

Abstract
Objective
Tumor necrosis factor (TNF) and interleukin‐17A (IL‐17A) may independently contribute to the pathophysiology of rheumatoid arthritis (RA). This study sought to evaluate the safety and efficacy of ABT‐122, a novel dual variable domain immunoglobulin targeting human TNF and IL‐17A, in patients with RA who have experienced an inadequate response to methotrexate.
Methods
Patients with active RA who were receiving treatment with methotrexate and had no prior exposure to biologic agents (n = 222) were enrolled in a 12‐week phase II randomized, double‐blind, active‐controlled, parallel‐group study. Patients were randomized to receive either ABT‐122 at dosages of 60 mg every other week, 120 mg every other week, or 120 mg every week or adalimumab at 40 mg every other week, administered subcutaneously. The primary efficacy end point was the proportion of patients achieving a ≥20% improvement response based on the American College of Rheumatology criteria for 20% improvement (ACR20) at week 12.
Results
Treatment‐emergent adverse events were similar across all treatment groups, with no serious infections or systemic hypersensitivity reactions reported with ABT‐122. ACR20 response rates at week 12 were 62%, 75%, and 80% with ABT‐122 60 mg every other week, 120 mg every other week, and 120 mg every week, respectively, compared with an ACR20 response rate of 68% with 40 mg adalimumab every other week. The corresponding response rates for ACR50 and ACR70 improvement in the ABT‐122 dose groups and adalimumab group were 35%, 46%, 47%, and 48%, respectively, and 22%, 18%, 36%, and 21%, respectively.
Conclusion
Over the 12‐week study period, dual inhibition of TNF and IL‐17A with ABT‐122 produced a safety profile consistent with that of adalimumb used for inhibition of TNF alone. The efficacy of ABT‐122 over 12 weeks at dosages of 120 mg every other week or 120 mg every week was not meaningfully differentiated from that of adalimumab at a dosage of 40 mg every other week in patients with RA receiving concomitant methotrexate.
The treatment goal for all patients with rheumatoid arthritis (RA) should be tailored to the individual patient, keeping in mind that low disease activity and disease remission are optimal aims 1. Because current therapies may fall short of these target goals 1, 2, 3 and fail to improve quality of life in some patients 4, novel pharmacologic therapies are needed to improve outcomes. RA is a complex disease involving numerous cell types and a variety of inflammation mediators operating in the innate and adaptive immune systems 5. Thus, therapies that simultaneously target different pathways involved in the pathogenesis of RA may enhance treatment responses in patients with RA.
Some patients fail to improve following treatment with tumor necrosis factor (TNF) inhibitors or these agents tend to lose effectiveness over time 6, 7. TNF inhibitor therapy in patients with RA was found to significantly increase the frequency of circulating Th17 cells in the peripheral blood 8, thereby enhancing the release of the proinflammatory cytokine interleukin‐17A (IL‐17A) 9. Moreover, in one study, patients with RA who failed to respond to TNF inhibition had elevated concentrations of circulating Th17 cells and increased serum levels of IL‐17 in comparison to patients who responded to TNF inhibition 10.
IL‐17A induces the release of a broad range of proinflammatory cytokines and chemokines and may be involved in cartilage destruction 9. TNF and IL‐17A may act synergistically to induce osteoclastogenesis via pathways involving RANKL, resulting in bone loss 11, 12. Serum and synovial concentrations of IL‐17A are elevated in patients with RA compared with age‐ and sex‐matched healthy individuals, and these raised levels are correlated with the severity of the disease 13. In a mouse collagen‐induced arthritis model, simultaneous neutralization of TNF and IL‐17 by 2 different selective monoclonal antibodies (mAb) reduced the arthritis severity scores to a greater extent than did neutralization by either mAb alone 14, 15. Thus, targeting both TNF and IL‐17A in a combined strategy of dual neutralization may lead to more effective and enhanced clinical responses in patients with RA compared with the response achieved by targeting TNF or IL‐17A alone. However, some studies reported an increased rate of serious adverse events (AEs), most notably serious infections, in patients with RA who received combination therapy with 2 biologic disease‐modifying antirheumatic drugs (bDMARDs) compared with patients who received single bDMARD therapy 16, 17, 18. Combination bDMARD therapy was associated with little or no incremental efficacy benefit when compared with a single biologic agent in patients with RA 16, 17, 18. Therefore, it is important to carefully assess the safety, tolerability, and efficacy of any combination biologic therapy.
ABT‐122 is a dual variable domain immunoglobulin (DVD‐Ig) targeting human TNF and IL‐17A 19. Each molecule of ABT‐122 has 2 sets of selective binding domains, with 1 pair targeting TNF and the other pair targeting IL‐17A. Thus, the ratio of TNF to IL‐17A binding sites in each molecule of ABT‐122 is inherently constant (ratio of 2:2) 20. In vitro surface plasmon resonance analysis revealed that ABT‐122 bound a similar amount of TNF with high affinity independent of the occupancy of IL‐17A binding sites, and vice versa 20. In fibroblast‐like synoviocytes from patients with RA, ABT‐122 fully inhibited TNF‐ and IL‐17A–induced production of IL‐6 20.
In addition, ABT‐122 rapidly modulates potential pathophysiologic pathways in patients with RA, including reducing the serum levels of chemokines CXCL9, CXCL10, and CCL23 21, all of which are involved in lymphocyte and myeloid cell recruitment into inflamed tissue. With single doses of up to 3 mg/kg subcutaneously (SC) and 10 mg/kg intravenously in healthy volunteers, ABT‐122 was well tolerated, and at these doses, target activity was demonstrated ex vivo 20. In phase I studies, multiple ascending SC doses of ABT‐122 were administered to patients with RA for 8 weeks 21, 22. ABT‐122 showed approximately dose‐proportional exposure at SC doses of >1 mg/kg 22. Peak ABT‐122 serum concentrations were observed within 2–4 days after SC dosing, with steady‐state levels obtained by 6 weeks of SC dosing. The effective half‐life of ABT‐122 was 10 days with dosing every other week, and 18 days with dosing every week. The results of these studies in patients with RA indicate that there are no meaningful safety signals with ABT‐122, and it has potential antiinflammatory effects and a pharmacokinetic profile favorable for SC administration every other week or every week.
The objective of the current study was to determine the safety, efficacy, and pharmacokinetics of ABT‐122 in patients with RA who had an inadequate response to methotrexate.
Patients and Methods
The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice, and adhered to the principles of the Declaration of Helsinki. An independent ethics committee or institutional review board at each center approved the protocol. All patients provided written informed consent.
Study design. The study was designed as a phase II randomized, double‐blind, active‐controlled, parallel‐group study in patients with active RA who had experienced an inadequate response to methotrexate, conducted at 46 sites in the United States, Bulgaria, the Czech Republic, Germany, Hungary, Poland, Romania, and New Zealand. After screening, eligible patients receiving weekly stable treatment with methotrexate were randomized 1:1:1:1 to receive either ABT‐122 at dosages of 60 mg every other week, 120 mg every other week, or 120 mg every week or adalimumab (Humira; AbbVie) at a dosage of 40 mg every other week, administered SC for 12 weeks.
Patients. Adult patients (ages ≥18 years) who had a diagnosis of RA for at least 3 months, with the diagnosis based on the American College of Rheumatology (ACR)/European League Against Rheumatism 2010 criteria 23, were eligible. All eligible patients had active RA, defined as the presence of ≥6 swollen joints and ≥6 tender joints (based on the 66/68‐joint counts at screening and baseline visits) and a high‐sensitivity C‐reactive protein (hsCRP) level higher than the upper limit of normal (cutoff 4.99 mg/liter) at screening or the presence of rheumatoid factor and anti–cyclic citrullinated peptide antibodies at screening. Eligible patients were those who had experienced an inadequate response to methotrexate after having received the treatment for ≥3 months (with no change in the route of administration) and after having received a stable dosage of ≥10 mg/week for at least 4 weeks before baseline.
Key exclusion criteria included the following: previous exposure to adalimumab, other TNF inhibitors, and other bDMARDs; current treatment with conventional DMARDs (except methotrexate, sulfasalazine, and hydroxychloroquine); having received stable oral doses of prednisone or its equivalent at >10 mg/day within 30 days of the first dose of study medication; having received treatment with intraarticular or parenteral glucocorticoids in the 4 weeks before the first dose of study medication; presence of active tuberculosis, chronic recurring infections, and/or active viral infections; and having received treatment with anti‐infective agents within 30 days (intravenously) or 14 days (orally) of the first dose of study medication. Patients could continue to receive stable treatment with oral prednisone or its equivalent (at a dosage of ≤10 mg/day) if they had begun the treatment at least 4 weeks before the first dose of study medication. Furthermore, use of inhaled glucocorticoids to treat stable medical conditions was allowed.
Safety assessments. Occurrence of AEs was monitored and recorded throughout the course of the study. AEs were coded using the preferred terms and system organ classes designated in the Medical Dictionary for Regulatory Activities, version 17.1. Treatment‐emergent AEs (TEAEs) were categorized by treatment and by severity. The severity of TEAEs was classified by investigators according to the Rheumatology Common Toxicity Criteria, version 2.0 24.
Efficacy assessments. The primary efficacy end point was the proportion of patients achieving a ≥20% improvement response based on the ACR criteria for 20% improvement (ACR20) 25 at week 12. Missing data were analyzed using nonresponder imputation (NRI) and also the last observation carried forward (LOCF) method. Secondary end points included the following: the proportion of patients achieving a ≥50% and ≥70% ACR improvement response (ACR50 and ACR70, respectively) at week 12, as well as change from baseline in the Disease Activity Score in 28 joints using hsCRP level (DAS28‐hsCRP) 26 at weeks 2, 4, 6, 8, and 12; the proportion of patients achieving a DAS28‐hsCRP score of <3.2 or Clinical Disease Activity Index (CDAI) score 27 of ≤10 at week 12; and the proportion of patients achieving a DAS28‐hsCRP score of <2.6 or CDAI score of ≤2.8 at week 12. Exploratory end points included ACR response rates at weeks 2, 4, 6, and 8 and changes in ACR components over time, including changes from baseline of at least −1.2 in the DAS28‐hsCRP and of at least −0.5 in the Health Assessment Questionnaire (HAQ) disability index (DI) 28.
Assessments of pharmacokinetics and immunogenicity. Serum concentrations of ABT‐122 were determined at baseline (before dosing), weekly during the 12‐week treatment period, and at a follow‐up visit 6 weeks after treatment ended. Immunogenicity was characterized by assessment of antidrug antibodies to ABT‐122 at baseline, every 2–3 weeks during the 12‐week treatment period, and at a follow‐up visit 6 weeks after treatment ended. Serum concentrations of ABT‐122 were determined using a validated chimeric electrochemiluminescence immunoassay, and the antidrug antibody assessment was performed using a validated bridging electrochemiluminescence immunoassay. The assay included an acid dissociation step to allow for dissociation of antibodies from the drug–antibody complex, thereby ensuring reduced drug interference. The details of the bioanalytic assays have been described previously 22, 29.
Statistical analysis. The safety analysis set included patients who received ≥1 dose of study drug. The full analysis set, defined as all randomized patients from the safety analysis set, was used for baseline and efficacy analyses. Summary statistics were provided by treatment group. Given a sample size of ~55 patients per group and an estimated ACR20 response rate of 60% for those treated with adalimumab, the study had 88% power to detect an ACR20 response rate of 85% among patients who were treated with ABT‐122, using a 1‐sided α level of 0.05, with comparisons between groups by Fisher's exact test. Missing categorical efficacy end points were imputed using NRI, whereas missing continuous efficacy end points were imputed using LOCF; safety data were not imputed. Statistical analyses were performed using SAS software (version 9.2 or higher; SAS Institute).
Results
Characteristics of the study patients. The demographic and clinical characteristics of the patients at baseline were comparable across the treatment groups (Table 1). Most of the patients were white (92%) and female (82%), with a mean duration of RA of ~8 years. Of the 222 randomized patients, 209 (94%) completed the study (Figure 1). Three patients discontinued because of TEAEs. The safety analysis set and the full analysis set comprised the same patients.
Table 1.
Demographic and baseline clinical characteristics of the patients in the full analysis seta
| Characteristic | Adalimumab 40 mg SC every other week (n = 56) | ABT‐122 60 mg SC every other week (n = 55) | ABT‐122 120 mg SC every other week (n = 56) | ABT‐122 120 mg SC every week (n = 55) |
|---|---|---|---|---|
| Women, no. (%) | 42 (75.0) | 45 (81.8) | 49 (87.5) | 45 (81.8) |
| White, no. (%) | 51 (91.1) | 52 (94.5) | 53 (94.6) | 49 (89.1) |
| Age, years | 57.6 ± 12.4 | 55.2 ± 11.8 | 53.5 ± 13.0 | 55.6 ± 12.3 |
| Weight, kg | 80.2 ± 17.6 | 78.4 ± 19.5 | 73.7 ± 16.5 | 77.3 ± 14.1 |
| BMI, kg/m2 | 29.3 ± 6.0 | 28.7 ± 5.9 | 27.5 ± 5.1 | 28.3 ± 4.7 |
| Duration of RA, years | 7.6 ± 7.8 | 7.0 ± 8.1 | 9.4 ± 9.2 | 6.8 ± 6.7 |
| MTX dose, mg/week | 16.8 ± 4.3 | 17.5 ± 4.9 | 17.1 ± 4.7 | 16.7 ± 4.7 |
| Prior use of non‐MTX DMARD, no. (%)b | 8 (14.3) | 12 (21.8) | 10 (17.9) | 8 (14.5) |
| 1 | 5 (8.9) | 10 (18.2) | 9 (16.1) | 6 (10.9) |
| 2 | 3 (5.4) | 1 (1.8) | 1 (1.8) | 2 (3.6) |
| ≥3 | 0 | 1 (1.8) | 0 | 0 |
| Prior use of systemic glucocorticoids, no. (%)b | 33 (58.9) | 36 (65.5) | 34 (60.7) | 30 (54.5) |
| hsCRP, mg/liter | 16.3 ± 24.0 | 13.8 ± 13.1 | 15.7 ± 19.3 | 17.3 ± 25.0 |
| hsCRP category, no. (%) | ||||
| ≤4.99 mg/liter | 16 (28.6) | 15 (27.3) | 18 (32.1) | 20 (36.4) |
| >4.99 mg/liter | 40 (71.4) | 40 (72.7) | 38 (67.9) | 35 (63.6) |
| DAS28‐hsCRP score | 5.8 ± 1.0 | 6.0 ± 0.8 | 5.6 ± 0.9 | 5.7 ± 0.9 |
| DAS28‐hsCRP category, no. (%) | ||||
| <5.1 | 12 (21.4) | 8 (14.5) | 14 (25.0) | 11 (20.0) |
| ≥5.1 | 44 (78.6) | 47 (85.5) | 42 (75.0) | 44 (80.0) |
Except where indicated otherwise, values are the mean ± SD. Patients were enrolled from sites in Poland (n = 112), the United States (n = 49), Bulgaria (n = 17), the Czech Republic (n = 17), Hungary (n = 11), New Zealand (n = 9), Romania (n = 6), and Germany (n = 1). SC = subcutaneous; BMI = body mass index; RA = rheumatoid arthritis; MTX = methotrexate; DMARD = disease‐modifying antirheumatic drug; hsCRP = high‐sensitivity C‐reactive protein; DAS28‐hsCRP = Disease Activity Score in 28 joints using hsCRP level.
Therapy was stopped before the first dose of study drug.
Figure 1.
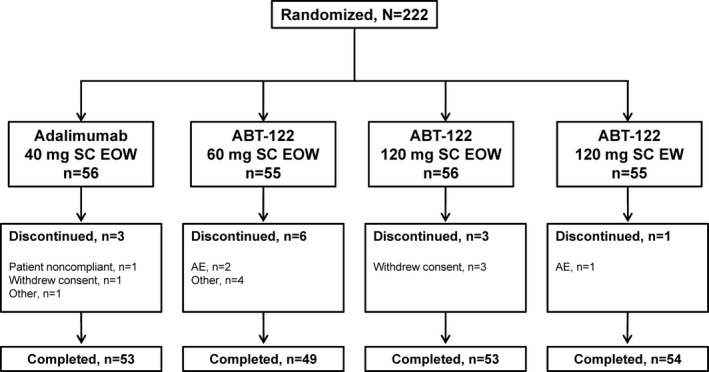
Disposition of the study patients. SC = subcutaneous; EOW = every other week; EW = every week; AE = adverse event.
Safety. The overall incidence of TEAEs was similar among the treatment groups, with most events being mild or moderate in severity, and without evidence of a relationship to dose (Table 2). There were very few severe TEAEs or TEAEs that led to discontinuation from the study. There were 4 serious AEs that occurred after treatment with ABT‐122, compared with no serious AEs after treatment with adalimumab, without any deaths. There were no reported serious infections. Two serious AEs (1 case of head injury and 1 case of an ovarian cyst) occurred in patients treated with ABT‐122 at 60 mg every other week. Two serious AEs occurred in patients treated with ABT‐122 at 120 mg every week (1 case of a tibia fracture, and 1 case of angina pectoris). The latter serious AE, angina pectoris, occurred in a 69‐year‐old male patient with a history of angina and hypertension, and resolved after 2 days; on follow‐up, the patient was found to have cardiac failure.
Table 2.
Frequency of TEAEs observed in patients in the safety analysis seta
| Event | Adalimumab 40 mg SC every other week (n = 56) | ABT‐122 60 mg SC every other week (n = 55) | ABT‐122 120 mg SC every other week (n = 56) | ABT‐122 120 mg SC every week (n = 55) |
|---|---|---|---|---|
| Any TEAE | 24 (42.9) | 23 (41.8) | 21 (37.5) | 20 (36.4) |
| Any serious AE | 0 | 2 (3.6) | 0 | 2 (3.6) |
| Any severe AE | 0 | 1 (1.8) | 0 | 0 |
| Any AE leading to discontinuation | 0 | 2 (3.6) | 0 | 1 (1.8) |
| Infectionb | 10 (17.9) | 8 (14.5) | 6 (10.7) | 13 (23.6) |
| Infections reported in ≥2 patientsb | ||||
| Urinary tract infection | 1 (1.8) | 5 (9.1) | 1 (1.8) | 1 (1.8) |
| Nasopharyngitis | 3 (5.4) | 2 (3.6) | 1 (1.8) | 3 (5.5) |
| Upper respiratory tract infection | 1 (1.8) | 1 (1.8) | 0 | 4 (7.3) |
| Lower respiratory tract infection | 0 | 0 | 2 (3.6) | 2 (3.6) |
| Serious infectionb | 0 | 0 | 0 | 0 |
| Ischemic eventb | 0 | 0 | 0 | 1 (1.8)c |
| Cardiac failureb | 0 | 0 | 0 | 1 (1.8) |
| Malignancyb | 0 | 1 (1.8)d | 0 | 0 |
| Systemic hypersensitivity reactionb | 1 (1.8) | 0 | 0 | 0 |
| Severe injection site reactionb | 0 | 0 | 0 | 0 |
| Demyelinating disorderb | 0 | 0 | 0 | 0 |
| Hematologic disorderb | 0 | 0 | 0 | 0 |
| Hepatic AEb | 0 | 0 | 0 | 0 |
Values are the number (%) of patients. Treatment‐emergent adverse events (TEAEs) were defined as an adverse event (AE) with an onset date that was on or after the first dose of study drug administration and no more than 70 days after the last dose of study drug administration, or an AE with an onset date before the first dose of study drug administration but with increased severity on or after the first dose of study drug administration and no more than 70 days after the last dose of study drug administration. SC = subcutaneous.
Based on a standardized search of AEs, using a set of terms from the Medical Dictionary for Regulatory Activities (version 17.1) or compiled by the study sponsor.
Angina pectoris.
Meningioma.
Twenty‐seven patients (16%) who received ABT‐122 reported experiencing ≥1 infection‐related TEAE during the study, with the highest proportion of patients (24%) being those who received ABT‐122 at 120 mg every week; 18% of patients who received adalimumab at 40 mg every other week reported experiencing ≥1 infection over the 12‐week treatment course (Table 2). The most frequently reported infections (≥2 patients in any treatment group) were urinary tract infection, nasopharyngitis, upper respiratory tract infection, and lower respiratory tract infection. All infections were mild or moderate in severity, were transient, and resolved with standard therapy. Meningioma was diagnosed in 1 patient who was receiving ABT‐122 at 60 mg every other week. Systemic hypersensitivity (urticaria) occurred in 1 patient who was being treated with adalimumab at 40 mg every other week; after a temporary treatment interruption, the patient completed the study.
Efficacy. At week 12, there was a dose‐related increase in the proportion of patients achieving the primary efficacy end point, the ACR20 response, among those receiving ABT‐122, as observed in NRI analyses (Table 3). The ACR20 treatment response rate was numerically higher, but not statistically significantly different, in patients treated with ABT‐122 at 120 mg every other week (75%) or 120 mg every week (80%) compared with those receiving adalimumab (68%) (Table 3). At week 12, the ACR50 treatment response rate was similar between patients treated with ABT‐122 at 120 mg every other week (46%) or 120 mg every week (47%) and those treated with adalimumab (48%), whereas the ACR70 response was numerically higher in patients treated with ABT‐122 at 120 mg every week (36%) compared with those who received ABT‐122 at 120 mg every other week (18%) or those who received adalimumab (21%) (Table 3). Similar results were obtained when missing data were imputed using LOCF (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40580/abstract).
Table 3.
Treatment response rates at week 12 in patients in the full analysis set with nonresponder imputationa
| Response at week 12 | Adalimumab 40 mg SC every other week (n = 56) | ABT‐122 60 mg SC every other week (n = 55) | ABT‐122 120 mg SC every other week (n = 56) | ABT‐122 120 mg SC every week (n = 55) |
|---|---|---|---|---|
| ACR20 | 38 (67.9) | 34 (61.8) | 42 (75.0) | 44 (80.0) |
| ACR50 | 27 (48.2) | 19 (34.5) | 26 (46.4) | 26 (47.3) |
| ACR70 | 12 (21.4) | 12 (21.8) | 10 (17.9) | 20 (36.4) |
| DAS28‐hsCRP <3.2 | 25 (44.6) | 18 (32.7) | 29 (51.8) | 30 (54.5) |
| DAS28‐hsCRP <2.6 | 17 (30.4) | 12 (21.8) | 21 (37.5) | 23 (41.8) |
| CDAI ≤10 | 22 (39.3) | 18 (32.7) | 24 (42.9) | 30 (54.5) |
| CDAI ≤2.8 | 4 (7.1) | 4 (7.3) | 6 (10.7) | 6 (10.9) |
Values are the number (%) of responders based on the American College of Rheumatology response criteria for improvement of 20% (ACR20), 50% (ACR50), and 70% (ACR70), 2 different cutoffs for scores on the Clinical Disease Activity Index (CDAI), and 2 different cutoffs for scores on the Disease Activity Score in 28 joints using high‐sensitivity C‐reactive protein level (DAS28‐hsCRP). SC = subcutaneous.
Mean improvements in the ACR score components from baseline to week 12 were numerically highest among patients who were treated with ABT‐122 at 120 mg every week compared with those who received the other treatments (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40580/abstract). Following initiation of ABT‐122, there was a time‐dependent increase in the proportion of patients achieving ACR20, ACR50, and ACR70 responses during the course of the 12‐week study (Figure 2).
Figure 2.
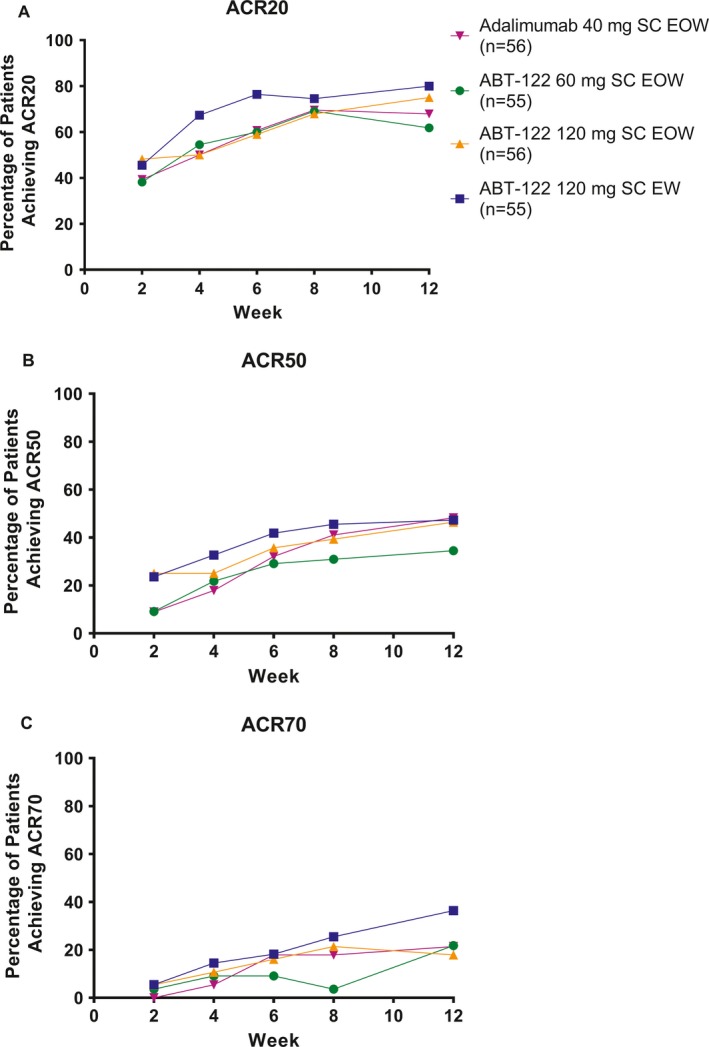
Percentage of patients in each treatment group achieving improvement according to the American College of Rheumatology response criteria for an improvement of at least 20% (ACR20) (A), 50% (ACR50) (B), and 70% (ACR70) (C) over the 12‐week treatment course (full analysis set, nonresponder imputation). SC = subcutaneous; EOW = every other week; EW = every week.
At week 12, measures of disease activity based on DAS28‐hsCRP cutoff scores of <3.2 and <2.6 showed a dependence on the ABT‐122 dose. The proportions of patients falling within these disease activity score ranges were numerically higher among those receiving ABT‐122 at 120 mg every week (55% for DAS28‐hsCRP <3.2 and 42% for DAS28‐hsCRP <2.6) compared with those receiving adalimumab (45% for DAS28‐hsCRP <3.2 and 30% for DAS28‐hsCRP <2.6), in analyses using NRI (Table 3).
At week 12, measures of disease activity based on CDAI cutoff scores of ≤10 and ≤2.8 also showed a dependence on the ABT‐122 dose. The proportions of patients falling in these score ranges were numerically higher among those receiving ABT‐122 120 mg every week (55% for CDAI ≤10 and 11% for CDAI ≤2.8) compared with those receiving adalimumab (39% for CDAI ≤10 and 7% for CDAI ≤2.8), in analyses using NRI (Table 3). These patterns of disease activity levels based on the DAS28‐hsCRP and CDAI scores were similar when determined with LOCF methods (see Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40580/abstract).
When the data were analyzed as forest plots, we found that the response rates at 12 weeks favored ABT‐122 over adalimumab in most of the comparisons of efficacy end points, although the 95% confidence intervals crossed zero for all parameters except for change from baseline in the HAQ DI scores when comparing the adalimumab group with the ABT‐122 120 mg every week group (Figure 3). However, differences considered to be clinically important between the treatment groups were few, and no statistically significant and clinically meaningful differences between the ABT‐122 groups and the adalimumab group were observed.
Figure 3.
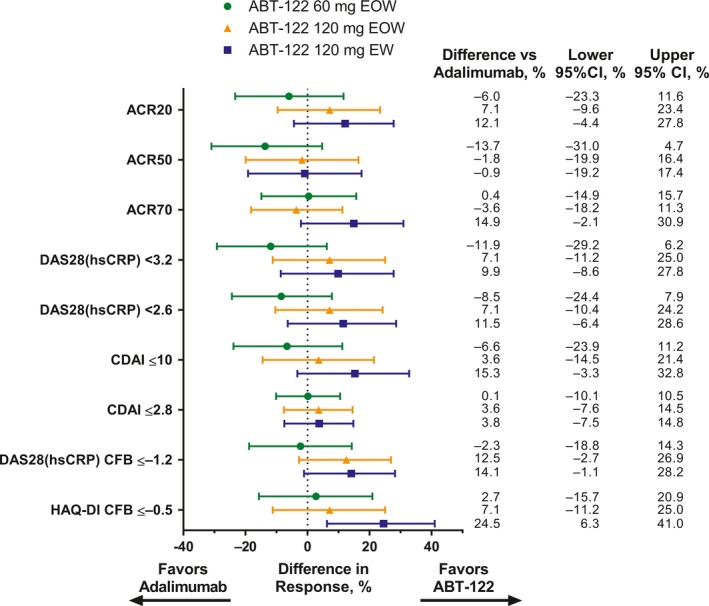
Differences in efficacy end points at 12 weeks between the ABT‐122 dosing groups compared with the adalimumab group. The end points include the American College of Rheumatology response criteria for an improvement of at least 20% (ACR20), 50% (ACR50), and 70% (ACR20), 2 score cutoffs for the Disease Activity Score in 28 joints based on high‐sensitivity C‐reactive protein level (DAS28‐hsCRP), 2 score cutoffs for the Clinical Disease Activity Index (CDAI), and change from baseline (CFB) of ≤−1.2 in the DAS28‐hsCRP score or ≤−0.5 on the disability index (DI) of the Health Assessment Questionnaire (HAQ). Bars show the difference in response with 95% confidence interval (95% CI). EOW = every other week; EW = every week.
Pharmacokinetics and immunogenicity. ABT‐122 serum concentrations increased in a dose‐dependent manner and reached steady state at ~6 weeks after initiation of dosing. The mean steady‐state serum concentrations (Ctrough) of ABT‐122 at week 12 were 1.4 μg/ml, 4.4 μg/ml, and 12.1 μg/ml in the 60 mg every other week, 120 mg every other week, and 120 mg every week treatment groups, respectively. The mean serum concentration at steady state for adalimumab at week 12 was 5.4 μg/ml. The average molar serum concentrations of ABT‐122 in the dosing interval at steady state at 60 mg every other week (15 nM), 120 mg every other week (34 nM), and 120 mg every week (60 nM) were approximately one‐half, comparable, and 2‐fold higher, respectively, compared with the average serum exposure at steady state for adalimumab 40 mg every other week (30 nM).
The proportions of patients with treatment‐emergent antidrug antibodies to ABT‐122 at week 12 were 56%, 20%, and 13% in the ABT‐122 60 mg every other week, 120 mg every other week, and 120 mg every week groups, respectively. During treatment with ABT‐122, some patients showed a decrease in ABT‐122 serum concentrations to below the lower limit of detection (13.6 ng/ml), which was likely the result of the development of antidrug antibodies. The proportions of patients with ABT‐122 serum concentrations below the lower limit of detection at week 12 were 36%, 13%, and 5% for ABT‐122 60 mg every other week, 120 mg every other week, and 120 mg every week, respectively. Details on the ABT‐122 pharmacokinetic and immunogenicity results are reported elsewhere 29. Overall, the frequency of TEAEs and clinical laboratory profiles in patients receiving ABT‐122 treatment were comparable between those who had detectable antidrug antibodies to ABT‐122 and those without antidrug antibodies to ABT‐122, indicating that the safety profile of ABT‐122 was not associated with the development of antidrug antibodies to ABT‐122 during the study.
Discussion
In this study utilizing dual inhibition of TNF and IL‐17A with a single immunoglobulin, there was no observable increase in safety findings with ABT‐122 compared with adalimumab. The safety profile of ABT‐122 was similar across all dosing regimens, with the possible exception of more frequent overall infections with ABT‐122 at 120 mg every week. Moreover, the safety profile of ABT‐122 was comparable to that of adalimumab and did not appear to be affected by the formation of antidrug antibodies. There were no serious infections or systemic hypersensitivity reactions with ABT‐122. This safety profile is of particular importance because in some studies 16, 17, 18, but not all 30, an increased rate of serious AEs, most notably serious infections, was observed in patients with RA who received combination therapy with 2 bDMARDs compared with patients with RA who received single bDMARD therapy. Although these studies targeted different mechanisms than those targeted by ABT‐122, and the results may not be directly comparable, it is reassuring, in this short‐term study, to note that the safety profile of ABT‐122, a novel bispecific DVD‐Ig construct, was comparable to that of adalimumab. The similarity of the safety profiles of adalimumab and ABT‐122 in the current study and its 24‐week open‐label extension 31 provides evidence that dual inhibition of TNF and IL‐17A with the use of a single agent that targets both of these inflammatory cytokines did not increase the incidence of AEs, a complication that is known to occur as a result of treatment with adalimumab.
ABT‐122 was also efficacious over 12 weeks in patients with RA who have demonstrated an inadequate response to methotrexate. A time‐ and dose‐dependent increase in the percentage of patients achieving the primary end point of an ACR20 response was observed over the 12‐week treatment period. Compared with patients receiving adalimumab at 40 mg every other week, the ACR20 response rate was numerically higher following treatment with ABT‐122 at 120 mg every week, and was comparable to that following treatment with ABT‐122 at 120 mg every other week.
Although the efficacy of ABT‐122 could not be consistently differentiated from adalimumab in the present study, ABT‐122 is known to be a suitable bispecific blocking antibody for clinical research, having demonstrated high affinity, high selectivity, and independent engagement of each targeted cytokine, with corresponding biologic activity 20, 21. Further, with the evaluated doses, ABT‐122 serum exposures approximated those of other biologic drugs, such as adalimumab and ixekizumab. The average molar serum concentration of ABT‐122 at a dosage of 120 mg every other week (34 nM) was comparable to that of adalimumab at a dosage of 40 mg every other week (30 nM), while the average molar serum concentration with the ABT‐122 dosage of 120 mg every week (60 nM) was ~2‐fold higher than that for adalimumab. Furthermore, the steady‐state Ctrough molar concentrations for the ABT‐122 120 mg every week dosage were comparable to that of the anti–IL‐17 mAb ixekizumab at 80 mg every other week 32, which has a similar anti–IL‐17A affinity.
The proportion of anti‐TNF and anti–IL‐17A functionalities in the ABT‐122 molecule is inherently constant because of the fixed 2:2 ratio of TNF to IL‐17A binding sites in the ABT‐122 molecule. These serum exposure comparisons suggest that exposure to the anti–IL‐17A and anti‐TNF components of ABT‐122 would be approximately in the same range as exposure to the anti‐TNF component of adalimumab or the anti–IL‐17A component of ixekizumab. Overall, the lack of evidence to indicate a consistently differentiable efficacy profile of ABT‐122 in comparison with adalimumab suggests that the anti–IL‐17A component of ABT‐122 did not significantly add to the efficacy at the evaluated dosages or exposure range. Results of mechanistic analyses in the present study are consistent with the notion of an anti‐TNF effect of ABT‐122 as the main driver of its efficacy 33. In addition, the results of recent studies published after the development of ABT‐122 suggest that IL‐17A may not be a major contributor to the pathophysiology of RA 34, 35, 36. This is consistent with the findings in studies showing no or only modest improvement in the efficacy of anti‐TNF therapy when coadministered with other biologic agents that target IL‐1 16, CD20‐positive B cells 30, or T cell activation 17 in patients with RA.
A strength of the present study was having a simultaneous cohort of patients treated with adalimumab, which allowed a direct comparison to the safety and efficacy of ABT‐122. However, a number of limitations should be noted. The lack of a placebo control group (i.e., patients who were receiving methotrexate only) did not allow for direct assessment of the magnitude of the response to ABT‐122. It is conceivable that a higher dose of ABT‐122 might have improved its efficacy; however, in a separate study conducted in patients with psoriatic arthritis, even ABT‐122 at a dosage of 240 mg every week was not associated with significant differentiation from adalimumab at 40 mg every other week 37. The proportions of anti‐TNF activity versus anti–IL‐17A activity of ABT‐122 are fixed, which is unavoidable with a dual inhibitor, thereby precluding the evaluation of different ratios of anti‐TNF and anti–IL‐17A activity on efficacy outcomes.
The duration of the study was 12 weeks, which did not allow for longer‐term assessment of the safety of ABT‐122 or the potential for identification of longer‐term efficacy. However, results of a 24‐week open‐label extension of this study found that the frequency of AEs was unchanged and the clinical effect was stable when all patients continued treatment with ABT‐122 at a dosage of 120 mg every other week 31.
Futhermore, the current study was limited to biologic‐naive patients; therefore, it could not be determined whether ABT‐122 might be of benefit in patients with a partial response to anti‐TNF therapy.
Finally, the size of the study precluded a robust subgroup analysis. This would have been helpful to investigate whether ABT‐122 might be more effective in certain subgroups of patients.
In conclusion, dual inhibition of TNF and IL‐17A with a DVD‐Ig at the dosages tested in this study provided efficacy with an acceptable safety profile in patients with RA who had an inadequate response to methotrexate. However, the clinical efficacy of ABT‐122 (120 mg SC every other week or every week) was comparable to, and not significantly distinguishable from, that of adalimumab (40 mg SC every other week). Therefore, based on the results of this study, it does not appear that a strategy of dual inhibition of TNF and IL‐17A with ABT‐122 in patients with RA is significantly different from that of TNF inhibition alone with adalimumab. Because of these findings, further development of ABT‐122 for the treatment of RA is not being pursued. It remains to be seen whether a therapeutic approach involving dual inhibition targeting TNF and IL‐17A might be more efficacious with the use of other molecules, or whether the efficacy of this approach might differ in other populations of RA patients.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Genovese had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Genovese, Weinblatt, Mansikka, Peloso, Chen, Othman, Khan, Padley.
Acquisition of data
Aelion, Mansikka, Peloso, Chen, Li, Padley.
Analysis and interpretation of data
Genovese, Weinblatt, Aelion, Mansikka, Peloso, Chen, Li, Othman, Khatri, Khan, Padley.
Role of the Study Sponsor
AbbVie funded the study, contributed to the study design, and was involved in the collection, analysis, and interpretation of the data and in the writing, review, and approval of the publication. All authors contributed to the development of the content. All authors and AbbVie reviewed and approved the final manuscript, but the authors maintained control over the final content. Medical writing support, provided by Richard M. Edwards, PhD, and Michael J. Theisen, PhD, of Complete Publication Solutions, LLC (North Wales, PA), was funded by AbbVie.
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supporting information
Acknowledgments
We thank all of the patients who participated in this clinical trial of an investigational product, and all study investigators for their contributions.
ClinicalTrials.gov identifier: NCT02141997.
Supported by AbbVie.
Dr. Genovese has received consulting fees from AbbVie, Eli Lilly, Janssen, Novartis, and UCB (less than $10,000 each) and has received research support from those companies. Dr. Weinblatt has received consulting fees from AbbVie, Amgen, Crescendo Bioscience, Janssen, Pfizer, and Roche (less than $10,000 each) and from Bristol‐Myers Squibb, Eli Lilly, and UCB (more than $10,000 each), and has received research support from Amgen, Bristol‐Myers Squibb, Crescendo Bioscience, DxTerity, and UCB. Dr. Aelion has received consulting fees from AbbVie, Boehringer Ingelheim, Celgene, and Eli Lilly (less than $10,000 each) and has received research support from AbbVie, Ardea Biosciences, AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Galapagos, GlaxoSmithKline, Janssen, Eli Lilly, Merck, Mesoblast, Novartis, Novo Nordisk, Pfizer, Roche, Sanofi‐Aventis, Takeda, and UCB. Drs. Mansikka, Peloso, Chen, Li, Othman, Khatri, Khan, and Padley own stock or stock options in AbbVie.
The copyright line for this article was changed on 09 August 2019 after original online publication.
References
- 1. Smolen JS, Breedveld FC, Burmester GR, Bykerk V, Dougados M, Emery P, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis 2016;75:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prince FH, Bykerk VP, Shadick NA, Lu B, Cui J, Frits M, et al. Sustained rheumatoid arthritis remission is uncommon in clinical practice. Arthritis Res Ther 2012;14:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tymms K, Zochling J, Scott J, Bird P, Burnet S, de Jager J, et al. Barriers to optimal disease control for rheumatoid arthritis patients with moderate and high disease activity. Arthritis Care Res (Hoboken) 2014;66:190–6. [DOI] [PubMed] [Google Scholar]
- 4. Taylor PC, Moore A, Vasilescu R, Alvir J, Tarallo M. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol Int 2016;36:685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011;365:2205–19. [DOI] [PubMed] [Google Scholar]
- 6. Emery P. Optimizing outcomes in patients with rheumatoid arthritis and an inadequate response to anti‐TNF treatment. Rheumatology (Oxford) 2012;51 Suppl 5:v22–30. [DOI] [PubMed] [Google Scholar]
- 7. Rubbert‐Roth A, Finckh A. Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther 2009;11 Suppl 1:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hull DN, Williams RO, Pathan E, Alzabin S, Abraham S, Taylor PC. Anti‐tumour necrosis factor treatment increases circulating T helper type 17 cells similarly in different types of inflammatory arthritis. Clin Exp Immunol 2015;181:401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furst DE, Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine‐associated cell targets. Rheumatology (Oxford) 2014;53:1560–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen DY, Chen YM, Chen HH, Hsieh CW, Lin CC, Lan JL. Increasing levels of circulating Th17 cells and interleukin‐17 in rheumatoid arthritis patients with an inadequate response to anti‐TNF‐α therapy. Arthritis Res Ther 2011;13:R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006;203:2673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zrioual S, Toh ML, Tournadre A, Zhou Y, Cazalis MA, Pachot A, et al. IL‐17RA and IL‐17RC receptors are essential for IL‐17A‐induced ELR+ CXC chemokine expression in synoviocytes and are overexpressed in rheumatoid blood. J Immunol 2008;180:655–63. [DOI] [PubMed] [Google Scholar]
- 13. Metawi SA, Abbas D, Kamal MM, Ibrahim MK. Serum and synovial fluid levels of interleukin‐17 in correlation with disease activity in patients with RA. Clin Rheumatol 2011;30:1201–7. [DOI] [PubMed] [Google Scholar]
- 14. Cuff C, Hsieh C, Mathieu S, Murtaza A, Hugunin M, Bryant S, et al. Dual neutralization of TNF and IL‐17 provides greater efficacy in collagen induced arthritis through regulation of a gene transcription program that includes CXCL1 and CXCL5 [abstract]. Arthritis Rheumatol 2013;65 Suppl:948. [Google Scholar]
- 15. Cuff C, Bryant S, Hugunin M, Kamath R, Voss J, Olson L, et al. Dual neutralization of TNF and IL‐17 with a DVD‐Ig protein is efficacious in collagen induced arthritis [abstract]. Ann Rheum Dis 2014;73 Suppl S2:S363. [Google Scholar]
- 16. Genovese MC, Cohen S, Moreland L, Lium D, Robbins S, Newmark R, et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum 2004;50:1412–9. [DOI] [PubMed] [Google Scholar]
- 17. Weinblatt M, Schiff M, Goldman A, Kremer J, Luggen M, Li T, et al. Selective costimulation modulation using abatacept in patients with active rheumatoid arthritis while receiving etanercept: a randomised clinical trial. Ann Rheum Dis 2007;66:228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinblatt M, Combe B, Covucci A, Aranda R, Becker JC, Keystone E. Safety of the selective costimulation modulator abatacept in rheumatoid arthritis patients receiving background biologic and nonbiologic disease‐modifying antirheumatic drugs: a one‐year randomized, placebo‐controlled study. Arthritis Rheum 2006;54:2807–16. [DOI] [PubMed] [Google Scholar]
- 19. Hsieh CM, Cuff C, Tarcsa E, Hugunin M. FRI0303 Discovery and characterization of ABT‐122, an anti‐TNF/IL‐17 DVD‐IG™ molecule as a potential therapeutic candidate for rheumatoid arthritis [abstract]. Ann Rheum Dis 2014;73 Suppl 2:495. [Google Scholar]
- 20. Mansikka H, Ruzek M, Hugunin M, Ivanov A, Brito A, Clabbers A, et al. FRI0164 Safety, tolerability, and functional activity of ABT‐122, a dual TNF‐ and IL‐17A–targeted DVD‐IG™, following single‐dose administration in healthy subjects [abstract]. Ann Rheum Dis 2015;74 Suppl 2:482–3. [Google Scholar]
- 21. Fleischmann RM, Wagner F, Kivitz AJ, Mansikka HT, Khan N, Othman AA, et al. Safety, tolerability, and pharmacodynamics of ABT‐122, a tumor necrosis factor– and interleukin‐17–targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol 2017;69:2283–91. [DOI] [PubMed] [Google Scholar]
- 22. Khatri A, Goss S, Jiang P, Mansikka H, Othman AA. Pharmacokinetics of ABT‐122, a TNF‐α‐ and IL‐17A‐targeted dual‐variable domain immunoglobulin, in healthy subjects and patients with rheumatoid arthritis: results from three phase I trials. Clin Pharmacokinet 2018;57:613–23. [DOI] [PubMed] [Google Scholar]
- 23. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 24. Woodworth T, Furst DE, Alten R, Bingham CO III, Yocum D, Sloan V, et al. Standardizing assessment and reporting of adverse effects in rheumatology clinical trials II: the Rheumatology Common Toxicity Criteria v. 2.0. J Rheumatol 2007;34:1401–14. [PubMed] [Google Scholar]
- 25. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 26. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 27. Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7:R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- 29. Khatri A, Othman AA. Population pharmacokinetics of the TNF‐α and IL‐17A dual‐variable domain antibody ABT‐122 in healthy volunteers and subjects with psoriatic or rheumatoid arthritis: analysis of phase 1 and 2 clinical trials. J Clin Pharmacol 2018;58:803–13. [DOI] [PubMed] [Google Scholar]
- 30. Greenwald MW, Shergy WJ, Kaine JL, Sweetser MT, Gilder K, Linnik MD. Evaluation of the safety of rituximab in combination with a tumor necrosis factor inhibitor and methotrexate in patients with active rheumatoid arthritis: results from a randomized controlled trial. Arthritis Rheum 2011;63:622–32. [DOI] [PubMed] [Google Scholar]
- 31. Genovese MC, Weinblatt ME, Mease PJ, Aelion JA, Peloso PM, Chen K, et al. Dual inhibition of tumour necrosis factor and interleukin‐17A with ABT‐122: open‐label long‐term extension studies in rheumatoid arthritis or psoriatic arthritis. Rheumatology (Oxford) 2018. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 32. Taltz (ixekizumab) prescribing information. Indianapolis (IN): Eli Lilly and Company; 2016. [Google Scholar]
- 33. Georgantas RW, Ruzek M, Davis JW, Hong F, Asque E, Idler K, et al. Genomic and epigenetic bioinformatics demonstrate dual TNF‐α and IL17a target engagement by ABT‐122, and suggest mainly TNF‐α–mediated relative target contribution to drug response in MTX‐IR rheumatoid arthritis patients [abstract]. Arthritis Rheumatol 2016;68 Suppl 10 URL: http://acrabstracts.org/abstract/genomic-and-epigenetic-bioinformatics-demonstrate-dual-tnf-%CE%B1-and-il17a-target-engagement-by-abt-122-and-suggest-mainly-tnf-%CE%B1-mediated-relative-target-contribution-to-drug-response-i/. [Google Scholar]
- 34. Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS, et al. A phase II randomized study of subcutaneous ixekizumab, an anti‐interleukin‐17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol 2014;66:1693–704. [DOI] [PubMed] [Google Scholar]
- 35. Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose‐finding, double‐blind, randomised, placebo controlled study. Ann Rheum Dis 2013;72:863–9. [DOI] [PubMed] [Google Scholar]
- 36. Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, Erondu N. A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J Rheumatol 2015;42:912–9. [DOI] [PubMed] [Google Scholar]
- 37. Mease PJ, Genovese MC, Weinblatt M, Peloso PM, Chen K, Othman AA, et al. Phase II study of ABT‐122, a tumor necrosis factor– and interleukin‐17A–targeted dual variable domain immunoglobulin, in patients with psoriatic arthritis with an inadequate methotrexate response. Arthritis Rheumatol 2018;70:1778–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials