

Abstract
Fluorinated organic molecules are pervasive within the pharmaceutical and agrochemical industries due to the range of structural and physicochemical properties that fluorine imparts. Currently, the most abundant methods for the synthesis of the aryl–CF2 functionality have relied on the deoxyfluorination of ketones and aldehydes using expensive and poorly atom economical reagents. Here, we report a general method for the synthesis of aryl–CF2R and aryl–CF2H compounds through activation of the corresponding trifluoromethyl arene precursors. This strategy is enabled by an endergonic electron transfer event that provides access to arene radical anions that lie outside of the catalyst reduction potential. Fragmentation of these reactive intermediates delivers difluorobenzylic radicals that can be intercepted by abundant alkene feedstocks or a hydrogen atom to provide a diverse array of difluoalkylaromatics.
Graphical Abstract
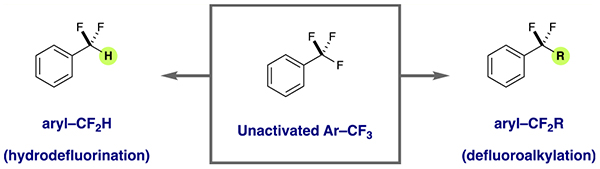
INTRODUCTION
The trifluoromethylaromatic (Ar–CF3) motif is routinely utilized in drug and agrochemical design, because its incorporation can significantly alter the properties of a given biologically active small molecule.1–4 Accordingly, a number of methods have been reported over the past decade that enable the efficient construction of Ar–CF3 compounds.5–10 Although trifluoromethyl units are traditionally unreactive, we recently became interested in selective manipulation of these compounds through a radical anion-based mechanism for carbon-fluorine (C–F) bond cleavage. We envisioned that the resulting radical intermediates could serve as precursors to a number of valuable fluorinated scaffolds, including difluoroalkyl aromatics (isosteres for aryl ethers and aryl ketones)11 and difluoromethyl arenes (lipophilic hydrogen bond donors).12–16 These difluorobenzylic arrays have emerged as important components of bioactive small molecules (two of which are shown in Figure 1). The direct molecular editing of trifluoromethylaromatics in this way would offer a valuable complement to technologies that grant access to these difluorinated motifs through construction of the CAryl–CF2R/H bond.17–23
Figure 1.

Molecular editing of Ar–CF3 groups via catalytic C–F functionalization.
Although reductive activation of the C–F bonds in this substrate class can be accomplished using a range of approaches (e.g., electrochemical reduction,24,25 low-valent metals,26–28 or frustrated Lewis pairs29,30), preventing exhaustive defluorination (thereby bypassing valuable di- and monofluorinated intermediates) remains a significant synthetic problem. This is because, in part, C–F bond strength decreases as defluorination proceeds.31 Troupel32 and Lalic33 reported two different systems for interception of anionic difluorobenzylic species through trapping with carbonyl-based electrophiles. In addition, Prakash reported a Mg0-based system for hydrodefluorination (HDF) of bis(trifluoromethyl)-benzene derivatives.26 Yoshida and Hosoya recently described an intermolecular coupling of trifluoromethyl arenes that contain ortho-silyl groups with allyl silanes, which operates through a cationic mechanism.34 While these systems allow for selective defluorofunctionalization of specific substrate classes, a general approach to Ar–CF3 functionalization remains elusive.
Our strategy is mechanistically founded on recent work from our group35 and König’s,36 where the use of highly reducing photoredox catalysts resulted in partial defluorination of activated trifluoromethylaromatic substrates (Figure 2), under irradiation with visible light. In our method, single electron transfer (SET) from an organic photoredox catalyst to the aryl substrate is followed by C–F cleavage to deliver difluorobenzylic radicals that display unique reactivity profiles. This system, ultimately propelled by oxidation of formate to CO2, effectively performs monodefluoroalkylation reactions with a wide range of unactivated olefins as alkyl sources. However, the utility of both of these systems is limited by the requirement for electronic activation of the substrates. More specifically, successful reaction has only been demonstrated for trifluoromethylaromatics that contain auxiliary electron-with-drawing groups (e.g., −CF3, −CN, −SO2NH2). Here, we describe the development of conditions that overcome this limitation, thus enabling controlled C–F functionalization of unactivated trifluoromethylaromatics. We show that these difluorobenzylic radical intermediates can be utilized selectively in both alkylation and reduction processes.
Figure 2.

Radical C–F functionalization of Ar–CF3 substrates via photoredox catalysis.
RESULTS AND DISCUSSION
We began our study by examining the reaction of benzotrifluoride (Ph–CF3) with 3-buten-1-ol to afford the defluoroalkylation (DFA) product 1 (shown in Scheme 1). After slight modification of our initially reported conditions, we evaluated a series of photoredox catalysts, a selection of which is shown in Scheme 1. While N-phenylphenothiazine (PTH, P1) was ineffective at room temperature, the use of P2–P4 afforded the desired product to varying degrees (up to 31% yield after 24 h). Increasing reaction temperature resulted in significantly improved yields of the desired linear alkylation product. The most effective catalyst (P3, developed by Miyake for organic atom-transfer radical polymerization)37 has been reported to be highly absorbent in the visible spectrum (λmax = 388 nm, εmax = 26 635 M−1 cm−1) with a 98% intersystem crossing (ISC) efficiency to a long-lived triplet excited state that is strongly reducing (E1/2* = desired products1.70 V vs standard calomel electrode (SCE)).38 The use of P3 (2 mol %) at 100 °C resulted in 89% yield of 1, as determined by NMR. Interestingly, for the organic SET catalysts (P1, P3, P4), reaction progress is not strongly correlated with reduction potential of the catalyst. Rather, conversion at higher temperatures is more closely related to excited-state lifetime. This effect is most pronounced through the use of P4 in this process, because it is significantly less reducing than P1 but equally effective at high temperature.
Scheme 1.

Activation of Stable C–F Bonds by Single-Electron Transfer using Photoredox Catalysisa
aReaction conditions: benzotrifluoride (0.25 mmol), 3-buten-1-ol (1.25 mmol), photoredox catalyst (P1: 10 mol %; P2–P4: 2 mol %), potassium formate (0.75 mmol), thiophenol (10 mol %), DMSO (2.5 mL), blue light, 24 h. Yields determined by 19F NMR analysis with internal standard.
As shown in Table 1, a collection of unactivated trifluoromethylaromatics could be engaged under these conditions. In addition to carbon and oxygen-substituted substrates, arylamine derivatives reacted smoothly to afford the desired products 2–10 in 44–92% yield. The benzylic carbinol was cleanly retained, giving alkylation product 11 in 82% yield. Pyridine substrates with trifluoromethyl groups at the 2- and 3-positions gave rise to 12–15 in 41–78% yield. These reactions were most effectively performed at room temperature, because the difluoroalkylpyridine products can be further reductively defluorinated. In fact, 4-trifluoromethylpyridine currently remains outside the scope of these processes; attempted alkylation reactions largely resulted in picoline production with only trace amounts of the desired product. Likewise, while this protocol is effective in activating electron-deficient aromatics (such as those described in our previous report35), the elevated temperature and more effective catalyst typically resulted in further defluorination of these substrates. The functionalization of marketed pharmaceuticals could also be performed under these conditions to afford derivatives 16–18 in 61%–73% yield, demonstrating that basic functions, alcohols, and internal alkenes are well-tolerated.
Table 1.
Catalytic Defluoroalkylation: Scope of Unactivated Trifluoromethylaromatic Substratesa
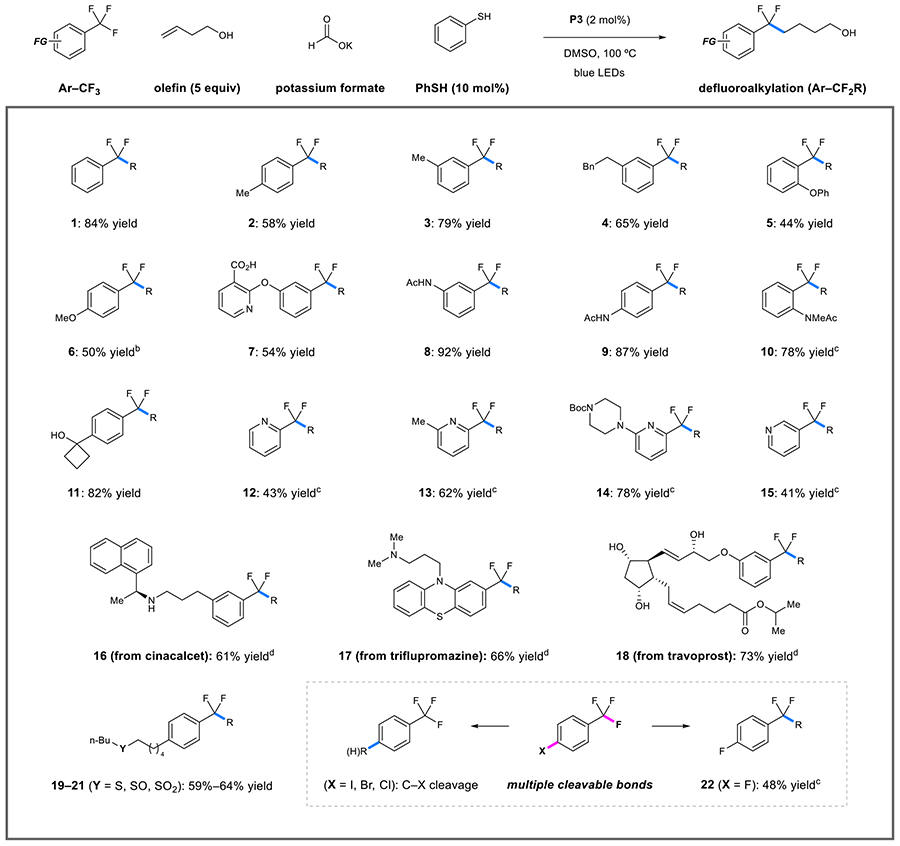
|
Reaction conditions: Ar–CF3 substrate (0.5 mmol), 3-buten-1-ol (2.5 mmol), P3 (2 mol %), potassium formate (1.5 mmol), thiophenol (10 mol %), DMSO (5.0 mL), blue light, 100 °C, 24 h, isolated yields given.
Reaction conducted with 2.5 mmol of potassium formate.
Reaction conducted at 23 °C.
Reaction conducted with 5.0 mmol of olefin.
Although these conditions are able to accomplish challenging redox events, we found that thioethers, sulfoxides, and sulfones alike were preserved throughout the course of the reaction without any observable alteration of the sulfur oxidation states (19–21, 59–64% yield). To probe the chemoselective nature of heterolytic C–X cleavage within a single π-system, we examined the reactivity of a series of 4-halogenated benzotrifluorides. From the corresponding radical anions, cleavage of the aryl C–I, C–Br, and C–Cl bonds (bond dissociation energy (BDE) = 71.6, 80.3, and 95.5 kcal/mol, respectively; see Supporting Information for details) occurred in preference to the stronger benzylic C(sp3)–F bond (BDE = 118.1 kcal/mol), resulting in mixtures of protodehalogenation and aryl radical alkylation products.39 In contrast, the aryl C–F bond (BDE = 127.5 kcal/mol) was completely preserved, affording 22 in 48% yield (where the mass balance was comprised of unreacted starting material). In addition to aryl halides, alkyl halides currently lie outside the scope of this process, where either reductive dehaogenation or substitution with formate are primary pathways of these substrates.
In line with our previous report, the olefin scope for coupling with unactivated trifluoromethylaromatics is broad, giving a range of difluoroalkylated benzene derivatives from benzotrifluoride. The use of benzotrifluoride as a precursor to difluoroalkyl benzene derivatives is attractive, because it is abundantly available at low cost (~$7/mol) when compared to conventional reagents that accomplish deoxyfluorination of aryl ketones (e.g., diethylaminosulfur trifluoride (DAST) or Fluolead).40 While the standard conditions (5 equiv of olefin with limiting Ar–CF3 substrate) were effective here (see Table 1 and Supporting Information for examples), we found that using a cosolvent quantity (50 equiv) of benzotrifluoride could be conveniently employed for coupling of simple olefins with acetate, acetal, or Weinreb amide groups (Table 2: 23–25, 64–78% yield). Other nucleophilic olefins including the vinyl ether, acetate, amides, and vinyl carbazole were also effective coupling partners under this protocol (26–30, 55–95%). β-Substitution of aliphatic olefins was also well-tolerated (31, 94% yield). Although these conditions operate at elevated temperature, decomposition through saponification or hydrolysis pathways was not observed. Ethylene, propylene, isobutylene, and 2-methyl-2-butene) are attractive two-, three-, four-, and five-carbon synthons that are produced on large scale from hydrocarbon cracking. Performing our reductive DFA method on Ph–CF3 under 25 psi of ethylene provided the desired product (33) in 65% yield (based on formate as limiting reagent). This result was replicated with propylene (34, 68% yield), isobutylene (35, 87% yield), and 2-methyl-2-butene (36, 46% yield).
Table 2.
Scope of Olefinic Coupling Partner for Photocatalytic Difluoroalkylaromatic Synthesisa
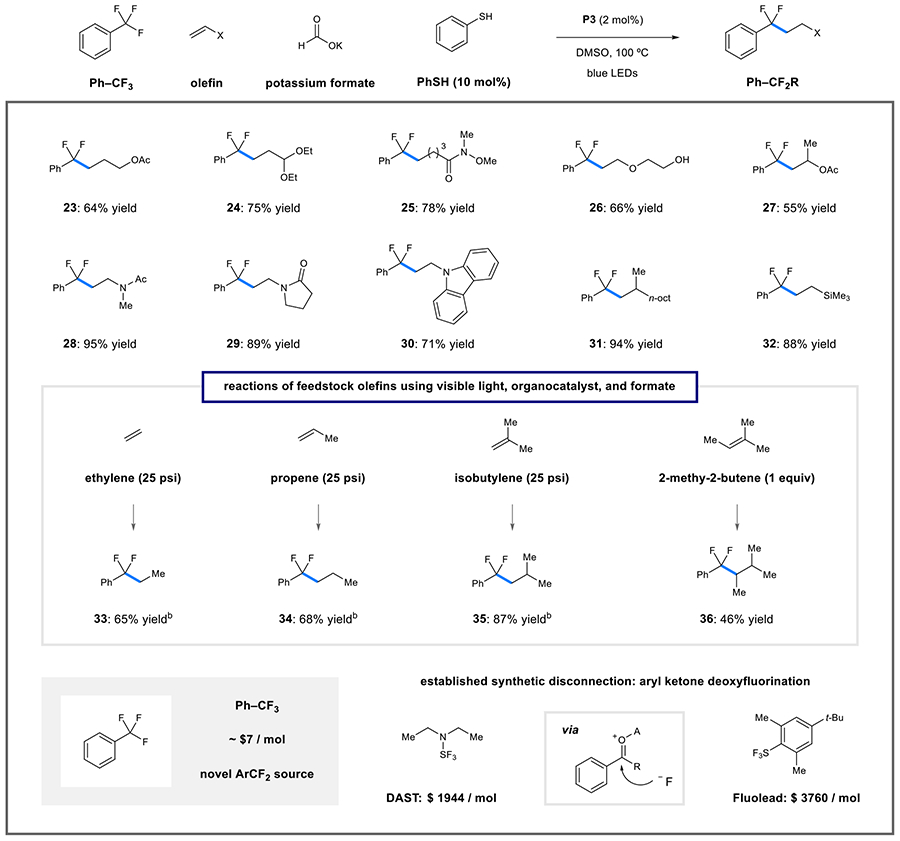
|
Reaction conditions: benzotrifluoride (3 mL), alkene (0.5 mmol), P3 (2 mol %), potassium formate (2.5 mmol), thiophenol (10 mol %), DMSO (5.0 mL), blue light, 100 °C, 24 h; isolated yields given.
Reaction conducted with potassium formate as limiting reagent, yields determined by 19F NMR analysis with an internal standard.
To further illustrate the value of this photocatalytic DFA in medicinal chemistry, we applied it in the formal syntheses of bioactive difluoroalkylaromatic molecules (as indicated in Scheme 2). The patent routes proceed through difluorobenzyl arene intermediates 37, 40, and 44, all of which were synthesized via dexoyfluorination of the corresponding carbonyl compounds. The reported preparation of growth hormone secretagogue 38 involves construction of a 1,3-dithiolane intermediate from 39 and 1,2-ethanedithiol. Subsequent desulfurative fluorination with nitrosonium cation and Olah’s reagent delivered the geminal difluoride motif. Ester reduction provided primary alcohol 37 in 45% yield over three steps.41 With this catalytic approach, reaction of Ph–CF3 (50 equiv) with allyl alcohol provided the same intermediate in a single step (63% yield). The reported synthesis of 41, an Retinoid-related orphan receptor gamma (RORγt) modulator, also involves a dithiolation/desulfurative fluorination sequence using ketone 43 as starting material. Palladium-catalyzed enolate coupling and carboxylic acid deprotection afforded 40 in 4% yield over four steps.42 Under our conditions, commercial trifluoromethyl arene 42 reacted with ethylene gas (25 psi) to afford the same intermediate, after ester saponification, in 63% overall yield. In the reported synthesis of amine 44, a precursor to β2 adrenergic receptor agonist 45, DAST was employed for direct deoxyfluorination of benzylic ketone 47. Phenethylamine 44 was then delivered via sequential alcohol deprotection, Mitsunobu etherification, and reduction (4% yield over five steps).43 Our alternative route to this intermediate involves reacting trifluorotoluene (50 equiv) with vinyl ether 46 on 12 mmol scale (1 mol % P3) to give the corresponding defluoroalkylation product in 92% yield (4 g). Acidic cleavage of the tert-butyloxycarbonyl (Boc) group gave 44 in 91% over two steps. While deoxyfluorination remains an effective tool for difluoroalkylaromatic production, these examples illustrate the potential of this catalytic protocol to streamline the preparation of medicinally relevant Ar–CF2R building blocks.
Scheme 2.

Catalytic Defluoroalkylation in the Synthesis of Medicinal Building Blocks
Scheme 3 shows our proposed mechanism for this transformation, where excitation of P3 with visible light (through irradiation with commercial blue light-emitting diodes (LEDs)) delivers the excited-state reductant P3*. Reversible single-electron transfer to the trifluoromethylarene substrate gives rise to the corresponding radical anion and the ground-state catalyst radical cation. Formation of the key difluorobenzylic radical intermediate is then accomplished via thermodynamically driven mesolytic C–F cleavage event. Regioselective intermolecular addition to the olefin substrate provides an alkyl radical adduct that undergoes polarity matched hydrogen atom transfer (HAT) with thiophenol to furnish the defluoroalkylation products. Regeneration of both the thiol and photoredox catalysts occurs with formate as stoichiometric reductant producing a metal fluoride salt and CO2 as byproducts.
Scheme 3.

Catalytic or Direct HAT Mechanisms for Defluorofunctionalization of Trifluoromethyl Arenes
The findings that are described in this paper were enabled by the discovery that P3 has the ability to engage unactivated trifluoromethylaromatics. This is mechanistically interesting because, in comparison to our initial report in this area (using P1 and activated Ar–CF3 substrates), this substrate set is more difficult to reduce, and the most effective catalyst (P3) is not as strongly reducing. Within this context, we propose that the reversible SET events that are operational here are endergonic in the forward direction. Consistent with this proposal are Stern–Volmer excited-state quenching studies, which revealed that quenching of P3* with benzotrifluoride occurs to a minute extent (KSV = 0.02; none of the other reaction components quench the excited state). Although the concentration of radical anion intermediate is low under these conditions, fluoride expulsion provides a thermodynamic driving force for radical formation. Indeed, the calculated thermodynamic profile (details given in the Supporting Information) indicates that C–F bond elongation from benzotrifluoride radical anion is essentially barrierless (<3 kcal/mol). Taken together, these results indicate that, although substrate reduction is a critical step within this pathway, mesolytic cleavage acts as the primary determinant in radical formation.
We questioned whether the radical intermediates that are accessible under this paradigm could be generally utilized in the formation of other bonds. Of particular interest to us was the idea that interception of this species with a hydrogen atom would grant access to the corresponding difluoromethyl arene (Ar–CF2H). Selective HDF of trifluoromethylaromatics in this manner would offer a novel approach to this high-value motif.
Summarized in Scheme 3 are existing strategies for trifluoromethylaromatic HDF. In 2016, Lalic reported a system that utilizes catalytic amounts of both palladium and copper in conjunction with triphenylsilane and potassium tert-butoxide to defluorinate a series of 4-trifluoromethyl biaryl compounds, generating presumed nucleophilic addition products to dimethylformamide (DMF).33 Reaction of these intermediates with tert-butanol resulted in selective production of the corresponding difluoromethylaromatics. Prakash described that Mg0 can be utilized in HDF processes of various bis(trifluoromethyl)benzene derivatives with varying selectivity for the corresponding reduction products.26 While appealing for its operational simplicity, this protocol is limited to bis(trifluoromethyl)benzene substrates, as benzotrifluoride was reported to be unreactive, and more electron-poor systems underwent exhaustive defluorination (complete reduction of trifluoromethylaromatics to the corresponding toluene derivatives). Exhaustive defluorination is the major outcome within the trifluoromethylaromatic HDF literature, where electrochemical reduction,25 alkali metal reduction,26–28 or Lewis acids29,30 have been reported to activate these strong C–F bonds.
To evaluate the feasibility of the proposed HDF pathway, we reacted 2-trifluoromethylacetanilide with P3 and a range of reductants in dimethyl sulfoxide (DMSO) under irradiation with blue LEDs. These studies (optimization details are given in the Supporting Information) revealed that formate reductants are uniquely effective for HDF and that there is a pronounced effect of the formate counterion on reactivity. The optimized conditions (indicated in Table 3) again involve the use of P3 (2 mol %) along with cesium formate (3 equiv) at 50 °C, which delivered the desired difluoromethylaromatic 48 in 75% isolated yield without detectable overreduction products. Similarly, the isomeric trifluoromethyl-bearing acetanilides were good substrates for this transformation, affording the corresponding products 49 and 50 in 55–70% yield. Additionally, the outlined conditions efficiently engaged compounds bearing only alkyl substituents in the para- (51, 64% yield), meta- (52, 48% yield), and ortho- (53, 50% yield), a significant advance over previously reported technologies. Other oxygen- or nitrogen-substituted substrates could be reductively defluorinated with high selectivity for the Ar–CF2H products (54–56, 45–65% yield).
Table 3.
Substrate Scope for Hydrodefluorinationa
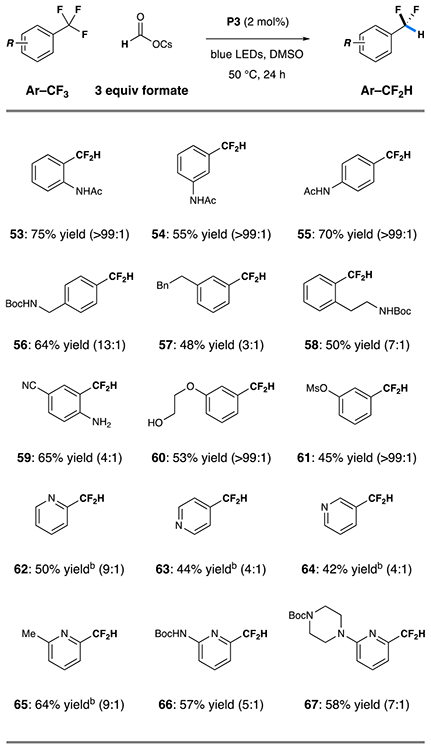
|
Reaction conditions: Ar–CF3 substrate (0.5 mmol), cesium formate (1.5 mmol), Miyake phenoxazine P3 (2 mol %), DMSO (5.0 mL), blue light, 50 °C, 24 h; isolated yields given; parentheses indicate the ratios of Ar–CF2H to Ar–CFH2 products, as determined by NMR analysis of the crude reaction mixtures.
Reaction run at 23 °C
Yield determined by 19F NMR with an internal standard.
Hydrodefluorination of 2-, 3-, and 4-trifluoromethyl substituted pyridines 57–59 was also successful under this protocol, giving the desired products in acceptable (42–50%) yield. Much like we observed under DFA conditions, yields of pyridine-based substrates were bolstered by substitution of electron-donating groups at the 2-position (60–62, 57–64% yield). Although this substrate set underwent HDF in moderate yield, these results demonstrate that significant structural diversity is tolerated by this protocol including acidic protons, heterocycles, and other reducible groups.
CONCLUSIONS
In summary, we have developed a robust method for the defluoroalkylation and hydrodefluorination of unactivated trifluorotoluene derivatives using the combination of two organocatalysts (no metal is required), inexpensive formate salts, and visible light. The use of Miyake’s phenoxazine, P3, was key in the development and generality of these methods. The reductive radical process, driven by the selective cleavage of a single benzylic C–F bond, can be employed to access a diverse range of Ar–CF2R and Ar–CF2H substrates. Mechanistic studies and the development of additional applications of difluorobenzylic radicals are underway in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by funds from the National Institutes of Health (GM129495), and NMR data were collected under support of the National Science Foundation (CHE-1531620).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b06004.
Experimental procedures, spectral data, and computational data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (2).Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- (3).Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- (4).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- (5).Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nagib DA; MacMillan DWC Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhu W; Wang J; Wang S; Gu Z; Aceña JL; Izawa K; Liu H; Soloshonok VA Recent Advances in the Trifluoromethylation Methodology and New CF3-Containing Drugs. J. Fluorine Chem 2014, 167, 37–54. [Google Scholar]
- (8).Morimoto H; Tsubogo T; Litvinas ND; Hartwig JF A Broadly Applicable Copper Reagent for Trifluoromethylations and Perfluoroalkylations of Aryl Iodides and Bromides. Angew. Chem., Int. Ed 2011, 50, 3793–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ji Y; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Innate C-H Trifluoromethylation of Heterocycles. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cho EJ; Senecal TD; Kinzel T; Zhang Y; Watson DA; Buchwald SL The palladium-catalyzed trifluoromethylation of aryl chlorides. Science 2010, 328, 1679–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Müller K; Faeh C; Diederich F Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (12).Erickson JA; McLoughlin JI Hydrogen Bond Donor Properties of the Difluoromethyl Group. J. Org. Chem 1995, 60, 1626–1631. [Google Scholar]
- (13).Sessler CD; Rahm M; Becker S; Goldberg JM; Wang F; Lippard SJ CF2H, a Hydrogen Bond Donor. J. Am. Chem. Soc 2017, 139, 9325–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amir D; Marciano D; Gershonov E; Saphier S Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]
- (15).Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (16).Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (17).Merchant RR; Edwards JT; Qin T; Kruszyk MM; Bi C; Che G; Bao D-H; Qiao W; Sun L; Collins MR; Fadeyi OO; Gallego GM; Mousseau JJ; Nuhant P; Baran PS Modular radical cross-coupling with sulfones enables access to sp3-rich (fluoro)alkylated scaffolds. Science 2018, 360, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fujiwara Y; Dixon JA; Rodriguez RA; Baxter RD; Dixon DD; Collins MR; Blackmond DG; Baran PS A New Reagent for Direct Difluoromethylation. J. Am. Chem. Soc 2012, 134, 1494–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bacauanu V; Cardinal S; Yamauchi M; Kondo M; Fernández DF; Remy R; MacMillan DWC Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem., Int. Ed 2018, 57, 12543–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xiao YL; Min QQ; Xu C; Wang RW; Zhang X Nickel-Catalyzed Difluoroalkylation of (Hetero)Arylborons with Unactivated 1-Bromo-1,1-Difluoroalkanes. Angew. Chem., Int. Ed 2016, 55, 5837–5841. [DOI] [PubMed] [Google Scholar]
- (21).Zhou Q; Ruffoni A; Gianatassio R; Fujiwara Y; Sella E; Shabat D; Baran PS Direct Synthesis of Fluorinated Hetero-arylether Bioisosteres. Angew. Chem., Int. Ed 2013, 52, 3949–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fier PS; Hartwig JF Copper-Mediated Difluoromethylation of Aryl and Vinyl Iodides. J. Am. Chem. Soc 2012, 134, 5524–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Prakash GKS; Ganesh SK; Jones J-P; Kulkarni A; Masood K; Swabeck JK; Olah GA Copper-Mediated Difluoromethylation of (Hetero)Aryl Iodides and β-Styryl Halides with Tributyl(Difluoromethyl)Stannane. Angew. Chem., Int. Ed 2012, 51, 12090–12094. [DOI] [PubMed] [Google Scholar]
- (24).Yamauchi Y; Fukuhara T; Hara S; Senboku H Electrochemical Carboxylation of α,α-Difluorotoluene Derivatives and Its Application to the Synthesis of α-Fluorinated Nonsteroidal Anti-Inflammatory Drugs. Synlett 2008, 2008, 438–442. [Google Scholar]
- (25).Lund H; Jensen NJ; et al. Electroorganic Preparations. XXXVI. Stepwise Reduction of Benzotrifluoride. Acta Chem. Scand 1974, 28B, 263–265. [Google Scholar]
- (26).Munoz SB; Ni C; Zhang Z; Wang F; Shao N; Mathew T; Olah GA; Prakash GKS Selective Late-Stage Hydrodefluorination of Trifluoromethylarenes: A Facile Access to Difluoromethylarenes. Eur. J. Org. Chem 2017, 2017, 2322–2326. [Google Scholar]
- (27).Fuchibe K; Ohshima Y; Mitomi K; Akiyama T Low-Valent Niobium-Catalyzed Reduction of α,α,α-Trifluorotoluenes. Org. Lett 2007, 9, 1497–1499. [DOI] [PubMed] [Google Scholar]
- (28).Amii H; Hatamoto Y; Seo M; Uneyama K A New C-F Bond-Cleavage Route for the Synthesis of Octafluoro[2.2]-paracyclophane. J. Org. Chem 2001, 66, 7216–7218. [DOI] [PubMed] [Google Scholar]
- (29).Stahl T; Klare HFT; Oestreich M Main-Group Lewis Acids for C-F Bond Activation. ACS Catal 2013, 3, 1578–1587. [Google Scholar]
- (30).Forster F; Metsänen TT; Irran E; Hrobárik P; Oestreich M Cooperative Al-H Bond Activation in DIBAL-H: Catalytic Generation of an Alumenium-Ion-Like Lewis Acid for Hydrodefluorinative Friedel-Crafts Alkylation. J. Am. Chem. Soc 2017, 139, 16334–16342. [DOI] [PubMed] [Google Scholar]
- (31).Radom L; Hehre WJ; Pople JA Molecular Orbital Theory of the Electronic Structure of Organic Compounds. VII. A Systematic Study of Energies, Conformations, and Bond Interactions. J. Am. Chem. Soc 1971, 93, 289–300. [Google Scholar]
- (32).Saboureau C; Troupel M; Sibille S; Perichon J Electro-reductive Coupling of Trifluoromethylarenes with Electrophiles: Synthetic Applications. J. Chem. Soc., Chem. Commun 1989, 1138–1139. [Google Scholar]
- (33).Dang H; Whittaker AM; Lalic G Catalytic Activation of a Single C-F Bond in Trifluoromethyl Arenes. Chem. Sci 2016, 7, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yoshida S; Shimomori K; Kim Y; Hosoya T Single C-F Bond Cleavage of Trifluoromethylarenes with an Ortho-Silyl Group. Angew. Chem., Int. Ed 2016, 55, 10406–10409. [DOI] [PubMed] [Google Scholar]
- (35).Wang H; Jui NT Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 163–166. [DOI] [PubMed] [Google Scholar]
- (36).Chen K; Berg N; Gschwind R; König B Selective Single C(sp3)-F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]
- (37).Pearson RM; Lim C-H; McCarthy BG; Musgrave CB; Miyake GM Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc 2016, 138, 11399–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Du Y; Pearson RM; Lim C-H; Sartor SM; Ryan MD; Yang H; Damrauer NH; Miyake GM Strongly Reducing, Visible-Light Organic Photoredox Catalysts as Sustainable Alternatives to Precious Metals. Chem. - Eur. J 2017, 23, 10962–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Boyington AJ; Seath CP; Zearfoss AM; Xu Z; Jui NT Catalytic Strategy for Regioselective Arylethylamine Synthesis. J. Am. Chem. Soc 2019, 141, 4147–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).For an alternative strategy to Ar-CF2R synthesis via deprotonation of difluoromethylaromatics, see:; Geri JB; Wade Wolfe MM; Szymczak NK The Difluoromethyl Group as a Masked Nucleophile: A Lewis Acid/Base Approach. J. Am. Chem. Soc 2018, 140, 9404–9408. [DOI] [PubMed] [Google Scholar]
- (41).Ewing W; Li J; Sulsky RB; Hernandez AS Preparation of Azoles as Growth Hormone Secretagogues US 20060079562, April 13, 2006.
- (42).Das S; Gharat LA; Harde RL; Shelke DE; Pardeshi SR; Thomas A; Khairatkar-Joshi N; Shah DM; Bajpai M Preparation of Carbocyclic Compounds as ROR Gamma Modulators WO 2017037595, March 9, 2017.
- (43).Tana JB; Crespo MIC; Duran CP; Roig SG; Munoz AO Derivatives of 4-(2-Amino-1-Hydroxyethyl)Phenol as Agonists of the B2 Adrenergic Receptor WO 2008046598, April 30, 2008.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.