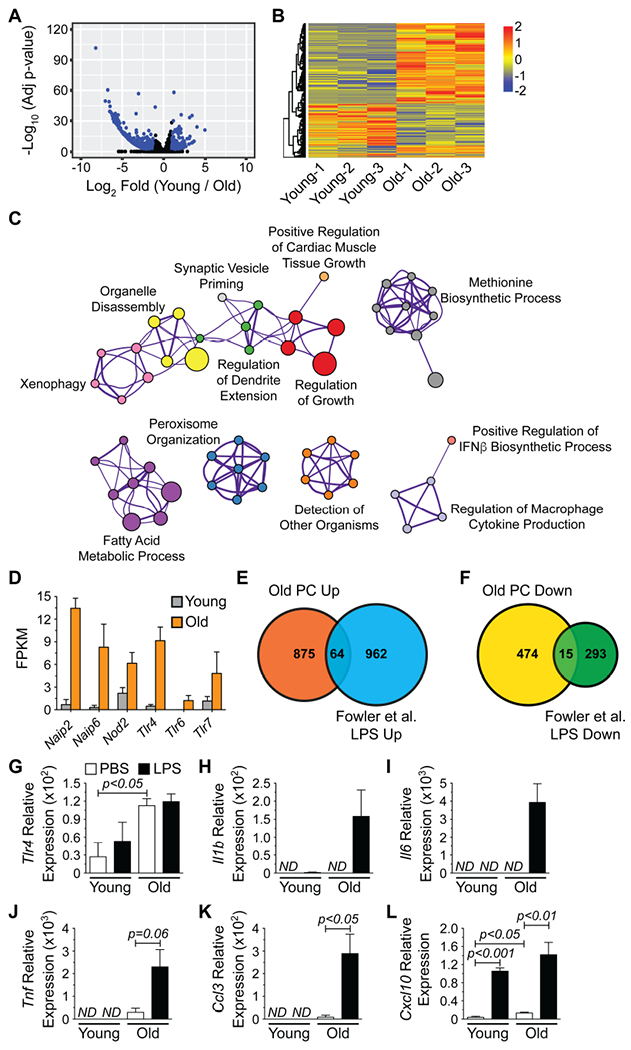
(A) Volcano plot depicting RNA-seq data from young and old PCs. All expressed genes are shown and blue dots indicate genes showing significant (adjusted p-value < 0.05, Log2 fold change > |1.0|) alterations in their expression levels. (B) Heatmap of genes significantly altered between young and old PCs. (C) Cytoscape-generated network diagram summarizing GO analysis performed on genes with increased expression in old PCs using Metascape. Nodes with the same color are specific ontologies in the same GO generic class and are labeled using a representative member. Node size is proportional to the number of genes per category. Edge thickness is proportional to between-node similarity (Kappa similarity >0.3, Metascape) and reflects the overlap between the gene sets annotated in both ontology terms. (D) Fragments per kilobase of transcript length per million reads (FPKM) for pathogen receptors with significantly increased expression in old PCs. Genes were identified from (C). Bars represent mean ± SEM derived from RNA-seq data. Venn diagrams illustrating the number of genes with (E) increased or (F) decreased expression in both old PCs and LPS-stimulated B cells from (Fowler et al., 2015). (G-L) qPCR analysis for (G) Tlr4, (H) Il1b (I) Il6, (J) Tnf, (K) Ccl3 and (L) Cxcl10 expression by young and old PCs treated with PBS or LPS (1 μg/mL) for 2 hours in vitro. Results are normalized to Gapdh expression. ND = not detected. Bars represent mean ± SEM from 3 independent experiments.
These data indicated that old PCs have the potential to respond to pathogen components such as bacterial secretion systems (Naip2) (Kofoed and Vance, 2011), flagellin (Naip6) (Kofoed and Vance, 2011), peptidoglycan (Nod2 and Tlr6) (Girardin et al., 2003; Nakao et al., 2005), lipopolysaccharide (LPS) (Tlr4) (Qureshi et al., 1999) and viral single-stranded ribonucleic acid (ssRNA) (Tlr7) (Lund et al., 2004). We further analyzed our data set for the presence of a TLR responsive gene signature and focused on TLR4 as LPS, a TLR4 ligand, increases in the circulation with age (Kim et al., 2016; Thevaranjan et al., 2017) and TLR4 was the most highly overexpressed TLR by old PCs. We compared our data to published lists of protein coding genes whose expression is increased and decreased in B cells following 2 hours of LPS stimulation (Fowler et al., 2015). Expression of sixty-four genes, including the chemokines Ccl3 and Cxcl10, was increased in both old PCs and LPS-stimulated B cells while expression of 15 genes was decreased in common (Figures 5E and 5F and Table S3). Additionally, we observed increased expression of LPS-inducible genes such as Ccl6 and Hdc (Das et al., 2017) in old PCs that did not overlap with the (Fowler et al., 2015) dataset (Tables S1 and S3). Overall, these data suggested that old PCs are more highly activated than young PCs due to TLR-driven responses.
We assessed expression of genes known to be activated by TLR4 signaling in PCs from both young and old mice following treatment in vitro with 1 μg/mL of LPS for 2 hours in order to provide additional support for the above conclusion. This treatment had no effect on Tlr4 levels (Figure 5G), but LPS treated old PCs exhibited increased expression of Il1b, Il6 and Tnf (Figures 5H–5J). Additionally, Ccl3 and Cxcl10, identified from the (Fowler et al., 2015) dataset were also LPS-inducible in old PCs (Figures 5K and 5L). Based on Tlr4 expression levels, we expected young PCs to be less responsive to LPS. This was indeed the case as young PCs only increased Cxcl10 levels following stimulation with LPS (Figures 5H–5L).
Old PCs regulate pro-inflammatory gene expression by BM stroma
The above data demonstrated that PCs have the potential to produce cytokines that may directly act on HSPCs (King and Goodell, 2011; Mirantes et al., 2014), but they could also alter the function of other BM microenvironmental constituents. We hypothesized that the BM stromal cells were a likely candidate as both HSPCs (Cordeiro Gomes et al., 2016; Sugiyama et al., 2006) and PCs (Minges Wols et al., 2002; Mokhtari et al., 2015; Zehentmeier et al., 2014) localize near stromal cells and are regulated by stromal-derived factors. Additionally, PCs can suppress human B lymphopoiesis in vitro via stromal interactions (Tsujimoto et al., 1996).
We repeated our PC depletion experiments (Figure 2A) and FACS purified the Lin™ CD45™ CD31™ stromal compartment from the BM of young, huCD20 and muCD138 treated old mice (Figure 6A). This purification strategy yields a heterogeneous mix of “stroma” inclusive of mesenchymal stem cells and osteoblast-lineage cells (Hu et al., 2016; Schepers et al., 2012). We then determined the transcriptional profiles for various cytokines and chemokines using an 84-gene Mouse Inflammatory Response and Autoimmunity PCR array. Six genes age-regulated independently of PCs were identified (Figure 6B). In contrast, 22 stromal genes that were PC-regulated were detected (Figure 6C). Hematopoietic regulatory factors whose increased expression required the presence of PCs included Csf1 (M-CSF) and Il1b, both of which are known to promote and suppress the development of myeloid and B lymphoid cells, respectively (Dorshkind, 1988a, b; Kennedy and Knight, 2015, 2017; Mossadegh-Keller et al., 2013; Pietras et al., 2016). In addition, we observed PC dependent expression of the inflammatory molecule Tnfsf14, or LIGHT (Kim et al., 2011; Shaikh et al., 2001). PCs were also obligate for increased stromal-derived expression of Il23 which regulates PC function (Cocco et al., 2011). Finally, we observed altered expression of Tirap, Ly96 (MD-2), Tlr4, Tlr5 and Tlr7 by stromal cells upon the removal of PCs suggesting that PCs regulated the capacity of the stroma to respond to pro-inflammatory stimuli (Zhou et al., 2016; Ziegler et al., 2016).
Figure 6. Old PCs Regulate Inflammatory Gene Expression in Stromal Cells.
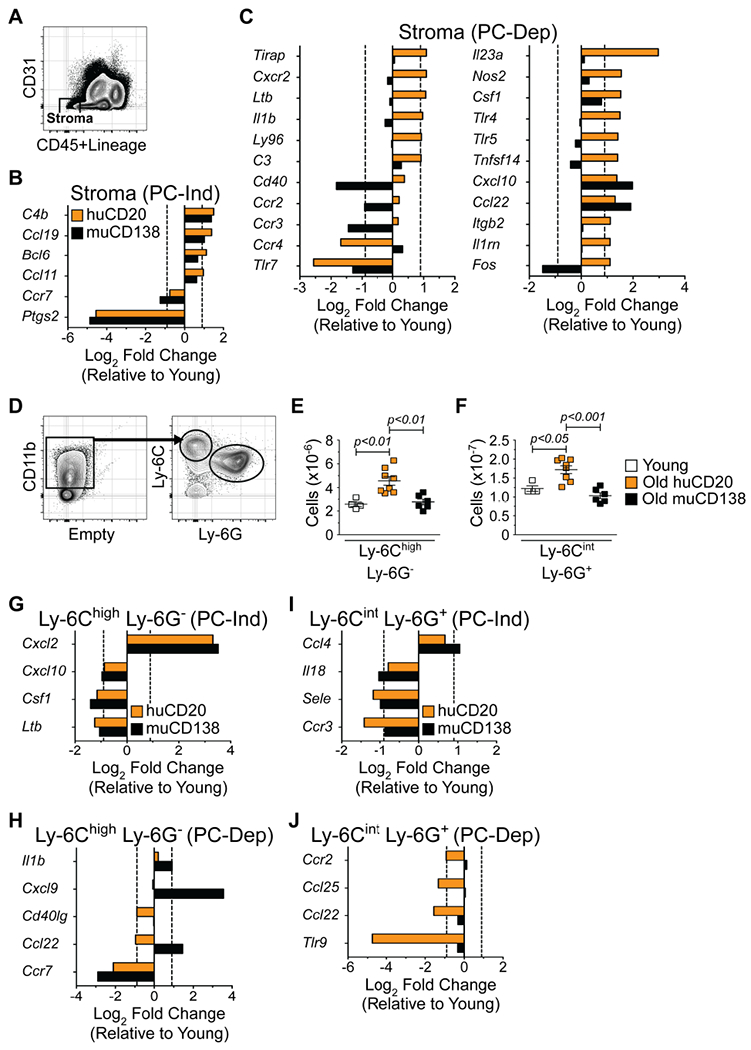
(A) Representative FACS plot depicting purification strategy of Lin™ CD45™ CD31™ bone marrow stromal cells. Genes differentially expressed by young and old stroma (B) independent of PCs (PC-Ind) and (C) dependent on PCs (PC-Dep). (D) Strategy for purification of BM Ly-6Chigh Ly-6G™ inflammatory monocytes and Ly-6Cint Ly-6G+ granulocytes. Numbers of (E) Ly-6Chigh Ly-6G™ inflammatory monocytes and (F) Ly-6Cint Ly-6G+ granulocytes in BM of young, huCD20 and muCD138 treated old mice. Each symbol represents an individual mouse. Animals used: Young = 4; huCD20 = 8; muCD138 = 6. Statistics: One-way ANOVA with Bonferroni’s correction. Genes differentially expressed by young and old Ly-6Chigh Ly-6G™ inflammatory monocytes (G) independent of PCs (PC-Ind) and (H) dependent on PCs (PC-Dep). Genes differentially expressed by young and old Ly-6Cint Ly-6G+ granulocytes (I) independent of PCs (PC-Ind) and (J) dependent on PCs (PC-Dep). Bars represent average of 2 independent experiments and show average Log2 fold changes in gene expression of samples isolated from huCD20 (orange) and muCD138 (black) treated old mice relative to their young counterparts. Sort purities of stromal cells, monocytes and granulocytes were routinely greater than 99%.
Recent reports suggest that mature myeloid cells such as macrophages (Luo et al., 2018) and granulocytes (Chen et al., 2017; Fang et al., 2018) can act on upstream progenitors. In addition, Ly-6Chigh inflammatory monocytes increase in number with age and contribute to the enhanced systemic levels of IL-6 and TNF-α (Puchta et al., 2016). We found that Ly-6Chigh Ly-6G™ inflammatory monocytes and Ly-6Cint Ly-6G+ granulocytes, which dominated the CD11b+ fraction within the BM (Figure 6D), increased with age and were significantly decreased upon the removal of PCs (Figures 6E and 6F). Of the 84 genes in our panel, only 9 were differentially expressed between young and old Ly-6Chigh Ly-6G™ monocytes and of these, 5 were PC regulated (Figures 6G and 6H). Eight genes were differentially expressed in young and old Ly-6Cint Ly-6G+ granulocytes and the expression of 4 of these was affected by PC depletion (Figures 6I and 6J). Thus, PCs did not regulate inflammatory cytokine gene expression in myeloid cells to the same degree as in the stroma.
Inflammatory cytokines stimulate myelopoiesis
The above results indicate that PCs are at the center of an inflammatory cytokine network and their depletion abrogated the enhanced myelopoiesis observed in old mice (Figure 7A). A basic tenet of this model is that the age-associated enhancement of myelopoiesis is driven by increased inflammation in the environment. In this case, a prediction is that young and old HSCs would exhibit similar myelopoietic capacity when old HSCs are removed from the BM. To test this, we FACS purified My-HSCs from the BM of young and old mice and performed colony assays. As predicted, young and old My-HSCs generated similar numbers of colonies (Figure 7B). We also examined the colony forming potential of young and old CMPs in view of the fact that they are under PC regulation (Figure 2I), and this revealed that old CMPs also did not exhibit enhanced myeloid potential in vitro (Figure 7C). In fact, they generated significantly fewer colonies compared to their young counterparts.
Figure 7. IL-1 and TNF-α Blockade Attenuates Myelopoiesis in Old BM.
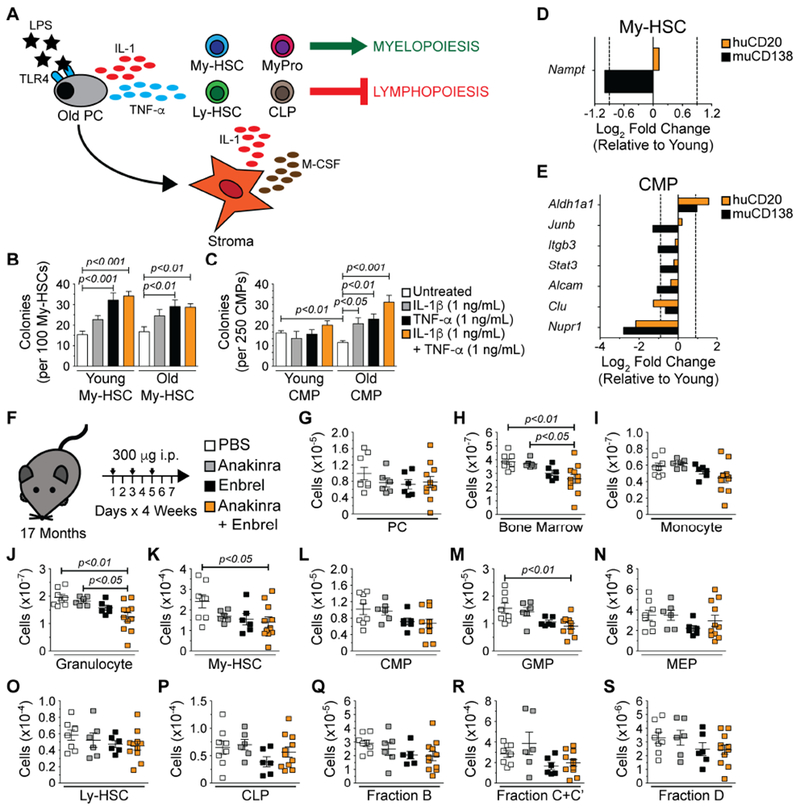
(A) Model depicting selected cytokines produced by old PCs and stromal cells and their potential hematopoietic regulatory roles. Numbers of colonies generated per (B) 100 young and old My-HSCs following 10 days in culture and (C) 250 young and old CMPs following 8 days in culture. Bars represent mean ± SEM of colonies counted per individual 3.5-cm2 dish. Data are derived from 2 independent experiments with at least 3-6 technical replicates per condition. Statistics: One-way ANOVA with Dunnett’s correction for the comparison of 3 or more groups. Unpaired Student’s t-Test for the comparison of 2 groups. Gene expression in (D) My-HSCs and (E) CMPs isolated from pools of 3-5 young, huCD2o and muCD138 treated old mice using a custom Qiagen PCR array. Bars represent average of 2 independent experiments and show average Log2 fold changes in gene expression of samples isolated from huCD20 (orange) and muCD138 (black) treated old mice relative to their young counterparts. (F) Schematic depicting experimental strategy to block IL-1 and TNF-α in old mice. Numbers of (G) PCs, (H) total BM cells, (I) monocytes, (J) granulocytes, (K) My-HSCs, (L) CMPs, (M) GMPs, (N) MEPs (O) Ly-HSCs, (P) CLPs (Q) Fraction B, (R) Fraction C+C’ and (S) Fraction D B lineage cells. Symbols in G-S represent analyses of individual mice. Animals used: PBS = 8; Anakinra = 6; Enbrel = 6; Anakinra + Enbrel = 11. Statistics: One-way ANOVA with Bonferroni’s correction.
If age-associated myeloid skewing is driven by inflammation (Figure 7A), then the addition of inflammatory cytokines to these cultures should enhance myelopoiesis. Low concentrations of IL-1β (1 ng/mL) and TNF-α (1 ng/mL) alone or in combination were included in order to model the inflammatory milieu present in the bone marrow of old mice (Henry et al., 2015). IL-1β increased the numbers of colonies generated by young and old My-HSCs, but this did not reach statistical significance (Figure 7B). In contrast, TNF-α alone or in combination with IL-1β significantly enhanced colony formation by young and old My-HSCs (Figure 7B). IL-1β and TNF-α alone or in combination had no effect on colony formation from young CMPs (Figure 7C), but old CMPs were highly responsive to these cytokines (Figure 7C).
Inflammation is known to initiate signaling cascades that affect gene expression in cells (Oduro et al., 2012; Pietras et al., 2016), and this raised the possibility that PC depletion would have such effects in stem and progenitor cells. We tested this by examining how muCD138 treatment of old mice affected expression of 28 genes involved in key stem cell functions such as growth, differentiation and cytokine signaling in My-HSCs and CMPs. Twenty-two of these genes were selected from a list of 148 genes, generated by performing RNA-seq on My-HSCs from young and old mice (Montecino-Rodriguez et al., 2019). These 22 genes exhibit robust differences in levels of expression between young and old My-HSCs. The remaining 6 genes are implicated in the response of HSPCs to inflammation (Baldridge et al., 2011; Oduro et al., 2012). Only 1 gene, Nampt, an essential regulator of stress granulopoiesis (Skokowa et al., 2009), was dependent on the presence of PCs for normal expression in My-HSCs (Figure 7D). The effects of PC depletion on gene expression were more pronounced in CMPs where expression levels of 7 genes (Aldh1a1, Junb, Itgb3, Stat3, Alcam, Clu and Nupr1) were altered (Figure 7E). These included key myeloid transcription factors such as Junb (Rosenbauer and Tenen, 2007) and Stat3 (Panopoulos et al., 2002; Panopoulos et al., 2006) which exhibited decreased expression. Furthermore, expression of Itgb3 and Alcam, which regulate interactions between CMPs and the extracellular matrix, was also reduced (Choi and Harley, 2017).
IL-1 and TNF-α regulate myelopoiesis in old BM
The above results showed that old PCs are a source of cytokines known to inhibit lymphopoiesis and stimulate myelopoiesis from HSPCs (Nagai et al., 2006; Ueda et al., 2005). Furthermore, PCs regulated inflammatory cytokine gene expression in BM stroma. As a final test of the model in Figure 7A, we determined how disruption of this circuitry affected patterns of hematopoiesis by treating old mice with Anakinra and Enbrel alone or in combination to inhibit IL-1 and TNF-α, respectively (Figure 7F). While this treatment did not significantly reduce the number of PCs (Figure 7G), mice treated with both drugs had a significant reduction in BM cellularity (Figure 7H). In view of this result, we surveyed myelopoiesis and lymphopoiesis in the Anakinra plus Enbrel treated mice to identify which hematopoietic populations were affected by cytokine neutralization (Figures 7I–7S). We observed significant declines in numbers of granulocytes (Figure 7J) as well as upstream My-HSCs (Figure 7K) and GMPs (Figure 7M). Similar to what was observed upon PC depletion (Figure 2), B lymphocyte development remained suppressed in mice receiving either drug alone or in combination (Figures 7P–7S).
Discussion
We found that PCs accumulated in aged BM and that PC depletion attenuated myelopoiesis. Our findings suggest that PCs are key members of the hematopoietic microenvironment and that their accumulation in the BM is responsible for age-related myeloid skewing. These results do not exclude the possibility that other cellular changes in the old environment contribute to the stimulation of myelopoiesis. However, we found that PCs played an obligate role in doing so, and that redundant mechanisms that sustain increases in myeloid cell production did not exist in their absence. While some age-related changes in HSC behavior may be cell intrinsic, our data also establish that myeloid skewing primarily results from stem and progenitor cell extrinsic events. Finally, the fact that PC depletion restored myelopoiesis but not lymphopoiesis to levels in young animals indicates that these two major changes during aging hematopoiesis are separable events. In particular, the results discount the possibility that when myelopoiesis expands, declines in lymphocyte production occur because the increased numbers of myeloid progenitors outcompete lymphoid progenitors for access to marrow niches.
PC depletion reduced My-HSC number in old mice. This result, in conjunction with the in vitro colony forming studies showing that inflammatory factors stimulate myelopoiesis, strongly suggests the expansion of My-HSCs, myeloid progenitors and mature myeloid cells in aging BM results from their response to inflammatory cytokines. In contrast, PC depletion in old mice did not rejuvenate lymphopoiesis. This was not due to the fact that longer times are needed for that process to re-establish as we have previously shown (Dorshkind, 1991). However, the in vitro studies showed that PCs may contribute to the inhibition of that process. This result is consistent with a report that PCs can inhibit human pre-B cell survival in short term cultures (Tsujimoto et al., 1996). Thus, in contrast to increases in myelopoiesis, redundant mechanisms that suppress lymphocyte development during aging exist, and rejuvenation of lymphocyte development in old mice may be dependent on addressing each of these. Serial treatment of old mice with a cocktail of antibodies to CD19, CD45R(B220) and CD20 can rejuvenate B cell development (Keren et al., 2011). However, this procedure may deplete PCs (Chernova et al., 2014) as well as other populations such as B220+ natural killer cells (King et al., 2009) and age-associated B cells (ABCs) (Hao et al., 2011; Ratliff et al., 2013) that can inhibit B lymphopoiesis.
The effects of PCs in old mice are not simply the result of their increased number, because young PCs did not stimulate myelopoiesis or inhibit lymphopoiesis in vitro. Differences between young and old PCs were apparent when their transcriptomes were compared. In this regard, old PCs had acquired a TLR responsive gene signature and heightened responses to TLR ligands in vitro, raising the possibility that their activity is influenced by systemic events. Levels of circulating TLR4 and TLR2 ligands such as LPS (Kim et al., 2016) and muramyl dipeptide (MDP) (Thevaranjan et al., 2017), respectively, increase with age as a consequence of breakdown in the regulation of intestinal permeability (Buford, 2017; Qi et al., 2017). Additionally, microbiota derived from old mice enhance levels of systemic TNF-α when transferred into young recipients (Thevaranjan et al., 2017). Thus, age-related events occurring in extramedullary tissues may have significant effects on microenvironmental cells in the BM.
The production of inflammatory cytokines and chemokines increases with age (Ergen et al., 2012; Henry et al., 2015), and these factors can bind to receptors on HSPCs and stimulate myelopoiesis and inhibit lymphopoiesis (Dorshkind, 1988a, b; Kennedy and Knight, 2015, 2017; Maeda et al., 2005; Pietras et al., 2016). Our results demonstrating that PCs express genes encoding cytokines that include IL-1, IL-6 and TNF-α indicated that they contribute to the process of inflammaging and that PC derived factors may directly promote myelopoiesis as well as inhibit lymphocyte development from HSPCs. However, our in vitro systems were dependent on the presence of stromal cells, raising the possibility that indirect effects mediated by other microenvironmental elements also occurred. In support of this, transcriptional analysis of stroma isolated from young, huCD20 and muCD138 treated old mice revealed an age dependent increase in the expression of myeloid promoting cytokines such as Il1b and Csf1 that was abolished upon PC depletion. Monocytes and granulocytes are also a source of inflammatory cytokines, but PC depletion had only a minimal effect on inflammatory gene expression in these populations. Thus, while PCs have the potential to regulate various cellular components of the aging BM environment, the degree to which they do so varies significantly according to cell type.
Our data suggest a model in which PCs secrete inflammatory cytokines and regulate their production from stromal cells. We validated aspects of this model by blocking IL-1 and TNF-α in old mice with Anakinra and Enbrel, respectively, and found a significant diminution of myelopoiesis. Not all Anakinra and Enbrel treated mice exhibited changes in hematopoiesis, suggesting that we did not fully disrupt the circuitry presented in Figure 7A. This is not unexpected given the wide array of inflammatory factors that exist such as IL-6 (Kayaba et al., 2018). Nevertheless, neutralization of only two inflammatory cytokines had significant effects on the age-related increase in myelopoiesis. Ultimately, these data provide support for inflammaging in regulating patterns of hematopoiesis, and myelopoiesis in particular, in the aged bone marrow environment (Kovtonyuk et al., 2016).
Studies showing that HSCs from old mice exhibit myeloid skewing and attenuated lymphopoiesis following transplantation into irradiated young recipients have led to the view that HSC intrinsic events underlie age-related changes in hematopoiesis (de Haan and Lazare, 2018). However, we recently reported that old Ly-HSCs exhibit normal lymphoid developmental potential and no myeloid skewing when transplanted into busulfan conditioned mice. The fact that irradiation induces high levels of inflammation while busulfan does not may explain why our results differ from previous studies as discussed (Montecino-Rodriguez et al., 2019). These recent observations indicate that changes in the old environment, rather than intrinsic stem cell defects, are primarily responsible for alterations in patterns of hematopoiesis with age. It is for these reasons that the current study is significant, because it identified a key age-related change in the microenvironment that alters hematopoietic development. Additional studies are needed to define how signals in the old PC inflammatory network, as well as other age-related changes in the hematopoietic microenvironment affect HSC expansion and differentiation. Our data also demonstrated that events acting at the level of lymphoid and myeloid progenitors also contribute to the decline in lymphopoiesis and increased myelopoiesis. Thus, a full understanding of how aging affects hematopoiesis will need to analyze a wide spectrum of HSPCs.
In summary, we found that the accumulation and functional alteration of PCs is a major age-related change in the hematopoietic microenvironment and that PCs played an obligate role in the stimulation of myelopoiesis and contributed to declines in lymphopoiesis. It will be important to determine if various interventions such as caloric restriction (Tang et al., 2016), CCL5 blockage (Ergen et al., 2012) and clearance of senescent cells (Chang et al., 2016), which reduce My-HSC number in old mice, act via effects on PC number and/or function. These issues aside, the data herein indicate that targeting extrinsic signals may be a relevant strategy in reversing some of the deleterious effects of aging on hematopoiesis. It is also relevant that genetic alterations in the BM niche can lead to the development of myeloproliferative disorders (Hoggatt et al., 2016; Walkley et al., 2007a; Walkley et al., 2007b). It will be of significant interest to determine if PCs play any role in this process.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth Dorshkind (kdorshki@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
All mice were of the C57BL/6J strain and CD45.2 positive unless otherwise noted. Young (2–3 months) and middle-aged (9-10 months) mice were purchased from the Jackson Laboratory or the UCLA Division of Laboratory Animal Medicine (DLAM). Old (17-19 months) mice were provided by the National Institute on Aging aged rodent colony. Young (2-3 months) B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were purchased from the Jackson Laboratory. Both males and females were used when possible. Animal care and use were conducted according to the guidelines of the UCLA Institutional Animal Care and Use Committee. All animals were housed and/or bred in the Division of Laboratory Animal Medicine vivarium at UCLA.
METHOD DETAILS
Isolation of Spleen and Bone Marrow Tissue
All tissues were processed and collected in calcium and magnesium-free 1× phosphate buffered saline (PBS). Spleens were dissected and crushed between the frosted ends of two slides. Bone marrow was isolated from both femurs and tibias by cutting off the end of bones and flushing the marrow from the shaft using a 23-gauge needle. Cell suspensions were centrifuged for 5 minutes at 4 °C and 400g. Red blood cells were lysed by suspending cells in 3 mL of 1× red blood cell lysis buffer on ice for 3 minutes. Lysis was stopped with the addition of 7 mL of 1× PBS. Cell suspensions were counted in a hemocytometer and filtered through 70-μm nylon mesh before use.
Immunostaining
All staining procedures were performed in 1× PBS. Samples were labeled with a CD16/32 blocking antibody to eliminate non-specific binding of antibodies to cells via Fc receptors. In those samples in which fluorescence-conjugated CD16/32 antibody was used to resolve myeloid progenitors, total mouse IgG was used as a blocking reagent. All antibodies utilized are listed in the Key Resources Table. For surface staining, cells were incubated on ice for 40 minutes with the appropriate antibodies. Unbound antibodies were washed from cells with PBS and cells were resuspended in an appropriate volume of 1× PBS for flow cytometric analysis.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified CD16/32 (Clone: 93) | Thermo Fisher Scientific | Cat#: 14-0161-86 |
| Unlabeled Mouse IgG | Southern Biotech | Cat#: 0107-01 |
| CD138-Biotin (Clone: 281-2) | BD Biosciences | Cat#: 553713 |
| CD135-Biotin (Clone: A2F10) | Thermo Fisher Scientific | Cat#: 13-1351-85 |
| CD11b-Biotin (Clone: M1/70) | Thermo Fisher Scientific | Cat#: 13-0112-85 |
| Ly-51(BP-1)-Biotin (Clone: 6C3) | BD Biosciences | Cat#: 553159 |
| CD48-FITC (Clone: HM48-1) | BioLegend | Cat#: 103404 |
| CD3ε-FITC (Clone: 145-2C11) | Thermo Fisher Scientific | Cat#: 11-0031-85 |
| CD8α-FITC (Clone: 53-6.7) | Thermo Fisher Scientific | Cat#: 11-0081-85 |
| TCRβ-FITC (Clone: H57-597) | Thermo Fisher Scientific | Cat#: 11-5961-85 |
| TCRγδ-FITC (Clone: UC7-13D5) | Thermo Fisher Scientific | Cat#: 11-5811-85 |
| NK1.1-FITC (Clone: PK136) | Thermo Fisher Scientific | Cat#: 11-5941-85 |
| Gr-1-FITC (Clone: RB6-8C5) | Thermo Fisher Scientific | Cat#: 11-5931-85 |
| Ter-119-FITC (Clone: TER-119) | Thermo Fisher Scientific | Cat#: 11-5921-85 |
| CD45R(B220)-FITC (Clone: RA3-6B2) | Thermo Fisher Scientific | Cat#: 11-0452-85 |
| IgM-FITC (Polyclonal) | Southern Biotech | Cat#: 1020-02 |
| IgA-FITC (Clone: mA-6E1) | Thermo Fisher Scientific | Cat#: 11-4204-83 |
| CD11b-FITC (Clone: M1/70) | Thermo Fisher Scientific | Cat#: 11-0112-85 |
| IgD-FITC (Clone: 11-26) | Southern Biotech | Cat#: 1120-02 |
| CD45-FITC (Clone: 30F-11) | Thermo Fisher Scientific | Cat#: 11-0451-85 |
| Ly-6C-FITC (Clone: AL-21) | BD Biosciences | Cat#: 553104 |
| IgM-PE (Polyclonal) | Southern Biotech | Cat#: 1020-09 |
| IgA-PE (Clone: mA-6E1) | Thermo Fisher Scientific | Cat#: 12-4204-83 |
| Gr-1-PE (Clone: RB6-8C5) | Thermo Fisher Scientific | Cat#: 12-5931-85 |
| CD16/32-PE (Clone: 93) | Thermo Fisher Scientific | Cat#: 12-0161-83 |
| CD150-PE (Clone: TC15-12F12.2) | BioLegend | Cat#: 115904 |
| CD45R(B220)-PerCP/Cy5.5 (Clone: RA3-6B2) | Thermo Fisher Scientific | Cat#: 45-0452-1631 |
| Sca-1-PerCP/Cy5.5 (Clone: D7) | Thermo Fisher Scientific | Cat#: 45-5981-82 |
| CD19-PE/Cy7 (Clone: eBio1D3) | Thermo Fisher Scientific | Cat#: 25-0193-82 |
| CD24(HAS)-PE/Cy7 (Clone: M1/69) | BD Biosciences | Cat#: 560536 |
| CD127-PE/Cy7 (Clone: eBioSB/199) | Thermo Fisher Scientific | Cat#: 25-1273-82 |
| Ly-6G-PE/Cy7 (Clone: 1A8) | BD Biosciences | Cat#: 560601 |
| CD117(c-Kit)-APC (Clone: ACK2) | Thermo Fisher Scientific | Cat#: 17-1172-83 |
| CD43-APC (Clone: S7) | BD Biosciences | Cat#: 560663 |
| CD45R(B220)-APC (Clone: RA3-6B2) | Thermo Fisher Scientific | Cat#: 17-0452-83 |
| Rat IgG2a, к-APC (Clone: RTK2758) | BioLegend | Cat#: 400511 |
| Blimp-1-APC (Clone: 5E7) | BioLegend | Cat#: 150007 |
| Streptavidin-APC | Thermo Fisher Scientific | Cat#: 17-4317-82 |
| Streptavidin-eFluor 450 | Thermo Fisher Scientific | Cat#: 48-4317-82 |
| CD45.2-eFluor 450 (Clone: 104) | Thermo Fisher Scientific | Cat#: 48-0454-80 |
| CD138-Brilliant Violet 421 (Clone: 281-2) | BD Biosciences | Cat#: 562610 |
| CD34-Brilliant Violet 421 (Clone: SA376A4) | BioLegend | Cat#: 152208 |
| CD11b-eVolve 605 | Thermo Fisher Scientific | Cat#: 83-0112-42 |
| Streptavidin-Brilliant Violet 605 | BioLegend | Cat#: 405229 |
| IgD-Brilliant Violet 605 (Clone: 11-26c.2a) | BioLegend | Cat#: 405727 |
| CD45R(B220)-Alexa Fluor 700 (Clone: RA3-6B2) | BioLegend | Cat#: 103232 |
| CD45.1-APC-eFluor 780 (Clone: A20) | Thermo Fisher Scientific | Cat#: 47-0453-82 |
| Unlabeled anti-mouse CD138 | Laboratory of Sherie L Morrison | N/A |
| Unlabeled anti-human CD20 | Laboratory of Sherie L Morrison | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant mouse SCF | Thermo Fisher Scientific | Cat#: PMC2115 |
| Recombinant mouse Flt-3L | R&D Biosystems | Cat#: 427-FL-025/CF |
| Recombinant mouse IL-3 | Thermo Fisher Scientific | Cat#: PMC0035 |
| Recombinant mouse TPO | Thermo Fisher Scientific | Cat#: PMC1144 |
| Recombinant mouse IL-7 | Biosource International | N/A |
| Recombinant mouse IL-6 | Thermo Fisher Scientific | Cat#: PMC0065 |
| Recombinant mouse IL-11 | R&D Biosystems | Cat#: 418-ML/CF |
| Recombinant mouse GM-CSF | R&D Biosystems | Cat#: 415-ML |
| Recombinant mouse IL-1β | Thermo Fisher Scientific | Cat#: PMC0816 |
| Recombinant mouse TNF-α | R&D Biosystems | Cat#: 410-MT |
| Anakinra (Kineret) | UCLA Pharmacy | N/A |
| Enbrel (Etanercept) | UCLA Pharmacy | N/A |
| MethoCult H4100 | STEMCELL Technologies | Cat#: 04100 |
| 4% Paraformaldehyde | Alfa Aesar | Cat#: J61899 |
| BD Perm/Wash | BD Biosciences | Cat#: 51-2091KZ |
| True-Nuclear Transcription Factor Buffer Set | BioLegend | Cat#: 424401 |
| TRIzol LS Reagent | Thermo Fisher Scientific | Cat#: 10296028 |
| LPS from Escherichia coli O55:B5 | Sigma-Aldrich | Cat#: L6529-1MG |
| Critical Commercial Assays | ||
| RNeasy Plus Micro Kit | Qiagen | Cat#: 74034 |
| RNeasy Micro Kit | Qiagen | Cat#: 74004 |
| RT2 First Strand Kit | Qiagen | Cat#: 330404 |
| RT2 Profiler PCR Array: Mouse Inflammatory Response and Autoimmunity | Qiagen | Cat#: PAMM-077Z |
| Custom RT2 Profiler PCR Array: | Qiagen | Cat#: CLAM30623 |
| TaqMan Universal PCR Master Mix, No AmpErase UNG | Thermo Fisher Scientific | Cat#: 4324018 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#: 170-8882 |
| SMARTer Stranded Total RNA-seq (Pico) Kit | Clontech | Cat#: 634411 |
| PROTOCOL Hema 3 manual staining system | Fisher Scientific | Cat#: 23-123869 |
| Deposited Data | ||
| RNA-Seq of Young and Old PCs | This paper | GEO: GSE112939 |
| RNA-Seq of Young and Old My-HSCs | Montecino-Rodriguez et al., 2019 | GEO: GSE112769 |
| RNA-Seq of In Vitro and In Vivo Derived B Cell and PC Subsets | Shi et al., 2015 | GEO: GSE60927 |
| RNA-Seq of PC Subsets | Lam et al., 2018 | GEO: GSE115858 |
| Experimental Models: Cell Lines | ||
| GFP-expressing OP9 Stromal Cells | Schmitt and Zuniga-Pflucker, 2002 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mice (Young and Middle-aged) | UCLA DLAM | N/A |
| C57BL/6J Mice (Young) | The Jackson Laboratory | JAX: 000664 |
| C57BL/6J Mice (Old) | National Institute on Aging (NIA) | N/A |
| B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) Mice (Young) | The Jackson Laboratory | JAX: 002014 |
| Oligonucleotides | ||
| Gapdh (Mm99999915_g1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Tlr4 (Mm00445273_m1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Tnf (Mm01161290_g1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Il1b (Mm00434228_m1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Il6 (Mm00446190_m1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Ccl3 (Mm00441259_g1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Cxcl10 (Mm00445235_m1) | Thermo Fisher Scientific | Cat#: 4331182 |
| Software and Algorithms | ||
| FlowJo (v10) | FlowJo | https://www.flowjo.com/ |
| Graphpad Prism (v5) | Graphpad |
https://www.graphpad.com/ scientific-software/prism/ |
| bcl2fastq2 (v2.17) | Illumina |
https://support.illumina.com/downloads/ bcl2fastq-conversion-software-v2-20.html |
| STAR ultrafast universal RNA-seq aligner v2.5.2b | Dobin et al., 2013 | N/A |
| RStudio | RStudio | https://www.rstudio.com/ |
| Metascape | Tripathi et al., 2015 |
http://metascape.org/gp/ index.html#/main/step1 |
| Cytoscape | Shannon et al., 2003 | http://www.cytoscape.org/ |
| SaVanT | Lopez et al., 2017 | N/A |
| Qiagen Data Analysis Center | Online |
https://www.qiagen.com/us/shop/ genes-and-pathways/data-analysis- center-overview-page/?akamai-feo=off |
For intracellular immunoglobulin staining, cells were stained for surface antigens and then fixed at room temperature for 20 minutes with 4% paraformaldehyde. Subsequently, cells were or were not permeabilized at room temperature for 30 minutes with 1× BD Perm/Wash Buffer in the presence of a CD16/32 blocking antibody and then stained for intracellular antigens at room temperature for 2 hours. Unbound antibodies were washed from cells with 1× BD Perm/Wash (2 times) followed by PBS (1 time) and cells were resuspended in an appropriate volume of 1× PBS for flow cytometric analysis.
For intracellular Blimp-1 staining, cells were stained for surface antigens and then fixed at room temperature for 1 hour with fixative provided in the BioLegend True-Nuclear Transcription Factor Buffer Set. Subsequently, cells were permeabilized at room temperature for 30 minutes with the provided permeabilization buffer in the presence of a CD16/32 blocking antibody and then stained for intracellular antigens at room temperature for 2 hours. Unbound antibodies were washed from cells with 1× PBS and cells were resuspended in an appropriate volume of 1× PBS for flow cytometric analysis.
Flow Cytometry and FACS-Sorting
Flow cytometry was performed on an LSRII (BD Biosciences) located in the Broad Stem Cell Research Center flow cytometry core at UCLA. Data were analyzed using FlowJo (v10) software. Cell sorting was performed with an Aria I (BD Biosciences) cell sorter located in the Jonsson Comprehensive Cancer Center flow cytometry core at UCLA. Prior to sorting, cells were resuspended at a final concentration of 2×107 cells per mL in αMEM supplemented with HEPES (25 mM), Penicillin-Streptomycin (100 U/mL), L-glutamine (2 mM), Gentamicin (50 μg/mL) and EDTA (2 mM). Cells were collected in RPMI 1640 supplemented with heat-inactivated fetal calf serum (10%), 2-ME (50 μM), Penicillin-Streptomycin (100 U/mL), L-glutamine (2 mM), Gentamicin (50 μg/mL), sodium pyruvate (1 mM), non-essential amino acids (1×) and vitamins (1×) (all from Gibco except for 2-ME which was from Sigma-Aldrich and EDTA which was from EMD Millipore).
Cytospin
FACS-sorted PCs were centrifuged onto glass slides at 350 rpm for 3 minutes using a Shandon Cytospin 3 cytocentrifuge. Slides were subsequently stained using the PROTOCOL Hema 3 manual staining system. Cells were visualized using a Leitz Laborlux D microscope equipped with a 50× H2O immersion objective and images were captured using an Olympus DP12 camera.
Liquid Cell Culture
All cell culture was conducted at 37 °C in a 5% CO2 and air incubator. During passaging, GFP-expressing OP9 (Schmitt and Zuniga-Pflucker, 2002) stromal cells were cultured in αMEM supplemented with heat-inactivated fetal calf serum (5%), Penicillin-Streptomycin (100 U/mL), L-glutamine (2 mM) and Gentamicin (50 μg/mL). All in vitro co-culture steps were carried out using RPMI 1640 supplemented with heat-inactivated fetal calf serum (10%), 2-ME (50 μM), Penicillin-Streptomycin (100 U/mL), L-glutamine (2 mM), Gentamicin (50 μg/mL), sodium pyruvate (1 mM), non-essential amino acids (1×) and vitamins (1×).
For PC:progenitor co-culture experiments, PCs were pre-incubated with 50 stem and/or progenitor cells at a 10:1 ratio for ~15 hours in wells of a 96-well U-bottom plate. Only freshly harvested PCs were used in these and all other experiments involving these cells. For My-HSCs, Ly-HSCs and myeloid progenitors, this step was performed in the presence of SCF (20 ng/mL, Thermo Fisher Scientific), Flt-3L (10 ng/mL, R&D Biosystems), IL-3 (20 ng/mL, Thermo Fisher Scientific) and TPO (10 ng/mL, Thermo Fisher Scientific). For CLPs, this step was performed in the presence SCF (20 ng/mL), Flt-3L (10 ng/mL), IL-3 (20 ng/mL) and IL-7 (10 ng/mL, Biosource International). Subsequently, the total contents of each well were transferred onto monolayers of OP9 stromal cells in 48-well plates. For long-term HSC cultures, the total contents of wells were transferred to 12-well and 6-well plates at 1 and 2 weeks post-culture initiation, respectively. Cultures initiated with Ly-HSCs were analyzed 3 weeks after initiation and cultures established with My-HSCs, myeloid progenitors and CLPs were assayed after 1 and/or 3 weeks in culture. Cultures were fed at days 2 and 4 of each week with RPMI 1640 (25% of total volume). Fold change was calculated using cell numbers derived from each culture condition (see Figure S4) as follows: (Progenitor + Old PC) / (Progenitor Only) = relative fold change.
For in vitro stimulation of PCs, 4×103 cells were resuspended at a density of 2×104 cells/mL in RPMI 1640 supplemented with PBS or LPS (1 μg/mL, Sigma-Aldrich) for 2 hours at 37 °C. Subsequently, cells were washed with RPMI 1640, centrifuged and cell pellets were flash frozen in liquid nitrogen and stored at −80 °C.
In Vitro Colony Forming Unit Assay
Colony forming potential of My-HSCs and CMPs FACS-purified from the bone marrow of young and old mice was assayed by mixing 100 My-HSCs or 250 CMPs in 1 mL of methylcellulose (MC) medium. MC medium was prepared by supplementing α-MEM with methylcellulose (1%, MethoCult H4100, STEMCELL Technologies), heat-inactivated fetal calf serum (30%), 2-ME (50 μM), Penicillin-Streptomycin (100 U/mL), L-glutamine (2 mM), Gentamicin (50 μg/mL), sodium pyruvate (1 mM), non-essential amino acids (1×) and vitamins (1×), SCF (50 ng/mL), IL-3 (30 ng/mL), IL-6 (10 ng/mL), IL-11 (10 ng/mL) and GM-CSF (20 ng/mL). In some instances, IL-1 β (1 ng/mL) and/or TNF-α (1 ng/mL) were included in the MC medium. The mixture was plated in nontissue culture treated 3.5-cm2 dishes (Becton Dickinson). Plates were incubated at 37 °C in a 5% CO2 and air incubator and colonies were counted following 8 days (CMPs) or 10 days (My-HSCs) of culture.
In Vivo Plasma Cell Depletion and Cytokine Blockade
Young and old mice were between 2-3 months and 17-19 months of age, respectively. For plasma cell depletion experiments, mice received a total of 300 μg of antibodies specific for either anti-human CD20 (negative control) or anti-mouse CD138. Administration was split evenly among six total intraperitoneal injections (50 μg per injection) over a two-week span.
For cytokine blockade experiments, animals received intraperitoneal injections of 300 μg of Anakinra (UCLA Pharmacy), Enbrel (UCLA Pharmacy) or Anakinra and Enbrel. An additional cohort of animals received injections of an equal volume of 1× PBS. Mice received twelve injections over the course of one month.
RNA-Seq
PCs were FACS-sorted from 3 pools of 2-4 young (2-3 months, female) and old (17-19 months, male) mice. Each pool consisted of ~5193 and ~10,903 PCs for young and old mice, respectively. Following isolation, cells were resuspended in 750 μL TRIzol LS Reagent, flash frozen in liquid N2 and stored at −80 °C until further processing. RNA isolation, preparation of sequencing libraries and sequencing was performed at the Technology Center for Genomics and Bioinformatics located at UCLA. RNa was isolated using a Qiagen RNeasy Plus Micro Kit and sequencing libraries were prepared with the Clontech SMARTer Stranded Total RNA-seq (Pico) Kit. The library preparation workflow consisted of first-strand cDNA synthesis, template switching, adaptor ligation and cleavage of ribosomal cDNA and PCR amplification. Unique adaptors were utilized for each library to facilitate multiplexing of samples. One hundred fifty base pair paired-end sequencing was performed using 3 lanes on an Illumina Hiseq3000. Data quality was verified with Illumina SAV and de-multiplexing was performed with Illumina bcl2fastq2 v2.17.
The STAR ultrafast universal RNA-seq aligner v2.5.2b (Dobin et al., 2013) was used to generate the genome index and perform paired-end alignments. Reads were aligned to a genome index that includes both the genome sequence (GRCm38 primary assembly) and the exon/intron structure of known gene models (Gencode M12 genome annotation). Alignment files were used to generate strand-specific, gene-level count summaries with the built-in gene counter in STAR. Technical replicates showed high reproducibility and were pooled. Only protein-coding genes in the Gencode M12 annotation were considered (85% of total counts on average). Independent filtering was applied as follows: genes with no counts in any sample, count outliers or low mappability were filtered out for downstream analysis (Casero et al., 2015). Counts were normalized per-sample in units of fragments per kilobase of transcript length per million reads (FPKM) after correcting for gene mappable length and sample total counts. Differential expression analysis was performed with DESeq2 (Love et al., 2014). Data are deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE112939. The table of expression estimates (FPKM) was used as input for SaVanT (Lopez et al., 2017) to compute enrichment scores on two different databases of mouse gene expression signatures (ImmGen (http://immgen.org) and Haemopedia (www.haemosphere.org)) as well as previously published RNA-seq analyses of in vitro-derived PCs (GSE60927) as well as FACS purified PCs (GSE115858) from the spleen and bone marrow (Lam et al., 2018; Shi et al., 2015). Non-default parameters for SaVanT were “Convert matrix values to ranks” and “Compute null distribution with 10000 iterations”.
Volcano plots (all expressed protein-coding genes minus immunoglobulin and Y-chromosome genes) and heatmaps (all significantly altered genes) were generated using R Studio. Gene ontology (GO) analysis of differentially expressed genes was performed using Metascape (Tripathi et al., 2015). Cytoscape (Shannon et al., 2003) was used to generate and visualize network interactions. Nodes with the same color are specific ontologies in the same GO generic class and are labeled using a representative member. Node size is proportional to the number of genes per category. Edge thickness is proportional to between-node similarity (Kappa similarity > 0.3, Metascape) and reflects the overlap between the gene sets annotated in both ontology terms.
qPCR
RNA was isolated from ~4×103 PCs using a Qiagen RNeasy Plus Micro Kit. Synthesis of cDNA was performed with the Qiagen RT2 First Strand Kit. Reactions were performed according to manufacturer instructions except for the extension step, which was performed at 42 °C for 1 hour. qPCR was performed according to manufacturer instructions using a TaqMan Universal PCR Mix with no AmpErase UNG (Thermo Fisher Scientific). Details for Gapdh, Tlr4, Tnf, Il1b, Il6, Ccl3 and Cxcl10 TaqMan primers can be found in the Key Resources Table. qPCR reactions were carried out on a Bio-Rad MyiQ Single-Color Real-Time PCR Detection System using iQ5 software. Reactions were run in triplicate in 96-well plates. Each plate was run in duplicate and PCR efficiencies were 95% to 105% as validated by a Gapdh standard curve. Data are presented as 2(GapdhCT – TargetCT).
For transcriptional profiling of bone marrow stroma (~1×106 cells), Ly-6Chigh Ly-6G™ inflammatory monocytes (~1×105 cells), Ly-6Cint Ly-6G+ granulocytes (~1×106 cells), My-HSCs (~2×103 cells) and CMPs (~4×103 cells), RNA and cDNA was generated as above. Stroma, Ly-6Chigh Ly-6G™ inflammatory monocytes and Ly-6Cint Ly-6G+ granulocytes were assayed with a Qiagen RT2 Profiler PCR Array for Mouse Inflammatory Response and Autoimmunity (Catalogue #: PAMM-077Z).
My-HSCs and CMPs were assayed with a custom Qiagen RT2 Profiler PCR Array that included probes for Mab21l2, Selp, Nuprl, Clu, Aldhlal, Plscrl, Itgb3, Sdpr, Cd74, Alcam, Plk2, Clecla, Cst3, Klhl4, Sultlal, Kctd12, Nampt, Tyk2, Socs3, Gata1, Vwf, Klf1, S100a8, Cd34, Tet1, Cd24a, Junb, Stat3, B2m (housekeeping) and Gapdh (housekeeping). qPCR was performed using iQ SYBR Green Supermix and reactions were carried out on a Bio-Rad MyiQ Single-Color Real-Time PCR Detection System using iQ5 software. Data were analyzed using the Qiagen online data resource center. Two biological replicates were performed per experiment and all data was normalized to B2m expression. Genes were considered significantly different if samples from either huCD20 or muCD138 treated old mice had a Log2 fold change ≥ |0.9| relative to samples derived from young mice. Furthermore, genes were considered to be PC-regulated if the ΔLog2 fold change (huCD20 – muCD138) ≥ |0.6|. Only genes that had consistent positive (or negative) signals for all replicates as well as a standard deviation of the ΔCT (B2m - gene of interest) < 2 for a particular cell subset were analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
The numbers of mice used or replicates performed per experiment are listed in the figure legends. Statistical analyses were performed using GraphPad Prism (v5) software. An unpaired Student’s t-Test or Wilcoxon matched-pairs signed rank test when appropriate was used for statistical comparisons between 2 groups. For comparisons among 3 or more groups, a one-way ANOVA with Bonferroni’s or Dunnett’s correction was utilized. Statistically significant p-values are shown within each figure.
Supplementary Material
Table S1 (Related to Figure 5). Protein Coding Genes Differentially Expressed Between Young and Old Plasma Cells
Table S2 (Related to Figure 5). Metascape Gene Ontology Analysis of Protein Coding Genes Differentially Expressed Between Young and Old Plasma Cells
Table S3 (Related to Figure 5). Protein Coding Genes Regulated in Common Between Old Plasma Cells and Lipopolysaccharide Stimulated B Cells
Highlights.
Inflammatory cytokine producing plasma cells (PCs) increase in number in old BM
Antibody-mediated depletion of PCs from old mice reduces myeloid cell production
PCs from old mice regulate inflammatory cytokine gene expression in BM stroma
Treatment of old mice with IL-1 and TNF-α inhibitors reduces myelopoiesis
Acknowledgements
This work was supported by NIH grant AG056480 (K.D.). S.L.M. was supported by NIH grant CA162964. P.D.P. was supported by NIH T32 training grants in Tumor Cell Biology (CA009056) and Hematology (HL086345). The Jonsson Comprehensive Cancer Center flow cytometry core is supported by NIH grants CA16042 and AI28697.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Alter-Wolf S, Blomberg BB, and Riley RL (2009). Old mice retain bone marrow B1 progenitors, but lose B2 precursors, and exhibit altered immature B cell phenotype and light chain usage. Mechanisms of ageing and development 130, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, and Goodell MA (2011). Inflammatory signals regulate hematopoietic stem cells. Trends in immunology 32, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratono SR, Chu N, Richman LP, and Behrens EM (2015). Toll-like receptor 9 and interferon-gamma receptor signaling suppress the B-cell fate of uncommitted progenitors in mice. European journal of immunology 45, 1313–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, and Rossi DJ (2010). Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proceedings of the National Academy of Sciences of the United States of America 107, 5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. (2012). Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell stem cell 10, 273–283. [DOI] [PubMed] [Google Scholar]
- Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, Singh AK, Khim S, Mucci J, Liggitt D, et al. (2013). Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORgammat and Ahr that leads to IL-17 production by activated B cells. Nature immunology 14, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulais PE, and Frenette PS (2015). Making sense of hematopoietic stem cell niches. Blood 125, 2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buford TW (2017). (Dis)Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome 5, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, Luong A, Parekh C, and Crooks GM (2015). Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nature immunology 16, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, and Goodell MA (2010). Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell stem cell 6, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, and Goodell MA (2007). Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS biology 5, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Deng H, Churchill MJ, Luchsinger LL, Du X, Chu TH, Friedman RA, Middelhoff M, Ding H, Tailor YH, et al. (2017). Bone Marrow Myeloid Cells Regulate Myeloid-Biased Hematopoietic Stem Cells via a Histamine-Dependent Feedback Loop. Cell stem cell 21, 747–760 e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernova I, Jones DD, Wilmore JR, Bortnick A, Yucel M, Hershberg U, and Allman D (2014). Lasting antibody responses are mediated by a combination of newly formed and established bone marrow plasma cells drawn from clonally distinct precursors. Journal of immunology 193, 4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, and Harley BA (2017). Marrow-inspired matrix cues rapidly affect early fate decisions of hematopoietic stem and progenitor cells. Science advances 3, e1600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, and Berek C (2013). The establishment of the plasma cell survival niche in the bone marrow. Immunological reviews 251, 177–188. [DOI] [PubMed] [Google Scholar]
- Cocco C, Morandi F, and Airoldi I (2011). Interleukin-27 and interleukin-23 modulate human plasmacell functions. Journal of leukocyte biology 89, 729–734. [DOI] [PubMed] [Google Scholar]
- Conley ME, and Cooper MD (1981). Immature IgA B cells in IgA-deficient patients. The New England journal of medicine 305, 495–497. [DOI] [PubMed] [Google Scholar]
- Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, Tani-Ichi S, Schlenner S, Richie E, Rodewald HR, et al. (2016). Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 45, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GM, Jeffery E, and Morrison SJ (2017). Adult haematopoietic stem cell niches. Nature reviews. Immunology 17, 573–590. [DOI] [PubMed] [Google Scholar]
- Dang VD, Hilgenberg E, Ries S, Shen P, and Fillatreau S (2014). From the regulatory functions of B cells to the identification of cytokine-producing plasma cell subsets. Current opinion in immunology 28, 77–83. [DOI] [PubMed] [Google Scholar]
- Das A, Arifuzzaman S, Yoon T, Kim SH, Chai JC, Lee YS, Jung KH, and Chai YG (2017). RNA sequencing reveals resistance of TLR4 ligand-activated microglial cells to inflammation mediated by the selective jumonji H3K27 demethylase inhibitor. Scientific reports 7, 6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan G, and Lazare SS (2018). Aging of hematopoietic stem cells. Blood 131, 479–487. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorshkind K (1988a). IL-1 inhibits B cell differentiation in long term bone marrow cultures. Journal of immunology 141, 531–538. [PubMed] [Google Scholar]
- Dorshkind K (1988b). Interleukin-1 inhibition of B lymphopoiesis is reversible. Blood 72, 2053–2055. [PubMed] [Google Scholar]
- Dorshkind K (1991). In vivo administration of recombinant granulocyte-macrophage colony-stimulating factor results in a reversible inhibition of primary B lymphopoiesis. Journal of immunology 146, 4204–4208. [PubMed] [Google Scholar]
- Dykstra B, and de Haan G (2008). Hematopoietic stem cell aging and self-renewal. Cell and tissue research 331, 91–101. [DOI] [PubMed] [Google Scholar]
- Dykstra B, Olthof S, Schreuder J, Ritsema M, and de Haan G (2011). Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. The Journal of experimental medicine 208, 2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergen AV, Boles NC, and Goodell MA (2012). Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 119, 2500–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax KA, Kallies A, Nutt SL, and Tarlinton DM (2008). Plasma cell development: from B-cell subsets to long-term survival niches. Seminars in immunology 20, 49–58. [DOI] [PubMed] [Google Scholar]
- Fang MM, Barman PK, Thiruppathi M, Mirza RE, McKinney RD, Deng J, Christman JW, Du X, Fukai T, Ennis WJ, et al. (2018). Oxidant Signaling Mediated by Nox2 in Neutrophils Promotes Regenerative Myelopoiesis and Tissue Recovery following Ischemic Damage. Journal of immunology 201, 2414–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ, Niedernhofer LJ, and Robbins PD (2017). Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-kappaB-dependent mechanism. Aging cell 16, 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler T, Garruss AS, Ghosh A, De S, Becker KG, Wood WH, Weirauch MT, Smale ST, Aronow B, Sen R, and Roy AL (2015). Divergence of transcriptional landscape occurs early in B cell activation. Epigenetics & chromatin 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, and De Benedictis G (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences 908, 244–254. [DOI] [PubMed] [Google Scholar]
- Frasca D, Landin AM, Lechner SC, Ryan JG, Schwartz R, Riley RL, and Blomberg BB (2008). Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. Journal of immunology 180, 5283–5290. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Rojas OL, Simard N, McCarthy DD, Hapfelmeier S, Rubino S, Robertson SJ, Larijani M, Gosselin J, Ivanov II, et al. (2011). Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger H, de Haan G, and Florian MC (2013). The ageing haematopoietic stem cell compartment. Nature reviews. Immunology 13, 376–389. [DOI] [PubMed] [Google Scholar]
- Geiger H, and Van Zant G (2002). The aging of lympho-hematopoietic stem cells. Nature immunology 3, 329–333. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, and Sansonetti PJ (2003). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. The Journal of biological chemistry 278, 8869–8872. [DOI] [PubMed] [Google Scholar]
- Guidi N, and Geiger H (2017). Rejuvenation of aged hematopoietic stem cells. Seminars in hematology 54, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi N, Sacma M, Standker L, Soller K, Marka G, Eiwen K, Weiss JM, Kirchhoff F, Weil T, Cancelas JA, et al. (2017). Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. The EMBO journal 36, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, O’Neill P, Naradikian MS, Scholz JL, and Cancro MP (2011). A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, and Hayakawa K (1991). Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. The Journal of experimental medicine 173, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, and Hayakawa K (1995). B-lineage differentiation stages resolved by multiparameter flow cytometry. Annals of the New York Academy of Sciences 764, 19–24. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Casas-Selves M, Kim J, Zaberezhnyy V, Aghili L, Daniel AE, Jimenez L, Azam T, McNamee EN, Clambey ET, et al. (2015). Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. The Journal of clinical investigation 125, 4666–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, Robson SC, Frenette PS, and Fujisaki J (2018). CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell stem cell 22, 445–453 e445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J, Kfoury Y, and Scadden DT (2016). Hematopoietic Stem Cell Niche in Health and Disease. Annual review of pathology 11, 555–581. [DOI] [PubMed] [Google Scholar]
- Hu X, Garcia M, Weng L, Jung X, Murakami JL, Kumar B, Warden CD, Todorov I, and Chen CC (2016). Identification of a common mesenchymal stromal progenitor for the adult haematopoietic niche. Nature communications 7, 13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen M, Nguyen H, Rapraeger A, Kurn N, and Bernfield M (1985). Heparan sulfate proteoglycans from mouse mammary epithelial cells: localization on the cell surface with a monoclonal antibody. The Journal of cell biology 101, 976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, and Rudolph KL (2007). Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nature medicine 13, 742–747. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kayaba A, Itoh-Nakadai A, Niibe K, Shirota M, Funayama R, Sugahara-Tobinai A, Wong YL, Inui M, Nakayama K, and Takai T (2018). Bone Marrow PDGFR+Sca-1+ Enriched Mesenchymal Stem Cells Support Survival of and Antibody Production by Plasma Cells in vitro through IL-6. International immunology. [DOI] [PubMed] [Google Scholar]
- Kennedy DE, and Knight KL (2015). Inhibition of B Lymphopoiesis by Adipocytes and IL-1-Producing Myeloid-Derived Suppressor Cells. Journal of immunology 195, 2666–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DE, and Knight KL (2017). Inflammatory Changes in Bone Marrow Microenvironment Associated with Declining B Lymphopoiesis. Journal of immunology 198, 3471–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, Schmidt-Supprian M, Lapidot T, and Melamed D (2011). B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood 117, 3104–3112. [DOI] [PubMed] [Google Scholar]
- Kim HM, Jeong CS, Choi HS, Kawada T, and Yu R (2011). LIGHT/TNFSF14 enhances adipose tissue inflammatory responses through its interaction with HVEM. FEBS letters 585, 579–584. [DOI] [PubMed] [Google Scholar]
- Kim KA, Jeong JJ, Yoo SY, and Kim DH (2016). Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC microbiology 16, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, and Kim TS (2014). IgA+ plasma cells in murine intestinal lamina propria as a positive regulator of Treg differentiation. Journal of leukocyte biology 95, 461–469. [DOI] [PubMed] [Google Scholar]
- King AM, Keating P, Prabhu A, Blomberg BB, and Riley RL (2009). NK cells in the CD19− B220+ bone marrow fraction are increased in senescence and reduce E2A and surrogate light chain proteins in B cell precursors. Mechanisms of ageing and development 130, 384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, and Goodell MA (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature reviews. Immunology 11, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofoed EM, and Vance RE (2011). Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtonyuk LV, Fritsch K, Feng X, Manz MG, and Takizawa H (2016). Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Frontiers in immunology 7, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisawa J, Gohda M, Hashimoto E, Ishikawa I, Higuchi M, Suzuki Y, Goto Y, Panea C, Ivanov II, Sumiya R, et al. (2013). Microbe-dependent CD11b+ IgA+ plasma cells mediate robust early-phase intestinal IgA responses in mice. Nature communications 4, 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WY, Jash A, Yao CH, D’Souza L, Wong R, Nunley RM, Meares GP, Patti GJ, and Bhattacharya D (2018). Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell reports 24, 2479–2492 e2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, and Hauser AE (2016). Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal immunology 9, 83–97. [DOI] [PubMed] [Google Scholar]
- Lo Celso C, and Scadden DT (2011). The haematopoietic stem cell niche at a glance. Journal of cell science 124, 3529–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Montoya D, Ambrose M, Lam L, Briscoe L, Adams C, Modlin RL, and Pellegrini M (2017). SaVanT: a web-based tool for the sample-level visualization of molecular signatures in gene expression profiles. BMC genomics 18, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, and Flavell RA (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America 101, 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shao L, Chang J, Feng W, Liu YL, Cottler-Fox MH, Emanuel PD, Hauer-Jensen M, Bernstein ID, Liu L, et al. (2018). M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood advances 2, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, Kincade PW, Sakaguchi N, and Coggeshall KM (2005). IL-6 blocks a discrete early step in lymphopoiesis. Blood 106, 879–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JP, and Allman D (2003). The decline in B lymphopoiesis in aged mice reflects loss of very early B-lineage precursors. Journal of immunology 171, 2326–2330. [DOI] [PubMed] [Google Scholar]
- Min H, Montecino-Rodriguez E, and Dorshkind K (2006). Effects of aging on the common lymphoid progenitor to pro-B cell transition. Journal of immunology 176, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Minges Wols HA, Underhill GH, Kansas GS, and Witte PL (2002). The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. Journal of immunology 169, 4213–4221. [DOI] [PubMed] [Google Scholar]
- Mirantes C, Passegue E, and Pietras EM (2014). Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Experimental cell research 329, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari Z, Mech F, Zehentmeier S, Hauser AE, and Figge MT (2015). Quantitative image analysis of cell colocalization in murine bone marrow. Cytometry. Part A : the journal of the International Society for Analytical Cytology 87, 503–512. [DOI] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Berent-Maoz B, and Dorshkind K (2013). Causes, consequences, and reversal of immune system aging. The Journal of clinical investigation 123, 958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Kong Y, Casero D, Rouault A, Dorshkind K, and Pioli PD (2019). Lymphoid-Biased Hematopoietic Stem Cells Are Maintained with Age and Efficiently Generate Lymphoid Progeny. Stem cell reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, Moore J, and Sieweke MH (2013). M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 497, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Sieburg CE, Cho RH, Karlsson L, Huang JF, and Sieburg HB (2004). Myeloid-biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood 103, 4111–4118. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, and Kincade PW (2006). Toll like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takubo K, Fujioka M, and Suda T (2014). Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochemical and biophysical research communications 454, 353–357. [DOI] [PubMed] [Google Scholar]
- Nakao Y, Funami K, Kikkawa S, Taniguchi M, Nishiguchi M, Fukumori Y, Seya T, and Matsumoto M (2005). Surface-expressed TLR6 participates in the recognition of diacylated lipopeptide and peptidoglycan in human cells. Journal of immunology 174, 1566–1573. [DOI] [PubMed] [Google Scholar]
- O’Reilly SM, and Moynagh PN (2003). Regulation of Toll-like receptor 4 signalling by A20 zinc finger protein. Biochemical and biophysical research communications 303, 586–593. [DOI] [PubMed] [Google Scholar]
- Oduro KA Jr., Liu F, Tan Q, Kim CK, Lubman O, Fremont D, Mills JC, and Choi K (2012). Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood 120, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, and Weissman IL (2011). Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proceedings of the National Academy of Sciences of the United States of America 108, 20012–20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, Bartos D, Zhang L, and Watowich SS (2002). Control of myeloid-specific integrin alpha Mbeta 2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. The Journal of biological chemistry 277, 19001–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, and Watowich SS (2006). STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 108, 3682–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nature cell biology 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, and Jarrossay D (2013). A functional BCR in human IgA and IgM plasma cells. Blood 121, 4110–4114. [DOI] [PubMed] [Google Scholar]
- Poulos MG, Ramalingam P, Gutkin MC, Llanos P, Gilleran K, Rabbany SY, and Butler JM (2017). Endothelial transplantation rejuvenates aged hematopoietic stem cell function. The Journal of clinical investigation 127, 4163–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchta A, Naidoo A, Verschoor CP, Loukov D, Thevaranjan N, Mandur TS, Nguyen PS, Jordana M, Loeb M, Xing Z, et al. (2016). TNF Drives Monocyte Dysfunction with Age and Results in Impaired Anti-pneumococcal Immunity. PLoS pathogens 12, e1005368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Goel R, Kim S, Richards EM, Carter CS, Pepine CJ, Raizada MK, and Buford TW (2017). Intestinal Permeability Biomarker Zonulin is Elevated in Healthy Aging. Journal of the American Medical Directors Association 18, 810 e811–810 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi ST, Gros P, and Malo D (1999). The Lps locus: genetic regulation of host responses to bacterial lipopolysaccharide. Inflammation research : official journal of the European Histamine Research Society ... [et al. ] 48, 613–620. [DOI] [PubMed] [Google Scholar]
- Ratliff M, Alter S, Frasca D, Blomberg BB, and Riley RL (2013). In senescence, age-associated B cells secrete TNFalpha and inhibit survival of B-cell precursors. Aging cell 12, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, Tiglao E, Figueiredo JL, Iwamoto Y, Theurl I, et al. (2012). Innate response activator B cells protect against microbial sepsis. Science 335, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Kuraoka M, and Kelsoe G (2015). Natural IgM is produced by CD5-plasma cells that occupy a distinct survival niche in bone marrow. Journal of immunology 194, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F, and Tenen DG (2007). Transcription factors in myeloid development: balancing differentiation with transformation. Nature reviews. Immunology 7, 105–117. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, and Weissman IL (2007). Hematopoietic stem cell aging: mechanism and consequence. Experimental gerontology 42, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, and Weissman IL (2005). Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America 102, 9194–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, and Pronk CJ (2016). Human and Murine Hematopoietic Stem Cell Aging Is Associated with Functional Impairments and Intrinsic Megakaryocytic/Erythroid Bias. PloS one 11, e0158369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K, Hsiao EC, Garg T, Scott MJ, and Passegue E (2012). Activated Gs signaling in osteoblastic cells alters the hematopoietic stem cell niche in mice. Blood 120, 3425–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, and Zuniga-Pflucker JC (2002). Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17, 749–756. [DOI] [PubMed] [Google Scholar]
- Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, Cheroutre H, and Ware CF (2001). Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. Journal of immunology 167, 6330–6337. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, and Calame K (2003). Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19, 607–620. [DOI] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, et al. (2014). IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nature immunology 16, 663–673. [DOI] [PubMed] [Google Scholar]
- Signer RA, Montecino-Rodriguez E, Witte ON, McLaughlin J, and Dorshkind K (2007). Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 110, 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, Brechlin AM, Schambach A, Hinrichsen L, Meyer G, et al. (2009). NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nature medicine 15, 151–158. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, and Nagasawa T (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. [DOI] [PubMed] [Google Scholar]
- Suzuki-Yamazaki N, Yanobu-Takanashi R, Okamura T, and Takaki S (2017). IL-10 production in murine IgM(+) CD138(hi) cells is driven by Blimp-1 and downregulated in class-switched cells. European journal of immunology 47, 493–503. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, and Akira S (1999). Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451. [DOI] [PubMed] [Google Scholar]
- Tang D, Tao S, Chen Z, Koliesnik IO, Calmes PG, Hoerr V, Han B, Gebert N, Zornig M, Loffler B, et al. (2016). Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. The Journal of experimental medicine 213, 535–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Koh LK, Jiang D, and Schwarz H (2013). CD137 ligand reverse signaling skews hematopoiesis towards myelopoiesis during aging. Aging 5, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, and Nutt SL (2012). High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. Journal of immunology 189, 3328–3338. [DOI] [PubMed] [Google Scholar]
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. (2017). Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell host & microbe 21, 455–466 e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell host & microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Lisukov IA, Huang N, Mahmoud MS, and Kawano MM (1996). Plasma cells induce apoptosis of pre-B cells by interacting with bone marrow stromal cells. Blood 87, 3375–3383. [PubMed] [Google Scholar]
- Tung JW, Mrazek MD, Yang Y, Herzenberg LA, and Herzenberg LA (2006). Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proceedings of the National Academy of Sciences of the United States of America 103, 6293–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Kondo M, and Kelsoe G (2005). Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. The Journal of experimental medicine 201, 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, and Purton LE (2007a). A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129, 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Shea JM, Sims NA, Purton LE, and Orkin SH (2007b). Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129, 1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani R, Azizi G, Abolhassani H, and Aghamohammadi A (2017). Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scandinavian journal of immunology 85, 3–12. [DOI] [PubMed] [Google Scholar]
- Zehentmeier S, Roth K, Cseresnyes Z, Sercan O, Horn K, Niesner RA, Chang HD, Radbruch A, and Hauser AE (2014). Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. European journal of immunology 44, 2306–2317. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dai J, Lu Y, Yao Z, O’Brien CA, Murtha JM, Qi W, Hall DE, Manolagas SC, Ershler WB, and Keller ET (2004). In vivo visualization of aging-associated gene transcription: evidence for free radical theory of aging. Experimental gerontology 39, 239–247. [DOI] [PubMed] [Google Scholar]
- Zhou A, Li M, He B, Feng W, Huang F, Xu B, Dunker AK, Balch C, Li B, Liu Y, and Wang Y (2016). Lipopolysaccharide treatment induces genome-wide pre-mRNA splicing pattern changes in mouse bone marrow stromal stem cells. BMC genomics 17 Suppl 7, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler P, Boettcher S, Takizawa H, Manz MG, and Brummendorf TH (2016). LPS-stimulated human bone marrow stroma cells support myeloid cell development and progenitor cell maintenance. Annals of hematology 95, 173–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Related to Figure 5). Protein Coding Genes Differentially Expressed Between Young and Old Plasma Cells
Table S2 (Related to Figure 5). Metascape Gene Ontology Analysis of Protein Coding Genes Differentially Expressed Between Young and Old Plasma Cells
Table S3 (Related to Figure 5). Protein Coding Genes Regulated in Common Between Old Plasma Cells and Lipopolysaccharide Stimulated B Cells