

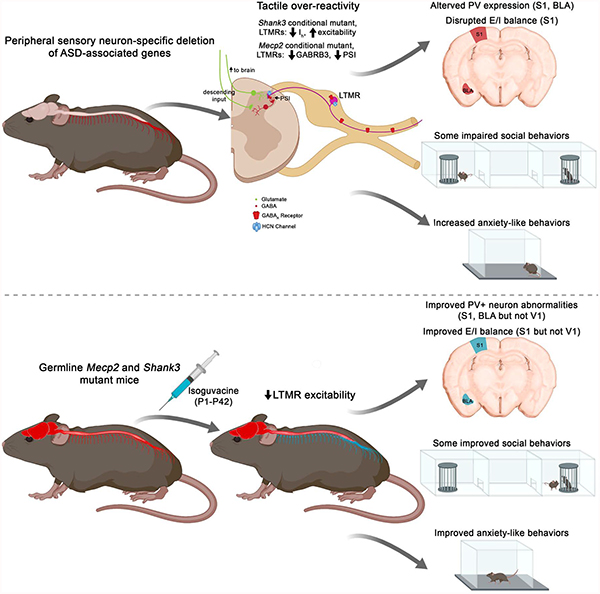
Summary
Somatosensory over-reactivity is common among patients with autism spectrum disorders (ASDs) and is hypothesized to contribute to core ASD behaviors. However, effective treatments for sensory over-reactivity and ASDs are lacking. We found distinct somatosensory neuron pathophysiological mechanisms underlie tactile abnormalities in different ASD mouse models and contribute to some ASD-related behaviors. Developmental loss of ASD-associated genes Shank3 or Mecp2 in peripheral mechanosensory neurons leads to region-specific brain abnormalities, revealing links between developmental somatosensory over-reactivity and the genesis of aberrant behaviors. Moreover, acute treatment with a peripherally-restricted GABAA receptor agonist that acts directly on mechanosensory neurons reduced tactile over-reactivity in six distinct ASD models. Chronic treatment of Mecp2 and Shank3 mutant mice improved body condition, some brain abnormalities, anxiety-like behaviors, and some social impairments, but not memory impairments, motor deficits or overgrooming. Our findings reveal a potential therapeutic strategy targeting peripheral mechanosensory neurons to treat tactile over-reactivity and select ASD-related behaviors.
Graphical Abstract
Treatment with a peripherally-restricted GABAA receptor agonist in multiple distinct autism spectrum disorder mouse models reveals a potential therapeutic strategy for select ASD-related behaviors.
Introduction
Autism spectrum disorders (ASDs) are a heterogeneous group of complex neurodevelopmental disorders characterized by impairments in social communication and interactions and restricted and repetitive behaviors. Although rates of ASD diagnosis are increasing, with approximately 1 in 59 people in the U.S. reported to be living with ASD, there are no FDA-approved treatments for core ASD symptoms (Baio et al., 2018). Furthermore, a majority of adults with ASD (60%) exhibit concerns about current medication options aimed at alleviating co-morbid ASD symptoms due to a lack of efficacy and adverse side effects, including fatigue and sedation (Howes et al., 2018). Of note, pharmacological modulators of GABA receptor signaling have gained attention for possible therapeutic utility in patients with ASD. Bumetanide (a selective chloride importer NKCC1 antagonist) and arbaclofen (a GABAB receptor agonist) are being explored as means of restoring GABAergic inhibition and may show promise in recent clinical trials with regards to improving social behaviors in children with ASD, although side effects of fatigue and irritability pose concerns (Erickson et al., 2014; Hadjikhani et al., 2018; James et al., 2018; Lemonnier et al., 2017; Veenstra-VanderWeele et al., 2017). New therapeutic strategies to treat symptoms observed in ASD patients and reduce off-target effects are clearly needed.
A major hurdle in developing effective treatments for ASD patients is that symptoms and severity are heterogeneous across individuals. In addition, identifying neurobiological underpinnings of behavioral and cognitive abnormalities in ASD, which are needed for rational drug design, has been challenging. Mounting evidence indicates that sensory processing impairments are a key feature of ASD, and aberrant sensory reactivity is now recognized as a core diagnostic symptom (DSM-V, 2013). Furthermore, regression modeling indicates that sensory over-responsivity is strongly correlated with anxiety and gastrointestinal dysfunction, which are frequently co-morbid in people with ASD (Mazurek et al., 2013). Of particular interest is an emerging body of literature indicating that abnormal responses to touch are highly correlated with- and predictive of- ASD severity. Children with ASD often exhibit greater sensitivity to light touch than children with other developmental disorders (Wiggins et al., 2009), and touch avoidance during infancy predicts deficits in social development and ASD diagnosis in young children (Mammen et al., 2015).
Recent work aimed at identifying the neurobiological basis of abnormal tactile sensitivity in ASD revealed that peripheral mechanosensory neurons, called low-threshold mechanoreceptor neurons (LTMRs), and their connections within the spinal cord (SC), are dysfunctional in Mecp2 and Gabrb3 ASD mouse models due to a loss of GABAA receptor (GABAAR)-dependent presynaptic inhibition (PSI) (Orefice et al., 2016). Moreover, aberrant tactile reactivity observed in both Mecp2 and Gabrb3 mutant mice contributes to a subset of ASD-associated behavioral phenotypes, including social impairments and anxiety-like behaviors (Orefice et al., 2016). It remains unclear, however, whether peripheral mechanosensory neuron abnormalities are common across additional pervasive developmental disorders associated with ASD, as well as idiopathic ASD. Furthermore, it is not known whether pharmacological strategies targeting peripheral mechanosensory neurons can be used to treat tactile over-reactivity and other ASD-related behaviors. Here, we report that tactile over-reactivity in different ASD models can result from disparate molecular and pathophysiological mechanisms, aberrant tactile reactivity leads to region-specific abnormal brain development, and a peripherally-restricted pharmacological approach to suppress tactile over-reactivity during early postnatal development has the potential to improve some behavioral abnormalities associated with ASD.
Results
Shank3 functions in peripheral sensory neurons for normal touch behaviors and some ASD-related behaviors
We previously found that Mecp2 and Gabrb3 function in peripheral somatosensory neurons for normal tactile behaviors (Orefice et al., 2016), however the extent to which dysfunction of the peripheral nervous system (PNS) neurons contributes to altered somatosensation in other genetic models for ASD is not known. Haploinsufficiency of the gene Shank3 causes Phelan-McDermid Syndrome, a neurodevelopmental disorder very often presenting with ASD symptoms (Phelan and McDermid, 2012). Patients with Phelan-McDermid syndrome often exhibit enhanced sensitivity to light touch stimuli and tactile defensiveness and, paradoxically, reduced responsivity to certain painful stimuli (Phelan and McDermid, 2012; Philippe et al., 2008). Mice with a germline loss-of-function mutation in Shank3 (Shank3B+/−) exhibit ASD-like traits (Jaramillo et al., 2017; Peca et al., 2011), including tactile over-reactivity (Orefice et al., 2016). SHANK3 is a synaptic protein that is expressed in both mouse and human DRG neurons (Ray et al., 2018; Usoskin et al., 2015), including at the presynaptic terminals of LTMRs that are responsible for transmitting light touch information to the SC dorsal horn (Figure S1A). We therefore asked whether peripheral mechanosensory neuron dysfunction in Shank3 mutant mice underlies abnormal innocuous touch behaviors. To address this, we generated a mouse line harboring a Shank3 floxed (Shank3f) allele (Figures S1B–D) to enable selective ablation of Shank3 in cells expressing Cre recombinase. Shank3f mice were crossed with mice expressing Cre recombinase below cervical level 2 [Cdx2Cre; Shank3f/+; (Akyol et al., 2008)] or all DRG, trigeminal and sympathetic ganglia neurons [AdvillinCre; Shank3f/+ or f/f; (Hasegawa et al., 2007) Figure S1A]. Of note, Shank3 is expressed at almost undetectable levels in sympathetic neurons (Furlan et al., 2016). These conditional mutants as well as mice with heterozygous germline Shank3 deletion (Shank3B+/−) and, for comparison, mice with sensory-neuron specific deletion of Mecp2 (AdvillinCre; Mecp2f/+ or f/y), were subjected to behavioral assays to assess tactile sensitivity and ASD-related behaviors. Consistent with previous findings, Shank3B+/−, and AdvillinCre; Mecp2f/+ or f/y mutant mice exhibited hairy skin hypersensitivity, measured by a tactile prepulse inhibition of an acoustic startle response assay (tactile PPI) and responsivity to an air puff stimulus alone delivered to back hairy skin [Figures 1A, 1B, Table S1; (Orefice et al., 2016)]. Shank3B+/−, and AdvillinCre; Mecp2f/+ or f/y mutant mice also displayed texture discrimination deficits, assessed using a textured novel object recognition test [“textured NORT”, Figure 1C, (Orefice et al., 2016)]. Loss of Shank3 in cells below the neck (Cdx2Cre; Shank3f/+) or in somatosensory neurons (AdvillinCre; Shank3f/+ or f/f) also led to an increase in tactile PPI, increased responsivity to an air puff stimulus, and deficits in texture discrimination (Figures 1A–C). Cdx2Cre; Shank3f/+ and AdvillinCre; Shank3f/+ or f/f mutant mice were overtly normal, however, and no differences in gross motor behaviors, acoustic PPI, or memory retention were observed among any of the groups (Figures S1E–J).
Figure 1. Shank3 functions cell-autonomously in peripheral somatosensory neurons for normal innocuous touch behaviors.

(A) Hairy skin sensitivity was measured using tactile PPI. Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff (250 ms ISI). Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(B) Response to a light air puff stimulus alone directed to the back hairy skin. Responses are expressed as percent of startle response to a 125 dB noise. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(C) Texture discrimination was measured using the textured NORT behavioral assay. A positive value indicates preference for the novel object, compared to the familiar object. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Open field (OF) test was used as a general measure of exploration and anxiety-like behavior. Shown are representative activity traces in the OF test for mutant mice and control littermates.
(E) Percent time spent in the center of the OF chamber. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) Percent time spent in the open arms of the EPM. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(G) Preference index for the percentage of time spent investigating a novel mouse, compared to a novel object, in the “Sociability” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(H) Preference index for the percentage of time spent investigating a novel mouse, compared to a familiar mouse, in the “Social Novelty Preference” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(I) Voltage step protocol used to activate HCN channels and elicit Ih during whole-cell voltage clamp recordings.
(J-K) Representative electrophysiological traces showing Ih during a hyperpolarizing voltage step protocol in large diameter DRG neurons cultured control and Shank3B+/− mutant mice, at baseline (K) and with a selective HCN-channel blocker, ZD-7288 (L).
(L) Quantification of Ih density at each voltage step for large diameter neurons cultured from DRGs of control and mutant mice. Two-way ANOVA with post-hoc Sidak’s test, [F (1,1470) =187.7; P < 0.0001] *, p < 0.05.
(M) Representative traces from large diameter DRG neurons cultured from control and Shank3B+/− mutant mice during whole cell current clamp recordings, in which the minimal amount of current required to elicit an action potential in each neuron (rheobase, Rh), was determined.
(N) Quantification of average R in large diameter DRG neurons cultured from control and Shank3B+/− mutant mice. Student’s unpaired t-test, *, p < 0.005.
Concomitant with altered reactivity to tactile stimuli, mice in which either Mecp2 or Gabrb3 was ablated in peripheral somatosensory neurons during embryonic development exhibited increased anxiety-like behaviors and abnormal social behaviors in adulthood [Figures 1D–H, S1K–M; (Orefice et al., 2016)]. We therefore hypothesized that altered tactile processing due to developmental loss of Shank3 in peripheral sensory neurons may also contribute to anxiety-like behaviors and abnormal social behaviors in adult mice. Consistent with prior findings, Shank3B+/− mice displayed anxiety-like behaviors in the open field test, elevated plus maze (EPM) and lack of habituation to an acoustic startle noise (Peca et al., 2011) (Figures 1D–F, S1K–M). Cdx2Cre; Shank3f/+, and AdvillinCre; Shank3f/+ or f/f mutant mice also exhibited anxiety-like behaviors (Figures 1D–F, S1K–M). Abnormal social interactions, neophobia, and over-grooming behaviors are additional features of Shank3B+/− mutant mice (Jaramillo et al., 2017; Peca et al., 2011). We implemented the three-chamber social interaction test to assess sociability and social novelty recognition preference (“social novelty preference”) in the conditional mutants (Silverman et al., 2010). Similar to Shank3B+/− mice, the Cdx2Cre; Shank3f/+ and AdvillinCre; Shank3f/+ or f/f mice did not show a preference for a novel mouse in either the sociability or social novelty preference portion of the test, although Cdx2Cre; Shank3f/+ and AdvillinCre; Shank3f/+ mutants did exhibit a trend towards preference in the sociability assay (Figures 1G, 1H, Table S2). Shank3+/− mice exhibited neophobia during a marble burying task, evidenced by a decreased number of marbles buried compared to controls (Jaramillo et al., 2017) (Figure S1N). A modest decrease in the number of marbles buried was also observed in Cdx2Cre; Shank3f/+ and AdvillinCre; Shank3f/+ or f/f mutant mice (Figure S1N). Finally, while Shank3B+/− mutants display an overgrooming behavior, this phenotype was not observed in either Cdx2Cre; Shank3f/+ or AdvillinCre; Shank3f/f mice (Figure S1O). These findings indicate that a subset of behavioral abnormalities observed in Shank3B+/− mice are also observed in mice lacking one or both Shank3 alleles in peripheral somatosensory neurons.
We next asked whether restoration of Shank3 expression selectively in peripheral somatosensory neurons might improve tactile deficits and other ASD-related phenotypes observed in Shank3 germline mutant mice. We employed a Cre-dependent genetic switch (FLEx) knock-in mouse, which enables conditional expression of Shank3 from its endogenous genomic locus in the presence of Cre recombinase (Mei et al., 2016) (Figure S1Q). Selective restoration of Shank3 in cells below the neck (Cdx2Cre; Shank3FX/+) or in somatosensory neurons (AdvillinCre; Shank3FX/+, Figures S1Q, S1R) normalized hairy skin hypersensitivity, textured discrimination deficits and anxiety-like behaviors observed in Shank3FX/+ mice (Figures 1A–F, S1L). Furthermore, while Shank3FX/+ mice did not show a preference for a novel mouse in either portion of the 3-chamber social interaction test, both Cdx2Cre; Shank3FX/+ and AdvillinCre; Shank3FX/+ rescue mice exhibited preference for the novel mouse in the sociability assay (Figures 1G, 1H, Table S2). We also observed an intermediate phenotype in the marble burying test: Cdx2Cre; Shank3FX/+ and AdvillinCre; Shank3FX/+ rescue mice buried more marbles than their Shank3FX/+ littermates, but fewer than control littermates (Figure S1N). Conversely, no improvements in memory deficits or overgrooming behaviors were observed in either Cdx2Cre; Shank3FX/+ or AdvillinCre; Shank3FX/+ rescue mice (Figures S1I, S1O, S1P). Therefore, as observed for Mecp2 and Gabrb3 (Orefice et al., 2016), Shank3 is required cell-autonomously in peripheral somatosensory neurons for normal tactile sensitivity and texture discrimination as well as a subset of ASD-related behaviors. Thus, dysfunction of mechanosensory neurons is a common feature of multiple ASD mouse models, despite disparate molecular properties and functions of ASD-associated genes.
Distinct mechanosensory neuron physiological dysfunctions underlie altered tactile processing across animal models for ASD
Prior work indicated that peripheral somatosensory neurons lacking Mecp2 or Gabrb3 exhibit decreased expression of GABRB3, the obligatory beta subunit of the GABAAR in these neurons (Orefice et al., 2016; Zimmerman et al., 2019). This leads to a deficit in GABAAR-mediated PSI of somatosensory neuron inputs to the SC, thus explaining tactile over-reactivity in Mecp2 and Gabrb3 mutants (Orefice et al., 2016). Therefore, we asked whether a similar pathophysiological mechanism could explain tactile over-reactivity observed in Shank3 mutant mice. However, no alteration in GABRB3 expression in LTMR central terminals of Shank3 mutant mice was observed (Figures S2E, S2F). Guided by recent observations that Shank3 ablation in hippocampal neurons causes a reduction of HCN channel expression leading to a loss of the current mediated by these channels, known as Ih, as well as increased excitability (Yi et al., 2016), we next asked whether loss of Shank3 leads to altered expression of HCN channels in sensory neurons. Indeed, fewer HCN1-containing puncta were detected at presynaptic terminals of Aβ- and Aδ-LTMRs in the SC dorsal horn of Shank3B+/− and AdvillinCre; Shank3f/+ or f/f mutant mice (Figures S2A, S2B), and a loss of HCN1 expression in large but not small diameter DRG cell bodies (Figures S2C, S2D). Consistent with this, whole-cell patch clamp recordings of large diameter sensory neurons cultured from Shank3 mutant mice revealed multiple physiological alterations, including increased input resistance, decreased Ih elicited during a hyperpolarizing voltage step protocol, and increased excitability (Figures 1I–N, S2G–I, S2N). No differences in capacitance or cell body size of recorded neurons were observed between groups (Figures S2O, S2P). In contrast to these alterations in large diameter neurons, a majority of which are mechanosensory neurons, no deficits in Ih or HCN1 expression were observed in small-diameter DRG neurons from Shank3 mutant mice, although these neurons exhibited a modest reduction in excitability (Figures S2C, S2D, S2J–M). For comparison, similar measurements were made using DRG neurons cultured from Mecp2 mutant mice. Neither large nor small diameter DRG neurons from Mecp2 mutant mice exhibited abnormalities in Ih, although there was a trend towards a small increase in excitability of large diameter neurons (Figures S2Q–S). Consistent with our findings that large diameter DRG neurons from Shank3B+/− mice display decreased Ih and altered excitability, Hcn1−/− mice exhibited enhanced hairy skin sensitivity and deficits in texture discrimination (Figures S2T–W). Together, these findings indicate that multiple ASD-associated genes function cell-autonomously in peripheral somatosensory neurons for normal tactile sensitivity, and at least two distinct pathophysiological mechanisms underlie tactile over-responsivity in ASD mouse models: loss of GABAAR-mediated PSI of LTMR inputs to the SC dorsal horn, as observed in Mecp2 and Gabrb3 mutants (Orefice et al, 2016), or decreased Ih and hyperexcitability of somatosensory neurons, observed in Shank3 mutants.
ASD-related gene mutations in somatosensory neurons lead to region-specific alterations in brain inhibitory interneurons and cortical microcircuit properties
We next explored the relationship between aberrant tactile reactivity, caused by ablation of either Mecp2 or Shank3 in sensory neurons, and brain development and cortical microcircuit properties. One clue that may help to explain any potential relationship stems from decades of research indicating that sensory experience guides development of neocortical areas where features of sensory stimuli are represented (Hubel and Wiesel, 1970; Simons and Land, 1987; Wiesel and Hubel, 1965). In line with this, postmortem analysis of brains from ASD patients as well as animal models for ASD (such as Mecp2 and Shank3 mutants) revealed abnormalities in parvalbumin-positive (PV+) inhibitory interneurons in multiple brain regions, including primary sensory cortices (Filice et al., 2016; Fukuda et al., 2005; Hashemi et al., 2017; Marin, 2012; Nelson and Valakh, 2015; Tomassy et al., 2014; Zikopoulos and Barbas, 2013). Cortical PV+ neurons are fast-spiking interneurons that contribute to feed-forward and feedback inhibition and modulate sensory responsiveness (Konig et al., 1996; Sohal et al., 2009; Womelsdorf et al., 2014). Therefore, we hypothesized that aberrant sensory inputs caused by peripheral somatosensory neuron dysfunction in ASD models may affect properties of PV+ inhibitory interneurons in brain circuits that process these inputs. In agreement with this idea, loss of Mecp2 in peripheral somatosensory neurons in AdvillinCre; Mecp2f/y mice caused an increased number of PV+ interneurons in primary somatosensory cortex (S1), but not primary visual cortex (V1), of adult mice compared to controls (Figures 2A, 2D, 2E, Table S1). However, loss of Shank3 in sensory neurons, in AdvillinCre; Shank3f/+ mice, led to a reduction in PV+ neurons in S1, but not V1, of adult mice (Figures 2B–2E). These region-specific alterations in PV+ interneurons in Shank3 and Mecp2 conditional mutants are in contrast to the more widespread alterations observed in the germline mutants: Mecp2STOP/y and Shank3FX/+ germline mutant mice exhibited alterations in PV+ neurons in both S1 and V1 (Figures 2G–J). We also observed a decrease in the number of PV+ neurons in the basolateral amygdala (BLA), a region of the brain that plays critical roles in anxiety and social behaviors, in both Shank3 and Mecp2 conditional mutants (Figures 2A, 2F). Conversely, developmental restoration of either Shank3 or Mecp2 in somatosensory neurons, in AdvillinCre; Shank3FX/+ and AdvillinCre; Mecp2STOP/y mice, respectively, improved the PV+ neuron abnormalities observed in Shank3FX/+ and Mecp2STOP/y mice in both S1 and BLA, but not in V1 (Figures 2G–K). Related to this, we also found that loss of either Mecp2 or Shank3 in somatosensory neurons led to decreased excitatory/inhibitory (E/I) ratios measured electrophysiologically in layer 2/3 pyramidal neurons using acute S1 slices, compared to control littermates (Figure 2R). Layer 2/3 pyramidal neurons from S1 slices of AdvillinCre2; Mecp2f/y mice exhibited an increase in spontaneous inhibitory postsynaptic current (sIPSC) frequency, with no differences observed in spontaneous excitatory postsynaptic current (sEPSC) or sIPSC amplitudes (Figures 2Q, 2S, 2T). On the other hand, while layer 2/3 pyramidal neurons from S1 slices of AdvillinCre; Shank3f/+ mice showed no difference in the frequency of sIPSCs, decreased sEPSC and sIPSC amplitudes were observed in these mutants (Figures 2P, 2S, 2T). No differences in either the amplitude or frequency of events were observed in V1 in any of the conditional mutants analyzed (Figures 2U, 2V). These analyses indicate that physiological dysfunction of peripheral somatosensory neurons caused by distinct ASD-associated gene mutations leads to functional changes in cortical microcircuit properties in a region-specific manner.
Figure 2. Loss of either Shank3 or Mecp2 in peripheral somatosensory neurons leads to abnormal brain interneuron development and microcircuit properties in a region-specific manner.

(A) Representative immunohistochemistry (IHC) images of parvalbumin (PV) immunoreactivity in control or AdvillinCre; Mecp2f/y mutant mice, showing full sagittal brain sections and corresponding magnified sections of primary somatosensory cortex (S1) and primary visual cortex (V1), or coronal sections of basolateral amygdala (BLA).
(B) Representative IHC images of coronal brain sections, showing PV immunoreactivity in control or AdvillinCre; Shank3f/+ mutant mice.
(C) Representative IHC images of coronal S1 brain sections, showing PV and GAD67 immunoreactivity in control or AdvillinCre; Shank3f/+ mutant mice.
(D-F) Quantification of the number of PV-positive (PV+) neurons in S1 (D), V1 (E), and BLA (F) in mutant mice and their control littermates. Student’s unpaired t-test, *, p < 0.05.
(G) Representative IHC images of coronal S1 brain sections, showing PV immunoreactivity in control, Shank3FX/+, or AdvillinCre; Shank3FX/+ mutant mice.
(H) Representative IHC images of coronal S1 brain sections, showing PV immunoreactivity in control, Mecp2STOP/y, or AdvillinCre; Mecp2STOP/y mutant mice.
(I-K) Quantification of the number of PV+ neurons in S1 (I), V1 (J), and BLA (K) in mutant mice and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(L) Example image of a coronal S1 slice with a layer 2/3 pyramidal neuron in whole cell patch clamp recording configuration.
(M) Example firing pattern of a layer 2/3 pyramidal neuron in whole cell patch clamp recording configuration during current injection steps.
(N) Example traces from a layer 2/3 pyramidal neuron showing spontaneous excitatory postsynaptic currents (sEPSCs) in normal bath solution or following application of NBQX (10 μM) to block AMPA receptors.
(O) Example traces from a layer 2/3 pyramidal neuron showing spontaneous inhibitory postsynaptic currents (sIPSCs) in normal bath solution or following application of gabazine (3 μM) to block GABAA receptors.
(P) Representative traces showing sEPSCS (−70 mv hold) and sIPSCs (0 mV hold) from S1 slices of control or AdvillinCre; Shank3f/+ mutant mice.
(Q) Representative traces showing sEPSCs (−70 mv hold) and sIPSCs (0 mV hold) from S1 slices of control or AdvillinCre; Mecp2f/y mutant mice.
(R) Quantification of excitatory/inhibitory (E/I) ratios in S1 or V1 slices from control and mutant mice. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(S-T) Quantification of sEPSC and sIPSC event frequency (S) and event amplitude (T) in S1 slices from control and mutant mice. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05; #, p < 0.10.
(U-V) Quantification of sEPSC and sIPSC event frequency (U) and event amplitude (V) in V1 slices from control and mutant mice. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
A critical window during which somatosensory neuron dysfunction influences cognitive and social behaviors
We next determined when during development ASD-associated genes function in peripheral somatosensory neurons to govern tactile reactivity, brain microcircuit development, and behavior. To address this, we used the tamoxifen-sensitive AdvillinCreERT2 mouse line (Lau et al., 2011) to delete either Shank3 or Mecp2 during different postnatal developmental stages (Figures S4A–D) and then assessed tactile reactivity, texture discrimination, PV+ interneurons in S1, V1 and BLA, and cognitive and social behaviors in young adult mice beginning at 6 weeks of age. Early postnatal deletion of either Shank3 or Mecp2, beginning at P5 (AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y), recapitulated the behavioral phenotypes observed in AdvillinCre; Shank3f/+ and AdvillinCre; Mecp2f/y mice (Figures 3, S3A, S3H–K, Tables S3, S4). Moreover, similar to AdvillinCre; Mecp2f/y mice, AdvillinCreERT2; Mecp2f/y mutant mice receiving P5 tamoxifen treatment exhibited more PV+ neurons in S1 (Figure S4G). Likewise, AdvillinCreERT2; Shank3f/+ mutant mice displayed fewer PV+ neurons in S1, which was similar to that observed in AdvillinCre; Shank3f/+ mice (Figure S4G). Decreased density of PV+ neurons in BLA was also observed in both AdvillinCreERT2; Shank3f/+ and AdvillinCreERT2; Mecp2f/y mutant mice receiving the P5 tamoxifen treatment (Figure S4I). In contrast, while deletion of either Mecp2 or Shank3 in somatosensory neurons (AdvillinCreERT2; Mecp2f/y or AdvillinCreERT2; Shank3f/+) at P28 led to abnormalities in tactile behaviors and PV+ neuron density in S1, these mice did not exhibit anxiety-like behaviors or neophobia, and their social impairments were considerably less severe than mice with embryonic or P5 deletion of either gene in peripheral sensory neurons (Figures 3, S3A, S3H–K, S4F, S4G, Table S3). Mice with P28 deletion also displayed no abnormalities in BLA PV+ neuron density (Figure S4I). Interestingly, when either Mecp2 or Shank3 was ablated in somatosensory neurons beginning at an intermediate time point, P10, mice exhibited a distinct set of behavioral alterations. In P10 deletions, tactile and social behavior abnormalities were observed, similar to embryonic deletions, but this was accompanied by hyperactivity and reduced anxiety-like behaviors (Figures 3, S3A, S3H–M, Table S3). Moreover, P10 ablations of Shank3 and Mecp2 in somatosensory neurons both lead to a reduction in PV+ neurons in S1, but increased density of PV+ neurons in the BLA (Figures S4G–I).
Figure 3. Normal tactile sensitivity is necessary during early postnatal periods for normal brain development and behavior.

(A) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in mutant mice and control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05. For experiments shown in this figure, tamoxifen was administered to all littermates, over a five-day period, beginning on the day noted for each condition. For all behavioral experiments in this figure, animals began testing at P42.
(B) Discrimination index for textured NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(C) Representative activity traces in the OF test for Shank3 mutant mice and control littermates. Tamoxifen (TAM) was administered over a five-day period, beginning on the day noted for each condition.
(D) Representative activity traces in the OF test for Mecp2 mutant mice and control littermates.
(E) Percent time spent in the center of the OF chamber. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) Percent time spent in the open arms of the EPM. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(G-H) Preference index for the percentage of time spent investigating the novel mouse in the “Sociability” (G) and “Social Novelty Preference” (H) portions of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
In related experiments, we asked whether postnatal restoration of Mecp2 or Shank3 function in somatosensory neurons might improve ASD-related phenotypes in mice. For this, AdvillinCreERT2 mice crossed to either Shank3FX/+ or Mecp2STOP/+ mice were used to restore Shank3 or Mecp2 expression, respectively, in somatosensory neurons following 5 days of tamoxifen administration beginning at P28 (Figure S4J). Here, restoration of either Shank3 or Mecp2 in somatosensory neurons, beginning at P28, was sufficient to normalize hairy skin hypersensitivity observed in Shank3FX/+ or Mecp2STOP/y littermates (Figures 3A, S3A). However, memory deficits were not improved in either AdvillinCreERT2; Shank3FX/+ or AdvillinCreERT2; Mecp2STOP/y P28 rescue mice (Figure S3F), and motor impairments typically observed in Mecp2 germline mutant mice were also not improved in AdvillinCreERT2; Mecp2STOP/y mice (Figures S3B). Furthermore, AdvillinCreERT2; Shank3FX/+ and AdvillinCreERT2; Mecp2STOP/y mice treated with tamoxifen beginning at P28 did not show improvements in texture discrimination deficits, anxiety-like behaviors or neophobia (Figures 3B–F, S3B–I). AdvillinCreERT2; Shank3FX/+ and AdvillinCreERT2; Mecp2STOP/y rescue mice showed modest improvements in social behaviors, with a significant preference for a novel mouse in the sociability assay, but not in the social novelty preference test (Figures 3G, 3H, Table S3). These findings indicate that normal tactile reactivity is necessary during postnatal development for the acquisition of normal brain microcircuit properties and cognitive behaviors.
Postnatal viral restoration of GABRB3 improves behavioral deficits in an Mecp2 mouse model of RTT/ASD
Our findings suggest that targeting the PNS may provide an opportunity for improving tactile reactivity, anxiety-like and some social behaviors in adult mice harboring ASD-associated gene mutations, with optimal intervention occurring early during postnatal life. Therefore, we considered whether enhancing GABAAR signaling in peripheral sensory neurons may provide a means of attenuating tactile over-reactivity in ASD models and, in doing so, improve anxiety-like behaviors and potentially other ASD-associated behaviors. The rationale for this idea stems from the finding that Mecp2 mutant mice exhibit decreased expression of the GABAAR obligatory subunit GABRB3 at presynaptic terminals of LTMRs in the SC, leading to functional deficits in PSI of mechanosensory neuron input to the SC and altered tactile processing (Orefice et al., 2016). Using conditional mouse genetics and a FLExed AAV delivery strategy, we were able to selectively restore GABRB3 expression in somatosensory neurons of Mecp2 mutant mice in a temporally precise manner. Mice harboring an arginine-to-cysteine missense mutation in Mecp2 (Mecp2R306C), which is a common allele found in patients with Rett Syndrome (RTT) (Lyst et al., 2013), were used in these studies. We used AdvillinCre mice and intraperitoneal (i.p.) injection of an AAV9.FLEx.GABRB3.mCHERRY virus at P5, to selectively express GABRB3 in peripheral sensory neurons. Thus, DRG neurons in mice harboring the AdvillinCre allele expressed functional GABRB3 following i.p. injection of P5 pups with AAV9.FLEx.GABRB3.mCHERRY (Figures 4A–C, S5A, S5B). Increased expression of GABRB3 in somatosensory neurons improved tactile behavior abnormalities in a dose-dependent manner: AdvillinCre; Mecp2C/y mice displayed significant improvements in hairy skin hypersensitivity and texture discrimination, compared to Mecp2C/y mice (Figures 4D–G, Table S5). Female ‘GABRB3 rescue’ mice, which are heterozygous for the Mecp2 mutation (AdvillinCre; Mecp2R/C), showed a normalization of somatosensory phenotypes (Figures 4D–G). Similarly, we found a significant correlation between both the number of DRG neurons transduced and the number of GABRB3 puncta in the dorsal horn and reduced responsivity to an air puff stimulus in Mecp2 mutants (Figures 4F and S5H). Restoration of GABRB3 expression in somatosensory neurons also improved anxiety-like behaviors, social impairments, and PV+ neuron abnormalities in S1 and BLA (but not V1), of both male and female rescue mice, with female AdvillinCre; Mecp2R/C mice exhibiting values similar to those of control littermates (Figures 4H–O, S5L–Q). Augmented GABRB3 expression, however, did not improve memory deficits, motor impairments or heightened acoustic PPI performance in mutant animals (Figures S5C–E, S5I, S5J). This proof-of-concept experiment suggests that increasing GABAAR function in somatosensory neurons during early postnatal development ameliorates tactile over-reactivity, altered S1 and BLA microcircuits, and at least some behavioral deficits observed in germline Mecp2 mutants.
Figure 4. Viral expression of GABRB3 in peripheral sensory neurons, beginning at P5, improves some tactile, brain and behavioral deficits observed in Mecp2R306C mutant mice.

(A) Intraperitoneal (i.p.) injection of AAV.FLEx.GABRB3.mCHERRY into AdvillinCre; Mecp2C/y hemizygous null mutant mice at P5 transduces peripheral sensory neurons, as evidenced by immunoreactivity for mCHERRY. Transduced large diameter neurons are immunoreactive for both mCHERRY and NF200. All littermates received i.p. injection of AAV.FLEx.GABRB3.mCHERRY at P5.
(B) IHC images of spinal cord (SC) dorsal horn lamina III/IV from male control, Mecp2C/y or AdvillinCre; Mecp2C/y mice, showing GABRB3 puncta at vGLUT1+ presynaptic terminals for Aα and Aδ LTMRs.
(C) Quantification of vGLUT1+ puncta co-labeled with GABRB3, relative to the total number of vGLUT1+ puncta visualized per image of SC dorsal horn. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.01.
(D) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in male control, Mecp2C/y, AdvillinCre; Mecp2C/y or female control, Mecp2R/C, and AdvillinCre; Mecp2R/C mice. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(E) Response to a light air puff stimulus alone. Responses are expressed as percent of startle response to a 125 dB noise. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) The percentage of vGLUT1+ puncta co-labeled with GABRB3, relative to the total number of vGLUT1+ puncta is negatively correlated with hairy skin sensitivity. Comparison of individual animals’ expression levels of GABRB3 at vGLUT1+ terminals, to their responses to a light air puff stimulus (50 ms, 0.9 PSI). Linear regression analysis, R2 = 0.8792.
(G) Discrimination index for textured NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(H) Representative activity traces in the OF test.
(I) Percent time spent in the center of the OF chamber. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05; #, p < 0.10.
(J) Percent time spent in the open arms of the EPM. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Preference index for the percentage of time spent investigating the novel mouse in the “Sociability” or “Social Novelty Preference” portion of the 3-chamber social interaction test. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(L) Representative IHC images of coronal S1, V1 and BLA brain sections, showing PV immunoreactivity in control, Mecp2C/y or AdvillinCre; Mecp2C/y mice.
(M-O) Quantification of the number of PV-positive (PV+) neurons in S1 (M), V1 (N), and BLA (O) in mutant, mutant rescues and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
Acute treatment with a peripherally-restricted GABAAR agonist improves hairy skin over-reactivity in six distinct, genetic and environmental models for ASD
Results of the GABAAR restoration experiments raised the possibility that pharmacological treatment with a GABAAR agonist may improve tactile reactivity in Mecp2 mutant mice. We therefore tested whether benzodiazepines, which are positive allosteric modulators (PAMs) of GABAARs, would attenuate hairy skin over-reactivity in Mecp2 mutant mice. As expected, AdvillinCre; Mecp2f/y mice receiving vehicle treatment showed tactile hypersensitivity compared to control littermates (Figures 5A, 5B, Table S5). Midazolam treatment attenuated tactile sensitivity in both Mecp2 mutants and controls (Figures 5A, 5B). However, this treatment also caused significant sedation in all mice tested (Figure 5C) complicating interpretations and limiting the utility of this approach. In line with this, traditional blood-brain barrier (BBB) penetrating GABAAR PAMs are useful for treating some ASD-related symptoms in patients, including acute anxiety, but adverse effects such as sedation and addictive potential are problematic (Gudex, 1991). Furthermore, long-term benzodiazepine use is linked to cognitive impairment across multiple domains, including short-term verbal memory and attention (Golombok et al., 1988; Tata et al., 1994). These considerations prompted us to ask whether peripherally restricted GABAAR agonists administered beginning at a young age could ameliorate tactile hypersensitivity in ASD models with fewer adverse effects than brain-penetrating benzodiazepines, including sedation and cognitive impairment, which are attributed to a central nervous system site of drug action. Isoguvacine is a potent and selective agonist for the GABAAR (Bowery et al., 1983; Hill and Bowery, 1981; Krogsgaard-Larsen and Johnston, 1978; Krogsgaard-Larsen et al., 1977). Due to its zwitterionic structure, isoguvacine is predicted to not cross the BBB (Krogsgaard-Larsen et al., 1981). In agreement with this, we found that isoguvacine administration (2 mg/kg, i.p.) did not cause sedation in mice, which contrasts with BBB-penetrating benzodiazepine midazolam treatment (Figure 5D). Moreover, bio-distribution analyses indicated that following i.p. administration of isoguvacine (20 mg/kg) the drug was detected in blood and liver, but was undetectable in the brain and cerebrospinal fluid, confirming that isoguvacine does not cross the BBB (Figures 5E–H). RNA deep sequence analysis of genetically labeled DRG neuron subtypes (Zheng et al., 2019) revealed that α1β3γ2- and α2β3γ2-containing GABAARs are the major holopentameric GABAARs expressed in LTMR subtypes, with lower levels of expression in nociceptors and proprioceptors (Figures 5J–L, S6H). Using a GABAAR chloride flux assay, isoguvacine was observed to potently activate both α1β3γ2- and α2β3γ2-containing GABAARs, with EC50 values of 19.7 μM and 18.7 μM, respectively (Figure 5I).
Figure 5. Acute administration of the peripherally-restricted GABAA receptor agonist isoguvacine improves tactile hypersensitivity in five genetic and one environmental model for ASD.

(A) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in mice following i.p. administration of either saline or 2 mg/kg midazolam treatment. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(B) Response to a light air puff stimulus alone in mice following i.p. administration of either saline or 2 mg/kg midazolam treatment. Responses are expressed as percent of startle response to a 125 dB noise. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05 for comparisons between mutant group with saline to control littermates with saline; #, p < 0.05, for comparisons between mutant group with saline to same mutant group with isoguvacine.
(C) Magnitude of startle response to a 125 dB noise in mice following i.p. administration of either saline or 2 mg/kg midazolam treatment. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05 for comparisons between mutant group with saline to control littermates with saline; #, p < 0.05, for comparisons between mutant group with saline to same mutant group with isoguvacine.
(D) Magnitude of startle response to a 125 dB noise in control mice following i.p. administration of saline, midazolam (2mg/kg) or a peripherally-restricted GABAA receptor agonist, isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(E-H) Liquid chromatography mass spectrometry (LC-MS) quantified isoguvacine concentrations in cerebrospinal fluid (CSF) (E), brain homogenate (F), plasma (G) or liver (H) samples of mice treated with: i.p. saline, i.p. isoguvacine (“Iso.”, 20 mg/kg, samples collected multiple time points post-injection), or intracerebral injection isoguvacine (20 mg/kg, 1 hour post-injection).
(I) Normalized peak chloride flux through α1β3γ2- and α2β3γ2-containing GABAA receptors in response to isoguvacine application. Response data has been normalized to the baseline peak current induced by addition of EC100 GABA (30μM) for 2 seconds during the assay
(J-L) GABAA receptor subunit RNA expression levels across peripheral somatosensory neuron subtypes, for the alpha (I), beta (J), and gamma (K) subunit types.
(M) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in control mice following i.p. administration of saline or isoguvacine at a range of concentrations. One-way ANOVA with post-hoc Sidak’s test [F (4, 113) = 8.005, P < 0.0001], above bars: *, p < 0.05 for comparisons between saline to each isoguvacine concentration; above brackets: *, p < 0.05, for comparisons between indicated conditions.
(N) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in mice following i.p. administration of either saline or 2 mg/kg isoguvacine. Two-way ANOVA with post-hoc Sidak’s test [F (7,277) = 16.44, P < 0.0001], *, p < 0.05 for comparisons between mutant group with saline to control littermates with saline; #, p < 0.05, for comparisons between mutant condition with saline to same mutant group with isoguvacine.
(O) Response to a light air puff stimulus alone in mice following i.p. administration of either saline or 2 mg/kg isoguvacine treatment. Responses are expressed as percent of startle response to a 125 dB noise. Two-way ANOVA with post-hoc Sidak’s test [F (7,277) = 9.650, P < 0.0001], *, p < 0.05 for comparisons between mutant group with saline to control littermates with saline; #, p < 0.05, for comparisons between mutant condition with saline to same mutant group with isoguvacine.
(P) Magnitude of startle response to a 125 dB noise in mice following i.p. administration of either saline or 2 mg/kg isoguvacine treatment.
We found that acute administration of isoguvacine (2 mg/kg, i.p.) reduced tactile reactivity in control, AdvillinCre; Mecp2f/y and AdvillinCre; Gabrb3f/+mutant mice (Figures 5M–O). These findings prompted us to ask whether this peripherally restricted GABAAR agonist may attenuate tactile over-reactivity in other ASD mouse models regardless of the pathophysiological mechanism of mechanosensory neuron dysfunction. Indeed, acute administration of isoguvacine also attenuated hairy skin hypersensitivity in Shank3, Fmr1, and Cntnap2 mutant mice (Figures 5N, 5O) as well as mice exposed to polyinosinic:polycytidylic acid (Poly I:C) at E12.5 (Figures 5N, 5O), which is a model of maternal immune activation-induced ASD in rodents (Choi et al., 2016). Of note, hairy skin hyposensitivity was observed in 16p11.2 deletion mutant mice (16p11.2+/−) and no alterations in tactile behaviors were observed in mice with a deletion spanning exons 4–9 in Shank3 (Shank3ex4−9, Figures S6I–N). Isoguvacine administration led to a further reduction in tactile PPI in 16p11.2+/−mice (Figure S6I). Consistent with a peripheral site of action, isoguvacine did not cause sedation in any animal cohorts tested, as measured by startle amplitude (Figure 5P).
Isoguvacine attenuates tactile sensitivity through reduced excitability of LTMRs
We next performed both in vitro and in vivo electrophysiological recordings to ask whether isoguvacine exerts its effects by acting directly on peripheral somatosensory neurons. Isoguvacine application reduced the hyperexcitability phenotype observed in large diameter neurons cultured from Mecp2 and Shank3 mutant mice, without affecting Ih (Figure S6A–G). We tested whether administration of isoguvacine would reduce mechanosensory neuron sensitivity in vivo by performing multi-unit electrode recordings in the L4 DRG of anesthetized mice (Figure 6A–E, See STAR Methods). Our findings indicate that while proprioceptor sensitivity and firing were unaffected, subcutaneous administration of isoguvacine increased response thresholds to light touch stimuli and reduced spiking in Aβ LTMRs in vivo (Figures 6F, 6G, S6P, S6Q). These results are consistent with our finding that while LTMRs exhibit high expression levels of GABAAR subunits, including GABRB3, proprioceptors do not (Figures 5J–L, S6H). To test whether the effects of isoguvacine on cutaneous LTMR firing properties and tactile sensitivity are mediated through its direct activation of GABAARs in somatosensory neurons, we next performed a similar set of in vivo DRG recordings in mice with somatosensory neuron specific, homozygous deletion of Gabrb3 (AdvillinCre; Gabrb3f/f) and control littermates (Gabrb3f/f). Since Gabrb3 encodes the principal obligatory beta subunit of the GABAAR in DRG neurons (Figures 5J–L, S6H), somatosensory neurons in AdvillinCre; Gabrb3f/f mutant mice are virtually devoid of GABAARs (Orefice et al., 2016; Zimmerman et al., 2019). Sixty minutes following subcutaneous injection of isoguvacine (2 mg/kg), tactile stimulus-evoked (brush or air puff stimulus) LTMR responsivity was decreased in control littermates, while no change in tactile-evoked LTMR responses were observed in control mice injected with saline (Figures 6F, 6G). Importantly, mice with somatosensory neuron specific homozygous deletion of Gabrb3 showed no changes in tactile-evoked LTMR spiking following administration of isoguvacine, indicating that isoguvacine exerts its effects by acting directly on GABRB3-containing GABAARs expressed on somatosensory neurons (Figures 6F–H). We next asked whether isoguvacine treatment would attenuate LTMR sensitivity in a mouse model in which deficits in GABAAR signaling is not the primary pathophysiological deficit. For this, Shank3B+/− and control littermates were subjected to the same in vivo DRG recording preparation. LTMRs from saline-injected Shank3B+/− mutant exhibited higher response rates to innocuous touch stimuli compared to LTMRs from control littermates injected with saline (Figures 6I, 6J), and isoguvacine attenuated firing in both Shank3B+/− and control littermates (Figures 6I, 6K, S6O). No differences in proprioceptor sensitivity were observed in any of the experiments (Figures S6P–R). Consistent with these electrophysiological measurements, mice with homozygous deletion of Gabrb3 in peripheral sensory neurons (AdvillinCre; Gabrb3f/f mice) exhibited no reduction in hairy skin sensitivity following isoguvacine treatment (Figures 6L, 6M). As above, startle amplitude was unaffected by isoguvacine (Figure 6N), indicating that isoguvacine was not sedating these mice. These findings indicate that a peripherally-restricted GABAAR agonist, isoguvacine, which attenuates tactile over-reactivity in several genetic and environmental ASD mouse models, exerts its effects through direct activation of GABAARs expressed on LTMRs.
Figure 6. Isoguvacine attenuates tactile sensitivity through reduced excitability of peripheral, low-threshold mechanosensory neurons.

(A) Diagram for in vivo dorsal root ganglion (DRG) multi-unit electrode recordings, showing tetrode placement into the left L4 ganglia.
(B) Example single unit identified during the spike sorting process. Average waveform at each electrode site.
(C) Example inter-spike interval for a single unit identified during the spike sorting process.
(D) Activity traces of two putative single units in response to a brush stimulus.
(E) Activity traces of a putative single unit in response to a light air puff stimulus (1 PSI).
(F) Representative activity raster plots for multiple putative LTMRs in multiple mice over the duration of a recording experiment in controls and AdvillinCre; Gabrb3f/f mice. Mice received a subcutaneous injection of either saline or isoguvacine (2 mg/kg) during the experiment, and activity of light-touch responsive units in response to a light brush stimulus was assessed over a 90-minute period. Arrows indicate time of injection.
(G) Relative firing frequency of LTMRs in response to a brush stimulus over the duration of each recording experiment, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3,136) = 9.326, P < 0.0001], *p < 0.05.
(H) Average baseline spike rate of LTMRs in response to an air puff stimulus (1 PSI), in control and AdvillinCre; Gabrb3f/f mice. Student’s t-test, *p = 0.490.
(I) Representative activity raster plots for putative LTMRs in multiple mice over the duration of recordings in controls and Shank3B+/− mice. Mice received a subcutaneous injection of either saline or isoguvacine (2 mg/kg) during the experiment, and activity of light-touch responsive units was assessed over a 90-minute period. Arrows indicate time of injection.
(J) Average baseline spike rate of LTMRs in response to an air puff stimulus (1 PSI), in control and Shank3B+/− mice. Student’s t-test, *p = 0.0291.
(K) Relative firing frequency of LTMRs in response to a brush stimulus over the duration of each recording experiment, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3, 216) = 22.69, P < 0.0001], *p < 0.05.
(L) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in control, AdvillinCre; Gabrb3f/+ and AdvillinCre; Gabrb3f/f mice following i.p. administration of 2 mg/kg isoguvacine (i.p., 2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05 for comparisons between mutant group to control littermates with saline; #, p < 0.05, for comparisons between mutant condition with saline to same mutant group with isoguvacine.
(M) Response to a light air puff stimulus alone in mice following i.p. administration of either saline or 2 mg/kg isoguvacine treatment. Responses are expressed as percent of startle response to a 125 dB noise. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05 for comparisons between mutant group to control littermates with saline; #, p < 0.05, for comparisons between mutant condition with saline to same mutant group with isoguvacine.
(N) Magnitude of startle response to a 125 dB noise in mice following i.p. administration of either saline or 2 mg/kg isoguvacine treatment. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
Chronic treatment with a peripherally-restricted GABAAR agonist improves some ASD phenotypes in two mouse models for ASD
Our results raised the possibility that peripherally-restricted GABAAR agonists may be useful for treating tactile over-reactivity, and potentially anxiety and social impairments in different ASD mouse models, if treatment is administered during early postnatal development. Therefore, we tested whether chronic treatment of Mecp2 and Shank3 germline mutant mice with isoguvacine beginning shortly after birth improves any of the ASD-related phenotypes commonly observed in adulthood. Following a six-week treatment regimen, isoguvacine was detected in the liver, but not brain or SC (Figure S7A). When isoguvacine-treated mice were first compared to saline-treated groups at weaning age (P21), we noticed an improvement in their overall body health, or phenotypic score (Figures 7A, 7B; See Star Methods). Moreover, while saline-treated Shank3B and Mecp2 mutant mice displayed reduced bodyweight at P21 compared to control littermates, isoguvacine-treated mutant mice were not different in weight from control mice (Figure 7C). Chronic isoguvacine administration also improved multiple ASD-associated behavioral phenotypes, including tactile over-reactivity, anxiety-like behaviors, social impairments, as well as PV+ neuron abnormalities in S1 and BLA and normalized E/I ratios in S1 of 8-week-old mice (Figures 7D–O, S7I–K, S7N–S, Table S6). Chronic isoguvacine administration however, did not improve texture discrimination deficits, increased acoustic PPI performance, memory impairments, PV+ neuron abnormalities in V1 or altered E/I ratios in V1 in either Shank3 or Mecp2 mutants (Figures S7C, S7E, S7H, S7T–V). Motor impairments were also not improved in Mecp2 mutants, nor was the overgrooming phenotype rescued in Shank3 mutants (Figures S7D, S7L, S7M). Taken together, treatment with a peripherally-restricted GABAAR agonist during early postnatal development may provide a therapeutic strategy for improving tactile over-reactivity and a subset of other key features of ASD, including anxiety, without causing sedation and other undesirable effects of activating brain GABAARs during development.
Figure 7. Chronic administration of isoguvacine improves tactile over-reactivity, region-selective cortical abnormalities, and some ASD-related behaviors in Mecp2 and Shank3 mutant mice.

(A) Representative images of P21 control and Mecp2C/y mutant mice treated daily from P1–21 with either saline or isoguvacine (2 mg/kg).
(B) Average phenotypic score of P21 Shank3B+/− or Mecp2C/y mutant mice and control littermates treated daily with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(C) Average bodyweight of P21 Shank3B+/− or Mecp2C/y mutant mice and control littermates treated daily with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(E) Response to a light air puff stimulus alone in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) Representative activity traces in the OF test.
(G) Percent time spent in the center of the OF chamber. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(H) Percent time spent in the open arms of the EPM. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(I-J) Preference index for the percentage of time spent investigating the novel mouse in the “Sociability” (I) or “Social Novelty Preference” (J) portion of the 3-chamber social interaction test. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Representative IHC images of coronal S1 brain sections, showing PV immunoreactivity in Shank3B+/− or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(L-M) Quantification of the number of PV-positive (PV+) neurons in S1 (L) and V1 (M). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(N) Representative traces showing sEPSCS (−70 mv hold) and sIPSCs (0 mV hold) from S1 slices from Shank3B+/− or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(O) Quantification of excitatory/inhibitory (E/I) ratio in S1 slices from control and mutant mice. Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
Discussion
Aberrant sensory reactivity is now regarded as a diagnostic feature of ASDs. The present study adds to a growing body of work demonstrating that somatosensory neurons are dysfunctional and contribute to behavioral phenotypes in a range of genetic and environmental models for ASD, including Mecp2, Gabrb3, Shank3, Cntnap2, Fmr1, and MIA mouse models [present study, (Bhattacherjee et al., 2017; Chen et al., 2014; Dawes et al., 2018; Han et al., 2016; Oginsky et al., 2017; Orefice et al., 2016; Perche et al., 2018; Price and Melemedjian, 2012)]. A main finding of the present work is that tactile over-reactivity in ASD models can arise from distinct cell-autonomous, pathophysiological mechanisms. Tactile over-reactivity may result from loss of GABAAR signaling and PSI in somatosensory neurons, as is the case for Mecp2 and Gabrb3 mutants, or loss of potassium channel function leading to somatosensory neuron hyper-excitability, as seen here in Shank3 mutants. It is noteworthy that mutations in Mecp2 and Shank3 may differentially affect sensitivity of LTMRs and small diameter nociceptive neurons, which may explain the seemingly paradoxical findings that at least some ASD patients can exhibit both hypersensitivity and aversion to light touch but also decreased responsiveness to noxious stimuli (Downs et al., 2010; Tomchek and Dunn, 2007). Consistent with this, reduced sensitivity to painful thermal and chemical stimuli is observed in mice with conditional deletion of Shank3 in sensory neurons (Han et al., 2016).
A second principle to emerge from the present work is that loss of either Mecp2 or Shank3 in peripheral sensory neurons leads to changes in neurochemical and functional properties of brain circuits. We found that PV+ interneurons in S1 and BLA are adversely affected in mice lacking Mecp2 or Shank3 in somatosensory neurons (Table S7). We speculate that alterations in the number of PV+ neurons or PV expression in cortex reflect homeostatic mechanisms for increasing inhibitory neuron response rates under conditions of enhanced sensory drive to the cortex and that these alterations differ depending on the developmental time point at which sensory drive is altered. This is consistent with prior findings that both increased PV+ neuron number and decreased PV expression are associated with enhanced inhibitory transmission and reduced cortical response rates to sensory stimuli (Volman et al., 2011; Vreugdenhil et al., 2003) as well as recent findings that changes in sensory cortex E/I balance observed in ASD models may reflect adaptations to altered sensory input from the periphery (Antoine et al., 2019). It is important to note, however, that while peripheral somatosensory neuron dysfunction contributes to altered cortical circuit development, loss of ASD-related genes within the brain is also likely to contribute to altered sensory cortex microcircuit function in ASD models. While our findings implicate Mecp2 and Shank3 function in peripheral sensory neurons for normal tactile, anxiety-like and some social behaviors, we also emphasize that loss of Mecp2 or Shank3 in peripheral sensory neurons does not recapitulate all ASD behavioral phenotypes observed in the germline mutation models. This includes memory impairments, motor deficits, respiratory function and early lethality in Mecp2 mutants, and memory impairments and overgrooming behaviors observed in Shank3 mutants.
A third general finding of the present work is the extent to which sensory neuron dysfunction contributes to aberrant behavior in disparate ASD models varies greatly with respect to the timing or developmental onset of sensory dysfunction. While early developmental restoration of either Mecp2 or Shank3 function in sensory neurons improves hairy skin sensitivity, some aspects of social behavior, and anxiety-like behaviors, P28 restoration improves hairy skin sensitivity and sociability but has no effect on social novelty recognition preference and anxiety-like behaviors. This is consistent with prior studies in which global restoration of Shank3 at P21 was sufficient to normalize sociability behaviors but not anxiety-like behaviors (Mei et al., 2016; Wang et al., 2017).
Our findings lead us to propose a pharmacological approach to augment GABAA receptor signaling in LTMRs during early postnatal development as a novel therapy for ASD. The logic behind the “LTMR GABAAR hypothesis” is based on the following observations: 1) ASD gene dysfunction in mechanosensory neurons causes altered physiological properties of LTMRs, including hypersensitivity and reduced PSI in the SC, which contribute to tactile over-reactivity in ASD mouse models (present study) (Orefice et al., 2016); 2) GABAARs are present all along myelinated axons of peripheral nerves (Zeilhofer et al., 2012), and peripheral release of GABA controls somatosensory neuron sensitivity (Carlton et al., 1999; Hanack et al., 2015; Obradovic et al., 2015); 3) the ASD-associated gene Gabrb3, which encodes the principal obligatory beta subunit of GABAARs in DRG neurons in both mice (present study) and humans (Flegel et al., 2015; Ray et al., 2018), functions cell autonomously in LTMRs to control tactile sensitivity (Orefice et al., 2016, present study); 4) GABA acts directly on DRG neurons to reduce excitability in vitro (present study) (Du et al., 2017); 5) administration of the peripherally-restricted GABA analog, isoguvacine, attenuates LTMR firing properties and tactile sensitivity in vivo in a manner that is dependent on GABAARs present on LTMRs (present study); 6) isoguvacine normalizes tactile over-reactivity in five genetic and one environmental model for ASD (present study); and 7) chronic treatment with isoguvacine beginning at early postnatal ages in two distinct models for ASD improves overall body condition, body weight, PV+ interneuron alterations in S1 and BLA, E/I ratios in S1, as well as anxiety-like behaviors and some social impairments in young adult mice (present study). In line with the LTMR GABAAR hypothesis, other new treatment strategies that show promise for ASD may also work through affecting peripheral nerve function, including bumetanide, which is largely peripherally-restricted (He et al., 2018; Romermann et al., 2017). We propose that a benefit of peripheral restriction is to avoid complications associated with direct brain actions and adverse effects on brain development. Consistent with this, peripheral administration of isoguvacine failed to penetrate the brain to an appreciable extent and did not lead to sedation, as compared to brain-penetrating GABAAR PAMs. Thus, we propose that GABAAR agonists, GABA reuptake inhibitors, or GABAAR PAMs that are peripherally-restricted may reduce tactile over-reactivity and improve brain microcircuit function and related ASD behaviors observed in certain patients with ASD, while minimizing or avoiding entirely potentially detrimental effects on brain development observed in clinical use of classical, FDA-approved GABAA drugs (Kodish et al., 2011), all of which penetrate the BBB (Groeneveld et al., 2016). Peripherally restricted methods for augmenting GABAAR signaling may also have applicability in other diseases and disorders in which touch over-reactivity is present, such as mechanical allodynia in neuropathic pain states, sensory processing disorder, and schizophrenia. Whether acute treatment of ASD patients with peripherally-restricted compounds to augment GABAAR signaling normalizes tactile over-reactivity, and whether chronic treatment beginning at early ages ameliorates some ASD-associated behaviors await the development of safe, peripherally-restricted compounds and their testing in patient trials.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Ginty (david_ginty@hms.harvard.edu).
Experimental Model and Subject Details
All procedures performed in this study were approved by the Harvard Medical School Institutional Animal Care and Use Committee (IACUC). Male and female mice of mixed genetic backgrounds (C57BL/6J, 129/SvEv, CD1) were used for these studies. The only exceptions were germline mutant mice (Mecp2R306C, Shank3B+/− and Shank3Δex4−9), which were backcrossed for at least 5 generations to a C57BL/6J background. Testing was done beginning at 6 weeks of age, and in most cases, behavioral testing was completed by 8 weeks of age. All behavioral testing performed in female mice was completed prior to the start of estrous cycles. The ages of mice used for histological and electrophysiological experiments are denoted in the appropriate Method Details and relevant Figure Legends. Mice were weaned and ear notched for genotyping at P21 (+/− 2 days). All animals were group housed, with control and mutant animals in the same litters and cages. Littermates from the same genetic crosses were used as controls for each group, to control for variability in mouse strains/backgrounds. No differences were observed between wild type animals and any single floxed or Cre control groups for any of the tests performed. Male and female mice of the same genotype were first analyzed separately to assess potential sex-related differences in behaviors. If no differences were observed, male and female mice of the same genotype were grouped together for final analyses. Behavioral and histological differences were found between hemizygous null male Mecp2C/y and heterozygous female Mecp2R/C mice. For this reason, these two groups were analyzed separately and compared to male and female littermate controls.
Generation of Shank3 conditional knockout mouse
The targeting vector flanked Exons 13–16 (PDZ domain) of the Shank3 gene with loxP sites and a NEO cassette. Chimeric mice were crossed to C57Bl/6J females from Jackson Labs. Germline transmission was assessed through genotyping PCR of mouse tail DNA, using primers pFW (Sh3cKO) Gen 1a and pRV (Sh3cKO) Gen 3b for the wild-type allele (positive band = 188 base pairs) and the floxed allele (positive band = 228 base pairs). The F1 hybrids were crossed to C57Bl/6J β-Actin Flp mice to excise the NEO cassette. The floxed mice were then backcrossed to C57Bl/6J mice for 5 generations. After the 5th generation, speed congenic genotyping PCRs were conducted to determine the approximate purity of the background. Only mice showing >95% C57Bl/6J background were used for subsequent matings. Backcrossed Shank3floxed mice were then bred with C57Bl/6J β-Actin Cre mice to produce germline knockouts of the floxed allele (termed Shank3floxed mice). Genotypes were determined by PCRs using the pFW (Sh3cKO) Gen 1a and pRV (Sh3cKO) Gen 3b for the knockout allele (positive band = 290 base pairs). pFW (Sh3cKO) Gen 1a forward primer: 5’- CAG CAT TTA TAC CTG ACT GTG AAG C −3’; reverse primer: 5’- GGG AGT AGA GCT CAG ATA ACC −3’.
Mouse Lines and Genotyping
Mice were group housed with littermates in standard housing on a 12-hour light/dark cycle. Tail biopsies were taken at weaning (P21, +/− 2 days), which were used for genetic identification.
Shank3B null mice were obtained from the Jackson Laboratory (017688) and were previously described (Peca et al., 2011). The following primers were used to identify the null allele: common forward 5’- GAG ACT GAT CAG CGC AGT TG −3’; wild type reverse 5’- TGA CAT AAT CGC TGG CAA AG −3’; mutant reverse 5’- GCT ATA CGA AGT TAT GTC GAC TAG G −3’.
Shank3Δex4−9 null mice were obtained from the Jackson Laboratory (017890) and were previously described (Bozdagi et al., 2010). The following primers were used to identify the null allele: common forward 5’- TGG GAT GTG AGA GTG ACC AG-3’; wild type reverse 5’- AGG AGG TCA GTG GCG TTG T −3’; mutant reverse 5’- AAT TCT GTT CAG TCC ACA CAG G −3’.
Fmr1 null mice were obtained from the Jackson Laboratory (003025) and were previously described (Bakker, 1994). The following primers were used to identify the null allele: mutant forward 5’- CAC GAG ACT AGT GAG ACG TG −3’; wild type forward 5’- TGT GAT AGA ATA TGC AGC ATG TGA-3’; common reverse 5’- CTT CTG GCA CCT CCA GCT T −3’.
16p11.2df null mice (16p11.2) were obtained from the Jackson Laboratory (013128) and were previously described (Horev et al., 2011). The following primers were used to identify the null allele: mutant forward 5’- ACC TGG TAT TGA ATG CTT GC −3’; wild type forward 5’- CCT GAG CCT CTT GAG TGT CC −3’; mutant reverse 5’- TGG TAT CTC CAT AAG ACA GAA TGC −3’; wild type reverse 5’- GTC GGT TCA GGT GGT AGA CG −3’.
Hcn1 null mice were obtained from the Jackson Laboratory (028301) and were previously described (Nolan et al., 2003). The following primers were used to identify the null allele: mutant forward 5’- AGA GAA ATC ATT CCC CGT GA-3’; wild type forward 5’- CAC CTG CTA CGC AAT GTT TG −3’; common reverse 5’- ATT GGG CAC TAC ACG CTA GG −3’.
Shank3FX mice were previously described (Mei et al., 2016). The following primers were used to identify the mutant allele: for the wildtype allele, forward 5′ - CGT TTG ACA CAC ATA AGC ACC −3′ and reverse 5′- CTC CAC CTA GCT GAA TTT CCC −3′ were used to produce a band of 340 bp. For the knockout (FX) allele, forward 5′- CGT TTG ACA CAC ATA AGC ACC-3′ and reverse 5′- GCT GAC ATC ACA TTG CTG CC −3′ were used to produce a band of 481 bp. For the rescue allele, forward 5′- CGT TTG ACA CAC ATA AGC ACC −3′ and reverse 5′- CTC CAC CTA GCT GAA TTT CCC −3ƍ) were used to produce a band of 408 bp.
Mecp2R306C mice were obtained from Michael Greenberg and were previously described (Lyst et al., 2013). The following primers were used to identify the mutant allele: forward 5’-GGA TTG TGG AAA AGC CAG-3’; and reverse 5’-ATG ACC TGG GCA GAT GTG GTA G-3’.
Mecp2 floxed mice were obtained from the Jackson Laboratory (006847). The floxed Mecp2 sequence was identified using the following primers: forward 5’-TGG TAA AGA CCC ATG TGA CCC AAG-3’ and reverse 5’-GGC TTG CCA CAT GAC AAG AC-3’. The following primers were used to identify the null allele (post Cre excision), as well as to check for off-target Cre expression in AdvillinCre mice: forward 5’-TGG TAA AGA CCC ATG TGA CCC AAG-3’; and post-Cre excision reverse 5’-TCC ACC TAG CCT GCC TGT ACT TTG-3’.
Mecp2STOP mice were obtained from the Jackson Laboratory (006849) and were previously described (Guy et al., 2007). The following primers were used to identify the mutant allele: forward common 5’- AAC AGT GCC AGC TGC TCT TC-3’; wildtype reverse 5’- CTG TAT CCT TGG GTC AAG CTG −3’ and mutant reverse 5’- GCC AGA GGC CAC TTG TGT AG −3’.
Gabrb3 floxed mice were obtained from the Jackson Laboratory (008310) and were previously described (Ferguson et al., 2007). The floxed Gabrb3 sequence was identified using the following primers: forward 5’- ATT CGC CTG AGA CCC GAC T −3’ and reverse 5’- GTT CAT CCC CAC GCA GAC −3’.
Cntnap2 null mice were obtained from the Jackson Laboratory (028635) and were previously described (Gordon et al., 2016). The mutant Cntnap2 sequence was identified using the following primers: wildtype forward 5’- TGC CCT CCT AGA AAG TAA ATG C T-3’; mutant forward 5’- GCC AGA GGC CAC TTG TGT AG −3’ and common reverse 5’- TCC TCT CTT CAT GCA CAC TAT GA −3’.
AdvillinCre mice were obtained from Fan Wang (Duke University) and were previously described (Hasegawa et al., 2007). The AdvillinCre transgene was identified using the following primers: 5’- CCC TGT TCA CTG TGA GTA GG −3’; reverse 5’- AGT ATC TGG TAG GTG CTT CCA G −3’; and internal control 5’- GCG ATC CCT GAA CAT GTC CAT C −3’.
AdvillinCreERT2 mice were obtained from John Wood and previously described (Lau et al., 2011). The AdvillinCreERT2 transgene was identified using the following primers: 5’- CCC TGT TCA CTG TGA GTA GG −3’; 5’- AGT ATC TGG TAG GTG CTT CCA G −3’; and 5’- GCG ATC CCT GAA CAT GTC CAT C −3’.
Cdx2Cre mice were obtained from Eric Fearon, and previously described (Akyol et al., 2008). The Cdx2Cre transgene was identified using the following primers: forward 5’- CTC GAC GTC TCC AAC CAT TG - 3’; and reverse 5’-ATC TTC AGG TTC TGC GGG AA −3’.
Proper expression of each floxed allele using each Cre transgene was assessed using PCR and histological verification. AdvillinCre or AdvillinCreERT2 animals with post-Cre excision expression in tail biopsy tissue or animals with transgene expression and recombination in the spinal cord or brain were excluded from analyses.
METHOD DETAILS
Generation and use of AAV.hSYN.FLEx.GABRB3.t2A.mCHERRY
AAV cloning and production: cDNA of mouse Gabrb3 was purchased from GE Healthcare. Gabrb3 cDNA and T2A-mCherry were cloned into an AAV2.9 plasmid that contains the hSyn-FLEx cassette (Addgene #84481) by In-Fusion cloning kit (Takara). AAV particles were produced by The Boston Children’s Hospital Viral Core, with a titer of 8E+12 vg/mL. 2 μl of AAV.hSYN.FLEx.GABRB3.t2A.mCHERRY (‘AAV.FLEx.GABRB3.mCHERRY’) were diluted into 18 μl of saline, and then injected i.p. into each pup at both P5 and P6. Pups were returned to their home cage each day following injections. Mice harboring an arginine-to-cysteine missense mutation in Mecp2 (Mecp2R306C), which is a common allele found in patients with Rett Syndrome (RTT) (Lyst et al., 2013), were used in these studies because Mecp2R306C mutant mice live longer than mice harboring the Mecp2 null allele, allowing a full two-week behavioral assessment at six to eight weeks of age prior to the development of severe breathing abnormalities. Mecp2R306C littermate mice lacking the AdvillinCre allele and injected with AAV.FLEx.GABRB3.mCHERRY served as a control, as did wildtype littermates with and without the AdvillinCre allele. We found that i.p. injection of AdvillinCre; Mecp2R306C mice, but not Mecp2R306C mice lacking the AdvillinCre allele, with AAV.FLEx.GABRB3.mCHERRY led to transduction of all types of DRG sensory neurons, a large fraction of which are LTMRs, and a dramatic increase in the amount of GABRB3 puncta at presynaptic terminals of Aβ-LTMRs and Aδ-LTMRs in the spinal cord (SC) dorsal horn (Figures 4A–C, S5A, S5B).
Tamoxifen Administration
Tamoxifen was administered to AdvillinCreERT2 mice to allow for the excision of floxed alleles (either Shank3f, Shank3FX, Mecp2f or Mecp2STOP) in peripheral somatosensory neurons of adult mice. Intraperitoneal injections of tamoxifen (1 mg per day, Toronto Research Chemicals) were administered to mice for 5 consecutive days, from either P5–9, P10–14 or P28–32. All mice in this study shown for AdvillinCreERT2 mouse lines, including AdvillinCreERT2 and floxed allele controls in these groups, received this tamoxifen regimen, and no changes in health or behaviors were observed in either sets of controls compared to non-tamoxifen treated animals. A five-day tamoxifen dosing regimen resulted in recombination of target genes in >90% of DRG neurons, while Shank3 and Mecp2 gene expression was unaltered in the brains or spinal cord of these mice (Figures S4C–E).
Poly I:C Administration
Administration of polyinosinic: polycytidylic acid (Poly I:C) was performed as previously described (Shin Yim et al., 2017). Timed-pregnant female mice were ordered from Charles River, and were verified to be positive for segmented filamentous bacteria. E12.5 pregnant female mice were weighed and injected with a single dose of poly(I:C) (20mg/kg i.p., Millipore) or saline as a vehicle control. Each dam was returned to its cage and left undisturbed until the birth of its litter.
Acute Drug Treatment to Assess Tactile Sensitivity
Mutant mice and their control littermates were subjected to two consecutive days of tactile PPI testing (see below for behavioral testing). Animals received i.p. injection of either vehicle or drug (midazolam 2mg/kg or isoguvacine 2mg/kg). After a mouse was injected, it was returned to their home cage for 30 minutes until they were subjected to tactile PPI. Litters were counterbalanced for treatment, such that half of the animals received saline on the first day and the other half of the litter received drug treatment on the first day. The following day, treatment regimens were reversed, and the mice were subjected to a second round of tactile PPI testing. For mutant animals of each line, percent PPI data are represented as percent of control littermate performance for saline treatment.
Chronic Isoguvacine Administration
Mecp2R306C and Shank3B+/− mice were chosen for these analyses because both are well-established models for ASD that exhibit behavioral abnormalities with high penetrance and severity. Shank3B+/− or Mecp2R/C female mice were bred to male c57Bl6/J mice. Once females were visibly pregnant (E17–20), they were singly-housed. On the day after each litter was born (P1), pups were weighed and administered i.p. injection of either isoguvacine (2mg/kg) or saline control. Pups were weighed and injected every day thereafter with the same treatment, ending on P42. At P21, pups were weighed, photographed, assessed for overall phenotype, and weaned. Females were counterbalanced such that half of the females had litters that received isoguvacine first, while the other half of the females’ litters received saline treatment first. Whole litters received the same treatment regimen. Once a litter was weaned, mothers were re-mated with the same c57Bl6/J male mouse, and the treatment condition of the subsequent litter was reversed. At P42, mice began behavioral testing.
Behavioral Testing
Male and female mice of mixed genetic backgrounds (C57BL/6J, 129/SvEv, CD1) were used for behavioral analyses. The only exceptions were germline mutant mice (Mecp2R306C, Shank3B+/− and Shank3Δex4−9), which were backcrossed for at least 5 generations to a C57BL/6J background. Testing was done beginning at 6 weeks of age, and in most cases, behavioral testing was completed by 8 weeks of age. All behavioral testing performed in female mice was completed prior to the start of estrous cycles. Mice were weaned and ear notched for genotyping at P21 (+/− 2 days). All animals were group housed, with control and mutant animals in the same litters and cages. Littermates from the same genetic crosses were used as controls for each group, to control for variability in mouse strains/backgrounds. No differences were observed between wild type animals and any single floxed or Cre control groups for any of the tests performed. Male and female mice of the same genotype were first analyzed separately to assess potential sex-related differences in behaviors. If no differences were observed, male and female mice of the same genotype were grouped together for final analyses. Prior to behavioral testing, animals were brought into the procedure room and allowed to habituate to the room for 30 minutes. Animal numbers per group for behavioral tests are indicated in figures and/or supplemental tables. All behavioral analyses were performed and analyzed by experimenters who were blinded to genotype and treatment.
Ear Notching
Ear notching was employed as the method of identification for all animals used in behavioral experiments, as toe tagging or metal ear tags can affect animals’ ambulatory behavior. Ear notching was performed using an ear punch device (Kent Scientific) on mice at weaning age (3 weeks of age). For this, mice were restrained by the scruff and the ear punch was used to create holes and/or notches in the ears, following the “Universal Mouse Numbering System”. This method of identification allowed experimenters to identify mice without unnecessary handling of the mouse.
Whisker Plucking
To avoid the confound of whisker sensing by mice during NORT experiments, bilateral removal of all mystacial pad vibrissae was performed 3 days prior to the start of experiments. Mice were placed under isoflurane anesthesia and all mystacial vibrissae were plucked with tweezers. Whisker removal caused a small but significant increase in grooming time during a 20-minute open field test, but no differences in speed of movement, ambulatory time or jump time were observed following whisker removal (Orefice et al., 2016). Animals with and without whiskers also performed comparably on the textured NORT test, but removal of whiskers did promote object investigation using the glabrous skin on paws over whisking/nose poke investigations (Orefice et al., 2016). Whisker removal also caused no changes in anxiety-like behavior, as measured by time spent in the open arms of an elevated plus maze (Orefice et al., 2016). Whiskers begin to regrow 8–12 days after plucking. NORT was completed in the first week following whisker removal, and therefore whiskers were only plucked once. Whiskers were allowed to regrow thereafter, and animals had regrown whiskers by the time social behavior tests were performed.
Open Field Test
On the first day of behavioral testing, animals were subjected to the open field test. During this test, an animal was placed in an empty plexiglass arena (40 cm × 40 cm × 40 cm) and allowed to explore for 10 minutes, under dim lighting. The outside walls of the chamber were opaque. The position of the mouse was tracked using custom Matlab scripts. A portion of videos were blindly hand-scored to verify accuracy. Distance traveled and time spent away from walls (time in center) were analyzed per 10-minute video.
Novel Object Recognition Test (NORT)
NORT testing was performed as previously described (Orefice et al., 2016). NORT was performed in the same plexiglass arena used for open field testing. The cube-shaped objects were designed to promote climbing, which enhances novel object recognition performance requiring texture discrimination via glabrous skin on the paws (Heyser and Chemero, 2012). In this test, mice were first habituated to an open field chamber by allowing free exploration of an empty chamber for 10 minutes during each of two consecutive days (Figure 1D). The first day of habituation was used as the open field test (see above for details.)
Each of the three subsequent testing days included two sessions. In the first session (learning phase), the mouse was placed in the testing arena, equidistant to, and facing away from, the two identical objects placed in the center of the arena. The two objects were positioned equidistant from the center of the arena, and equidistant from the walls of the arena. Each mouse was allowed to explore the objects for 10 minutes. Animals were then removed from the testing arena and placed in the home cage for 5 minutes. During this time, the arena was cleaned with 70% ethanol, and one of the objects was replaced with a novel object. The mouse was then placed back into the chamber and allowed to explore objects for 10 minutes (testing phase). The amount of time the mouse spent physically investigating (touching) each of the objects was assessed during both the learning and testing phases. If an animal did not physically touch both objects during the learning phase, it was excluded from NORT analysis.
Animals were also subjected to a ‘control’ NORT on the third day, where the test objects differed in color and shape, but not texture (5 minute shape NORT). In the learning phase of this assay, the animal was presented with two objects of identical texture, color and shape and was allowed to freely explore the objects for ten minutes. After a 5 minute retention period in a the home cage, the animal was placed back into the test chamber, with one of the objects exchanged for a novel object of a different color and shape. Since mice prefer novel stimuli, an animal that can discriminate between the textures on the objects spends more time investigating the novel object (Heyser and Chemero, 2012), whereas an animal that does not discriminate novelty is expected to investigate the objects equally.
On the fourth day, a texture-specific NORT (textured NORT) was performed: in the learning phase, the animal was presented with two cubes of identical texture and was allowed to freely explore the objects for ten minutes. After a 5 minute retention period in a separate holding cage, the animal was placed back into the test chamber, with one of the objects exchanged for a novel textured object. Since mice prefer novel stimuli, an animal that can discriminate between the textures on the objects spends more time investigating the novel textured object (Heyser and Chemero, 2012), whereas an animal that cannot discriminate between the textures is expected to investigate the objects equally (Orefice et al., 2016).
Lastly, a second control NORT was implemented on the fifth day, but with a one hour retention period between exploration and testing phases to assess memory performance (1 hour retention NORT).
For textured NORT, one object was replaced with a second, novel object that differed only in texture. Textured objects (either smooth or rough) were 4 cm on each side and constructed of plexiglass. All sides of the textured objects had ridges etched into the plexiglass using a laser cutter. For rough objects these ridges faced outwardly, while for smooth objects, the ridges pointed inward so that the objects appear visually identical but differed only in their textures. For the 5 minute shape and 1 hour retention versions of NORT, small wooden blocks were utilized. Blocks differed in shape, size and color, but overall volumes were similar (~6 cm × 3 cm × 3 cm). For each test, the placement and object used as the novel object was pseudorandomized and counterbalanced between groups.
To avoid confounding whisker movements and sensations, whiskers were plucked three days prior to the start of habituation (see above section on whisker plucking). Under these conditions, animals showed no adverse behavioral effects and used glabrous skin on their paws during more than 90% of object exploration (Orefice et al., 2016).
The position of the mouse was tracked using custom Matlab scripts. Whisking, nose pokes and investigation using the paws were all included in the time spent investigating objects, though over 90% of the time investigating objects was performed using the glabrous skin on paws (Orefice et al., 2016). All three versions of NORT were analyzed using the same object exploration criteria. All groups exhibited a preference for the novel object during the 5 minute color/shape NORT, and the animal numbers per group are the same as the animal numbers for the textured NORT.
Startle Reflex/Prepulse Inhibition (PPI)
PPI testing was performed as previously described (Orefice et al., 2016). The response of mice to tactile and acoustic startle stimuli, and the ability of a tactile or acoustic stimulus to inhibit startle to a loud acoustic stimulus was measured using a San Diego Instruments startle reflex system (SR-LAB™ Startle Response System). While the startle response to tactile stimuli has been previously investigated (Logue et al., 1997; Taylor et al., 1991; Torkamanzehi et al., 2008), we sought to determine whether mice exhibit specific tactile sensorimotor gating deficits by employing a PPI assay in which the pre-stimulus is an air puff (0.9 PSI, 50ms), followed by a startle tone stimulus (125 dB, 20ms). Air puffs were administered to the back of the mouse to assess hairy skin sensitivity. The 0.9 PSI prepulse stimulus strength was chosen because control animals showed a minimal, but statistically significant response to the stimulus alone, compared to baseline movement in the chamber without any stimulus (average response in controls, 5.89% +/− 2.15). In addition, this prepulse intensity was chosen because it produced significant inhibition of startle reflex at multiple interstimulus intervals (ISIs). A tone pre-stimulus (ranging from 68 dB to 80 dB, for 20ms), followed by startle tone stimulus (120 dB, 20ms) version of the PPI assay (acoustic PPI) was done as a control.
For testing sessions, animals were placed into a ventilated, cylindrical holder on a platform within a soundproof chamber. Protocols consisted of an acclimation phase, block I, block II, block III and block IV trials. Each time an animal was tested, it first underwent an acclimation phase to acclimate to the animal holder, startle box and background noise. Each mouse was placed in the chamber for a 5 minute acclimation period, during which constant background noise of broadband white noise was presented. Background noise for the acoustic PPI testing sessions was 65 dB. Background noise for the tactile PPI testing sessions was increased to 75 dB, to ensure that that the animal could not hear the air puff prepulse.
Block I consisted of 5 startle stimuli alone (120 or 125 dB broadband white noise for acoustic and tactile PPI testing sessions, respectively), to measure the initial startle reflex. Block II consisted of 5 prepulse stimuli alone (either 80 dB broadband white noise or a 0.9 PSI air puff for acoustic and tactile PPI testing sessions, respectively), to measure response to the prepulse stimulus alone. Block III incorporated prepulse/pulse, pulse alone and no stimulation trials that were pseudorandomized. Block IV consisted of 5 startle stimuli alone, to measure habituation to the startle stimuli over the testing session. Inter-trial intervals were varied from 10 seconds to 50 seconds (average 30 seconds). Acoustic PPI and tactile PPI sessions were run on separate days.
For acoustic PPI, the prepulse was 20ms in duration and presented 100 ms before the startle pulse (100 ms ISI). For tactile PPI, the prepulse intensity remained constant (0.9 PSI, 20 ms), and the ISI was varied from 50 ms to 1 second in duration. Whole body flinch, or startle reflex, was quantified using an accelerometer sensor measuring the amplitude of movement of the animal within the cylindrical holder.
Elevated Plus Maze
The elevated plus maze was used to measure anxiety-like behavior (Moy et al., 2007). The elevated plus maze was a custom-built acrylic chamber. The arms were each 30 cm long × 5 cm wide, with two of the opposing arms having walls that were 15 cm high. The maze stood 40 cm above the ground. Testing occurred in a dimly lit room with a camera overhead for tracking animal movement. The animal was placed into the center of the maze and allowed to freely explore for 10 minutes. The time spent in either the center, closed arms or open arms of the maze during each test session was quantified using a custom Matlab script. The percentage of time each mouse spent in the open arms, compared to the closed arms and center of the chamber was calculated and expressed as a percent time spent in the open arms of the maze.
3-Chamber Social Interaction Test
The 3-chamber social interaction assay is a commonly used method of assessing both sociability and social recognition/preference in rodents, and is often used to measure ASD-like behavioral deficits in mice (Moy et al., 2007). Testing occurred in three sessions within a three-chambered box with openings between the chambers (each compartment is 20 cm wide × 40 cm long × 22 cm high). The outside walls of the chamber were opaque, while the inner dividers were clear plexiglass. After a 5 minute habituation period in the empty chamber, the test mouse was moved into the empty center chamber with partitions in place. A wire mesh cup containing a novel mouse was placed on one side of the chamber and an empty mesh cup was placed on the other side during the “sociability” session. The partitions were then lifted, and the test mouse was free to explore all three sections of the chamber. After 10 minutes, the test mouse was moved into the empty center chamber with partitions in place. A second novel mouse was placed under the empty cup, partitions were removed, and the test mouse was allowed to freely explore the chamber during the social novelty recognition preference session (“Social Novelty Preference”) for 10 minutes. The time spent in each of the three chambers during each test session was quantified using custom Matlab scripts. Control mice show a significant preference for the chamber containing the novel mouse in each portion of the test, indicating a preference for novel mice compared to novel objects as well as a preference (and recognition of) a novel mouse versus a familiar mouse.
Marble Burying
The marble burying test is used to measure both restricted repetitive behaviors and neophobia in mice (Jaramillo et al., 2017). Each mouse is placed into the center of a separate, clean cage with 2 inches of normal bedding and 12 marbles on top of the bedding arranged in three even rows. Mice are left alone in these individual cages for 20 minutes, during this time mice typically explore the new environment and dig in the bedding ultimately burying marbles in the process. After 20 minutes, the experimenter re-entered the room and removed each mouse from its testing cage. Images of each cage were acquired, and the number of marbles buried (denoted by greater than 2/3 of the marble not being visible) was quantified per mouse.
Phenotypic Scoring
Mice were observed for indicators of general well-being such as coat condition, eyes, and body stance as previously described (Guy et al., 2007). A score of ‘0’ indicated a clean shiny coat, clear eyes and normal stance. A score of ‘1’ indicated the appearance of dull eyes, dull/ungroomed coat and somewhat hunched stance. A score of ‘2’ denoted narrowed or crusted eyes, piloerection and a hunched posture. A score of ‘3’ indicated severe eye narrowing, profound piloerection or loss of fur, and severely hunched posture.
Immunohistochemistry
For histology, mutant and control littermates were sacrificed in pairs or groups. Mice (P42–P84) were anesthetized with isoflurane and transcardially perfused with 20 mL of PBS, following by 25 mL of 4% paraformaldehyde (PFA) in PBS at room temperature (RT). Brains, spinal cords and dorsal root ganglia (DRG) were dissected from perfused mice and post-fixed overnight in 4% PFA at 4°C. Tissue was then washed 3 times in 1X PBS for 1–2 hours each, then brain, spinal cord and attached DRGs were finely dissected out of the vertebral column. Tissue was cryoprotected in 30% sucrose in 1X PBS overnight at 4°C, then embedded in NEG50 and frozen at −20°C. Tissue was stored at −70°C until it was sectioned at 30ȝm using a cryostat.
Sections on slides were dried at RT for 1 hour, then washed 3×10 minutes in 1X PBS. Tissue was then blocked for 1–2 hours in 1X PBS containing 0.3% Triton-X 100 and 5% normal donkey or goat serum (Jackson Immuno, 005-000-121). Sections were incubated with primary antibodies diluted in blocking solution (5% normal donkey serum, no detergent) at 4°C for 1–2 days. After which, sections were washed with 1X PBS containing 0.02% Tween 20 (PBST) and then incubated with secondary antibodies diluted 1:500 in blocking solution, at room temperature for 2 hours. IB4 was diluted at 1:500 and applied with secondary antibodies when needed. Following secondary antibody incubation, tissue was washed 3×10 minutes with PBST. One wash of PBST contained a 1:10,000 dilution of Hoechst solution. Sections were washed one more time with PBST, then mounted with fluoromount-G (Southern Biotech). Primary antibodies used: mouse anti-Shank3 (ab193307, Abcam, 1:1000); guinea pig anti-vGLUT1 (AB5905, Millipore, 1:1000); rabbit anti-HCN1 (APC-056, Alomone Labs, 1:500); rabbit anti-KCNQ2 (ab22897, Abcam, 1:500); goat anti-GABRB3 (PA5–19060, Life Technologies, 1:250); rabbit anti-PARVALBUMIN (PV-27, Swant, 1:1000); goat anti-PARVALBUMIN (PVG-213, Swant, 1:1000); rabbit anti-MECP2 (provided by Michael Greenberg, 1:1000); mouse anti-GAD67 (MAB5406, Fisher Scientific, 1:1000); rabbit anti-dsRed (632496, Clontech, 1:1000); and Alexa 647-conjugated IB4 (I6520–100, Life Technologies, 1:1000). Secondary antibodies include: Alexa 488, 546 or 647 conjugated donkey or goat anti-mouse, rabbit, goat or guinea pig. All secondary antibodies were purchased from Life Technologies or Jackson Immuno Research and used at a 1:500 dilution.
Puncta Analysis
Z-stack images of spinal cord slices were taken on a Zeiss LSM 700 confocal microscope using a 63X oil-immersion lens (Zeiss; Plan-Apochromat 63X/NA 1.40). Images were taken in lamina III/IV of the dorsal horn, which was identified by immunostaining with IB4 to delineate lamina IIi. The percent of SHANK3, GABRB3, HCN1 or KCNQ2 puncta that were colocalized with vGLUT1 puncta were analyzed using NIH ImageJ software with a custom ImageJ plugin (Schneider et al., 2012). Colocalization was analyzed per 0.5-micron section thickness of tissue, with a total of ten continuous sections. At least three images from three to seven animals per genotype were analyzed for each experiment. Experimenters were blind to genotype and treatment conditions during both experiment and analysis portions of the experiments.
Parvalbumin+ Neuron Analyses
Z-stack images of coronal brain sections were taken on a Zeiss LSM 700 confocal microscope using a 63X oil-immersion lens (Zeiss; Plan-Apochromat 10X/NA 0.45 and 20X/NA0.8). Images were taken in trunk primary somatosensory cortex (S1), primary visual cortex (V1), and basolateral amygdala (BLA). Cell bodies of parvalbumin-positive (PV+) neurons in each of these regions were traced using ImageJ. The number, size and fluorescence intensity of PV+ neurons in 1 mm2 region were analyzed per image, and 2–3 images were analyzed per region of interest. Three to seven animals were analyzed per genotype per experiment, by an experimenter blind to both genotype and treatment conditions.
Cultured Dorsal Root Ganglia Electrophysiology
6–8 week old mice were euthanized via CO2 narcosis, and dorsal root ganglia (DRGs) were rapidly dissected out on ice. DRGs were digested in collagenase (2 mg/mL)/dispase (5 mg/mL) solution at 37°C with constant rotation for 45 minutes. Following enzymatic digesting, tissue was centrifuged, washed three times with 1X HBSS and then mechanically triturated using a glass Pasteur pipette. Dissociated cells were plated onto coverslips coated with poly-D-lysine (50 μg/mL) and laminin (1 μg/mL). Cells were allowed to attach to coverslips for 30 minutes, and then high glucose DMEM supplemented with penicillin, streptomycin and fetal bovine serum was added to each well. Patch-clamp recording of DRG neurons was performed 1–2 days post plating.
Coverslips with attached, cultured dorsal root ganglion neurons were stored in a submerged recording chamber at room temperature and continuously perfused with extracellular solution containing 132 mM NaCl, 3 mM KCl, 2.5 mM CaCl2, 10 mM HEPES, and 10 mM glucose, saturated with 95% O2, 5% CO2 at a rate of ~2 ml/min (pH 7.4). Cells were visualized using infrared differential interference contrast microscopy. Whole cell voltage-clamp and current-clamp recordings from neurons were obtained under visual guidance using a 40x objective. Patch electrodes (3.0–4.0 MΩ) were filled with a KCl-based internal solution containing 134 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, 10 mM EGTA, 4 mM ATP-Mg2+ and 0.4 mM GTP-Na+ (pH 7.4, 300 mOsm). Neurons were voltage clamped and subjected to a hyperpolarization voltage-step protocol to asses Ih and Im. Neurons were held in current clamp and excitability was assessed using increasing steps of current (100 pA) to determine the threshold at which each neuron would fire an action potential. In a subset of experiments, 10 μM isoguvacine was bath applied and the same experiments were repeated. At the end of an experiment, 100 μM ZD-7288 was bath applied, followed by bath application of 5mM CsCl2, to block HCN channels. Images of each recorded neuron were acquired and the size of each cell body was recorded. Data were acquired using a Multiclamp amplifier, a Digidata 1440A acquisition system, and pClamp10 software (Molecular Devices). Sampling rate was 10 kHz. No correction for junction potential was applied. Cells were discarded if membrane potential (Vm) at break-in was >−60 mV, Rin was < 75 MΩ, residual uncompensated Rs was >20 MΩ. Input resistance and access resistance were monitored continuously throughout each experiment and cells were excluded from analysis if these values changed by more than 10% during the experiment.
For each culture and recording session, a control and mutant littermate were prepared at the same time, with the experimenter blind to genotype. Electrophysiology recordings alternated between coverslips of the two different conditions for the duration of the experimental session. The experimenter was blind to genotype and treatment conditions during both experiment and analysis portions of the experiments.
Brain Slice Electrophysiological Recordings
8–10 week old mice were deeply anesthetized and rapidly transcardially perfused with choline chloride solution (92 mM choline chloride, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 2 mM thiourea, 3 mM sodium pyruvate, 10 mM MgSO4.7H20, 0.5 mM CaCl2.2H20). Coronal brain sections (300 μm) were cut anterior to posterior, including sections containing primary somatosensory cortex and primary visual cortex. Slices recovered for 25 minutes at 35°C in HEPES recovery solution (76 mM Tris-HCl, 19.5 mM Tris base, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 2 mM thiourea, 3 mM sodium pyruvate, 10 mM MgSO4.7H2O, 0.5 mM CaCl2.2H2O). After 25 minutes at 35°C, slices were kept at room temperature in HEPES recovery solution until recording experiments began. Brain slices were stored in a submerged recording chamber at room temperature and continuously perfused with ACSF containing 2.5 mM CaCl2·2H20, 1 mM NaH2PO4·H20, 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4·7H2O, 26 mM NaHCO3, 25 mM dextrose, and 1.3 mM Na L-ascorbate (pH 7.4, 305–310 mOsm), saturated with 95% O2, 5% CO2 at a rate of ~2 ml/min. Cells were visualized using infrared differential interference contrast microscopy. Whole cell voltage-clamp and current-clamp recordings from layer 2/3 pyramidal neurons in trunk S1 or V1 were obtained under visual guidance using a 40x objective. Patch electrodes (3.5–4.5 MΩ) were filled with a CsCl-based internal solution containing 117 mM CsCl, 87 mM CsOH, 2.8 mM NaCl, 5 mM MgCl2, 2 mM ATP-Na+, 0.3 mM GTP-Na+, 0.6 mM EGTA, and 20 mM HEPES (pH 7.25–7.28, 275–285 mOsm), and neurons were voltage clamped at −70 mV and then at 0 mV. Spontaneous excitatory postsynaptic currents (sEPSCs, −70 mV hold) and inhibitory postsynaptic currents (sIPSCs, 0mV hold) were each measured during a 5–10 minute period. At the end of an experiment, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX; 10 μM) was bath-applied to block AMPA receptors, followed by bath-applied 3 μM gabazine to block GABAA receptors. For a separate set of neurons, patch electrodes (3.5–4.5 MΩ) were filled with a K-gluconate-based internal solution containing 150 mM K-gluconate, 3 mM KCl, 3 mM ATP-Mg2+, 0.5 mM GTP-Na+, 0.5 mM EGTA, 5 mM phosphocreatine-tris2, 5 mM phosphocreatine-Na2 and 10 mM HEPES (pH 7.2, 290–300 mOsm), and held in current clamp to confirm excitatory pyramidal neuron firing properties. Data were acquired using a Multiclamp amplifier, a Digidata 1440A acquisition system, and pClamp10 software (Molecular Devices). Sampling rate was 10 kHz, and data were low-pass filtered at 3 kHz. No correction for junction potential was applied. Cells were discarded if membrane potential (Vm) at break-in was >−50 mV, or Rin was < 75 MΩ, residual uncompensated Rs was > 20 MΩ. Input resistance and access resistance were monitored continuously throughout each experiment and cells were excluded from analysis if these values changed by more than 10% during the experiment. Four animals were included per genotype per experiment, with 2–4 neurons analyzed per brain region per mouse (S1 or V1). The experimenter was blind to genotype and treatment conditions during both experiment and analysis portions of the experiments.
In Vivo DRG Multi-Unit Electrode Recordings
In vivo recordings were performed on mutant mice and their control littermates at 8–12 weeks of age, using a preparation modified from prior studies (Ma and LaMotte, 2007). This preparation allows measurement of action potentials evoked by cutaneous stimulation for up to four hours in vivo. Mice were administered dexamethasone 1 hour prior to the start of surgery. Each animal was anesthetized using a combination of urethane (1mg/g) and isoflurane (2%), and then surgery was performed to expose the left, L4 DRG. Temperature was monitored and maintained at 35.5–37.5°C with a temperature controller (TC-344B, Warner Instruments) and thermoelectric heater (C3200–6145, Honeywell) embedded in castable cement (Aremco). At the beginning of the DRG exposure, an incision was made over the spine (T10 - L6) and the overlaying tissue retracted to expose the vertebral column. The spine was secured with custom spinal clamps (Mike’s Machine, Attleboro MA) and bone dorsal to the target DRG removed with rongeurs.
Following surgery, and during the entire recording preparation, animals were maintained on 1% isoflurane. A NeuroNexus tetrode was inserted into the DRG under visual guidance using a 10x objective. Light touch responsive units were identified as units that fired action potentials in response light brush and air puff stimuli. Units were characterized as glabrous or hairy skin innervating Aβ RA1-LTMRs, Aβ SA1-LTMRs or Aβ field-LTMRs, or proprioceptors, based on firing rate, and optimal stimulus type (Figures 6D, 6E). In this preparation, multiple single units (2–12 per experiment; example of typical single unit in Figures 6B–C) with receptive fields on the thigh or hind paw could be identified following skin stimulation with innocuous mechanical stimuli.
Once LTMR units were identified, animals were administered a subcutaneous injection of either saline vehicle or isoguvacine (2 mg/kg), and sensitivity and firing frequency in response to tactile stimuli were monitored over a 90-minute period for each identified unit. Data was digitized at 40 kHz by a 16-bit A/D converter (USB-6259, National Instruments), low pass filtered at 10 kHz using the amplifier’s internal four-pole Bessel filter. Units were sorted and classified as cell types using JRClust (Figures 6B–E) (Jun et al., 2017). Firing properties were analyzed using custom code. The experimenter was blind to genotype and treatment conditions during both experiment and analysis portions of the experiments.
Liquid Chromatography Mass Spectrometry
All samples were harvested from 8–12 week old male mice. For cerebrospinal fluid (CSF) and plasma sample preparation: 5 μl of sample (if less was present, water was added to reach 5 μl and the concentrations were calculated appropriately) was mixed with 20 μL of a 150 nM solution of nipecotic acid in methanol (Sigma Aldrich). The samples were incubated at −20°C for 2 hours to precipitate the proteins. After centrifugation at 10000 g for 35 minutes, the supernatants were transferred to microinserts, dried under nitrogen flow and resuspended in 15 μL of water. A set of calibration standards (1/5 dilution series, starting at 50 μM) is prepared the same way as the fluid samples.
Tissue sample preparation: weighed pieces of frozen tissue were transferred to bead beater tube containing 1mL of 50 nM nipecotic acid (internal standard) in cold methanol and garnet pieces (SPEX SamplePrep tubes). Samples were homogenized for 10 minutes at 50 Hz in a TissueLyser LT (Biorad). Homogenized samples were transferred to 8 mL glass vial. The Bead Beater vials were washed with 1 mL methanol, and the methanol were combined with the samples. 4 mL of chloroform was added and the samples were incubated 1 hour at room temperature. Phase separation was then initiated by adding 2 mL water to each sample. After vortexing, the samples were centrifuged at 2500 g for 50 minutes at 4°C. The aqueous phases were then transferred to new vial, dried under nitrogen flow, and resuspended in 100 μL of water. A set of calibration standards was also prepared using the method.
Isoguvacine concentration was quantified on a Thermo Q-Exactive plus mass spectrometer coupled with an Ultimate 3000 LC (both Thermo Fisher). 8 μlof fluid samples or 5 μlof tissue samples were injected on a PFPP column (Phenomenex, 150×2mm). Mobile phase A was water with 1% acetic acid and mobile phase B was Acetonitrile with 1% acetic acid. The elution gradient was as follow: 3min with 0% B, then to 100%B in 2 min, followed by 3 minutes at 100%B. the column was then re-equilibrated with 0% B for 4 min. The flow rate was 0.2 mL/min. The MS was run in positive ion mode at 70000 resolution in full MS and in PRM mode. Quantification was achieved by integrating the signal of the transition 128.07061 to 82.06570 for isoguvacine, 130.08626 to 112.0760 for Nipecotic acid (internal standard), all at a normalized collision energy of 80 and a 1m/z isolation window. A ten-point calibration curve was used ranging from 25 pM to 50 μM. The detection limits for isoguvacine were 640 pM in the fluid samples and 16 nM in the tissue samples, and 128 pM for diazepam. Blank samples were run in between each sample to insure no carry over interferences.
GABAA Receptor Agonist Assay
Isoguvacine agonism for the GABAA receptor was tested using the IonFlux HTTM Automated Patch Clamp System (Eurofins). HEK293T cells transfected with human GABAA α1β3γ2 or α21β3γ2 subunits were subjected to patch clamp electrophysiology in the presence of varying concentrations (6-point concentration response) of isoguvacine for two seconds during each application. The maximal Cl-ion current elicited during each drug application was recorded. Six-point concentration response of GABA was tested to serve as an agonist control. Eight replicates were performed for each drug at each concentration. DMSO application for 2 seconds served as a time matched vehicle control. Peak isoguvacine-evoked ion currents were normalized to the peak Cl-ion current evoked by EC100 GABA for 2 seconds.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all figures and tables, data are expressed as the mean values +/− standard error of the mean (SEM). Bartlett’s test for equal variance was applied to all data sets, and the variances among groups were not shown to be statistically different from each other. The number of animals per group used in each experiment is denoted within the bar for that group in each panel.
For NORT, three-chamber social interaction tests, and habituation to startle noise experiments, each group was analyzed compared to a chance level using a one sample t-test (chance level: 0% preference or 0% habituation), with statistical significance being denoted as a symbol directly above the bar being analyzed. Comparisons between groups in all experiments were performed using Student’s t-test (in the case of two groups in one condition), one-way ANOVA (in the case of three or more groups in one condition), or two-way ANOVA (in the case of at least two groups with multiple conditions or timepoints). Comparisons between groups with significant differences are indicated using a bracket above the appropriate groups. Asterisks indicate p values for Student’s t-tests, unless otherwise noted in the figure legends. Unless otherwise noted, *, p < 0.05 and #, p < 0.10. Main effects from one-way and two-way ANOVAs are expressed as an F-statistic and P-value within brackets of the figure or denoted in Supplemental Tables. For one-way and two-way ANOVAs, post-hoc comparisons were performed using the post-hoc test indicated in the figure legend. The p values of post-hoc comparisons are represented with asterisks in figures. All statistics were performed using GraphPad Prism.
Supplementary Material
Figure S1. Shank3 functions cell-autonomously in peripheral somatosensory neurons for normal innocuous touch behaviors, related to Figure 1.
(A) IHC images of spinal cord (SC) dorsal horn lamina III/IV from control, Shank3B−/− or AdvillinCre; Shank3f/f mice, showing SHANK3 expression at vGLUT1+ presynaptic terminals for Aβ and Aδ LTMRs.
(B) Schematic describing strategy to create Shank3B conditional knockout (Shank3f) mouse. Mice containing targeted allele were crossed to Flp mice to remove the Neo cassette and β-ActinCre mice to excise Exons 13–16.
(C) PCR genotyping confirms successful deletion of Exons 13–16 from genome of Shank3f.
(D) Western blot of whole brain lysates from Shank3B−/− and β-ActinCre; Shank3f mice shows deletion of protein products from full-length isoform (white arrows).
(E) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates.
(F) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse (80 dB, ‘acoustic PPI’) in mutant mice and control littermates.
(G) Response to a non-startling acoustic noise (80 dB, 20 ms), mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(H) Discrimination index for 5 minute shape NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(I) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(J) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates.
(K) Average total distance traveled in the open field chamber for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05
(L) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(M) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(N) Average number of marbles buried (out of 12) during a twenty-minute assay for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Average amount of time spent grooming during a 10-minute open field assessment for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Representative images of control, Shank3FX/+ and AdvillinCre; Shank3FX/+ mice, showing excessive grooming and loss of fur in the Shank3FX/+ and AdvillinCre; Shank3FX/+ mice.
(Q) IHC images of SC dorsal horn lamina III/IV from control, Shank3FX/FX or AdvillinCre; Shank3FX/FX mice, showing SHANK3 expression at vGLUT1+ presynaptic terminals.
(R) IHC images of SC dorsal horn lamina III/IV from control, Shank3FX/FX or AdvillinCre; Shank3FX/FX mice, showing HCN1 expression at vGLUT1+ presynaptic terminals.
Figure S2. Loss of Shank3 leads to increased excitability of large diameter peripheral somatosensory neurons, related to Figure 1.
(A) IHC images of SC dorsal horn lamina III/IV from HCN1−/−, Shank3B−/−, AdvillinCre; Shank3f/f mice and their control littermates, showing HCN1 expression at vGLUT1+ presynaptic terminals.
(B) Quantification for the number of HCN1 puncta that co-localize with vGLUT1+ puncta in lamina III/IV of the spinal cord dorsal horn of mutants and control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, [for germline mutants: F (2,6) = 24.76, P = 0.0013; for conditional mutants: F (2,6) = 14.25, P = 0.0053], *, p < 0.05.
(C) Immunocytochemistry images of cultured DRG neurons from Shank3+/−mice and their control littermates, showing HCN1 expression.
(D) HCN1 expression in small diameter (<30 μm diameter) and large diameter (>40 μm diameter) cultured DRG neuron cell bodies, for control and Shank3+/−mutant mice. One-way ANOVA with post-hoc Tukey’s test [F (2,165) = 5.868, P = 0.0165], *, p < 0.05.
(E) IHC images of spinal cord (SC) dorsal horn lamina III/IV from AdvillinCre; Mecp2f/y and Shank3−/−mice and their control littermates, showing GABRB3 expression at vGLUT1+ presynaptic terminals.
(F) Quantification for the number of GABRB3 puncta that co-localize with vGLUT1+ puncta in lamina III/IV of the spinal cord dorsal horn of mutants and control littermates. Student’s unpaired t-test, *, p < 0.05.
(G) Voltage step protocol used to activate HCN channels and elicit Ih during whole-cell voltage clamp recordings.
(H) Whole-cell voltage clamp trace showing Ih during a hyperpolarizing voltage step protocol in a large diameter neuron cultured from mouse DRGs, which is blocked following application of 5 mM CsCl2.
(I) Representative traces from large diameter DRG neurons cultured from a control and Shank3B+/− mice, showing Ih during a hyperpolarizing voltage step protocol, with and without application of CsCl2 to block HCN channels.
(J) Representative traces from small diameter DRG neurons cultured from a control and Shank3B+/− mice, showing Ih during a hyperpolarizing voltage step protocol.
(K) Quantification of total Ih density at each hyperpolarizing voltage step for small diameter neurons cultured from DRGs of control and Shank3B+/− mutant mice. Repeated measures two-way ANOVA [F (1,532) = 0.001563; P = 0.9685], not significant.
(L) Representative traces from small diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole cell current clamp recordings, in which the minimal amount of current required to elicit an action potential in each neuron (rheobase, Rh), was determined.
(M) Average Rh in small diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell current clamp recordings. Student’s t-test, p < 0.05.
(N) Average input resistance (Rin) of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, p < 0.05.
(O) Average capacitance (Cm) of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, not significant.
(P) Average cell body size of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, not significant.
(Q) Quantification of total Ih density at each hyperpolarizing voltage step for large diameter neurons cultured from DRGs of control and Mecp2STOP/y mutant mice. Repeated measures two-way ANOVA [F (1,980) = 3.474, P = 0.0626], not significant.
(R) Quantification of total Ih density at each hyperpolarizing voltage step for small diameter neurons cultured from DRGs of control and Mecp2STOP/y mutant mice. Repeated measures two-way ANOVA [F (1,240) = 3.147, P = 0.0773], not significant.
(S) Average Rh in large and small diameter neurons cultured from DRGs of control and Mecp2STOP/y mice during whole-cell current clamp recordings. Student’s t-test, p < 0.05.
(T) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff (‘tactile PPI’), or tone prepulse (‘acoustic PPI’) in mutant mice and control littermates. One-way ANOVA with post-hoc Tukey’s test [for tactile PPI: F (2,33) = 7.064, P = 0.0028; for acoustic PPI: F (2,28) = 3.999, P = 0.0297], *, p < 0.05.
(U) Response to a light air puff stimulus (0.9 PSI, 50 ms) or non-startling tone prepulse (80dB, 20 ms) alone. Responses are expressed as percent of startle response to a 125 dB noise. One-way ANOVA with post-hoc Tukey’s test [F (2,33) = 0.2048, P = 0.8158], not significant.
(V) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates.
(W) Discrimination index for textured NORT, 5 minute shape NORT and 1 hour retention NORT. One-way ANOVA with post-hoc Tukey’s test [for textured NORT: F (2,33) = 7.064, P = 0.0028; for 1 hour retention NORT: F (2,33) = 6.308, P = 0.0048], *, p < 0.05.
Figure S3. Normal tactile sensitivity is necessary during early postnatal periods for normal behaviors in adult mice, related to Figure 3.
(A) Response to a light air puff stimulus alone mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB noise. Mice received five days of tamoxifen (TAM) treatment, beginning at either P5, P10 or P28. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(B) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(C) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in mutant mice and control littermates.
(D) Response to a non-startling acoustic noise (80 dB, 20 ms) in mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(E) Discrimination index for 5 minute shape NORT.
(F) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(G) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates.
(H) Average total distance traveled in the open field chamber for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(I) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(J) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average number of marbles buried (out of 12) during a twenty-minute assay for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
Figure S4. Loss of Mecp2 or Shank3 in peripheral somatosensory neurons during early postnatal periods impacts brain development, related to Figure 3.
(A) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from a control mouse showing MeCP2 expression (top panels) or MeCP2, Hoechst and IB4 expression (bottom panels).
(B) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from an AdvillinCreERT2; Mecp2f/y mouse that received TAM treatment, showing MeCP2 expression (top panels) and MeCP2, Hoechst and IB4 expression (bottom panels). Note lack of MeCP2 expression in the DRG, while MeCP2 expression is normal in S1 and spinal cord sections.
(C) Average percentage of MeCP2+ DRG neurons in control mice or mice with sensory-neuron specific deletion of Mecp2 (AdvillinCreERT2; Mecp2f/y), receiving five days of TAM beginning at P5, P10 or P28. Three to five DRGs were analyzed per mouse, and three to five mice per genotype and TAM date were included in the analysis. Student’s unpaired t-test, *, p < 0.001.
(D) Average percentage of SHANK3+, vGLUT1+ puncta in spinal cord sections from control mice or mice with sensory-neuron specific deletion of Shank3 (AdvillinCreERT2; Shank3f/f), receiving five days of TAM beginning at P5, P10 or P28. Three to five spinal cord sections were analyzed per mouse, and three to five mice per genotype and TAM date were included in the analysis. Student’s unpaired t-test, *, p < 0.001.
(E) IHC images of transverse spinal cord (SC) and DRG sections from a control mouse showing SHANK3 expression (top panels) or SHANK3 and vGLUT1 expression (bottom panels) in control mice. Note lack of SHANK3 expression in control SC at P7, and robust SHANK3 expression in DRG at P7.
(F) Representative IHC images of coronal S1 brain sections, showing PV immunoreactivity in control, AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. TAM was administered from P28–32 to all littermates.
(G) Quantification of the number of PV+ neurons in S1 of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(H) Quantification of the number of PV+ neurons in V1 of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(I) Quantification of the number of PV+ neurons in BLA of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(J) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from control and AdvillinCreERT2; Mecp2STOP/y mice showing MeCP2, Hoechst and IB4 expression with and without the TAM administration (‘+ TAM’, ‘-TAM’). TAM was administered from P28–32.
(K) Quantification of the number of PV+ neurons in S1 of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 6.206, P = 0.0202; for Mecp2 groups: F (2,10) = 30.95, P < 0.0001], *, p < 0.05.
(L) Quantification of the number of PV+ neurons in V1 of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 5.794, P = 0.0241; for Mecp2 groups: F (2,10) = 30.45, P < 0.0001], *, p < 0.05.
(M) Quantification of the number of PV+ neurons in BLA of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 25.88, P = 0.0002; for Mecp2 groups: F (2,10) = 13.24, P < 0.0015], *, p < 0.05.
Figure S5. Viral expression of GABRB3 in peripheral sensory neurons, beginning at P5, improves some tactile, brain and behavioral deficits observed in Mecp2R306C mutant mice, related to Figure 4.
(A) Percentage of transduced DRG neurons (mCherry+ neurons) in mice that received P5 i.p. injection of AAV.FLEx.GABRB3.mCHERRY
(B) Percentage of mCherry+ DRG neurons that are NF200+ neurons in mice that received P5 i.p. injection of AAV.FLEx.GABRB3.mCHERRY
(C) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in male and female control (Mecp2R/y or R/R), mutant (Mecp2C/y or R/C) and mutant rescue (AdvillinCre; Mecp2C/y or R/C) mice. All littermates received an i.p. injection of AAV.FLEx.GABRB3.mCHERRY at P5. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Response to a non-startling acoustic noise (80 dB, 20 ms) in male and female control, mutant and mutant rescue mice. Responses are expressed as percent of startle response to a 125 dB startle noise.
(E) Magnitude of startle response to a 125 dB noise in male and female control, mutant and mutant rescue mice. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in Mecp2R/y and AdvillinCre; Mecp2R/y control mice. One-way ANOVA, not significant.
(G) Response to a light air puff stimulus alone in Mecp2R/y or R/R and AdvillinCre; Mecp2R/y or R/R control mice. Responses are expressed as percent of startle response to a 125 dB noise. One-way ANOVA, not significant.
(H) Percent of DRG neurons transduced with AAV.FLEx.GABRB3.mCHERRY, plotted against response to a light air puff stimulus alone for AdvillinCre; Mecp2C/y mice.
(I) Discrimination index for 5 minute shape NORT.
(J) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates. Bars with no lines: 5 minute shape NORT; bars with horizontal lines: textured NORT; bars with vertical lines: 1 hour retention NORT.
(L) Average total distance traveled in the open field chamber for mutant, mutant rescue mice and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(M) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(N) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant, mutant rescue mice and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Preference index for the percentage of time spent investigating a novel mouse, compared to a novel object, in the “Sociability” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Preference index for the percentage of time spent investigating a novel mouse, compared to a familiar mouse, in the “Social Novelty Preference” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
Figure S6. Isoguvacine attenuates tactile sensitivity through reduced excitability of peripheral, low-threshold mechanosensory neurons but does not affect proprioceptor firing rates, related to Figures 5 and 6.
(A-B) Representative traces from large diameter neurons cultured from DRGs of control (A) and Mecp2STOP/y (B) mice during whole cell current clamp recordings, in which the minimal amount of current required to elicit an action potential in each neuron (rheobase, Rh), was determined in the absence or presence of 10 μM isoguvacine.
(C) Quantification of average Rh in large diameter neurons cultured from DRGs of control mice during a baseline period and then in presence of 10 μM isoguvacine. Paired t-test, *, p < 0.0005.
(D) Quantification of average Rh in large diameter neurons cultured from DRGs of control mice during a baseline period, followed application of 10 μM isoguvacine, then washout period. Repeated measures one-way ANOVA, post-hoc Sidak’s test [F (5,10) = 20.63, P < 0.0001], *, p < 0.05.
(E) Quantification of average Rh in large diameter neurons cultured from DRGs of control and Mecp2STOP/y mice during a baseline period and then in presence of 10 μM isoguvacine. Repeated measures two-way ANOVA, post-hoc Sidak’s test [F (11,11) = 20.63, P < 0.0001], *, p < 0.05.
(F) Quantification of average Rh in large diameter neurons cultured from DRGs of control and Shank3B+/−mice during a baseline period and then in presence of 10 μM isoguvacine. Repeated measures two-way ANOVA, post-hoc Sidak’s test [F (15,15) = 4.545, P = 0.0029], *, p < 0.05.
(G) Quantification of total Ih density at each hyperpolarizing voltage step for large diameter neurons cultured from DRGs of Mecp2STOP/y mutant mice and control littermates, in the presence of vehicle or 10 μM isoguvacine. Repeated measures two-way ANOVA [F (3, 1280) = 1.953, P = 0.1193], not significant.
(H) GABAA receptor subunit RNA expression levels across peripheral somatosensory neuron subtypes, for the delta, epsilon, pi, and theta subunit types.
(I) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in 16p11.2 deletion mutant mice (16p11.2+/−) or heterozygous Shank3Δex4−9 null mice and their control littermates. 16p11.2+/−mice received i.p. administration of either saline (‘SAL’) or 2 mg/kg isoguvacine (‘ISO’). Shank3Δex4−9 mice received no treatment (‘No txt’). For 16p11.2+/−mice: repeated measures, two-way ANOVA with post-hoc Tukey’s test [F (2,36) = 14.86, P < 0.0001], *, p < 0.05. For Shank3Δex4−9 mice: Student’s unpaired t-test, not significant.
(J) Response to a light air puff stimulus alone in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB noise in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(K) Magnitude of startle response to a 125 dB noise in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(L) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse (80 dB, ‘acoustic PPI’) in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(M) Response to a non-startling acoustic noise (80 dB, 20 ms), in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(N) Discrimination index for textured, 5 minute shape and 1 hour retention NORT in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(O) Average firing frequency of LTMRs in response to an air puff stimulus over the duration of each recording experiment, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3, 108) = 16.02, P < 0.0001], *, p < 0.05.
(P) Representative activity raster plots for multiple putative proprioceptors in multiple mice over the duration of a recording experiment in controls and Shank3B+/−mice. Mice received a subcutaneous injection of isoguvacine (2 mg/kg) during the experiment, and activity of proprioceptive units in response to a brush stimulus was assessed over a 90-minute period. Arrows indicate time of injection.
(Q) Average firing frequency of proprioceptors in response to a brush stimulus over the duration of each recording experiment in controls and Shank3B+/−mice, following subcutaneous injection isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3, 100) = 0.01423, P = 0.9053], not significant. (R) Average brush stroke frequency over the duration of each recording experiment in all controls and mutant mice, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, one-way ANOVA with post-hoc Dunnett’s test [F (3,44) = 0.3736, P = 0.7725], not significant.
Figure S7. Chronic administration of isoguvacine improves tactile over-reactivity, region-specific cortical abnormalities, and some ASD-related behaviors in Mecp2 and Shank3 mutant mice, related to Figure 7.
(A) Liquid chromatography mass spectrometry (LC-MS) quantified isoguvacine concentrations in brain homogenate (forebrain and hindbrain), spinal cord and liver samples of mice treated with isoguvacine (2 mg/kg, i.p.) from P1–42. Samples were obtained six hours following the final day of isoguvacine administration.
(B) Percent of pups surviving to P21 in litters from Shank3B+/− or Mecp2R/C female mice, in which litters were treated daily with either saline or isoguvacine (2 mg/kg).
(C) Discrimination index for textured NORT. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Magnitude of startle response to a 125 dB noise in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(E) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(F) Response to a non-startling acoustic noise (80 dB, 20 ms) in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Responses are expressed as percent of startle response to a 125 dB startle noise.
(G) Discrimination index for 5 minute shape NORT in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(H) Discrimination index for 1 hour retention NORT in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(I) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(J) Average total distance traveled in the open field chamber for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average number of EPM arm entries during the 10-minute assay, for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(L) Average number of marbles buried (out of 12) during a twenty-minute assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(M) Average amount of time spent grooming during a 10-minute open field assessment for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(N) Time spent per compartment in the 3-chamber social interaction test during the “Sociability” portion of the assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Time spent per compartment in the 3-chamber social interaction test during the “Social Novelty Preference” portion of the assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(Q) Quantification of the number of PV+ neurons in BLA of Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Unpaired Student’s t-test, *, p < 0.05.
(R) Quantification of sEPSC and sIPSC event amplitude in S1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(S) Quantification of sEPSC and sIPSC event frequency in S1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(T) Quantification of excitatory/inhibitory (E/I) ratios in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Unpaired Student’s t-test, *, p < 0.05.
(U) Quantification of sEPSC and sIPSC event amplitude in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(V) Quantification of sEPSC and sIPSC event frequency in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti GABRB3 | Life Technologies | PA5–19060 |
| Mouse anti GAD67 | Fisher Scientific | MAB5406 |
| Rabbit anti HCN1 | Alomone Labs | APC-056 |
| Alexa 647-conjugated IB4 | Life Technologies | I16520–100 |
| Rabbit anti MeCP2 | Michael Greenberg | n/a |
| Goat anti PARVALBUMIN | Swant | PVG-213 |
| Rabbit anti PARVALBUMIN | Swant | PV-27 |
| Rabbit anti dsRED | Clontech | 632496 |
| Mouse anti Shank3 | Abcam | Ab193307 |
| Guinea pig anti vGLUT 1 | Millipore | AB5905 |
| Bacterial and Virus Strains | ||
| AAV2.9 hSyn-FLEX | Addgene | #84481 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dexamethasone | Henry Schein | Cat. # 2459 |
| Isoguvacine | Sigma Aldrich | Cat. #G002 |
| Isoflurane (Isothesia) | Henry Schein | Cat. # 029405 |
| Midazolam | Henry Schein | Cat. # 054788 |
| Nipecotic Acid | Sigma Aldrich | Cat. # 211672 |
| Polyinosinic: polycytidylic acid | Millipore | Cat. # 528906 |
| T amoxifen | Toronto Research Chemicals | Cat. # 10540-29-1 |
| Urethane | Sigma Aldrich | Cat. # U2500 |
| Critical Commercial Assays | ||
| IonFlux HTTM Automated Patch Clamp System | Eurofins | n/a |
| In-Fusion® HD Cloning Plus Kit | Takara | 638911 |
| Experimental Models: Organisms/Strains | ||
| AdvillinCre | Fan Wang | Hasegawa et al., 2007 |
| 16p11.2df null | Jackson Laboratory | 013128 |
| AdvillinCreERT2 | John Wood | Lau et al., 2011 |
| Cdx2Cre | Eric Fearon | Akyol et al., 2008 |
| Cntnap2 null | Jackson Laboratory | 028635 |
| Fmr1 null | Jackson Laboratory | 003025 |
| Gabrb3floxed | Jackson Laboratory | 008310 |
| Mecp2floxed | Jackson Laboratory | 006847 |
| Mecp2R306C | Michael Greenberg | Lyst et al., 2013 |
| Mecp2STOP | Jackson Laboratory | 006849 |
| Shank3B Null | Jackson Laboratory | 017688 |
| Shank3Δex4-y null | Jackson Laboratory | 017890 |
| Shank3floxed | This paper | n/a |
| Shank3FX | Guoping Feng | Mei et al., 2016 |
| Oligonucleotides | ||
| Primers for mouse genotyping (see Supplemental Table S8) | This paper | n/a |
| Recombinant DNA | ||
| AAV.hSYN.FLEx.GABRB3.t2A.mCHERRY | This paper | n/a |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| JRClust | Jun et al., 2017 | https://github.com/JaneliaSciComp/JRCLUST |
| Other | ||
Highlights:
Distinct pathophysiologies cause peripheral sensory neuron dysfunction in ASD models
Peripheral somatosensory neuron dysfunction alters brain development and behavior
A peripherally-restricted GABAAR agonist, isoguvacine, reduces tactile sensitivity
Chronic isoguvacine treatment improves a subset of ASD-related phenotypes in mice
Acknowledgements
We thank Bruce Bean, Irit Ben-Chelouche, Christopher Harvey, Curtis Keith, Zoe Klein, Michal Preminger, Yang Zheng and members of the Ginty lab for discussions and comments on the manuscript. We thank the Harvard Small Molecule Mass Spectrometry Facility for assistance with the biodistribution studies, and Ofer Mazor and Pavel Gorelik of the Harvard Research Instrumentation Core for help with electrophysiology and behavioral assay development (Core Grant for Vision Research EY012196). This work was supported by a Goldenson Postdoctoral Fellowship (LLO), a SFARI Research Award from the Simons Foundation (DDG), the Harvard Blavatnik Accelerator and QFASTR (LLO and DDG), and NIH grants K99 NS101057 (LLO), F31 MH098641A (MFW), T32 HL110852 (RMF), R01 NS088566 (MKL), R01 MH097104 (GF), NIMH Conte Center grant P50 MH094271 (GF) and R35 NS097344 (DDG). MKL is a New York Stem Cell Foundation – Robertson Investigator. DDG is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
LLO and DDG have patents pending on the use of peripherally restricted GABAA receptor drugs for the treatment of tactile over-reactivity in ASD and other disorders.
References
- Akyol A, Hinoi T, Feng Y, Bommer GT, Glaser TM, and Fearon ER (2008). Generating somatic mosaicism with a Cre recombinase-microsatellite sequence transgene. Nature methods 5, 231–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine MW, Langberg T, Schnepel P, and Feldman DE (2019). Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 101, 648–661 e644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, Kurzius-Spencer M, Zahorodny W, Robinson Rosenberg C, White T, et al. (2018). Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ 67, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, and Oostra BA (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33. [PubMed] [Google Scholar]
- Bhattacherjee A, Mu Y, Winter MK, Knapp JR, Eggimann LS, Gunewardena SS, Kobayashi K, Kato S, Krizsan-Agbas D, and Smith PG (2017). Neuronal cytoskeletal gene dysregulation and mechanical hypersensitivity in a rat model of Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America 114, E6952–E6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG, Hill DR, and Hudson AL (1983). Characteristics of GABAB receptor binding sites on rat whole brain synaptic membranes. Br J Pharmacol 78, 191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, and Coggeshall RE (1999). Peripheral GABA(A) receptors: evidence for peripheral primary afferent depolarization. Neuroscience 93, 713–722. [DOI] [PubMed] [Google Scholar]
- Chen JT, Guo D, Campanelli D, Frattini F, Mayer F, Zhou L, Kuner R, Heppenstall PA, Knipper M, and Hu J (2014). Presynaptic GABAergic inhibition regulated by BDNF contributes to neuropathic pain induction. Nature communications 5, 5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, and Huh JR (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes JM, Weir GA, Middleton SJ, Patel R, Chisholm KI, Pettingill P, Peck LJ, Sheridan J, Shakir A, Jacobson L, et al. (2018). Immune or Genetic-Mediated Disruption of CASPR2 Causes Pain Hypersensitivity Due to Enhanced Primary Afferent Excitability. Neuron 97, 806–822 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J, Geranton SM, Bebbington A, Jacoby P, Bahi-Buisson N, Ravine D, and Leonard H (2010). Linking MECP2 and pain sensitivity: the example of Rett syndrome. Am J Med Genet A 152A, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DSM-V (2013). Diagnostic and statistical manual of mental disorders : DSM-5 (Fifth edition Arlington, VA: : American Psychiatric Publishing, [2013] ©2013). [Google Scholar]
- Du X, Hao H, Yang Y, Huang S, Wang C, Gigout S, Ramli R, Li X, Jaworska E, Edwards I, et al. (2017). Local GABAergic signaling within sensory ganglia controls peripheral nociceptive transmission. J Clin Invest 127, 1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Veenstra-Vanderweele JM, Melmed RD, McCracken JT, Ginsberg LD, Sikich L, Scahill L, Cherubini M, Zarevics P, Walton-Bowen K, et al. (2014). STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J Autism Dev Disord 44, 958–964. [DOI] [PubMed] [Google Scholar]
- Ferguson C, Hardy SL, Werner DF, Hileman SM, Delorey TM, and Homanics GE (2007). New insight into the role of the beta3 subunit of the GABAA-R in development, behavior, body weight regulation, and anesthesia revealed by conditional gene knockout. BMC Neurosci 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filice F, Vorckel KJ, Sungur AO, Wohr M, and Schwaller B (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol Brain 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegel C, Schobel N, Altmuller J, Becker C, Tannapfel A, Hatt H, and Gisselmann G (2015). RNA-Seq Analysis of Human Trigeminal and Dorsal Root Ganglia with a Focus on Chemoreceptors. PLoS One 10, e0128951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Yamashita Y, Nagamitsu S, Miyamoto K, Jin JJ, Ohmori I, Ohtsuka Y, Kuwajima K, Endo S, Iwai T, et al. (2005). Methyl-CpG binding protein 2 gene (MECP2) variations in Japanese patients with Rett syndrome: pathological mutations and polymorphisms. Brain Dev 27, 211–217. [DOI] [PubMed] [Google Scholar]
- Furlan A, La Manno G, Lubke M, Haring M, Abdo H, Hochgerner H, Kupari J, Usoskin D, Airaksinen MS, Oliver G, et al. (2016). Visceral motor neuron diversity delineates a cellular basis for nipple- and pilo-erection muscle control. Nature neuroscience 19, 1331–1340. [DOI] [PubMed] [Google Scholar]
- Golombok S, Moodley P, and Lader M (1988). Cognitive impairment in long-term benzodiazepine users. Psychol Med 18, 365–374. [DOI] [PubMed] [Google Scholar]
- Gordon A, Salomon D, Barak N, Pen Y, Tsoory M, Kimchi T, and Peles E (2016). Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol Cell Neurosci 70, 42–53. [DOI] [PubMed] [Google Scholar]
- Groeneveld GJ, Hay JL, and Van Gerven JM (2016). Measuring blood-brain barrier penetration using the NeuroCart, a CNS test battery. Drug Discov Today Technol 20, 27–34. [DOI] [PubMed] [Google Scholar]
- Gudex C (1991). Adverse effects of benzodiazepines. Soc Sci Med 33, 587–596. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, and Bird A (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Asberg Johnels J, Lassalle A, Zurcher NR, Hippolyte L, Gillberg C, Lemonnier E, and Ben-Ari Y (2018). Bumetanide for autism: more eye contact, less amygdala activation. Sci Rep 8, 3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Kim YH, Wang X, Liu D, Zhang ZJ, Bey AL, Lay M, Chang W, Berta T, Zhang Y, et al. (2016). SHANK3 Deficiency Impairs Heat Hyperalgesia and TRPV1 Signaling in Primary Sensory Neurons. Neuron 92, 1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanack C, Moroni M, Lima WC, Wende H, Kirchner M, Adelfinger L, Schrenk-Siemens K, Tappe-Theodor A, Wetzel C, Kuich PH, et al. (2015). GABA blocks pathological but not acute TRPV1 pain signals. Cell 160, 759–770. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Abbott S, Han BX, Qi Y, and Wang F (2007). Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 14404–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemi E, Ariza J, Rogers H, Noctor SC, and Martinez-Cerdeno V (2017). The Number of Parvalbumin-Expressing Interneurons Is Decreased in the Medial Prefrontal Cortex in Autism. Cereb Cortex 27, 1931–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Arroyo ED, Smukowski SN, Xu J, Piochon C, Savas JN, Portera-Cailliau C, and Contractor A (2018). Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ, and Chemero A (2012). Novel object exploration in mice: not all objects are created equal. Behavioural processes 89, 232–238. [DOI] [PubMed] [Google Scholar]
- Hill DR, and Bowery NG (1981). 3H-baclofen and 3H-GABA bind to bicuculline-insensitive GABA B sites in rat brain. Nature 290, 149–152. [DOI] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proceedings of the National Academy of Sciences of the United States of America 108, 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Rogdaki M, Findon JL, Wichers RH, Charman T, King BH, Loth E, McAlonan GM, McCracken JT, Parr JR, et al. (2018). Autism spectrum disorder: Consensus guidelines on assessment, treatment and research from the British Association for Psychopharmacology. J Psychopharmacol 32, 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, and Wiesel TN (1970). The period of susceptibility to the physiological effects of unilateral eye closure in kittens. The Journal of physiology 206, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James B, MA G, and BJ G (2018). Bumetanide for Autism Spectrum Disorder in Children: A Review of Randomized Controlled Trials. Annals of Pharmacotherapy 10.1177/1060028018817304. [DOI] [PubMed] [Google Scholar]
- Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Escamilla CO, Weaver TP, Liu S, Filonova I, and Powell CM (2017). Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res 10, 42–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JJ, Mitelut C, Lai C, Gratiy S, Anastassiou C, and Harris TD (2017). Real-time spike sorting platform for high-density extracellular probes with ground-truth validation and drift correction. 101030. [Google Scholar]
- Kodish I, Rockhill C, Ryan S, and Varley C (2011). Pharmacotherapy for anxiety disorders in children and adolescents. Pediatr Clin North Am 58, 55–72, x. [DOI] [PubMed] [Google Scholar]
- Konig P, Engel AK, and Singer W (1996). Integrator or coincidence detector? The role of the cortical neuron revisited. Trends Neurosci 19, 130–137. [DOI] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, and Johnston GA (1978). Structure-activity studies on the inhibition of GABA binding to rat brain membranes by muscimol and related compounds. J Neurochem 30, 1377–1382. [DOI] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, Johnston GA, Lodge D, and Curtis DR (1977). A new class of GABA agonist. Nature 268, 53–55. [DOI] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, Schultz B, Mikkelsen H, Aaes-Jorgensen T, and Bogeso KP (1981). THIP, isoguvacine, isoguvacine oxide, and related GABA agonists. Adv Biochem Psychopharmacol 29, 69–76. [PubMed] [Google Scholar]
- Lau J, Minett MS, Zhao J, Dennehy U, Wang F, Wood JN, and Bogdanov YD (2011). Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible Advillin-Cre-ERT2 recombinase mouse. Mol Pain 7, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier E, Villeneuve N, Sonie S, Serret S, Rosier A, Roue M, Brosset P, Viellard M, Bernoux D, Rondeau S, et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 7, e1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Owen EH, Rasmussen DL, and Wehner JM (1997). Assessment of locomotor activity, acoustic and tactile startle, and prepulse inhibition of startle in inbred mouse strains and F1 hybrids: implications of genetic background for single gene and quantitative trait loci analyses. Neuroscience 80, 1075–1086. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature neuroscience 16, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, and LaMotte RH (2007). Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 14059–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen MA, Moore GA, Scaramella LV, Reiss D, Ganiban JM, Shaw DS, Leve LD, and Neiderhiser JM (2015). Infant Avoidance during a Tactile Task Predicts Autism Spectrum Behaviors in Toddlerhood. Infant Ment Health J 36, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O (2012). Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 13, 107–120. [DOI] [PubMed] [Google Scholar]
- Mazurek MO, Vasa RA, Kalb LG, Kanne SM, Rosenberg D, Keefer A, Murray DS, Freedman B, and Lowery LA (2013). Anxiety, sensory over-responsivity, and gastrointestinal problems in children with autism spectrum disorders. J Abnorm Child Psychol 41, 165–176. [DOI] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, and Feng G (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, et al. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behavioural brain research 176, 4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, and Valakh V (2015). Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, et al. (2003). The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115, 551–564. [DOI] [PubMed] [Google Scholar]
- Obradovic AL, Scarpa J, Osuru HP, Weaver JL, Park JY, Pathirathna S, Peterkin A, Lim Y, Jagodic MM, Todorovic SM, et al. (2015). Silencing the alpha2 subunit of gamma-aminobutyric acid type A receptors in rat dorsal root ganglia reveals its major role in antinociception posttraumatic nerve injury. Anesthesiology 123, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oginsky MF, Cui N, Zhong W, Johnson CM, and Jiang C (2017). Hyperexcitability of Mesencephalic Trigeminal Neurons and Reorganization of Ion Channel Expression in a Rett Syndrome Model. J Cell Physiol 232, 1151–1164. [DOI] [PubMed] [Google Scholar]
- Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, and Ginty DD (2016). Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 166, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perche O, Felgerolle C, Ardourel M, Bazinet A, Paris A, Rossignol R, Meyer-Dilhet G, Mausset-Bonnefont AL, Hebert B, Laurenceau D, et al. (2018). Early Retinal Defects in Fmr1(-/y) Mice: Toward a Critical Role of Visual Dys-Sensitivity in the Fragile X Syndrome Phenotype? Front Cell Neurosci 12, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, and McDermid HE (2012). The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol Syndromol 2, 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe A, Boddaert N, Vaivre-Douret L, Robel L, Danon-Boileau L, Malan V, de Blois MC, Heron D, Colleaux L, Golse B, et al. (2008). Neurobehavioral profile and brain imaging study of the 22q13.3 deletion syndrome in childhood. Pediatrics 122, e376–382. [DOI] [PubMed] [Google Scholar]
- Price TJ, and Melemedjian OK (2012). Fragile X mental retardation protein (FMRP) and the spinal sensory system. Results Probl Cell Differ 54, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, Lam T, Kim JY, Kim TH, Zhang MQ, et al. (2018). Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 159, 1325–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romermann K, Fedrowitz M, Hampel P, Kaczmarek E, Tollner K, Erker T, Sweet DH, and Loscher W (2017). Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 117, 182–194. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, Yeon Kim J, Kim S, Kim H, Waisman A, et al. (2017). Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DJ, and Land PW (1987). Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature 326, 694–697. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, and Deisseroth K (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata PR, Rollings J, Collins M, Pickering A, and Jacobson RR (1994). Lack of cognitive recovery following withdrawal from long-term benzodiazepine use. Psychol Med 24, 203–213. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Casto R, and Printz MP (1991). Dissociation of tactile and acoustic components in air puff startle. Physiology & behavior 49, 527–532. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Morello N, Calcagno E, and Giustetto M (2014). Developmental abnormalities of cortical interneurons precede symptoms onset in a mouse model of Rett syndrome. J Neurochem 131, 115–127. [DOI] [PubMed] [Google Scholar]
- Tomchek SD, and Dunn W (2007). Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am J Occup Ther 61, 190–200. [DOI] [PubMed] [Google Scholar]
- Torkamanzehi A, Boksa P, and Joober R (2008). Prepulse inhibition (PPI) of tactile startle response in recombinant congenic strains of mice: QTL mapping and comparison with acoustic PPI. J Genet Genomics 35, 139–151. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nature neuroscience 18, 145–153. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Cook EH, King BH, Zarevics P, Cherubini M, Walton-Bowen K, Bear MF, Wang PP, and Carpenter RL (2017). Arbaclofen in Children and Adolescents with Autism Spectrum Disorder: A Randomized, Controlled, Phase 2 Trial. Neuropsychopharmacology 42, 1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Behrens MM, and Sejnowski TJ (2011). Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 18137–18148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreugdenhil M, Jefferys JG, Celio MR, and Schwaller B (2003). Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. Journal of neurophysiology 89, 1414–1422. [DOI] [PubMed] [Google Scholar]
- Wang W, Li C, Chen Q, van der Goes M-S, Hawrot J, Yao AY, Gao X, Lu C, Zang Y, Zhang Q, et al. (2017). Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. The Journal of Clinical Investigation 127, 1978–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1965). Extent of recovery from the effects of visual deprivation in kittens. Journal of neurophysiology 28, 1060–1072. [DOI] [PubMed] [Google Scholar]
- Wiggins LD, Robins DL, Bakeman R, and Adamson LB (2009). Brief report: sensory abnormalities as distinguishing symptoms of autism spectrum disorders in young children. J Autism Dev Disord 39, 1087–1091. [DOI] [PubMed] [Google Scholar]
- Womelsdorf T, Valiante TA, Sahin NT, Miller KJ, and Tiesinga P (2014). Dynamic circuit motifs underlying rhythmic gain control, gating and integration. Nature neuroscience 17, 1031–1039. [DOI] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, and Sudhof TC (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Wildner H, and Yevenes GE (2012). Fast synaptic inhibition in spinal sensory processing and pain control. Physiological reviews 92, 193–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, and Barbas H (2013). Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci 7, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman AL, Kovatsis EM, Pozsgai RY, Tasnim A, Zhang Q, and Ginty DD (2019). Distinct Modes of Presynaptic Inhibition of Cutaneous Afferents and Their Functions in Behavior. Neuron 102, 420–434.e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Shank3 functions cell-autonomously in peripheral somatosensory neurons for normal innocuous touch behaviors, related to Figure 1.
(A) IHC images of spinal cord (SC) dorsal horn lamina III/IV from control, Shank3B−/− or AdvillinCre; Shank3f/f mice, showing SHANK3 expression at vGLUT1+ presynaptic terminals for Aβ and Aδ LTMRs.
(B) Schematic describing strategy to create Shank3B conditional knockout (Shank3f) mouse. Mice containing targeted allele were crossed to Flp mice to remove the Neo cassette and β-ActinCre mice to excise Exons 13–16.
(C) PCR genotyping confirms successful deletion of Exons 13–16 from genome of Shank3f.
(D) Western blot of whole brain lysates from Shank3B−/− and β-ActinCre; Shank3f mice shows deletion of protein products from full-length isoform (white arrows).
(E) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates.
(F) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse (80 dB, ‘acoustic PPI’) in mutant mice and control littermates.
(G) Response to a non-startling acoustic noise (80 dB, 20 ms), mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(H) Discrimination index for 5 minute shape NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(I) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(J) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates.
(K) Average total distance traveled in the open field chamber for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05
(L) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(M) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(N) Average number of marbles buried (out of 12) during a twenty-minute assay for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Average amount of time spent grooming during a 10-minute open field assessment for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Representative images of control, Shank3FX/+ and AdvillinCre; Shank3FX/+ mice, showing excessive grooming and loss of fur in the Shank3FX/+ and AdvillinCre; Shank3FX/+ mice.
(Q) IHC images of SC dorsal horn lamina III/IV from control, Shank3FX/FX or AdvillinCre; Shank3FX/FX mice, showing SHANK3 expression at vGLUT1+ presynaptic terminals.
(R) IHC images of SC dorsal horn lamina III/IV from control, Shank3FX/FX or AdvillinCre; Shank3FX/FX mice, showing HCN1 expression at vGLUT1+ presynaptic terminals.
Figure S2. Loss of Shank3 leads to increased excitability of large diameter peripheral somatosensory neurons, related to Figure 1.
(A) IHC images of SC dorsal horn lamina III/IV from HCN1−/−, Shank3B−/−, AdvillinCre; Shank3f/f mice and their control littermates, showing HCN1 expression at vGLUT1+ presynaptic terminals.
(B) Quantification for the number of HCN1 puncta that co-localize with vGLUT1+ puncta in lamina III/IV of the spinal cord dorsal horn of mutants and control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, [for germline mutants: F (2,6) = 24.76, P = 0.0013; for conditional mutants: F (2,6) = 14.25, P = 0.0053], *, p < 0.05.
(C) Immunocytochemistry images of cultured DRG neurons from Shank3+/−mice and their control littermates, showing HCN1 expression.
(D) HCN1 expression in small diameter (<30 μm diameter) and large diameter (>40 μm diameter) cultured DRG neuron cell bodies, for control and Shank3+/−mutant mice. One-way ANOVA with post-hoc Tukey’s test [F (2,165) = 5.868, P = 0.0165], *, p < 0.05.
(E) IHC images of spinal cord (SC) dorsal horn lamina III/IV from AdvillinCre; Mecp2f/y and Shank3−/−mice and their control littermates, showing GABRB3 expression at vGLUT1+ presynaptic terminals.
(F) Quantification for the number of GABRB3 puncta that co-localize with vGLUT1+ puncta in lamina III/IV of the spinal cord dorsal horn of mutants and control littermates. Student’s unpaired t-test, *, p < 0.05.
(G) Voltage step protocol used to activate HCN channels and elicit Ih during whole-cell voltage clamp recordings.
(H) Whole-cell voltage clamp trace showing Ih during a hyperpolarizing voltage step protocol in a large diameter neuron cultured from mouse DRGs, which is blocked following application of 5 mM CsCl2.
(I) Representative traces from large diameter DRG neurons cultured from a control and Shank3B+/− mice, showing Ih during a hyperpolarizing voltage step protocol, with and without application of CsCl2 to block HCN channels.
(J) Representative traces from small diameter DRG neurons cultured from a control and Shank3B+/− mice, showing Ih during a hyperpolarizing voltage step protocol.
(K) Quantification of total Ih density at each hyperpolarizing voltage step for small diameter neurons cultured from DRGs of control and Shank3B+/− mutant mice. Repeated measures two-way ANOVA [F (1,532) = 0.001563; P = 0.9685], not significant.
(L) Representative traces from small diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole cell current clamp recordings, in which the minimal amount of current required to elicit an action potential in each neuron (rheobase, Rh), was determined.
(M) Average Rh in small diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell current clamp recordings. Student’s t-test, p < 0.05.
(N) Average input resistance (Rin) of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, p < 0.05.
(O) Average capacitance (Cm) of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, not significant.
(P) Average cell body size of large diameter neurons cultured from DRGs of control and Shank3B+/−mice during whole-cell patch clamp recordings. Student’s t-test, not significant.
(Q) Quantification of total Ih density at each hyperpolarizing voltage step for large diameter neurons cultured from DRGs of control and Mecp2STOP/y mutant mice. Repeated measures two-way ANOVA [F (1,980) = 3.474, P = 0.0626], not significant.
(R) Quantification of total Ih density at each hyperpolarizing voltage step for small diameter neurons cultured from DRGs of control and Mecp2STOP/y mutant mice. Repeated measures two-way ANOVA [F (1,240) = 3.147, P = 0.0773], not significant.
(S) Average Rh in large and small diameter neurons cultured from DRGs of control and Mecp2STOP/y mice during whole-cell current clamp recordings. Student’s t-test, p < 0.05.
(T) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff (‘tactile PPI’), or tone prepulse (‘acoustic PPI’) in mutant mice and control littermates. One-way ANOVA with post-hoc Tukey’s test [for tactile PPI: F (2,33) = 7.064, P = 0.0028; for acoustic PPI: F (2,28) = 3.999, P = 0.0297], *, p < 0.05.
(U) Response to a light air puff stimulus (0.9 PSI, 50 ms) or non-startling tone prepulse (80dB, 20 ms) alone. Responses are expressed as percent of startle response to a 125 dB noise. One-way ANOVA with post-hoc Tukey’s test [F (2,33) = 0.2048, P = 0.8158], not significant.
(V) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates.
(W) Discrimination index for textured NORT, 5 minute shape NORT and 1 hour retention NORT. One-way ANOVA with post-hoc Tukey’s test [for textured NORT: F (2,33) = 7.064, P = 0.0028; for 1 hour retention NORT: F (2,33) = 6.308, P = 0.0048], *, p < 0.05.
Figure S3. Normal tactile sensitivity is necessary during early postnatal periods for normal behaviors in adult mice, related to Figure 3.
(A) Response to a light air puff stimulus alone mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB noise. Mice received five days of tamoxifen (TAM) treatment, beginning at either P5, P10 or P28. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(B) Magnitude of startle response to a 125 dB noise in mutant mice and control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(C) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in mutant mice and control littermates.
(D) Response to a non-startling acoustic noise (80 dB, 20 ms) in mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(E) Discrimination index for 5 minute shape NORT.
(F) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(G) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates.
(H) Average total distance traveled in the open field chamber for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(I) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(J) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average number of marbles buried (out of 12) during a twenty-minute assay for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
Figure S4. Loss of Mecp2 or Shank3 in peripheral somatosensory neurons during early postnatal periods impacts brain development, related to Figure 3.
(A) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from a control mouse showing MeCP2 expression (top panels) or MeCP2, Hoechst and IB4 expression (bottom panels).
(B) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from an AdvillinCreERT2; Mecp2f/y mouse that received TAM treatment, showing MeCP2 expression (top panels) and MeCP2, Hoechst and IB4 expression (bottom panels). Note lack of MeCP2 expression in the DRG, while MeCP2 expression is normal in S1 and spinal cord sections.
(C) Average percentage of MeCP2+ DRG neurons in control mice or mice with sensory-neuron specific deletion of Mecp2 (AdvillinCreERT2; Mecp2f/y), receiving five days of TAM beginning at P5, P10 or P28. Three to five DRGs were analyzed per mouse, and three to five mice per genotype and TAM date were included in the analysis. Student’s unpaired t-test, *, p < 0.001.
(D) Average percentage of SHANK3+, vGLUT1+ puncta in spinal cord sections from control mice or mice with sensory-neuron specific deletion of Shank3 (AdvillinCreERT2; Shank3f/f), receiving five days of TAM beginning at P5, P10 or P28. Three to five spinal cord sections were analyzed per mouse, and three to five mice per genotype and TAM date were included in the analysis. Student’s unpaired t-test, *, p < 0.001.
(E) IHC images of transverse spinal cord (SC) and DRG sections from a control mouse showing SHANK3 expression (top panels) or SHANK3 and vGLUT1 expression (bottom panels) in control mice. Note lack of SHANK3 expression in control SC at P7, and robust SHANK3 expression in DRG at P7.
(F) Representative IHC images of coronal S1 brain sections, showing PV immunoreactivity in control, AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. TAM was administered from P28–32 to all littermates.
(G) Quantification of the number of PV+ neurons in S1 of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(H) Quantification of the number of PV+ neurons in V1 of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(I) Quantification of the number of PV+ neurons in BLA of AdvillinCreERT2; Shank3f/+ or AdvillinCreERT2; Mecp2f/y mutant mice and their control littermates. Mice received five days of TAM treatment, beginning at either P5, P10 or P28. Student’s t-test, *, p < 0.05.
(J) IHC images of coronal primary somatosensory cortex (S1), spinal cord, or DRG sections from control and AdvillinCreERT2; Mecp2STOP/y mice showing MeCP2, Hoechst and IB4 expression with and without the TAM administration (‘+ TAM’, ‘-TAM’). TAM was administered from P28–32.
(K) Quantification of the number of PV+ neurons in S1 of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 6.206, P = 0.0202; for Mecp2 groups: F (2,10) = 30.95, P < 0.0001], *, p < 0.05.
(L) Quantification of the number of PV+ neurons in V1 of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 5.794, P = 0.0241; for Mecp2 groups: F (2,10) = 30.45, P < 0.0001], *, p < 0.05.
(M) Quantification of the number of PV+ neurons in BLA of control, mutant and mutant rescue mice. Mice received five days of TAM treatment, beginning at P28. One-way ANOVA with post-hoc Tukey’s test, [for Shank3 groups: F (2,9) = 25.88, P = 0.0002; for Mecp2 groups: F (2,10) = 13.24, P < 0.0015], *, p < 0.05.
Figure S5. Viral expression of GABRB3 in peripheral sensory neurons, beginning at P5, improves some tactile, brain and behavioral deficits observed in Mecp2R306C mutant mice, related to Figure 4.
(A) Percentage of transduced DRG neurons (mCherry+ neurons) in mice that received P5 i.p. injection of AAV.FLEx.GABRB3.mCHERRY
(B) Percentage of mCherry+ DRG neurons that are NF200+ neurons in mice that received P5 i.p. injection of AAV.FLEx.GABRB3.mCHERRY
(C) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in male and female control (Mecp2R/y or R/R), mutant (Mecp2C/y or R/C) and mutant rescue (AdvillinCre; Mecp2C/y or R/C) mice. All littermates received an i.p. injection of AAV.FLEx.GABRB3.mCHERRY at P5. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Response to a non-startling acoustic noise (80 dB, 20 ms) in male and female control, mutant and mutant rescue mice. Responses are expressed as percent of startle response to a 125 dB startle noise.
(E) Magnitude of startle response to a 125 dB noise in male and female control, mutant and mutant rescue mice. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(F) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in Mecp2R/y and AdvillinCre; Mecp2R/y control mice. One-way ANOVA, not significant.
(G) Response to a light air puff stimulus alone in Mecp2R/y or R/R and AdvillinCre; Mecp2R/y or R/R control mice. Responses are expressed as percent of startle response to a 125 dB noise. One-way ANOVA, not significant.
(H) Percent of DRG neurons transduced with AAV.FLEx.GABRB3.mCHERRY, plotted against response to a light air puff stimulus alone for AdvillinCre; Mecp2C/y mice.
(I) Discrimination index for 5 minute shape NORT.
(J) Discrimination index for 1 hour retention NORT. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests in mutant mice and their control littermates. Bars with no lines: 5 minute shape NORT; bars with horizontal lines: textured NORT; bars with vertical lines: 1 hour retention NORT.
(L) Average total distance traveled in the open field chamber for mutant, mutant rescue mice and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(M) Average number of EPM arm entries during the 10-minute assay, for mutant mice and their control littermates. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(N) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for mutant, mutant rescue mice and their control littermates. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Preference index for the percentage of time spent investigating a novel mouse, compared to a novel object, in the “Sociability” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Preference index for the percentage of time spent investigating a novel mouse, compared to a familiar mouse, in the “Social Novelty Preference” portion of the 3-chamber social interaction test. Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
Figure S6. Isoguvacine attenuates tactile sensitivity through reduced excitability of peripheral, low-threshold mechanosensory neurons but does not affect proprioceptor firing rates, related to Figures 5 and 6.
(A-B) Representative traces from large diameter neurons cultured from DRGs of control (A) and Mecp2STOP/y (B) mice during whole cell current clamp recordings, in which the minimal amount of current required to elicit an action potential in each neuron (rheobase, Rh), was determined in the absence or presence of 10 μM isoguvacine.
(C) Quantification of average Rh in large diameter neurons cultured from DRGs of control mice during a baseline period and then in presence of 10 μM isoguvacine. Paired t-test, *, p < 0.0005.
(D) Quantification of average Rh in large diameter neurons cultured from DRGs of control mice during a baseline period, followed application of 10 μM isoguvacine, then washout period. Repeated measures one-way ANOVA, post-hoc Sidak’s test [F (5,10) = 20.63, P < 0.0001], *, p < 0.05.
(E) Quantification of average Rh in large diameter neurons cultured from DRGs of control and Mecp2STOP/y mice during a baseline period and then in presence of 10 μM isoguvacine. Repeated measures two-way ANOVA, post-hoc Sidak’s test [F (11,11) = 20.63, P < 0.0001], *, p < 0.05.
(F) Quantification of average Rh in large diameter neurons cultured from DRGs of control and Shank3B+/−mice during a baseline period and then in presence of 10 μM isoguvacine. Repeated measures two-way ANOVA, post-hoc Sidak’s test [F (15,15) = 4.545, P = 0.0029], *, p < 0.05.
(G) Quantification of total Ih density at each hyperpolarizing voltage step for large diameter neurons cultured from DRGs of Mecp2STOP/y mutant mice and control littermates, in the presence of vehicle or 10 μM isoguvacine. Repeated measures two-way ANOVA [F (3, 1280) = 1.953, P = 0.1193], not significant.
(H) GABAA receptor subunit RNA expression levels across peripheral somatosensory neuron subtypes, for the delta, epsilon, pi, and theta subunit types.
(I) Percent inhibition of the startle response to a 125 dB noise, when the startle noise is preceded by a light air puff in 16p11.2 deletion mutant mice (16p11.2+/−) or heterozygous Shank3Δex4−9 null mice and their control littermates. 16p11.2+/−mice received i.p. administration of either saline (‘SAL’) or 2 mg/kg isoguvacine (‘ISO’). Shank3Δex4−9 mice received no treatment (‘No txt’). For 16p11.2+/−mice: repeated measures, two-way ANOVA with post-hoc Tukey’s test [F (2,36) = 14.86, P < 0.0001], *, p < 0.05. For Shank3Δex4−9 mice: Student’s unpaired t-test, not significant.
(J) Response to a light air puff stimulus alone in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB noise in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(K) Magnitude of startle response to a 125 dB noise in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(L) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse (80 dB, ‘acoustic PPI’) in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(M) Response to a non-startling acoustic noise (80 dB, 20 ms), in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates. Responses are expressed as percent of startle response to a 125 dB startle noise.
(N) Discrimination index for textured, 5 minute shape and 1 hour retention NORT in 16p11.2+/− or heterozygous Shank3Δex4−9 mutant mice and their control littermates.
(O) Average firing frequency of LTMRs in response to an air puff stimulus over the duration of each recording experiment, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3, 108) = 16.02, P < 0.0001], *, p < 0.05.
(P) Representative activity raster plots for multiple putative proprioceptors in multiple mice over the duration of a recording experiment in controls and Shank3B+/−mice. Mice received a subcutaneous injection of isoguvacine (2 mg/kg) during the experiment, and activity of proprioceptive units in response to a brush stimulus was assessed over a 90-minute period. Arrows indicate time of injection.
(Q) Average firing frequency of proprioceptors in response to a brush stimulus over the duration of each recording experiment in controls and Shank3B+/−mice, following subcutaneous injection isoguvacine (2 mg/kg). Repeated measures, two-way ANOVA with post-hoc Dunnett’s test [F (3, 100) = 0.01423, P = 0.9053], not significant. (R) Average brush stroke frequency over the duration of each recording experiment in all controls and mutant mice, following subcutaneous injection of either saline or isoguvacine (2 mg/kg). Repeated measures, one-way ANOVA with post-hoc Dunnett’s test [F (3,44) = 0.3736, P = 0.7725], not significant.
Figure S7. Chronic administration of isoguvacine improves tactile over-reactivity, region-specific cortical abnormalities, and some ASD-related behaviors in Mecp2 and Shank3 mutant mice, related to Figure 7.
(A) Liquid chromatography mass spectrometry (LC-MS) quantified isoguvacine concentrations in brain homogenate (forebrain and hindbrain), spinal cord and liver samples of mice treated with isoguvacine (2 mg/kg, i.p.) from P1–42. Samples were obtained six hours following the final day of isoguvacine administration.
(B) Percent of pups surviving to P21 in litters from Shank3B+/− or Mecp2R/C female mice, in which litters were treated daily with either saline or isoguvacine (2 mg/kg).
(C) Discrimination index for textured NORT. One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(D) Magnitude of startle response to a 125 dB noise in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(E) Percent inhibition of the startle response to a 125 dB noise (pulse), when the startle noise is preceded by tone prepulse in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(F) Response to a non-startling acoustic noise (80 dB, 20 ms) in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Responses are expressed as percent of startle response to a 125 dB startle noise.
(G) Discrimination index for 5 minute shape NORT in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(H) Discrimination index for 1 hour retention NORT in Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(I) Average amount of time (seconds) spent physically interacting with both the familiar and novel object in the NOR tests Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(J) Average total distance traveled in the open field chamber for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(K) Average number of EPM arm entries during the 10-minute assay, for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Student’s unpaired t-test or one-way ANOVA with post-hoc Tukey’s test, not significant.
(L) Average number of marbles buried (out of 12) during a twenty-minute assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(M) Average amount of time spent grooming during a 10-minute open field assessment for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test or unpaired Student’s t-test, *, p < 0.05.
(N) Time spent per compartment in the 3-chamber social interaction test during the “Sociability” portion of the assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(O) Time spent per compartment in the 3-chamber social interaction test during the “Social Novelty Preference” portion of the assay for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). One-way ANOVA with post-hoc Tukey’s test, *, p < 0.05.
(P) Percent decrease in startle response to a 125 dB noise during a 30-minute tactile PPI session, when comparing the first five startle responses to the last five responses to a 125 dB noise for Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg).
(Q) Quantification of the number of PV+ neurons in BLA of Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Unpaired Student’s t-test, *, p < 0.05.
(R) Quantification of sEPSC and sIPSC event amplitude in S1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(S) Quantification of sEPSC and sIPSC event frequency in S1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(T) Quantification of excitatory/inhibitory (E/I) ratios in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Unpaired Student’s t-test, *, p < 0.05.
(U) Quantification of sEPSC and sIPSC event amplitude in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.
(V) Quantification of sEPSC and sIPSC event frequency in V1 slices from Shank3B+/−, Mecp2R/C or Mecp2C/y mutant mice and control littermates treated daily from P1–42 with either saline or isoguvacine (2 mg/kg). Two-way ANOVA with post-hoc Sidak’s test, *, p < 0.05.