

Abstract
Standard therapy of osteosarcoma (OS) and Ewing sarcoma (EW) rests on cytotoxic regimes, which are largely unsuccessful in advanced patients. Preclinical models are needed to break this impasse. A panel of patient-derived xenografts (PDX) was established by implantation of fresh, surgically resected osteosarcoma (OS) and Ewing sarcoma (EW) in NSG mice. Engraftment was obtained in 22 of 61 OS (36%) and 7 of 29 EW (24%). The success rate in establishing primary cell cultures from OS was lower than the percentage of PDX engraftment in mice, whereas the reverse was observed for EW; the implementation of both in vivo and in vitro seeding increased the proportion of patients yielding at least one workable model. The establishment of in vitro cultures from PDX was highly efficient in both tumor types, reaching 100% for EW. Morphological and immunohistochemical (SATB2, P-glycoprotein 1, CD99, caveolin 1) studies and gene expression profiling showed a remarkable similarity between patient’s tumor and PDX, which was maintained over several passages in mice, whereas cell cultures displayed a lower correlation with human samples. Genes differentially expressed between OS original tumor and PDX mostly belonged to leuykocyte-specific pathways, as human infiltrate is gradually replaced by murine leukocytes during growth in mice. In EW, which contained scant infiltrates, no gene was differentially expressed between the original tumor and the PDX. A novel therapeutic combination of anti-CD99 diabody C7 and irinotecan was tested against two EW PDX; both drugs inhibited PDX growth, the addition of anti-CD99 was beneficial when chemotherapy alone was less effective. The panel of OS and EW PDX faithfully mirrored morphologic and genetic features of bone sarcomas, representing reliable models to test therapeutic approaches.
Subject terms: Bone cancer, Sarcoma
Introduction
Osteosarcoma (OS) and Ewing sarcoma (EW), the two most common primary tumors of bone, are high-grade malignant neoplasms with very aggressive behavior and high tendency to form metastasis; they arise frequently in children and remain prominent among teenagers and young adults1–4.
Patients are still treated with conventional therapies comprising a combination of high-dose multidrug chemotherapy associated with local control of the tumor by surgery and/or radiotherapy1,3,5. As a consequence of this multimodal treatment, patients with localized disease at diagnosis have a 5-year survival rate of nearly 65% for OS and 70% for EW4,6. However, patients with disseminated disease at diagnosis and patients who fail first-line treatment have survival rates as low as 30–35%2,7. Furthermore, heavy side effects severely compromise the quality of life in these young patients. There is a strong demand from patients, families and oncologists of therapies with improved efficacy and reduced side effects.
Any further improvement in the design of innovative therapeutic approaches requires a better understanding of tumor evolution, development of drug resistance and the testing of new compounds in appropriate experimental models8–12. Recently the development of patient-derived xenografts (PDXs) obtained by direct implant of surgically resected tumors in immunodeficient mice, has offered a more accurate and reliable preclinical model of cancer9,13–18. We present here a new large panel of OS and EW PDX and primary cell cultures obtained from patients treated at IRCCS Istituto Ortopedico Rizzoli, and we show that PDX faithfully mirror the molecular and cellular phenotype of the original human tumor.
Methods
Tissue sampling
Tumor samples were obtained from surgical specimens under sterile conditions. Whenever sample size was deemed to be sufficient, the available material was split into four parts and processed as follows: (1) the tissue to be implanted in immunodeficient mice for the generation of PDX was placed in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% Fetal Bovine Serum (FBS) (Euroclone) and antibiotics (penicillin, streptomycin), hereafter referred to as complete medium; (2) tissue for genetic analyses was frozen in liquid nitrogen and stored at −80 °C; (3) tissue for histopathology and immunohistochemistry was fixed in a 10% formalin solution; (4) any remaining tissue was used for in vitro cultures (see below).
Mice and establishment of PDX
Immunodeficient NOD Scid gamma (NSG) mice were bred under sterile conditions in our animal facilities from founders originally obtained from Charles River, Italy. To generate PDXs, a fresh tumor specimen measuring approximately 4 mm3 was implanted subcutaneously (s.c.) at the level of trans-scapular brown fat of 5–11-week-old NSG male mice within an average of 1–2 hours following patient’s surgery. Tumor growth was monitored at least twice weekly using calipers until it reached a maximal volume of 2.5 cm3, then the mouse was sacrificed by CO2 inhalation and cervical dislocation, the tumor was removed, and an accurate necropsy was performed to assess metastatic spread. The tumor was minced with scissors and tumor fragments were implanted in NSG mice; the remaining fragments were immersed in 90% FBS + 10% DMSO for viable storage in liquid nitrogen or used for histopathological and molecular analyses.
Growth of established PDX in different immunodeficient mice
Established PDXs were also implanted in 5-11-week-old BALB/c Rag2−/−; Il2rg−/− (hereafter referred to as RGKO) mice bred in our animal facilities from founders kindly given to us by Drs. T. Nomura and M. Ito of the Central Institute for Experimental Animals (Kawasaki, Japan)19. Equal amounts of PDX fragments, obtained as described above from tumors grown in NSG mice, were implanted in parallel in RGKO mice and in NSG mice. Tumor growth was measured as described above.
Primary patient-derived or PDX-derived cell lines
Tissue samples obtained from the patient or from a PDX were minced into small pieces and placed in complete medium into 60-mm dishes (Falcon) incubated at 37 °C in a 5% CO2 humified atmosphere. When the outgrowth cultures formed a confluent monolayer, the cells were sub-cultured after enzymatic removal with 0.05% trypsin-EDTA and maintained in vitro for at least 10 passages, before being processed for in vitro studies. Cell lines were authenticated through STR analysis (PowerPlex ESX Fast System, Promega) in comparison to the profile of the original surgical specimen and of the PDX when appropriate, moreover the human origin of in vitro cultures was confirmed by PCR analysis with species-specific primers.
Histopathology and immunohistochemistry
The tissues were fixed in 10% buffered formalin, routinely processed, and embedded in paraffin. Serial, 3-μm-thick, paraffin sections mounted on pre-coated slides were processed according to standardized automated procedures (Ventana Medical Systems, Tucson AZ, USA), and immunostained with the following antibodies: CD99 (Ventana, Mouse Monoclonal antibody O13, pre-diluted), SATB2 (Santa Cruz Biotechnology, Mouse Monoclonal antibody SATBA4B10, 1:200 dilution), RUNX2 (Santa Cruz Biotechnologies, SC-101145 27-K 1:10 dilution), caveolin-1 (BD - Transduction Labs, 610058 1:500 dilution), anti MDR1 P-gp (ABCB1), clone JSB-1 (Monosan - MON9011-1 1:50 diluition), or with buffer alone (negative control). Pretreatment for antigen retrieval was performed at 95 °C with Tris-EDTA, pH 8.00 for 20 minutes. Staining was performed with the UltraView Universal DAB Detection Kit (Ventana Medical Systems, Tucson AZ, USA). Appropriate positive and negative controls were included in each run, furthermore all stained sections included non-tumor mouse cells, such as endothelial cell, myopericytes and fibroblasts which were invariably negative (see Fig. 1). For morphological analyses the slides were stained with haematoxylin, rehydrated and coverslipped.
Figure 1.

Histologic and immunohistochemical features of patients’ tumors and corresponding PDX at different in vivo passages. OS sections were stained with hematoxylin and eosin (H&E) or with antibodies against OS biomarkers SATB2 and PGP. PDXs closely resembles patient’s tumor, including the production of neoplastic bone and the presence of anaplastic cells. SATB2 and PGP expression of PDX mirrored that of patient’s tumor. EW sections were stained with H&E or with antibodies against EW biomarkers CD99 and CAV-1. EW PDXs consist of small round cell sheets, closely packed and without matrix, resembling patient’s tumors. CD99 and CAV-1 expression of PDX mirrored that of patient’s tumor. Bar: 50 μm.
Whole gene expression analysis
RNA from 9 EW and 11 OS samples was hybridized on Agilent whole human genome microarray (#G4851C, Agilent Technologies), which represents 60k unique human transcripts. Gene expression analysis was conducted on PDXs that were representative of the major clinical variables under study, i.e. pediatric and adult cases, treated and untreated cases, primary, relapsed and metastatic cases, in all instances selecting a PDX that had yielded an in vitro culture (see Supplementary Table 1). One-color gene expression was performed according to the manufacturer’s procedure. Briefly, RNA quality was assessed by Agilent Bioanalyzer to have a RIN (RNA integrity number) higher than 7. Labeled cRNA was synthesized from 100 ng of total RNA using the Low Input Quick-Amp Labeling Kit, one color (Agilent Technologies) in the presence of cyanine 3-CTP. Hybridization was performed at 65 °C for 17 hours in a rotating oven. Images at 5 µm (3 µm) resolution were generated by Agilent scanner, Feature Extraction 10.7.3.1 software (Agilent Technologies) was used to obtain the microarray raw-data. Data are deposited in the ArrayExpress database (accession E-MTAB-7568).
Bioinformatic data analysis
Data were normalized and analyzed using GeneSpring GX v.14.8 software (Agilent Technologies). Data transformation was applied to set all the negative raw values at 1.0, then the quintile normalization was applied. The probes detected in at least one sample were used for statistical analyses. Unsupervised principal component analysis and correlation analysis (Pearson’s correlation) were performed to assess sample similarity and to assess the global gene expression profile of PDX models. Differentially expressed genes were selected to have a ≥2-fold expression difference between matching PDX and primaries and an adjusted p-value ≤ 0.05 at paired t-test, with Benjamini and Hoechberg correction for false positive reduction. Hierarchical clustering was performed for OS samples with GeneSpring clustering tool using the list of differentially expressed genes and the Manhattan correlation as a measure of similarity. Pathway and network analysis of differentially expressed genes was determined using the web-based software MetaCore (GeneGo, Thomson Reuters).
Sanger analysis of TP53 mutational status and assessment of fusion transcripts
DNA or RNA was extracted from the original tumor or from PDX samples using standard DNAzol or TRIzol procedure (Thermo Fisher Scientific, Foster City, CA, USA). Nucleic acid quality and concentration were evaluated by Nanodrop (Thermo Fisher Scientific). DNA aliquots of 20 µl at the concentration of 12 ng/µl for each sample were used for genotyping analysis of the TP53 mutational status, performed by mean of Sanger Sequencing on an ABI3130xl platform using the BigDye Terminator 3.1 technology (Life Technologies, Carlsbad, CA, USA) amplifying exons together with exon-intron boundaries. The obtained sequence was compared to the NCBI RefSeq (NT_010718) using CodonCode Aligner software (CodonCode Corporation, Centerville, MA, USA) and manual reading. For identification of EWS-ETS fusion transcripts, 500 ng of total RNA was reverse transcribed according the manufacturer’s protocol (High Capacity cDNA Archive Kit, Life Technologies, Carlsbad, CA, USA). cDNA was used as template to amplify EWS-ETS fusion transcripts as previously described20. All primer sequences are available upon request.
In vivo therapy of PDX-bearing mice
Freshly obtained fragments of established EW PDX were implanted s.c. in the scapular region of 6–11-week-old immunodeficient male mice. Pharmacological treatments started when tumors reached a volume of 10 mm3, i.e. a mean diameter of 2.7 mm. Animals were randomized to receive two cycles of anti-CD99 diabody C7 (dAbd C7)21 peritumorally (Diatheva srl, 1 mg per injection, 5 days/week for 2 consecutive weeks followed by one week of rest) plus irinotecan intraperitoneally (Selleckchem, 0.5 mg/kg, 5 days/week for 1 week, starting after the first week of treatment with dAbd C7); control mice were not treated. Tumor size was measured with calipers; tumor volumes were calculated according to the formula π [√(a × b)]3/6, where a = maximal tumor diameter and b = tumor diameter perpendicular to a. Mouse body weights and tumor volumes were measured at least once a week. Experimental humane endpoint was a tumor maximum volume of 3 cm3; as soon as an experimental group (usually untreated controls) overcame this threshold, all other groups were sacrificed to evaluate metastatic spread under comparable conditions. To compare the slopes of tumor growth curves, regression coefficients of linear regressions were calculated and compared pairwise by means of the Prism v 7.03 software (GraphPad Software, San Diego, CA).
Ethics approval and consent to participate
The collection of human tumor tissue was approved by the ethical committee of the IRCCS Istituto Ortopedico Rizzoli (project #0038254, approval with protocol 0009323) and patient-informed consent forms were obtained for the establishment of PDX models; all methods were performed in accordance with institutional guidelines and Italian law. All animal procedures were done in accordance with European directive 2010/63/UE and Italian Law (DL 26/2014); experimental protocols were reviewed and approved by the institutional animal care and use committee (“Comitato per il Benessere Animale”) of the University of Bologna and by the Italian Ministry of Health with letters 782/2015-PR, 208/2017-PR and 755/2018-PR.
Results
Establishment of bone sarcoma PDXs
We implanted in immunodeficient mice 90 primary or metastatic tumor samples, 61 from OS and 29 from EW patients (analytical data are shown in Supplementary Table 1). Successful engraftment was obtained in 36% of OS and 24% of EW samples (Table 1). Of note, extraskeletal OS, which have a poorer prognosis than bone OS22, yielded PDX establishment with 100% efficiency, as compared to 30% for bone OS (Table 1).
Table 1.
Establishment of bone sarcoma PDX.

♣OS vs EW, χ2 with Yates’s correction.
‡χ2, Yates’s correction.
●Fisher’s exa ct test.
†χ2, 3 × 2 contingency table.
◊OS vs EW, Wilcoxon’s non-parametric test.
§Student’s t test.
*Patient numbers as per “Neo-adjuvant therapy” line above.
No significant difference was observed in engraftment efficiency between OS primary tumors (32%), local relapses (57%) and metastases (35%); analogous conclusions held true for EW, even though limited numbers of relapses and metastases were available (Table 1). In five OS cases we received more than one specimen from the same patient, which included at least one metastatic sample (one primary tumor + metastasis, two cases of local relapse + metastasis, two cases of multiple metastases). Only in one case both specimens yielded a PDX, whereas in four cases one grew as PDX and the other(s) did not (Supplementary Table 1), leading to the conclusion that the ability to engraft was specimen-specific, rather than patient-specific.
Our clinical series contained a mix of pediatric and adult cases, reflecting the cohort of bone sarcoma patients admitted to our Institution, thus we compared their engraftment efficiencies. No significant difference was found between pediatric and adult tumors (Table 1).
After implantation of the surgical sample in mice, the time required for the appearance of a sizeable tumor was highly variable, raging between one week and one year (Table 1). OS patient’s samples generally grew in mice significantly faster than EW (9 vs. 27 weeks), however the difference disappeared in subsequent in vivo passages, which converged to shorter latency times of the order of 4–5 weeks (Table 1).
We feared that neo-adjuvant cytotoxic therapy, which is frequently administered to OS and EW patients, could jeopardize PDX establishment23. We found an opposite trend between the two tumor types. In primary OS the specimens obtained after neoadjuvant therapy gave rise to PDX with a lower efficiency than those obtained in the absence of therapy (Table 1). On the contrary, no EW specimen from untreated patients produced a PDX (Table 1). However, it should be noted that most untreated EW specimens were biopsies, which also failed to yield PDXs (Supplementary Table 1), therefore at least two variables (untreated vs. treated and bioptical vs. surgical) were at play here, but the numerosity was insufficient for a meaningful stratification of patients.
The level of necrosis induced by therapy was routinely evaluated in all treated patients. In OS patients there was no significant difference in the level of necrosis between cases that gave rise to a PDX and cases which failed to engraft. In contrast, a significant difference was found between those EW patients from which a PDX was obtained, which had a low level of necrosis, and patients which failed to produce a PDX, which had a much higher level of necrosis (Table 1).
A notable difference between our sarcoma series and most carcinoma PDX studies was that we did not observe any human lymphoma development, a frequent event occurring in one-fourth to one-third of mice receiving implants of human carcinomas23–25. As human lymphomas of mice implanted with human solid tumors arise from EBV-immortalized human infiltrating lymphocytes growing in the immunodeficient host25, the lack of lymphomas in our series could be attributed both to the extreme scarcity of infiltrating lymphocytes in bone sarcomas26 and to the young age of patients, which in Western countries have a lower prevalence of EBV positivity than adults27.
We took advantage of the availability in our animal facilities of two popular immunodeficient knockout mice, i.e. NSG and RGKO, to directly compare their permissivity to the growth of established human PDX. Overlapping growth rates were observed when equal amounts of the same in vivo passage of six OS and three EW PDX were implanted in parallel both in NSG and in RGKO (Supplementary Fig. 1), leading to the conclusion that bone sarcoma PDX established in NSG mice grow equally well in RGKO mice. Other Authors used NSG mice to establish human PDX, then switched to nude mice for subsequent passages28. The use of a more robust (and in some instances less expensive) mouse host for experiments entailing the use of large numbers of mice could be advantageous.
Establishment of bone sarcoma cell cultures from clinical samples and from PDXs
We had the opportunity to obtain tumor material also to seed in vitro primary cultures from most clinical specimens (73 of 90 cases), thus enabling us to directly compare the two major ways to establish human tumor models10,17,29.
We found that the rate of success in establishing primary cell cultures from OS tumor samples was lower than the percentage of successful PDX engraftment in mice, whereas the reverse was observed for EW (Table 2). In both tumor types primary cultures from PDX were established with a significantly higher efficiency than from the original specimen, reaching 100% for EW (Table 2).
Table 2.
PDX versus in vitro cell cultures.

†χ2, 3 × 2 contingency table.
The overall proportion of clinical specimens yielding primary cultures (24/73, 33%) overlapped that of PDX (25/73, 34%), however we found several discordant cases. On the whole, it can be said that the simultaneous use of both approaches raised the proportion of cases yielding a viable model (either in vitro or in vivo) from one-third to almost one-half (34/73, 47%). The analysis of concordant and discordant cases showed a strong imbalance between tumor types. All cases which gave rise to a PDX while failing to produce a primary culture were OS (Table 3), thus confirming the propensity of OS to grow in vivo rather than in vitro.
Table 3.
Concordance between successful in vitro culture from patient specimen and successful PDX, by tumor type*.
| Successful culture | Successful PDX | OS + EW cases | OS cases (%) | EW cases (%) |
|---|---|---|---|---|
| Yes | Yes | 15 | 8 (57%) | 7 (43%) |
| No | No | 39 | 24 (62%) | 15 (38%) |
| Yes | No | 9 | 5 (56%) | 4 (44%) |
| No | Yes | 10 | 10 (100%) | 0 (0%) |
| TOTAL | 73 | 47 (64%) | 26 (36%) |
*Each line adds to 100%.
Fidelity and stability of bone sarcoma PDX
To compare PDX models and patient’s tumors we performed histological and molecular profiling at different in vivo passages in mice.
A comparative morphological analysis of all xenografts showed that PDX maintained a striking similarity of the histological features with those of the patient’s tumors at least until the third transplant generation (see Fig. 1 for representative pictures). The expression of relevant molecular biomarkers of both tumor types, analyzed in all PDXs, was also faithfully mirrored by PDX, in particular SATB2 and PGP in OS PDX (Fig. 1), CD99 and caveolin 1 in EW PDX (Fig. 1).
Fusion transcripts of EW PDX also mirrored those found in patients (Supplementary Table 2). As TP53 mutations are relatively rare (<10%) in EW30, we compared gene sequences of the patient’s sample and of the PDX, because TP53 alterations can arise during the adaptation to in vitro culture of human sarcoma cells, resulting in a much higher proportion of cell lines harboring TP53 mutations than actual human tumors31. The results showed that de novo TP53 mutations did not arise in EW PDX (Supplementary Table 2), further confirming that PDX reliably reproduce the molecular features of the original tumor.
Gene expression profiling
To gain a better insight into the similarity of the PDX with the original tumor, we performed a global gene expression correlation analysis between gene expression profiles of primary OS and EW samples and the corresponding PDX. Unsupervised clustering using all (40 k) genes and all samples showed that each tumor histotype formed a separate cluster, and all samples matching tumors and PDXs derived from the same patient clustered together (Fig. 2A). Indeed, the correlation between primary tumors and their PDXs was extremely high for both tumor types (Pearson’s r range r = 0.94–0.96), thus confirming the close resemblance of PDXs to the original human neoplasm. We included in the analysis two OS PDX at the sixth in vivo passage, also in these cases we observed a strong correlation with both the original specimen and the first in vivo passage (Fig. 2B,C).
Figure 2.

Unsupervised clustering using all (40 K) genes and all specimens (A). Correlation between OS (B) and EW (C) samples calculated using the whole gene expression profile obtained from microarray analysis. Correlation indexes (Pearson’s r) was used to perform the hierarchical clustering of samples (Euclidean distance). Abbreviations: PC, primary in vitro culture; F0, patient’s specimen; F1, PDX at first in vivo passage; F6, PDX at sixth in vivo passage.
For EW, the comparison of original patient samples with PDXs did not reveal any significant differentially expressed gene (fold change >2, adjusted p < 0.05 at paired t-test). On the contrary, the comparison between OS primaries and PDX yielded 397 differentially expressed genes (Fig. 3A and Supplementary Table 3).
Figure 3.

(A) Heatmap of OS samples obtained using the list of 397 genes (see Supplementary Table 3) that are differentially expressed (adjusted p < 0.05) between primary tumors and PDX. Genes (columns) and samples (rows) were grouped by hierarchical clustering (Manhattan correlation). High- and low- expression is normalized to the average expression across all samples. (B) Map of “Immune response_Antigen presentation by MHC class I” pathway, which is the top scored (lowest p value) map based on Genego pathway enrichment analysis. Experimental data (OS PDX/primary tumor ratio) from microarray experiments are visualized on the map as thermometer-like figures. Significantly upregulated genes show upward, red bars, while down-regulated genes show downward, blue bars.
A Pathway Enrichment Analysis (Metacore software) showed that the genes that differentiate human OS from their PDX belong to immune functional categories (Fig. 3B, Supplementary Tables 4 and 5), in line with the idea that, upon engraftment in the mouse, human leukocytes, which also include lymphocytes, are gradually replaced by leukocytes of the immunodeficient host, lacking T, B and NK cells32.
Cell cultures displayed a reduced correlation with the primary tumor if compared to PDX (r = 0.90–0.93, Fig. 2), moreover an unsupervised principal Component Analysis revealed a marked difference between OS- and EW-derived samples, with a major distance within each group for the primary cell line sample (Supplementary Fig. 2), thus indicating that in vitro adaptation has a higher impact on the molecular profile than in vivo growth10. Primary cell cultures appeared less reliable than PDX models, a relevant feature especially in OS, that has a high level of genetic heterogeneity33,34.
Novel chemo-immunotherapeutic combination for the treatment of Ewing sarcoma
PDX are an ideal model to test therapeutic approaches. We have recently developed dAbd C7, a therapeutic bivalent antibody (diabody) against CD9921,35,36, and we were interested in testing combinations with effective drugs in clinical use, such as irinotecan37. PDX-EW#3 and PDX-EW#2 were thus treated with dAbd C7, irinotecan or both. While both dAbd C7 and irinotecan effectively hampered tumor growth of both PDXs, the combined treatment provided a definite advantage in comparison to single treatments in PDX-EW#3 (Fig. 4). In PDX-EW#2, which displayed a faster growth rate than PDX-EW#3, the high effectiveness of irinotecan alone was not significantly enhanced by the combined treatment (Fig. 4). Interestingly, we found that 40% (2 of 5) untreated mice bearing PDX-EW#3 tumors had lung metastases, whereas all treated mice were metastasis-free, thus suggesting that this PDX could be further developed to analyze the effect of therapeutic treatments on metastatic spread of EW.
Figure 4.
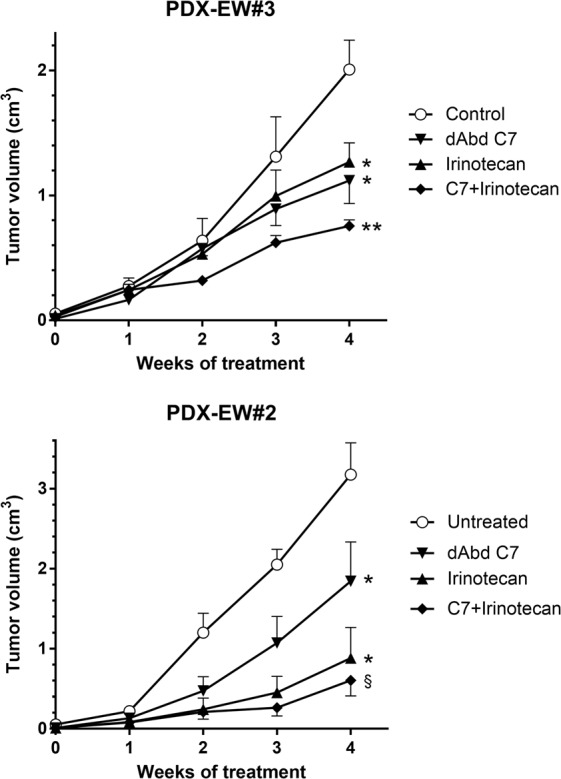
Inhibition of PDX-EW#3 and PDX-EW#2 (as indicated) tumor growth by a combination of anti-CD99 diabody C7 and irinotecan. Groups of 5–6 mice were treated as described under Material and Methods. Statistical comparisons: *slope of linear regression significantly different from untreated, p < 0.05; **slope of linear regression significantly different from all other groups, p < 0.01 at least; §slope of linear regression significantly different from untreated and from C7, p < 0.01 at least, p = 0.17 versus irinotecan.
Discussion
We generated a large panel of bone sarcoma PDX, namely OS and EW, comprising a sizeable number of PDX from pediatric patients, thus creating a powerful tool for future prognostic and sensitivity analyses. All the PDXs described in this work are available for distribution, within the limits of the informed consent of patients, upon the establishment of a standard material transfer agreement.
PDX have been so far successfully established and characterized for many different cancer types, such as colorectal38,39, pancreatic40, lung41, breast42–44, ovarian45,46 and endometrial cancer47. Together with other bone tumor PDX series17,28,48–53, the systematic collection of PDX from bone sarcomas, including EW, allows the generation of a wider repository of reliable models for testing drug sensitivity, biomarkers evaluation, or tuning of a personalized therapeutical schedule. A comparison of our series of bone sarcoma PDXs with those recently published by Rainusso et al.53 and Stewart et al.28 shows a lower (but not statistically significant) global percentage of engraftment (32% vs 54% vs 45%, respectively). These differences might result both from differences in the techniques of tumor implantation and from differences in the clinical series. For what concerns the former, Stewart et al.28 used enzymatic dissociation of clinical samples to obtain a cellular suspension that was then injected orthotopically in mice together with an extracellular matrix preparation, whereas in the paper by Rainusso et al.53 and in the present work tumor fragments were implanted subcutaneously. Regarding clinical series, both Rainusso et al.53 and Stewart et al.28 had a higher percentage of OS than our series, and in all cases OS showed a higher engraftment rate than EW. However, the relative proportion of OS vs EW within each clinical series does not seem to explain the higher engraftment rate reported by Rainusso et al.53 and Stewart et al.28, because they had higher engraftment rates than ours, in particular within the OS group. A further relevant aspect that might contibute to the observed differences was the different proportion of clinical samples from treated OS patients in each clinical series. As therapeutic treatments and patient’s responses were difficult to compare among the three series, the most straightforward comparison is based on untreated patients. Under this respect, both Rainusso et al.53 and our series showed comparable engraftment rates (42% vs 50%), whereas the series of Stewart et al.28 was not comparable because it contained only one untreated OS patient. A further element of difference among the three clinical series was the proportion of pediatric vs adult cases. Both Rainusso et al.53 and Stewart et al.28 only studied pediatric bone sarcomas, whereas our series included a mix of pediatric and adult cases, both among OS and EW. However, this difference should not affect the differences in engraftment, because we found that pediatric and adult bone sarcomas did not significantly differ in the engraftment rates (Table 1).
Two important properties of our PDX panel were revealed by in-depth molecular and morphological studies: a faithful reproduction of the phenotypic features of the human tumor of origin and a considerable stability at least until the 6th in vivo generation. A direct comparison of the gene expression profiles of the PDX and of the cell culture obtained from the same patient showed that the PDX better reflected the molecular features of the human tumor than the cell culture. Although the underlying cause of cell lines limited predictive value is not fully understood10,54,55, evidence suggests that the process of generating in vitro cancer cultures results in major and irreversible alterations of biological properties, including gain and loss of genetic information, alteration in growth and invasion properties, and loss of specific cell populations10,56,57.
The faithfulness of preclinical models has been considerably debated in recent times as one of the reasons of the poor translatability of preclinical endeavors into effective therapeutic approaches14,57,58. This problem is particularly evident in OS and EW, in which therapeutic options are often inadequate in relapsed disease, but clinical evidence of substantial advancements is lacking1–3. The problem is further worsened by the rarity of these two malignancies, which slows down any potential scientific improvement resulting from basic or translational research and results in neglect by big pharma companies. The availability of both pediatric and adult PDX will allow the evaluation of therapeutic regimes tailored to pediatric tumors, which are currently treated with scaled-down protocols originally designed for adult tumors59,60. Some recent findings report new drugs sensitivity tested also in EW and OS PDX models57,61–65, further sustaining the potential of these models. We investigated here a combination of irinotecan and dAbd C7 treatment in two EW-PDX. Our results suggest that the addition of the anti-CD99 treatment could be beneficial when irinotecan alone is less effective, possibly depending on tumor growth rate. As we tested only two PDX, these conclusions will need to be confirmed in a larger series of preclinical models.
Conclusion
In conclusion, expandable, shareable and reliable preclinical models are a highly desirable tool for the identification of predictive biomarkers and for the evaluation of effective treatment strategies.
Supplementary information
Acknowledgements
This work was supported by the Italian Association for Cancer Research - AIRC (grant numbers IG18451 to KS; IG15324 to P-LL), Ministero della Salute (RF-2016-02361373; 5 × 1000 Anno 2016 Redditi 2015, contributions to the IRCCS Istituto Ortopedico Rizzoli), The European Union (TRANSCAN-2_TORPEDO ER-2015-2360405 to KS); Alleanza Contro il Cancro (ACC Genomics-WG Sarcoma to KS), the University of Bologna, Italy (“Pallotti” fund to P-LL and PN). The materials presented and views expressed here are the responsibility of the authors only. The sponsor takes no responsibility for any use made of the information set out.
Author Contributions
P.N., P.L.L. and K.S. coordinated the research project, designed experiments and wrote the manuscript. P.P. provided clinical interface and coordination. L.L. and G.N. coordinated in vivo studies, acquired and interpreted data, revised the manuscript. M.C.M. coordinated in vitro and molecular studies, acquired and interpreted data, and revised the manuscript. A.R. performed pathological studies and interpreted pathological data. APalladini, M.I., F.R. and V.G. acquired and interpreted in vivo data. C.C., M.P., AParra, and M.C. acquired and interpred in vitro and molecular data. M.F. performed gene expression analysis. D.M.D. provided clinical coordination and support. M.M. provided materials.
Data Availability
The datasets generated during and/or analysed during the current study are available in the ArrayExpress repository (accession E-MTAB-7568).
Competing Interests
M. Magnani holds shares in Diatheva SrL. The company has an exclusive licence on a patent protecting dAbd C7 antibody. All other authors declare no potential conflict of interests.
Footnotes
on behalf of ACC-SARCOMA Working Group
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Patrizia Nanni, Lorena Landuzzi and Maria Cristina Manara contributed equally
Pier-Luigi Lollini and Katia Scotlandi jointly supervised this work.
Contributor Information
Pier-Luigi Lollini, Email: pierluigi.lollini@unibo.it.
Katia Scotlandi, Email: katia.scotlandi@ior.it.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48634-y.
References
- 1.Reed DR, et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. 2017;123:2206–18. doi: 10.1002/cncr.30589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arndt CAS, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475–87. doi: 10.1016/j.mayocp.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hattinger CM, et al. Advances in emerging drugs for osteosarcoma. Expert Opin Emerg Drugs. 2015;20:495–514. doi: 10.1517/14728214.2015.1051965. [DOI] [PubMed] [Google Scholar]
- 4.Gaspar N, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol. 2015;33:3036–46. doi: 10.1200/JCO.2014.59.5256. [DOI] [PubMed] [Google Scholar]
- 5.Pappo AS, Dirksen U. Rhabdomyosarcoma, Ewing Sarcoma, and Other Round Cell Sarcomas. J Clin Oncol. 2018;36:168–79. doi: 10.1200/JCO.2017.74.7402. [DOI] [PubMed] [Google Scholar]
- 6.Picci P, et al. Survival in high-grade osteosarcoma: Improvement over 21 years at a single institution. Ann Oncol. 2010;21:1366–73. doi: 10.1093/annonc/mdp502. [DOI] [PubMed] [Google Scholar]
- 7.Tirtei E, et al. Survival after Second and Subsequent Recurrences in Osteosarcoma: A Retrospective Multicenter Analysis. Tumori. 2017;104:202–206. doi: 10.1177/0300891617753257. [DOI] [PubMed] [Google Scholar]
- 8.Byrne AT, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17:254–68. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- 9.Hidalgo M, et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillet J-P, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA. 2011;108:18708–13. doi: 10.1073/pnas.1111840108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grünewald TGP, Fulda S. Editorial: Biology-Driven Targeted Therapy of Pediatric Soft-Tissue and Bone Tumors: Current Opportunities and Future Challenges. Front Oncol. 2016;6:39. doi: 10.3389/fonc.2016.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ordóñez JL, Osuna D, Herrero D, Alava Ede, Madoz-Gúrpide J. Advances in Ewing’s sarcoma research: Where are we now and what lies ahead? Cancer Res. 2009;69:7140–50. doi: 10.1158/0008-5472.CAN-08-4041. [DOI] [PubMed] [Google Scholar]
- 13.Jung J, Seol HS, Chang S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res Treat. 2018;50:1–10. doi: 10.4143/crt.2017.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ledford H. US cancer institute to overhaul tumour cell lines. Nature. 2016;530:391. doi: 10.1038/nature.2016.19364. [DOI] [PubMed] [Google Scholar]
- 15.Siolas D, Hannon GJ. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–9. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pompili L, Porru M, Caruso C, Biroccio A, Leonetti C. Patient-derived xenografts: A relevant preclinical model for drug development. J Exp Clin Cancer Res. 2016;35:189. doi: 10.1186/s13046-016-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W, Chao T, Ruiqi C, Juan S, Zhihong L. Patient-derived xenograft models in musculoskeletal malignancies. J Transl Med. 2018;16:107. doi: 10.1186/s12967-018-1487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izumchenko E, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595–605. doi: 10.1093/annonc/mdx416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nomura T, Tamaoki N, Takakura A, Suemizu H. Basic concept of development and practical application of animal models for human diseases. Curr Top Microbiol Immunol. 2008;324:1–24. doi: 10.1007/978-3-540-75647-7_1. [DOI] [PubMed] [Google Scholar]
- 20.Scotlandi K, et al. Identification of EWS/FLI-1 transcripts in giant-cell tumor of bone. Int J Cancer. 2000;87:328–35. doi: 10.1002/1097-0215(20000801)87:3<328::AID-IJC4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Moricoli D, et al. Process development of a human recombinant diabody expressed in E. coli: Engagement of CD99-induced apoptosis for target therapy in Ewing’s sarcoma. Appl Microbiol Biotechnol. 2016;100:3949–63. doi: 10.1007/s00253-015-7226-5. [DOI] [PubMed] [Google Scholar]
- 22.McCarter MD, Lewis JJ, Antonescu CR, Brennan MF. Extraskeletal osteosarcoma: Analysis of outcome of a rare neoplasm. Sarcoma. 2000;4:119–23. doi: 10.1080/13577140020008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu J, et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017;19:130. doi: 10.1186/s13058-017-0920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bondarenko G, et al. Patient-Derived Tumor Xenografts Are Susceptible to Formation of Human Lymphocytic Tumors. Neoplasia. 2015;17:735–41. doi: 10.1016/j.neo.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen K, Ahmed S, Adeyi O, Dick JE, Ghanekar A. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein-Barr virus. PLoS ONE. 2012;7:e39294. doi: 10.1371/journal.pone.0039294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vakkila J, Jaffe R, Michelow M, Lotze MT. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: A major nosologic difference with adult tumors. Clin Cancer Res. 2006;12:2049–54. doi: 10.1158/1078-0432.CCR-05-1824. [DOI] [PubMed] [Google Scholar]
- 27.Fourcade G, et al. Evolution of EBV seroprevalence and primary infection age in a French hospital and a city laboratory network, 2000–2016. PLoS ONE. 2017;12:e0175574. doi: 10.1371/journal.pone.0175574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart E, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. 2017;549:96–100. doi: 10.1038/nature23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dienstmann R, Tabernero J. Cancer: A precision approach to tumour treatment. Nature. 2017;548:40–1. doi: 10.1038/nature23101. [DOI] [PubMed] [Google Scholar]
- 30.Tirode F, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–53. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowalewski AA, Randall RL, Lessnick SL. Cell Cycle Deregulation in Ewing’s Sarcoma Pathogenesis. Sarcoma. 2011;2011:598704. doi: 10.1155/2011/598704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao C, et al. Patient-derived Xenografts from Colorectal Carcinoma: A Temporal and Hierarchical Study of Murine Stromal Cell Replacement. Anticancer Res. 2017;37:3405–12. doi: 10.21873/anticanres.11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smida J, et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int J Cancer. 2017;141:816–28. doi: 10.1002/ijc.30778. [DOI] [PubMed] [Google Scholar]
- 34.Rickel K, Fang F, Tao J. Molecular genetics of osteosarcoma. Bone. 2017;102:69–79. doi: 10.1016/j.bone.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerzoni C, et al. CD99 triggering in Ewing sarcoma delivers a lethal signal through p53 pathway reactivation and cooperates with doxorubicin. Clin Cancer Res. 2015;21:146–56. doi: 10.1158/1078-0432.CCR-14-0492. [DOI] [PubMed] [Google Scholar]
- 36.Gellini M, et al. Generation of human single-chain antibody to the CD99 cell surface determinant specifically recognizing Ewing’s sarcoma tumor cells. Curr Pharm Biotechnol. 2013;14:449–63. doi: 10.2174/1389201011314040011. [DOI] [PubMed] [Google Scholar]
- 37.Palmerini E, et al. Irinotecan and temozolomide in recurrent Ewing sarcoma: An analysis in 51 adult and pediatric patients. Acta Oncol. 2018;57:958–64. doi: 10.1080/0284186X.2018.1449250. [DOI] [PubMed] [Google Scholar]
- 38.Julien S, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314–28. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 39.Bertotti A, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 40.Jung J, et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget. 2016;7:62533–46. doi: 10.18632/oncotarget.11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X-c, et al. Establishment of patient-derived non-small cell lung cancer xenograft models with genetic aberrations within EGFR, KRAS and FGFR1: Useful tools for preclinical studies of targeted therapies. J Transl Med. 2013;11:168. doi: 10.1186/1479-5876-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z-H, et al. Antitumor effects of a novel histone deacetylase inhibitor NK-HDAC-1 on breast cancer. Oncol Rep. 2013;30:499–505. doi: 10.3892/or.2013.2434. [DOI] [PubMed] [Google Scholar]
- 43.Marangoni E, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13:3989–98. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–97. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricci F, et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014;74:6980–90. doi: 10.1158/0008-5472.CAN-14-0274. [DOI] [PubMed] [Google Scholar]
- 46.Topp MD, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol. 2014;8:656–68. doi: 10.1016/j.molonc.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Depreeuw J, et al. Characterization of patient-derived tumor xenograft models of endometrial cancer for preclinical evaluation of targeted therapies. Gynecol Oncol. 2015;139:118–26. doi: 10.1016/j.ygyno.2015.07.104. [DOI] [PubMed] [Google Scholar]
- 48.Stewart E, et al. The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev Biol. 2016;411:287–93. doi: 10.1016/j.ydbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bruheim S, Bruland OS, Breistol K, Maelandsmo GM, Fodstad O. Human osteosarcoma xenografts and their sensitivity to chemotherapy. Pathol Oncol Res. 2004;10:133–41. doi: 10.1007/BF03033741. [DOI] [PubMed] [Google Scholar]
- 50.Monsma DJ, et al. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J Transl Med. 2012;10:125. doi: 10.1186/1479-5876-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stewart E, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9:829–41. doi: 10.1016/j.celrep.2014.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blattmann C, et al. Establishment of a patient-derived orthotopic osteosarcoma mouse model. J Transl Med. 2015;13:136. doi: 10.1186/s12967-015-0497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rainusso N, et al. Generation of patient-derived tumor xenografts from percutaneous tumor biopsies in children with bone sarcomas. Pediatr Blood Cancer. 2019;66:e27579. doi: 10.1002/pbc.27579. [DOI] [PubMed] [Google Scholar]
- 54.Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013;93:970–82. doi: 10.1038/labinvest.2013.92. [DOI] [PubMed] [Google Scholar]
- 55.Daniel VC, et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69:3364–73. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hausser H-J, Brenner RE. Phenotypic instability of Saos-2 cells in long-term culture. Biochem Biophys Res Commun. 2005;333:216–22. doi: 10.1016/j.bbrc.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 57.Willyard C. The mice with human tumours: Growing pains for a popular cancer model. Nature. 2018;560:156–7. doi: 10.1038/d41586-018-05890-8. [DOI] [PubMed] [Google Scholar]
- 58.Ben-David U, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567–75. doi: 10.1038/ng.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Editorial Children first. Nat Med. 2017;23:1005. doi: 10.1038/nm.4404. [DOI] [PubMed] [Google Scholar]
- 60.Schäfer BW, Koscielniak E, Kovar H, Fulda S. ESF-EMBO Symposium “Molecular Biology and Innovative Therapies in Sarcomas of Childhood and Adolescence” Sept 29–Oct 4. Polonia Castle Pultusk, Poland. Front Oncol. 2013;3:142. doi: 10.3389/fonc.2013.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ambati SR, et al. BO-1055, a novel DNA cross-linking agent with remarkable low myelotoxicity shows potent activity in sarcoma models. Oncotarget. 2016;7:43062–75. doi: 10.18632/oncotarget.9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ordóñez JL, et al. The PARP inhibitor olaparib enhances the sensitivity of Ewing sarcoma to trabectedin. Oncotarget. 2015;6:18875–90. doi: 10.18632/oncotarget.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xian M, et al. Bortezomib sensitizes human osteosarcoma cells to adriamycin-induced apoptosis through ROS-dependent activation of p-eIF2α/ATF4/CHOP axis. Int J Cancer. 2017;141:1029–41. doi: 10.1002/ijc.30792. [DOI] [PubMed] [Google Scholar]
- 64.Stebbing J, et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer. 2014;120:2006–15. doi: 10.1002/cncr.28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manara MC, et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol Cancer Ther. 2018;17:1881–92. doi: 10.1158/1535-7163.MCT-17-0818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available in the ArrayExpress repository (accession E-MTAB-7568).