

Abstract
Background:
The significant proportion of schizophrenia patients refractory to treatment, primarily directed at the dopamine system, suggests that multiple mechanisms may underlie psychotic symptoms. Reinforcement learning tasks have been employed in schizophrenia to assess dopaminergic functioning and reward processing, but these have not directly compared groups of treatment-refractory and non-refractory patients.
Methods:
In the current fMRI study 21 patients with treatment resistant schizophrenia (TRS), 21 patients with non-treatment resistant schizophrenia (NTR), and 24 healthy controls (HC) performed a probabilistic reinforcement learning task, utilising emotionally valenced face stimuli which elicit a social bias toward happy faces. Behavior was characterized with a reinforcement learning model. Trial-wise reward prediction error (RPE)-related neural activation and the differential impact of emotional bias on these reward signals were compared between groups.
Results:
Patients showed impaired reinforcement learning relative to controls, while all groups demonstrated an emotional bias favouring happy faces. The pattern of RPE signaling was similar in the HC and TRS groups, whereas NTR patients showed significant attenuation of RPE-related activation in striatal, thalamic, precentral, parietal, and cerebellar regions. TRS patients, but not NTR patients, showed a positive relationship between emotional bias and RPE signal during negative feedback in bilateral thalamus and caudate.
Conclusion:
TRS can be dissociated from NTR on the basis of a different neural mechanism underlying reinforcement learning. The data support the hypothesis that a favourable response to antipsychotic treatment is contingent on dopaminergic dysfunction, characterized by aberrant RPE signaling, whereas treatment resistance may be characterized by an abnormality of a non-dopaminergic mechanism-a glutamatergic mechansim would be a possible candidate.
Keywords: Treatment resistant schizophrenia, psychosis, antipsychotics, prediction error, reward learning, dopamine, Schizophrenia, reinforcement learning
Introduction
Antipsychotic medication has been used to treat the symptoms of schizophrenia since the early 1950s. The mode of action for all currently licensed antipsychotics is via their action on dopamine D2 receptors (Kapur and Seeman, 2001; Seeman and Lee, 1975; Seeman et al., 1976). However, approximately one third of patients with a diagnosis of schizophrenia - (Lindenmayer, 2000; Mortimer et al., 2010) – fail to respond adequately to a trial of antipsychotic medication at recommended doses and duration; surprisingly, this occurs despite adequate D2 receptor occupancy (Coppens et al., 1991; Wolkin et al., 1989). The implication is that these occurrences of “treatment resistant” schizophrenia (TRS) are either characterized by a distinct neurochemical deficit, reflecting the heterogeneous nature of schizophrenia, or that the dopaminergic dysfunction is markedly more severe in TRS, sufficient that modulating the dopaminergic system with standard dopamine blocking antipsychotics is not enough to alleviate symptoms in these complex cases.
Schizophrenia has frequently been studied within a framework of reinforcement learning given the involvement of dopamine function in reward prediction (Deserno et al., 2016). Reinforcement learning is driven by midbrain dopamine neurons encoding violations of expected reward outcomes (Schultz, 1998), known as reward prediction error (RPE) signals. Specifically, unexpected reward elicits a phasic increase in firing of dopamine neurons, whereas omission of an expected reward results in a phasic decrease in firing. Midbrain RPE signals are thought to act as a learning signal which is fed through fronto-cortical basal ganglia loops in order to adjust behaviour accordingly. Functional magnetic resonance imaging (fMRI) of brain regions which are densely innervated by dopamine neurons, particularly the striatum and aspects of the prefrontal cortex, typically show activation reflective of an RPE response, in line with the notion that the blood oxygen level dependent (BOLD) signal likely reflects the information an area is receiving and processing. A recent meta-analysis of neuroimaging studies of prediction error during reinforcement learning confirmed robust prediction error activation in both ventral and dorsal aspects of the striatum as well as cortical regions including medial prefrontal, inferior and superior frontal, inferior parietal, and occipital cortex (Garrison et al., 2013). Consistent with pathologically increased tonic striatal dopamine in schizophrenia, phasic RPE signaling in the striatum has been shown to be reduced in schizophrenia patients (Schlagenhauf et al., 2014; Murray et al., 2008; Waltz et al., 2009), a finding attributed to “drowning” of these phasic signals due to elevated presynaptic dopamine. As the primary target of dopaminergic neurons, the ventral striatum has been a major region of interest for reinforcement learning studies in schizophrenia; however, impaired RPE signaling has also been detected in patients in additional areas such as prefrontal cortex (Corlett et al., 2007; Koch et al., 2010), parietal cortex (Waltz et al., 2009), thalamus (Murray et al., 2008; Gradin et al., 2011), and cerebellum (Waltz et al., 2009). Furthermore, there is evidence that reward feedback processing and RPE signaling in schizophrenia is selectively impaired for reward outcomes, but largely intact for loss outcomes, typically consisting of omission of expected reward (Gold et al., 2012; Waltz et al., 2007; Dowd et al., 2016; Waltz et al., 2009; Koch et al., 2010; Waltz et al., 2010; Simon et al., 2010). While meta-analytic findings have shown some overlap of neural regions processing reward and punishment in healthy individuals including in the striatum und medial frontal cortex, encoding of prediction errors during gain and loss outcomes appears to be spatially segregated in temporal and occipital regions (Garrison et al., 2013). This supports the possibility that the reward processing network could be selectively impaired in schizophrenia.
The question of whether a common dopaminergic abnormality underlies both treatment responsive and treatment resistant schizophrenia remains largely unresolved. Recent evidence suggests that elevated striatal dopamine synthesis capacity is specific to treatment responsive schizophrenia, whereas anterior cingulate glutamate levels may be selectively increased in TRS (Demjaha et al., 2012; Demjaha et al., 2014). However, the neural activation associated with dopamine functioning in the context of reinforcement learning has not been explicitly compared between these patient groups. Given the link between dopamine and RPE activation, a normal RPE signature would be expected in TRS if dopamine function is indeed unimpaired in this group. In contrast, treatment responsive patients would be expected to exhibit the abnormal RPE activation typically associated with schizophrenia. Note that behavior may be similarly impaired in the two groups if distinct nodes of the same reward network are differentially impaired. Reinforcement learning relies not only on striatal dopamine function, but also on complex fronto-striatal interactions regulating related processes such as cognitive control, goal maintenance and planning, as well as action value and effort computations (Barch and Dowd, 2010; Frank et al., 2001; Frank and Claus, 2006). As bottom-up learning signals are utilized to update a model of the surrounding environment, it is necessary to exert top-down cognitive control-particularly in the presence of persistent cognitive or behavioral bias-in order to optimise task focused learning. As such, it is possible that even with intact RPE signaling, a lack of cognitive control modulating learning processes could lead to a disruption of reinforcement learning. Notably, glutamatergic dysfunction may be associated with these cognitive control deficits in schizophrenia (Falkenberg et al., 2012; Taylor et al., 2015), providing a useful explanatory mechanisms for potential deficits in TRS.
In this study, we aimed to tap into these processes by quantifying cognitive bias in a reinforcement learning task and observing its modulation of RPE signaling. We compared treatment resistant and treatment responsive patients with a diagnosis of schizophrenia using fMRI while investigating 1) neural correlates of RPEs during wins and losses and 2) the association of cognitive bias with these learning signals. Cognitive bias was induced with a probabilistic reinforcement learning task using faces with varying expressions (Averbeck and Duchaine, 2009), which is known to elicit a bias towards happy faces in both healthy controls and patients with schizophrenia (Evans et al., 2011b). We examined RPE signaling separately for wins and losses on this task both because dissociable systems have been suggested for prediction error signaling of rewards and losses (Yacubian et al., 2006; Garrison et al., 2013) and due to evidence that reward and loss processing may be differentially impacted in schizophrenia (Chang et al., 2016; Reinen et al., 2016; Waltz et al., 2007; Waltz et al., 2011). In addition we anticipated that this would more closely reflect variabilities in prediction errors rather than effects of outcome itself.
Based on the theory that treatment responsive schizophrenia, but not TRS, is characterized by an abnormal dopaminergic signature, we tested the hypothesis that responsive patients would show reduced RPE signaling compared to healthy controls and TRS patients. This effect was expected to be particularly pronounced for win outcomes in areas typically associated with RPE signaling and dysfunctions in schizophrenia such as the striatum and thalamus. An additional exploratory analysis examined whether emotional bias would differentially modulate the neural RPE response in TRS patients compared with both responsive patients and controls.
Methods and materials
Participants
The study recruited 42 individuals with a diagnosis of schizophrenia (according to ICD-10 criteria) and 24 healthy controls matched for age, sex, and socioeconomic background consented to participate in this study. The patient sample included 21 with treatment resistant schizophrenia (TRS), based on persistent psychotic symptoms as defined as a score of at least 4 (moderate) on at least two positive symptom items of the Positive and Negative Syndrome Scale (PANSS) (Kay et al., 1987), at least two prior drug trials of 4–6 weeks duration with no clinical improvement, and persistence of illness for longer than five years with no period of good social or occupational functioning. The latter two criteria were ascertained by reviewing patients’ medical records and self-report of occupational status. The remaining 21 patients (NTR) fulfilled criteria for being in symptomatic remission, as defined by a score of 3 or less on all items of the PANSS (Conley and Kelly, 2001), these symptoms having been stable for at least 6 months (Andreasen et al., 2005) and prescribed a stable dosage of antipsychotic for the previous 6 months. Current clozapine use was an exclusion criterion for all patients. Exclusion criteria for all subjects were a history of neurological illness, current major physical illness, and drug dependency over the last six months. Exclusion criteria for HC were a history of psychiatric illness and a first-degree relative having suffered from a psychotic illness. All subjects had normal hearing and normal or corrected-to-normal vision. The two patient groups were matched for age, sex, duration of illness, medication type and dosage. Intelligence quotient was measured with the two-item Wechsler Abbreviated Scale of Intelligence (WASI; Wechsler, 1999). Chlorpromazine (CPZ) equivalent doses of medications were calculated using conversion tables (Bazire, 2005; Woods, 2003). Ethical approval was provided by the London Camberwell St Giles Research and Ethics Committee. All participants provided informed written consent and were compensated for their time and travel.
fMRI procedure
A schematic of a trial sequence is shown in Supplementary Figure 1. Subjects underwent a reward learning paradigm consisting of choosing between two simultaneously presented faces, and over a series of iterative trials, learning to identify which of the faces was associated with a higher reward probability. Subjects were given the task of maximizing the reward (10p per correct choice) achieved during the task. The task screen was viewed via a head-mounted mirror inside the MRI scanner and response selection was via a button box operated by the right index and middle fingers.
The task consisted of four blocks of 30 trials each, during which two faces were presented side by side. One face was associated with a 60% reward probability and the other with a 40% reward probability. Faces within a block differed either in emotional expression (blocks 1 and 3) or identity (blocks 2 and 4), as described previously (Evans et al., 2011a). In brief, emotional blocks consisted of one happy and one angry face with the same identity. Neutral blocks consistent of two faces with different identities but with neutral expressions. Combinations of identities and reward contingencies were counterbalanced across blocks and subjects.
Each trial began with a period of 1000 ms during which a white central fixation cross was presented against a dark background. This was followed by two faces being presented to the right and left of the fixation cross for 4500 ms. Within this time window subjects were required to select one of the faces by pressing the corresponding button with their right hand. The selected face was highlighted by a yellow square surrounding it. Feedback was then presented on the screen for 1500 ms. The task had a total duration of approximately 15 minutes.
Scanning parameters
Functional scans were acquired using a T2* echo planar sequence (430 volumes, TR=2000 ms, TE=35 ms, field of view = 24 cm, slice thickness = 3 mm, matrix = 64 × 64, flip angle = 75°) sensitive to blood oxygenation level-dependent (BOLD) contrast on a 3T GE Excite II MR scanner (GE Healthcare, USA). A structural image was acquired for each subject with a T1-weighted magnetization prepared rapid acquisition gradient echo (MP RAGE) sequence (TR=7321 ms, TE=3 ms, TI = 400 ms, field of view = 240, slice thickness = 1.2 mm, 196 slices).
Reinforcement learning model
The behavioural data was modelled using a “double update” reinforcement learning model (Schlagenhauf et al., 2014). Choice probability for choosing option 1 on trial t was computed on each trial using the softmax function
where the inverse temperature β determines the randomness of the subject’s choice, and Q1(t) denotes the action value, or expected reward, for choice 1 on trial t. The action value for the chosen option is updated on a trial-by-trial basis using the reward prediction error, defined as the difference between the expected reward Q and obtained reward R on trial t, scaled by the learning rate parameter α.
The action value for the unchosen option 2 was additionally updated on each trial, using the inverse reward value and identical learning rate parameter:
This model reflects the symmetry of choice outcomes, whereby feedback associated with a chosen option is also informative of the unchosen option (e.g., if stimulus 1 lost, stimulus 2 would have won).
The two free parameters β and α were estimated for each group separately by minimizing the negative log likelihood of the observed data pooled across all subjects within the group.
Behavioural analysis
Choices were defined as ideal if the action value (computed by the model) of the chosen option was greater than that of the unchosen option. Subjects’ proportions of ideal choices were analysed using a linear mixed effects model including the predictors group (HC vs. NTR vs. TRS) and condition (emotional vs. neutral)
Emotional bias was defined as the difference between the proportion of choices for the happy face when the angry face would have been an ideal choice, and proportion of choices for the angry face when the happy face would have been the ideal choice. Emotional bias was compared between groups using one-way ANOVA.
fMRI preprocessing and analysis
The fMRI data were preprocessed and analyzed using the FEAT tool from the FMRIB Software Library (FSL, http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/, Smith et al. 2004). Functional and structural brain images were extracted from non-brain tissue using FSL’s brain extraction tool (BET), and EPI images were realigned using MCFLIRT to correct effects of head motion. A 100-s temporal high-pass filter was applied and data were spatially smoothed using a Gaussian kernel of 5mm FWHM.
The functional MRI data were analyzed using the general linear model as implemented in FSL FEAT. For the first level analysis, the phases of the task (face presentation, choice, win outcome, and loss outcome) were modelled separately for emotional and neutral trials, resulting in eight unmodulated regressors. In addition, the win outcome and loss outcome phases were parametrically modulated with the trial-by-trial RPE values, again separately for emotional and neutral trials, resulting in four additional parametric regressors.
Each regressor was modelled with a delta function of zero duration and convolved with a canonical hemodynamic response function and its temporal derivative. Six standard motion parameters as well as a motion artefact confound matrix, which identified motion-corrupted volumes, were added as regressors of no interest. Volumes detected as corrupted were calculated by DVARS (Power et al., 2012) as implemented by FSL Motion Outliers. Percentage of corrupted volumes did not differ between groups, F(2,60) = 0.166, p > .848 (HC: N = 24; M = 0.4%, SD = 0.2%; NTR: N = 21; M = 0.4%, SD = 0.2%; TRS: N = 18; M = 0.4%, SD = 0.3%).
Contrasts of interest were constructed using the RPE regressors of win and loss outcomes separately. The first two contrasts averaged across the emotional and neutral conditions, resulting in the contrasts of interest: 1) win RPE and 2) loss RPE. The following two contrasts were constructed to detect activation which was greater in the emotional condition compared to the neutral condition: 3) win RPE [emotional > neutral] and 4) loss RPE [emotional > neutral].
At the group level, contrasts were submitted to separate mixed effects analyses (FLAME1), modelling the effect of group (HC, NTR, or TRS) on BOLD signal. Whole-brain activation differences between groups were tested for win RPE and loss RPE. In order to detect subcortical RPE activation we conducted an ROI analysis using a binary subcortical mask consisting of the bilateral striatum and thalamus (anatomically defined from the probabilistic Harvard Oxford Subcortical Structural Atlas thresholded at 30%). Broad inclusion of all structures of the striatum as well as the thalamus was based on the fact that subcortical RPE signaling was detected in each of these regions in a meta-analysis (Garrison et al., 2013) and dysfunctions in schizophrenia have also been observed in both striatum and thalamus (Gradin et al., 2011).
In order to assess the differential effect of emotional bias on RPE-related signal, analyses of the win RPE [emotional > neutral] and loss RPE [emotional > neutral] contrasts included emotional bias as a covariate, and group × bias interaction effects were assessed. Significant clusters were determined by a voxelwise z-threshold of 2.3 and a cluster significance threshold of p=0.05 (whole-brain family wise error corrected for multiple comparisons).
Correlation analyses were conducted between key positive symptoms (delusion and hallucinations) and significant clusters of RPE-related activation detected in the subcortical ROI analysis, and are reported where significant.
Results
Demographic characteristics the studied samples are presented in Table 1. The TRS patients showed higher scores on all PANSS symptom dimensions compared to NTR patients.
Table 1.
Means (M) and standard deviations (SD) of demographic and clinical variables per group
| HC |
NTR |
TRS |
||||||
|---|---|---|---|---|---|---|---|---|
| M | SD | M | SD | M | SD | Group statistics | ||
| χ2(2) | P | |||||||
| Female (%) | 25 | 14 | 14 | 1.18 | .555 | |||
| Smokers (%) | 17 | 67 | 62 | 14.0 | <.001 | |||
| F(2,63) | P | |||||||
| Age | 38.4 | 10.0 | 41.3 | 10.4 | 41.5 | 10.6 | 0.67 | .515 |
| WASI | 115.8 | 11.7 | 91.86 | 14.8 | 97.1 | 16.4 | 16.8 | <.001 |
| NS-SEC | 3.13 | 1.62 | 3.74 | 1.88 | 3.39 | 1.76 | 0.65 | .525 |
| t(40) | P | |||||||
| Onset age (years) | 27.7 | 6.2 | 26.0 | 7.7 | 0.80 | .431 | ||
| Illness duration (years) | 14.1 | 10.1 | 15.5 | 8.8 | 0.46 | .650 | ||
| CPZ equivalents | 280.3 | 147.1 | 383.5 | 236.5 | 1.67 | .103 | ||
| PANSS score | ||||||||
| Positive symptoms | 10.7 | 2.1 | 20.5 | 3.1 | 12.10 | <.001 | ||
| Negative symptoms | 13.1 | 4.6 | 19.5 | 4.6 | 4.08 | <.001 | ||
| General symptoms | 23.6 | 5.1 | 34.9 | 9.2 | 5.91 | <.001 | ||
| Total score | 46.9 | 10.3 | 76.2 | 10.6 | 9.14 | <.001 | ||
Abbreviations: HC, heathy controls; NTR, non-treatment resistant; TRS, treatment resistant schizophrenia; WASI, Wechsler Abbreviated Scale of Intelligence; NS-SEC, National Statistics Socio-economic Classification; CPZ, Chlorpromazine; PANSS, Positive and Negative Symptom Scale.
Behavioural results
The proportion of ideal choices differed significantly between the three groups, F(2,63) = 3.69, p = .031, with HC (M = 0.63, SD = 0.13) making significantly more ideal choices compared to NTR patients (M = 0.55, SD = 0.13), p = .037, and marginally more compared to TRS patients (M = 0.57, SD = 0.11), p = .062. There was no significant main effect of (emotional vs. neutral) condition, and no group × condition interaction.
All groups showed an emotional bias towards choosing the happy over the angry face, which did not differ significantly between groups, p > .05 (HC: M = 0.06, SD = 0.13; NTR: M = 0.13, SD = 0.22; TRS: M = 0.04, SD = 0.16).
Neuroimaging results
RPE signaling for wins and losses
HC showed RPE-related activation in response to win outcomes of the bilateral dorsolateral prefrontal cortices, superior frontal cortex, parietal cortices and visual cortex as well as cerebellum (see Figure 1A). TRS patients showed a similar activation pattern (Figure 1C). In contrast, NTR patients showed no supra-threshold RPE-related activation. Group comparisons showed that NTR patients had significantly reduced RPE-related activation in precentral gyrus compared to TRS, in angular gyrus compared to HC, as well as in cerebellum compared to both HC and TRS (Figure 2, Supplementary Table 1). The subcortical ROI analysis revealed a significant effect of group (p < .05 uncorrected), with NTR patients showing reduced RPE-related activation in bilateral thalamus and caudate head compared to both HC and TRS (Figure 3A).
Figure 1.
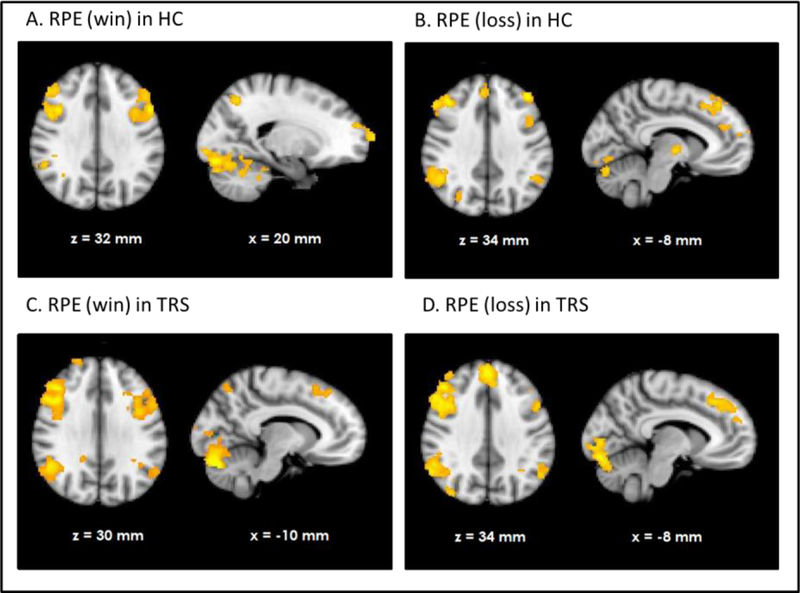
Neural response correlating with reward prediction error (RPE) in healthy controls (HC) during wins (A) and losses (B) and well as in treatment resistant schizophrenia (TRS) during wins (C) and losses (D).
Figure 2.
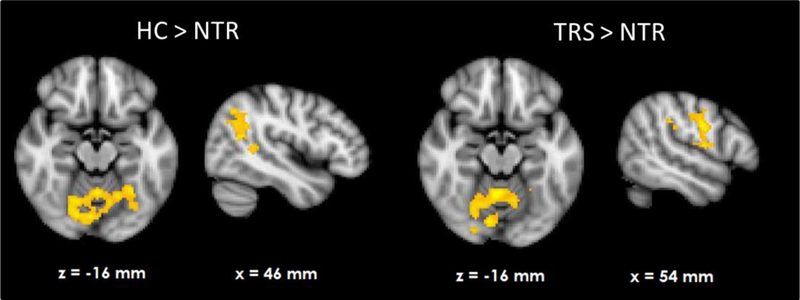
Group differences in neural reward prediction error related activation during wins.
Figure 3.
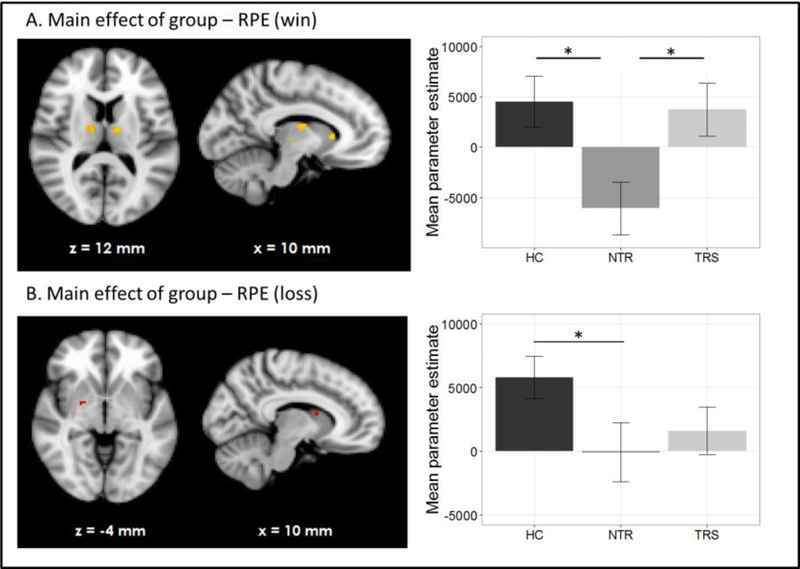
Subcortical region of interest analysis (p < .05, uncorrected) of group differences in reward prediction error signal during wins (A) and losses (B).
Loss-related RPE response was observed in a widespread network in both HC and TRS, similar to that during win outcomes (Figures 1B and 1D). Due to the negative sign of loss-related RPE, this signal reflects a negative RPE signal, with greater prediction errors resulting in greater deactivation in these areas. The NTR group showed no significant supra-threshold RPE related signal, with no significant group differences at whole-brain level. The subcortical ROI analysis revealed reduced RPE-related signal in bilateral pallidum and caudate in NTR compared to HC (p < .05 uncorrected) and no significant difference between TRS and either of the other two groups (Figure 3B).
Emotional bias × group interaction on RPE signal
During the emotional (versus neutral) loss trials, the whole-brain analysis showed a significant group × emotional bias interaction on RPE signal in bilateral thalamus and caudate nucleus, indicating a differential correlation in TRS and NTR patients (Figure 4, Supplementary Table 2). In TRS patients, a stronger emotional bias was associated with increased RPE signal in this region (R = 0.58, p = .006). In contrast, in NTR patients, the opposite was the case (R = −0.56, p = .008). This negative correlation in NTR was no longer significant after excluding one outlier, however the difference between correlation coefficients in the two groups remained significant (Fisher’s R to Z = 2.69, two-tailed p = .007). Interestingly, RPE signal in this region was significantly correlated with delusion severity in TRS patients, with stronger RPE signaling associated with more severe symptoms of delusions (R = 0.48, p = .027). This interaction was not evident in the emotional (versus neutral) win trials.
Figure 4.
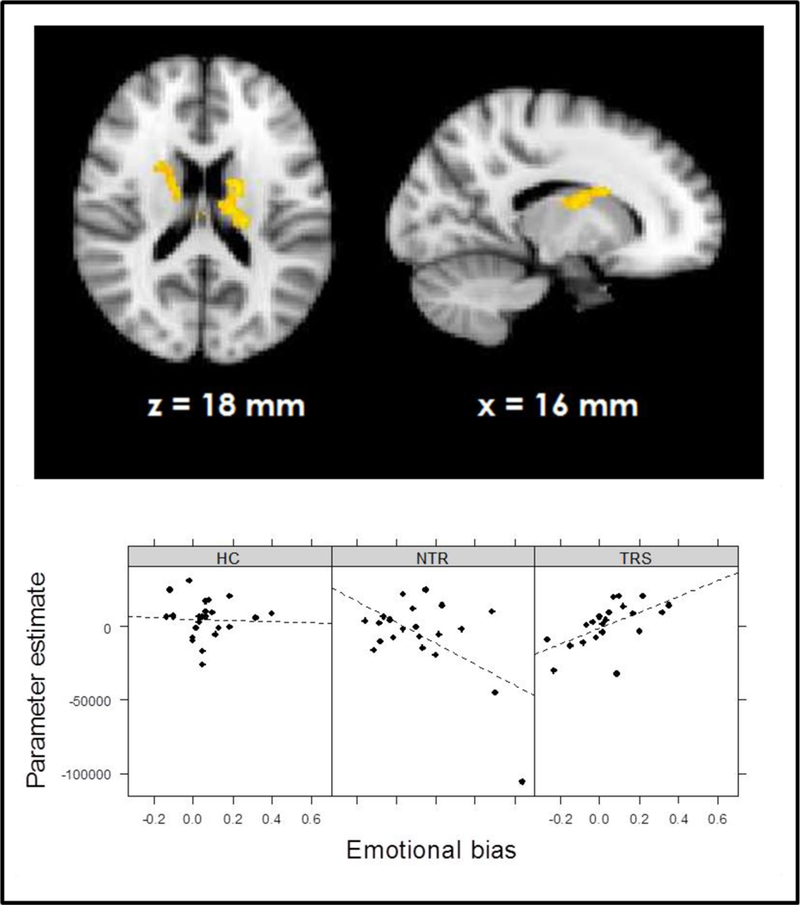
Group × emotional bias interaction in prediction error signal during losses.
Discussion
We used a probabilistic reward learning task to assess differences in neural mechanisms underlying reinforcement learning in patients with schizophrenia who were either treatment resistant (TRS) or non-treatment resistant (NTR), relative to a healthy control (HC) group. Our findings support the hypothesis that NTR patients show abnormal prediction error related activation compared to both HC and TRS, consistent with the theory that this patient group is characterized by a greater disruption of dopaminergic functioning. We also found that underlying cognitive bias differentially modulated learning processes in the two patient groups.
We found that HC and TRS patients showed similar patterns of prediction error signaling both during wins and losses. RPE activation was evident in a widespread network in these groups, consistent with the notion that reward processing is almost ubiquitous in the brain (Vickery et al., 2011). The observed regions of activation, including medial, superior and dorsolateral frontal cortex as well as visual areas and parietal cortex, are largely in line with the human cortical substrate of prediction error reported elsewhere (Schultz and Dickinson, 2000; Garrison et al., 2013). In contrast, NTR patients did not exhibit the same activation pattern. During receipt of rewarding outcomes, a whole-brain analysis showed reduced activation in the cerebellum in NTR compared to both HC and TRS patients; in parietal cortex compared to HC; and precentral gyrus compared to TRS. An ROI analysis revealed reduced activation in NTR in the thalamus and caudate compared to both HC and TRS. Reduced RPE-related activation in the thalamus and caudate in schizophrenia patients has been previously reported and linked with dopaminergic dysfunction (Gradin et al., 2011). Moreover, a further study found attenuated responses to unexpected reward, but intact responses to omission of expected reward, in several overlapping regions including the striatum, precentral gyrus, parietal cortex and cerebellum in schizophrenia (Waltz et al., 2009). In line with this, group differences with respect to loss outcomes in our study were less widespread, with NTR patients showing attenuated RPE signaling only in the pallidum and caudate compared to HC. The findings support previous suggestions that prediction error related reinforcement learning deficits in schizophrenia stem primarily from abnormal processing of rewarding, rather than aversive, outcomes (Gold et al., 2012; Waltz et al., 2007; Dowd et al., 2016).
Encoding of prediction errors during reinforcement learning is extensively driven by dopaminergic function (Schultz, 1998). Although not all the regions found to encode prediction error in our study are densely innervated by dopaminergic projections, it is possible that a “global reinforcement signal” which is elicited by firing of dopamine neurons and broadcast through other regions of the brain (Schultz, 2002) indirectly modulates activation of structures with fewer direct connections to the dopamine system. An important criterion determining whether prediction error activation might reflect dopaminergic activity is a sign change for negative outcomes (Niv and Schoenbaum, 2008; Schultz, 2002), which was indeed observed in this study. The observed activation is therefore unlikely to reflect simple attentional or surprise processing. Group differences observed in the ROI analyses are highly likely to reflect dopaminergic functioning, given that the striatum and thalamus receive dense dopamine projections from the midbrain (Garcia et al., 2015; Groves et al., 1995; Schultz, 2002). Our findings thus imply that putatively dopamine-driven mechanisms underlying reinforcement learning in response to reward feedback are selectively disrupted in NTR. In contrast, the similar RPE-related activation pattern in TRS patients and HC suggests that reinforcement learning deficits in this patient group do not stem from dopaminergically driven RPE signaling dysfunctions. The data are consistent with the notion that TRS patients do not respond to dopaminergic antipsychotic medication because a dopaminergic abnormality is not the primary cause of symptoms in this subgroup (Demjaha et al., 2012). Importantly, medication dosage did not significantly differ between the two patient groups in our sample. Non-response to medication in the TRS group is unlikely to arise from a lower prescribed medication dosage compared to NTR patients as CPZ equivalent dosages were descriptively higher in the TRS group. However, due to the illness chronicity of patients included in our sample it was not possible to exhaustively ascertain the exact dosage and duration of all previous medication trials, thus cumulative medication exposure remains as a potential confound in this study.
Interestingly, groups did not differ in terms of their bias towards choosing the happy face over the angry face on emotional trials. However, there was a significant difference between TRS and NTR patients in how this bias was associated with RPE signal in the thalamus and caudate during loss processing. In NTR patients, a strong emotional bias was associated with further attenuation of the RPE signal. By comparison, emotional bias in TRS was associated with an increased RPE signal. In turn, RPE signal in this region was positively related to delusional symptom severity specifically in the TRS group. This is surprising as striatal RPE signal has previously been reported to be negatively linked with symptom severity in schizophrenia (Gradin et al., 2011; Culbreth et al., 2016; Schlagenhauf et al., 2009; Corlett et al., 2007); in line with the view that hyperdopaminergia-reflected in reduced RPE signaling-drives psychosis (Kapur, 2003). Our findings suggest that this relationship may be inverted in TRS patients in the thalamus and caudate. Increased RPE signaling specifically on loss trials may reflect less accurate predictions, resulting in greater prediction errors when the outcome is negative. As such, a strong social bias in TRS may lead to worse predictions about outcomes but an intact subcortical response to prediction error, which in turn is not adequately utilized to update predictions. In contrast, in NTR the prediction error response itself seems to be impaired, an effect which is further augmented in the presence of cognitive bias.
These data support a putative model of TRS whereby the central dysfunction lies not in the subcortical dopamine system itself, but in the implementation of cognitive control mechanisms interacting with this system. This control could be contributed to by glutamatergic mechanisms (Falkenberg et al., 2012; Taylor et al., 2015). The striatum and cortex are interconnected by multiple partially overlapping circuits subserving learning and flexible cognition (Kehagia et al., 2010). The ability to maintain behavioural goals in the presence of interference, uncertainty, or bias-broadly the definition of cognitive control-is an integral aspect of feedback learning (Collins and Frank, 2013; Ridderinkhof et al., 2004). A breakdown of this system may not only lead to reinforcement learning deficits, but also psychotic symptoms such as delusions as control processes are not adequately exerted in order to update internal models of the environment (Adams et al., 2013). Control-related regions such as prefrontal cortex, which also shows strong functional connectivity with the striatum (Di Martino et al., 2008), may indeed be involved in delusion formation and maintenance (Heinz and Schlagenhauf, 2010). Arguably, in the absence of an adequate cognitive control mechanism regulating bias, solely targeting subcortical dopamine with antipsychotics may not suffice to alleviate symptoms. In contrast, NTR patients may have sufficient cognitive control such that alleviating the striatal dysfunction is sufficient to reduce symptoms adequately.
Our study offers the first task-related neuroimaging evidence for differential caudate function in chronic TRS and NTR patients. It has been suggested that metabolic as well as anatomical abnormalities in the basal ganglia including the caudate nucleus are involved in TRS and may also be associated with clozapine response. For example, clozapine responders show hypermetabolism in the thalamus and basal ganglia, which is reduced following successful clozapine treatment (Rodriguez et al., 1996; Rodríguez et al., 1997). A reduction of metabolism specifically in the caudate after clozapine response was observed more recently (Molina et al., 2007) and clozapine administration is associated with a reduction of caudate volume (Chakos et al., 1995; Frazier et al., 1996; Scheepers et al., 2001a; Scheepers et al., 2001b). Notably, treatment responsive patients were found to have increased dopamine synthesis capacity compared to TRS (Demjaha et al., 2012), a finding which was most strong in the caudate nucleus. Thus the caudate may constitute an interesting target for further investigation of TRS in studies stratifying patient subgroups by response.
The study has certain limitations common to fMRI studies of a potential selection bias in medicated patients suitable for scanning; however, there are scant studies comparing TRS and NTR patients and withdrawal from medication for the purposes of imaging is not ethical. We did not include patients treated with clozapine in order to maintain the homogeneity of the patient sample and TRS patients fulfilled the standard criteria for treatment resistance-thus avoiding the introduction of sub-groups of patients refractory to clozapine (super-resistant patients). The differences in striatal RPE activation between groups are apparent at a liberal statistical threshold uncorrected for multiple comparisons; however, the consistent pattern of hypoactivation in NTR patients across the network lends support to this finding as a true positive. Subcortical dysfunctions in reward processing in NTR may be particularly hard to detect given that these may be attenuated in chronic patients after antipsychotic medication (Culbreth et al., 2016).
In summary, the data suggest that while the behavioral output during reward learning of patients with treatment resistant and treatment responsive schizophrenia appears to be similar, it is underpinned by different neural systems. The data support the idea that TRS may represent a different disease from treatment responsive schizophrenia; confirming the evidence from clinical observation that TRS does not fit well into the contemporary dopaminergic dysfunction model of schizophrenia. Despite extensive research on task-related neural activity in schizophrenia, studies typically do not use key stratifiers to reduce the heterogeneity of the sample and are likely combining neurobiologically distinct subtypes of schizophrenia. This not only clouds studies of mechanism, but potentially also of treatment trials; missing effects that are specific to one or the other subset of patients (Joyce et al., 2017). There is an urgent need for stratification of patients by response; both at the chronic stage of the illness and in patients suffering a first episode of psychosis. Indeed recent data following up first episode samples of patients with schizophrenia suggests that over 70% of treatment resistant cases are apparent at onset (Lally et al., 2016). The separation of schizophrenia subgroups will allow the development of clearer hypotheses into the neural mechanisms underlying antipsychotic treatment response and potentially move us closer to being able to use these biomarkers to tailor treatment in a more personalized and effective manner.
Supplementary Material
Acknowledgments
We thank the radiographic team at the Centre for Neuroimaging Sciences for their support, and Felix Dransfield, Christiana Ilesanmi, Valentina Forassi and Juliet Gillam for assistance with fMRI scanning and behavioral testing.
Financial Support
This research was funded by a European Research Council Grant to SSS (grant number 311686), who is supported by the National Institute for Health Research (NIHR) Mental Health Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London and a joint infrastructure grant from Guy’s and St Thomas’ Charity and the Maudsley Charity. LDV is supported by a Medical Research Council studentship.
Footnotes
Conflict of interest
None
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first Authors.
References
- Adams RA, Stephan KE, Brown HR, Frith CD & Friston KJ (2013). The computational anatomy of psychosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Carpenter WT Jr, Kane JM, Lasser RA, Marder SR & Weinberger DR (2005). Remission in schizophrenia: proposed criteria and rationale for consensus. American Journal of Psychiatry 162, 441–449. [DOI] [PubMed] [Google Scholar]
- Averbeck BB & Duchaine B (2009). Integration of social and utilitarian factors in decision making. Emotion 9, 599. [DOI] [PubMed] [Google Scholar]
- Barch DM & Dowd EC (2010). Goal representations and motivational drive in schizophrenia: the role of prefrontal-striatal interactions. Schizophrenia Bulletin 36, 919–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazire S (2005). Psychotropic drug directory. The professionals’ pocket handbook and aide memoire, Drug treatment options in psychiatric illness. Dementia, 40–48. [Google Scholar]
- Chakos M, Lieberman J, Alvir J, Bilder R & Ashtari M (1995). Caudate nuclei volumes in schizophrenic patients treated with typical antipsychotics or clozapine. The Lancet 345, 456–457. [DOI] [PubMed] [Google Scholar]
- Chang WC, Waltz JA, Gold JM, Chan TCW & Chen EYH (2016). Mild Reinforcement Learning Deficits in Patients With First-Episode Psychosis. Schizophrenia Bulletin, sbw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AG & Frank MJ (2013). Cognitive control over learning: creating, clustering, and generalizing task-set structure. Psychological Review 120, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley RR & Kelly DL (2001). Management of treatment resistance in schizophrenia. Biological Psychiatry 50, 898–911. [DOI] [PubMed] [Google Scholar]
- Coppens HJ, Slooff CJ, Paans AM, Wiegman T, Vaalburg W & Korf J (1991). High central D 2-dopamine receptor occupancy as assessed with positron emission tomography in medicated but therapy-resistant schizophrenic patients. Biological Psychiatry 29, 629–634. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Murray GK, Honey GD, Aitken MR, Shanks DR, Robbins TW, Bullmore ET, Dickinson A & Fletcher PC (2007). Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain 130, 2387–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbreth AJ, Westbrook A, Xu Z, Barch DM & Waltz JA (2016). Intact ventral striatal prediction error signaling in medicated schizophrenia patients. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging 1, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM & Mcguire PK (2014). Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biological Psychiatry 75, e11–e13. [DOI] [PubMed] [Google Scholar]
- Demjaha A, Murray RM, Mcguire PK, Kapur S & Howes OD (2012). Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. American Journal of Psychiatry. [DOI] [PubMed] [Google Scholar]
- Deserno L, Schlagenhauf F & Heinz A (2016). Striatal dopamine, reward, and decision making in schizophrenia. Dialogues in Clinical Neuroscience 18, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino A, Scheres A, Margulies DS, Kelly A, Uddin LQ, Shehzad Z, Biswal B, Walters JR, Castellanos FX & Milham MP (2008). Functional connectivity of human striatum: a resting state FMRI study. Cerebral Cortex 18, 2735–2747. [DOI] [PubMed] [Google Scholar]
- Dowd EC, Frank MJ, Collins A, Gold JM & Barch DM (2016). Probabilistic reinforcement learning in patients with schizophrenia: Relationships to anhedonia and avolition. Biological psychiatry: cognitive neuroscience and neuroimaging 1, 460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S, Fleming SM, Dolan RJ & Averbeck BB (2011a). Effects of emotional preferences on value-based decision-making are mediated by mentalizing and not reward networks. Journal of Cognitive Neuroscience 23, 2197–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S, Shergill SS, Chouhan V, Bristow E, Collier T & Averbeck B (2011b). Patients with schizophrenia show increased aversion to angry faces in an associative learning task. Psychological Medicine 41, 1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg LE, Westerhausen R, Specht K & Hugdahl K (2012). Resting-state glutamate level in the anterior cingulate predicts blood-oxygen level-dependent response to cognitive control. Proceedings of the National Academy of Sciences 109, 5069–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ & Claus ED (2006). Anatomy of a decision: striato-orbitofrontal interactions in reinforcement learning, decision making, and reversal. Psychological Review 113, 300. [DOI] [PubMed] [Google Scholar]
- Frank MJ, Loughry B & O’reilly RC (2001). Interactions between frontal cortex and basal ganglia in working memory: a computational model. Cognitive, Affective, & Behavioral Neuroscience 1, 137–160. [DOI] [PubMed] [Google Scholar]
- Frazier JA, Giedd JN, Kaysen D, Albus K, Hamburger S, Alaghband-Rad J, Lenane MC, Mckenna K, Breier A & Rapoport JL (1996). Childhood-onset schizophrenia: brain MRI rescan after 2 years of clozapine maintenance treatment. American Journal of Psychiatry 153, 564–566. [DOI] [PubMed] [Google Scholar]
- Garcia GJ, Chagas MH, Silva CH, Machado-De-Sousa JP, Crippa JA & Hallak JE (2015). Structural and functional neuroimaging findings associated with the use of clozapine in schizophrenia: a systematic review. Revista Brasileira de Psiquiatria 37, 71–79. [DOI] [PubMed] [Google Scholar]
- Garrison J, Erdeniz B & Done J (2013). Prediction error in reinforcement learning: a meta-analysis of neuroimaging studies. Neuroscience and Biobehavioral Reviews 37, 1297–1310. [DOI] [PubMed] [Google Scholar]
- Gold JM, Waltz JA, Matveeva TM, Kasanova Z, Strauss GP, Herbener ES, Collins AG & Frank MJ (2012). Negative symptoms and the failure to represent the expected reward value of actions: behavioral and computational modeling evidence. Archives of General Psychiatry 69, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradin VB, Kumar P, Waiter G, Ahearn T, Stickle C, Milders M, Reid I, Hall J & Steele JD (2011). Expected value and prediction error abnormalities in depression and schizophrenia. Brain, awr059. [DOI] [PubMed] [Google Scholar]
- Groves PM, Garcia-Munoz M, Linder JC, Manley MS, Martone ME & Young SJ (1995). Elements of the Intrinsic Organization. Models of information processing in the basal ganglia, 51. [Google Scholar]
- Heinz A & Schlagenhauf F (2010). Dopaminergic dysfunction in schizophrenia: salience attribution revisited. Schizophrenia Bulletin 36, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce DW, Kehagia AA, Tracy DK, Proctor J & Shergill SS (2017). Realising stratified psychiatry using multidimensional signatures and trajectories. Journal of Translational Medicine 15, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S (2003). Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. American Journal of Psychiatry. [DOI] [PubMed] [Google Scholar]
- Kapur S & Seeman P (2001). Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics?: A new hypothesis. American Journal of Psychiatry 158, 360–369. [DOI] [PubMed] [Google Scholar]
- Kay SR, Flszbein A & Opfer LA (1987). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 13, 261. [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Murray GK & Robbins TW (2010). Learning and cognitive flexibility: frontostriatal function and monoaminergic modulation. Current Opinion in Neurobiology 20, 199–204. [DOI] [PubMed] [Google Scholar]
- Koch K, Schachtzabel C, Wagner G, Schikora J, Schultz C, Reichenbach JR, Sauer H & Schlösser RG (2010). Altered activation in association with reward-related trial-and-error learning in patients with schizophrenia. Neuroimage 50, 223–232. [DOI] [PubMed] [Google Scholar]
- Lally J, Ajnakina O, Di Forti M, Trotta A, Demjaha A, Kolliakou A, Mondelli V, Marques TR, Pariante C & Dazzan P (2016). Two distinct patterns of treatment resistance: clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychological Medicine 46, 3231. [DOI] [PubMed] [Google Scholar]
- Lindenmayer J-P (2000). Treatment refractory schizophrenia. Psychiatric Quarterly 71, 373–384. [DOI] [PubMed] [Google Scholar]
- Molina V, Sanz J, Sarramea F & Palomo T (2007). Marked hypofrontality in clozapine-responsive patients. Pharmacopsychiatry 40, 157–162. [DOI] [PubMed] [Google Scholar]
- Mortimer A, Singh P, Shepherd C & Puthiryackal J (2010). Clozapine for treatment-resistant schizophrenia: National Institute of Clinical Excellence (NICE) guidance in the real world. Clinical Schizophrenia & Related Psychoses 4, 49–55. [DOI] [PubMed] [Google Scholar]
- Murray G, Corlett P, Clark L, Pessiglione M, Blackwell A, Honey G, Jones P, Bullmore E, Robbins T & Fletcher P (2008). Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Molecular Psychiatry 13, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niv Y & Schoenbaum G (2008). Dialogues on prediction errors. Trends in cognitive sciences 12, 265–272. [DOI] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL & Petersen SE (2012). Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 59, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinen JM, Van Snellenberg JX, Horga G, Abi-Dargham A, Daw ND & Shohamy D (2016). Motivational Context Modulates Prediction Error Response in Schizophrenia. Schizophrenia Bulletin, sbw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridderinkhof KR, Van Den Wildenberg WP, Segalowitz SJ & Carter CS (2004). Neurocognitive mechanisms of cognitive control: the role of prefrontal cortex in action selection, response inhibition, performance monitoring, and reward-based learning. Brain and Cognition 56, 129–140. [DOI] [PubMed] [Google Scholar]
- Rodriguez VM, Andree RM, Castejön M & Garcia EC (1996). SPECT study of regional cerebral perfusion in neuroleptic-resistant schizophrenic patients who responded or did not respond to clozapine. The American journal of psychiatry 153, 1343. [DOI] [PubMed] [Google Scholar]
- Rodríguez VM, Andrée RM, Castejön MJP, Zamora MLC, Alvaro PC, Delgado JLC & Vila FJR (1997). Fronto-striato-thalamic perfusion and clozapine response in treatment-refractory schizophrenic patients. A 99m Tc-HMPAO study. Psychiatry Research: Neuroimaging 76, 51–61. [DOI] [PubMed] [Google Scholar]
- Scheepers FE, De Wied CCG, Pol HEH, Van De Flier W, Van Der Linden JA & Kahn RS (2001a). The effect of clozapine on caudate nucleus volume in schizophrenic patients previously treated with typical antipsychotics. Neuropsychopharmacology 24, 47–54. [DOI] [PubMed] [Google Scholar]
- Scheepers FE, Gispen De Wied CC, Hulshoff Pol HE & Kahn RS (2001b). Effect of clozapine on caudate nucleus volume in relation to symptoms of schizophrenia. American Journal of Psychiatry 158, 644–646. [DOI] [PubMed] [Google Scholar]
- Schlagenhauf F, Huys QJ, Deserno L, Rapp MA, Beck A, Heinze H-J, Dolan R & Heinz A (2014). Striatal dysfunction during reversal learning in unmedicated schizophrenia patients. Neuroimage 89, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagenhauf F, Sterzer P, Schmack K, Ballmaier M, Rapp M, Wrase J, Juckel G, Gallinat J & Heinz A (2009). Reward feedback alterations in unmedicated schizophrenia patients: relevance for delusions. Biological Psychiatry 65, 1032–1039. [DOI] [PubMed] [Google Scholar]
- Schultz W (1998). Predictive reward signal of dopamine neurons. Journal of Neurophysiology 80, 1–27. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002). Getting formal with dopamine and reward. Neuron 36, 241–263. [DOI] [PubMed] [Google Scholar]
- Schultz W & Dickinson A (2000). Neuronal coding of prediction errors. Annual Review of Neuroscience 23, 473–500. [DOI] [PubMed] [Google Scholar]
- Seeman P & Lee T (1975). Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188, 1217–1219. [DOI] [PubMed] [Google Scholar]
- Seeman P, Lee T, Chau-Wong M & Wong K (1976). Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 261, 717–719. [DOI] [PubMed] [Google Scholar]
- Simon JJ, Biller A, Walther S, Roesch-Ely D, Stippich C, Weisbrod M & Kaiser S (2010). Neural correlates of reward processing in schizophrenia—relationship to apathy and depression. Schizophrenia Research 118, 154–161. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I & Flitney DE (2004). Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23, S208–S219. [DOI] [PubMed] [Google Scholar]
- Taylor R, Neufeld RW, Schaefer B, Densmore M, Rajakumar N, Osuch EA, Williamson PC & Théberge J (2015). Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color-word Stroop task. NPJ schizophrenia 1, 15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery TJ, Chun MM & Lee D (2011). Ubiquity and specificity of reinforcement signals throughout the human brain. Neuron 72, 166–177. [DOI] [PubMed] [Google Scholar]
- Waltz JA, Frank MJ, Robinson BM & Gold JM (2007). Selective reinforcement learning deficits in schizophrenia support predictions from computational models of striatal-cortical dysfunction. Biological Psychiatry 62, 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz JA, Frank MJ, Wiecki TV & Gold JM (2011). Altered probabilistic learning and response biases in schizophrenia: behavioral evidence and neurocomputational modeling. Neuropsychology 25, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz JA, Schweitzer JB, Gold JM, Kurup PK, Ross TJ, Salmeron BJ, Rose EJ, Mcclure SM & Stein EA (2009). Patients with schizophrenia have a reduced neural response to both unpredictable and predictable primary reinforcers. Neuropsychopharmacology 34, 1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz JA, Schweitzer JB, Ross TJ, Kurup PK, Salmeron BJ, Rose EJ, Gold JM & Stein EA (2010). Abnormal responses to monetary outcomes in cortex, but not in the basal ganglia, in schizophrenia. Neuropsychopharmacology 35, 2427–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D (1999). WASI manual. San Antonio, Psychological Corporation. [Google Scholar]
- Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue C-Y, Cooper TB & Brodie JD (1989). Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. American Journal of Psychiatry 146, 905–908. [DOI] [PubMed] [Google Scholar]
- Woods SW (2003). Chlorpromazine equivalent doses for the newer atypical antipsychotics. Journal of Clinical Psychiatry 64, 663–667. [DOI] [PubMed] [Google Scholar]
- Yacubian J, Gläscher J, Schroeder K, Sommer T, Braus DF & Büchel C (2006). Dissociable systems for gain-and loss-related value predictions and errors of prediction in the human brain. The Journal of Neuroscience 26, 9530–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.