

SUMMARY
Macrophage plasticity is critical for normal tissue repair to ensure transition from the inflammatory to the proliferative phase of healing. We examined macrophages isolated from wounds of patients afflicted with diabetes and of healthy controls and found differential expression of the methyltransferase Setdb2. Myeloid-specific deletion of Setdb2 impaired the transition of macrophages from an inflammatory phenotype to a reparative one in normal wound healing. Mechanistically, Setdb2 trimethylated histone 3 at NF-κB binding sites on inflammatory cytokine gene promoters to suppress transcription. Setdb2 expression in wound macrophages was regulated by interferon (IFN) β, and under diabetic conditions, this IFNβ-Setdb2 axis was impaired, leading to a persistent inflammatory macrophage phenotype in diabetic wounds. Setdb2 regulated the expression of xanthine oxidase and thereby the uric acid (UA) pathway of purine catabolism in macrophages, and pharmacologic targeting of Setdb2 or the UA pathway improved healing. Thus, Setdb2 regulates macrophage plasticity during normal and pathologic wound repair and is a target for therapeutic manipulation.
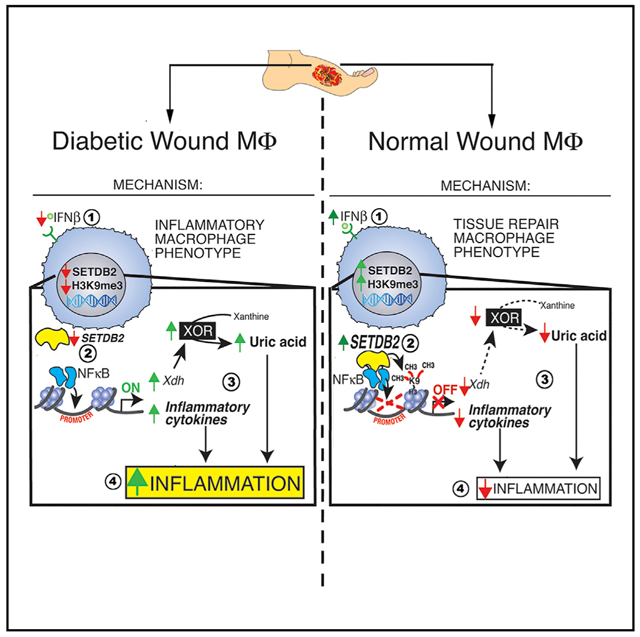
Graphical Abstract
In Brief
Kimball and Davis et al. reveal that IFN-I induces the expression of the methyltransferase Setdb2 in wound macrophages, which in turn regulates macrophage plasticity to promote wound repair. This axis is dysfunctional in diabetic wounds, where decreased Setdb2 expression results in increased NF-κB-mediated inflammation and production of uric acid.
INTRODUCTION
Non-healing wounds impair quality of life and are associated with increased mortality in patients afflicted with type 2 diabetes (T2D) (Falanga, 2005; Ghanassia et al., 2008). One-third of the cost of T2D treatment is related to non-healing peripheral wounds (Hicks et al., 2016). Complications from diabetic foot wounds represent the leading cause of lower extremity amputation in the United States; these interventions have a survival rate of 50% at 5 years (Faglia et al., 2001; Izumi et al., 2009). Chronic dysregulated inflammation, and the associated impairment in tissue repair, is a hallmark of diabetic wounds. Macrophage plasticity, allowing transition in macrophages from an inflammatory to a reparative phenotype, is critical for normal wound healing, but there is limited understanding of the molecular mechanisms that program these transitions and sustain reparative phenotypes in wounds.
Wound repair is a complex process that occurs in overlapping stages of coagulation, inflammation, proliferation, and remodeling (Eming et al., 2007). During the inflammatory phase, macrophage plasticity is essential for the repair and remodeling of wounds. The inflammatory phase (days 1–5 post-injury) is divided into an early phase, wherein macrophages and neutrophils promote inflammation and tissue destruction, and a late phase, in which macrophages promote tissue repair and allow for transition to the proliferative phase. Macrophages in the early inflammatory phase exhibit increased production of inflammatory cytokines and enhanced pathogen killing capacity (Porcheray et al., 2005), whereas late inflammatory phase macrophages produce transforming growth factor b (TGF-b), interleukin (IL) 10, and other mediators important in the transition from the inflammatory to the proliferative phase of wound healing (Gallagher et al., 2015; Martinez et al., 2008; Mirza et al., 2013, 2015). The predominance of these phenotypically distinct macrophages at specific times during healing facilitates the development of a tailored macrophage-dependent response.
Epigenetic regulation of gene expression plays a major role in the phenotype and function of immune cells in both normal and pathologic conditions by controlling downstream protein expression patterns (Hewagama and Richardson, 2009). We and others have shown that histone methylation regulates immune-mediator expression in in vitro and in vivo macrophages (Hersh et al., 2007; Kimball et al., 2017). Despite this, there remains a paucity of data on epigenetic mechanisms that regulate macrophage phenotypes following injury.
Here, we examined macrophages isolated from wounds in diabetic subjects and healthy controls. Gene expression analyses revealed differential expression of Setdb2, encoding the histone methyltransferase Setdb2, in normal versus diabetic wounds. Setdb2 specifically trimethylates lysine 9 (K9) on histone 3 (H3) (H3K9me3) and keeps chromatin in a conformation in which the promoter is not accessible for transcription factor binding, effectively silencing gene transcription (Schliehe et al., 2015). Experiments in mouse models (Setdb2LacZ; Setdb2f/fLyz2Cre+) revealed that Setdb2 facilitated the transition of macrophages from an inflammatory phenotype to a reparative one in normal wound healing. Increased expression of Setdb2 in wound macrophages post-injury led to higher amounts of repressive H3K9me3 at nuclear factor κB (NF-κB) binding sites on inflammatory cytokine gene promoters, resulting in the cessation of inflammation and the transition to a reparative phenotype. Setdb2 expression in wound macrophages was regulated by interferon (IFN) β via the janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) pathway. Under diabetic conditions, this IFNβ-Setdb2 axis was impaired in macrophages, leading to a persistent inflammatory macrophage phenotype in diabetic wounds. Setdb2 regulated the expression of xanthine oxidase (XO), and thus the uric acid (UA) pathway of purine catabolism in macrophages, providing mechanistic insight into the increased UA seen diabetic tissue. Our findings have therapeutic implications for abrogating dysregulated inflammation in diabetic wounds.
RESULTS
Inflammatory Cytokine Production Is Increased in Diabetic Wound Macrophages
Given the critical role that macrophages play in the switch from the inflammatory to the reparative processes in normal wounds and the established link between obesity-T2D and chronic systemic inflammation, we examined macrophages isolated from wounds of control and pre-diabetic or diet-induced obese (DIO) mice for inflammatory cytokine expression. Macrophages (CD3−/CD19−/NK1.1−/Ly6G−/CD11b+) sorted from DIO and control wounds on day 5 post-injury revealed increased ∥1b, ∥12, and Tnfa transcript and protein in DIO macrophages compared with controls (Figures 1A and 1B). Pro-inflammatory Ly6CHi monocytes-macrophages are recruited to DIO wounds at later points following injury (Kimball et al., 2018). Intracellular flow cytometry analyses revealed that DIO wound Ly6CHi cells produced more IL-1 β and tumor necrosis factor alpha (TNF-α) than Ly6CLo monocytes-macrophages (Figures 1C and 1D). Peripheral blood CD14+ monocytes from matched T2D patients with non-healing wounds and non-diabetic control subjects exhibited increased IL-1β in monocytes from T2D patients (Figure 1E). This increased inflammatory signature in diabetes suggests a failure of the pro- to anti-inflammatory macrophage phenotype switch that is necessary for transition from the inflammatory to the proliferative phase of healing.
Figure 1. Inflammatory Cytokine Production Is Increased in Diabetic Wound Macrophages.

(A) ∥1b, ∥12, and Tnfa expression levels in macrophages isolated via sorting from diet-induced obese (DIO) mice and controls on day 5 post-wounding (n = 3 mice per group, replicated in triplicate).
(B) Supernatant protein levels of IL-1β, IL-12, and TNF-α from wound macrophages from DIO and control mice (n = 3 mice per group, replicated in triplicate).
(C) Gating strategy to isolate Ly6CHi and Ly6CLo monocytes-macrophages as a percentage of live, ly6G−, CD11b+ cells by flow cytometry at day 5 post-injury.
(D) IL-1β and TNF-α in Ly6CHi versus Ly6CLo gates expressed as a percentage of live, Ly6G−, CD11b+ cells by flow cytometry in DIO wound monocytes-macrophages at day 5 post-injury (n = 4 mice per group, replicated in five time).
(E) IL1β expression in CD14+ monocytes from matched patients with T2D and non-T2D controls (n = 3 nondiabetic and 9 diabetic patients, replicated in triplicate).
*p < 0.05, **p < 0.01, ***p < 0.001. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used.
Differential Expression of Setdb2 in Macrophages during Normal and T2D Wound Healing
Epigenetic-based histone modifications regulate macrophage phenotypes by controlling downstream gene expression (Gallagher et al., 2015; Ishii et al., 2009; Porta et al., 2015). To examine the kinetics of chromatin-modifying enzyme (CME) expression during wound healing, we isolated normal murine wound macrophages (CD3−/CD19−/Ly6G−/NK1.1−/CD11b+) by cell sorting on days 3 and 5 post-wounding during the macrophage transition from an inflammatory to a reparative phenotype. These macrophages were analyzed using a gene array, probing the expression patterns of 96 distinct CMEs. Of these genes, Setdb2 expression was significantly increased in wound macrophages at day 5 (Table S1). Setdb2 plays a vital role in the regulation of macrophage phenotype and function following viral infection (Kroetz et al., 2015), and given the critical nature of macrophage inflammation to normal and pathologic wound healing (Boniakowski et al., 2017; Carson et al., 2017; Davis et al., 2019), we investigated the overall impact of Setdb2 on wound healing. Human wound tissue was examined from patients with non-healing wounds and T2D, and significantly less SETDB2 transcript and SETDB2 were found on histologic assessment in wounds from T2D patients compared with controls (Figure 2A). To establish a time course of Setdb2 expression in normal wound healing, we sorted wound macrophages (CD3−/CD19−/Ly6G−/NK1.1−/CD11b+) on days 0, 1, 3, 5, and 7 following injury and analyzed for Setdb2 expression (Figure 2C). We found increased Setdb2 expression at day 5 post-injury that paralleled increased SETDB2 protein on day 5 as measured by western blot (Figures 2C and 2D).
Figure 2. Differential Expression of Setdb2 in Macrophages during Normal and T2D Wound Healing.

(A) Peripheral blood (30 mL) was collected from patients with T2D and control subjects without diabetes. Peripheral blood mononuclear cells underwent Ficoll separation. CD14+ monocytes were then positively selected by MACS, and SETDB2 gene expression was measured by qPCR (n = 11 control and 8 diabetic patients, replicated in triplicate).
(B) SETDB2 mRNA expression (normalized to 18 s rRNA) in human wounds from matched T2D (n = 5) and non-T2D (n = 3) patients (RNA integrity number [RIN] score > 8). Immunohistochemistry performed on human wounds from T2D and non-T2D patients for SETDB2.
(C) Setdb2 expression in C57BL/6 murine wound macrophages on days 0-7 post-injury (n = 6 mice per time point, replicated in triplicate).
(D) SETDB2 protein measured from wound lysates on days 0 and 5 post-injury (n = 7 mice per group).
(E) LacZ expression over time post-injury measured by flow cytometry from wound macrophages isolated from Setdb2LacZ mice (n = 8 mice per time point).
(F) LacZ cells expressed as a percentage of wound macrophages (CD11b+, Ly6G−), wound monocytes (CD11b+, Ly6C+), T cells (CD3+), and B cells (CD19+) by flow cytometry isolated at day 5 post-injury in Setdb2LacZ mice (n = 4 mice per group, replicated twice).
(G) Setdb2 expression in CD11b+ cells compared to Mertk+, CD64+, or F4/80+ cells at day 5 post-injury (n = 3 mice per group, replicated in triplicate).
*p < 0.05, **p < 0.01. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups, one-way ANOVA followed by a Newman-Keuls post hoc test was used.
See also Figure S1.
To track Setdb2 expression and determine the phenotypic effects of Setdb2 deficiency during wound healing, we used Setdb2LacZ reporter mice that possess the lacZ reporter at the promoter for Setdb2 as a surrogate marker of Setdb2 expression. Wound cells from Setdb2LacZ reporter mice were interrogated for lacZ expression by flow cytometry on days 1–6 post-injury. LacZ expression was increased over time post-injury in wound macrophages, peaking between days 5 and 6 (Figure 2E). Furthermore, the predominant Setdb2-expressing cell type was of myeloid lineage, with other lymphocyte populations demonstrating significantly less Setdb2 expression (Figure 2F). Regarding wound macrophage markers, CD11b+ cells expressed the highest level of Setdb2, while cells with other common macrophage markers (CD64, F4/80, and Mertk) expressed lower levels of Setdb2 (Figure 2G; Figure S1). Altogether, these data suggest that Setdb2 is increased in normal wound macrophages and may initiate the transition of wound macrophages from an inflammatory to a reparative phenotype during normal wound healing.
Depletion of Setdb2-H3K9me3 Increased Inflammation and Impaired Wound Healing
Because we found that Setdb2 expression is dynamic in wound macrophages during normal healing, we examined the effect of macrophage-specific loss of Setdb2 on wound healing. To examine the function of Setdb2 in the macrophage-specific innate immune response during wound healing, we generated a myeloid-specific Setdb2-deficient mouse using a Setdb2-floxed mouse (Clausen et al., 1999; Kroetz et al., 2015) crossed with a Lyz-M Cre mouse (Setdb2f/fLyz2Cre+). Confirmation of reduced Setdb2 expression in these mice was determined in bone marrow-derived macrophages (BMDMs) isolated from Setdb2f/fLyz2Cre+ and their littermate controls (Figure 3A). Setdb2f/fLyz2Cre+ mice and their littermate controls were wounded with a 4-mm punch biopsy, with wound healing rates monitored over time. We identified that mice lacking Setdb2 in their monocytes-macrophages (Setdb2f/fLyz2Cre+) had significantly delayed wound healing compared with littermate controls, specifically during the inflammatory resolution phase of healing. Histologic assessment of wounds revealed a decrease in re-epithelialization and collagen deposition in the Setdb2f/fLyz2Cre+ mice without a significant change in angiogenesis or scar formation (Figure 3B; Figure S2).
Figure 3. Depletion of Setdb2-H3K9me3 Increased Inflammation and Impaired Wound Healing.

(A) Setdb2 expression measured in BMDMs from Setdb2f/fLyz2Cre mice and littermate controls on day 5 post-injury (n = 6 mice per group, replicated in triplicate).
(B) Wound healing curve for Setdb2f/fLyz2Cre+ mice and littermate controls (n = 16 mice per group). Wounds were stained with Masson’s trichrome stain. Percentage of re-epithelialization was calculated. Representative images are shown in 4x magnification, with the black bar in the lower right corner specifying 200 μm. The black bar above the wound represents the entire wound distance, and the arrowheads denote the epithelial tongues for re-epithelialization (n = 5 mice per group).
(C) IL-1β, IL-6, IL-12, MIP1α, TNF-α, and MIP1β were measured by Bioplex from wound macrophages from Setdb2f/fLyz2Cre mice and littermate controls on day 5 post-injury (stimulated ex vivo for 2 h with 10 ng/mL LPS and supernatants collected) (n = 6 mice per group).
(D) Flow cytometry analysis of Ly6CHi and Ly6CLo wound monocytes-macrophages (live, Ly6G−, CD11b+) from Setdb2f/fLyz2Cre mice and littermate controls on day 5 (n = 6 mice per group).
(E) ChIP analysis of H3K9me3 on the IL1β, TNFα, and NOS2 promoters of wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls on day 5 post-injury (n = 4 mice per group).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are presented as the mean ± SEM. All data are representative of 2-4 independent experiments. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups, ANOVA followed by Newman-Keuls post hoc test was used.
The control of gene expression by Setdb2 in vivo is of interest, because it sets a repressive H3K9me3 mark that renders the promoter inaccessible to transcription factor (such as NF-κB) binding, resulting in gene silencing. To determine the transcriptional effects of Setdb2 deficiency on NF-κB-mediated inflammatory gene expression in in vivo wound macrophages, we wounded our Setdb2f/fLyz2Cre+ mice and littermate controls and isolated wound macrophages (CD3−/CD19−/NK1.1−/Ly6G−/CD11b+). We found increased protein levels of IL-1 β, TNF-α, and other NF-κB-mediated inflammatory cytokines-che-mokines (IL-12, IL-6, MIP1α, and MIP1β) by Bioplex in Setdb2-deficient wound macrophages compared with controls (Figure 3C). Similarly, we performed flow cytometry on wounds from our Setdb2f/fLyz2Cre+ mice and controls and found that inflammatory recruited monocytes-macrophages were increased in the Setdb2-deficient wounds (Figure 3D; Figure S3). These data identified that Setdb2 is important in myeloid cells for normal tissue repair and that in vivo, wound macrophage NF-κB-mediated inflammatory gene expression is controlled, at least partly, by Setdb2.
The finding that key inflammatory cytokines involved in wound repair, such as IL-1β and TNF-α, are increased in Setdb2-deficient wound macrophages suggests that Setdb2 epigenetic regulation of inflammatory gene promoters may control gene expression at critical macrophage transition points in the wound healing cascade. IL-1β is a major macrophage-derived cytokine that drives tissue inflammation in wounds, and its pathologic expression is increased in several diabetic mouse models and human T2D wounds (Mirza et al., 2015; Zhou et al., 2010). Furthermore, TNF-a and NOS2 are inflammatory mediators that play significant roles in wound repair (Ashcroft et al., 2012; Sen, 2009). To examine the role of Setdb2 on transcriptional regulation in wound macrophages, we isolated wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls on day 5 post-injury and performed chromatin immunoprecipitation (ChIP) analysis using primers for the NF-κB binding site on the IL1β, TNFα, and NOS2 promoters. We found significantly reduced levels of H3K9me3 in the Setdb2f/fLyz2Cre+ wound macrophages at the NF-κB binding site of the IL1β, TNFα, and NOS2 promoters compared with littermate controls (Figure 3E). Because Setdb2 mechanistically silences gene expression through trimethylation of H3K9, these data suggest that increased repressive H3K9me3 by Setdb2 in normal wound macrophages decreases transcription of IL1β, TNFα, and NOS2 at day 5, at a key transition time in wound healing, in which macrophages switch from an inflammatory to a reparative phenotype. Thus, Setdb2 is important for promoting resolution of inflammation during wound repair.
Setdb2 Is Significantly Reduced in Diabetic Wound Macrophages
Given that Setdb2 is critical for decreasing macrophage inflammatory cytokine production during the transition from the inflammatory to the reparative phase in normal wound healing and that diabetic wound macrophages fail to transition from an inflammatory state, we examined the role of Setdb2 in diabetic wound macrophages following injury. Diabetic and control wound macrophages (CD3−/CD19−/NK1.1−/Ly6G−/CD11b+) were isolated from DIO mice and normal-diet controls on day 5, and we found that DIO wound macrophages had significantly less Setdb2 transcript and protein compared with control wound macrophages (Figures 4A and 4B). Because we have previously identified that DIO wound macrophages make increased NF-κB-mediated inflammatory cytokines, we sought to determine whether Setdb2 was directly responsible for the increased inflammatory cytokines. Hence, we performed a ChIP assay for Setdb2 and H3K9me3 on DIO and control wound macrophages isolated on day 5. Setdb2 and the repressive H3K9me3 were significantly reduced at the NF-κB binding site on the IL1β, TNFα, and NOS2 promoters in DIO wound macrophages compared with controls (Figure 4C). In normal wounds, increased expression of Setdb2 serves as a brake in inflammation in which the inflammatory transcriptional program gets turned off and macrophages switch to a reparative phenotype; however, this process did not occur in diabetic wound macrophages, allowing unrestricted inflammatory gene expression.
Figure 4. Setdb2 Is Significantly Reduced in Diabetic Wound Macrophages.

(A) Setdb2 expression in macrophages from wounds of DIO and control mice at day 5 post-injury (n = 10 per group).
(B) SETDB2 protein in wounds from DIO and control mice on days 0 and 5 post-injury via western blot (n = 10 mice per group). Immunohistochemistry was performed for SETDB2 in DIO and control wounds. Representative slides are shown.
(C) ChIP analysis of Setdb2 and H3K9me3 at the IL1β, TNFα and NOS2 promoters of wound macrophages from DIO and control mice at day 5 post-injury (n = 12 mice per group).
*p < 0.05, **p < 0.01. All data represent 3 independent experiments. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups, ANOVA followed by Newman-Keuls post hoc test was used.
IFNβ/JAK/STAT1 Pathway-Induced Setdb2-H3K9me3 and IFNβ Signaling Is Impaired in Diabetic Wound Macrophages
Type I INFs serve as cell signaling molecules that bind to cell surface receptors and ultimately phosphorylate tyrosine kinases, inducing specific transcription factors to activate inflammatory genes (Rayamajhi et al., 2010). Although type I INFs (IFN-I) has been well-studied in viral disorders and autoimmune diseases, little is known about the role of IFN-I in both normal and pathologic tissue repair (Fislová et al., 2005; Shahangian et al., 2009). Our group found that IFNβ production during influenza infection drives expression of Setdb2 in lung tissue (Kroetz et al., 2015); however, it is unknown whether IFNβ plays a role in wound healing.
To determine whether IFNβ increased production of SETDB2 in wound macrophages, we isolated wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls, stimulated them ex vivo with IFNβ, and analyzed them for Setdb2 gene expression. In the control (Setdb2f/fLyz2Cre−) wound macrophages, Setdb2 was significantly increased following IFNβ stimulation, whereas in wound macrophages lacking Setdb2, IFNβ had no influence on Setdb2 expression (Figure 5A). Next, we harvested normal wound macrophages, stimulated them ex vivo with IFNβ, and performed ChIP for Setdb2-H3K9me3 on the NF-κB binding site on the IL1β promoter. We found that Setdb2 and its repressive H3K9me3 mark were increased significantly at the NF-κB binding site on the promoter in wound macrophages following stimulation with IFNβ (Figure 5B). As a translational corollary, we isolated and cultured human monocyte-derived macrophages from peripheral blood and stimulated them with IFNβ (100 U). We found that human monocyte-derived macrophages stimulated with IFNβ significantly increased SETDB2 expression compared with unstimulated controls (Figure 5C). Altogether, these results show that IFNβ increases expression of Setdb2 in wound macrophages, resulting in the macrophage phenotype switch seen during the transition to the proliferative phase.
Figure 5. IFNβ/JAK/STAT1 Pathway-Induced Setdb2-H3K9me3 and IFNβ Signaling Is Impaired in Diabetic Wound Macrophages.

(A) Setdb2 expression in wound macrophages from Setdb2f/fLyz2Cre+ and littermate controls on day 5 post-wounding, treated ex vivo with IFNβ for 8 h (n = 6 mice per group).
(B) ChIP analysis of wound macrophages from controls treated ex vivo with IFNβ for 6 h and analyzed for Setdb2 and H3K9me3 at the NF-κB binding site on the IL1β promoter (n = 6 mice per group).
(C) SETDB2 expression in human monocyte-derived macrophages (MoM) treated ex vivo with IFNβ for 24 h (n = 5 patients per group).
(D) Wound healing curve for Ifnar−/− and Ifnar+/+ mice (n = 16 mice per group). Wounds were stained with Masson’s trichrome stain. Percentage of re-epithelialization was calculated, and representative images are shown in 2x magnification (n = 10 mice). The black bar above the wound represents the entire wound distance, and the arrowheads denote the epithelial tongues for re-epithelialization (n = 5 mice per group). Intracellular flow cytometry was performed on a subset of mice on day 6 for IL-1β, TNF-α, and NOS2 in wound monocytes-macrophages isolated from wounds of Ifnar−/− and Ifnar+/+ mice (n = 5 mice per group).
(E) ChIP analysis for H3K9me3 and Setdb2 at the NF-κB binding site on the IL1β promoter in wound macrophages from Ifnar−/− mice and Ifnar+/+ controls (n = 6 mice per group).
(F) BMT was performed by injecting 5 × 106 cells from Ifnar−/− or Ifnar+/+ control mice into irradiated Ifnar+/+ control mice. Following reconstitution, a wound curve was performed (n = 11 mice per group).
(G) Setdb2 expression in BMDMs treated with IFNβ ± tofacitinib (JAK inhibitor) (n = 3 mice per group).
(H) Wound healing curve for Stat1−/− and Stat1+/+ mice (n = 15 mice per group). Wounds were harvested on day 6, paraffin embedded, and stained with Masson’s trichrome stain. Percentage of re-epithelialization was calculated, and representative images are shown in 2x magnification. The black bar above the wound represents the entire wound distance, and the arrowheads denoting the epithelial tongues for re-epithelialization (n = 5 mice per group).
(I) ChIP analysis of wound macrophages from Stat1−/− and Stat1+/+ mice for Setdb2 and H3K9me3 at the NF-κB binding site on the IL1β promoter (n = 6 mice per group).
(J) IFNβ in whole wound lysates measured via western blot in DIO and control wounds on days 0-7 post-injury (n = 25 mice per group).
All data are representative of 2-4 independent experiments. *p < 0.05, **p < 0.01. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups, ANOVA followed by Newman-Keuls post hoc test was used.
See also Figures S4 and S5.
Next, to determine whether IFNβ signaling affects wound healing, we wounded IFN-I receptor-deficient mice (Ifnar−/−) and analyzed their healing rates compared with matched controls (Ifnar+/+). Ifnarmice had significantly impaired wound healing and decreased re-epithelialization at multiple time points compared with controls (Figure 5D). Furthermore, when we examined the wounds by flow cytometry on day 5, we found that Ifnar+/+ wound macrophages (live, lin−, NK1.1−, Ly6G−, CD11b+) made significantly fewer ∥1b, Tnf, and Nos2 inflammatory cytokines compared with age-matched Ifnar−/−controls. To confirm that loss of IFNβ signaling decreases Setdb2-H3K9me3 at the promoter or promoters of inflammatory genes in wound macrophages, we isolated wound macrophages from Ifnar−/− mice and Ifnar+/+ controls on day 5 and examined via ChIP H3K9me3 and Setdb2 at the NF-κB binding site on the IL1b, TNFa, and NOS2 promoters. H3K9me3 and Setdb2 were significantly reduced in the Ifnar−/− wound macrophages compared with matched controls (Figure 5E; Figure S4). To determine whether absence of IFNβ signaling impaired wound healing, we performed a bone marrow transplant (BMT) in which bone marrow (BM) was harvested from Ifnar−/−and Ifnar+/+ donor mice and infused by tail vein injection into lethally irradiated Ifnar+/+ recipients. Complete immune reconstitution is achieved 5 weeks following the infusion of 5 × 106 whole BM cells into total body irradiation recipients. Ifnar+/+ mice with BM from Ifnar−/− demonstrated impaired healing at later points compared with Ifnar+/+ mice with BM from Ifnar+/+ mice, suggesting IFN-I signaling is at least partially responsible for wound repair (Figure 5F). Furthermore, ChIP analysis of wound macrophages from BMT animals revealed that Setdb2 and H3K9me3 were markedly reduced in wound macrophages from Ifnar+/+ mice with BM from Ifnar−/−(Figure S5).
IFN-I signals through the JAK/STAT1 pathway to promote gene transcription during viral infection; however, the role of IFN-I in wound repair is unknown. We isolated BMDMs and stimulated them with IFNβ (100 U) with or without the JAK1 inhibitor tofacitinib (50 nM). Setdb2 was significantly decreased in BMDMs treated with the JAK1 inhibitor following stimulation with IFNβ (Figure 5G). To further investigate JAK/STAT1 downstream signaling, we wounded Stat1−/− mice and matched controls (Stat1+/+) and analyzed wound repair over time. Although not as significant as the Ifnar−/− mice, Stat1−/− mice demonstrated delayed healing and impaired re-epithelialization (Figure 5H). Furthermore, ChIP analysis of Stat1−/− wound macrophages revealed decreased H3K9me3-Setdb2 at the NF-κB binding site on the IL1β promoter (Figure 5I). Altogether, these findings demonstrate that IFNβ signaling via the JAK/STAT1 pathway is essential for normal wound healing and that the IFN-I pathway is critical for increased Setdb2 expression in wound macrophages.
Next, to determine the kinetic regulation of IFNβ in normal and diabetic wounds, we examined protein levels of IFNβ by western blot. IFNβ was significantly increased in normal wounds on day 5 post-injury, correlating with the increased expression of Setdb2 in normal wound macrophages at day 5 (Figure 5J). In DIO mice, however, there was significantly decreased IFNβ protein on days 3 and 5 post-injury, correlating with the lack of Setdb2 expression in DIO wound macrophages at these time points. These data suggest that IFNβ is important for increasing Setdb2 expression and that decreased IFNβ signaling in diabetic wound tissue may lead to decreased Setdb2 with the associated failure of wound macrophages to transition to a reparative phenotype.
Loss of Setdb2 Altered UA Metabolism by Regulating XO Production
The underlying link between the innate immune system and metabolic function has been well documented in the context of cancer and more recently in metabolic diseases such as T2D (Gut and Verdin, 2013). The initial trigger for inflammatory changes accompanying a metabolic derangement has not been identified; however, it is likely that epigenetic alterations lie upstream of these events. Thus, epigenetics plays a critical role in the complex interplay between genes and environmental cues that drive inflammation (Barres et al., 2013; Vander Heiden et al., 2009). Because we identified Setdb2 as a regulator of macrophage-mediated inflammation in wound repair, we examined whether Setdb2 had any effect on metabolic function promoting dysregulated inflammation in wounds. We performed liquid chromatography-mass spectrometry (LC-MS) on BMDMs isolated from Setdb2f/fLyz2Cre+ and littermate control mice. We then analyzed the data using bioinformatics software to assess for differences among 55 cell metabolites. We identified that Setdb2 deficiency in macrophages decreased levels of the metabolite xanthine, which is involved in purine catabolism, with few differences seen in other metabolites (Figure 6A). Specifically, xanthine is a substrate that is converted to UA by the xanthine oxo-reductase (XOR) enzyme, encoded for by the xanthine dehydrogenase (Xdh) gene. This is relevant because soluble UA has both direct and indirect pro-inflammatory effects in macrophages and is elevated in T2D, driving low-grade inflammation (Braga et al., 2017). Specifically, UA increases NF-κB-dependent gene transcription and increases activation of the nod-like receptor protein-3 (NLRP3) inflammasome, resulting in overabundance of functional IL-1β (Martinon et al., 2006; Uratsuji et al., 2012). It is unknown what regulates UA in diabetic tissue or whether it is important in propagating excess inflammation in diabetic wound repair.
Figure 6. Loss of Setdb2 Altered UA Metabolism by Regulating XO Production.

(A) Xanthine levels in BMDMs isolated from Setdb2f/fLyz2Cre+ mice and littermate controls stimulated for 6 h with LPS, measured by liquid chromatography-mass spectrometry (LC-MS) (n = 5 mice per group).
(B) UA levels in cultured monocytes isolated from Setdb2f/fLyz2Cre+ mice and littermate controls (n = 10 mice per group).
(C) UA levels in wound macrophages isolated on day 5 from Setdb2ff/Lyz2Cre+ and littermate controls (n = 10 mice per group).
(D) XO levels in wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls by western blot on day 5 post-injury (n = 8 mice per groups).
(E) ChIP analysis of H3K9me3 at the NF-κB binding site on the Xdh promoter in day 5 wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls stimulated ex vivo with IFNβ (n = 6 mice per group).
(F) UA levels in wound macrophages isolated on day 5 from control mice stimulated ex vivo for 6 h with IFNβ (n = 6 mice per group).
(G) Sequential ChIP was performed at the Xdh promoter on ex vivo wound macrophages treated for 6 h with IFNβ. DNA bound to Setdb2 was isolated by ChIP, followed by a second immunoprecipitation with antibody to NF-κB or IgG (n = 6 mice per group).
(H) Representative picture of the regulation of Xdh gene expression and UA production during a diabetic state when there is decreased Setdb2.
All experiments ae representative of 3 independent experiments. *p < 0.05. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups,ANOVA followed by Newman-Keuls post hoc test was used.
See also Figure S6.
To examine the effects of Setdb2 on UA production, we isolated peripheral blood monocytes from Setdb2f/fLyz2Cre+ and littermate control mice and found a significant increase in UA in the Setdb2-deficient cells (Figure 6B). The increased production of UA was also seen in Ifnar-deficient cells (Figures S6). Next, we verified these findings in wound macrophages and found that UA is increased in wound macrophages fromSetdb2f/fLyz2Cre+ mice (Figure 6C).
Through a rate-limiting reaction, increased XOR activity results in decreased xanthine and increased UA. Given our results, we examined whether Setdb2 regulates Xdh expression and thus controls XOR enzyme production. Wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls were isolated, and levels of XOR were quantified by western blot. The Setdb2-deficient wound macrophages generated significantly more XOR with respect to their littermate controls (Figure 6D). To more fully examine the mechanism, we isolated wound macrophages from Setdb2f/fLyz2Cre+ mice and littermate controls, stimulated them ex vivo with IFNβ, and analyzed them via ChIP for the presence of H3K9me3 at the NF-κB binding site on the Xdh promoter. Setdb2f/fLyz2Cre+ wound macrophages had decreased repressive H3K9me3 at the Xdh promoter, resulting in increased Xdh transcription and XOR production (Figure 6E). Furthermore, when we isolated wound macrophages from control mice, stimulated with IFNβ, increased Setdb2 expression resulted in a reciprocal decrease in UA production (Figure 6F). To determine the mechanism for Setdb2 localization to the NF-βB binding site on the Xdh promoter, we performed sequential ChIP (ChIP-reChIP) to help identify co-localization of proteins interacting with a specific DNA sequence (i.e., NF-κB binding site of the Xdh promoter) using double and independent rounds of ChIP, as previously detailed (Beischlag et al., 2018). In wound macrophages treated with IFNβ, we first isolated DNA bound to Setdb2, followed by a second immunoprecipitation with antibody to NF-κB or immunoglobulin G (IgG). We found that the Xdh promoter was co-occupied by Setdb2 and NF-κB, which led to transcriptional silencing of Xdh by Setdb2-H3K9me3 in wound macrophages (Figure 6G). These findings suggest that Setdb2 plays a role in regulating UA production and that manipulation of this pathway may be a viable therapeutic target (Figure 6H).
Diabetic Macrophages Demonstrate Increased XO and UA, while Blockade of XO Decreased Inflammation and Improved Healing
Because both IFNβ and Setdb2 are decreased in diabetic wounds during the critical period when macrophages transition from an inflammatory to a reparative phenotype, we examined DIO wound macrophages for xanthine, XO, and UA. Consistent with the Setdb2f/fLyz2Cre+ wound macrophages, DIO wound macrophages had significantly less Setdb2 at the NF-κB binding site on the Xdh promoter when examined by ChIP (Figure 7A). Increased levels of XO protein were identified in DIO wound macrophages by western blot (Figure 7B). Finally, commensurate with the increased production of XO in DIO wound macrophages, there was decreased xanthine in BMDMs from DIO mice, as well as increased UA in DIO peripheral blood monocytes and wound macrophages (Figures 7C-7E). Altogether, these results suggest that the diabetic wound macrophages function similar to the Setdb2-deficient macrophages, in which they make more XO and UA, likely through a Setdb2-dependent mechanism.
Figure 7. Diabetic Macrophages Demonstrate Increased XO and UA, while Blockade of XO Decreased Inflammation and Improved Healing.

(A) ChIP analysis of Setdb2 at the NF-κB binding site on the Xdh promoter in DIO and control wound macrophages at day 5 post-injury (n = 6 mice per group).
(B) XO in wound macrophages from control and DIO wounds at day 5 post-injury via western blot (n = 8 mice per group).
(C) Xanthine levels in BMDMs isolated from DIO and control mice stimulated for 6 h with LPS, measured by liquid chromatography-mass spectrometry (LC-MS) (n = 5 mice per group).
(D) UA levels in cultured monocytes isolated from DIO and control mice (n = 10 mice per group).
(E) UA levels in wound macrophages isolated on day 5 from DIO and control mice (n = 6 mice per group).
(F) IL-1β in DIO BMDMs treated with UA (50, 150, and 450 mg/mL) for 24 h and analyzed by western blot (n = 3 mice per group).
(G) ∥1b expression in DIO BMDMs stimulated with allopurinol (450 nM) for 6 h (n = 3 mice per group).
(H) UA in DIO wound macrophages isolated on day 5 and stimulated ex vivo with allopurinol (450 nM) for 6 h (n = 6 mice per group).
(I) IL-1β in DIO wound macrophages isolated on day 5 and stimulated ex vivo with allopurinol (450 nM) for 6 h (n = 6 mice per group).
(J) Wound healing curve for DIO mice following daily injection of DMSO or allopurinol (20 mg/kg) starting on day 3 post-injury (n = 15 mice per group).
All data are representative of 2–4 independent experiments. *p < 0.05, **p < 0.01. Data are presented as the mean ± SEM. Data were first analyzed for normal distribution, and if data passed the normality test, two-tailed Student’s t test was used. For comparison among multiple groups, ANOVA followed by Newman-Keuls post hoc test was used.
UA directly increases IL1β expression and indirectly increases activation of the NLRP3 inflammasome, which in turn converts pro-interleukin 1β (pro-IL-1β) to active IL-1β (Martinon et al., 2006; Mirza et al., 2014). Given that Setdb2 controls UA production via Xdh-XO, we examined the effects of UA on DIO macrophages. First, DIO BMDMs were stimulated with increasing concentrations of UA (50–450 nM), and cell supernatants were collected at 24 h and analyzed for IL-1β protein by western blot. Following UA stimulation of DIO macrophages, IL-1β increased in a dose-dependent fashion (Figure 7F). Given the effect of UA on IL-1β, we next assessed the ability of allopurinol, an XOR inhibitor that blocks conversion of xanthine to UA, to decrease the expression of IL1β in the setting of diabetes. DIO BMDMs were stimulated with lipopolysaccharide (LPS) with or without allopurinol, and ∥1b gene expression was determined. Allopurinol-treated DIO BMDMs had a significant decrease in ∥1b expression (Figure 7G). We harvested DIO wound macrophages and treated them ex vivo with allopurinol to determine in vivo whether allopurinol can alter UA production. DIO wound macrophages treated with allopurinol displayed decreased UA and IL-1β production (Figures 7H and 7I). To translate these findings in vivo, DIO mice were wounded and injected with allopurinol. Injection of allopurinol in the DIO mice starting on day 3 resulted in a significant improvement in healing in the DIO mice through day 7 (Figure 7J). These results suggest that XO antagonists may represent a target for the abrogation of inflammation in diabetic wounds.
DISCUSSION
In this study, we identified that the CME Setdb2 is crucial for the transition from the inflammatory to the proliferative phase of wound healing. Specifically, at the end of the inflammatory phase, increased IFNβ signals through the JAK/STAT1 pathway to increase Setdb2 expression. Setdb2 localized to NF-κB binding sites on the promoters of XO and other inflammatory genes, where it trimethylated H3K9 and suppressed transcription. Here, we demonstrated that diabetic wound macrophages failed to have a spike in IFNβ at the time of the inflammatory phase transition. Hence, diabetic wound macrophages had decreased Setdb2 localized on inflammatory cytokine promoters, resulting in unregulated transcription of XO and inflammatory genes. Furthermore, we define the mechanism or mechanisms for increased UA in diabetic wounds, where Setdb2 directly regulated macrophage UA production. Thus, the manipulation of this pathway in wound macrophages, either by the use of IFNβ-induced increased expression of Setdb2 or downstream XO inhibition, offers promise as a translational therapy.
It is well established that diabetic wounds fail to heal secondary to an impaired resolution of tissue inflammation driven by wound monocyte-macrophage phenotypes (Boniakowski et al., 2017; Kimball et al., 2018; Shoelson et al., 2006; Wynn and Vannella, 2016). The literature identifies that diabetics have a predisposition to harbor a resilient pro-inflammatory macrophage phenotype beyond the early inflammatory phase of healing (Gallagher et al., 2015; Kimball et al., 2017, 2018; Mantovani et al., 2013; Mirza and Koh, 2011; Sica and Mantovani, 2012). The etiology behind this resilient pro-inflammatory macrophage population remains unclear. Here, we identified Setdb2 as a regulator of macrophage phenotype in both normal and diabetic wounds. Furthermore, we established the regulation of Setdb2 during this process via the IFNβ/JAK/STAT1 pathway and identified two other mechanisms of impaired wound healing in diabetes: failure of the acute increase of IFNβ in wound tissue and increased levels of UA produced by wound macrophages.
There is a paucity of literature regarding the role of IFN-I in wounds. IFN-Is (IFNα, IFNα, and IFNK) are known to be secreted by many cell types, including plasmacytoid dendritic cells in peripheral blood (Siegal et al., 1999). IFN-Is control early inflammatory responses through regulation of cytokine and T cell immune phenotype (Gregorio et al., 2010). Because IFN-I is present in wound tissue and regulates Setdb2 in vitro, we evaluated IFNβ in normal and diabetic wound tissue. In this study, we found that IFNβ is increased at the end of the inflammatory phase and increased Setdb2 expression via the JAK/STAT1 pathway. This in turn decreased inflammation via selective H3K9me3 at inflammatory and metabolic gene promoters. This was confirmed with BMT studies that showed wild-type mice transplanted with Ifnar−/− BM had impaired wound healing, and as expected, macrophages isolated from these wounds demonstrated decreased Setdb2-H3K9me3 at the IL-1β promoter. Furthermore, the impact of Setdb2 absence is implicated beyond macrophage inflammation, because Setdb2f/fLyz2Cre+ mice demonstrated reduced collagen deposition, suggesting the persistent macrophage inflammatory phenotype can influence surrounding cell function, particularly fibroblasts. In addition, we found that levels of IFNβ are significantly decreased in diabetic wounds, suggesting that a lack of IFN-I at the end of the inflammatory phase of wound repair in diabetics may contribute to excess inflammation through a lack of Setdb2 expression. In this regard, our research identified that direct stimulation of Setdb2 via the IFN-I/JAK/STAT1 pathway with local administration of recombinant IFNβ is a therapeutic option for diabetic wounds. It is important to emphasize the significance of the timing of administration of this potential therapy, because our data would suggest that the kinetics are important and the administration should not occur so early that it disturbs the necessary early inflammatory response for the protection of host defense and clearance of cellular debris.
Given the metabolic effects of diabetes, its relationship to epigenetics, and its association with a chronic systemic inflammatory state, there has been considerable interest in defining the link of metabolism, epigenetics, and inflammation. Here, using LC-MS, we identified a significant change in the metabolite xanthine in macrophages isolated from Setdb2-deficient mice and diabetic mice. Given that xanthine is a precursor for UA production via purine catabolism, we examined that pathway and found Setdb2-mediated increased production of the XO enzyme that regulated UA production. Hence, Setdb2 directly altered UA metabolism via H3K9me3 at the NF-κB binding site on the Xdh gene promoter. Because the Xdh gene encodes XO, decreased H3K9me3 results in increased XO, increased substrate drive through the purine UA pathway, and excess production of UA. This finding is of significant clinical importance, because it has long been known that serum UA levels are positively associated with T2D and lead to excess inflammation through activation of the NLRP3 inflammasome, as well as direct increased expression of pro-IL-1β transcription (Kodama et al., 2009). Furthermore, it has been speculated that UA plays a key role in the excess inflammation seen in diabetic foot ulcers; however, the etiology of this overproduction of UA in T2D patients is poorly understood. In this study, we found that UA is not only increased in wounds but also contributes to increased inflammation by overproduction of IL-1β, likely through direct effects on the NLRP3 inflammasome (Martinon et al., 2006; Mirza et al., 2014). Finally, we defined XO inhibitors as potential practical therapy and demonstrated that allopurinol reduced the production of IL-1β and improved healing in wounds from diabetic mice.
In conclusion, we identified that Setdb2 is a regulator of macrophage polarity during the inflammatory phase of wound healing, which mediates the transition from an inflammatory to a reparative phenotype. In diabetic wounds, Setdb2 expression failed to increase and macrophages remained in an inflammatory state, resulting in defective tissue repair. Through IFN-I signaling, Setdb2 regulated inflammatory cytokines and UA production via a H3K9me3 mechanism. This offers the opportunity to therapeutically target this pathway in a cell-specific manner for treatment of non-healing diabetic wounds. It will be important to further explore the expression and function of IFNβ and Setdb2 in chronic wounds, because IFNβ may be an attractive therapeutic target because of its capacity to increase Setdb2 expression, alleviate inflammation, and promote tissue repair.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katherine A. Gallagher (kgallag@med.umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were maintained at the University of Michigan Biomedical Sciences and Research Building in the Unit for Laboratory and Animal Medicine (ULAM). Mouse experiments were conducted with approval from our Institutional Animal Care and Use Committee (IACUC) and all regulatory and safety standards were strictly adhered to. C57BL/6 mice (RRID: IMSR_JAX:000664) were obtained at 6-7 weeks age from Jackson Laboratory (Bar Harbor, ME), and were maintained in breeding pairs in the ULAM facilities. Setdb2LacZ reporter mice and Setdb2f/f were created as previously published by our lab (Kroetz et al., 2015).Setdb2f/f mice were then bred with B6.129P2-Lyz2tm1(Cre)Ifo/J (Lyz2Cre) mice from the Jackson Laboratory to obtain mice deficient in Setdb2 in monocytes, macrophages and granulocytes (Clausen et al., 1999). Genotyping of floxed-cre mice was performed regularly after birth with custom primers. Ifnar−/−, Statl−/− and BALB/c control mice were obtained from Christiane Wobus, Ph.D. and maintained in breeding pairs at the ULAM facilities.
To induce a ‘pre-diabetic’ state, male C57BL/6 mice were maintained on a standard normal rodent diet (13.5% kcal saturated fat, 28.5% protein, 58% carbohydrate; Lab Diet) or standard high-fat diet (60% kcal saturated fat, 20% protein, 20% carbohydrate; Research Diets, Inc) for 12-18 weeks to induce the diet-induced obese (DIO) model of type 2 diabetes mellitus (Corredor et al., 2003; Toye et al., 2005). After the appropriate time period, HFD-fed (DIO) mice develop obesity and insulin resistance with fasting blood sugars in the mid-200 s and elevated insulin levels. All DIO/control animals underwent procedures at 20-32 weeks of age with Institutional Animal Care and Use Committee approval. For these experiments only male mice were used as female mice do not develop DIO.
Human Wound Isolation
All experiments using human samples were approved by the IRB at the University of Michigan and were conducted in accordance with the principles in the Declaration of Helsinki. For description of patient cohort see Table S2. Briefly, wounds were isolated from male age-matched patients with or without T2D who were undergoing amputation for medical reasons. Co-morbid conditions were not statistically different between the groups. Wounds were obtained from the lateral edge of wound specimens using an 8mm punch biopsy tool. Wounds were then processed for RT-PCR as described for the murine wounds. RNA with RIN scores of greater than 8 were used and all values were done with comparison to 28 s/18 s ratios and other housekeeping genes.
METHOD DETAILS
Wound Healing
For all surgical procedures, mice were anesthetized with 80mg/kg I.P. injection of Ketamine (Hospira) and 20mg/kg Xylazine (Llyod). Number of mice used per experiment can be found in the figure legend of each corresponding experiment.
Wound model
Peripheral wounds were generated by anesthetizing the mice with ketamine, removing the dorsal fur with hair removal cream (Veet), rinsing with sterile water, and creating two full-thickness skin wounds in the mid-back with a 4-mm punch biopsy.
Assessment of Wound Healing
Digital photography utilizing an 8mp iPad camera was used to assess wound healing over time, as previously described (Nathan, 2002). Wound areas were calculated using an internal scale and National Institutes of Health ImageJ software (RRID: SCR_003070). Wound photographs were then obtained each day post-injury and wound closure was calculated each day as a percentage of initial wound area. All images were evaluated by two independent-blinded observers. In allopurinol wound experiments local injection with allopurinol (20mg/kg) or vehicle control was performed at four points along the wound edge (Gallagher et al., 2007).
Wound Cell Isolation
Wounds were harvested with a 6mm punch biopsy. Wounds were then minced finely with sharp scissors and suspensions were placed in a Liberase TL (Sigma-Aldrich, St. Louis, MO, cat. No. 5101020001) and DNase I (Sigma-Aldrich, St. Louis, MO, cat. No. 9003-98-9) solution at 37°C for 30 minutes for enzymatic digestion. RPMI + FBS was then added to stop the reaction and the resultant solution was gently agitated to encourage cell separation, and transferred through a 100μm filter to result in a single cell suspension and processed for RT-PCR.
Histology
Whole wounds were excised from mice or humans using a 6-8 mm punch biopsy. Wound sections were fixed in 10% formalin overnight before embedding in paraffin. 5 μM sections were stained with Mason’s Trichrome for evaluation of reepithelialization, granulation and collagen deposition. For immunohistochemistry, paraffin embedded tissue sections were heated at 60C for 30 minutes, de-paraffinized, and rehydrated. Slides were placed in Ph9 antigen retrieval buffer and heated at 95C for 20 minutes in a hot water bath. After cooling, slides were treated with 3% H2O2 (5 minutes) and blocked using 10% goat serum (30 minutes). Overnight incubation (4C) was then performed using first antibody at a working concentration. Slides were then washed, treated with secondary antibody, peroxidase (30 minutes) and diaminobenzidine substrate. Antibodies used were CD31 (Abcam, Cat.No: ab182981, RRID:AB_2756834, working concentration: 2.5ug/ml); Pro-collagen I (Lifespan Biosciences, Cat.No: LS-C372629/123774, working concentration: 2.5ug/ml); Pro-collagen III (Abbexa, Cat.No: abx132179, working concentration: 5ug/ml); SDF-1 (St John’s laboratory, RRID:AB_1191731, Cat.No: STJ98786, working concentration: 5ug/ml); MKI67 / Ki67 Antibody (LifeSpan Biosciences, Cat. No. LS-C141898-100, RRID:AB_1061197, working concentration: 2.5ug/ml); mouse anti-SETDB2 (Yali Dou, University of Michigan) and human anti-SETDB2 (Applied Biosystems, RRID:AB_10982748). Images were quantified ImageScope software and ImageJ at 20 X magnification. Percent reepithelialization was calculated by measuring distance traveled by epithelial tongues on both sides of wound divided by total distance (Nishiyama et al., 2011; Schrementi et al., 2008).
Magnetic-Activated Cell Sorting (MACs) of Murine Wound and Human Monocyte Cell Isolates
MACs sorting of wound cell isolates was performed (Mirza et al., 2013). Briefly, wound cell isolates were incubated with fluorescein isothiocyanate (FITC)-labeled anti-mouse anti-CD3 (RRID:AB_312660), anti-NK1.1 (RRID:AB_448547), anti-CD19 (RRID:AB_2629813), and anti-Ly6G (Biolegend, RRID:AB_470400) monoclonal antibodies conjugated to FITC. Wound isolates were then washed and incubated with anti-FITC microbeads (Miltenyi Biotec, RRID:AB_244371, cat No. 130-049-601) and passed through a MACs column (Miltenyi Biotec). The resultant eluent was then incubated with anti-mouse anti-CD11b microbeads, (Miltenyi Bioter, cat No. 130-049-601). The remaining cell population was analyzed by flow cytometry and found to be 97% macrophages consistent with previous literature (Gallagher et al., 2015; Mirza et al., 2013). For human monocyte isolation, peripheral blood was collected and subjected to RBC lysis and Ficoll separation (GE healthcare). Cell suspensions were then treated with anti-human CD14 microbeads. Magnetic separation yielded 95% purity by flow cytometry.
RNA Extraction
Total RNA extraction was performed with Trizol using manufacturer’s directions. RNA was extracted using chloroform, isopropanol, and ethanol. iScript (BioRad) or Superscript III Reverse Transcriptase (ThermoFisher Scientific) kits were used to synthesize cDNA from extracted RNA. cDNA primers for IL1β, Setdb2, IFNβ, TNFα, NOS2, human Setdb2 were purchased from Applied Biosciences. RT-PCR was performed with 2x taqman PCR mix and run on a 7500 Real-Time PCR System (Applied Biosciences), and data was then reviewed in a relative quantification analysis to the 18 s ribosomal RNA (2ΔCt). All samples were assayed in triplicate. Data was then compiled in Microscoft Excel (Microsoft) and presented using Prism software (GraphPad).
PCR arrays
The murine chromatin modifying enzyme array was purchased from SABiosciences. RNA was DNase digested using the RNAeasy mini kit and reverse transcription RT2 first strand kit (QIAGEN). RT-PCR was performed according to manufacturers’ instructions and gene expression was normalized to multiple housekeeping genes.
Chromatin Immunoprecipitation (ChIP)
In brief, cross-linking was performed in a final concentration of 1% formaldehyde for 10 min at RT and cell pellets were stored at −80°C until analyzed. Cells were lysed for 10 min on ice in SDS lysis buffer supplemented with a protease inhibitor cocktail (Sigma Aldrich), syringe passed, and sonicated 3 x for 10 s using a Branson Sonifier 450 (Branson Ultrasonics) to obtain DNA fragments ranging from 200 - 1000 bp length. Five percent of the total chromatin volume was put aside for the input control. The rest of the chromatin was then incubated with antibodies against trimethylated H3K4, trimethylated H3K27 or rabbit polyclonal IgG (Millipore) overnight at 4°C (Kittan et al., 2013). The following antibodies were used for immunoprecipitation: anti-H3k9me3 (RRID:AB_306848) and anti- NF-κB (Abcam, Cambridge, MA), anti-SETDB2 (kind gift from Dr. Yali Dou, University of Michigan) and Rabbit IgG (Millipore). Immune complexes were collected after incubation with a salmon sperm DNA/agarose A mix (Invitrogen) for 1h at 4°C. The pellet was washed and the chromatin was eluted 2 x for 15 min at room temperature with 5 min at 65°C at the end of the second elution. The combined eluates were reverse cross-linked for 5h at 65°C. Samples were stored overnight at −20°C, followed by a proteinase K digestion for 1h at 45°C. DNA was recovered by a phenol/chloroform/isoamyl alcohol extraction followed by an ethanol precipitation. Precipitated DNA was analyzed using quantitative real-time PCR on a TaqMan 7500 sequence detection system (Applied Biosystems). The following primers were used to amplify DNA in samples: IL1β: 5’ - GCAGGAGTGGGTGGGTGAGT- 3’ and 5’ -CAGTCTGATAATGCCAGGGTGC- 3’, NOS2: 5’ -GTCCCAGTTTTGAAGTGACTACG - 3’ and 5’ -GTTGTGACCCTGGCAGCAG- 3’, TNFα: 5’ - TCCTGATTGGCCCCAGATTG - 3’ and 5’ -TAGTGGCCCTACACCTCTGT - 3’. Xdh - 5’ - AAGTAACAGCAGAGCGTCATCA -3’ and 5’ - GTCTCAACCCTGGCATTTACTC-3’.
Bone Marrow Derived Macrophage Culture
Femurs and tibias of mice were flushed with RPMI and bone marrow derived macrophages were cultured (Gallagher et al., 2015). After initial cell counting and plating in RPMI, FBS, L-cell supernatant, Glutamine, P/S cells were cultured for 7 days. Briefly, on day 7, cells were replated in triplicate (3×105cells/well). When indicated, BMDMs were stimulated with/without IFNβ (10U/ml) (PBL Assay Science, catalog 12400-01) or Allupurinol (50-450nM) (Sigma-Aldrich, St. Louis, MO). For JAK1, 3 inhibition, cells were treated with 50nM tofacitinab (Cayman Chemicals) at the time of stimulation with IFNβ. For human monocyte-derived macrophages, CD14+ monocytes were cultures in complete media supplemented with 50ng/ml of M-CSF (R & D Systems) for 1 week. Adherent cells were washed and harvested with trypsin/EDTA (Lonza).
Western Blot
For western blot, whole wounds were collected from mice and ground using a tissue homogenizer to obtain a homogeneous protein suspension. Protein suspensions were then standardized for protein concentrations using a Bradford protein assay (BioRad). Equal amounts of protein were mixed with loading buffer and subjected to 4%-12% SDS gel electrophoresis under reducing conditions. Proteins were then transferred to nitrocellulose membranes and probed with primary antibodies (anti-mouse Setdb2, Xor (Abcam, Inc, RRID:AB_11154903), Ifnb (Abcam, Inc, RRID:AB_297431), ∥1 β (R&D Systems, RRID:AB_2049773)) diluted 1:500 in 5% non-fat dried milk powder in Tris-buffered saline with Tween buffer. Nitrocellulose membranes were then washed and incubated with HRP-secondary antibody (Cell Signaling, Inc) and visualized with timed chemiluminescence (Thermofisher Scientific).
Metabolite Measurement
Briefly, cell plates were rapidly rinsed with water and quenched with liquid nitrogen. Metabolites were extracted with 75% 9:1 methanol:chloroform/25% water and assayed by high performance liquid chromatography with time-of-flight mass spectrometry (HPLC-TOF-MS). Chromatographic separations were performed with an Agilent Technologies (Santa Clara, CA) 1200 HPLC system equipped with a Phenomenex (Torrance, CA) column using the following conditions: mobile phase A was 100% acetonitrile (ACN); mobile phase B was 100% 5mM ammonium acetate pH 9.9 with ammonium hydroxide; gradient program was (time, %B, flow rate) 0 min, 20%, 200 ul/min, 20 min, 100%, 200ul/min, 20.1 min, 100%, 300 ul/min; column temperature was 35°C; injection volume was 80 ul; autosampler temperature was 4°C. Lipids were separated on a C18 Capcell column (2mm bore by 150 mm long packed with 3 um particles). Mobile phases and gradient were used as described (Sato et al., 2010). Detection was performed on an Agilent Technologies LC/MSD TOF using a dual electrospray ionization (ESI) source in negative-ion mode. Directed data processing was performed as described. Metabolites previously implicated in GSIS (e.g., glycolytic and TCA cycle intermediates) were identified using standards, accurate mass, and isotope ratios to confirm peak assignments (Lorenz et al., 2011). For entire list of identified metabolites see Table S3.
Flow Cytometry
For surface staining, wound cell isolates were collected either directly from wounds or after ex vivo stimulation. Two wounds were collected per mouse and three mice were pooled for one biological sample. Following red cell lysis and Ficoll separation, cells were processed for surface staining (Kimball et al., 2018). Briefly, cells were stained with a Fixable LIVE/DEAD Yellow (Molecular Probes by Life Technologies; Ref. L34959; 1:1,000 dilution). FcR-receptors were then blocked with anti-CD16/32 (BioXCell, RRID:AB_2687830, Cat. CUS-HB-197, 1:200 dilution) for 10 minutes. Monoclonal antibodies for surface staining included: anti-CD3 (Biolegend, Cat. 100304, RRID:AB_314058, 1:400 dilution), anti-CD19-Biotin (Biolegend, Cat. 115504, RRID:AB_313638, 1:400 dilution), anti-Ter-119-Biotin (Biolegend, Cat. 116204, RRID:AB_466797, 1:400 dilution), anti-NK1.1-Biotin Cat. 108704, RRID:AB_2539358, 1:400 dilution), anti-Ly6G-Biotin (Biolegend, Cat. 127604, RRID:AB_470753, 1:400 dilution), anti-CD11b-PerCP (Biolegend, Cat. 101230, RRID:AB_2129374, 1:400 dilution) and anti-Ly6C-BV605 (Biolegend, Cat. 128035, RRID:AB_2562352, 1:400 dilution). Following surface staining, cells were washed twice, and biotinylated antibodies were labeled with streptavidin-APC-Cy7 (Biolegend, Cat. 405208, RRID:AB_2801672, 1:1,000 dilution). Next, cells were either washed and acquired for surface-only flow cytometry, or were fixed with 2% formaldehyde and then washed/permeablized with BD perm/wash buffer (BD Biosciences, Ref. 00-8333-56) for intracellular flow cytometry. After permeablization, intracellular stains included: anti-IL1β-Pro-PE Cy7 (eBioscience, Ref. 25-7114-82, RRID:AB_2573526, 1:200 dilution) and anti-TNFα-APC (Biolegend, Cat. 506308, RRID:AB_315429, 1:200 dilution). To characterize lacZ expression, the Fluoreporter LacZ antibody kit was used (Life Technologies). Samples were acquired on a 3-Laser Novocyte Flow Cytometer (Acea Biosciences) or FACs sorted on a FACsAria III Flow Sorter. FACs sorting was performed with FACsDiva Software (BD Biosciences), analysis was performed using FlowJo software version 10.0 (Tree Star), and data was compiled using Prism software (GraphPad). For the usual yield for sorted wound macrophages we obtain 1×10^5 cells for 2 wounds per mouse and pool 3 mice into one biological sample. These biological samples are then replicated 3-5 times depending on experiment with data reported as the mean and standard error of the mean among the biological samples. All populations were routinely back-gated to verify gating and purity.
Bone Marrow Transplantation
Bone marrow was harvested from C57BL/6 or Ifnar−/− donor mice and infused by tail vein injection into lethally irradiated Ifnar−/− recipients. Ablation of recipient-derived HSCs was achieved by the administration of a fractional 13 Gy dose of total body irradiation (TBI) from an X-ray orthovoltage source. Complete immune reconstitution is achieved five weeks following infusion of 5 × 106 whole bone marrow cells into TBI recipients (Coomes et al., 2010; Hubbard et al., 2008). As defined by our group, the percentage of donor-derived cells is approximately 95% +/−1% in the spleen and 82% +/−2% in the lung at this time point (Hubbard et al., 2008). Mice were recovered and wounded 6 weeks following transplantation.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism software (RRID:SCR_002798) version 6.0 was used to analyze the data. Data were analyzed for normal distribution and then statistical significance between multiple groups was determined using a one-way analysis of variance test followed by Newman-Keuls post hoc test. For all single group comparisons, if data passed normality test, we used a two-tailed Student’s t test. Otherwise data were analyzed using the Mann-Whitney U-test. All data are representative of at least two independent experiments as detailed in the figure legends. A P value of less than or equal to 0.05 was significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD16/32 | BioXCell | CUS-HB-197, RRID:AB_2687830 |
| FITC anti-mouse CD3 | Biolegend | Cat#100204, RRID:AB_312660 |
| FITC anti-mouse CD19 | Biolegend | Cat#115506 RRID:AB_2629813 |
| FITC anti-mouse Ly6G | Biolegend | Cat#127606, RRID:AB_470400 |
| FITC anti-mouse NK-1.1 | Biolegend, | Cat#108706, RRID:AB_448547 |
| FITC anti-mouse CD11c | Biolegend | Cat#117306, RRID:AB_313774 |
| Anti-Histone H3(tri methyl K9) | Abcam | ab8898, RRID:AB_306848 |
| Anti-Setdb2 | Gift of Dr. Yali Dou, University of Michigan | |
| Anti-interferon beta | Abcam | Ab140211, RRID:AB_297431 |
| Anti-xanthine oxidase | Abcam | Ab176165, RRID:AB_11154903 |
| Anti-CD31 | Abcam | Ab182981, RRID:AB_2756834 |
| Anti-MKI67/Ki67 | Lifespan Biosciences | LS-C141898-100, RRID:AB_1061197 |
| Anti-pro-collagen III | Abbexa | Abx132179, RRID:AB_2801669 |
| Anti-SDF-1 | St John's laboratory | STJ98786, RRID:AB_1191731 |
| Anti-IL1 beta | R&D Systems | AF-401-NA, RRID:AB_41668 |
| Anti-CD3 | Biolegend | 100304, RRID:AB_314058 |
| Anti-CD19-Biotin | Biolegend | 115504, RRID:AB_313638 |
| Anti-Ter-119-Biotin | Biolegend | 116204, RRID:AB_466797 |
| Anti-NK1.1-Biotin | Biolegend | 108704, RRID:AB_2539358 |
| Anti-Ly6G-Biotin | Biolegend | 127604, RRID:AB_470753 |
| Anti-CD11b-PerCP | Biolegend | 101230, RRID:AB_2129374 |
| Anti-Ly6C-BV605 | Biolegend | 128035, RRID:AB_2562352 |
| Streptavidin-APC-Cy7 | Biolegend | 40520, RRID:AB_2801672 |
| Anti-IL1β-Pro-PE Cy7 | eBioscience | 25-7114-82, RRID:AB_2573526 |
| Anti-TNFα-APC | Biolegend | 506308, RRID:AB_315429 |
| Biological Samples | ||
| Human Wound punch biopsy | University of Michigan Health System | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tofacitinib CP690,550 | Cayman Chemical | Cat#11598 |
| Allopurinol | Sigma-Aldrich | Cat#A8003 |
| Recombinant Mouse IFN-β | PBL Assay Science | Cat#12400-1 |
| DNase I | Sigma-Aldrich | D5025 |
| Liberase™ TM Research Grade | Sigma-Aldrich | 05401127001 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596026 |
| SuperScriptIIITM First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18080051s |
| Critical Commercial Assays | ||
| RT2 Profiler™ PCR Array Mouse Epigenetic | Qiagen | Cat#PAMM-085Z |
| Cd11b MicroBeads | Miltenyi Biotec | Cat#130-049-601 |
| Anti-FITC MicroBeads | Miltenyi Biotec | Cat#130-048-701 |
| Anti-Rabbit HRP-DAB Cell & Tissue Staining Kit | R&D | Cat#CTS005 |
| Bio-Plex Pro™ Mouse Cytokine Assay | Bio-Rad | Cat#171304070M |
| DuoSet ELISA Mouse IL-1β | R&D | Cat# DY401-05 |
| Amplex Red Uric Acid Assay | Thermo Fisher Scientific | Cat#A22181 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock#000664, RRID: IMSR_JAX:000664 |
| Mouse:Balb/cJ | The Jackson Laboratory | Stock#000651, RRID:IMSR_JAX:000651 |
| Mouse: Setdb2flox/flox Lyz2Mcre | University of Michigan, Department of Pathology | https://journals.plos.org/plospathogens/article/authors?id=10.1371/journal.ppat.1005338 |
| Mouse: Setdb2flox/flox Lyz2Mcre | University of Michigan, Department of Pathology | https://journals.plos.org/plospathogens/article/authors?id=10.1371/journal.ppat.1005338 |
| Mouse: IFNAR−/− | Gift from Dr. Joan Durbin, Rutgers-New Jersey Medical School | |
| Mouse: STAT1−/− | Gift from Dr. Christiane Wobus, University of Michigan | |
| Oligonucleotides | ||
| SETDB2 gene expression assay (human) | Thermo Fisher Scientific | HS01126262_m1 |
| Il1b gene expression assay (mouse) | Thermo Fisher Scientific | Mm00434228_m1 |
| Setdb2 gene expression assay (mouse) | Thermo Fisher Scientific | Mm01318752_m1 |
| Ifnb gene expression assay (mouse) | Thermo Fisher Scientific | Mm00439552_s1 |
| Tnfa gene expression assay (mouse) | Thermo Fisher Scientific | Mm00443258_m1 |
| Nos2 gene expression assay (mouse) | Thermo Fisher Scientific | Mm00440502_m1 |
| Flox Primer Forward | 5′ CAA TGG CCA AAA CGA TAT GA 3′ | |
| Flox Primer Reverse | 5′ CTG AGC TCG CCA TCA GGC 3′ | |
| Software and Algorithms | ||
| Flowjo V10.4 | FlowJo LLC, RRID:SCR_008520 | https://www.flowjo.com/ |
| GraphPad Prizm 7 | GraphPad Software, Inc., RRID:SCR_002798 | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH, RRID:SCR_003070 | https://imagej.nih.gov/ij/ |
Highlights.
Setdb2 regulates macrophage plasticity during normal and pathologic wound repair
Setdb2 expression in wound macrophages depends on IFN-I/JAK/STAT1 signaling
This pathway is altered in chronic diabetic wounds because of failed surge of IFN-I
Elevated uric acid in diabetic wounds is driven by Setdb2 regulation of Xdh
ACKNOWLEDGMENTS
We thank Robin G. Kunkel, research associate in the Pathology Department, University of Michigan, for her artistic work. This study was supported by NIH (RO1-HL137919, NIH-T32 HL076123, and NIH-F32 DK-117545-01) and the Doris Duke Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2019.06.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ashcroft GS, Jeong M-J, Ashworth JJ, Hardman M, Jin W, Moutsopoulos N, Wild T, McCartney-Francis N, Sim D, McGrady G, et al. (2012). Tumor necrosis factor-alpha (TNF-α) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen. 20, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R, Kirchner H, Rasmussen M, Yan J, Kantor FR, Krook A, Näslund E, and Zierath JR (2013). Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 3, 1020–1027. [DOI] [PubMed] [Google Scholar]
- Beischlag TV, Prefontaine GG, and Hankinson O (2018). ChIP-re-ChIP: Co-occupancy Analysis by Sequential Chromatin Immunoprecipitation. Methods Mol. Biol 1689, 103–112. [DOI] [PubMed] [Google Scholar]
- Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, and Gallagher KA (2017). Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J. Immunol 199, 17–24. [DOI] [PubMed] [Google Scholar]
- Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, Castoldi A, Hiyane MI, Davanso MR, Latz E, et al. (2017). Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep 7, 39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WF 4th, Cavassani KA, Soares EM, Hirai S, Kittan NA, Schaller MA, Scola MM, Joshi A, Matsukawa A, Aronoff DM, et al. (2017). The STAT4/MLL1 Epigenetic Axis Regulates the Antimicrobial Functions of Murine Macrophages. J. Immunol 199, 1865–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Förster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Coomes SM, Wilke CA, Moore TA, and Moore BB (2010). Induction of TGF-beta 1, not regulatory T cells, impairs antiviral immunity in the lung following bone marrow transplant. J. Immunol 184, 5130–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, and Polk DB (2003). Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am. J. Physiol. Cell Physiol 284, C953–C961. [DOI] [PubMed] [Google Scholar]
- Davis FM, Kimball A, denDekker A, Joshi AD, Boniakowski AE, Nysz D, Allen RM, Obi A, Singer K, Henke PK, et al. (2019). Histone Methylation Directs Myeloid TLR4 Expression and Regulates Wound Healing following Cutaneous Tissue Injury. J. Immunol 202, 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Krieg T, and Davidson JM (2007). Inflammation in wound repair: molecular and cellular mechanisms. J. Invest. Dermatol 127,514–525. [DOI] [PubMed] [Google Scholar]
- Faglia E, Favales F, and Morabito A (2001). New ulceration, new major amputation, and survival rates in diabetic subjects hospitalized for foot ulceration from 1990 to 1993: a 6.5-year follow-up. Diabetes Care 24, 78–83. [DOI] [PubMed] [Google Scholar]
- Falanga V (2005). Wound healing and its impairment in the diabetic foot. Lancet 366, 1736–1743. [DOI] [PubMed] [Google Scholar]
- Fislová T, Sládková T, Gocník M, Mucha V, Varecková E, and Kostolanský F (2005). Differences in antibody responses of mice to intranasal or intraperitoneal immunization with influenza A virus and vaccination with sub-unit influenza vaccine. Acta Virol. 49, 243–250. [PubMed] [Google Scholar]
- Gallagher KA, Joshi A, Carson WF, Schaller M, Allen R, Mukerjee S, Kittan N, Feldman EL, Henke PK, Hogaboam C, et al. (2015). Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 64, 1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KA, Liu Z-J, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, and Velazquez OC (2007). Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Invest 117, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanassia E, Villon L, Thuan Dit Dieudonné J-F, Boegner C, Avignon A, and Sultan A (2008). Long-term outcome and disability of diabetic patients hospitalized for diabetic foot ulcers: a 6.5-year follow-up study. Diabetes Care 31, 1288–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, and Gilliet M (2010). Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med 207, 2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, and Verdin E (2013). The nexus of chromatin regulation and intermediary metabolism. Nature 502, 489–498. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh CP, Raby BA, Soto-Quirós ME, Murphy AJ, Avila L, Lasky-Su J, Sylvia JS, Klanderman BJ, Lange C, Weiss ST, and Celedón JC (2007). Comprehensive testing of positionally cloned asthma genes in two populations. Am. J. Respir. Crit. Care Med 176, 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewagama A, and Richardson B (2009). The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 33, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks CW, Selvarajah S, Mathioudakis N, Sherman RE, Hines KF, Black JH 3rd, and Abularrage CJ (2016). Burden of Infected Diabetic Foot Ulcers on Hospital Admissions and Costs. Ann. Vasc. Surg 33, 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard LLN, Ballinger MN, Wilke CA, and Moore BB (2008). Comparison of conditioning regimens for alveolar macrophage reconstitution and innate immune function post bone marrow transplant. Exp. Lung Res 34, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Wen H, Corsa CAS, Liu T, Coelho AL, Allen RM, Carson WF 4th, Cavassani KA, Li X, Lukacs NW, et al. (2009). Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Satterfield K, Lee S, Harkless LB, and Lavery LA (2009). Mortality of first-time amputees in diabetics: a 10-year observation. Diabetes Res. Clin. Pract 83, 126–131. [DOI] [PubMed] [Google Scholar]
- Kimball A, Schaller M, Joshi A, Davis FM, denDekker A, Boniakowski A, Bermick J, Obi A, Moore B, Henke PK, et al. (2018). Ly6CHi Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol 38,1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball AS, Joshi A, Carson WF 4th, Boniakowski AE, Schaller M, Allen R, Bermick J, Davis FM, Henke PK, Burant CF, et al. (2017). The Histone Methyltransferase MLL1 Directs Macrophage-Mediated Inflammation in Wound Healing and Is Altered in a Murine Model of Obesity and Type 2 Diabetes. Diabetes 66, 2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, Carson WF 4th, Mukherjee S, Grembecka J, Cierpicki T, et al. (2013). Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 8, e78045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama S, Saito K, Yachi Y, Asumi M, Sugawara A, Totsuka K, Saito A, and Sone H (2009). Association between serum uric acid and development of type 2 diabetes. Diabetes Care 32, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroetz DN, Allen RM, Schaller MA, Cavallaro C, Ito T, and Kunkel SL (2015). Type I Interferon Induced Epigenetic Regulation of Macrophages Suppresses Innate and Adaptive Immunity in Acute Respiratory Viral Infection. PLoS Pathog. 11, e1005338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MA, Burant CF, and Kennedy RT (2011). Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem 83, 3406–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Biswas SK, Galdiero MR, Sica A, and Locati M (2013). Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol 229, 176–185. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, and Locati M (2008). Macrophage activation and polarization. Front. Biosci 13, 453–461. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Goutassociated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- Mirza R, and Koh TJ (2011). Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 56, 256–264. [DOI] [PubMed] [Google Scholar]
- Mirza RE, Fang MM, Ennis WJ, and Koh TJ (2013). Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes 62, 2579–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, and Koh TJ (2014). Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 63, 1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza RE, Fang MM, Novak ML, Urao N, Sui A, Ennis WJ, and Koh TJ (2015). Macrophage PPARγ and impaired wound healing in type 2 diabetes. J. Pathol 236, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C (2002). Points of control in inflammation. Nature 420, 846–852. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Kii I, Kashima TG, Kikuchi Y, Ohazama A, Shimazaki M, Fukayama M, and Kudo A (2011). Delayed re-epithelialization in periostin-deficient mice during cutaneous wound healing. PLoS ONE 6, e18410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcheray F, Viaud S, Rimaniol A-C, Léone C, Samah B, Dereuddre-Bosquet N, Dormont D, and Gras G (2005). Macrophage activation switching: an asset for the resolution of inflammation. Clin. Exp. Immunol 742, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Riboldi E, Ippolito A, and Sica A (2015). Molecular and epigenetic basis of macrophage polarized activation. Semin. Immunol 27, 237–248. [DOI] [PubMed] [Google Scholar]
- Rayamajhi M, Humann J, Kearney S, Hill KK, and Lenz LL (2010). Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence 1, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Nakamura T, Aoshima K, and Oda Y (2010). Quantitative and wide-ranging profiling of phospholipids in human plasma by two-dimensional liquid chromatography/mass spectrometry. Anal. Chem 82, 9858–9864. [DOI] [PubMed] [Google Scholar]
- Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, Bauer L, Kandasamy RK, Griesshammer IM, Kosack L, et al. (2015). The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat. Immunol 16, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrementi ME, Ferreira AM, Zender C, and DiPietro LA (2008). Site-specific production of TGF-beta in oral mucosal and cutaneous wounds. Wound Repair Regen. 16, 80–86. [DOI] [PubMed] [Google Scholar]
- Sen CK (2009). Wound healing essentials: let there be oxygen. Wound Repair Regen. 17, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, and Deng JC (2009). Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest 119, 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, and Goldfine AB (2006). Inflammation and insulin resistance. J. Clin. Invest 116, 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, and Mantovani A (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, and Liu YJ (1999). The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837. [DOI] [PubMed] [Google Scholar]
- Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, et al. (2005). A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686. [DOI] [PubMed] [Google Scholar]
- Uratsuji Hau., Tada Y, Kawashima T, Kamata M, Hau CS, Asano Y, Sugaya M, Kadono T, Asahina A, Sato S, and Tamaki K (2012). P2Y6 receptor signaling pathway mediates inflammatory responses induced by monosodium urate crystals. J. Immunol 188, 436–444. [DOI] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, and Tschopp J (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol 11, 136–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.