

Abstract
The Zika virus (ZIKV) became a major worldwide public concern in 2015 due to the congenital syndrome which presents the highest risk during the first trimester of pregnancy and includes microcephaly and eye malformations. Several cellular, genetic and molecular studies have shown alterations in metabolic pathways, endoplasmic reticulum (ER) stress, immunity and dysregulation of RNA and energy metabolism both in vivo and in vitro. Here we summarise the main metabolic complications, with a particular focus on the possibility that brain energy metabolism is altered following ZIKV infection, contributing to developmental abnormalities. Brain energetic failure has been implicated in neurological conditions such as autism disorder and epilepsy, as well as in metabolic diseases with severe neurodevelopmental complications such as Glut‐1 deficiency syndrome. Therefore, these energetic alterations are of wide‐ranging interest as they might be directly implicated in congenital ZIKV syndrome. Data showing increased glycolysis during ZIKV infection, presumably required for viral replication, might support the idea that the virus can cause energetic stress in the developing brain cells. Consequences may include neuroinflammation, cell cycle dysregulation and cell death. Ketone bodies are non‐glycolytic brain fuels that are produced during neonatal life, starvation or fasting, ingestion of high‐fat low‐carbohydrate diets, and following supplementation with ketone esters. We propose that dietary ketones might alter the course of the disease and could even provide some degree of prevention of ZIKV‐associated abnormalities and potentially related neurological conditions characterised by brain glucose impairment.
Keywords: congenital Zika syndrome, diet, glucose, ketone bodies, metabolism, microcephaly, Zika virus
Introduction
Zika viruses (ZIKV), positive single‐stranded RNA [(+)ssRNA] mosquito‐transmitted viruses of the genus Flavivirus, were first isolated in 1947 from a febrile rhesus macaque in Uganda (Dick et al. 1952; Platt & Miner, 2017; Wen et al. 2017). Outbreaks in 2007 (Pacific islands), 2013 (French Polynesia; Cao‐Lormeau et al. 2014; Duffy et al. 2009; Musso et al. 2015) and 2015 (Brazil), were associated with an increase in birth abnormalities (Heymann et al. 2016; Wen et al. 2017) and other neurological disorders. Several studies have shown the propensity of ZIKV to infect neuronal tissue (Garcez et al. 2016; Petersen et al. 2016; Qian et al. 2016; Souza et al. 2016; Tang et al. 2016; Zhang et al. 2016; Bhatnagar et al. 2017; Li et al. 2017), hence associating it with neurological complications such as Guillain‐Barré syndrome, meningoencephalitis and myelitis (Blázquez & Saiz, 2016; Heymann et al. 2016; Pinheiro et al. 2016; Wen et al. 2017; Uncini et al. 2018).
Congenital microcephaly was principally observed when ZIKV infection occurred during the first and second trimesters of pregnancy (Cauchemez et al. 2016; Kleber de Oliveira et al. 2016; Pacheco et al. 2016), potentially due to vertical transmission through the placenta (Brasil et al. 2016; Calvet et al. 2016; de Noronha et al. 2016; de Paula Freitas et al. 2016; Ventura et al. 2016; Bhatnagar et al. 2017; Platt & Miner, 2017). ZIKV‐infection studies, both in vitro and in vivo, have shown that neuronal death, dysregulation of apoptosis and neurogenesis, and a decrease in brain size are common outcomes (Cugola et al. 2016; Rossi & Vasilakis, 2016; Tang et al. 2016; Garcez et al. 2017; Platt & Miner, 2017; Wen et al. 2017).
Brain reduction and microcephaly are phenotypes also exhibited by genetic mutations and/or energy impairment during neurodevelopment (Woods, 2004; Faheem et al. 2015). Microcephaly and Glut‐1 deficiency syndrome, where glucose uptake in the brain is decreased due to lack of expression of glucose receptors (Klepper et al. 2004; Jensen et al. 2006; Klepper, 2008; Tang et al. 2017), highlights the importance of energy supply for brain growth and function, as the brain is a sensitive organ with complex energy pathways and interactions (Owen et al. 1967; Dienel & Hertz, 2001; Cahill, 2006; Schönfeld & Reiser, 2013; Schousboe et al. 2014; Falkowska et al. 2015; Hofmann et al. 2017). In this review, we summarise data regarding the cellular processes exhibited during ZIKV infection and suggest that a potential cellular energetic failure underlies most of the cellular hallmarks, thus providing new insights for possible treatments.
Energy metabolism in the developing and adult brain
Brain fuels required for development and function
Neurons and glia have distinct requirements for energy substrates, particularly during activation (Schönfeld & Reiser, 2013; Tracey et al. 2018), yet the interactions between the metabolism of the different cell types remain unclear. To date, it has been hypothesised that the metabolism of neurons is more dependent on oxygen compared with glia metabolism (Itoh et al. 2003; Herrero‐Mendez et al. 2009).
Glucose is considered to be the primary fuel for the brain and is essential for normal placental and fetal growth (Hay, 2006). The glucose absorption for adequate brain development and activity (Devraj et al. 2011; Tang et al. 2017) is mainly mediated by glucose transporters (GLUTs), Glut‐1, Glut‐3 and Glut‐5, which are expressed across the placenta, blood‐brain barrier and neural progenitors, differentiated neuronal tissue, and microglia, respectively (Fig. 1) (Maher et al. 1991; Payne et al. 1997; Jurcovicova, 2014).
Figure 1.
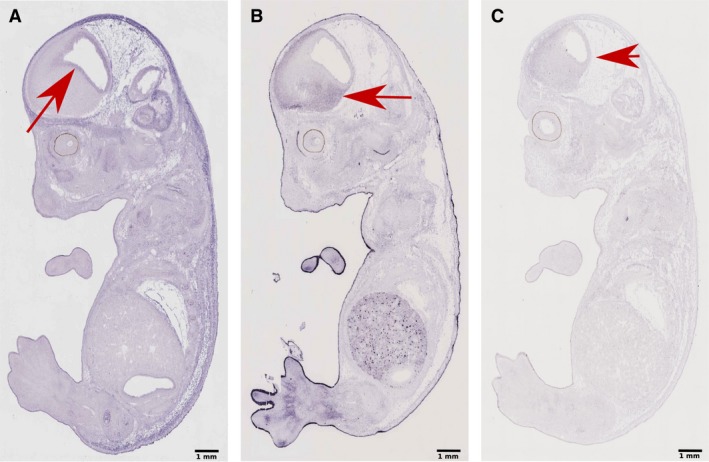
Glucose receptors in the embryonic mouse brain. genepaint sagittal plane sectioning image data of E14.5 Strain C57BL/6. (A) In situ hybridisation of the solute carrier family 2 (facilitated glucose transporter) member 1 (SLC2A1), mainly located in the ventricular zone at the neural progenitor region of the cerebral cortex and the ganglionic eminence (red arrow) (genepaint set ID: EH739). (B) In situ hybridisation of the solute carrier family 2 (facilitated glucose transporter) member 3 (SLC2A3) mainly located in postmitotic and differentiated neuronal populations in the primordial plexiform zone or preplate of the cortex and the striatum (red arrow) (genepaint set ID: EH4603). (C) In situ hybridisation of the solute carrier family 2 (facilitated glucose transporter) member 5 (SLC2A5) widely spread across the brain (red arrow) (genepaint set ID: EH4766). Scale bar: 1 mm.
During fetal growth, the placenta regulates nutrient absorption (Fig. 2). In sheep, placental glucose uptake was directly correlated with the fetal artery concentration, independently of maternal plasma glucose concentration (Hay et al. 1990). Similarly, human studies showed that uteroplacental glucose absorption is exerted in both fetal and maternal sides of the placenta yet is slightly higher on the maternal side (Holme et al. 2015). Hence, transplacental glucose uptake is not exclusively related to the maternal glucose levels but is mostly determined by the fetal venous‐arterial glucose concentration (Hay et al. 1990; Schneider et al. 2003; Hay, 2006).
Figure 2.
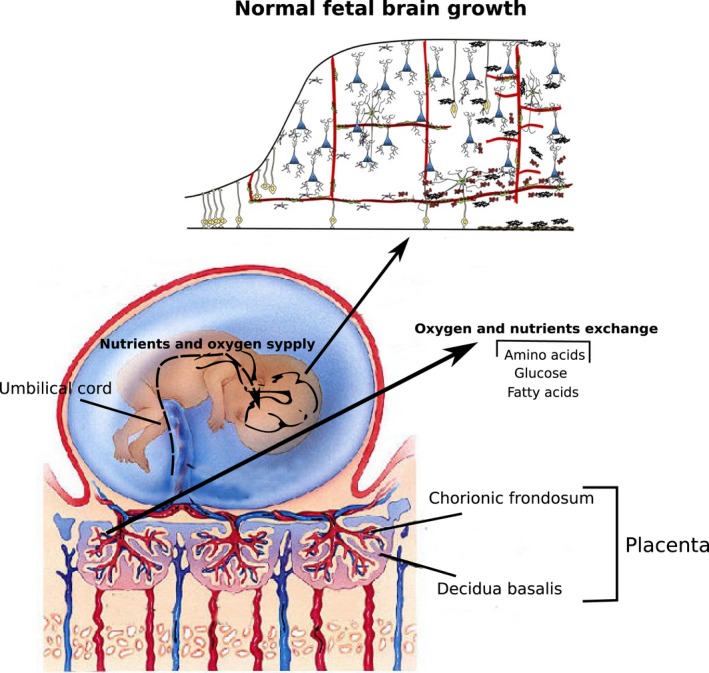
Nutrient delivery through the interactions of the maternal and fetal circulatory system for fetal growth. The maternal and fetal circulatory systems interact in the placenta to deliver the nutrients to the fetus. The main macro‐nutrients, glucose, amino acids and fatty acids, cross the placenta by means of their respective transporters (e.g. Glut‐1 and Glut‐3 for glucose molecules) and provide the energetic and structural requirements for normal fetal growth. During brain development, glucose is considered to provide energy for cell division and differentiation, amino acids contribute to the homeostasis and fatty acids are mainly used to generate myelin sheets during myelination. Modified from www.differencebetween.net (Copyright © The McGraw‐Hill Companies, Inc.), schoolbag.info and Stolp et al. (2012).
Neurons are able to convert glucose into Acetyl‐CoA for the production of substrates and the generation of ATP by oxidative phosphorylation, whereas glial cells, despite the presence of oxygen, preferentially convert glucose to lactate via cytosolic ‘aerobic glycolysis” (Schönfeld & Reiser, 2013; Camandola & Mattson, 2017); in a similar phenomenon to the ‘Warburg effect’ reported in oncology. Thus, it is hypothesised that the lactate produced by glial cells can also be metabolised by differentiated neurons for mitochondrial respiration and generation of ATP; potentially in a preferable way to glucose (Itoh et al. 2003). This complementary function of neurons and glia, known as the astrocyte‐neuron lactate shuttle (ANLS; Dienel & Hertz, 2001; Falkowska et al. 2015; Thevenet et al. 2016), potentially suggests that glial aerobic glycolysis may act as a fundamental mechanism to support neuronal metabolism (Fig. 3).
Figure 3.
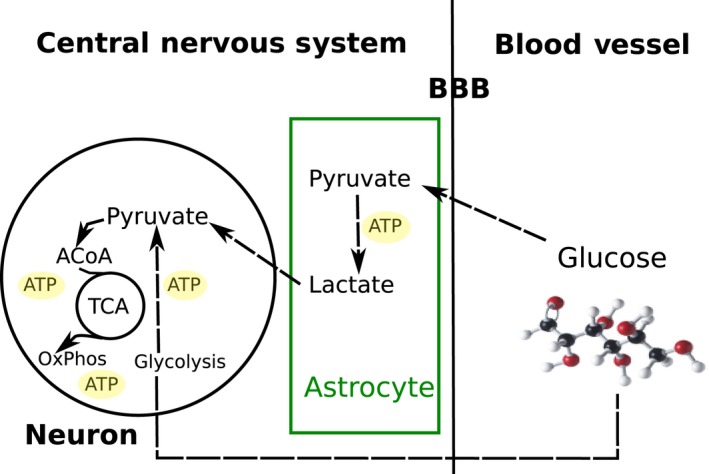
Glucose metabolism in astrocytes and neuronal cells. Glucose molecules available in the blood vessels cross the blood‐brain barrier (BBB) and are taken up by astrocytes and neurons. Astrocytes use aerobic glycolysis, despite the presence of oxygen, to produce ATP and Lactate (Warburg‐like effect). Neurons take up glucose directly from the bloodstream, which is converted to pyruvate via glycolysis. Lactate shuttled by astrocytes is also taken up by neurons and converted into pyruvate. Pyruvate inside the neuronal cytosol is converted into Acetyl‐CoA and enters the Krebs cycle, releasing by‐products for OxPhos to produce sufficient ATP, required for neuronal activity.
When exposed to the synaptic release of glutamate, astrocytes cycle glutamate/GABA to glutamine‐producing anaplerotic substrates that can feed and support the Krebs cycle maintenance (Dienel & Hertz, 2001; Schousboe et al. 2014) and hence oxidative phosphorylation. Astrocytes can also oxidise fatty acids to Acetyl‐CoA (Pellerin & Magistretti, 1994; Magistretti & Pellerin, 1999); therefore, upon activation such as during pre‐ and post‐synaptic processes, fatty acids metabolism in astrocytes is suggested to co‐occur together with cytosolic glucose oxidation to shuttle lactate for neuronal energy production while producing sufficient adenosine triphosphate (ATP) in the mitochondria to maintain the glutamate/glutamine cycle and astrocytic functions (Panov et al. 2014).
Moreover, oxygen is a limiting factor for brain activity. Higher oxygen consumption, particularly during synaptic activity, releases reactive oxygen species (ROS), to which brain cells are sensitive. Metabolism of ascorbic acid, the main brain antioxidant, is hypothesised to play a key role in the control of ROS toxicity (Castro et al. 2007, 2008). In brief, it has been suggested that ascorbic acid released from glial cells to the synaptic cleft and taken up by neurons, is oxidised to dehydroascorbic acid during ROS scavenging and released to be subsequently absorbed by astrocytes and re‐reduced to ascorbic acid to restart the cycle (Covarrubias‐Pinto et al. 2015).
Because of the sensitivity of neurons to oxidative stress, it is expected that glial cells, rather than neurons, would oxidize fatty acids (Bélanger et al. 2011; Panov et al. 2014; Romano et al. 2017), yet medium‐chain fatty acids (MCFA) can be metabolised as an additional energy substrate in both glial cells and neurons (Augustin et al. 2018) (Fig. 4). Interestingly, in astrocytes, it has been shown that MCFA decrease the mitochondrial respiratory chain capacity without decreasing the intracellular levels of ATP, potentially by promoting an increase in aerobic glycolysis and/or ketogenesis (Thevenet et al. 2016).
Figure 4.
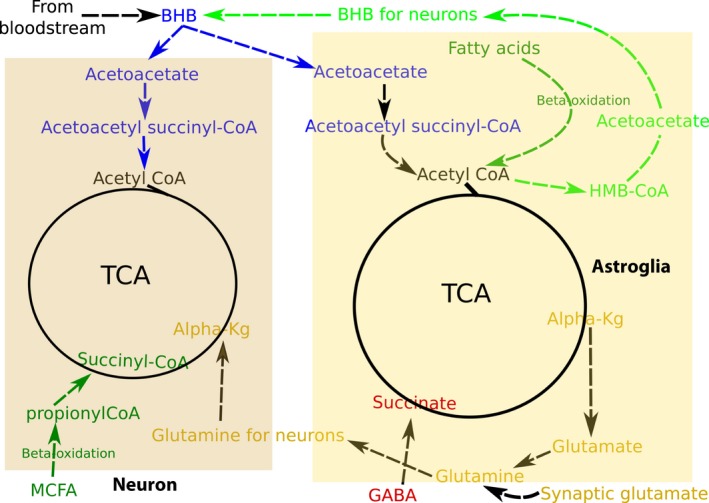
Model of non‐glycolytic metabolism interaction in astroglial and neuronal cells. In the presence of glucose, free fatty acids in the brain are taken up and oxidised by astroglial and neuronal cells to produce ATP via Krebs cycle/OxPhos as a supplementary energy source. Potentially, MCFA are mainly oxidised in neurons and other fatty acids in astroglial cells (dark green pathways). Under glucose deprivation: (1) astroglial cells can conduct ketogenesis to provide fast energetic substrates to neurons (light green pathway) and (2) ketone bodies from the bloodstream are taken up and oxidised by brain cells, providing up to ~70% of the energetic requirements (blue pathways). (3) Glutamate/glutamine is necessary to regulate the Krebs cycle, playing a major role in the communication of astroglia and neurons by processing excessive synaptic glutamate and providing amino acid glutamine. Also, but to a lesser extent, astroglial produced alpha‐ketoglutarate can be converted into glutamate to promote neurotransmitter and/or neuronal energetic by‐product release (mustard coloured pathways). (5). Neurotransmitter GABA can also be converted into a Krebs cycle metabolite depending on the needs of the mitochondrial Krebs cycle (red coloured pathway).
Despite β‐oxidation of fatty acids providing a higher yield of ATP than glucose metabolism when considering the net ATP production per available carbon bond (7.1 mol ATP in palmitate compared with 6.4 in glucose), neuronal cells might not favour this pathway: (1) it requires more oxygen due to a higher number of Acetyl‐CoA molecules, (2) it produces higher levels of ROS during oxidative phosphorylation when NADH and FADH2 are oxidised, which has been associated with neuronal damage and death (Liu et al. 2017) and (3) the rate of ATP production from blood glucose matches the fast requirements of the neurons (Schönfeld & Reiser, 2013).
During neonate life, while breastfeeding, mammalians are exposed to a particular condition where carbohydrates, previously supplied by the maternal circulation, became restricted, and the diet is mainly constituted on fats (Bougneres et al. 1986; Gustafsson, 2009; Cotter et al. 2011). Ketone bodies, mainly synthesised in the liver when only low carbohydrates are available, are distributed, absorbed and utilised as an energy fuel in other organs, particularly the brain (Grabacka et al. 2016; Le Foll & Levin, 2016). Once carbohydrates become available in the diet, the previously described process can only occur under particular physiological stress conditions such as starvation, fasting and ingestion of high‐fat low‐carbohydrate diets. Ketone bodies in the brain, presumably exclusively produced in astrocytes to a lesser extent when high rates of free fatty acids are observed (Le Foll & Levin, 2016), are mainly diffused from the bloodstream and account as the major substrate to provide energy (~ 70%) under glucose deprivation (Fig. 4) (Owen et al. 1967; Morris, 2005; Cahill, 2006; Owen, 2006; Klepper, 2008).
Ketone body metabolism is commonly studied based on the most abundant blood ketone body, β‐hydroxybutyrate (BHB), but two other ketone bodies (acetoacetate and acetone) are produced as result of ketogenesis. While BHB and acetoacetate are used by the cells to produce energy, acetone is released when breathing, serving as an indicator of the ketotic state (Musa‐Veloso et al. 2002). As energy substrate, BHB is converted into acetoacetate by 3‐hydroxybutyrate dehydrogenase; acetoacetate molecules are converted to acetoacetyl‐CoA by acetoacetyl succinyl CoA transferase to produce two molecules of acetyl‐CoA through the effects of acetoacetyl‐CoA thiolase. Molecules of acetyl‐CoA enter the Krebs cycle and respiratory chain to produce ATP (Grabacka et al. 2016).
ZIKV emergence and importance
The worldwide emergence of ZIKV became a major public health concern in 2015 when the epidemic in Brazil was shown to be highly correlated with an increase in abnormalities in newborn children (Musso et al. 2015; Heymann et al. 2016; Coelho & Crovella, 2017). Brazil was the first country in the American continent to suffer from the ZIKV epidemic, but it is now widespread on the entire continent (Pacheco et al. 2016; Colón‐González et al. 2017), with the potential of compromising currently unaffected countries where there is no herd immunity and where the environmental conditions allow the reproduction of mosquitos and viral transmission (Messina et al. 2016; Samy et al. 2016; Colón‐González et al. 2017).
The public concern about ZIKV disease and the neurological complications (Araujo et al. 2016; Blázquez & Saiz, 2016; Rabaan et al. 2017) relies heavily on the potential routes of transmission and infection (Gebre et al. 2016). ZIKV is mainly transmitted by infected Aedes mosquitos, sexual relations and vertical (fetal‐placental) transmission (Araujo et al. 2016; Brasil et al. 2016; Calvet et al. 2016; Cugola et al. 2016; Kleber de Oliveira et al. 2016; Bhatnagar et al. 2017; Weaver, 2017; Shi et al. 2018), and, according to other research, it may potentially be transmitted by blood transfusions and breastfeeding (Gebre et al. 2016; Colt et al. 2017; Deng et al. 2017; Sharma & Lal, 2017).
The complications due to ZIKV infections are variable, being either asymptomatic (~ 80% of the cases) or symptomatic (Gebre et al. 2016; Rabaan et al. 2017). Symptomatic infections exhibit a generalised mild fever, myalgia, arthralgia and headache, barely distinguishable from other diseases such as influenza. However, additional symptoms can include rash and conjunctivitis (Duffy et al. 2009; Hayes, 2009; Chen & Hamer, 2016; Rabaan et al. 2017), similar to those seen with other flaviviruses such as dengue (DENV) and chikungunya viruses (Mlakar et al. 2016). As both prevention of infection and early and specific diagnosis are challenging, it is highly desirable to develop post‐infection treatments to prevent risks to pregnancy, birth defects and adult complications (Krow‐Lucal et al. 2018).
ZIKV infection: fetal abnormalities and potential long‐term neurological effects
The Zika virus has been shown to infect different tissues such as decidua, fetal placenta and the umbilical cord (El Costa et al. 2016) which support vertical transmission; however, the virus has also shown a marked preference for neuronal tissue (Garcez et al. 2016; Petersen et al. 2016; Qian et al. 2016; Souza et al. 2016; Tang et al. 2016; Zhang et al. 2016; Bhatnagar et al. 2017; Li et al. 2017). It has been shown that ZIKV‐related fetal neurological complications are highest during the first trimester of pregnancy (El Costa et al. 2016; Bhatnagar et al. 2017; Honein et al. 2017). However, findings suggest that these may also continue to occur during the second (Lin et al. 2017) and third trimesters (Hayes, 2009; Shapiro‐Mendoza, 2017) to a lesser extent (Bhatnagar et al. 2017).
The Zika virus preferential infection for neural stem cells and progenitors is more marked than for other flaviviruses (Brault et al. 2016; El Costa et al. 2016; Miner & Diamond, 2017; Ming et al. 2016; de Noronha et al. 2016; Martines et al. 2016; Souza et al. 2016; Zhang et al. 2016), potentially explaining why adult symptomatology is mostly mild with no further complications but causing severe neurological complications in the fetus and newborns (Brault et al. 2016; Hughes et al. 2016).
Among the ZIKV fetal/newborn complications, a reduction in the head and brain size, called microcephaly, together with eye malformations are the most relevant abnormalities (Araujo et al. 2016; Mlakar et al. 2016; Sarno et al. 2016; Merfeld et al. 2017). However, the infection can also be the cause of long‐term mental health conditions such as cognitive impairment, behavioural and neurological complications in newborns with no evident phenotype after maternal immune activation due to viral, including flaviviral, and bacterial infections (Verma et al. 2011; Stolp et al. 2012; Estes & McAllister, 2016; Lombardo et al. 2018).
Microcephaly and eye malformations
Microcephaly in newborns is characterised by a head circumference of 2 SD smaller than the local average. Microcephaly can vary broadly in clinical severity with differences in the brain mass morphology (Woods, 2004; Adachi et al. 2014), underlying severe neuronal loss in the cerebral cortex (Azevedo et al. 2009; Merfeld et al. 2017) and/or reduction in the population of glial cells (Lin et al. 2017). Microcephaly is characterised by abnormalities in the production/differentiation of neural progenitor cells (NPC; Gilmore & Walsh, 2013) potentially through alterations in the endothelial cells of the brain blood vessels that directly alter NPC homeostasis by decreasing nutrients flow and trophic factors (Shen et al. 2004; Garcez et al. 2018). Primary microcephaly is the disease type caused by diverse genetic and epigenetic factors, including maternal viral infections (Woods, 2004; Faheem et al. 2015; Zhang et al. 2016; Merfeld et al. 2017) with an estimated rate of 2–12 cases per 10 000 births which increased up to ~ 20‐fold in early 2016 in Brazil due to ZIKV infections (Duffy et al. 2009; Cao‐Lormeau et al. 2014; Heymann et al. 2016; Mlakar et al. 2016; Coelho & Crovella, 2017; Cunha et al. 2017; Krow‐Lucal et al. 2018), with fewer neurological complications in other countries (Pacheco et al. 2016; Colón‐González et al. 2017; Honein et al. 2017; Rick et al. 2017).
ZIKV‐related microcephaly has been intensively studied since the outbreak in Brazil, showing negative effects on neurogenesis (Wen et al. 2017) and microgliogenesis (Li et al. 2018). Microgliogenesis is a process that occurs alongside neurogenesis and is of high relevance during development, as a close interaction between microglia and neuronal cells is maintained in the developing and adult CNS in both health and disease (Nayak, Roth, & McGavern, 2014), potentially suggesting a major role of microglia activation/death in ZIKV‐microcephaly.
As previously stated, cell and animal model studies of ZIKV‐microcephaly have been widely investigated to reveal the potential causes of the neurological complications showing dysregulation of different genes related to neuronal differentiation, neuronal growth, cellular metabolic function and cell death (Tang et al. 2016; Zhang et al. 2016; Devhare et al. 2017; Garcez et al. 2017; Lin et al. 2017), providing insights into potential prevention mechanisms.
The Zika virus congenital eye malformations are yet to be fully determined due to the poor knowledge of the full spectrum of ocular lesions (Jampol & Goldstein, 2016; de Paula Freitas et al. 2017). Nevertheless, reports of infants’ studies exhibited focal pigment mottling of the retina, atrophy in the chorioretina, alteration in the retinal vasculature, optic nerve abnormalities, bilateral iris coloboma and lens subluxation (de Paula Freitas et al. 2016; Garcez et al. 2018) and, in the fetus, loss of retinal epithelium, thin choroid, perivascular choroidal inflammatory infiltrate and atrophy of the optic nerve (Fernandez et al. 2017).
Cellular changes, damage and death in the CNS during ZIKV infection
ZIKV compromises neuronal and glial cell homeostasis
Recent research focused on the cellular and molecular dysregulation during ZIKV infections has provided insights into different potential biological processes responsible for neurological damage and microcephaly (Tang et al. 2016; Merfeld et al. 2017).
In a similar fashion to DENV (Savidis et al. 2016), ZIKV replication might activate early glycolysis and late fatty acids β‐oxidation; inducing a change in lipid metabolism. Observed lipid droplet formation, autophagy (Samsa et al. 2009; McLean et al. 2011; Tiwari et al. 2017; Tongluan et al. 2017; Lee et al. 2018) and enhanced higher intracellular ATP levels (Samsa et al. 2009; Heaton & Randall, 2010, 2011; Vidali et al. 2015) could be supported by lipid β‐oxidation processes.
Of relevance, ZIKV infection causes ER stress (Blázquez et al. 2014; Gladwyn‐Ng et al. 2018) and cell death (Fig. 5). ER stress occurs when an accumulation of unfolded proteins causes perturbation of the ER (Hetz, 2012; Cornejo et al. 2013). In response to this, a mechanism mainly conducted by three proteins named inositol‐required enzyme 1 (IRE1α), protein kinase RNA‐like ER kinase (PERK) and activating transcription factor 6 (ATF6) potentially alleviates the stress via decreasing mRNA translation and/or by degradation of unfolded proteins (Hetz, 2012; Cornejo et al. 2013; Sano & Reed, 2013).
Figure 5.
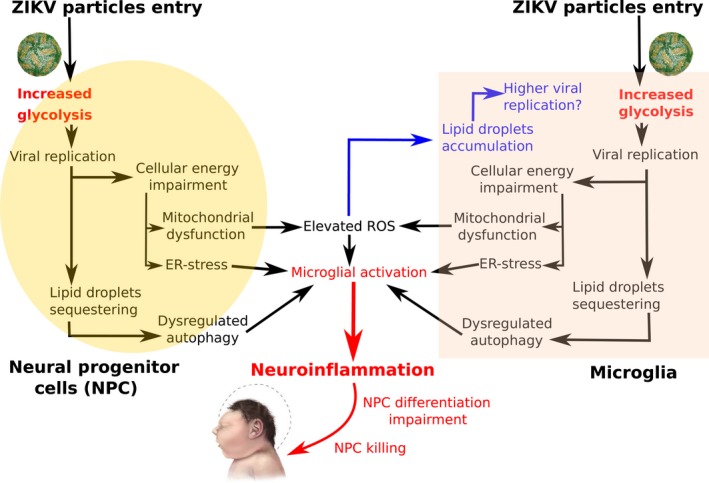
Hypothesised key role of glycolysis and neuroinflammation in congenital ZIKV syndrome. Increased glycolysis following ZIKV infection and replication in the endoplasmic reticulum (ER) of both neural progenitor (NPC) and microglial cells potentially causes (1) cellular energy impairment with further mitochondrial dysfunction and ER stress, and (2) sequestration of cytoplasmic lipid droplets required for final replication (pathways highlighted in black). Elevated reactive oxygen species (ROS) due to mitochondrial dysfunction cause an accumulation of lipid droplets in microglia, potentially favouring a higher viral replication rate than in neurons (pathway highlighted in blue). (3) Microglial activation by dysregulated autophagy, ER stress and elevated ROS contribute to neuroinflammation, impairing NPC differentiation and promoting NPC killing mechanisms (pathway highlighted in red); a possible explanation for brain growth impairment and congenital ZIKV syndrome.
In a normal cellular state, PERK and ATF6 remain inactive and bound to the ER, while IRE1α appears to be activated only when unfolded proteins bind to it (Gardner & Walter, 2011). Interestingly, PERK is upregulated in the brains of ZIKV‐infected humans, and this is replicated in both murine models and human cultured neural stem cells challenged with ZIKV in which downregulation of disturbed PERK pathway prevents microcephaly, potentially highlighting mechanisms for therapy (Gladwyn‐Ng et al. 2018).
In most viral infections, the innate immune system is highly responsible for countering viral replication (Xie et al. 2016). Interferon (IFN) activation in response to infection plays a key part of this response that restricts virus replication, depending on the viral strain (Evans et al. 2011; Lubick et al. 2015; Xia et al. 2016). ZIKV infection particularly alters the IFNAR1, by showing a decrease on the mRNA expression levels (Tang et al. 2016; Tiwari et al. 2017); suggesting a diminished innate immune response, potentially favouring viral replication.
To this end, several in vivo murine models systems of Ifnar1 are used to study ZIKV disease phenotypes such as microcephaly and ocular abnormalities (Bayer et al. 2016; Cugola et al. 2016; Morrison & Diamond, 2017), yet the inoculation route and mice strains should be considered. For example, when infecting –C57BL/6 mice subcutaneously, intravenously or intraperitoneally, weight loss, paralysis and high rates of mortality (20–100%) are observed; however, intrauterine growth impairment is not (Lazear et al. 2016; Ma et al. 2016; Miner et al. 2016), and when infecting subcutaneously on Ifnar1−/− mated with WT C57BL/6, the common phenotypes are placental injury, resorption and intrauterine growth impairment (Miner et al. 2016; Sapparapu et al. 2016; Yockey et al. 2016; Morrison & Diamond, 2017).
Furthermore, evidence of a major role for microglia‐mediated neuroinflammation in ZIKV‐associated phenotypes (Fig. 5) includes the observation that ZIKV infection of microglia inhibits the differentiation of neural precursors (Mesci et al. 2018; Wang et al. 2018) and leads to the death of damaged neurons (Brown & Vilalta, 2015). The downregulation of immunological genes in ZIKV‐infected microglia (Tiwari et al. 2017) suggests that the virus has evolved sophisticated strategies for modulating the innate antiviral response.
Glycolytic and cellular energetic dysregulation during ZIKV
Edward Blonz hypothesised that, similar to DENV and other viruses, ZIKV replication might alter brain cell glucose uptake by close interaction with the Glut‐1 receptor (Yu et al. 2011a,b; Fontaine et al. 2014). In this setting, brain energetic failure could potentially be the cause of ZIKV congenital malformations, as similar phenotypes are exhibited in Glut‐1 deficiency syndrome (Jensen et al. 2006; Blonz, 2016; Solomon et al. 2016; Tang et al. 2017).
In this context, research has shown that, as with DENV (Fontaine et al. 2014; Jordan & Randall, 2016; Rothan et al. 2018), glycolysis is used to produce energy, potentially for optimal ZIKV replication (Tiwari et al. 2017). Increased glycolysis evokes cellular energy impairment, which contributes to ER stress via the PERK pathway (de la Cadena et al. 2014), mitochondrial dysfunction and ROS production, all contributing to neurodegeneration (Rossignol & Frye, 2012; Liu et al. 2017).
Moreover, elevated ROS production in the brain contributes to lipid droplet accumulation, particularly in glial cells (Liu et al. 2015). Lipid droplets are intracellular storages with a crucial role in fatty acid trafficking and energy homeostasis, while also being a source of lipid metabolism in viral immunity (Welte, 2015). ZIKV, similar to DENV, potentially triggers lipid droplet accumulation for (1) final replication of the viral capsid and (2) increasing ATP availability in the cell via beta‐oxidation of fatty acids (Samsa et al. 2009; Heaton & Randall, 2010, 2011), causing dysregulated autophagy and, together with elevated ROS and ER stress, neuroinflammation, potentially explaining neural progenitors death, increased blood‐brain barrier permeability and brain growth impairment (Fig. 5).
Despite the higher complexity in brain energetics, which might be specific to brain regions, cellular subtypes and activity, glucose metabolism plays a major role as the main energetic substrate, and protein degradation and fatty acid oxidation are hypothesised to be conducted principally in glial cells (Dienel & Hertz, 2001; Panov et al. 2014; Schousboe et al. 2014; Romano et al. 2017). For these reasons, glucose metabolism could be implicated as the potential main cause for congenital ZIKV syndrome.
Geographical distribution can play a role in congenital ZIKV syndrome, with the highest prevalence of the epidemic being observed in the North‐East region of Brazil, a population characterised by poverty (Jimena Barbeito et al. 2018bb; Krow‐Lucal et al. 2018), presenting with comorbidities such as malnutrition. Therefore, this review considers that specific dietary patterns favouring the activation of certain metabolic pathways might play an important role in the susceptibility or protection against ZIKV virulence (Jimena Barbeito et al. 2018a).
Potential nutritional intervention to prevent ZIKV replication and fetal abnormalities
Ketones induce positive effects in brain homeostasis under glucose deprivation
Energetic deficit due to glucose impairment has a direct negative impact on human brain development and is therefore of potential consideration in ZIKV infection, particularly during the first trimester of pregnancy. As Blonz (2016) hypothesised, a high‐fat metabolism might prevent congenital abnormalities due to glucose impairment. Here we highlighted a potential link between cellular glucose impairment and different cellular alterations exhibited during ZIKV infection, emphasising that a ketone metabolism, as an efficient brain alternative fuel with positive effects in human physiology (Klepper et al. 2005; Morris, 2005; Owen Oliver, 2006), might prevent ZIKV‐related congenital phenotypes.
In addition to the positive effects of ketone metabolism in restoring glucose energetic impairment (Owen, 2006), ketones have been shown: (1) to contribute to brain homeostasis under glucose deprivation by reducing neuroinflammation via the restoration of mitochondria energetics (Vidali et al. 2015) and regulating autophagic flux to prevent neuronal death (Camberos‐Luna et al. 2016); (2) to decrease ROS release (Maalouf et al. 2007; Kim et al. 2010); (3) to alleviate energetic failure‐related ER stress (Bae et al. 2016; Soejima et al. 2018); (4) to promote efficient immune response (Kono et al., 2004; Rhyu & Cho, 2014).
The above raises the possibility that ketone metabolism, by either ingestion of a ketogenic diet or ketone ester supplementation, might prevent ZIKV‐congenital neurological phenotypes (1) by an energetic metabolic switch potentially preventing/decreasing ZIKV replication, via blocking glycolysis (Randle, 1998; Hue & Taegtmeyer, 2009), if it is essential for viral replication, and/or (2) by providing additional fuel to regulate cell functioning and homeostasis, promoting an efficient brain immune response against ZIKV infection. Finally, it is relevant to highlight that the neurological benefits of a ketone metabolism need to be further investigated in ZIKV infection, as it might ameliorate related adult mental/cognitive conditions and potential late‐onset complications in cases with no exhibited congenital syndrome.
Declaration of interest
The authors declare no conflicts of interest and that no interests of any person/organisation are affected by the information presented in the present manuscript. J.G.‐J. held a scholarship from the Ecuadorian National Government. The Molnár Laboratory held an MRC Rapid Response Grant in collaboration with Dr Patricia Garcéz (Federal University of Rio de Janeiro, Brazil).
Acknowledgements
The authors wish to thank the Anatomical Society for the Summer Meeting on Human Brain Development hosted by St John's College, Oxford. This review contributes to the special issue of JOA associated with this meeting. We are grateful to Mr Matthew Kerr, Mr Andrew Tyler and Miss Daria Iakovleva for their valuable comments on an earlier version of this manuscript and for their assistance with the figures.
References
- Adachi Y, Mochida G, Walsh C, et al. (2014) Posterior fossa in primary microcephaly: relationships between forebrain and mid‐hindbrain size in 110 patients. Neuropediatrics 45(2), 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo AQC, Silva MTT, Araujo APQC (2016) Zika virus‐associated neurological disorders: a review. Brain 139(8), 2122–2130. [DOI] [PubMed] [Google Scholar]
- Augustin K, Khabbush A, Williams S, et al. (2018) Mechanisms of action for the medium‐chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol 17(1), 84–93. [DOI] [PubMed] [Google Scholar]
- Azevedo FAC, Carvalho LRB, Grinberg LT, et al. (2009) Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled‐up primate brain. J Comp Neurol 513(5), 532–541. [DOI] [PubMed] [Google Scholar]
- Bae HR, Kim DH, Park MH, et al. (2016) β‐Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 7(41), 66444–66454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, et al. (2016) Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19(5), 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte‐neuron metabolic cooperation. Cell Metab 14(6), 724–738. [DOI] [PubMed] [Google Scholar]
- Bhatnagar J, Rabeneck DB, Martines RB, et al. (2017) Zika virus RNA replication and persistence in brain and placental tissue. Emerg Infect Dis 23(3), 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez A‐B, Saiz J‐C (2016) Neurological manifestations of Zika virus infection. World J Virol 5(4), 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez A‐B, Escribano‐Romero E, Merino‐Ramos T, et al. (2014) Stress responses in flavivirus‐infected cells: activation of unfolded protein response and autophagy. Front Microbiol 5, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonz ER (2016) Zika virus and GLUT1. Lancet Infect Dis 16(6), 642. [DOI] [PubMed] [Google Scholar]
- Bougneres PF, Lemmel C, Ferré P, et al. (1986) Ketone body transport in the human neonate and infant. J Clin Invest 77(1), 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil P, Pereira JP, Moreira ME, et al. (2016) Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 375(24), 2321–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault J‐B, Khou C, Basset J, et al. (2016) Comparative analysis between flaviviruses reveals specific neural stem cell tropism for Zika virus in the mouse developing neocortex. EBioMedicine 10, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Vilalta A (2015) How microglia kill neurons. Brain Res 1628, 288–297. [DOI] [PubMed] [Google Scholar]
- de la Cadena SG, Hernández‐Fonseca K, Camacho‐Arroyo I, et al. (2014) Glucose deprivation induces reticulum stress by the PERK pathway and caspase‐7‐ and calpain‐mediated caspase‐12 activation. Apoptosis, 19(3), 414–427. [DOI] [PubMed] [Google Scholar]
- Cahill GF (2006) Fuel metabolism in starvation. Annu Rev Nutr 26, 1–22. [DOI] [PubMed] [Google Scholar]
- Calvet G, Aguiar RS, Melo ASO, et al. (2016) Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16(6), 653–660. [DOI] [PubMed] [Google Scholar]
- Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration. EMBO J 36(11), 1474–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camberos‐Luna L, Gerónimo‐Olvera C, Montiel T, et al. (2016) The ketone body, β‐hydroxybutyrate stimulates the autophagic flux and prevents neuronal death induced by glucose deprivation in cortical cultured neurons. Neurochem Res 41(3), 600–609. [DOI] [PubMed] [Google Scholar]
- Cao‐Lormeau V‐M, Roche C, Teissier A, et al. (2014) Zika virus, French Polynesia, South Pacific, 2013. Emerg Infect Dis 20(6), 1084–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro MA, Pozo M, Cortés C, et al. (2007) Intracellular ascorbic acid inhibits transport of glucose by neurons, but not by astrocytes. J Neurochem 102(3), 773–782. [DOI] [PubMed] [Google Scholar]
- Castro MA, Angulo C, Brauchi S, et al. (2008) Ascorbic acid participates in a general mechanism for concerted glucose transport inhibition and lactate transport stimulation. Pflugers Arch 457(2), 519–528. [DOI] [PubMed] [Google Scholar]
- Cauchemez S, Besnard M, Bompard P, et al. (2016) Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet 387(10033), 2125–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LH, Hamer DH (2016) Zika virus: rapid spread in the Western Hemisphere. Ann Intern Med 164(9), 613. [DOI] [PubMed] [Google Scholar]
- Coelho AVC, Crovella S (2017) Microcephaly prevalence in infants born to Zika virus‐infected women: a systematic review and meta‐analysis. Int J Mol Sci 18(8), 1–10. 10.3390/ijms18081714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colón‐González FJ, Peres CA, Steiner SãoBernardo C, et al. (2017) After the epidemic: Zika virus projections for Latin America and the Caribbean. PLoS Negl Trop Dis 11(11), e0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colt S, Garcia‐Casal MN, Peña‐Rosas JP, et al. (2017) Transmission of Zika virus through breast milk and other breastfeeding‐related bodily fluids: a systematic review. PLoS Negl Trop Dis 11(4), e0005528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo VH, Pihán P, Vidal RL, et al. (2013) Role of the unfolded protein response in organ physiology: lessons from mouse models. IUBMB Life 65(12), 962–975. [DOI] [PubMed] [Google Scholar]
- Cotter DG, d'Avignon DA, Wentz AE, et al. (2011) Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J Biol Chem 286(9), 6902–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias‐Pinto A, Acuña AI, Beltrán FA, et al. (2015) Old things new view: ascorbic acid protects the brain in neurodegenerative disorders. Int J Mol Sci 16(12), 28194–28217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, et al. (2016) The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534(7606), 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha AJLA, de Magalhães‐Barbosa MC, Lima‐Setta F, et al. (2017) Microcephaly case fatality rate associated with Zika virus infection in Brazil: current estimates. Pediatr Infect Dis J 36(5), 528–530. [DOI] [PubMed] [Google Scholar]
- Deng Y‐Q, Zhang N‐N, Li X‐F, et al. (2017) Intranasal infection and contact transmission of Zika virus in guinea pigs. Nat Commun 8(1), 1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devhare P, Meyer K, Steele R, et al. (2017) Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis 8(10), e3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devraj K, Klinger ME, Myers RL, et al. (2011) GLUT‐1 Glucose transporters in the blood‐brain barrier: differential phosphorylation. J Neurosci Res 89(12), 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GW, Kitchen S, Haddow A. (1952) Zika Virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg 46(5), 509–520. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L (2001) Glucose and lactate metabolism during brain activation. J Neurosci Res 66(5), 824–838. [DOI] [PubMed] [Google Scholar]
- Duffy MR, Chen T‐H, Hancock WT, et al. (2009) Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360(24), 2536–2543. [DOI] [PubMed] [Google Scholar]
- El Costa H, Gouilly J, Mansuy J‐M, et al. (2016) ZIKA virus reveals broad tissue and cell tropism during the first trimester of pregnancy. Sci Rep 6, 35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK. (2016) Maternal immune activation: implications for neuropsychiatric disorders. Science (New York, N.Y.), 353(6301), 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Crown RA, Sohn JA, et al. (2011) West Nile virus infection induces depletion of IFNAR1 protein levels. Viral Immunol 24(4), 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faheem M, Naseer MI, Rasool M, et al. (2015) Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 8(Suppl 1), S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowska A, Gutowska I, Goschorska M, et al. (2015) Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int J Mol Sci 16(11), 25959–25981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MP, Parra Saad E, Ospina Martinez M, et al. (2017) Ocular histopathologic features of congenital Zika syndrome. JAMA Ophthalmol 135(11), 1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine KA, Sanchez EL, Camarda R, et al. (2014) Dengue virus induces and requires glycolysis for optimal replication. J Virol 89(4), 2358–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, et al. (2016) Zika virus impairs growth in human neurospheres and brain organoids. Science 352(6287), 816–818. [DOI] [PubMed] [Google Scholar]
- Garcez Patricia P, Nascimento JM, de Vasconcelos JM, et al. (2017) Zika virus disrupts molecular fingerprinting of human neurospheres. Sci Rep 7, 40780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Stolp HB, Sravanam S, et al. (2018) Zika virus impairs the development of blood vessels in a mouse model of congenital infection. Sci Rep 8(1), 12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Walter P. (2011) Unfolded proteins are Ire1‐activating ligands that directly induce the unfolded protein response. Science (New York, N.Y.), 333(6051), 1891–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebre Y, Forbes N, Gebre T (2016) Zika virus infection, transmission, associated neurological disorders and birth abnormalities: a review of progress in research, priorities and knowledge gaps. Asian Pac J Trop Biomed 6(10), 815–824. [Google Scholar]
- Gilmore EC, Walsh CA (2013). Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Dev Biol 2(4): 461–478. 10.1002/wdev.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwyn‐Ng I, Cordón‐Barris L, Alfano C, et al. (2018) Stress‐induced unfolded protein response contributes to Zika virus–associated microcephaly. Nat Neurosci 21(1), 63–71. [DOI] [PubMed] [Google Scholar]
- Grabacka M, Pierzchalska M, Dean M, et al. (2016) Regulation of Ketone body metabolism and the role of PPARα. Int J Mol Sci 17(12), 1–24. 10.3390/ijms17122093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson J (2009) Neonatal energy substrate production. Indian J Med Res 130(5), 618–623. [PubMed] [Google Scholar]
- Hay W W (2006) Placental‐fetal glucose exchange and fetal glucose metabolism. Trans Am Clin Climatol Assoc 117, 321–340. [PMC free article] [PubMed] [Google Scholar]
- Hay WW, Molina RA, DiGiacomo JE, et al. (1990) Model of placental glucose consumption and glucose transfer. Am J Physiol 258(3 Pt 2), R569–R577. [DOI] [PubMed] [Google Scholar]
- Hayes EB (2009) Zika virus outside Africa. Emerg Infect Dis 15(9), 1347–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Randall G (2010) Dengue virus induced autophagy regulates lipid metabolism. Cell Host Microbe 8(5), 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Randall G (2011) Dengue virus and autophagy. Viruses 3(8), 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero‐Mendez A, Almeida A, Fernández E, et al. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C‐Cdh1. Nat Cell Biol 11(6), 747–752. [DOI] [PubMed] [Google Scholar]
- Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2), 89–102. [DOI] [PubMed] [Google Scholar]
- Heymann DL, Hodgson A, Sall AA, et al. (2016) Zika virus and microcephaly: why is this situation a PHEIC? Lancet 387(10020), 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Rodriguez‐Rodriguez R, Gaebler A, et al. (2017) Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci Rep 7(1), 10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme AM, Roland MCP, Lorentzen B, et al. (2015) Placental glucose transfer: a human in vivo study. PLoS ONE 10(2), e0117084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honein MA, Dawson AL, Petersen EE, et al., US Zika Pregnancy Registry Collaboration (2017) Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. JAMA 317(1), 59–68. [DOI] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H (2009) The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297(3), E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes BW, Addanki KC, Sriskanda AN, et al. (2016) Infectivity of immature neurons to Zika virus: a link to congenital Zika syndrome. EBioMedicine 10, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, et al. (2003) Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo . Proc Natl Acad Sci U S A 100(8), 4879–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jampol LM, Goldstein DA (2016) Zika virus infection and the eye. JAMA Ophthalmol 134(5), 535–536. [DOI] [PubMed] [Google Scholar]
- Jensen PJ, Gitlin JD, Carayannopoulos MO (2006) GLUT1 deficiency links nutrient availability and apoptosis during embryonic development. J Biol Chem 281(19), 13382–13387. [DOI] [PubMed] [Google Scholar]
- Jimena Barbeito A, Pezzuto P, Higa LM, et al. (2018. a) Exploring brain phenotypic outcomes when Zika virus and protein undernutrition interact during early development, 165(1), 18–19.
- Jimena Barbeito A, Schuler‐Faccini L, Garcez PP (2018b) Why is congenital Zika syndrome asymmetrically distributed among human populations? PLoS Biol 16(8), e2006592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan TX, Randall G (2016) Flavivirus modulation of cellular metabolism. Curr Opin Virol 19, 7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurcovicova J (2014) Glucose transport in brain ‐ effect of inflammation. Endocr Regul 48(1), 35–48. [DOI] [PubMed] [Google Scholar]
- Kim DY, Vallejo J, Rho JM (2010) Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J Neurochem 114(1), 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber de Oliveira W, Cortez‐Escalante J, De Oliveira WTGH, et al. (2016). Increase in reported prevalence of microcephaly in infants born to women living in areas with confirmed Zika virus transmission during the first trimester of pregnancy – Brazil, 2015. MMWR Morb Mortal Wkly Rep 65(9), 242–247. [DOI] [PubMed] [Google Scholar]
- Klepper J (2008) Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 49(s8), 46–49. [DOI] [PubMed] [Google Scholar]
- Klepper J, Diefenbach S, Kohlschütter A, et al. (2004) Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids 70(3), 321–327. [DOI] [PubMed] [Google Scholar]
- Klepper J, Scheffer H, Leiendecker B, et al. (2005) Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2‐ to 5‐year follow‐up of 15 children enrolled prospectively. Neuropediatrics 36(5), 302–308. [DOI] [PubMed] [Google Scholar]
- Kono H, Fujii H, Asakawa M, et al. (2004) Medium‐chain triglycerides enhance secretory IgA expression in rat intestine after administration of endotoxin. Am J Physiol Gastrointest Liver Physiol 286(6), G1081–G1089. [DOI] [PubMed] [Google Scholar]
- Krow‐Lucal ER, de Andrade MR, Cananéa JNA, et al. (2018) Association and birth prevalence of microcephaly attributable to Zika virus infection among infants in Paraíba, Brazil, in 2015–16: a case‐control study. Lancet Child Adolesc Health 2(3), 205–213. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, et al. (2016) A mouse model of Zika virus pathogenesis. Cell Host Microbe 19(5), 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll C, Levin BE (2016) Fatty acid‐induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol 310(11), R1186–R1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y‐R, Kuo S‐H, Lin C‐Y, et al. (2018) Dengue virus‐induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo . Sci Rep 8(1), 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Deng Y‐Q, Wang S, et al. (2017) 25‐hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 46(3), 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Cui, Wang Q, Jiang Y, et al. (2018) Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Discov 4(1), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HZ, Tambyah PA, Yong EL, et al. (2017) A review of Zika virus infections in pregnancy and implications for antenatal care in Singapore. Singapore Med J 58(4), 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, et al. (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhou T, Ziegler AC, et al. (2017) Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med Cell Longev 2017, 2525967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MV, Moon HM, Su J, et al. (2018) Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol Psychiatry 23(4), 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubick KJ, Robertson SJ, McNally KL, et al. (2015) Flavivirus antagonism of type I interferon signaling reveals prolidase as a regulator of IFNAR1 surface expression. Cell Host Microbe 18(1), 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Li S, Ma S, et al. (2016) Zika virus causes testis damage and leads to male infertility in mice. Cell 167(6), 1511–1524.e10. [DOI] [PubMed] [Google Scholar]
- Maalouf M, Sullivan PG, Davis L, et al. (2007) Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145(1), 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L (1999) Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc B Biol Sci 354(1387), 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher F, Davies‐Hill TM, Lysko PG, et al. (1991) Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron‐specific expression of GLUT3. Mol Cell Neurosci 2(4), 351–360. [DOI] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, de Oliveira Ramos AM, et al. (2016). Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388(10047): 898–904. 10.1016/S0140-6736(16)30883-2 [DOI] [PubMed] [Google Scholar]
- McLean JE, Wudzinska A, Datan E, et al. (2011) Flavivirus NS4A‐induced autophagy protects cells against death and enhances virus replication. J Biol Chem 286(25), 22147–22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merfeld E, Ben‐Avi L, Kennon M, et al. (2017) Potential mechanisms of Zika‐linked microcephaly. Wiley Interdiscip Rev Dev Biol 6(4), e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesci P, Macia A, LaRock CN, et al. (2018) Modeling neuro‐immune interactions during Zika virus infection. Hum Mol Genet 27(1), 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina JP, Kraemer MU, Brady OJ, et al. (2016) Mapping global environmental suitability for Zika virus. ELife 5, 1–19. 10.7554/eLife.15272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Diamond MS (2017) Zika virus pathogenesis and tissue tropism. Cell Host Microbe 21(2), 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Sene A, Richner JM, et al. (2016) Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 16(12), 3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G, Tang H, Song H (2016) Advances in Zika virus research: stem cell models, challenges, and opportunities. Cell Stem Cell 19(6), 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, et al. (2016) Zika virus associated with microcephaly. N Engl J Med 374(10), 951–958. [DOI] [PubMed] [Google Scholar]
- Morris AAM (2005) Cerebral ketone body metabolism. J Inherit Metab Dis 28(2), 109–121. [DOI] [PubMed] [Google Scholar]
- Morrison TE, Diamond MS (2017) Animal models of Zika virus infection, pathogenesis, and immunity. J Virol 91(8), e00009–e00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa‐Veloso K, Likhodii SS, Cunnane SC (2002). Breath acetone is a reliable indicator of ketosis in adults consuming ketogenic meals. Am J Clin Nutr 76(1): 65–70. 10.1093/ajcn/76.1.65 [DOI] [PubMed] [Google Scholar]
- Musso D, Roche C, Nhan T‐X, et al. (2015) Detection of Zika virus in saliva. J Clin Virol 68, 53–55. [DOI] [PubMed] [Google Scholar]
- Nayak D, Roth TL, McGavern DB (2014). Microglia Development and function. Annu Rev Immunol 32: 367–402. 10.1146/annurev-immunol-032713-120240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Noronha L, Zanluca C, Azevedo MLV, et al. (2016) Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem Inst Oswaldo Cruz 111(5), 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen OE (2006) Ketone bodies as a fuel for the brain during starvation. Biochem Mol Biol Educ 33(4), 246–251. [Google Scholar]
- Owen OE, Morgan AP, Kemp HG, et al. (1967) Brain metabolism during fasting. J Clin Invest 46(10), 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco O, Beltrán M, Nelson CA, et al. (2016) Zika virus disease in Colombia – preliminary report. N Engl J Med. 1–10. 10.1056/NEJMoa1604037. [DOI] [PubMed] [Google Scholar]
- Panov A, Orynbayeva Z, Vavilin V, et al. (2014) Fatty acids in energy metabolism of the central nervous system. Biomed Res Int 2014, 472459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paula Freitas B, de Oliveira Dias JR, Prazeres J, et al. (2016) Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil. JAMA Ophthalmol 134(5), 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paula Freitas B, Ventura CV, Maia M, et al. (2017) Zika virus and the eye. Curr Opin Ophthalmol 28(6), 595–599. [DOI] [PubMed] [Google Scholar]
- Payne J, Maher F, Simpson I, et al. (1997) Glucose transporter Glut 5 expression in microglial cells. Glia 21(3), 327–331. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91(22), 10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen E, Wilson ME, Touch S, et al. (2016) Rapid spread of Zika virus in The Americas – implications for public health preparedness for mass gatherings at the 2016 Brazil Olympic games. Int J Infect Dis 44, 11–15. [DOI] [PubMed] [Google Scholar]
- Pinheiro TJ, Guimarães LF, Silva MTT, et al. (2016) Neurological manifestations of Chikungunya and Zika infections. Arq Neuropsiquiatr 74(11), 937–943. [DOI] [PubMed] [Google Scholar]
- Platt DJ, Miner JJ (2017) Consequences of congenital Zika virus infection. Curr Opin Virol 27, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, et al. (2016) Brain‐region‐specific organoids using mini‐bioreactors for modeling ZIKV exposure. Cell 165(5), 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaan AA, Bazzi AM, Al‐Ahmed SH, et al. (2017) Overview of Zika infection, epidemiology, transmission and control measures. J Infect Public Health 10(2), 141–149. [DOI] [PubMed] [Google Scholar]
- Randle PJ (1998) Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14(4), 263–283. [DOI] [PubMed] [Google Scholar]
- Rhyu H, Cho S‐Y (2014) The effect of weight loss by ketogenic diet on the body composition, performance‐related physical fitness factors and cytokines of Taekwondo athletes. J Exer Rehabil 10(5), 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rick A‐M, Domek G, Cunningham M, et al. (2017) Baseline microcephaly prevalence in rural Guatemala: implications for neonatal screening for congenital Zika virus infection. Lancet Glob Health 5, S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A, Koczwara JB, Gallelli CA, et al. (2017) Fats for thoughts: an update on brain fatty acid metabolism. Int J Biochem Cell Biol 84, 40–45. [DOI] [PubMed] [Google Scholar]
- Rossi SL, Vasilakis N (2016) Modeling Zika virus infection in mice. Cell Stem Cell 19(1), 4–6. [DOI] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE (2012) Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta‐analysis. Mol Psychiatry 17(3), 290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan HA, Fang S, Mahesh M, et al. (2018). Zika virus and the metabolism of neuronal cells. Mol Neurobiol, in press. 10.1007/s12035-018-1263-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa MM, Mondotte JA, Iglesias NG, et al. (2009) Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5(10), e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samy AM, Thomas SM, Wahed AAE, et al. (2016) Mapping the global geographic potential of Zika virus spread. Mem Inst Oswaldo Cruz 111(9), 559–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC (2013) ER stress‐induced cell death mechanisms. Biochim Biophys Acta 1833(12), 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapparapu G, Fernandez E, Kose N, et al. (2016) Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540(7633), 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno M, Sacramento GA, Khouri R, et al. (2016) Zika virus infection and stillbirths: a case of hydrops fetalis, hydranencephaly and fetal demise. PLoS Negl Trop Dis 10(2), e0004517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, et al. (2016) Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16(1), 232–246. [DOI] [PubMed] [Google Scholar]
- Schneider H, Reiber W, Sager R, et al. (2003) Asymmetrical transport of glucose across the in vitro perfused human placenta. Placenta 24(1), 27–33. [DOI] [PubMed] [Google Scholar]
- Schönfeld P, Reiser G (2013) Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab 33(10), 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A, Scafidi S, Bak LK, et al. (2014) Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol 11, 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro‐Mendoza CK (2017) Pregnancy outcomes after maternal Zika virus infection during pregnancy – U.S. Territories, January 1, 2016–April 25, 2017. MMWR Morb Mortal Wkly Rep 66, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lal SK (2017) Zika virus: transmission, detection, control, and prevention. Front Microbiol 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, et al. (2004) Endothelial cells stimulate self‐renewal and expand neurogenesis of neural stem cells. Science 304(5675), 1338. [DOI] [PubMed] [Google Scholar]
- Shi Y, Li S, Wu Q, et al. (2018) Vertical transmission of the Zika virus causes neurological disorders in mouse offspring. Sci Rep 8(1), 3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima E, Ohki T, Kurita Y, et al. (2018) Protective effect of 3‐hydroxybutyrate against endoplasmic reticulum stress‐associated vascular endothelial cell damage induced by low glucose exposure. PLoS ONE 13(3), e0191147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon T, Baylis M, Brown D (2016) Zika virus and neurological disease – approaches to the unknown. Lancet Infect Dis 16(4), 402–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza BSF, Sampaio GLA, Pereira CS, et al. (2016) Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 6(1), 39775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolp H, Neuhaus A, Sundramoorthi R, et al. (2012) The long and the short of it: gene and environment interactions during early cortical development and consequences for long‐term neurological disease. Front Psychiatry 3, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, et al. (2016) Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18(5), 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Gao G, Rueda CB, et al. (2017) Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter‐1 protein. Nat Commun 8, 14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenet J, De Marchi U, Domingo JS, et al. (2016) Medium‐chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte‐neuron lactate and ketone body shuttle systems. FASEB J 30(5), 1913–1926. [DOI] [PubMed] [Google Scholar]
- Tiwari SK, Dang J, Qin Y, et al. (2017) Zika virus infection reprograms global transcription of host cells to allow sustained infection. Emerg Microbes Infect 6(4), e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongluan N, Ramphan S, Wintachai P, et al. (2017) Involvement of fatty acid synthase in dengue virus infection. Virol J 14, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey TJ, Steyn FJ, Wolvetang EJ, et al. (2018) Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uncini A, González‐Bravo DC, Acosta‐Ampudia YY, et al. (2018) Clinical and nerve conduction features in Guillain−Barré syndrome associated with Zika virus infection in Cúcuta, Colombia. Eur J Neurol 25(4), 644–650. [DOI] [PubMed] [Google Scholar]
- Ventura CV, Maia M, Ventura BV, et al. (2016) Ophthalmological findings in infants with microcephaly and presumable intra‐uterus Zika virus infection. Arq Bras Oftalmol 79(1), 1–3. [DOI] [PubMed] [Google Scholar]
- Verma R, Sharma P, Garg RK, et al. (2011) Neurological complications of dengue fever: experience from a tertiary center of north India. Ann Indian Acad Neurol 14(4), 272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidali S, Aminzadeh S, Lambert B, et al. (2015) Mitochondria: the ketogenic diet – A metabolism‐based therapy. Int J Biochem Cell Biol 63, 55–59. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu J, Zhou R, et al. (2018) Zika virus infected primary microglia impairs NPCs proliferation and differentiation. Biochem Biophys Res Commun 497(2), 619–625. [DOI] [PubMed] [Google Scholar]
- Weaver SC (2017) Emergence of epidemic Zika virus transmission and congenital Zika syndrome: are recently evolved traits to blame? MBio 8(1), e02063–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA (2015) Expanding roles for lipid droplets. Curr Biol 25(11), R470–R481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Song H, Ming G (2017) How does Zika virus cause microcephaly? Genes Dev 31(9), 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG (2004) Human microcephaly. Curr Opin Neurobiol 14(1), 112–117. [DOI] [PubMed] [Google Scholar]
- Xia C, Vijayan M, Pritzl CJ, et al. (2016) Hemagglutinin of influenza A virus antagonizes type I interferon (IFN) responses by inducing degradation of type I IFN receptor 1. J Virol 90(5), 2403–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Shan C, Shi P‐Y (2016) Restriction of Zika virus by host innate immunity. Cell Host Microbe 19(5), 566–567. [DOI] [PubMed] [Google Scholar]
- Yockey LJ, Varela L, Rakib T, et al. (2016) Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166(5), 1247–1256.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Clippinger AJ, Alwine JC (2011a) Viral affects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol 19(7), 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maguire TG, Alwine JC (2011b) Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol 85(4), 1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hammack C, Ogden SC, et al. (2016) Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res 44(18), 8610–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]