

Abstract
Individuals at high risk for the future development of seropositive rheumatoid arthritis (RA) can be readily identified for translational research and disease prevention studies through the presence of highly informative and predictive patterns of RA-related autoantibodies, especially anti-citrullinated protein antibodies (ACPA), in the serum. Recent findings in serologically positive individuals without arthritis, designated ACPA+ At-Risk, have demonstrated the presence of mucosal inflammatory processes associated with the presence of local ACPA production. In other At-Risk populations, local RA-related autoantibody production is present even in the absence of serum autoantibodies. Additionally, a proportion of At-Risk individuals exhibit in the lung local mucosal ACPA production, radiographic small airways disease and sputum hyper-cellularity as well as increased neutrophil extracellular trap (NET) formation. Other mucosal sites in At-Risk individuals also exhibit autoantibody production, inflammation and/or evidence of dysbiosis. As the proportion of individuals who exhibit such localized inflammation-associated ACPA production is substantially higher than the likelihood of an individual developing future RA, this raises the hypothesis that these antibodies are generated in the mucosa to play biologically relevant protective roles. Identifying the mechanisms that drive both the generation and loss of externally focused mucosal ACPA production and promote systemic autoantibody expression and ultimately arthritis development should provide insights into new therapeutic approaches to prevent RA.
Introduction
It is now known that seropositive rheumatoid arthritis (RA), which occurs in individuals who have both genetic and familial risk factors for this disease1–4, begins as a prolonged multi-year period of serologically-detectable RA-related autoimmunity that is associated with systemic inflammatory biomarkers but the absence of clinical or histologically defined inflammatory arthritis5–9. Serum autoantibodies present at this time include multiple isotypes of both anti-citrullinated peptide/protein antibodies (ACPA) and Fc domain-recognizing rheumatoid factors (RF) in patterns highly predictive of the future onset of joint disease5,6,9,10. This period can be defined retrospectively as the “pre-clinical” period of RA in subjects who eventually develop the disease11 and an At- Risk status in the population when studied in a cross-sectional or longitudinal manner prior to arthritis. This condition is then followed by the development of early clinically detectable inflammatory arthritis (IA) and/or classified RA, the latter of which itself proceeds as a chronic arthritis and disease process that can also involve the lung and other target organs.
Epidemiologic and translational research studies of patients with early (< 1 year from diagnosis) or longer standing active RA suggest that mucosal exposures and/or dysbiosis may have played causal roles during the At-Risk state in the development of RA2,12–16. However, despite the support for a “mucosal origins hypothesis”, ie that RA development begins at mucosal sites and then transitions to involve the synovial joints, these study designs are limited in that they do not allow one to identify the specific mucosal processes present at the prior time point(s) during which such exposures would have influenced the concurrent autoimmune phenotypes as well as subsequent disease evolution and arthritis development. In this regard, it is also often assumed that a key factor in early RA development is the loss of self-tolerance to citrullinated self-antigens. However, studies of At-Risk as well as control populations strongly suggest that mucosal, and likely systemic, IgA isotype ACPA and RF generation is a rather common finding and is associated with local mucosal inflammation17,18. These findings suggest that the likely most important early event in the pre-clinical development of RA is not a loss of tolerance to self-antigens but rather the loss of the mucosal barrier function and systemic spread of an IgG ACPA response. With those issues in mind, and to address the mucosal origins hypothesis in the context of findings in subjects At-Risk for future RA, this manuscript will review the available evidence linking the presence of RA-related autoimmunity in specific At-Risk populations with the concurrent presence of local mucosal inflammation and autoantibody production, mucosal dysbiosis and/or systemic immune dysregulation consistent with mucosal inflammation.
Development of seropositive RA
RA is an autoimmune disease that evolves over decades and is manifest as both a locally joint destructive and systemic illness4,19. The disease is characterized by widespread inflammation as well as local joint-based signs and symptoms ranging from arthralgias, a term which has recently been specifically defined for research purposes to encompass seven factors (symptom duration <1 year, symptoms of metacarpophalangeal (MCP) joints, morning stiffness duration ≥60 minutes, most severe symptoms in early morning, first-degree relative (FDR) with RA, difficulty with making a fist, and a positive squeeze test of MCP joints)20, to swelling, pain and inflammatory synovitis that are clinically definable through techniques such as physical examination, imaging and synovial biopsy [reviewed in19,21]. Osteopenia and local erosions commonly occur in RA, likely through inflammatory and remodeling processes that alter relative rates of bone formation and degradation by osteoblasts and osteoclasts, respectively22.
RA exhibits a prevalence of 0.5–0.8% in the general population, and an ~3–5-fold increase in FDRs [reviewed in23] which appears to be due to both genetic and environmental influences24–26. Patients receive a diagnosis of RA based on the 1987 revised ACR or 2010 ACR/EULAR classification criteria27,28. In some patients RA first appears clinically as an undifferentiated inflammatory arthritis (IA)29,30.
The term ‘RA’ encompasses two major subsets of disease, seropositive and seronegative, that exhibit overlapping but individually distinct pathogenic mechanisms and clinical courses [reviewed in21,26,31]. Seropositive individuals exhibit substantial overlap of expression of two primary autoantibody systems: 1) ACPAs, with this posttranslational modification found in an antigenic form on fibrinogen, vimentin, type II collagen, histones and enolase32,33, and 2) antibodies to the Fc domains of self Ig molecules, designated rheumatoid factors (RF)34. Additional autoantibodies to modified protein antigens (AMPA) [reviewed in 35], such as anti-carbamylated protein antibodies36 and antibodies to malondialdehyde-acetaldehyde modified epitopes37, have been identified in patients with RA, although the specificity for RA of AMPA and at what point in the pre-clinical to clinically apparent disease transitions AMPA develop are less well defined features38,39.
RA can be driven by a variety of genetic and environmental factors that lead to a heterogeneous disease process with variable autoantibody positivity. In terms of genetic factors, there are many genes which associate with RA, including high risk HLA-DR alleles containing the shared epitope (SE)40 and PTPN2241, as well as many other genes with low relative risk variants42. Systemic targeting of citrullinated proteins is likely facilitated by the preferential interactions of peptides bearing citrulline with SE-containing DR molecules43–45. Heterogeneity in RA disease phenotypes is also likely to be influenced by environmental factors25,46. In addition, there are epigenetic changes that are associated with RA and affect lymphocytes, fibroblast like synoviocytes (FLS) and other cell populations3,4,47,48. These epigenetic changes represent key means by which environmental factors could influence gene expression and disease heterogeneity.
In subjects who eventually develop seropositive RA, there is a prolonged asymptomatic pre-clinical period without evidence of joint involvement that is characterized by the presence of serologically detected ACPA, measured through clinical tests designated anti-CCP (cyclic citrullinated peptide) antibodies, and RF in a process that typically occurs for 3–5 years prior to the onset of clinically apparent and classified disease [reviewed in49] (FIG. 1). Shortly before the onset of clinically apparent arthritis, systemic levels of cytokines/chemokines increase in a very heterogenous manner, both with regard to the number and the pattern of individual mediators that are elevated in an individual subject10. An increase in epitope spreading also occurs which stably encompasses a wider variety of citrullinated targets9,50, and avidity maturation of autoantibodies is present51.
Figure 1. Biomarker studies relevant to pre-clinical RA pathogenesis.
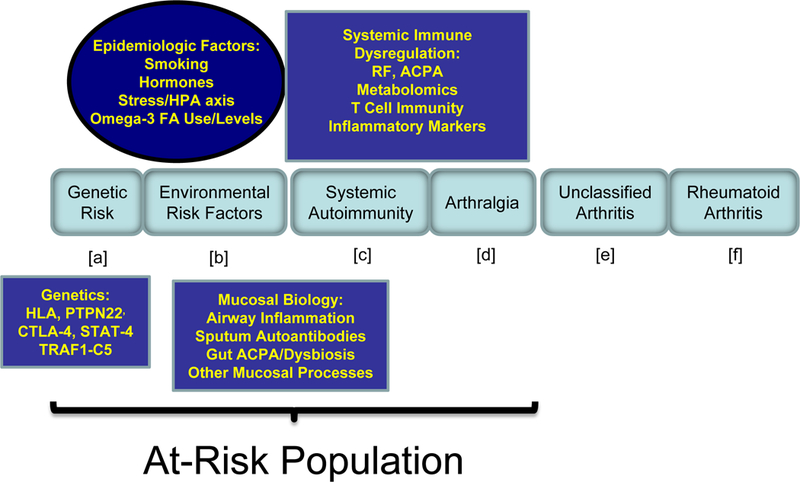
Shown in the middle are EULAR recommendations for definition of disease stages (a – f) in the natural history of RA, with the pre-clinical stages bracketed. Boxes above and below the natural history stages include experimental approaches that are discussed herein and utilized through the pre-clinical period to understand the causal drivers of pre-clinical disease. Study populations include retrospective serum samples in addition to analyses of biomarkers in At-Risk populations.
In addition, effector functions of ACPA evolve during the development of RA51,52. For example, it has been shown that altered ACPA Fc domain galactosylation and fucosylation53, as well as excessive Fab glycosylation54, is a common finding in patients with RA and those who are transitioning from the arthralgia state to IA and classified RA. Notably, recent studies have suggested that IL-23 and Th17 cells regulate the β-galactoside α2,6-sialyltransferase 1 activity in B cells that controls the extent of glycosylation of Ig molecules and their pathogenic potential, and that changes in these cytokines drive the disease-enhancing changes in secreted autoantibodies during the clinical transition to arthritis55. Importantly, autoantibodies have also been identified in patients with RA that demonstrate effector functions related to osteoclastogenesis56 and joint pain secondary to chemokine release57, although the point during disease development during which these appear is less certain. Perhaps most intriguingly relative to mucosal studies, it has been found that IgA ACPA and RF isotypes long precede the development of classified RA58, although the relative timing of appearance of each serum RA-related autoantibody isotype is not yet clearly defined. In addition, IgA RF contains in patients with RA a high proportion of mucosally-related secretory chain59, although the time point of appearance of this feature is again uncertain.
Serologic immune alterations during disease evolution
Studies in the pre-clinical time period have primarily relied on two experimental approaches. The first has been to utilize annotated sera that have been collected previously for other purposes, the origin of which includes blood banks and population studies5,6,8,60 as well as the United States Department of Defense Serum Repository7. As summarized above, despite the inherent limitations of subjects not being studied and classified with regard to arthritis status at the same time as the sample acquisition, many insights have been generated from these approaches. However, a more recent approach has been to perform studies of individuals with an ACPA+ At-Risk status, which is a method that has increasingly helped to define how the earliest immunologic abnormalities develop and what may be the early “drivers” of disease. These individuals are recruited through many different means, including recruitment of higher risk first-degree relatives (FDRs)61,62, outreach at community health fairs or other events to identify serum ACPA+ At-Risk subjects63 and the creation of referral networks to specifically identify subjects in primary care with arthralgias suggestive of pre-clinical RA who are subsequently followed for IA and classified RA evolution64. Ongoing population recruitment efforts include screening of biobanks and electronic health records, social media outreach, and the increasing referrals to rheumatologists of patients without IA or RA who are found to be ACPA+.
This heterogeneity in sources of samples may lead to some differences in the findings from retrospective studies. Nevertheless, a number of important observations have been made using these approaches (BOX 1). Key findings in serum samples from studies of At-Risk populations include the presence of elevated chemokines/cytokines/inflammatory biomarkers62,65,66 and altered T cell phenotypes67,68 as well as expanded autoantibodies61,65,69–71. Importantly, many of these biomarker patterns mirror those found in the serum from retrospectively studied populations and strongly support that these individuals are indeed in a pre- clinical period of RA development. Results of additional phenotypic studies are described below.
Box 1. Findings in At-Risk Populations Supportive of the Mucosal Origins Chronic Inflammation Hypothesis
Serologic:
-
-
Elevated levels of, and expanded epitope recognition by, IgA and IgG ACPA
-
-
Elevated levels of cytokines and chemokines
-
-
Elevated CRP
-
-
Elevated antibodies to P. gingivalis
Peripheral Blood Cell Phenotypes:
-
-
Elevated proportion of circulating IgA-encoding plamablasts
-
-
Elevated citH3, stimulated IL-17 and IL-2 levels, and decreased stimulated ATM and PFKFB3 levels
Findings at Mucosal Sites:
-
-
Lung High Resolution Computerized Tomography demonstrating small airways diseases
-
-
Elevated RA-related autoantibodies in sputum of At-Risk individuals
-
-
Hypercellularity and increased NET remnants in sputum of At-Risk individuals
-
-
Periodontal production of ACPA in patients with periodontitis
-
-
Cervicovaginal ACPA production
Key Points
Patients who eventually develop seropositive RA pass through a period of RA-related autoantibody positivity that is associated with elevated cytokines and chemokines
There are multiple means by which mucosal processes can influence the development of systemic immunity and autoimmunity
Individuals who are at high risk for the future development of RA demonstrate evidence of chronic systemic and mucosal inflammation
Rather than reflecting a loss of citrullinated antigen tolerance, evidence suggests that IgA ACPA are normally produced locally, and a systemic IgG response results from loss of externally focused compartmentalization
Ongoing studies are linking the development of mucosal dysbiosis, inflammation and autoantibody production to the next stages of development of systemic autoimmunity
Novel autoimmune-promoting processes are likely to be identified that act primarily, or exclusively, in the pre-clinical period of RA and could be the targets for new prevention strategies
Stage-specific environmental factors
The potential roles of environmental exposures in the development of RA have been extensively reviewed23,25. With regard to the At-Risk population, though, very few long-term prospective studies have been performed whose goal would be to understand how stage-specific exposures influence the initial pre-clinical development of RA-related autoimmunity and subsequent clinical and/or immune transitions. Nevertheless, a number of informative cross-sectional and short-term longitudinal studies have been performed in these subjects (TABLE 1).
Table 1. Environmental factors affecting the natural history of RA.
Environmental exposures and effects that have been shown to impact the total and/or stage-specific course of RA development.
| Environmental exposure | Patients with RA/Predominant Direction of Risk |
Subjects At-Risk of developing RA and/or with pre-clinical RA/Predominant Direction of Risk |
|---|---|---|
| Smoking | Yes/Increase | Yes/Increase |
| Omega-3 FA use | Yes/Decrease | Yes/Decrease |
| Oral contraceptive use | Yes/Decrease | Yes/Decrease |
| Breast-feeding | Yes/Decrease | Uncertain |
| Earlier age at menarche | Yes/Variable | Uncertain |
| Body mass index | Yes/Increase | Yes/Increase |
| Decaf coffee consumption | Yes/Increase | Uncertain |
| Silica dust, solvents, mineral oil, textile dust | Yes/Increase | Yes/Increase |
| Physical workload | Yes/Increase | Uncertain |
| Proximity to road/Air pollution | Yes/Increase | No |
| UV Light | Yes/Decrease | Uncertain |
| Alcohol | Yes/Decrease | Uncertain |
A well-accepted association in RA is the relationship between heavy smoking and the risk of development of seropositive RA, with the predominant risk being in subjects with SE26. The exact stage- specific timing of smoking effects during the pre-clinical to classified arthritis natural history of RA is not well understood, but several studies have provided insights. For example, initial studies of At-Risk subjects in a well studied cohort evaluated in real time have revealed that heavy smoking is associated with asymptomatic RF positivity72. In a Swedish twin study, heavy smoking impared a higher risk of being ACPA+ without RA, and without a relationship to SE in the preclinical period73. In addition, in a large (>40,000 subjects) population study in the Netherlands, an ACPA+ status was associated with smoking as well as older age and joint symptoms74. Further support for a role of smoking exposure in the future development of RA was provided by a Swedish population study where smoking was associated with an increased risk of development of both seropositive and seronegative RA75. Similarly, in a Japanese non-RA cohort, a history of smoking was associated with a positive RA-related autoantibody status, but interesting only when both ACPA and RF were present76. In a further analysis of the same population, ACPA and RF were shown to associate with each other, and smoking was associated with a higher level of ACPA, but the presence of SE was not associated with an ACPA+ status, and no interaction was found between SE, smoking and autoantibody positivity in the non-RA population77. Thus, it appears that smoking plays an important role in the initial development of ACPA, but without a clear relationship to SE status, which itself appears to play a more important role in the transition to classified RA.
With regard to other factors, a history of birth control pill use is inversely associated with the presence of RF positivity72. These results are consistent with findings reported when studying patients with classified RA and suggest that these environmental exposures can play key early roles in pre-clinical RA.
Conversely, particulate air pollution was not found to be associated with RA-related autoimmunity in At-Risk subjects78 in the same manner as patients with classified RA79, suggesting that this exposure may play a later role in the development of RA, perhaps promoting the transition from RA-related autoantibodies alone to the initial synovitis. Additional results from the Nurses Health Studies have suggested that an increased body mass index (BMI) in conjunction with expanded ACPA repertoire is associated with elevated future risk of developing RA and accelerated transition to arthritis80. Studies from the Epidemiologic Investigation of RA (EIRA) have also suggested that exposure to silica increases the risk of future development of RA81. Other occupational exposures, hormonal factors, dietary intake and alcohol use have demonstrated variable results, and the roles of these exposures in the pre-clinical stages of diseases are less well understood [reviewed in25].
An intriguing finding has been an inverse relationship between the presence of RA-related autoantibodies in At-Risk populations and intake of omega-3 fatty acids as well as lipid biomarkers that measure levels of these key anti-inflammatory molecules. Because of the potential relationship of this exposure to the chronic local inflammation that underlies the mucosal origins hypothesis, these findings are reviewed in more depth. Notably, there is both established and emerging evidence that poly-unsaturated fatty acids (PUFAs) derived from the omega-3 FA biosynthesis pathway may play an important role in RA treatment and prevention. Omega-3 PUFA supplementation in patients with classified RA has been shown to reduce signs and symptoms of inflammation and lead to decreased use of other therapeutic agents82,83. Population-based approaches have demonstrated that increased intake of fish rich in omega-3 PUFAs or direct supplementation with PUFAs are associated with a reduced risk for developing RA84. With regard to At-Risk subjects, it has recently been shown that increased omega-3 FA intake as well as higher levels of the red blood cell (RBC) omega-3 FA pathway eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are associated with a decreased risk of RA-related autoantibody positivity85. Specifically, in those studies the likelihood of anti-CCP2 positivity in subjects was inversely associated with total omega-3 FA% in RBCs (odds ratio: 0.47; 95% CI 0.24, 0.92, for a one standard deviation increase), and anti-CCP2 positive cases were less likely than controls to report omega-3 FA supplement use (odds ratio: 0.14; 95% CI 0.03, 0.68). Intriguingly, the strongest effect was found in SE+ individuals86. In further observational studies of ACPA+ subjects without synovitis at baseline, lower levels of RBC omega-3 FAs were associated with increased risk for progression to IA87.
Mucosal origins hypothesis
The mucosal origins hypothesis suggests that the disease process in subjects who eventually develop RA originates at one or more sites in the mucosa. The theory historically derived from an early suggestion that RA was an infectious illness, a conclusion that was subsequently been shown to be incorrect. Later suggestions included the possibility that RA developed through a cross-reactivity to bacterial, viral and/or mycobacterial antigens carried in such organisms as Proteus [reviewed in88]. More recently, the finding that Porphyromonas gingivalis, a major cause of periodontitis, encodes an enzyme capable of citrullinating proteins, including enolase, and cross-reactivity of these targets with patient ACPA suggested another means by which mucosal dysbiosis could drive or influence disease development89. Nevertheless, the hypothesis remains unproven, although an increasing number of studies are consistent with a major role for the microbiome in RA development.
The microbiome and autoimmunity
Increasing evidence from both murine and human translational research studies points to the important role of the microbiome that is present in the intestine and other mucosal as well as epithelial sites in modulating the immune, as well as autoimmune, response [reviewed in90]. Food intake and other environmental exposures play major roles in the development and maintenance of microbial kingdoms, including bacterial, fungal, helminth and viral organisms. Major effects on the immune system are played by mucosal exposure to specific microbial antigens as well as metabolic products of biosynthesis and energy generation/utilization2,91.
There are many challenges to the study of the microbiome in humans, including lack of direct access to most mucosal sites, difficulty in isolating and expanding many species in a manner that replicates the local mucosal ecosystem, an incomplete understanding of microbial genotype:phenotype relationships, substantial changes introduced to the microbiome by unrealized food exposures such as dietary emulsifiers and non-caloric sweeteners, long-term imprinting by the fetal microbiome whose effects cannot be discerned years later, cultural differences that affect microbial communities, and the rapid changes that can ensue following alterations in the environment which are perhaps best demonstrated by the effects of changing food intake on the microbial community in the gastrointestinal tract90. Nevertheless, studies of murine models of disease and an emerging number of human translational research studies have pointed to a causal or modulating role for the microbiome in human autoimmune disease.
Cross-talk with the local immune system
Although they may vary from one organ to another, there are a number of means by which the local mucosal immune system can interact with, respond to and constrain the microbiome (FIG. 2). These include induction and inhibition mechanisms that are strongly influenced by cells of the adaptive immune system as well as distinct types of mononuclear phagocytes90. An important feature of the cross-talk process between the immune system and the microbiome is the mucosal barrier, which is created not only by inter-epithelial cell tight junctions but also by the integrity of the mucous layer and serves as a component of the “mucosal firewall” [reviewed in 92]. Additional components of the firewall include anti-microbial peptides, encompassing in part the microbicidal protein Regllly and related families93, antibodies, macrophages, dendritic cells, Treg and Th17 T cells. Also important in mucosal defense from infection are neutrophils, which are attracted to the lumen through polarized secretion by epithelial cells of IL-8 as well as bioactive lipids, especially hepoxilin A3 [reviewed in94].
Figure 2. Local immune system and microbial factors that interact in the mucosal environment.
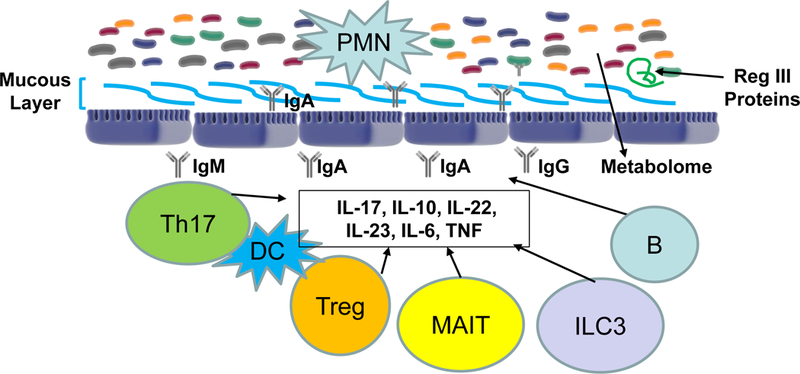
Illustrated are components of the bi-directional regulation between the luminal microbiome and the cells within the mucosa itself, with several cell types contributing individually to produce local cytokines. Specific examples are described in the text, but for example MAIT cells produce IL-17 while ILC3 are responsible for elaborating IL-22. PMN: polymorphonuclear neutrophils.
CD4 and CD8 T lymphocytes also continuously surveil antigens in this region, responses to which are regulated by cytokines including IL-17, IL-10, IL-22, IL-23, IL-6 and TNF family members, in addition to other populations such as Treg cells. In addition, specialized T lymphocytes defined as MAIT (mucosal associated invariant T) cells, whose innate immune functions and activation state are intimately tied to MR1 ligands that are elaborated during mucosal infection and inflammation, are present and produce IL-17 and Th1 cytokines95. Type 3 innate lymphoid cells (ILC3) contribute to the mucosal immune response through production of IL-22, a cytokine that stimulates production of RegIII lectins in response to dysbiosis96 as well as controls epithelial cell fucosylation and through that mechanism provides a carbohydrate source utilized by certain microbial subsets97. Also present are mucosal homing receptor98 α4β7high CD4+ T cells that are CCR5high and CXCR4low and which undergo antigen-specific activation in the development of mucosal inflammation99.
With regard to antibodies, IgA is the major Ig isoform produced at most mucosal surfaces, and while the circulating blood levels are low, IgA is the most highly produced Ig isoform in the body100. Importantly, both T-dependent and T-independent means regulate the production of this mucosal isoform101. IgA exists as monomeric and polymeric forms of IgA1 and IgA2 sub-isotypes as well as polymeric IgA linked by the J chain. The IgA receptor (IgAR1) is found on neutrophils, monocytes, eosinophils and a subset of dendritic cells, and IgA functions to enhance the respiratory burst and inhibit bacterial adhesion.
There are several contrasting roles ascribed to this isotype. Its primary role is to recognize and neutralize pathogens and exotoxins at the mucosal surface, and in that way to regulate the microbiome, maintain homeostasis and protect intestinal and other barriers from pathogens. A key mechanism is to block infection and invasion of the mucosa through exclusion from transport across the epithelium and clearance by peristalsis in the gut102. IgA plasmablasts are primarily generated in Peyer’s Patches and can circulate to secrete Ab at other mucosal sites. While IgA is view primarily in the context of mucosal immunity, when present systemically at high levels in immune complexes, it can damage target tissues, including the joint103.
In another sense, secreted IgA antibodies can be considered to be non-inflammatory as they are typically not strongly complement activating and are resistant themselves to proteolytic degradation104. Thus, the pro- versus anti-inflammatory roles of IgA must be considered to be context-dependent, and likely co-existing. Beyond IgA, there are prominent roles for IgM antibodies as well as IgG antibodies and their bidirectional transport by the neonatal Fc receptor FcRn in the regulation of the microbiome and control of local pathogens, perhaps most prominently in the cervicovaginal fluid [reviewed in105]. Notably, bacteria which penetrate the mucosal “demilitarized zone” are recognized by locally produced IgA isotype antibodies that are typically polyreactive106, and if they avoid neutralization and elimination can more readily cross the epithelial layer and have antigens be presented by dendritic cells107.
In addition to bacteria, viruses and other species can profoundly influence the mucosal immune response90. Examples include both bacteriophage as well as DNA and RNA viruses108. Other organisms including mycobacteria, fungi and helminths can play key roles in regulating local and systemic immune responses90. To date, little information is known regarding the potential influence of non-bacterial microbiome on the pre-clinical initiation or evolution of autoimmune diseases.
Another important feature of the mucosal immune response is the regulatory effect of the microbiome on local cells, as demonstrated by the generation of bacterial metabolites that modulate differentiation and effector functions. For example, the microbe-derived short chain fatty acids butyrate, propionate and acetate all strongly affect the differentiation of mucosal Treg cells109.
Evidence of disease modulation from experimental models
Several murine models of arthritis have been studied in order to define what changes occur to the mucosal microbiome during the evolution of experimental arthritis and conversely how those can affect the development of systemic autoimmunity and synovitis. These studies have in aggregate strongly supported a role for microbiome influences on arthritis development through several mechanisms.
In the type 2 collagen (CII)-induced arthritis (CIA) model, previous studies have defined important pathogenic roles for the ACPA response and the potential beneficial clinical and autoimmune outcomes following modulation of that response using toleragenic approaches to decrease the level of pathogenic ACPA110. More recent studies have demonstrated dysbiosis when evaluated at the terminal point of study in the CIA model and that transfer of fecal material from these arthritic as compared to non-arthritic mice accelerates the development of arthritis in recipient germ free mice111. Additionally, increases in IL-17 and splenic CD8 and Th17 lymphocytes were seen with this treatment, in contrast to decreases in dendritic, B and Treg cells. Other studies have demonstrated that, while modulation of the microbiota through the use of broad spectrum antibiotics prior to the induction of CIA resulted in a modest reduction in disease severity, antibiotic use during the late phase of CIA resulted in nearly complete suppression of arthritis112. The later effect was in part due to a significant impairment in the ability of anti-CII antibodies to activate complement ex vivo, while the earlier depletion was associated with decreased circulating inflammatory cytokines and anti-CII antibodies along with delayed IL-17A and IL-22 production in the intestine. Perhaps most notably, these data establish that the microbiome can exert profoundly different effects at time points during the pre-clinical to clinical evolution of arthritis, a finding which is likely quite germane to the study of the potential roles of the human microbiome during initiation of autoimmunity and through the multiple stages of the natural history of RA. In addition, this result suggests that all of the effects of the mucosal immune cell interactions with the microbiome may not be manifest only locally, as subsets of lymphocytes in the mucosa are also known to re-circulate and populate other sites, and in that process influence the immune and autoimmune response in other tissues113. Indeed, it may be that “licensing” of T and B cells to become pathogenic must occur through either local activation or passage of these cells through mucosal sites, especially the gut but perhaps also the lung, following which they can then elaborate their pathogenic potential in target tissues during autoimmune disease processes.
Other models of RA have demonstrated features that may also be relevant to the roles of microbiota in human disease. For example, the K/BxN TCR transgenic mouse is a model in which CD4 T cell driven autoantibody recognition of the ubiquitous autoantigen glucose-6-phosphate isomerase (GPI) results in an immune complex-driven arthritis114. In a germ free environment, transgenic mice exhibit a delayed onset of arthritis and reduced severity, while segmented filamentous bacteria (SFB) are capable to modulate the development of autoantibodies to GPI by activating and recruiting of Th17 and Tfh cells within the intestine115,116. In addition, studies of the human DR4/DQ8 humanized mice have demonstrated that treatment with a human commensal organism, Prevotella histicola, suppressed the development of CIA117. Mechanistically, this treatment altered CD103 dendritic cells, myeloid suppressors and Treg cells in the intestinal mucosa, resulting in suppression of antigen-specific Th17 responses as well as increases in antimicrobial peptides and tight junction proteins.
Inflammation in At-Risk populations
There are several systemic immune abnormalities found in retrospective serum studies as well as present in ACPA+ and other At-Risk populations that suggest the presence of ongoing mucosal inflammation (FIG. 3). One is the predominance of IgA isotype ACPA and RF in retrospectively studied preclinical samples58 as well as the presence of serum IgA ACPA and RF in At-Risk populations18. More compelling, however, are data obtained from an analysis of plasmablasts from At-Risk populations. These are relevant cells to study as during an immune response, peripheral blood plasmablasts arise from both naïve and memory B cells that are activated by inciting antigens, including those at mucosal sites. To evaluate these cells, plasmablasts derived from RA-related autoantibody positive At-Risk subjects as well as matched controls and individuals with early RA were sequenced and characterized utilizing a barcode-enabled single cell antibody methodology118. Notably, while total plasmablast levels were similar in RA-related autoantibody positive At-Risk individuals and controls, a markedly increased frequency of IgA+ as compared to IgG+ plasmablasts was observed in the At- Risk individuals as compared to the other groups, including patients with early RA119. Consistent with these findings, At-Risk individuals also demonstrated serum IgA anti-CCP and IgA ACPA peptide-specific antibodies119.
Figure 3. Representations of immune alterations in At-Risk individuals.
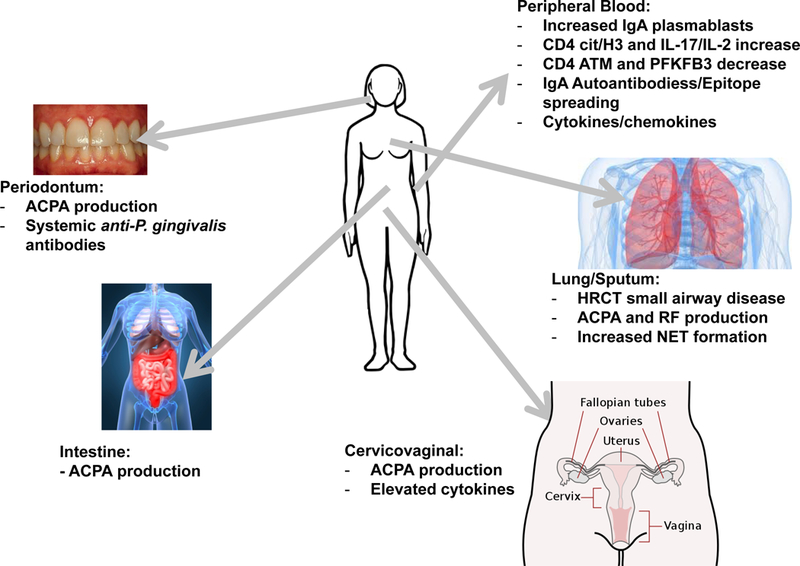
Studies of At-Risk individuals demonstrate immune alterations, both systemically and within local mucosal sites, consistent with the presence of chronic mucosal inflammation. Shown are findings found in these At-Risk populations. Not included are findings from patients with classified RA, as it is unknown as to whether those factors play a role in pre-clinical RA.
Beyond this, plasmablasts from a sub-group of At-Risk subjects underwent paired heavy and light chain repertoire sequencing. Phylogenetic trees of paired IgG and IgA sequences were generated118–120. From these data, clonal families of antibodies were identified that use the same heavy-chain V and J gene segments, as well as the same light-chain V and J gene segments, consistent with their inter-relatedness during development. Combined IgG + IgA phylogenetic trees were also generated to demonstrate the relationship between sequences across these isotypes, and remarkably a substantial number of dual IgG/IgA cross-isotype clonal families of varying sizes were observed119. These data are consistent with an ongoing immune response that has a pronounced mucosal origin and/or contribution.
Other studies of At-Risk individuals have also demonstrated findings consistent with a state of chronic immune stimulation and inflammation121, which in the absence of another tissue or organ site of origin is a finding very consistent with mucosal inflammation. For example, studies of peripheral blood cells exhibited unique phenotypes in CD4+ peripheral T cells consisting of elevated citrullinated histone H3 (cit-H3) as well as increased stimulated production of IL-2 and Th17 cytokines but fewer Th2 cytokines121. With regard to the underlying immune mechanisms of these particular findings, the abnormal features appear to be due to the impaired induction of PTPN22 in T cells. PTPN22 is a phosphatase that also suppresses citrullination independently of its phosphatase activity and when expressed as an RA-associated variant in neutrophils promotes accelerated NETosis122.
In addition, in the same study of At-Risk individuals attenuated phosphatase activity of PTPN22 was found to result in aberrant expression of IL-2, ataxia telangiectasia mutated (ATM), and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), which is associated with a hyper-proliferative phenotype121. Furthermore, diminished non-phosphatase activity of PTPN22 led to hyper-citrullination mediated by PADs and reduction of expression of Th2 cytokines. Notably, these T cell phenotypic changes identified in At-Risk subjects closely mirror prior results obtained in patients with longstanding RA, studies which showed that patients with active arthritis also exhibit pronounced changes in the metabolic activity of CD4+ T cells where they do not metabolize glucose as efficiently as controls, resulting in less intracellular ATP and enhanced sensitivity to apoptosis123,124. Mechanistically, the defect was related to a lower induction of PFKFB3, with resultant metabolic shifts by hypoglycolytic T cells through diversion of glucose toward the pentose phosphate pathway, more NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) and utilization of intracellular reactive oxygen species (ROS)123. In aggregate, these changes left the T cells energy deprived, apoptosis sensitive and autophagy-resistant. Subsequent studies demonstrated that insufficient activation of ATM drove T cells towards a hyper-inflammatory Th17 and Th1 lineage, bypassing of the G2M checkpoint and hyper-proliferation124. Notably, correction of the ROS changes reversed these defects both in vitro and in an in vivo model. Finally, one additional finding in support of a chronic infection, likely viral in nature, being relevant at an early this point in disease is the finding of an elevated type I interferon score in individuals who have transitioned from a late pre-clinical phase to classified RA64.
In sum, several lines of evidence from peripheral blood and serum studies have strongly suggested the presence of an ongoing inflammation, likely mucosal in origin, and an immune response that is associated with a state of heightened immune reactivity and inflammation. As the At-Risk individuals do not manifest evidence of arthritis, as assessed by MRI, physical exam and even synovial biopsy, the presence of an asymptomatic chronic mucosal process associated with these peripheral blood changes is very feasible.
Tissue-specific mucosal Inflammation
Also relevant in support of the mucosal origins hypothesis in RA development are studies in At-Risk populations demonstrating the concurrent presence of inflammation and RA-related autoantibody production in informative mucosal sites. To that end, this section will review available evidence in tissue sites when assessing both the presence of informative site-specific changes in early RA as well as what is known in the same organ when one is studying At-Risk populations.
Intestinal Biomarkers
With regard to RA, seminal studies performed in patients with new-onset rheumatoid arthritis (NORA) demonstrated a relative expansion of fecal Prevotella copri in a substantial number of patients13. Although no microbiome-based replication studies have been published, findings in a separate study population have demonstrated an enhanced IgG and IgA humoral and Th1 T cell immune response to this organism in a subset of patients with established RA125. Another study of patients with established RA identified alterations in fecal Lactobacilli15, changes which partially respond to drug treatment, and in a separate cohort study an expansion of rare species with the potential to modify experimental RA16 was found in patients. Notably, it is unknown as to whether the changes noted in these sites in patients with active disease are causal or secondary to inflammation or drug therapy, and why the microbiome findings differ from one research group to another. No comprehensive studies of the fecal or intestinal microbiome have been reported in RA At-Risk populations, but such studies are underway and have suggested the presence of local ACPA production and dysbiosis.
Lung Biomarkers
In addition to the risk factors described above that have been largely identified in epidemiologic studies of subjects with RA, and the demonstration that ACPA and RF are produced in inducible bronchial-associated lymphatic tissue (iBALT) in patients with chronic RA126, there have been compelling studies of early RA that have supported the presence of substantial pulmonary inflammation in conjunction with arthritis. These findings have included the presence of histologic structural abnormalities as well as inflammation and ACPA production of a similar nature as in the joint in lung biopsies and BAL fluid from early RA cases127,128. Microbiome studies of the BAL of similar early RA patients demonstrated that the microbiota was significantly less diverse and abundant when compared to matched healthy controls studied in parallel, and that the changes were quite similar to patients with sarcoidosis12. The dysbiosis could be attributed to the reduced presence of several genus and taxa. In addition, several clades correlated with the levels of autoantibodies both in the BAL and systemically, and the genus Pseudonocardia was the only taxa over-represented. Whether these same dysbiotic changes are seen in At-Risk subjects has not been reported.
To assess the presence of lung abnormalities and local RA-related autoantibody production in At-Risk populations, though, two techniques have primarily been used. The first was high resolution computerized tomography (HRCT), which demonstrated the presence in ~2/3 of At-Risk subjects of small airways abnormalities in the absence of arthritis that were consistent with airway inflammation, with the joints being assessed and negative for inflammatory arthritis by both clinical examination and in a subset of subjects by MRI129. Notably, the presence by lung HRCT of small airways abnormalities (bronchial wall thickening, bronchiectasis, bronchiolitis and air trapping) was not dependent upon a history of current or prior smoking, which suggested that other etiologic factors must be involved. In addition, subjects were not aware of any pulmonary problems and did not demonstrate evidence of abnormalities by pulmonary function testing.
The second technique utilized in At-Risk populations has been induced sputum collection, a validated methodology that importantly assesses changes in the same small airways as identified by HRCT18,130. This technique has demonstrated the presence of IgA and IgG RA-related autoantibodies in the lung of At-Risk subjects even in the absence of serum ACPA or RF18. Analysis of the ratios of specific to non-specific IgA and IgG demonstrated that the RA-related autoantibodies were produced locally. Importantly, local IgA anti-CCP generation was also closely associated with the concurrent presence of elevated inflammatory cells (neutrophils and macrophages) and increased endogenous NET formation in the sputum, as assessed by the detection and quantitation of NET remnants130. In aggregate, ~25% of FDRs or individuals with serum autoantibody positivity appeared to demonstrate the presence of local RA-related autoantibody production. Also, a subset of subjects demonstrated systemic ACPA but no pulmonary inflammation, suggesting that a separate mucosal site might be involved in those individuals in the generation of systemic ACPA.
In addition, sputum studies comparing individual ACPA reactivities identified an increased number of sputum ACPA peptide reactivities in RA subjects compared to At-Risk subjects131. While cross-sectional, this finding suggests that epitope spreading, which is well described in the blood during the preclinical period leading to RA onset, may also occur at mucosal sites. Furthermore, in serum anti-CCP positive subjects with lung disease but no joint disease, lymphoid aggregates consistent with iBALT were highly prevalent, further supporting RA-related immune reactivity in the lung in At-Risk individuals132.
Periodontal Biomarkers
Periodontal inflammation and chronic periodontitis, Porphyormonas (P.) gingivalis infection and a relationship to citrullinated enolase reactivity have long been associated with RA and RA severity [reviewed in133]. Although the relationship between RA and periodontitis and has been challenged through population-based studies, such as those within the EIRA cohort134, there remain substantial data supporting a close relationship between the ACPA response, the specific P. gingivalis peptidyl arginine deiminase, and RA risk.
Other factors have also been implicated in linking disease to periodontitis and ACPA production, including an elevated abundance of Anaeroglobus geminatus in periodontal fluid in patients with NORA that was correlated with the presence of ACPA and RF, and an increase in Prevotella and Leptotrichia species in patients with NORA irrespective of the presence of periodontitis 14. In other studies, Aggregatibacter actinomycetemcomitans has been associated with periodontitis in vivo and the capacity for hyper-citrullination ex vivo135.
As opposed to lung studies, there is a more limited understanding of the relationship between the RA At-Risk state and periodontitis, as studies of that population are just being completed. Prior studies have demonstrated an increased prevalence of antibodies to P. gingivalis in At-Risk populations136. In addition, initial metagenome sequencing reports from the periodontum of At-Risk subjects have begun to emerge, although no conclusions have been made available137. Studies of subjects with periodontitis without RA have demonstrated the presence of citrullinated proteins as well peptidyl arginine deiminase enzymes 2 and 4, with both correlated to the level of inflammation, and as well ACPA in a subset of periodontal fluids from patients138. Nevertheless, no comprehensive studies of At-Risk populations have been reported that can address the longitudinal relationships between local dysbiosis in the periodontum and the future development of RA. Thus, it remains uncertain as to whether, and in which direction, a causal relationship might exist in the At-Risk and established RA populations.
Other Mucosal Site Biomarkers: Salivary, Urinary and Cervicovaginal
Comprehensive studies of other mucosal sites have not been reported in At-Risk subjects, but are also underway. Salivary IgA ACPA levels have been previously studied in a small cohort of patients with RA, and it was found that ~20% of individuals expressed this phenotype139. Notably, in that setting IgA anti-CCP was found to be inversely associated with several poor patient outcomes, thus suggesting that the non-inflammatory, protective functions of the IgA isotype described above may be the predominant effect of expression at this site. To our knowledge, there are no similar studies of At-Risk populations, although in a small set of samples there were marked differences between ACPA levels in induced sputum and saliva of At- Risk subjects130, suggesting that the two sites operate independently and consistent with the understanding that sputum ACPA levels reflect lung and not oral sources. Additional studies demonstrated that IgA RF was likely to be produced locally within the saliva and tears in patients with RA, but no studies of At-Risk populations are available140.
With regard to urinary biomarkers, it is uncertain as to whether ACPA are present in either patients with RA or At-Risk populations. There have been postulated roles in the development of RA for urinary tract infections, perhaps most notably Proteus species [reviewed in141], but no studies of At-Risk populations have emerged that directly address this relationship. Opportunities for study of the urinary microbiome do exist, however142.
Finally, the cervicovaginal site has been previously evaluated with regard to the mucosal immune response and susceptibility to transmission of infections such as HIV143. Of interest, similar to findings discussed above demonstrating ACPA in the sputum of subjects At-Risk for RA, antigen-specific antibodies for HIV can be found in cervicovaginal fluid in the absence of serum antibodies consistent with a local-limited protective antibody response144. While published studies of cervicovaginal-specific biomarkers and ACPA production are lacking, our group has identified elevated levels of cervicovaginal fluid anti-CCP-IgG in premenopausal women with RA as well as a portion of subjects in the At-Risk state145. A portion of healthy controls in that study were also identified who were serum anti-CCP negative but had high levels of cervicovaginal fluid anti-CCP that strongly correlated with signs of local inflammation as measured by cytokine elevations. These findings further support the hypothesis that localized and externally focused ACPA production occurs at mucosal sites in response to inflammation and that identifying the mechanisms leading to a loss of externally focused ACPA production will improve our understanding of the development of systemic ACPA in individuals who develop RA.
One limitation to interpretation of the current studies, especially outside of the lungs, is a lack of understanding on a larger population basis what are the levels of mucosal ACPA, how these change over time, and what additional phenotypes and factors associated with antibody status. This is an important area for future investigation.
Potential disease mechanisms
After validation through a series of initial publications that deep phenotyping of RA At-Risk populations is informative, the study of mucosal sites in At-Risk subjects is only now starting to accelerate. No comprehensive longitudinal mucosal studies of At-Risk populations have been reported. Nevertheless, the conjunction of mechanistic studies in other settings with cross-sectional translational human studies in At-Risk subjects provides a conceptual framework in which to consider and then study this process.
Potential protective role of ACPA production
What is perhaps most striking from initial work is that local mucosal IgA and IgG ACPA and RF production is not an infrequent finding, ranging from 15–25% when evaluating a single isotype or autoantibody specificity in cross-sectional studies of the lung in At-Risk populations to ~40% when considering all reactivities. In addition, local lung mucosal RA-related autoantibody production is not a fixed phenotype, as we have observed in preliminary studies that individuals in the At-Risk state both gain and lose anti-CCP antibodies over time (unpublished observations).
With these observations and the knowledge gained from studies of the protective role of mucosal antibodies, the following hypotheses can be put forward (FIG. 4). First, local ACPA production does not reflect an inherent loss of tolerance to self-antigens but is rather a hard-wired form of natural IgA production, perhaps T cell-independent, that is normally externally directed into the mucosal fluid where it likely binds to citrullinated antigens that are released from the neutrophils undergoing NETosis and macrophages which are present as part of a chronic inflammatory state in a mucosal site. Second, multiple mucosal sites have demonstrated the presence of local ACPA production. This includes the lung and periodontum, and as well in preliminary studies the intestinal and CV sites. These findings suggest that RA does not have to begin in a single mucosal region, but may rather originate in one of many that vary from subject to subject. This finding may underlie the very heterogenous manner in which the autoimmune process develops in the pre-clinical period, with neither specific ACPA peptide reactivities9 nor individual or combinations of cytokines10 being either necessary or sufficient for the transition to classified RA. Third, it is likely that a major driver which results in the loss of the mucosal barrier and development of a systemic IgG ACPA response is an unresolved state of inflammation in the mucosa. This process could be driven by dysbiosis of bacteria or other microbiota, the nature of which may be similar from one individual to another, or alternately vary substantially across At-Risk subjects. It is important to note, though, that to further validate this proposed process going forward, a clear understanding of the relationships between systemic and mucosal B cells and the BCR sequences that encode ACPA must be generated through paired tissue and peripheral blood studies.
Figure 4. Loss of the mucosal firewall allows systemic spread of ACPA response.
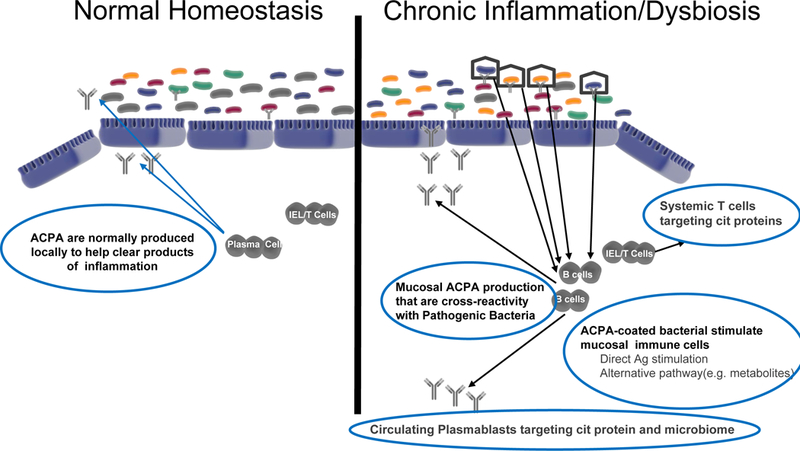
Hypothetical model for RA initiation as chronic mucosal inflammation and then transition to systemic autoimmune disease, focusing on the potential roles for normal and dysregulated local mucosal ACPA generation. Depicted at left is the reversible production of IgA ACPA in individuals as part of normal surveillance or who have transient mild inflammation. This process is common and part of normal homeostasis at the mucosal surface. At right is the postulated status of individuals in the pre-clinical RA state where persistent inflammation leads to increased ACPA production and systemic spread of an autoimmune process through multiple potential processes described herein. The drivers of this local and then systemic process could include cytokines, metabolomic changes, cross-reactive ACPA with bacterial antigens, or the presence of other antigen-specific cells that are expanded. Regardless of the mechanisms involved, these antibodies and cells begin to circulate as a reflection of the chronic inflammation and are detectable through serologic and other biomarker studies.
T cells and molecular mimicry as drivers of autoimmunity
There are certainly other factors that should be considered in the process of expansion of local autoantibody production to systemic spread and ultimately development of arthritis. These include the strong association of RA with SE, which implicates antigen presentation and CD4 T cells in its pathogenesis. Studies have demonstrated that CD4 T cells specific for joint derived citrullinated peptides are present in increased numbers and with distinct phenotypes in RA44,45. These CD4 T cells likely play a role in the evolution of RA by both promoting tissue specific inflammation and providing B cell help for the formation of high avidity pathogenic ACPA. Molecular mimicry has been proposed as one source of failed tolerance in RA and provides a framework with which to link the microbiome with genetic risk by identifying CD4 T cells that respond to microbial and self-antigens that share similar motifs.
Several approaches are being used to address the molecular mimicry hypothesis. For example, motif analysis has be utilized to identify alpha-viral peptides that have similar motif to type II collagen, a known autoantigen in RA146. A peptide sequence motif (DERAA) contained within HLA DRB1*13, a protective allele in RA, has been shown to mediate thymic tolerance in the context of HLA-DQ, but when this molecule is absent, the peptide sequence can be recognized by CD4 T cells_and is cross-reactive with microbial derived epitopes and the self-antigen vinculin147. This finding suggests a mechanism by which an initial failure of thymic tolerance is followed by activation with a microbial antigen that then supports a T and B cell response to citruillated-vinculin147. Most recently, proteomic tools have been used to perform epitope discovery through elution of peptides from HLA Class II molecules found in the synovium of RA patients and mass spectroscopy based characterization. This approach has identified novel autoantigens that are cross reactive with microbial proteins for which T and B cell responses are increased in RA patients125. As we increase our understanding of the microbial species that participate in the development of RA and determine which autoantigens are early targets of the immune response, the role of molecular mimicry in the development of RA should be further clarified.
It is of course conceivable that molecular mimicry on top of the hard wired IgA response to citrullinated products of inflammation are both necessary to develop classified RA. To address this question, regardless of whether the local mucosal ACPA immune response is meant to primarily react with citrullinated products of dead cells, the potential for molecular mimicry as a driver of an expanded pathogenic mucosal and then ACPA immune response, as recently demonstrated in type 1 Diabetes148 and consistent with plasmablast studies in patients with chronic RA that recognize citrullinated targets as well as P. gingivalis149, should be further evaluated. In this regard, it is also notable that in subjects with early RA, IgA ACPA are associated with smoking while IgG ACPA are associated with SE, supporting a possible distinction between the local and systemic processes150.
To better understand these causal mechanisms, and in a similar fashion as above related to the need to address the question of the relationship between systemic and locally produced ACPA, characterizing mucosal T cells and their autoreactivity is essential to further evaluating the potential role of autoreactive T cells in the initial and subsequent development of ACPA.
Other potential inter-related mechanisms
Other inter-relationships may also exist, the first being those that modify local ACPA generation and the second related to the potential ways by which ACPA originating at different mucosal sites, or through distinct inflammatory processes, might influence the subsequent arthritis phenotypes. For instance, alternate primary drivers of autoimmunity may also play key roles, such as inadequate local T cell immunoregulation influenced by genetic or environmental factors, aberrant expression of adhesion molecules and/or cytokines/chemokines, activation of non-toleragenic antigen presenting cells, or engagement of non-traditional APCs such as fibroblasts. Additional modulators of the process may include metabolomic changes that promote systemic spread of the ACPA response, a physical or physiological break in the mucosal barrier, direct or indirect activation of transiting autoimmune cells through enhancement of antigen presentation or alteration of dendritic cell function to expand a response, and elaboration of innate immune factors that promote activation such as LPS, HMGB, CpG or other danger-recognition molecules. In addition, acute infection that is operating on either a normal mucosal or on top of chronic local ACPA production may play an important role in triggering additional pathogenic changes.
Finally, one could ask how chronic inflammation might relate mechanistically to epidemiologic factors such as smoking or lower levels of omega-3 PUFA. Prior studies have suggested that smoking is associated with increased citrullinated proteins in the lung151. With regard to the omega-3 PUFA levels and exposures, recent studies have shown that molecules derived from this pathway, especially bioactive lipid mediators including resolvins and maresins that are biosynthesized in vivo from EPA and DHA, are key factors in resolving or reducing inflammation152. The observation that RBC levels of omega-3 fatty acids are lower in those progressing to IA suggest that these factors are either present in insufficient quantities or are consumed as part of the excessive mucosal inflammation that is present, and that these two processes, local inflammation and a lack of appropriate levels of omega3 fatty acids, may be both necessary for continued evolution of disease. These anti-inflammatory compounds decrease inflammation through mechanisms including blocking neutrophil and other cell chemotaxis/recruitment, increasing neutrophil apoptosis, and promoting cellular immune switching to regulatory phenotypes (reviewed in153). Furthermore, the actions of agents such as resolvins have been shown to reduce inflammation at mucosal surfaces in the lung as well as elsewhere (e.g. gut)154,155, and to reduce cell activation and immune-complex mediated inflammation in a variety of diseases including IgA nephropathy153,156,157. Importantly, as described above, all of these are pathways may be critical to the initiation and propagation of autoimmunity and the ultimate development of arthritis.
As to the second issue, ie the potential influence of mucosal inflammation at specific sites on subsequent arthritis generation, recently distinct synovial subtypes have been identified in RA, including subsets of patients with high-grade synovial infiltrates, vs. a fibrous synovium, vs. other synovial features158. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. It is possible that the specific mucosal trigger and IgA plasmablast response, either in general or from specific mucosal sites, could promote a transition to the high-grade synovial inflammatory subtype of RA and that other mechanisms promote transition of the fibrous synovial subtype. It is also of course striking that what are two distinct autoantibody systems, ACPA and RF, can come together and accelerate and amplify joint damage in an experimental model of RA159, again pointing to the possibility that separate mucosal processes drive distinct autoantibody phenotypes and then the inter-connectedness of the responses drives arthritis.
Therapeutic implications
Although the exact roles of mucosal inflammation in the development of RA are still uncertain, current findings do provide an impetus to consideration of specific mechanism-based therapeutic approaches. For one, while PUFA supplementation has not yet been formally tested in a clinical trial in pre-clinical RA, other trials in animals and humans across a range of diseases (including classified RA) have demonstrated that supplementation does improve clinical parameters 153,160, strongly suggesting that supplementation can result in biologic changes which may prevent development of clinically apparent RA. Additionally, although targeting IL-17 has not proven consistently beneficial in patients with established RA [reviewed in161], findings in At-Risk population described above, along with the known role of IL-17 in promoting mucosal inflammation162, suggest that therapeutic approaches directed at IL-17, or targeting Th17 cells through IL-12/23, may be particularly effective in pre-clinical RA. It is possible that Th17 cells contribute to the evolution of pre-clinical RA via pathways and mediators other than IL-17, including IL-6, IL-21, IL-22 and TNF-α163. Other pathways that play roles in epitope spreading following the breaking of tolerance, including B7/CD28 and CD40:CD40L, also bear strong consideration. The success of co-stimulatory pathway modulation in cancer immunotherapy highlights the importance of these pathways, and we anticipate that key T cell and other co-stimulatory pathways will be identified in autoimmunity164. If so, therapeutically targeting these pathways in pre-clinical RA could provide a powerful approach to abrogate the development of RA. Current clinical trials include methotrexate as well as hydroxychloroquine, the latter of which has been shown to exhibit important features such as inhibiting the transition of palindromic rheumatism to classified RA165 as well as being approved for monotherapy in RA166 and demonstrated the ability to block several key steps in antigen presentation. And of course, if a specific microbial species or metabolomic signaling pathway167 is identified as causal in pre-clinical RA, targeting of these processes would be especially informative.
Future directions
In order to test the hypotheses that are laid out above, a number of studies and experimental approaches must continue to be pursued. It is perhaps most important that longitudinal studies of the microbiome, local mucosal phenotypes at multiple sites and systemic RA-related autoantibody production should be pursued in order to determine whether potential causal relationships exist that could drive relevant phenotypic changes in the pre-clinical to clinical transitions. In a related fashion, understanding of the causes of local mucosal inflammation and local ACPA generation should be a research goal. Additionally, exploring the relationships between local and systemic IgA and IgG ACPA production at the molecular and Ig sequence level over time will allow one to understand how these two systems are inter-related. Also, although it is firmly established that local inflammation in the lung is associated with ACPA production, it is uncertain yet whether biomarkers of mucosal inflammation, such as fecal calprotectin, or responses to bacteria, such as increased expression of RegIII proteins, are altered in At-Risk subjects. Beyond these considerations, although studies of single mucosal sites have been performed in At-Risk subjects, a more comprehensive approach to simultaneously sampling multiple mucosal sites would allow one to understand whether changes are unique to one site or are shared across multiple sites, and then whether one site is sufficient to lead to systemic spread of ACPA generation. To address inter-relationships between local mucosal findings and systemic immune changes, additional studies of T cell antigen-specific or innate immune reactivity should be evaluated, both systemically and at mucosal sites. To further expand our understanding, a determination of whether other RA-related autoantibodies, including AMPA, are similarly generated locally in the mucosa as part of the hard wired innate immune system and then spread systemically should be undertaken. Likewise, understanding how systemic immune and metabolomics changes related to mucosal inflammation and ACPA production will provide additional insights. And finally, it would be important to determine whether this concept of “hard-wired mucosal autoimmunity” playing important roles in normal mucosal immune responses is relevant to the initial pre-clinical stages of other autoimmune diseases, such as SLE, where antibodies to intracellular proteins are prominent and it is uncertain as to why these intra-cellular antigens that are released during normal inflammatory processes are “chosen” as targets of autoantibodies in these conditions.
Conclusions
Prior to the onset of the first clinical signs and symptoms of seropositive RA, most patients will have passed through a prolonged period of serologic RA-related autoantibody positivity. Herein the current understanding of the relationships during this pre-clinical period between mucosal alterations, including dysbiosis and inflammation, and the initiation of local and systemic ACPA production has been reviewed. Mucosal ACPA production in the lung, and likely other sites, is associated with the presence of local inflammation and elevated NET formation. Mucosal IgA ACPA production appears to be a hard-wired and common phenotype, suggesting that this factor plays an important role in the local protective immune response outside of its eventual causal relationship with inflammatory arthritis. There are many potential factors in the mucosa, as well as systemic alterations, that could influence both the local initiation and systemic expansion of ACPA expression. Further study of these processes should provide both additional insights into very early disease pathogenesis in RA as well as the identification of novel therapeutic strategies.
Acknowledgements
Studies described herein have been supported by: U01 AI101981 (VMH), T32 AR07534 (VMH), UH2 AR067681 (VMH), UM1 AI110503 (VMH), Rheumatology Research Foundation (VMH, KD), R01 AR051394 (VMH), U19 AI50864 (VMH), K08 DK107905, Pfizer ASPIRE (KAK).
Glossary terms
- Dysbiosis
A general term used to indicate a change in the normal bacterial ecology, usually with a potential for association with a disease state
- Shared Epitope
A set of HLA antigens that are preferentially found in patients with RA, and which are characterized by conserved amino acids within the peptide-binding groove
- Epitope spreading
A process whereby the epitopes (target antigen shapes or sequences recognized by B and T cell receptors) that are distinct from and non-cross-reactive with an earlier epitope become major targets of an ongoing immune response
- Avidity maturation
A process whereby the accumulated strength of multiple affinities of individual non-covalent binding interactions between antigens and antibodies increases over time
- NETosis
A unique form of neutrophil extracellular trap (NET)-associated cell death characterized by nuclear condensation and extrusion of chromatic and granular contents in a manner especially conducive to capturing pathogens
- Molecular mimicry
The development of autoreactivity commonly considerd to be through the recognition of a microbial or other foreign pathogen peptide that is structurally similar to self-antigen
Footnotes
Competing interests
The authors declare no competing financial interests.
Key References
- 1.Catrina AI, Svensson CI, Malmström V, Schett G & Klareskog L Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nature Rev. Rheum. 13, 79–86 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Catrina AI, Deane KD & Scher JU Gene, environment, microbiome and mucosal immune tolerance in rheumatoid arthritis. Rheumatology 55, 391–402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viatte S, Plant D & Raychaudhuri S Genetics and epigenetics of rheumatoid arthritis. Nature Rev. Rheum. 9, 141–153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Firestein GS & McInnes IB Immunopathogenesis of rheumatoid arthritis. Immunity 46, 183–196 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielen MMJ et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis. Arth. Rheum. 50, 380–386 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Rantapaa Dahlqvist S et al. Antibodies against citrullinated peptides (CCP) predict the development of rheumatoid arthritis. Arth. Rheum 48, 2701–2705 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Majka DS et al. Duration of preclinical rheumatoid arthritis-related autoantibody positivity increases in subject with older age at time of disease diagnosis. Ann. Rheum. Dis. 67, 801–807 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karlson EW et al. Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arth. Rheum. 60, 641–652 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokolove J et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One 7, e35296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deane KD et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arth. Rheum. 62, 3161–3172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerlag DM et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann. Rheum. Dis. 71, 638–641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher JU et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scher JU et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife doi: 10.7554/eLife.01202. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher JU et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arth. Rheum. 64, 3083–3094 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature Med. 21, 895–905 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Chen J et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. doi: 10.1186/s13073-016-0299-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demoruelle MK et al. Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arth. Rheum. 69, 1165–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis VC et al. Sputa autoantibodies in patients with established rheumatoid arthritis and subjects at- risk for future clinically apparent disease. Arth. Rheum. 65, 2545–2554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee DM & Weinblatt ME Rheumatoid arthritis. Lancet 359, 903–911 (2001). [DOI] [PubMed] [Google Scholar]
- 20.van Steenbergen HW et al. EULAR definition of arthralgia suspicious for progression to rheumatoid arthritis. Ann. Rheum. Dis. 76, 491–496 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Fuchs HA & Sergent JS in Arthritis and Allied Conditions: A Textbook of Rheumatology (ed Koopman WJ) Ch. 54, 1041–1070 (Williams and Wilkins, 1997). [Google Scholar]
- 22.Schett G & Gravallese EM Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nature Rev. Rheum. 8, 656–664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silman AJ Epidemiology of rheumatoid arthritis. APMIS 102, 721–728 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Karlson EW & Costenbader KH Epidemiology: Interpreting studies of interactions between RA risk factors. Nat. Rev. Rheum. 6, 72–73 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Karlson EW & Deane KD Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum. Dis. Clin. North Am. 38, 405–426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klareskog L, Malmstrom V, Lundberg K, Padyukov L & Alfredsson L Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Seminars in Immunology 23, 92–98 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Aletaha D & al e. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arth. Rheum. 62, 2569–2581 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Arnett FC et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arth. Rheum. 31, 314–324 (1998). [DOI] [PubMed] [Google Scholar]
- 29.de Rooy DPC, van der Linden MPM, Knevel R, Huizinga TW & van der Helm-van Mil AHM Predicting arthritis outcomes-what can be learned from the Leiden Early Arthritis Clinic? Rheumatology 50, 93–100 (2011). [DOI] [PubMed] [Google Scholar]
- 30.ven der Helm-vanMihl AHM et al. A prediction rule for disease outcome in patients with recent- onset undifferentiated arthritis: how to guide individual treatment decisions. Arth. Rheum. 56, 433–440 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Padyukov L, Silva C, Stolt P, Alfredsson L & Klareskog L A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arth. Rheum. 50, 3085–3092 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Schellekens GA, de Jong BAW, van den Hoogen FHJ, van de Putte LBA & van Venrooij WJ Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis- specific autoantibodies. J. Clin. Invest. 101, 273–281 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Venrooij WJ & Pruijn GJM Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arth. Res. Ther. 2, 249–251 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bas S, Genevay S, Meyer O & Gabay C Anti-cyclic citrullinated peptide antibodies, IgM and IgA rheumatoid factors in the diagnosis and prognosis of rheumatoid arthritis. Rheumatology 42, 677–680 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Trouw LA, Rispens T & Toes REM Beyond citrullination: other post-translational protein modifications in rheumatoid arthritis. Nature Rev. Rheum. 13, 331–339 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Shi J et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. PNAS 108, 17372–17377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiele GM et al. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arth. Rheum. 67, 645–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pecani A et al. Prevalence, sensitivity and specificity of antibodies against carbamylated proteins in a monocentric cohort of patients with rheumatoid arthritis and other autoimmune rheumatic diseases. Arth. Res. Ther. 18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan RW et al. Anti-carbamylated protein antibodies are present prior to rheumatoid arthritis and are associated with its future diagnosis. J. Rheumatology 42, 572–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nepom GT & Nepom BS Prediction of susceptibility to rheumatoid arthritis by human leukocyte antigen phenotyping. Rheum. Dis. Clin. North Am. 18, 785–792 (1992). [PubMed] [Google Scholar]
- 41.Begovich AB et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. American J. Human Genetics 75, 330–337 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viatte S & Barton A Genetics of rheumatoid arthritis susceptibility, severity, and treatment response. Semin. Immunopathol. 39, 395–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill JA et al. The conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-BRB1*0401 MHC Class II molecule. J. Immunol. 171, 538–541 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Law SC et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arth. Res. Ther. 14, R118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.James E et al. Citrulline specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arth. Rheum. DOI: 10.1002/art.38637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klareskog L et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arth. Rheum. 54, 38–46 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Nakano K, Whitaker JW, Boyle DL, Wang W & Firestein GS DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 72, 110–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rhead B et al. Rheumatoid arthritis naive T cells share hypermethylation sites with synoviocytes. Arth. Rheum. 69, 550–559 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deane KD, Norris JM & Holers VM Pre-clinical rheumatoid arthritis: identification, evaluation and future directions for investigation. Rheum. Dis. Clin. North Am, 236–241 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brink M et al. Multiplex analyses of antibodies against citrullinated peptides in individuals prior to the development of rheumatoid arthritis. Arth. Rheum. 65, 899–910 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Suwannalai P et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arth. Rheum. 64, 1323–1328 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Trouw LA et al. Anti-cyclic peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arth. Rheum. 60, 1923–1931 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Rombouts Y et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 74, 234–241 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Rombouts Y et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 75, 578–585 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Pfeifle R et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nature Immunol. 18, 104–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harre U et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Invest. 122, 1791–1802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wigerblad G et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 75, 730–738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kokkonen H et al. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arth. Res. Ther. 13, R13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jorgensen C, Moynier M, Bologna C, Youinou P & Sany J Rheumatoid factor associated with a secretory component in rheumatoid arthritis. Br. Med. J. 34, 236–240 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Aho K, Heliovaara M, Maatela J, Tuomi T & Palosuo T Rheumatoid factors antedating clinical rheumatoid arthritis. J. Rheum. 18, 1282–1284 (1991). [PubMed] [Google Scholar]
- 61.Kolfenbach JR et al. A prospective approach to investigating the natural history of pre-clinical rheumatoid arthritis (RA) using first-degree relatives of probands with RA. Arth. Care Res. 61, 1735–1741 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Gabalawy HS et al. Familial clustering of the serum cytokine profile in the relatives of rheumatoid arthritis patients. Arth. Rheum. 64, 1720–1729 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Deane KD et al. Identification of undiagnosed inflammatory arthritis in a community health fair screen. Arth. Care Res. 61, 1642–1649 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lubbers J et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann. Rheum. Dis. 72, 776–780 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Young KA et al. Relatives without rheumatoid arthritis show reactivity to anti-citrullinated protein/peptide antibodies that are associated with arthritis-related traits: studies of the etiology of rheumatoid arthritis. Arth. Rheum. 65, 1995–2004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hughes-Austin J et al. Multiple cytokines and chemokines are associated with rheumatoid arthritis- related autoimmunity in first-degree relatives without rheumatoid arthritis: Studies of the Aetiology of Rheumatoid Arthritis (SERA). Ann. Rheum. Dis. 72, 901–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aslam A et al. Emergence of proinflammatory autoreactive T-cell responses in preclinical rheumatoid arthritis. Lancet S22. doi: 10.1016/S0140-6736(14)60285-3 (2014). [DOI] [Google Scholar]
- 68.Hunt L et al. T cell subsets: an immunological biomarker to predict progression to clinical arthritis in ACPA-positive individuals. Ann. Rheum. Dis. 75, 1884–1889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nielen MMJ et al. Increased levels of C-Reactive Protein in serum from blood donors before the onset of rheumatoid arthritis. Arth. Rheum. 50, 2423–2427 (2004). [DOI] [PubMed] [Google Scholar]
- 70.van de Stadt LA et al. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann. Rheum. Dis. 70, 128–133 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Arkema EV et al. Anti-citrullinated peptide autoantibodies, human leukocyte antigen shared epitope and risk of future rheumatoid arthritis: a nested case-control study. Arth. Res. Ther. 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhatia SS et al. Rheumatoid factor seropositivity is inversely associated with oral contraceptive use in women without arthritis. Ann. Rheum. Dis. 66, 267–269 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hensvold AH et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann. Rheum. Dis. 74, 375–380 (2015). [DOI] [PubMed] [Google Scholar]
- 74.van Zanten A et al. Presence of anticitrullinated protein antibodies in a large population-based cohort from the Netherlands. Ann. Rheum. Dis. 76, 1184–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hedstrom AK, Stawiarz L, Klareskog L & Alfredsson L Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur. J. Epidemiol. 33, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Wesemael TJ et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: a multicenter cohort study. Arth. Res. Ther. 18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terao C et al. Effects of smoking and shared epitope on the production of anti-citrullinated peptide antibody in a Japanese adult population. Arth. Care Res. 66, 1818–1827 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Gan RW et al. Relationship between air pollution and positivity of RA-related autoantibodies in individuals without established RA: a report on SERA. Ann. Rheum. Dis. 72, 2002–2005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hart JE, Laden F, Puett RC, Costenbader KH & Karlson EW Exposure to traffic pollution and increased risk of rheumatoid arthritis. Environ. Health. Perspect. 117, 1065–1069 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tedeschi SK et al. Elevated BMI and antibodies to citrullinated proteins interact to increase rheumatoid arthritis risk and shorten time to diagnosis: A nested case-control study of women in the Nurses’ Health Studies. Semin. Arth. Rheum. 46, 692–698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stolt P et al. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann. Rheum. Dis. 64, 582–586 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dawczynski C et al. Docosahexaenoic acid in the treatment of rheumatoid arthritis: A double-blind, placebo-controlled, randomized cross-over study with microalgae vs. sunflower oil. Clin. Nutr. 37, 494–504 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Souza PR & Norling LV Implications for eicosapentaenoic acid-and docosahexaenoic acid-derived resolvins as therapeutics for arthritis. Eur. J. Pharmacol. 785, 165–173 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Di Giuseppe D, Crippa A, Orsini N & Wolk A Fish consumption and risk of rheumatoid arthritis: a dose-response meta-analysis. Arthritis Res. Ther. doi: 10.1186/s13075-014-0446-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gan RW et al. Lower omega-3 fatty acids are associated with the presence of anti-cyclic citrullinated peptide autoantibodies in a population at risk for future rheumatoid arthritis: a nested case-control study. Rheumatology 55, 367–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gan RW et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope positive subjects at risk for rheumatoid arthritis. Ann. Rheum. Dis. 76, 147–152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gan RW et al. The association between omega-3 fatty acid biomarkers and inflammatory arthritis in an anti-citrullinated protein antibody positive population. Rheumatology 56, 2229–2236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li S, Yu Y, Yue Y, Zhang Z & Su K Microbial infection and rheumatoid arthritis. J. Clin. Cell. Immunol. 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lundberg K, Wegner N, Yucel-Lindberg T & Venables PJ Periodontitis in RA-the citrullinated enolase connection. Nature Rev. Rheum. 6, 727–730 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Longman RS & Littman DR The functional impact of the intestinal microbiome on mucosal immunity and systemic autoimmunity. Curr. Opin. Rheum. 27, 381–387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scher JU, Littman DR & Abramson SB Review: Microbiome in inflammatory arthritis and human rheumatic diseases. Arth. Rheum. 68, 35–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Belkaid Y & Hand TW Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cash HL, Whitham CV, Behrendt CL & Hooper LV Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szabady RL & McCormick BA Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front. Immunol. 10.3389/fimmu.2013.00220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Howson LJ, Salio M & Cerundolo V MR1-restricted mucosal-associated invariant T cells and their activation during infectious diseases. Front. Immunol., doi: 10.3389/fimmu.2015.00303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA & Artis D CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goto Y et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345, DOI: 10.1126/science.1254009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.von Andrian U & Mackay CR T-cell function and migration. Two sides of the same coin. NEJM 343, 1020–1034 (2000). [DOI] [PubMed] [Google Scholar]
- 99.Kurmaeva E et al. T cell-associated a4b7 but not a4b1 integrin is required for the induction and perpetuation of chronic coliti. Mucosal Immunol. 7, 1354–1365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Macpherson AJ, McCoy KD, Johansen FE & Brandtzaeg P The immune geography of IgA induction and function. Mucosal Immunol. 11, 11–22 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Fagarasan S, Kawamoto S, Kanagawa O & Suzuki K Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Ann. Rev. Immunol. 28, 243–273 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Corthésy B Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. doi: 10.3389/fimmu.2013.00185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aleyd E, Al M, Tuk CW, van der Laken CJ & van Egmond M IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcαRI. J. Immunol. 197, 4552–4559 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Russell MW, Sibley DA, Nikolova EB, Tomana M & Mestecky J IgA antibody as a non-inflammatory regulator of immunity. Biochem. Soc. Trans. 25, 466–470 (1997). [DOI] [PubMed] [Google Scholar]
- 105.Baker K, Lencer WI & Blumberg RS Beyond IgA: the mucosal immunoglobulin alphabet. Mucosal Immunol. 3, 324–325 (2010). [Google Scholar]
- 106.Bunker JJ et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science doi: 10.1126/science.aan6619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vaishnava S et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Virgin HW The virome in mammalian physiology and disease. Cell 157, 142–150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuhn KA et al. Antibodies to citrullinated proteins enhance tissue injury in experimental arthritis. J. Clin. Invest. 116, 961–973 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu X et al. Role of the gut microbiome in modulating arthritis progression in mice. Sci. Rep. doi: 10.1038/srep30594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jubair WK et al. Modulation of inflammatory arthritis by gut microbiota through mucosal inflammation and autoantibody generation Arth. Rheum. doi: 10.1002/art.40490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacques P & Elewaut D Joint expedition: linking gut inflammation to arthritis. Mucosal Immunol. 1, 364–371 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Ji H et al. Arthritis critically dependent on innate immune system players. Immunity 16, 157–168 (2002). [DOI] [PubMed] [Google Scholar]
- 115.Block KE, Zheng Z, Dent AL, Kee BL & Huang H Gut microbiota regulates K/BxN autoimmune arthritis through follicular helper T but not Th17 cells. J. Immunol. 196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu HJ et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marietta EV et al. Suppression of inflammatory arthritis by human gut-derived Prevotella histicola in humanized mice. Arth. Rheum. 68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tan YC et al. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin. Immunol. and Immunopathol 151, 55–66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kinslow JD et al. IgA plasmablasts are elevated in subjects at risk for future rheumatoid arthritis. Arth. Rheum. 68, 2372–2383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan YC et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arth. Rheum. 66, 2706–2715 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang HH et al. A molecular timeline of preclinical rheumatoid arthritis defined by dysregulated PTPN22, hypercitrullination, aberrant cytokine and metabolic profiles in PBMC of at-risk individuals. J. Clin. Invest. Insight doi: 10.1172/jci.insight.90045 (2016). [DOI] [Google Scholar]
- 122.Chang HH, Dwivedi N, Nicholas AP & Ho IC The W620 polymorphism in PTPN22 disrupts its interaction with peptidylarginine deiminase type 4 and enhances citrullination and NETosis. Arth. Rheum. 67, 2323–2334 (2015). [DOI] [PubMed] [Google Scholar]
- 123.Yang Z., Fujii H., Mohan SV., Goronzy JJ. & Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp. Med. 210, 2119–-2134. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang Z et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci.Trans.Med. 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pianta A et al. Evidence of the immune relevance of Prevotella copri, a gut microbe, in patients With rheumatoid arthritis. Arth. Rheum. 69, 964–975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rangel-Moreno J et al. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Invest. 116, 3183–3194 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reynisdottir G et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann. Rheum. Dis. 75, 1722–1727 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Ytterberg AJ et al. Identification of shared citrullinated immunological targets in the lungs and joints of patients with rheumatoid arthritis. Ann. Rheum. Dis, 1772–1777 (2012). [DOI] [PubMed] [Google Scholar]
- 129.Demoruelle MK et al. Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arth. Rheum. 64, 1756–1761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Demoruelle MK et al. Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arth. Rheum. 69, 1165–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Demoruelle MK et al. Antibody responses to citrullinated and non-citrullinated antigens in the sputum of subjects with and at-risk for rheumatoid arthritis. Arth. Rheum. doi: 10.1002/art.40401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fischer A et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Resp. Med. 106, 1040–1047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bingham CO & Moni M Periodontal disease and rheumatoid arthritis: the evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheum. 25, 345–353 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Eriksson K et al. Prevalence of periodontitis in patients with established rheumatoid arthritis: a Swedish population based case-control study. PLoS One doi: 10.1371/journal.pone.0155956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Konig MF et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Trans. Med. Dec 14;8(369):369ra176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mikuls TR et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arth. Rheum. 64, 3522–3530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cheng Z et al. The subgingival microbiomes in periodontitis and health of individuals with rheumatoid arthritis and at risk of developing rheumatoid arthritis. J. Oral Microbiol. doi: 10.1080/20002297.2017.1325216 (2017). [DOI] [Google Scholar]
- 138.Harvey GP et al. Expression of peptidylarginine deiminase-2 and −4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J. Periodontal Res. 48, 252–261 (2013). [DOI] [PubMed] [Google Scholar]
- 139.Svard A, Kastbom A, Sommarin Y & Skogh T Salivary IgA antibodies to cyclic citrullinated peptides (CCP) in rheumatoid arthritis. Immunobiology 218, 232–237 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Otten HG et al. IgA rheumatoid factor in mucosal fluids and serum of patients with rheumatoid arthritis: immunological aspects and clinical significance. Clin. Exp. Immunol. 90, 256–259 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ebringer A & Rashid T Rheumatoid arthritis is caused by a Proteus urinary tract infection. APMIS 122, 363–368 (2013). [DOI] [PubMed] [Google Scholar]
- 142.Whiteside SA, Razvi H, Dave S, Reid G & Burton JP The microbiome of the urinary tract--a role beyond infection. Nat. Rev. Urol. 12, 81–90 (2015). [DOI] [PubMed] [Google Scholar]
- 143.Gosmann C et al. Lactobacillus-deficient cervicovaginal bacterial communities are associated with increased HIV acquisition in young south African women. Immunity 46, 29–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tudor D et al. HIV-1 gp41-specific monoclonal mucosal IgAs derived from highly exposed but IgG-seronegative individuals block HIV-1 epithelial transcytosis and neutralize CD4(+) cell infection: an IgA gene and functional analysis. Mucosal Immunol 2, 412–426, doi: 10.1038/mi.2009.89 (2009). [DOI] [PubMed] [Google Scholar]
- 145.Khatter S, et al. Anti-CCP antibody levels are elevated in cervicovaginal fluid in association with local inflammation in premenopausal women without RA [abstract]. Arth. Rheum. 69 (2017). [Google Scholar]
- 146.Hogeboom C Peptide motif analysis predicts alphaviruses as triggers for rheumatoid arthritis. Mol. Immunol. 68, 465–475 (2015). [DOI] [PubMed] [Google Scholar]
- 147.van Heemst J et al. Crossreactivity to vinculin and microbes provides a molecular basis for HLA-based protection against rheumatoid arthritis. Nature Commun. doi: 10.1038/ncomms7681 (2015). [DOI] [PubMed] [Google Scholar]
- 148.Cole DK et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivit. J. Clin. Invest. 126, 2191–2204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li S et al. Autoantibodies from single circulating plasmablasts react with citrullinated antigens and Porphyromonas gingivalis in rheumatoid arthritis. Arth. Rheum. 68, 614–626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Svärd A et al. Associations with smoking and shared epitope differ between IgA-and IgG-class antibodies to cyclic citrullinated peptides in early rheumatoid arthritis. Arth. Rheum. 67, 2032–2037 [DOI] [PubMed] [Google Scholar]
- 151.Makrygiannakis D et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis 67, 1488–1492 (2008). [DOI] [PubMed] [Google Scholar]
- 152.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Souza PR & Norling LV Implications for eicosapentaenoic acid-and docosahexaenoic acid-derived resolvins as therapeutics for arthritis. Eur J Pharmacol 785, 165–173, doi: 10.1016/j.ejphar.2015.05.072 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Campbell EL et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J 21, 3162–3170, doi: 10.1096/fj.07-8473com (2007). [DOI] [PubMed] [Google Scholar]
- 155.Uddin M & Levy BD Resolvins: natural agonists for resolution of pulmonary inflammation. Prog Lipid Res 50, 75–88, doi: 10.1016/j.plipres.2010.09.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Norling LV et al. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight 1, e85922, doi: 10.1172/jci.insight.85922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Donadio JV & Grande JP The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin Nephrol 24, 225–243 (2004). [DOI] [PubMed] [Google Scholar]
- 158.Dennis GJ et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arth. Res. Ther. 16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sokolove J et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody mediated inflammation in rheumatoid arthritis. Arth. Rheum. 66, 813–821 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dawczynski C et al. Docosahexaenoic acid in the treatment of rheumatoid arthritis: A double-blind, placebo-controlled, randomized cross-over study with microalgae vs. sunflower oil. Clin Nutr, doi: 10.1016/j.clnu.2017.02.021 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Kunwar S, Dahal K & Sharma S Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol. Intl. 36, 1065–1075. [DOI] [PubMed] [Google Scholar]
- 162.Yen D et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Azizi G, Jadidi-Niaragh F & Mirshafiey A Th17 Cells in Immunopathogenesis and treatment of rheumatoid arthritis. Intl. J. Rheum. Dis. 16, 243–253 (2013). [DOI] [PubMed] [Google Scholar]
- 164.van der Vlist M, Kuball J, Radstake TR & Meyaard L Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nature Rev. Rheum. 12, 593–604 (2016). [DOI] [PubMed] [Google Scholar]
- 165.Gonzalez-Lopez L, Gamez-Nava JI, Jhangri G, Russell AS & Suarez-Almazor ME Decreased progression to rheumatoid arthritis or other connective tissue diseases in patients with palindromic rheumatism treated with antimalarials. J. Rheum. 27, 41–46 (2000). [PubMed] [Google Scholar]
- 166.Group HS A randomized trial of hydroxychloroquine in early rheumatoid arthritis: the HERA Study. Am. J. Med. 98, 156 (1995). [DOI] [PubMed] [Google Scholar]
- 167.O’Neill LA, Kishton RJ & Rathmell J A guide to immunometabolism for immunologists. Nature Rev. Immunol. 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]