

Abstract
Congenital extrahepatic portosystemic shunt (CEPS), also known as Abernethy malformation, is a rare condition in which the splenomesenteric blood drains directly into a systemic vein, bypassing the liver through a complete or partial shunt. The diagnosis is frequently made during childhood in the setting of neonatal cholestasis, hypergalactosemia, failure to thrive, mental retardation or other congenital defects. In adulthood, CEPS is usually found incidentally during diagnostic work-up for abdominal pain, liver test abnormalities, liver nodules, portopulmonary hypertension, portopulmonary syndrome or portosystemic encephalopathy. The diagnosis depends on imaging and portal venography, but sometimes only liver biopsy can be resolutive, demonstrating the absence of venules within the portal areas. Here we report four recent cases of Abernethy malformation diagnosed in young adults, in which ultrasound (US) was the initial imaging technique and allowed to suspect the diagnosis. Furthermore, we reviewed clinical presentations, associated anomalies and treatment of the 310 cases of CEPS previously reported in the literature.
Keywords: Congenital extrahepatic portosystemic shunt, Abernethy malformation, Ultrasound, Nodular regenerative hyperplasia, Liver vascular malformation
Sommario
Lo shunt portosistemico extraepatico congenito (CEPS), anche noto come malformazione di Abernethy, è una rara condizione patologica in cui il sangue venoso refluo dal distretto spleno-mesenterico drena direttamente nel circolo venoso sistemico, bypassando il fegato attraverso un shunt completo o parziale. La diagnosi viene posta frequentemente durante l’infanzia in presenza di colestasi neonatale, ipergalattosemia, scarso accrescimento o altri difetti congeniti. Durante l’età adulta la diagnosi di CEPS è spesso incidentale nel corso di accertamenti diagnostici per dolore addominale, alterazioni degli indici di funzionalità epatica, riscontro di lesioni focali epatiche, ipertensione porto-polmonare, sindrome porto-polmonare ed encefalopatia porto-sistemica. Le tecniche di imaging o la porto-venografia consentono solitamente di porre la diagnosi, ma in alcuni casi solo la biopsia epatica risulta dirimente, documentando l’assenza di strutture venose a livello degli spazi portali. Nel presente lavoro vengono presentati 4 casi di malformazione di Abernethy diagnosticati in giovani adulti, nei quali l’ultrasonografia (US) è stata la prima tecnica di immagine utilizzata consentendo di porre il sospetto diagnostico. Inoltre, abbiamo analizzato la presentazione clinica, le anomalie associate e le opzioni terapeutiche dei 310 casi di CEPS riportati in letteratura.
Introduction
Congenital extrahepatic portosystemic shunt (CEPS) is a rare condition in which the splenomesenteric blood drains directly into a systemic vein, bypassing the liver through a complete (type 1) or partial (type 2) extrahepatic shunt.
The clinical importance of this vascular abnormality relies on its wide spectrum of symptoms and complications, which ranges from asymptomatic cases, discovered incidentally, to various degrees of liver or metabolic dysfunction until severe clinical scenarios of portopulmonary hypertension (PPH), portopulmonary syndrome (PPS), portosystemic encephalopathy (PSE) and liver tumors. These latter comprise benign lesions such as hemangiomas, adenomas, nodular regenerative hyperplasia (NRH) and focal nodular hyperplasia (FNH) as well as malignant ones, such as hepatocellular carcinoma (HCC) and hepatoblastoma. Ascites and other signs of portal hypertension are not characteristic of isolated CEPS, while they are typical of secondary portosystemic shunts in the context of liver cirrhosis or portal vein occlusion.
Here we report four cases of Abernethy malformation type 1 evaluated at our Institutions from 2010 to 2017. In all cases, US abdomen study was the initial imaging technique and allowed to suspect the final diagnosis. Case 1 and 2 were diagnosed incidentally during diagnostic work-up for benign hepatic nodules and were managed with medical therapy; case 3 was associated with unresectable NRH leading to mild liver and progressive cardiac dysfunction, thus liver transplantation (LT) was required; case 4 was diagnosed in the setting of a complex association of musculoskeletal and cardiovascular anomalies, called Goldenhar syndrome, thus cardiac surgery was mandatory. We also reviewed clinical presentation, liver anomalies, associated defects and treatment options of the previous 310 cases of CEPS published in literature [1–39].
Case report 1
A 17-year-old boy was admitted for the occurrence of acute abdominal pain in the right hypochondrium, fever and dyspnea after an episode of alcohol abuse. The patient’s medical history included a patent foramen ovale with surgical repair at 2 years of age. Laboratory evaluation revealed mild hyperammonemia in the absence of clinical signs of encephalopathy and normal liver enzymes and function. Chest X-ray showed moderate right pleural effusion with suspected apical consolidation, which was treated with antimicrobial therapy and temporary indwelling chest tube. An urgent abdominal US examination showed several hyperechoic nodules in the liver, without signs of cirrhosis; furthermore, the extrahepatic portal trunk was not clearly visualized. Computed tomography (CT) with contrast enhancement confirmed the presence of multiple hypodense nodular lesions in the liver, the main one of 3.5 cm of diameter, suspected for NRH based on dynamic behavior, and displayed a splenomesenteric trunk that through the hepato-gastric ligament entered abruptly into the left renal vein. In the absence of the main portal vein (PV) and its intrahepatic branches, liver blood supply was provided by a hypertrophic hepatic artery. Contrast-enhanced abdominal magnetic resonance imaging (MRI) and liver biopsy confirmed the diagnosis of NRH. Other investigations showed arachnodactyly, mental impairment, dolicocephalia, alterations of optical nerve, cardiac multivalvular insufficiency, severe osteoporosis. The patient was started on lactulose achieving a normalization of ammonia levels. At 4 years of follow-up, he is doing well and hepatic lesions are stable on liver US.
Case report 2
A 23-year-old girl, without relevant medical history, was admitted for acute paraparesis and urinary retention secondary to a spinal epidural abscess, treated successfully with neurosurgical intervention and antimicrobial therapy. During hospitalization, abdominal US incidentally highlighted hepatosplenomegaly, multiple patchy hypoechoic liver nodules with well-defined borders or lobulated contours, the main one of 6.3 cm of diameter in the III–IV segment, absence of main intrahepatic portal branches with a tortuous splenomesenteric trunk which seemed to drain directly into inferior vena cava (IVC), hepatic artery hypertrophy (Fig. 1a, b, c, d). Laboratory tests showed pancytopenia (Hb 11.7 g/dL, WBC 1570 cells/µL, PLT 82,000 cells/µL), normal liver function tests and alfa-fetoprotein (AFP) serum levels. Contrast-enhanced MRI confirmed the presence of several nodules in the liver, the main one of 6.4 cm in diameter, some of them with radiologic findings suggestive for FNH, others suspected for NRH. In addition, it showed that the main portal trunk drained directly into a dilated right renal vein in the absence of intrahepatic PV branches (Fig. 2a, b). Upper gastrointestinal endoscopy ruled out signs of portal hypertension. The patient began treatment with lactulose for grade 1 hepatic encephalopathy. At 6 months of follow-up, she is asymptomatic and hepatic findings are stable on US.
Fig. 1.

Case 2 patient. Color-Doppler Ultrasound of the liver showing multiple patchy hypoechoic liver nodules, the main one of 6.3 cm of diameter in the III–IV segment (a), a tortuous splenomesenteric trunk (arrow) with a turbulent flow apparently directed towards inferior vena cava (IVC) in the absence of main intrahepatic portal branches (b, c), hepatic artery hypertrophy (1D)
Fig. 2.
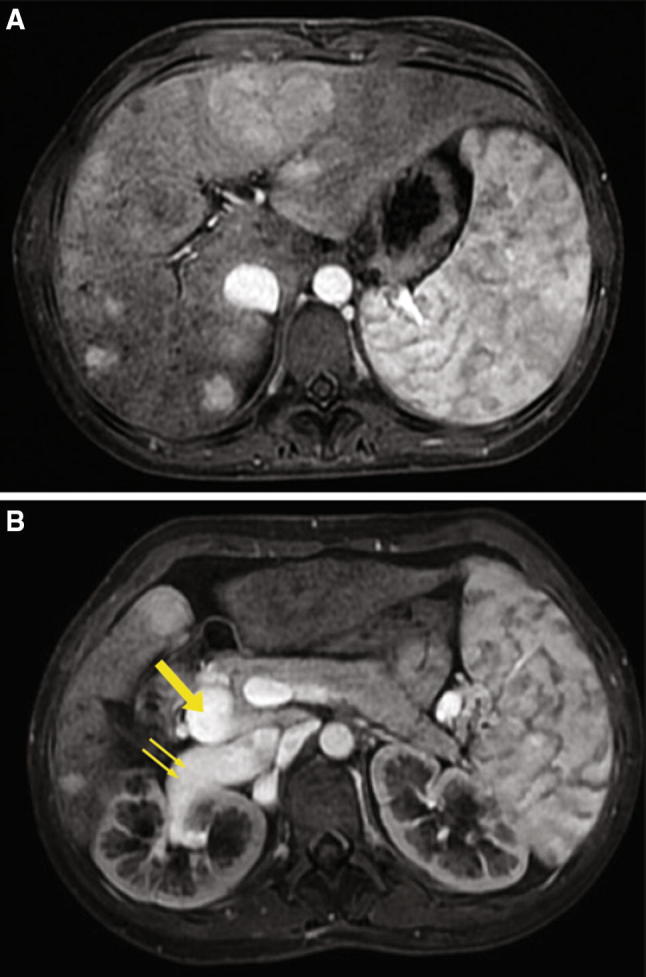
Case 2 patient. T1-Weighted fat-saturated magnetic resonance images obtained during the portal venous phase showing intensely enhanced nodules throughout the liver and the absence of intrahepatic portal branches (a), a splenomesenteric trunk (arrow) draining into a dilated right renal vein (double arrow) (b)
Case report 3
A 30-year-old man was admitted for massive subversion of liver parenchyma by multiple confluent nodules at abdominal US. He reported a 4-year history of mild liver enzymes elevations and moderate rise in cholestasis without clinical symptoms. He had no history of liver disease and alcohol use and denied taking over the counter medications and herbals. Medications included inhalation therapy for bronchial asthma. Screening for viral and autoimmune hepatitis and tumor markers (including AFP) resulted negative. The patient underwent liver US which confirmed the presence of multiple nodules throughout the liver, the main one of 9 cm with areas of internal colliquation in the II segment, and raised suspicion of a shunt between the PV and IVC without detectable intrahepatic portal branches (Fig. 3a, b, c, d). On contrast-enhanced total body CT scan the liver was nodular shaped, with a voluminous and patchy lesion in the left, enlarged, lobe and multiple hypodense areas in the right, lobe. A shunt between the main portal trunk and the IVC was evident, with no signs of intrahepatic portal flow and compensatory dilation of hepatic artery, determining a significant hyperafflux to the right heart and consequent dilatation of pulmonary arteries. Contrast-enhanced abdominal MRI confirmed the presence of the complex vascular malformation and showed that the multiple hepatic nodules, including the expansive plurinodular mass of the left lobe (diameter 13 cm) with necrotic areas inside were suggestive for NRH based on dynamic behavior (Fig. 4a, b, c, d). The patient underwent liver biopsy which confirmed features of adenomatous-like hepatocellular hyperplasia without foci of dysplasia and portal tracts. Cardiac US revealed mild pulmonary hypertension, biatrial enlargement and mild tricuspidal insufficiency; upper gastrointestinal endoscopy ruled out portal hypertension. Therefore, the diagnosis of NRH in the setting of type 1 Abernethy syndrome was made. As hepatic lesions were judged unresectable the patient underwent liver transplantation (LT) to prevent further deterioration of liver and right heart cardiac function. Orthotopic LT was performed using a deceased liver donor without complications. The liver was implanted using the piggyback technique, with side-to-side cavo-cavostomy and hepatic artery reconstruction. Recipient PV was detached from the IVC and anastomosed to the graft PV in an end-to-end fashion, without interposition of a vascular or synthetic conduit. After 4 years of follow-up, laboratory tests are in normal range without recurrent focal liver lesions in the graft.
Fig. 3.

Case 3 patient. Color-Doppler Ultrasound of the liver showing direct communication between portal vein and inferior vena cava (a) with phasic portal venous flow pattern secondary to porto-systemic shunt (b); nodular lesion of 10 cm of diameter with area of internal colliquation (arrow) in the II segment (c) and multiple nodular lesions of right hepatic lobe (3d)
Fig. 4.

Case 3 patient. T1-Weighted fat-saturated magnetic resonance images obtained during the portal venous phase showing a tortuous vascular shunt (arrow) between the main portal trunk (PV) and the inferior vena cava (IVC) in the absence of intrahepatic PV branches (a); an expansive multinodular mass of the left lobe with areas of internal colliquation (arrows) (b) and multiple nodules up to 5 cm in the right lobe with contrast enhancement persistent in late phase (c). T1-weighted fat-saturated hepatobiliary phase magnetic resonance image showing that hepatic lesions are hyperintense or have a hyperintense peripheral rim (d)
Case report 4
A 34-year-old male was referred for a 12-months history of vague left upper abdominal pain. He reported no history of abdominal trauma, no other gastrointestinal symptoms. Abdomen examination showed a tender palpable mass and a loud machinery murmur over left upper quadrant. Abdominal US showed a spleno-mesenteric trunk draining into a dilated extra-hepatic PV, which in turn, was draining into the IVC. Intra-hepatic branches of PV were absent. IVC was double, with the left one arising from the two renal veins. US also revealed two large splenic venous aneurysms close to the splenic hilum secondary to coexisting arteriovenous fistula. Liver parenchyma showed multiple hyperechoic lesions. Contrast-enhanced CT scan confirmed absence of intra-hepatic portal flow, dilation of extra-hepatic PV (diameter 2.2 cm) showing early enhancement on arterial phase denoting arterio-portal shunt, multiple hepatic lesions which appeared isodense at baseline scan, with sharp contrast enhancement, except for a hypodense central scar on arterial phase and hypo-isodensity to the liver on portal venous phase. Upper gastrointestinal endoscopy ruled out portal hypertension. Liver function tests were normal except for low serum albumin (2.8 mg/dl). Screening for viral hepatitis was negative. Furthermore, the medical history was characterized by several other abnormalities: gross facial asymmetry and severe midface and mandible retrusion; subvalvular aortic stenosis and left ventricular outflow tract obstruction with a dilated left atrium; vertebral body defects (hemivertebrae and posterior fusion defects of C3-4) and thoracic dextroscoliosis.
Thus, diagnosis of Goldenhar syndrome in the setting of Abernethy type 1 was made. The patient is waiting for aortic valve replacement to prevent further deterioration of cardiac function due to the right atrium hyperflow and left ventricular outflow tract obstruction. Before cardiac surgery, he underwent endovascular embolization of the large high-flow splenic aneurysms by Amplatzer vascular plug without complications.
Discussion
CEPS was first reported in 1793 by John Abernethy, who described the termination of PV in IVC at the level of the renal veins and other multiple congenital defects during postmortem examination of a 10-month-old girl died of unknown causes [1]. Therefore, CEPS is also known as Abernethy malformation.
Two variants of CEPS have been described according to the absence (type 1) or presence (type 2) of intrahepatic PV supply. In type 1 CEPS, there is a complete diversion of the portal blood into the systemic circulation through an end-to-side PV-IVC shunt with apparent absence of any portal branches in the liver. In type 2 CEPS, just some of the portal flow is diverted from intrahepatic PV branches into a systemic vein through a side-to-side shunt. The drainage vessels are mainly the IVC or the renal veins, as in our cases; other possible sites are iliac veins, directly or via the inferior mesenteric vein, the azygos vein or the right atrium. As a consequence, the liver is supplied by a hypertrophic hepatic artery [2, 23].
Further classifications of these 2 CEPS variants have been proposed over time to help in shunt description and to guide in management decisions. Type 1 CEPS have been classified by Morgan and Superina into type 1a, in which the splenic vein (SV) and superior mesenteric vein (SMV) drain separately into a systemic vein, and type 1b, in which SV and SMV join in a common trunk before shunting [3]. More recently, Kobayashi et al. identified 3 subtypes according to the site of shunt: the IVC in type A, the renal vein in type B, and the iliac vein through a dilated inferior mesenteric vein (IMV) in type C, respectively [4]. Type 2 CEPS has been classified by Lautz et al. according to the origin of the shunt into type 2a, which involves right or left PV (including patent ductus venosus), type 2b, with the shunt arising from the main PV at bifurcation or splenomesenteric confluence, and type 2c, involving the SMV, SV or gastric vein [5]. Kanazawa et al. also identified a mild, moderate and severe type 2 CEPS, based on the severity of the intrahepatic portal hypoplasia and portal pressure values at shunt occlusion [6]. These classifications, although useful, are not completely valid for several reasons. First of all, in some cases only an occlusion test during venography or at liver biopsy can ultimately help distinguishing between type 1 and 2 CEPS. Furthermore, successful portal revascularization of the liver after shunt closure in children makes the difference between these 2 entities less prominent. Finally, patent ductus venosus, cannot fit entirely into extrahepatic or intrahepatic shunt definition and must be considered apart [7].
Since CEPS was first described, the number of cases reported in the literature has progressively increased due to advances in imaging techniques and neonatal screening, and 310 cases have been reported so far (178 type 1, 132 type 2). The age at presentation is highly variable; ranging from prenatal diagnosis to 61 years of age for type 1 and prenatal diagnosis to 76 years for type 2, but up to 70% of cases are diagnosed in patients aged 18 years or less. There is no clear sex preponderance.
Clinical manifestations
CEPS can cause a broad spectrum of clinical manifestations. Diagnosis is frequently made during childhood in the setting of neonatal cholestasis, hypergalactosemia, failure to thrive, psychomotor delay or other congenital defects. Prenatal diagnosis is increasingly being reported and is suspected by an abnormal connection between the portal and peripheral venous system or an enlarged umbilical vein on prenatal US [8, 15, 21, 38]. Sometimes CEPS is incidentally found at abdominal US or during the diagnostic work-up for abdominal pain, liver tests abnormalities, liver nodules, PPH, PPS or PSE. Unusual presentations include gastrointestinal bleeding from colonic and rectal varices secondary to peripheral shunt draining into the iliac vein via the IMV [9, 10, 34], vaginal bleeding and hematuria [5, 9].
Congenital defects are rather common in CEPS. Cardiac anomalies are found up to 29% of type 1 and 22% of type 2 CEPS, and consist mainly in ventricular and atrial septal defect, patent ductus arteriosus, patent foramen ovale, coarctation of the aorta, tetralogy of Fallot, and other congenital valvulopathies. This association suggests either a prenatal insult on both heart and abdominal venous system development or an adaptive response of the heart to the hyperdynamic effect of the portosystemic shunt. Polysplenia or other heterotaxy syndromes and biliary atresia are found in 8% and 5% of type 1 patients. Rare congenital syndromes have been described in patients with CEPS as a result of multiple coexisting developmental disorders during embryogenesis. Our patient n.4 presented with craniofacial, vertebral and cardiac defects and was diagnosed with Goldenhar syndrome or oculoauriculovertebral dysplasia, and to our knowledge this is the third case of association with type 1 Abernethy syndrome reported in the literature [11, 12]. Other developmental defects encountered in type 1 and 2 CEPS are summarized in Table 1.
Table 1.
Anomalies associated with congenital portosystemic shunts. Number of cases reported in type 1 and 2 Abernethy syndrome
| Type of alteration | Abernethy type 1 (182 cases) | Abernethy type 2 (132 cases) |
|---|---|---|
| Cardiac anomalies | ||
| Atrial or ventricular or atrioventricular septal defect | 29 | 21 |
| Patent foramen ovale or patent ductus arteriosus | 9 | 11 |
| Dextrocardia or mesocardia | 2 | – |
| Congenital valvulopathy | 7 | 5 |
| Transposition of the great arteries or Tetralogy of Fallot | 6 | 1 |
| Coarctation of the aorta | 6 | 1 |
| Others (endocardial cushion defects, interauricular communication, hypertrophic cardiomyopathy unspecified) | 6 | 3 |
| Associated liver diseases | ||
| Biliary atresia | 9 | 1 |
| Congenital choledochal cyst/Caroli’s disease | 1 | 3 |
| Congenital hepatic fibrosis | 1 | 1 |
| Esophageal varices, splenomegaly | 7 | 2 |
| Intrahepatic gallbladder | – | 1 |
| Hepatic nodules | ||
| Focal nodular hyperplasia | 40 | 10 |
| Liver cell adenoma | 14 | 4 |
| Nodular regenerative hyperplasia | 24 | 7 |
| Hemangioma | 5 | 1 |
| Hepatoblastoma | 4 | 3 |
| Hepatocellular carcinoma | 13 | 5 |
| Hepatocellular-cholangiocarcinoma | 1 | – |
| Unspecified | 8 | 6 |
| Shunt complication | ||
| Portopulmonary hypertension | 15 | 11 |
| Portopulmonary syndrome | 10 | 22 |
| Portosystemic encephalopathy | 15 | 16 |
| Portosystemic collateral veins (gastrointestinal bleeding, hematuria, vaginal bleeding) | 8 | 7 |
| Endocrino-metabolic disorders | ||
| Hyperinsulinemic hypoglycemia | 5 | 1 |
| Early pubarche, primary amenorrhoea, virilization | 4 | – |
| Congenital adrenal hyperplasia | 1 | – |
| Hypergalactosemia | 9 | 1 |
| Hypothyroxinemia | 1 | 1 |
| Other developmental anomalies | ||
| Polysplenia/asplenia | 15 | 8 |
| Situs inversus/heterotaxia | 9 | 2 |
| Other vascular anomalies (double, left-sided, agenesia or stenosis of inferior vena cava; azygos/hemiazygos continuation of the inferior vena cava, 2 splenic veins, double aortic arch, pulmonary artery stenosis, intrapulmonary shunts, artery aneurism) | 15 | 19 |
| Gastrointestinal anomalies (intestinal malrotation, esophageal and anal atresia, tracheoesophageal fistula) | 7 | 8 |
| Urogenital system anomalies (cystic kidney dysplasia, ureteropelvic obstruction, crossed renal ectopia with vesicoureteral reflux, hypospadias, renal agenesis, hydronephrosis, varicocele) | 9 | 8 |
| Skeletal anomalies (short 5th fingers and toes, hypoplasia of the thumb, absence of the first metacarpophalangeal complex, polydactyly, arachnodactyly, clinodactyly, sacral anomalies, facial dysmorphism, dolicocephalia, tarsal synostosis, clavicle agenesis, hip dysplasia, scoliosis, hemivertebra) | 18 | 1 |
| Bronchopulmonary malformations (bronchomalacia, bronchial stenosis, laryngomalacia, tracheal diverticulum, lobar pulmonary sequestration) | 5 | – |
| Skin hemangiomas/telangiectasias | 4 | 3 |
| Mental retardation, microcephaly | 14 | 12 |
| Growth retardation | 2 | 1 |
| Genetic diseases | ||
| Down syndrome | 3 | 2 |
| Turner syndrome | 5 | – |
| Goldenhar syndrome | 3 | – |
| Others (Costello syndrome, Trisomy 8, Ataxia-telangiectasia syndrome, Holt–Oram syndrome, Noonan syndrome, Klippel–Trenaunay–Weber syndrome, Adams–Oliver syndrome) | 3 | 4 |
One of the most relevant consequence of CEPS is to reduce the hepatic metabolism of hormones and toxic compounds derived from the splenomesenteric circulation. This can lead to: (a) hypergalactosemia without uridine diphosphate enzyme deficiency [13, 29] (b) postprandial hypoglycemia, secondary to hypersecretion of insulin induced by high levels of prandial glucose and reduced hepatic insulin clearance [14] (c) endocrine disturbances, such as hypothyroidism due to thyroxin-binding globulin deficiency and hyperandrogenemia, which in turn leads to primary amenorrhoea, early pubarche and signs of virilization [14]. Conversely, hyperammonemia is not a universal finding and, when present, it is of mild entity, possibly as a result of adaptative mechanisms of intestinal flora. Hyperammonemia is often asymptomatic in patients with CEPS, but may sometimes be responsible of low-grade PSE in children [5, 15] or overt PSE in about 10% of patients, mainly in the elderly, or when the shunt ratio exceeds 60% [16, 35].
Patients with CEPS can also present with dyspnea on exertion or at rest, cyanosis and digital clubbing, as a consequence of PPH (8% of cases) and PPS (10% of cases), which occur in the absence of liver dysfunction and portal hypertension. It has been hypothesized that PPH may result from recurrent pulmonary microembolism from the mesenteric circulation via the portosystemic shunt and usually carries a poor prognosis [17]. PPS is secondary to the inflow of vasoactive mediators from the portal into the systemic circulation, leading to intrapulmonary vessels dilation, ventilation–perfusion mismatch and, ultimately, to hypoxemia. Furthermore, in some reports PPS was complicated by brain abscesses, probably due to the Gram-negative bacteria and endotoxin-contaminated splanchnic blood bypassing both liver and lung control [18, 19].
Single or multiple hepatic nodules are encountered in approximately 40% of the patients with CEPS and consist of FNH, NRH, adenomas, hemangiomas, HCC and hepatoblastoma. They occur more frequently in type 1 than in type 2 CEPS (50% vs 22%, respectively), and in older patients than in younger ones. Such lesions are thought to be a result of the alteration in local hemodynamics and of elevated circulating levels of hepatic growth factors (e.g. insulin, glucagon, hepatocyte growth factor) [20]. Such lesions may increase in number and size during follow-up, and become stable or even decrease after shunt closure [20, 21]. Malignant lesions can occur alone or in association with benign ones. Different variants of beta-catenin mutations have been reported in hepatocellular nodules of patients with CEPS irrespective of their morphological appearance, suggesting that hepatic lesions developing in the context of Abernethy malformation should be considered at risk of malignant progression independent of their size and radiological appearance [22]. Thus, long-term follow-up of benign nodules in patients with CEPS is mandatory, and radiological changes or increase in tumor markers should lead to suspicion of malignant transformation.
Ultrasound examination in the diagnosis of CEPS
Abdomen US is often the first imaging technique performed in these patients and in most cases Doppler US allows a good quality study of all vascular components of the liver and of their anatomic variants [40, 41]. Indeed, in all the four reported cases, US was the initial imaging technique and played a key role in suspecting CEPS based on the presence of vascular abnormalities and focal changes in liver parenchyma. First of all, showing the anatomy of the splenomesenteric venous system, portal venous trunk and its main branches, US can raise suspicion of CEPS in case of absence/hypoplasia of the PV, absence of intrahepatic portal signals, and evidence of spontaneous porto-systemic shunt. Furthermore, it rules out in most cases acquired causes of lack of visualization of the PV such as PV cavernoma and thrombosis, shows hepatic artery hypertrophy, which is an indirect sign of CEPS, and displays focal hepatic lesions, which are encountered in most of these patients [42, 43]. The main advantages of US are that it is a non-invasive, readily available, not expensive and radiation-free technique, therefore, it is well suited for early identification of these anomalies and for hepatic nodules surveillance. Furthermore ,US with the addition of contrast enhancement (CEUS) improves diagnostic accuracy in the evaluation of focal hepatic lesions compared to grey scale and Doppler US, showing a comparable accuracy to contrast-enhanced CT and MR imaging [44]. On CEUS benign lesions such as FNH, haemangiomas and hepatic adenomas, show typical and diagnostic appearances, whereas hepatic malignancies can be suspected according to their dynamic behavior characterized by arterial enhancement followed by portal/venous washout. CEUS is also superior to unenhanced Doppler US in detecting malignant PV thrombosis and cavernous transformation of the PV due to dynamic enhancement information [43, 45]. Since it is now approved by EFSUMB guidelines for the assessment of focal liver lesions also in the pediatric population, incorporation of CEUS into US evaluation of Abernethy patients may help to reduce the need of cross-sectional imaging in the young CEPS population [44].
However, US is operator-dependent and it lacks panoramicity, providing only a partial view of the liver and abdomen anatomy. As a consequence, the diagnosis should always be clarified with cross-sectional imaging methods such as MRI or CT which can accurately describe the anatomy, size and site of the shunt, and characterize liver lesions and coexisting congenital defects. In those cases, when the detection of tiny intrahepatic portal venous vessels is beyond the resolution limits of the available imaging methods, invasive tests are necessary for a definitive diagnosis. They include occlusive mesenteric portovenography, which allows a detailed evaluation of the intrahepatic PV system anatomy and shunt pressure measurement, and liver biopsy, which can help in differentiating between the complete absence of portal venules and various degrees of hypoplasia, and in characterizing hepatic nodules [23].
Treatment
There is currently no standard therapeutic approach for CEPS. Treatment strategy is decided on a case-by-case basis according to the shunt type and location, symptoms severity and related complications.
Shunt closure, through either interventional embolization with plug, stent or coils or surgical ligation, results in rapid amelioration of symptoms, normalization of ammonia levels, and regression of liver lesions [21, 24]. It is mandatory in type 2 CEPS whenever a complication is present and should be considered early in all the other cases, to prevent the occurrence of complications. Early intervention is necessary, as (a) the regression of PH is not certain once irreversible lesions of the pulmonary arteries develop, (b) chronic hyperammonemia, even if asymptomatic, may have adverse effects on brain, especially during childhood, (c) plasticity of the intrahepatic portal system in young children allows revascularization of the liver even when the intrahepatic portal branches are not detectable on imaging studies [7]. However, shunt closure may carry some complications: the native intrahepatic PV system may fail to expand after shunt closure, leading to severe portal hypertension, gut damage and gastrointestinal bleeding [25, 26]; the opening of new spontaneous intrahepatic shunts secondary to the sudden increase of the PV inflow may lead to pulmonary hypertension or recurrent hyperammonemia [27, 28]; coils or plugs migration in case of large and short shunts can cause obstruction of a systemic vessel. At this purpose, Franchi-Abella et al. proposed an occlusion test to estimate the portal pressure trend after temporary closure of the shunt, to decide whether to perform the closure of the shunt in one or more stages to favor the adaptation of the intrahepatic portal system to the new hemodynamic condition [21].
Conservative management including protein restriction, lactulose and non-absorbable antibiotics administration and a clinical, biochemical and imaging follow-up should be reserved to type 1 CEPS with no or mild symptoms. Shunt occlusion is not a treatment option in type 1 CEPS, since shunt represents the only drainage route for the mesenteric blood. Hence, the only therapeutic option relies on LT, although indications remain controversial. PSE and pulmonary complications, biliary atresia and hypergalactosemia represent the most common indications for LT. Other indications include worsening hepatic function, liver tumors or anemia secondary to gastrointestinal bleeding. To our knowledge, our case no. 3 is the 49th LT performed for complicated CEPS and the 9th LT performed in a patient with more than 18 years of age [3, 13, 18, 21, 22, 27, 29–36]. Five CEPS patients underwent an auxiliary portal orthotopic liver transplant (APOLT), which is the transplantation of a partial graft while preserving part of the native liver [29, 31]. Almost all cases reported complete resolution of symptoms after LT. The main challenge relies on technical difficulties of the procedure, since in most cases, the complex portal system anatomy requires vein graft interposition during PV reconstruction.
Conclusions
Although rare conditions, CEPS are being diagnosed with increased frequency as imaging techniques have become more refined and in many cases diagnosis is made incidentally during abdomen US study. We think that the concomitant occurrence of the following US signs should raise the suspicion of CEPS:
Presence of solid focal lesions in the liver parenchyma.
Absence/hypoplasia of the portal trunk.
Absence of intrahepatic portal vessels and flow signals.
Detection of porto/systemic shunts.
Detection of hepatic artery hypertrophy.
Compliance with ethical standards
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from participants included in the study.
References
- 1.Abernethy J. Account of two instances of uncommon formation in the viscera of the human body. Phil Trans R Soc Lond B Biol Sci. 1793;83:295–299. [Google Scholar]
- 2.Hao Y, Hong X, Zhao X. Congenital absence of the portal vein associated with focal nodular hyperplasia of the liver and congenital heart disease (Abernethy malformation): a case report and literature review. Oncol Lett. 2015;9(2):695–700. doi: 10.3892/ol.2014.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan BG, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portosystemic vascular anomalies. J Pediatr Surg. 1994;29:1239–1241. doi: 10.1016/0022-3468(94)90812-5. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi N, Niwa T, Kirikoshi H, et al. Clinical classification of congenital extrahepatic portosystemic shunts. Hepatol Res. 2010;40:585. doi: 10.1111/j.1872-034X.2010.00667.x. [DOI] [PubMed] [Google Scholar]
- 5.Lautz TB, Tantemsapya N, Rowell E, et al. Management and classification of type II congenital portosystemic shunts. J Pediatr Surg. 2011;46(2):308–314. doi: 10.1016/j.jpedsurg.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Kanazawa H, Nosaka S, Miyazaki O, et al. The classification based on intrahepatic portal system for congenital portosystemic shunts. J Pediatr Surg. 2015;50(4):688–695. doi: 10.1016/j.jpedsurg.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Bernard O, Franchi-Abella S, Branchereau S, et al. Congenital portosystemic shunts in children: recognition, evaluation, and management. Semin Liver Dis. 2012;32(4):273–287. doi: 10.1055/s-0032-1329896. [DOI] [PubMed] [Google Scholar]
- 8.Venkat-Raman N, Murphy KW, Ghaus K, et al. Congenital absence of portal vein in the fetus: a case report. Ultrasound Obstet Gynecol. 2001;17:71–75. doi: 10.1046/j.1469-0705.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- 9.Jiang C, Ye W, Liu C, et al. Surgical ligation of portosystemic shunt to resolve severe hematuria and hemafecia caused by type II abernethy malformation. Ann Vasc Surg. 2015;29(5):1020.e11–1020.e16. doi: 10.1016/j.avsg.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Gong Y, Zhu H, Chen J, et al. Congenital portosystemic shunts with and without gastrointestinal bleeding—case series. Pediatr Radiol. 2015;45(13):1964–1971. doi: 10.1007/s00247-015-3417-6. [DOI] [PubMed] [Google Scholar]
- 11.Kalifa G, Brunelle F, Chaumont P. Fistule porto-cavale congenitale. Ann Radiol. 1978;21:183–186. [PubMed] [Google Scholar]
- 12.Morse SS, Taylor KJW, Strauss EB, et al. Congenital absence of the portal vein in oculoauriculovertebral dysplasia (Goldenhar syndrome) Pediatr Radiol. 1986;16:437–439. doi: 10.1007/BF02386831. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto S, Shigeta T, Fukuda A, et al. The role of liver transplantation for congenital extrahepatic portosystemic shunt. Transplantation. 2012;93(12):1282–1287. doi: 10.1097/TP.0b013e318250c157. [DOI] [PubMed] [Google Scholar]
- 14.Bas S, Guran T, Atay Z, et al. Premature pubarche, hyperinsulinemia and hypothyroxinemia: novel manifestations of congenital portosystemic shunts (Abernethy Malformation) in Children. Horm Res Paediatr. 2015;83(4):282–287. doi: 10.1159/000369395. [DOI] [PubMed] [Google Scholar]
- 15.Sokollik C, Bandsma RH, Gana JC, et al. Congenital portosystemic shunt: characterization of a multisystem disease. J Pediatr Gastroenterol Nutr. 2013;56(6):675–681. doi: 10.1097/MPG.0b013e31828b3750. [DOI] [PubMed] [Google Scholar]
- 16.Uchino T, Matsuda I, Endo F. The long-term prognosis of congenital portosystemic venous shunt. J Pediatr. 1999;135(2 Pt 1):254–256. doi: 10.1016/s0022-3476(99)70031-4. [DOI] [PubMed] [Google Scholar]
- 17.Ohno T, Muneuchi J, Ihara K, et al. Pulmonary hypertension in patients with congenital portosystemic venous shunt: a previously unrecognized association. Pediatrics. 2008;121(4):e892–e899. doi: 10.1542/peds.2006-3411. [DOI] [PubMed] [Google Scholar]
- 18.Ohnishi Y, Ueda M, Doi H, et al. Successful liver transplantation for congenital absence of the portal vein complicated by intrapulmonary shunt and brain abscess. J Pediatr Surg. 2005;40(5):E-3. doi: 10.1016/j.jpedsurg.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez AE, Ribeiro AF, Hessel G, et al. Abernethy malformation: one of the etiologies of hepatopulmonary syndrome. Pediatr Pulmonol. 2002;34:391–394. doi: 10.1002/ppul.10182. [DOI] [PubMed] [Google Scholar]
- 20.Pupulim LF, Vullierme MP, Paradis V, et al. Congenital portosystemic shunts associated with liver tumours. Clin Radiol. 2013;68(7):e362–e369. doi: 10.1016/j.crad.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 21.Franchi-Abella S, Branchereau S, Lambert V, et al. Complications of congenital portosystemic shunts in children: therapeutic options and outcomes. J Pediatr Gastroenterol Nutr. 2010;51(3):322–330. doi: 10.1097/MPG.0b013e3181d9cb92. [DOI] [PubMed] [Google Scholar]
- 22.Sorkin T, Strautnieks S, Foskett P, et al. Multiple β-catenin mutations in hepatocellular lesions arising in Abernethy malformation. Hum Pathol. 2016;53:153–158. doi: 10.1016/j.humpath.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 23.Lisovsky M, Konstas AA, Misdraji J. Congenital extrahepatic portosystemic shunts (Abernethy malformation): a histopathologic evaluation. Am J Surg Pathol. 2011;35(9):1381–1390. doi: 10.1097/PAS.0b013e3182230ce4. [DOI] [PubMed] [Google Scholar]
- 24.Bruckheimer E, Dagan T, Atar E, et al. Staged transcatheter treatment of portal hypoplasia and congenital portosystemic shunts in children. Cardiovasc Intervent Radiol. 2013;36(6):1580–1585. doi: 10.1007/s00270-013-0581-7. [DOI] [PubMed] [Google Scholar]
- 25.Kanamori Y, Hashizume K, Kitano Y, et al. Congenital extrahepatic portocaval shunt (Abernethy type 2), huge liver mass, and patent ductus arteriosus–a case report of its rare clinical presentation in a young girl. J Pediatr Surg. 2003;38(4):E15. doi: 10.1053/jpsu.2003.50153. [DOI] [PubMed] [Google Scholar]
- 26.Tercier S, Delarue A, Rouault F, et al. Congenital portocaval fistula associated with hepatopulmonary syndrome: ligation vs liver transplantation. J Pediatr Surg. 2006;41:e1. doi: 10.1016/j.jpedsurg.2005.10.071. [DOI] [PubMed] [Google Scholar]
- 27.Iida T, Ogura Y, Doi H, et al. Successful treatment of pulmonary hypertension secondary to congenital extrahepatic portocaval shunts (Abernethy type 2) by living donor liver transplantation after surgical shunt ligation. Transpl Int. 2010;23(1):105–109. doi: 10.1111/j.1432-2277.2009.00964.x. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Ma Z, Xie Y, et al. Recurrent hyperammonemia after abernethy malformation type 2 closure: a case report. Ann Hepatol. 2017;16(3):460–464. doi: 10.5604/16652681.1235492. [DOI] [PubMed] [Google Scholar]
- 29.Özden İ, Yavru A, Güllüoğlu M, et al. Transplantation for large liver tumors in the setting of Abernethy malformation. Exp Clin Transplant. 2017;15(Suppl 2):82–85. doi: 10.6002/ect.TOND16.L23. [DOI] [PubMed] [Google Scholar]
- 30.Benedict M, Rodriguez-Davalos M, Emre S, et al. Congenital extrahepatic portosystemic shunt (Abernethy malformation type Ib) with associated hepatocellular carcinoma. Pediatr Dev Pathol. 2017;20(4):354. doi: 10.1177/1093526616686458. [DOI] [PubMed] [Google Scholar]
- 31.Brasoveanu V, Ionescu MI, Grigorie R, et al. Living donor liver transplantation for unresectable liver adenomatosis associated with congenital absence of portal vein: a case report and literature review. Am J Case Rep. 2015;19(16):637–644. doi: 10.12659/AJCR.895235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanada Y, Mizuta K, Niki T, et al. Hepatocellular nodules resulting from congenital extrahepatic portosystemic shunts can differentiate into potentially malignant hepatocellular adenomas. J Hepatobiliary Pancreat Sci. 2015;22(10):746–756. doi: 10.1002/jhbp.277. [DOI] [PubMed] [Google Scholar]
- 33.Yilmaz C, Onen Z, Farajov R, et al. Live donor liver transplantation for a child presented with severe hepatopulmonary syndrome and nodular liver lesions due to Abernethy malformation. Pediatr Transplant. 2017;21(2):e12874. doi: 10.1111/petr.12874. [DOI] [PubMed] [Google Scholar]
- 34.Murray CP, Yoo SJ, Babyn PS. Congenital extrahepatic portosystemic shunts. Pediatr Radiol. 2003;33(9):614–620. doi: 10.1007/s00247-003-1002-x. [DOI] [PubMed] [Google Scholar]
- 35.Ringe K, Schirg E, Melter M, et al. Congenital absence of the portal vein (CAPV). Two cases of Abernethy malformation type 1 and review of the literature (in German) Radiologe. 2008;48:493–502. doi: 10.1007/s00117-007-1561-1. [DOI] [PubMed] [Google Scholar]
- 36.Stewart JK, Kuo WT, Hovsepian DM, et al. Portal venous remodeling after endovascular reduction of pediatric autogenous portosystemic shunts. J Vasc Interv Radiol. 2011;22(8):1199–1205. doi: 10.1016/j.jvir.2011.01.438. [DOI] [PubMed] [Google Scholar]
- 37.Mistinova J, Valacsai F, Varga I. Congenital absence of the portal vein–Case report and a review of literature. Clin Anat. 2010;23(7):750–758. doi: 10.1002/ca.21007. [DOI] [PubMed] [Google Scholar]
- 38.Athanasiadis A, Karavida A, Chondromatidou S, et al. Prenatal diagnosis of Abernethy malformation by three-dimensional ultrasonography. Ultrasound Obstet Gynecol. 2015;46(5):638–639. doi: 10.1002/uog.14923. [DOI] [PubMed] [Google Scholar]
- 39.Mi XX, Li XG, Wang ZR, et al. Abernethy malformation associated with Caroli’s syndrome in a patient with a PKHD1 mutation: a case report. Diagn Pathol. 2017;12(1):61. doi: 10.1186/s13000-017-0647-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Draghi F, Rapaccini GL, Fachinetti C, et al. Ultrasound examination of the liver: normal vascular anatomy. J Ultrasound. 2007;10(1):5–11. doi: 10.1016/j.jus.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Battaglia S, Fachinetti C, Draghi F, et al. Ultrasound examination of the liver: variations in the vascular anatomy. J Ultrasound. 2010;13(2):49–56. doi: 10.1016/j.jus.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puccia F, Lombardo V, Giannitrapani L, et al. Case report: acute portal vein thrombosis associated with acute cytomegalovirus infection in an immunocompetent adult. J Ultrasound. 2017;20(2):161–165. doi: 10.1007/s40477-016-0227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricci P, Cantisani V, Biancari F, Drud FM, et al. Contrast-enhanced color Doppler US in malignant portal vein thrombosis. Acta Radiol. 2000;41(5):470–473. doi: 10.1080/028418500127345703. [DOI] [PubMed] [Google Scholar]
- 44.Sidhu PS, Cantisani V, Deganello A, et al. Role of contrast-enhanced ultrasound (CEUS) in paediatric practice: an EFSUMB position statement. Ultraschall Med. 2017;38(1):33–43. doi: 10.1055/s-0042-110394. [DOI] [PubMed] [Google Scholar]
- 45.Hwang M, Thimm MA, Guerrerio AL. Detection of cavernous transformation of the portal vein by contrast-enhanced ultrasound. J Ultrasound. 2018;21(2):153–157. doi: 10.1007/s40477-018-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]