

Abstract
The well‐established Hoppe–Beak chemistry, which involves enantioselective generation of organolithium compounds in the presence of (−)‐sparteine, was revisited and applied to unprecedented acylations with Weinreb amides to access highly enantioenriched α‐oxyketones and cyclic α‐aminoketones. Recycling of the sustainable solvent cyclopentyl methyl ether, sparteine, and the released Weinreb “amine” [HNMe(OMe)] was possible through a simple work‐up procedure that enabled full recovery of these precious materials. The methodology features a robust scope and flexibility, thus allowing the enantioselective preparation of scaffolds amenable of further derivatization.
Keywords: acylation, asymmetric chemistry, chemoselectivity, green solvents, organolithium
Introduction
Construction of new carbon–carbon bonds with full control of stereochemistry is highly demanded in organic synthesis.1 The conceptual simplicity of transferring an enantioenriched nucleophile to a given electrophile has emerged over the years as a reliable technique to accomplish this goal.2 In the beginning of the 1990s, the pioneering studies by Hoppe et al.3 and Beak et al.4 culminated in the development of strategies for asymmetric generation of highly nucleophilic organolithium species, which could be delivered to a plethora of electrophiles with excellent stereofidelity (Scheme 1 a).5 The combination of the Lupinus mutabili alkaloid (−)‐sparteine6 and sBuLi to provide a chiral base for the deprotonation event, and the use of an apolar solvent (e.g., diethyl ether) often ensures the preservation of the stereochemical information contained in the nucleophile.7 Unfortunately, both factors pose severe limitations on the sustainability of the processes.
Scheme 1.
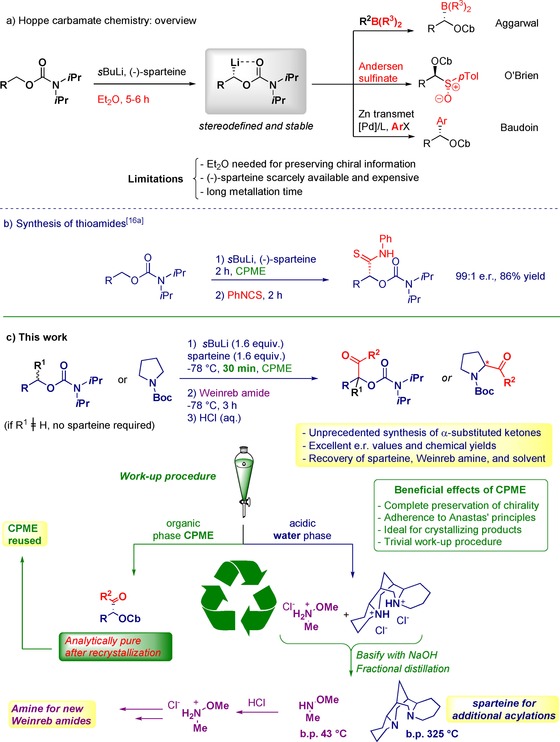
General context of the work.
The full potential of this versatile methodology is limited by the scarce commercial availability of sparteine in both enantiomeric forms8 and the inherent issues for its total synthesis methodologies.9 Additionally, the hazards of diethyl ether (flammability, low boiling point, peroxide formation, etc.)10 make this tactic hardly attractive from a green chemistry perspective, clearly violating the 3rd, 4th, 5th, and 7th Anastas’ Principles.11 Notwithstanding, the sparteine‐mediated methodology is quite flexible and adaptable to conceptually different chemistries to access highly versatile enantioenriched building blocks (Scheme 1 a), for example, Aggarwal's homologated boronates,12 O'Brien's chiral sulfoxides,13 and Baudoin's arenes,14 which can be further elaborated, for example, in Aggarwal's assembly‐line synthesis.15
During a recent study on the synthesis of thioamides from isothiocyanates and organolithium reagents,16 we noticed that the combination of sparteine and the sustainable solvent cyclopentyl methyl ether (CPME)17 effectively achieved a high level of stereocontrol (Scheme 1 b). Additionally, this solvent promoted fast lithiations of hindered benzoate esters, as observed by Aggarwal during the synthesis of (−)‐stemaphylline in the presence of the (+)‐sparteine surrogate.18
Surprisingly, the Hoppe–Beak approach did not find application to synthesize ketones, and thus its full preparative potential was underestimated.19 This is quite astounding because enantiomerically enriched α‐substituted ketones constitute privileged frameworks across the chemical sciences. Based on our experience of chemoselective acylations of α‐substituted carbanions with Weinreb amides,20 we devised an enantio‐ and chemoselective methodology to reach a wide scope of α‐oxyketones and cyclic α‐aminoketones in high yields, without the need for column chromatography (Scheme 1 c). Furthermore, a practical method for the sustainable recovery of solvent, sparteine, and Weinreb amine was devised, and the recovered materials were demonstrated to be equally efficient in subsequent transformations.
Results and Discussion
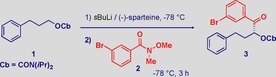
Hoppe's carbamate 1 5a and the challenging Weinreb amide 2, featuring a potentially exchangeable bromine substituent, were selected as the model substrates (Table 1). The use of 1.3 equiv. sBuLi and 1.3 equiv. (−)‐sparteine resulted in a satisfactory isolated yield and enantiomeric ratio (e.r.=97:3) of ketone 3 after a lithiation time of 2 h (entry 1). Increased reagent amounts only slightly improved the yield (entries 2 and 3), suggesting that the reagent loading does not play a crucial role in the process. We were delighted to find that reduced lithiation time increased the efficiency of the reaction, and an excellent 95 % yield of 3 was observed after only 30 min (entries 4 and 5). Shorter lithiation times proved insufficient, and a mixture of ketones 3 and different unidentified byproducts were formed, presumably because of attack of sBuLi on the Weinreb amide (entry 6). The reaction proved less efficient with 1.3 equiv. of the reagents, even if 30 min lithiation time was used (entry 7).
Table 1.
Optimization of the reaction.
| Entry | Solvent | 1/sBuLi/(−)‐sparteine [equiv.] | Lithiation time [min] | Yield of 3 [a] [%] | e.r. of 3 |
|---|---|---|---|---|---|
| 1 | CPME | 1.0/1.3/1.3 | 120 | 65 | 97:3 |
| 2 | CPME | 1.5/1.6/1.6 | 120 | 70 | 97:3 |
| 3 | CPME | 1.9/2.0/2.0 | 120 | 72 | 97:3 |
| 4 | CPME | 1.5/1.6/1.6 | 60 | 78 | 97:3 |
| 5 | CPME | 1.5/1.6/1.6 | 30 | 95 | 97:3 |
| 6 | CPME | 1.5/1.6/1.6 | 15 | 45[b] | – |
| 7 | CPME | 1.0/1.3/1.3 | 30 | 81 | 97:3 |
| 8 | THF | 1.5/1.6/1.6 | 30 | 55 | 60:40 |
| 9 | toluene | 1.5/1.6/1.6 | 30 | 30 | 80:20 |
| 10 | Et2O | 1.5/1.6/1.6 | 30 | 21[b] | – |
| 11[c] | CPME | 1.5/1.6/1.6 | 30 | – | – |
| 12[d] | CPME | 1.5/1.6/1.6 | 30 | 18 | 94:7 |
| 13[e] | CPME | 1.5/1.6/1.6 | 30 | 91 | 97:3 |
[a] Isolated yields. [b] Byproducts were observed. [c] The corresponding ester was employed instead of 2. [d] The corresponding acid chloride was used instead of 2. [e] (−)‐Sparteine recovered from the reaction in entry 5 was employed.
Solvents were subsequently screened, and THF dramatically induced racemization (Table 1, entry 8), in agreement with previous studies.5a This was true also for toluene (entry 9), whereas lithiation in the commonly employed Et2O was very limited within 30 min (entry 10). Clearly, the chemical stability of the lithiated carbamate is the main factor governing the transformation and its prompt generation in CPME ensures the success of the process. This solvent very efficiently preserves the stereochemical information of the nucleophile during the substitution event. CPME is a promising, apolar solvent,21 which is highly stable under strongly basic conditions, features a low peroxide‐formation tendency, and a narrow explosion range, thus fulfilling the 3rd, 4th, 5th, and 12th Green Chemistry Principles.11 Furthermore, the use of CPME was essential to avoid chromatographic purifications. Weinreb amides proved to be crucial as acylating agents for the reaction outcome,22 as demonstrated by the lack of reactivity of the corresponding ester (entry 11) and by the limited yield with the corresponding acid chloride (entry 12).
The sustainability elements of the process make this efficient transformation highly attractive: a) Recovering the precious (−)‐sparteine according to the Aggarwal procedure15 proved easily applicable to the presented methodology, and neither chemical yield nor e.r. were affected if it was reused in another reaction (Table 1, entry 13). b) Analogously, the N,O‐dimethylhydroxylamine fragment of the Weinreb amide, which is released at the end of the sequence, can easily be recycled and reused for the preparation of additional Weinreb amides.23 c) The substantial immiscibility of CPME with water allowed avoidance of classical organic solvents (e.g., EtOAc, Et2O) during the work‐up and, more importantly, the analytically pure ketone 3 was obtained after simple recrystallization from the reaction medium. d) The CPME could be recovered by a simple distillation.
With the optimized conditions in hand, the scope of the reaction was investigated by using a range of Weinreb amides (Scheme 2). High chemoselectivity was uniformly observed, and aromatic (3–10), heteroaromatic (11–12), conjugated (13, 14), and aliphatic (15–17) ketones were formed in excellent yields and enantiomeric ratios. Substitution across the aromatic moieties of the Weinreb amides was well tolerated, except in the case of the ortho‐fluoro analogue 6, which resulted in a somewhat lower e.r. value. Sterically congested amides, as well as α‐substituted amides, could also be employed (15–17). The protocol manifests genuine 1,2‐selectivity with unsaturated Weinreb amides (13, 14), which in principle could suffer from conjugate addition (see below). Gratifyingly, both enantiomers of a given ketone could be prepared in comparable efficiency and enantiopurity by selecting (−)‐ or (+)‐sparteine. Recovered (average 80 %) sparteine was employed in several experiments to demonstrate the efficiency of this process. Furthermore, the protocol allowed us to recover with comparable efficiency CPME and N,O‐dimethylhydroxylamine, which could be advantageously reused whenever necessary. Unambiguous proof of the absolute stereochemistry of 3 was ascertained through X‐ray crystallographic analysis.
Scheme 2.
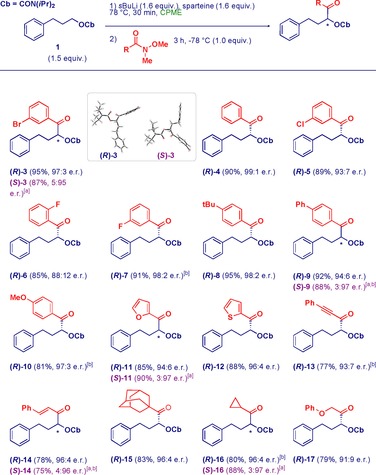
Reaction scope. [a] Reaction with (+)‐sparteine. [b] Reaction with recovered sparteine. See the Supporting Information for full details.
Cognizant of the stereochemical integrity of enantioenriched α‐oxy‐α‐methylbenzyllithium reagents,24 we were pleased to find that such trisubstituted carbanions were suitable for our methodology (Scheme 3). Upon deprotonation of enantiopure α‐methyl carbamate 18 with sBuLi in CPME, a series of α‐quaternary carbonyl compounds could be prepared in excellent yields and enantiomeric ratios starting from the R or S carbamates (sparteine was not required for deprotonating these chiral starting materials). The trapping with a formyl Weinreb amide provided the corresponding α‐oxyaldehyde 19, and aromatic (20, 21), aliphatic (22), or propargylic (23) analogues yielded the corresponding ketones.
Scheme 3.
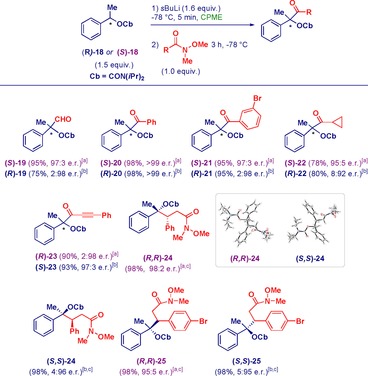
Reactivity of α‐oxy‐α‐methylbenzyllithiums with Weinreb amides. [a] Reaction performed with (S)‐18. [b] Reaction performed with (R)‐18. [c] Only one diastereoisomer observed.
The stereochemical course of the transformation is mainly controlled by the ability of the expelled leaving group to coordinate the lithium cation, and/or by the lowest unoccupied molecular orbital (LUMO) involved.24 Based on the assumption that NMe(OMe) efficiently coordinates Li, in analogy to a general RO group of an ester, it is possible to affirm that Weinreb amides prefer a suprafacial attack with retention of configuration. The absolute configuration of the ketones has been assigned based on comparison with literature data for compounds 19 and 20.
The corresponding ketone 23 was formed through a stereoinversion sequence, presumably owing to the energetically low LUMO of propargylic systems. Reactions with α,β‐unsaturated Weinreb amides surprisingly resulted in selective 1,4‐addition to yield amides 24 and 25 with full diastereo‐ and enantiocontrol, instead of the commonly observed 1,2‐addition.25 As noticed in the case of product 23, inversion of configuration occurred at the quaternary lithiated center, whereas the configuration of the tertiary stereocenter created during the reaction was determined by the steric hindrance of the carbamate.
We have recently reported the isolation of tetrahedral intermediates formed upon the addition of racemic functionalized organolithium reagents to Weinreb amides.26 To our delight, the reaction with an optically active organolithium reagent to form product (R)‐4 could be intervened by addition of trimethylsilyl‐imidazole (ImTMS). Thus, the enantiomerically enriched tetrahedral intermediate could, for the first time, be isolated as O‐TMS hemiaminals 27 and 28 (Scheme 4 a). However, the high chemical instability of this species did not allow an efficient separation of the diastereomers (40:60 d.r. mixture) by flash chromatography or preparative HPLC.
Scheme 4.
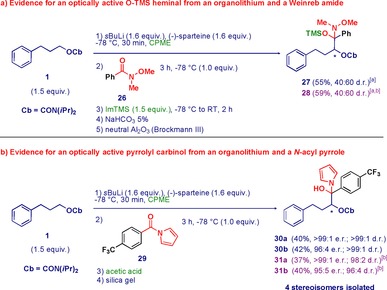
Isolation of hemiaminal‐type optically active tetrahedral intermediates. [a] Diastereomeric ratio determined by 1H NMR analysis. [b] Reaction with (+)‐sparteine.
N‐Acylpyrroles27 form highly stable carbinols upon reactions with carbon nucleophiles according to studies by Evans et al.28 and Brandänge et al..29 Pleasingly, the reaction of the lithiated Hoppe's carbamate with N‐acylpyrrole 29 resulted in the isolable, diastereopure tetrahedral intermediates 30 and 31 with excellent e.r. and d.r. values (Scheme 4 b). By performing the reaction in the presence of either enantiomer of sparteine, four stereoisomers were formed and easily separated through chromatography on silica gel. Subsequent HPLC analysis of each of the stereoisomers confirmed the enantiopurity of each single stereoisomer.
To further expand the synthetic potential of the methodology, it was applied to the synthesis of chiral α‐pyrrolidynyl ketones, which also constitute biologically active substances.30 Enantioselective lithiation of N‐Boc pyrrolidine31 (32) in CPME followed by treatment with a range of Weinreb amides delivered the targeted scaffolds with excellent enantiocontrol (Scheme 5). The scope includes (hetero)aromatic (33–35), unsaturated (36, 37), and aliphatic (38, 39) α‐aminoketones. Remarkable chemoselectivity was observed in reactions with Weinreb amides presenting additional electrophilic sites, leading to products with cyano (34), cinnamoyl (36), and propargyl (37) substituents. Aliphatic Weinreb amides with an acidic α‐proton are also suitable substrates for the transformation, delivering cyclic α‐aminoketones 38 and 39. The latter compound was obtained with perfect diastereoselectivity starting from an optically active Weinreb amide, and the stereochemistry was unambiguously assigned by X‐ray analysis. Again, the opposite enantiomers were prepared with equally excellent stereofidelity by use of (+)‐sparteine.
Scheme 5.
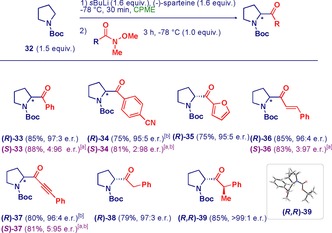
Enantioselective synthesis of α‐pyrrolydinyl ketones. [a] Reaction with (+)‐sparteine. [b] Reaction with recovered sparteine. See the Supporting Information for full details.
The synthetic potential of selected compounds is illustrated in Scheme 6. The reduction of the enantiomerically pure α‐oxyketone (R)‐7 with l‐selectride efficiently afforded the monoprotected 1,2‐diol 40 (Scheme 6 a). Subsequent removal of the carbamate moiety under O'Brien's conditions32 delivered the challenging 1,2‐diol (R,R)‐41 as a single diastereoisomer with excellent enantiopurity. The relative configuration was determined to be syn by diagnostic nuclear overhauser enhancement and exchange spectroscopy (NOESY) experiments on the corresponding acetonide.33 Analogously, the reaction of Hoppe's carbamate with the chiral Weinreb amide 42 yielded a diastereopure α,α′‐disubstituted ketone 43, which was directly subjected to reduction with l‐selectride. Alcohol 44 was obtained as a single diastereoisomer in excellent yield with very high enantiopurity (Scheme 6 b).
Scheme 6.
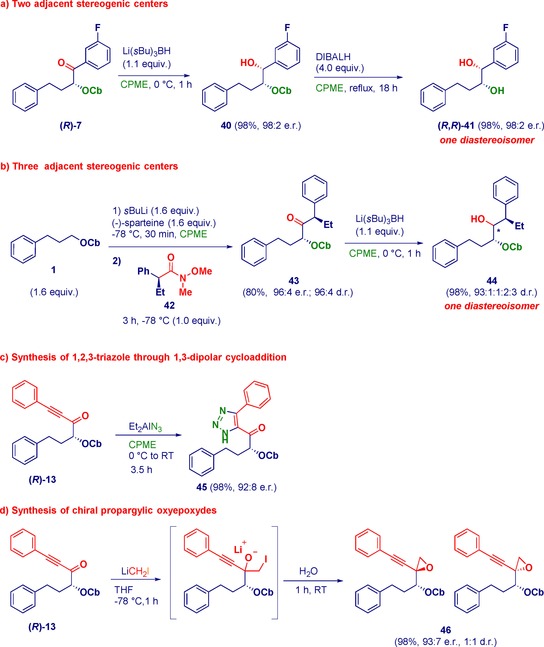
Synthetic manipulations of enantiopure α‐oxyketones.
The propargylic ketone (R)‐13 was used as a platform for the asymmetric construction of 1,2,3‐triazole 45 through a 1,3‐dipolar cycloaddition in the presence of diethyl aluminumazide34 in CPME (Scheme 6 c). Moreover, the same ketone underwent an efficient chemoselective homologation with a lithium carbenoid to produce the α‐oxyepoxydes 46 (1:1 d.r.) with excellent enantiopurity (Scheme 6 d).35
Conclusions
We have disclosed a conceptually new, flexible, versatile, and high‐yielding acylation of enantiopure organolithium reagents (Hoppe's carbamates and N‐Boc pyrrolidine) en route to new α‐oxyketones and cyclic α‐aminoketones. The combination of Weinreb amides and cyclopentyl methyl ether (CPME) as an environmentally benign reaction medium enables access to the targeted compounds with excellent stereofidelities within short reaction times. Both enantiomers of a given scaffold are accessible by simply selecting the appropriate sparteine enantiomer. Analogously, configurationally stable α‐oxy‐α‐methylbenzyllithiums, prepared without requirement of sparteine, can be added to Weinreb amides for enantioselective synthesis of α‐quaternary ketones.
Furthermore, several sustainable features of these new transformations have been developed to meet the drawbacks related to the use of the necessary substrates and reagents. The precious sparteine ligand was efficiently recycled (80 % average) in a practical fashion that also enabled recovery of the Weinreb amine and the solvent. The beneficial effect of CPME is further showcased by its excellent ability as a recrystallization medium to obtain analytically pure compounds, thus avoiding purification by column chromatography.
Experimental Section
General procedure for synthesis of compound 3
A solution of 3‐phenylpropyl diisopropylcarbamate (1, 0.077 g, 0.294 mmol, 1.5 equiv.) and (−)‐sparteine (0.073 g, 0.313 mmol, 1.6 equiv.) under argon in anhydrous CPME (2.0 mL) was cooled to −78 °C. A solution of sBuLi (1.4 m in cyclohexane/hexane 92:8, 0.022 mL, 0.313 mmol, 1.6 equiv.) was added dropwise and the reaction mixture was stirred for 30 min, then 3‐bromo‐N‐methoxy‐N‐methylbenzamide (2, 0.048 g, 0.196 mmol, 1.0 equiv.) in CPME was added slowly. The reaction mixture was quenched after 3 h with HCl (1 n solution, 5 mL) and stirred 1–2 min before the two phases were separated. The pure compound was recrystallized from the same solvent. Compound 3 was obtained in 95 % yield (0.087 g) as a white solid (m.p.: 75–82 °C). CPME (b.p.: 106 °C) was redistilled and could be reused. The acidic aqueous layer was basified with NaOH (20 % aq.) and extracted with CPME (5 mL). The organic layer was distilled to recover N,O‐dimethylhydroxylamine (b.p.: 43 °C), CPME (b.p.: 106 °C), and sparteine (137–138 °C, 1.33 mbar). Recovery data for CPME, sparteine, and N,O‐dimethylhydroxylamine are presented in Tables S1, S2, and S3 in the Supporting Information.
Crystallography
CCDC 1871507 [(S)‐3], 1871508 [(R)‐3], 1871505 [(R,R)‐24], 1571506 [(S,S)‐24], and 1871509 [(R,R)‐39] contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the University of Vienna and Stockholm University for generous support. S.M. acknowledges the University of Vienna for a Uni:docs doctoral fellowship. We thank F. Pisapia and K. Dimov for experimental assistance. We thank Albemarle Corporation and ABCR Germany for the generous gift of chemicals.
S. Monticelli, W. Holzer, T. Langer, A. Roller, B. Olofsson, V. Pace, ChemSusChem 2019, 12, 1147.
Contributor Information
Serena Monticelli, https://drugsynthesis.univie.ac.at/.
Berit Olofsson, Email: berit.olofsson@su.se.
Vittorio Pace, Email: vittorio.pace@univie.ac.at.
References
- 1.See, for example:
- 1a. Asymmetric Synthesis: The Essentials (Eds.: M. Christmann, S. Bräse), Wiley-VCH, Weinheim, 2007; [Google Scholar]
- 1b. Carreira E., Kvaerno L., Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, 2008; [Google Scholar]
- 1c. Quasdorf K. W., Overman L. E., Nature 2014, 516, 181–191; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Das J. P., Marek I., Chem. Commun. 2011, 47, 4593–4623; [DOI] [PubMed] [Google Scholar]
- 1e. Marek I., Minko Y., Pasco M., Mejuch T., Gilboa N., Chechik H., Das J. P., J. Am. Chem. Soc. 2014, 136, 2682–2694; [DOI] [PubMed] [Google Scholar]
- 1f. Bruffaerts J., Pierrot D., Marek I., Nat. Chem. 2018, 10, 1164–1170; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Castoldi L., Pace V., Nat. Chem. 2018, 10, 1081–1082. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Basu A., Thayumanavan S., Angew. Chem. Int. Ed. 2002, 41, 716–738; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 740–763; [Google Scholar]
- 2b. Wang C.-Y., Derosa J., Biscoe M. R., Chem. Sci. 2015, 6, 5105–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoppe D., Hintze F., Tebben P., Angew. Chem. Int. Ed. Engl. 1990, 29, 1422–1424; [Google Scholar]; Angew. Chem. 1990, 102, 1455–1456. [Google Scholar]
- 4.
- 4a. Beak P., Kerrick S. T., Wu S., Chu J., J. Am. Chem. Soc. 1994, 116, 3231–3239; [Google Scholar]
- 4b. Kerrick S. T., Beak P., J. Am. Chem. Soc. 1991, 113, 9708–9710. [Google Scholar]
- 5.For authoritative reviews, see:
- 5a. Hoppe D., Hense T., Angew. Chem. Int. Ed. Engl. 1997, 36, 2282–2316; [Google Scholar]; Angew. Chem. 1997, 109, 2376–2410; [Google Scholar]
- 5b. Hoppe D., Marr F., Brüggemann M. in Organolithiums in Enantioselective Synthesis (Ed.: D. M. Hodgson), Springer, Heidelberg, 2003, pp. 61–138; [Google Scholar]
- 5c. Clayden J., Organolithiums: Selectivity for Synthesis, Pergamon, Oxford, 2002; [Google Scholar]
- 5d. Lithium Compounds in Organic Synthesis: From Fundamentals to Applications (Eds.: R. Luisi, V. Capriati), Wiley-VCH, Weinheim, 2014. [Google Scholar]
- 6.
- 6a. Hoppe D., Morgan B. J., Kozlowski M. C., (−)-Sparteine in Encyclopedia of Reagents for Organic Synthesis (Eds.: L. A. Paquette, D. Crich, P. L. Fuchs, G. A. Molander), John Wiley & Sons, Hoboken, 2007; [Google Scholar]
- 6b. Breuning M., Steiner M., Synthesis 2008, 2841–2867; [Google Scholar]
- 6c. Kizirian J.-C., Chem. Rev. 2008, 108, 140–205; [DOI] [PubMed] [Google Scholar]
- 6d. Dewick P. M., Medicinal Natural Products: A Biosynthetic Approach, Wiley, Chichester, 2009. [Google Scholar]
- 7.For leading studies on enantiopurity/solvent relationships, see:
- 7a. Hoppe D., Christoph G. in The Chemistry of Organolithium Compounds, Vol. 2 (Eds.: Z. Rappoport, I. Marek), Wiley, Chichester, 2004; [Google Scholar]
- 7b. Kizirian J.-C. in Topics in Stereochemistry: Stereochemical Aspects of Organolithium Compounds, Vol. 26 (Eds.: R. E. Gawley, J. S. Siegel), Wiley-VCH, Weinheim, 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Salomone A., Perna F. M., Falcicchio A., Lill S. O. N., Moliterni A., Michel R., Florio S., Stalke D., Capriati V., Chem. Sci. 2014, 5, 528–538; [Google Scholar]
- 7d. Perna F. M., Salomone A., Dammacco M., Florio S., Capriati V., Chem. Eur. J. 2011, 17, 8216–8225; [DOI] [PubMed] [Google Scholar]
- 7e. Mansueto R., Perna F. M., Salomone A., Florio S., Capriati V., Chem. Commun. 2013, 49, 4911–4913. [DOI] [PubMed] [Google Scholar]
- 8. Ritter S. K., Chem. Eng. News 2017, 95, 18–20. [Google Scholar]
- 9.
- 9a. O'Brien P., Chem. Commun. 2008, 655–667; [DOI] [PubMed] [Google Scholar]
- 9b. Foley V. M., Cano R., McGlacken G. P., Tetrahedron: Asymmetry 2016, 27, 1160–1167; [Google Scholar]
- 9c. Firth J. D., Canipa S. J., Ferris L., O'Brien P., Angew. Chem. Int. Ed. 2018, 57, 223–226; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 229–232; [Google Scholar]
- 9d. Dearden M. J., Firkin C. R., Hermet J.-P. R., O'Brien P., J. Am. Chem. Soc. 2002, 124, 11870–11871; [DOI] [PubMed] [Google Scholar]
- 9e. Norcross N. R., Melbardis J. P., Solera M. F., Sephton M. A., Kilner C., Zakharov L. N., Astles P. C., Warriner S. L., Blakemore P. R., J. Org. Chem. 2008, 73, 7939–7951; [DOI] [PubMed] [Google Scholar]
- 9f. Smith B. T., Wendt J. A., Aubé J., Org. Lett. 2002, 4, 2577–2579; [DOI] [PubMed] [Google Scholar]
- 9g. Hermet J.-P. R., McGrath M. J., O'Brien P., Porter D. W., Gilday J., Chem. Commun. 2004, 1830–1831. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Henderson R. K., Jimenez-Gonzalez C., Constable D. J. C., Alston S. R., Inglis G. G. A., Fisher G., Sherwood J., Binks S. P., Curzons A. D., Green Chem. 2011, 13, 854–862; [Google Scholar]
- 10b. Constable D. J. C., Jimenez-Gonzalez C., Henderson R. K., Org. Process Res. Dev. 2007, 11, 133–137; [Google Scholar]
- 10c. Prat D., Pardigon O., Flemming H.-W., Letestu S., Ducandas V. R., Isnard P., Guntrum E., Senac T., Ruisseau S. P., Cruciani P., Org. Process Res. Dev. 2013, 17, 1517–1525; [Google Scholar]
- 10d. Jessop P. G., Green Chem. 2011, 13, 1391–1398; [Google Scholar]
- 10e. Sheldon R. A., Green Chem. 2005, 7, 267–278; [Google Scholar]
- 10f. Scott J. L., Sneddon H. F. in Green Techniques for Organic Synthesis and Medicinal Chemistry (Eds.: W. Zhang, B. W. Cue), Wiley-VCH, Weinheim, 2018, pp. 21–42. [Google Scholar]
- 11. Anastas P., Eghbali N., Chem. Soc. Rev. 2010, 39, 301–312. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Stymiest J. L., Dutheuil G., Mahmood A., Aggarwal V. K., Angew. Chem. Int. Ed. 2007, 46, 7491–7494; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7635–7638; [Google Scholar]
- 12b. Leonori D., Aggarwal V. K., Acc. Chem. Res. 2014, 47, 3174–3183; [DOI] [PubMed] [Google Scholar]
- 12c. Rasappan R., Aggarwal V. K., Nat. Chem. 2014, 6, 810–814; [DOI] [PubMed] [Google Scholar]
- 12d. Balieu S., Hallett G. E., Burns M., Bootwicha T., Studley J., Aggarwal V. K., J. Am. Chem. Soc. 2015, 137, 4398–4403. [DOI] [PubMed] [Google Scholar]
- 13. Rayner P. J., O'Brien P., Horan R. A. J., J. Am. Chem. Soc. 2013, 135, 8071–8077. [DOI] [PubMed] [Google Scholar]
- 14. Royal T., Baumgartner Y., Baudoin O., Org. Lett. 2017, 19, 166–169. [DOI] [PubMed] [Google Scholar]
- 15. Burns M., Essafi S., Bame J. R., Bull S. P., Webster M. P., Balieu S., Dale J. W., Butts C. P., Harvey J. N., Aggarwal V. K., Nature 2014, 513, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Pace V., Castoldi L., Monticelli S., Safranek S., Roller A., Langer T., Holzer W., Chem. Eur. J. 2015, 21, 18966–18970; For a perspective, see: [DOI] [PubMed] [Google Scholar]
- 16b. Pace V., Monticelli S., de la Vega-Hernandez K., Castoldi L., Org. Biomol. Chem. 2016, 14, 7848–7854. [DOI] [PubMed] [Google Scholar]
- 17.For reviews, see:
- 17a. Azzena U., Carraro M., Pisano L., Monticelli S., Bartolotta R., Pace V., ChemSusChem 2019, 12, 40–70; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Watanabe K., Yamagiwa N., Torisawa Y., Org. Process Res. Dev. 2007, 11, 251–258; for interesting examples in organolithium chemistry, see: [Google Scholar]
- 17c. Mallardo V., Rizzi R., Sassone F. C., Mansueto R., Perna F. M., Salomone A., Capriati V., Chem. Commun. 2014, 50, 8655–8658; [DOI] [PubMed] [Google Scholar]
- 17d. Sassone F. C., Perna F. M., Salomone A., Florio S., Capriati V., Chem. Commun. 2015, 51, 9459–9462. [DOI] [PubMed] [Google Scholar]
- 18. Varela A., Garve L. K. B., Leonori D., Aggarwal V. K., Angew. Chem. Int. Ed. 2017, 56, 2127–2131; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2159–2163. [Google Scholar]
- 19.
- 19a. Menges M., Brückner R., Eur. J. Org. Chem. 1998, 1023–1030; [Google Scholar]
- 19b. Hense T., Hoppe D., Synthesis 1997, 1394–1398; [Google Scholar]
- 19c. Carbone G., O'Brien P., Hilmersson G., J. Am. Chem. Soc. 2010, 132, 15445–15450. [DOI] [PubMed] [Google Scholar]
- 20.For recent works on Weinreb amides from the Pace group, see:
- 20a. Senatore R., Castoldi L., Ielo L., Holzer W., Pace V., Org. Lett. 2018, 20, 2685–2688; [DOI] [PubMed] [Google Scholar]
- 20b. Senatore R., Ielo L., Urban E., Holzer W., Pace V., Eur. J. Org. Chem. 2018, 2466–2470; [Google Scholar]
- 20c. Castoldi L., Monticelli S., Senatore R., Ielo L., Pace V., Chem. Commun. 2018, 54, 6692–6704; [DOI] [PubMed] [Google Scholar]
- 20d. Castoldi L., Ielo L., Hoyos P., Hernáiz M. J., De Luca L., Alcántara A. R., Holzer W., Pace V., Tetrahedron 2018, 74, 2211–2217; [Google Scholar]
- 20e. Parisi G., Colella M., Monticelli S., Romanazzi G., Holzer W., Langer T., Degennaro L., Pace V., Luisi R., J. Am. Chem. Soc. 2017, 139, 13648–13651; [DOI] [PubMed] [Google Scholar]
- 20f. Pace V., Murgia I., Westermayer S., Langer T., Holzer W., Chem. Commun. 2016, 52, 7584–7587; [DOI] [PubMed] [Google Scholar]
- 20g. Pace V., Holzer W., De Kimpe N., Chem. Rec. 2016, 16, 2061–2076; for a rare case of unreactivity of Weinreb amides in the presence of organolithiums, see: [DOI] [PubMed] [Google Scholar]
- 20h. Ielo L., Touqeer S., Roller A., Langer T., Holzer W., Pace V., Angew. Chem. Int. Ed. 2019, 58, 2479–2484; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2501–2506; for seminal work, see: [Google Scholar]
- 20i. Nahm S., Weinreb S. M., Tetrahedron Lett. 1981, 22, 3815–3818; for a review on the addition of organometallics to amides, see: [Google Scholar]
- 20j. Pace V., Holzer W., Olofsson B., Adv. Synth. Catal. 2014, 356, 3697–3736. [Google Scholar]
- 21.
- 21a. Watanabe K., Molecules 2013, 18, 3183–3194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Antonucci V., Coleman J., Ferry J. B., Johnson N., Mathe M., Scott J. P., Xu J., Org. Process Res. Dev. 2011, 15, 939–941. [Google Scholar]
- 22.For an authoritative discussion on the development of novel acylating agents, see: Shi S., Nolan S. P., Szostak M., Acc. Chem. Res. 2018, 51, 2589–2599. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Pace V., Castoldi L., Alcantara A. R., Holzer W., RSC Adv. 2013, 3, 10158–10162; see also: [Google Scholar]
- 23b. Kokotos C. G., Baskakis C., Kokotos G., J. Org. Chem. 2008, 73, 8623–8626. [DOI] [PubMed] [Google Scholar]
- 24. Carstens A., Hoppe D., Tetrahedron 1994, 50, 6097–6108. [Google Scholar]
- 25.
- 25a. Pace V., Pelosi A., Antermite D., Rosati O., Curini M., Holzer W., Chem. Commun. 2016, 52, 2639–2642; [DOI] [PubMed] [Google Scholar]
- 25b. Tsubogo T., Saito S., Seki K., Yamashita Y., Kobayashi S., J. Am. Chem. Soc. 2008, 130, 13321–13332; [DOI] [PubMed] [Google Scholar]
- 25c. Saito S., Tsubogo T., Kobayashi S., J. Am. Chem. Soc. 2007, 129, 5364–5365. [DOI] [PubMed] [Google Scholar]
- 26. Castoldi L., Holzer W., Langer T., Pace V., Chem. Commun. 2017, 53, 9498–9501. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Goldys A. M., McErlean C. S. P., Eur. J. Org. Chem. 2012, 1877–1888; [Google Scholar]
- 27b. Meng G., Szostak R., Szostak M., Org. Lett. 2017, 19, 3596–3599. [DOI] [PubMed] [Google Scholar]
- 28. Evans D. A., Borg G., Scheidt K. A., Angew. Chem. Int. Ed. 2002, 41, 3188–3191; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3320–3323. [Google Scholar]
- 29.
- 29a. Brandänge S., Holmgren E., Leijonmarck H., Rodriguez B., Acta Chem. Scand. 1995, 49, 922–928; [Google Scholar]
- 29b. Brandänge S., Rodriguez B., Acta Chem. Scand. Ser. B 1987, 41, 740–744. [DOI] [PubMed] [Google Scholar]
- 30. Ferraris D., Ko Y.-S., Calvin D., Chiou T., Lautar S., Thomas B., Wozniak K., Rojas C., Kalish V., Belyakov S., Bioorg. Med. Chem. Lett. 2004, 14, 5579–5583. [DOI] [PubMed] [Google Scholar]
- 31. Mitchell E. A., Peschiulli A., Lefevre N., Meerpoel L., Maes B. U. W., Chem. Eur. J. 2012, 18, 10092–10142. [DOI] [PubMed] [Google Scholar]
- 32. McGrath M. J., O'Brien P., Synthesis 2006, 2233–2241. [Google Scholar]
- 33.See the Supporting Information for full details.
- 34.
- 34a. Monticelli S., Pace V., Aust. J. Chem. 2015, 68, 703–706; [Google Scholar]
- 34b. Stadler M., Monticelli S., Seidel T., Luger D., Salzer I., Boehm S., Holzer W., Schwarzer C., Urban E., Khom S., Langer T., Pace V., Hering S., J. Med. Chem. 2019, 62, 317–341. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Pace V., Castoldi L., Monticelli S., Rui M., Collina S., Synlett 2017, 28, 879–888; [Google Scholar]
- 35b. Pace V., Castoldi L., Holzer W., Adv. Synth. Catal. 2014, 356, 1761–1766; [Google Scholar]
- 35c. Pace V., Castoldi L., Mamuye A. D., Langer T., Holzer W., Adv. Synth. Catal. 2016, 358, 172–177; interestingly, vinyl epoxides undergo a Meinwald-type rearrangement to aldehydes, see: [Google Scholar]
- 35d. Pace V., Castoldi L., Mazzeo E., Rui M., Langer T., Holzer W., Angew. Chem. Int. Ed. 2017, 56, 12677–12682; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12851–12856. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary